 Nadja Hilger1,2*†
Nadja Hilger1,2*† Claudia Mueller1†
Claudia Mueller1† Lilly Stahl1
Lilly Stahl1 Anne M. Mueller1Bianca Zoennchen1Sarah Dluczek1Christoph Halbich1Claudia Wickenhauser3
Anne M. Mueller1Bianca Zoennchen1Sarah Dluczek1Christoph Halbich1Claudia Wickenhauser3 Dennis Gerloff4
Dennis Gerloff4 Alexander A. Wurm5Gerhard Behre5Anna Kretschmer1‡Stephan Fricke1‡
Alexander A. Wurm5Gerhard Behre5Anna Kretschmer1‡Stephan Fricke1‡- 1Immune Tolerance, Immunology, Fraunhofer Institute for Cell Therapy and Immunology, Leipzig, Germany
- 2Institute for Clinical Immunology, University of Leipzig, Leipzig, Germany
- 3Institute for Pathology, Halle University Hospital, Halle, Germany
- 4Department of Dermatology and Venereology, University Hospital Halle, Halle, Germany
- 5Division of Hematology and Medical Oncology, Leipzig University Hospital, Leipzig, Germany
Despite the constant development of innovative therapeutic options for hematological malignancies, the gold-standard therapy regimen for curative treatment often includes allogeneic hematopoietic stem cell transplantation (HSCT). The graft-vs.-leukemia effect (GVL) is one of the main therapeutic goals that arises from HSCT. On the other hand, graft-vs.-host disease (GVHD) is still one of the main and most serious complications following allogeneic HSCT. In acute myeloid leukemia (AML), HSCT together with high-dose chemotherapy is used as a treatment option. An aggressive progression of the disease, a decreased response to treatment, and a poor prognosis are connected to internal tandem duplication (ITD) mutations in the Fms like tyrosine kinase 3 (FLT3) gene, which affects around 30% of AML patients. In this study, C3H/HeN mice received an allogeneic graft together with 32D-FLT3ITD AML cells to induce acute GVHD and GVL. It was examined if pre-incubation of the graft with the anti-human cluster of differentiation (CD) 4 antibody MAX.16H5 IgG1 prevented the development of GVHD and whether the graft function was impaired. Animals receiving grafts pre-incubated with the antibody together with FLT3ITD AML cells survived significantly longer than mice receiving untreated grafts. The observed prolonged survival due to MAX.16H5 incubation of immune cell grafts prior to transplantation may allow an extended application of additional targeted strategies in the treatment of AML.
Introduction
Allogeneic hematopoietic stem cell transplantation (HSCT) remains a standard therapy regimen for the curative treatment of hematological malignancies (1). The graft-vs.-leukemia effect (GVL) is the main therapeutic goal that arises from HSCT [reviewed in (2)]. The leading cause of morbidity and mortality following allogeneic HSCT is graft-vs.-host disease (GVHD) (3–5), which is characterized by an imbalance of effector and regulatory mechanisms of the generated immune response (6). Alloreactive αβ T cells in the graft are regarded as the major cause for the development of GVHD in certain transplantation settings (6). Additionally, natural killer (NK) cells and/or a failure of regulatory immune pathways are potentially involved in GVHD [reviewed in (7, 8)]. Although T cell depletion prevents GVHD development, the increased risk for infections and host-vs.-graft reaction, defective cytokine production, and reduced engraftment lead to an attenuated graft-vs.-leukemia effect (GVL) [(6), reviewed in (9)]. The GVL effect can be observed together with GVHD or can occur independently (10), also due to minor histocompatibility antigens [summarized in (11)].
It is known that T and NK cells are able to recognize and destroy tumor cells by binding to various immunologically important surface markers (12). Cytotoxic T lymphocytes [CTLs, cluster of differentiation (CD) 8+] play an important role in anti-tumor activity by the detection of endogenous tumor antigens presented by major histocompatibility complexes (MHC) class I leading to an activation of CTLs into effector cells which promote tumor lysis [reviewed in (13)]. Anti-tumor activity of T helper cells (CD4+) is often impaired since the expression of MHC class II is restricted to professional antigen presenting cells, e.g., dendritic cells, macrophages, and B cells [reviewed in (14)]. Despite an expression of MHC class II on hematological cancer cells [e.g., B cell malignancies (15)], the majority of solid cancers lack MHC class II expression completely or can escape the recognition by the immune system using immune editing mechanisms (16–18). Although CD4+ T cell induced expression of cytokines was shown to support CTL activation [summarized in (19, 20)], immune escape and tumor progression is promoted by CD4+ T cell anergy (21, 22). Besides MHC class I and MHC class II (23), surface antigens like CD40, CD80, and CD86 are widely expressed on both healthy (antigen presenting cells) and malignant cells where their co-stimulatory function triggers T cells and T cell driven immune responses [reviewed in (24, 25)].
In previous work, our data showed that the short-term ex vivo incubation of an allogeneic graft with the non-depleting anti-human CD4 antibody MAX.16H5 IgG1 (murine) led to a significant GVHD reduction without negatively influencing the induced GVL effect (26). Additionally, NOD.Cg-Prkdcscid IL-2rgtm1Wjl/SzJ (NSG) recipient mice showed a significantly increased survival after xenogeneic transplantation of human peripheral blood mononuclear cells when the graft was pre-treated with the anti-human CD4 antibody MAX.16H5 IgG1 ex vivo (27). Possible side effects emerging from the antibody treatment did not occur, most likely because a systemic administration of MAX.16H5 IgG1 was not required to achieve treatment success. The observation that a single administration of an anti-human CD4 antibody can downregulate GVHD development is challenging the accepted theory and practice of long-term continuous T cell suppression by systemic immunosuppressant drugs. The described anti-human CD4 antibody recognizes the first domain (D1) of the CD4 molecule, which is an Ig-like V-type domain and contains three CDR-like regions (CDR1, CDR2, CDR3) (28). In previous studies, we provided evidence that the GVHD development was significantly downregulated by using the MAX.16H5 IgG1 antibody (27, 29). The anti-tumor effect of MAX.16H5 IgG1 incubated grafts was shown to be concurrently unaffected in a murine mastocytoma model (BALB/c) (26).
Regarding these promising results, we decided to investigate whether the antibody-induced GVHD prevention and retained anti-tumor effect can be translated into an Fms like tyrosine kinase 3 (FLT3, CD135) internal tandem duplication (ITD) positive acute myeloid leukemia (AML) C3H mouse model since acute GVHD affects 45–53% of AML patients carrying FLT3 mutations (30, 31). FLT3 is involved in proliferation, survival, and differentiation processes of hematopoietic cells and in the development of B and T cells [reviewed in 32)]. The most frequent mutation detected in AML patients (approximately 30%) is the ITD mutation, which affects the juxtamembrane domain of the FLT3 receptor (class I mutation) [reviewed in 32, 33)]. Several studies connected the FLT3ITD mutation to a decreased response to treatment and a poor prognosis (34–37). The significance of the FLT3 receptor and its downstream signaling pathways in AML led to the development of several inhibitory drugs (e.g., Sorafenib®, Quizartinib®, Midostaurin®) that are currently under investigation in different clinical trials [(38), reviewed in (39, 40)] or that are already EMA and FDA approved for the treatment of FLT3-positive AML (41, 42).
In this study, we investigated whether the transplantation of anti-CD4 antibody (MAX.16H5 IgG1) pre-incubated grafts (of CD4/DR3 transgenic donor mice) leads to an attenuated GVHD in a full murine MHC mismatch FLT3ITD positive AML model. We further analyzed if the MAX.16H5 IgG1 antibody incubation negatively influences the graft function.
Materials and Methods
Animals
This study was carried out in accordance with the recommendations of the guideline of the University of Leipzig animal care committee. The protocol was approved by the regional board of animal care for the district of Leipzig (State Directorate Saxony, Leipzig). For transplantation experiments, C3H/HeN and CD4/DR3 [murine (mu) CD4 knockout, human (hu) CD4, human leukocyte antigen isotype DR3 (HLA-DR3); C57Bl/6 background (43)] mice were used. C3H/HeN (male) recipient mice were purchased from Charles River, Sulzfeld Germany. CD4/DR3 donor mice were bred at the Max-Bürger-Forschungszentrum, University of Leipzig under standardized conditions. After irradiation, C3H/HeN mice were treated with antibiotics for 14 days (Baytril® 2.5% ad us. vet., Bayer Animal Health GmbH, Leverkusen, Germany). Survival, general condition [e.g., structure of fur, behavior (44)], and weight of the recipient animals were determined.
Preparation of the Immune Cell Graft and Transplantation Procedure
Bone Marrow Cells and Splenocytes
Bone marrow cells (BMCs) and splenocytes (SpC) were dissected from CD4/DR3 mice as previously described (26). BMCs and splenocytes were either incubated with 800 μg MAX.16H5 IgG1 (1mg/mL, Biotest) per 1.4 × 108 cells or without antibody. Antibody incubation was performed in 30 mL serum free media (RPMI 1640, GlutaMAX™-I, Gibco™, ThermoFisher SCIENTIFIC, Darmstadt, Germany) and samples were incubated in the dark at room temperature for 1 to 2 h. After incubation, BMCs and splenocytes were washed twice with phosphate buffered saline (1 × PBS, Gibco™, ThermoFisher SCIENTIFIC, Darmstadt, Germany), centrifuged at 300 × g for 10 min and finally resuspended in 1 × PBS.
Tumor Cells
The myeloblast-like murine cell line 32D Clone 3 (ATCC® CRL-11346™) expressing either human FLT3wt (wildtype) or human FLT3ITD as described elsewhere (45) was kindly provided by the group of Prof. G. Behre (University of Leipzig). 32D-FLT3ITD cells show human FLT3 expression with the ITD mutation. The cells were cultured as previously described (46).
Transplantation Procedure
Before transplantation, all recipient C3H/HeN animals received a total body irradiation of 8 Gy (X-Ray, D3225, Orthovoltage; Gulmay Medical, Camberley, UK) as previously described (47). After 4 h, the mice underwent allogeneic BMC and splenocyte transplantation. Mice received either 2 × 107 BMCs and 2 × 107 splenocytes or 1 × 107 BMCs and 3 × 107 splenocytes from CD4/DR3 mice with or without previous ex vivo incubation with anti-human CD4 antibody MAX.16H5 IgG1 (murine). For co-transplantation experiments, 5 × 103 32D-FLT3wt or 5 × 103 32D-FLT3ITD tumor cells were added to the graft immediately before transplantation. All cells were mixed in a final volume of 150 μL sterile 0.9% NaCl (B. Braun Melsungen AG, Germany) and immediately injected intravenously into the lateral tail vein by using a syringe with integrated needle (0.3 × 8 mm, Omnican® 20, U-40-Insulin, 0.5 mL/20 I.U., B. Braun Melsungen AG, Germany). The engraftment of the transplanted cells was observed up to 56 days.
Flow Cytometry and Blood Sample Analysis
For all flow cytometry experiments, a BD FACSCantoTM II device was used as previously described (29). All grafts were analyzed by flow cytometry prior to transplantation. 1 × 106 BMCs, splenocytes, and 32D-FLT3wt or 32D-FLT3ITD cells were stained according to the manufacturer's protocols using specific antibodies as described in the following: Staining protocol for bone marrow (BM) and splenocytes: muCD3-FITC, muCD4-PE-Cy7, muCD8-PerCP, muCD19-APC-Cy7, huCD4-APC, HLA-DR3-FITC, muMHCI H-2K(b)-PE, non-vital staining: 7AAD. Staining protocol for 32D-FLT3ITD AML cells: huCD135-PE, muCD11b-APC-Cy7, F4/80-PE-Cy7, Gr1-APC, non-vital staining: 7AAD. Staining protocol for 32D-FLT3wtAML cells: huCD135-PE, muCD11b-APC-Cy7, non-vital staining: 7AAD. All antibodies were purchased from BD Biosciences, Heidelberg, Germany. After antibody incubation, the cells were washed twice with 1 × PBS and analyzed by flow cytometry.
Immune reconstitution and tumor growth were analyzed by flow cytometry and impedance measurement. Murine blood samples were collected weekly and supplemented with 10 μL Heparin (Heparin-Natrium-25000-ratiopharm®, 25,000 I.U./5 mL). The differential blood cell count was measured using the scil Vet abc hematology analyzer (scil animal care company, Viernheim, Germany). After plasma collection, the blood pellets were diluted in 100 μL 1 × PBS and incubated with antibodies (muCD3-FITC, muCD4-PE-Cy7, muCD8-PerCP, muCD19-APC-Cy7, huCD4-APC, HLA-DR3-FITC, muMHCI H-2K(b)-PE, huCD135-PE) according to the manufacturer's protocols. After incubation, each sample was incubated with 1 mL BD FACS™ lysing solution (BD Biosciences, Heidelberg, Germany) for 10 minutes to remove erythrocytes. Subsequently, the cells were washed twice with 1 × PBS. The cell pellets were resuspended in 100 μL 1 × PBS and analyzed by flow cytometry. All samples were measured and analyzed using FACS Canto BDTM II and BD FACSDIVATM Software (BD Biosciences, Heidelberg, Germany) as previously described (26).
TUNEL Assay
The detection of apoptotic cells was performed using the TdT-mediated dUTP-biotin nick end labeling (TUNEL) assay (ApopTag®, Merck KGaA, Darmstadt, Germany) for light microscopy as already described in previous studies (27) and by Loo et al. (48). Accurate cell numbers in TUNEL slides were determined by image thresholding using ImageJ software (v1.46r, NIH, USA). The channels for red, blue and green were separated for better detection of apoptotic cells. The red area in the blue channel was analyzed with a pre-determined threshold for all images. The ratio of the counts in the red area (i.e., structures connected over several pixels indicating apoptotic cell nuclei) to the total marked area (counts/cm2) was analyzed. Five individual visual fields of one histological slide of the gut of each mouse were randomly chosen and microscopically recorded in 10 × magnification as previously described (27).
Data Acquisition and Statistical Tests
BD FACSCanto™ II Flow Cytometry System with FACS Diva 6.1.3 software (BD Biosciences Heidelberg, Germany), scil Vet abc (scil animal care co. GmbH, Viernheim, Germany), ImageJ 1.46 (NIH, open source), and AU480® Chemistry Analyzer (Beckman Coulter, Krefeld, Germany) were used to acquire the data. Limit values stated in the following comply with scilVet standards. Weight and clinical score were determined by blinded employees of the Medical Experimental Center of University of Leipzig. Statistical analyzes and graphics were created using SigmaPlot 11.0/SYSTAT 3.5 software (Systat Software GmbH, Erkrath, Germany) and GraphPad Prism 5 (v5.02, GraphPad Software Inc., La Jolla, CA, USA). The results are represented as mean ± standard deviation or median and percentiles. P-values were determined by Wilcoxon rank sum test or Student's t-test depending on prior Shapiro–Wilk test for normality and Levene test for equal variances (49) with a type one error α = 0.05 using SigmaPlot/SYSTAT and R version 3.4.2 (50) if not marked otherwise. Log-Rank tests and pair-wise multiple comparisons of means (Holm-Sidak method) were used for the analysis of Kaplan-Meier survival curves. A P-value < 0.05 indicates statistical significant differences. The term “significant” stands for “statistically significant” in all cases. Adjustment for multiple comparisons was performed using the Bonferroni method controlling the family–wise error rate if not stated otherwise. The apoptotic cell count analysis was performed using multifactorial variance analysis in R version 3.4.2 (50) with the packages multcomp (51) and userfriendlyscience for Games–Howell tests (52), and GraphPad Prism 5 (two-way ANOVA with Tukey post-hoc test). Partial eta-squared, calculated with the DescTools package (53), and f were used as parameters for the effect size. Nonparametric post-hoc testing after Kruskal-Wallis test was performed with Nemenyi tests for manual comparisons, designed using packages coin (54, 55) and multcomp (51), in the case of at least 4 measurements per group and time point or Dunn's test using the FSA package (56) otherwise. P-values were adjusted according to the conservative Bonferroni method adapting for the large number of comparisons. Group differences in the figures are represented by asterisks with *meaning P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Prolonged Survival of Mice by Pre-Incubation of Allogeneic Transplants With the Anti-Human CD4 Antibody MAX.16H5 IgG1
The development of GVHD and the effects of the anti-human CD4 antibody MAX.16H5 IgG1 were compared between recipient mice receiving 1 × 107 BMCs and 3 × 107 splenocytes incubated with MAX.16H5 IgG1 (n = 5) and mice receiving a control graft, which was not incubated with the antibody (n = 5) (Figure 1). At the end of the experiment (day 56 following transplantation), the survival rate of recipient animals receiving a MAX.16H5 IgG1 pre-incubated graft was 40% (n = 2/5) (Figure 1A). Recipient animals receiving an untreated graft died significantly earlier (n = 0/5, P = 0.003, Log-Rank test) between days 8 and 14 after transplantation (Figure 1A). The analyses of leukocytes (white blood cells, WBCs), lymphocytes, monocytes and granulocytes were performed at different time points throughout the experimental course and results are shown in Table 1 and Figure S1. Decreased leukocyte counts were observed in animals receiving untreated grafts (Figure 1C and Table 1). The WBC counts in animals receiving a MAX.16H5 IgG1 pre-incubated graft dropped after transplantation (day −2 vs. day 14, P = 0.015; day −2 vs. day 20, P = 0.020; Nemenyi tests) and subsequently increased until the end of the experiment (day 56; day 14 vs. day 28, P = 0.042, Nemenyi test). Mice receiving a graft without antibody pre-incubation showed decreased lymphocyte (Figure S1A), monocyte (Figure S1B), and granulocyte counts (Figure S1C). From day 14 on, lymphocyte, monocyte, and granulocyte cell counts were below the systems detection limit. In summary, transplanted mice receiving the MAX.16H5 IgG1 pre-incubated graft initially showed a decrease in lymphocyte, monocyte, and granulocyte counts followed by their recovery until the end of the experiment (Table 1, Figure S1). Blood sample analysis was also performed at the respective endpoints but no changes in the tendency of these parameters were obtained (data not shown).
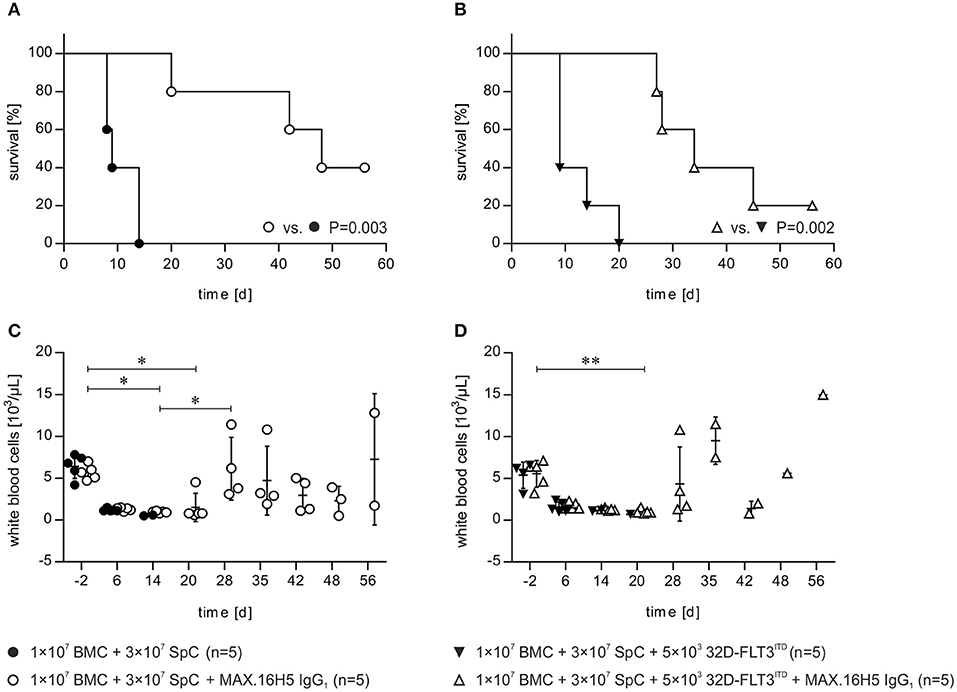
Figure 1. Short-term pre-incubation of bone marrow and spleen cell grafts with MAX.16H5 IgG1 antibody prolonged survival of C3H/HeN recipient mice after allogeneic transplantation either with or without co-transplantation of 32D-FLT3ITD AML cells. All C3H/HeN mice received 1 × 107 bone marrow cells (BMC) and 3 × 107 spleen cells (SpC) as an allogeneic transplant in these experiments. (A) Survival of recipient mice after allogeneic transplantation. The grafts were pre-incubated either with MAX.16H5 IgG1 or without antibody before administration to the recipients. C3H/HeN animals receiving a MAX.16H5 IgG1 pre-incubated graft (n = 5, open circles) showed a significantly prolonged survival compared to animals receiving an untreated graft (n = 5, closed circles, P = 0.003; Log-Rank test). (B) Survival of C3H/HeN recipient mice receiving short-term pre-incubated (with or without MAX.16H5 IgG1 antibody) grafts together with 5 × 103 32D-FLT3ITD AML cells. C3H/HeN animals receiving a MAX.16H5 IgG1 pre-incubated graft together with 32D-FLT3ITD cells (n = 5, open triangle) showed a statistically significant prolonged survival compared to animals receiving an antibody untreated graft (n = 5, closed triangle, P = 0.002; Log-Rank test). (C) Reconstitution of white blood cells in the blood of C3H/HeN mice which received either MAX.16H5 IgG1 pre-incubated grafts (n = 5, open circle) or grafts without prior antibody incubation (n = 5, closed circles). (D) White blood cell counts in the blood of C3H/HeN mice receiving bone marrow and spleen cell grafts either pre-incubated with MAX.16H5 IgG1 antibody (n = 5, open triangle) or without antibody pre-incubation (n = 5, closed triangles) is depicted. The blood sample data in (C,D) were obtained by electrical impedance measurement using a hematology analyzer. The error bars represent time-point specific means ± standard deviations. Nemenyi post-hoc tests with Bonferroni correction were used for comparative statistics. *P < 0.05, **P < 0.01.
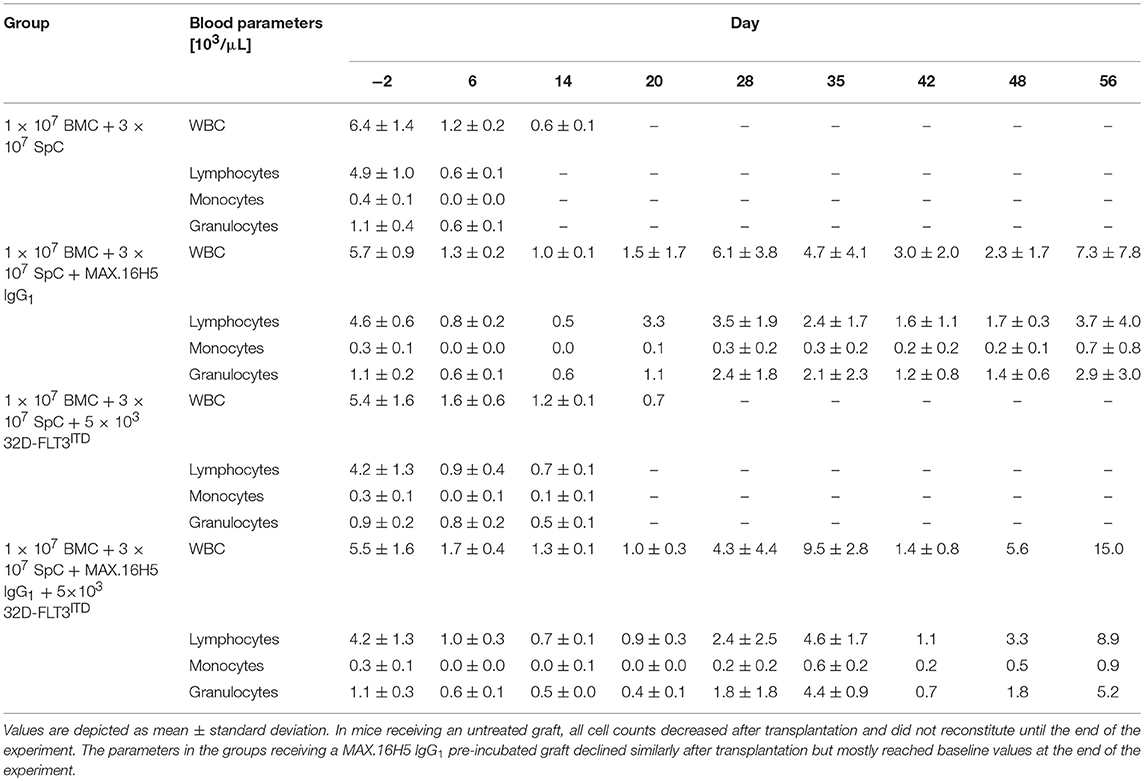
Table 1. Blood sample analysis (WBCs, lymphocytes, monocytes, and granulocytes) of C3H/HeN mice after transplantation of 1 × 107 bone marrow cells (BMC) and 3 × 107 spleen cells (SpC) with or without MAX.16H5 IgG1 pre-incubation in combination with or without 5 × 103 32D-FLT3ITD cells at different time points.
Furthermore, it was examined whether the ex vivo treatment of BMCs and splenocytes with MAX.16H5 IgG1 prevents GVHD and prolongs the survival of recipient animals in a mouse model of FLT3ITD positive AML. In initial experiments, the survival rate was 80% (n = 4/5) when 2 × 107 BMCs and 2 × 107 splenocytes were pre-incubated with MAX.16H5 IgG1 and co-transplanted with 5 × 103 32D-FLT3wt AML cells (FLT3 wild type) (Figure S2A). All mice transplanted with an untreated graft and 5 × 103 32D-FLT3wt AML cells survived (n = 5/5), but in two out of five animals, increased human CD135+ counts indicated tumor cell engraftment (Figure S2B). In contrast, no tumor engraftment was observed in mice receiving the antibody pre-incubated graft together with 32D-FLT3wt AML cells over the experimental period (Figure S2B). Next, it was examined if MAX.16H5 IgG1 pre-incubation of an allogeneic graft improves the survival in an FLT3ITD mutated AML setting. The mice were transplanted with 2 × 107 BMCs and 2 × 107 splenocytes incubated with or without MAX.16H5 IgG1 antibody and 5 × 103 32D-FLT3ITD mutated AML cells. The mice receiving an antibody pre-incubated graft survived until day 35, whereas the mice without a pre-treated graft died within 18 days after transplantation (Figure S2A). On day 5 after allogeneic transplantation, the flow cytometric analysis revealed increased human CD135+ counts in both groups' blood samples with one mouse transplanted with an untreated graft showing the highest number. The human CD135+ counts in the blood samples from all animals decreased to their initial value until day 13 after transplantation (Figure S2B).
To investigate if increased human CD4+ T cell counts result in a prolonged survival in FLT3ITD AML co-transplanted mice, splenocyte graft cell concentrations were increased from 2 × 107 in the initial experiments to 3 × 107. Additionally, the mice received 1 × 107 BMCs, and the graft was either pre-incubated with or without MAX.16H5 IgG1 anti-human CD4 antibody. The survival in the group receiving a graft pre-incubated with MAX.16H5 IgG1 was significantly prolonged compared to the control group (P = 0.002, Log Rank test, Figure 1B). Data of WBC, lymphocyte, monocyte and granulocyte counts obtained from blood samples of mice is shown in Table 1. Blood sampling and parameter determination was performed throughout the whole experimental course and is presented as mean ± standard deviation (SD) per group (Table 1). The WBC counts in the control group decreased after transplantation and did not reach their initial values until the end of the experiment (Figure 1D, Table 1, not significant in Nemenyi test). The WBC counts in the group receiving a MAX.16H5 IgG1 pre-incubated graft deteriorated until day 20 and increased until the end of the experiment (Figure 1D, Table 1, day 20 vs. day −2, P = 0.003, Nemenyi test). The control animals showed decreased lymphocyte, monocyte, and granulocyte values until they reached termination criteria (Table 1, Figure S3). In the animals receiving a graft pre-incubated with MAX.16H5 IgG1, lymphocyte, monocyte, and granulocyte counts dropped initially but exceeded their initial values at the end of the experiment (Table 1, Figure S3).
GVHD Induction and Immune Cell Reconstitution After Transplantation of Allogeneic Bone Marrow and Spleen Cells
Immune cell reconstitution after allogeneic transplantation of 1 × 107 BMCs and 3 × 107 splenocytes was investigated by flow cytometry of heparinized blood samples from recipient animals. In the experimental panel, the following markers were examined: murine CD3, murine CD4, human CD4, murine CD8, murine CD19, and HLA-DR3.
A detailed analysis was performed at four time points and exemplary scatter plots of flow cytometry measurements for one animal each receiving grafts with or without MAX.16H5 IgG1 pre-incubation are shown in Figures 2A,B and Figure S4. The data indicated as “endpoint” is a collection of measurements combining samples of mice, which were analyzed as soon as they had to be taken out of the experiment or as soon as they died which was not the exact same day in all cases. Detailed flow cytometry datasets of all analyzed mice are summarized in Table S1. Recipient animals, which received an untreated graft, showed a decreasing number of murine CD4+ events from 42.3 ± 3.2% on day −2 (n = 5/5) to 0.1 ± 0.2% on day 14 (n = 2/5) (Table S1A). In animals whose transplanted graft was pre-incubated with MAX.16H5 IgG1 murine CD4+ events dropped from 40.5 ± 3.9% on day −2 (n = 5/5) to 0.4 ± 0.4% on day 20 (n = 5/5) and to 1.1 ± 0.1% on day 56 (n = 2/5) (Table S1B), respectively. In the control animals, an increase in human CD4+ events from 0.2 ± 0.0% (day −2, n = 5/5) to 24.1 ± 3.1% (day 6, n = 5/5) and to 7.0 ± 4.3% (endpoint, day 14, n = 2/5) (Figures 2A,C and Table S1) was observed (day −2 vs. day 6, P = 0.003; day −2 vs. endpoint, P = 0.019; Nemenyi test). Animals receiving MAX.16H5 IgG1 pre-incubated grafts showed a delayed increase of human CD4+ events from 0.2 ± 0.1% (day −2, n = 5/5) and 0.3 ± 0.3% (day 6, n = 5/5) to 19.4 ± 2.3% (day 56, n = 2/5) (Figures 2B,C, Table S1, day 6 inter-group comparison, P = 0.003, Nemenyi test). On day 48, animals that received grafts pre-incubated with MAX.16H5 IgG1 showed a constant engraftment of human CD4+ cells (Table S1B). Additionally, increased numbers of CD8+ cytotoxic T cells were observed in both groups (Table S1). Only one mouse receiving a MAX.16H5 IgG1 pre-incubated graft showed constant murine CD19+ signals from day 20 after transplantation onwards (7.5%, Table S1B). Since no AML cells were co-transplanted in these experiments, similar percentages of human CD135+ events were detected in the blood from control animals transplanted with untreated grafts (5.0 ± 3.8%, n = 5/5, endpoint) and mice receiving MAX.16H5 IgG1 pre-incubated grafts (6.7 ± 2.3%, n = 5/5, endpoint) at the end of the experiment as expected (Figures 2A,B,D and Table S1). Still, an increase of CD135+ events toward the end of the experiment was observed in the group subjected to MAX.16H5 IgG1 pre-incubated transplants (day −2 vs. day 20, P = 0.001; day-2 vs. endpoint, P = 0.003; Nemenyi tests). Rank correlation tests (Kendall's tau) against the exact day after transplantation showed that CD135+ event rates increased in both groups with time (rτ = 0.482, 95% confidence interval 0.057–0.907 without MAX.16H5 IgG1 pre-incubation; rτ = 0.688, 95% confidence interval 0.505–0.870 with MAX.16H5 IgG1 pre-incubation). The confidence intervals might indicate a higher correlation in the MAX.16H5 IgG1 pre-treated group; however, possible between-group differences remain elusive.
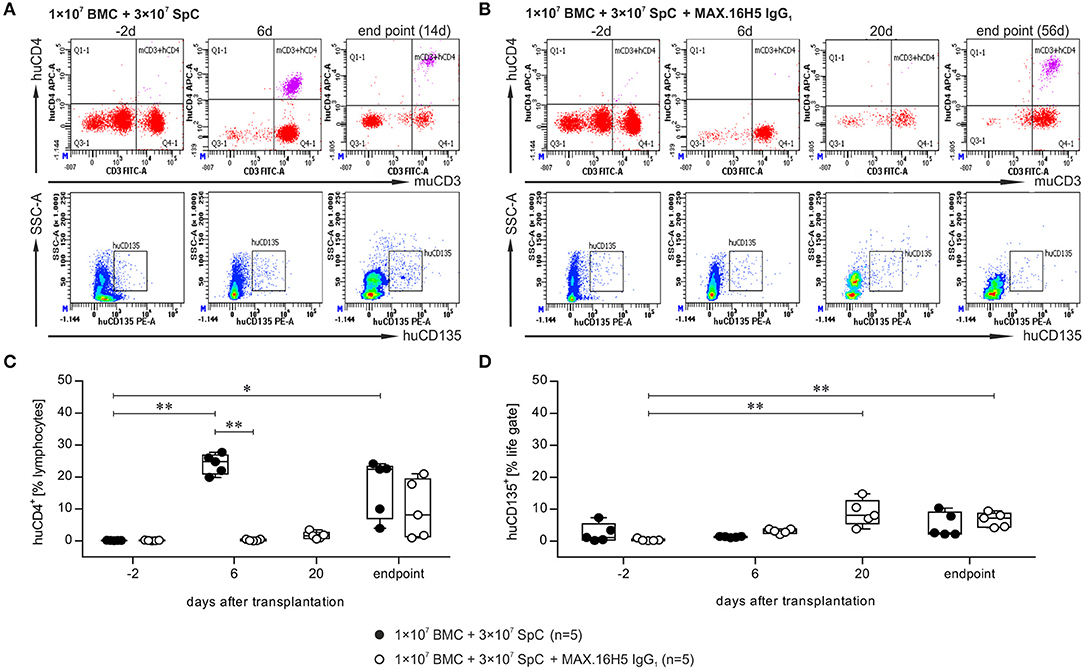
Figure 2. Flow cytometric analysis of the immune reconstitution (murine CD3+/human CD4+) as well as tumor growth (human CD135+) after transplantation of bone marrow and spleen cell grafts either pre-incubated with MAX.16H5 IgG1 or without antibody pre-treatment. All recipient animals received 1 × 107 bone marrow cells (BMC) and 3 × 107 splenocytes (SpC) in these experiments. (A,B) Representative plots obtained from flow cytometric analyses after transplantation of (A) one animal with untreated or (B) one animal with MAX.16H5 IgG1 pre-treated grafts at different time points (in (A): day −2, day 6, endpoint; in (B): day −2, day 6, day 20, endpoint) using specific antibodies against murine CD3/human CD4 or human CD135. In the lower rows, human CD135+ signals are depicted together with side-scattered light signals. (C) Human CD4+ events as percentages of lymphocyte gates in the blood samples of recipient mice. Every symbol indicates one animal at a defined time point. Engraftment and proliferation of human CD4+ cells were detected in five out of five animals receiving an untreated graft and in three out of five animals receiving the MAX.16H5 IgG1 pre-incubated grafts. The immune cell engraftment was delayed in animals receiving the MAX.16H5 IgG1 pre-incubated grafts. (D) Human CD135+ control staining of blood samples on labeled time points. (C,D) Nemenyi tests with Bonferroni correction computed the comparative statistics. *P < 0.05, **P < 0.01.
GVHD Induction, Immune Cell Reconstitution, and Tumor Growth After Transplantation of Allogeneic Bone Marrow and Spleen Cells in Co-Transplantation With 32D-FLT3ITD Cells
The flow cytometric analysis of blood samples of one representative animal collected from mice, which received a graft with or without pre-incubation with the antibody MAX.16H5 IgG1 in co-transplantation with 32D-FLT3ITD cells, is shown in Figures 3A,B and Figure S5. A detailed analysis is shown at four time points (day −2, 6, 20 and endpoint). The data indicated as “endpoint” is a collection of measurements combining samples of mice, which were analyzed as soon as they reached termination criteria or as soon as they died which was not the exact same day in all cases. Detailed flow cytometry datasets of all analyzed animals are summarized in Table S2. Additionally, tumor growth was monitored by flow cytometric measurement of human CD135. The murine CD4+ counts in the blood samples from animals receiving an untreated graft decreased from 38.4 ± 7.9% (day −2, n = 5) to 0.2% (day 20, n = 1) and from 40.4 ± 3.8% (day −2, n = 5) to 4.2% (day 56, n = 1) in the groups receiving MAX.16H5 IgG1 pre-incubated grafts (Tables S2A,B). The engraftment of human CD4+ cells was delayed in those animals transplanted with MAX.16H5 IgG1 incubated grafts and 32D–FLT3ITD AML cells (mean of human CD4+ events: 0.2 ± 0.0% (day −2, n = 5); 0.2 ± 0.1% (day 6, n = 5, inter-group comparison P = 0.006, Nemenyi test); 42.3% [endpoint, day 56, n = 1, day −2 vs. endpoint, P = 0.039, Nemenyi test)]. In the further course of the experiment however, CD4+ cell levels were higher than those of the corresponding control group [0.2 ± 0.0% (day −2, n = 5); 25.4 ± 1.2% (day 6, n = 5, day −2 vs. day 6, P = 0.003, Nemenyi test); 13.3% (day 20, n = 1)] (Figures 3A–C). The amount of CD8+ events was comparable in both groups (n = 5, Table S2). The CD19+ counts did not recover in the mice receiving an untreated graft (day 20: 0.6%, n = 1) such as in mice transplanted with an antibody treated graft (1.7 ± 1.5%, day 20, n = 5) (Table S2, Figure S5B). The 32D-FLT3ITD tumor engraftment was detected by analysis of the blood samples for human CD135 using flow cytometry. The human CD135+ counts from mice transplanted with 1 × 107 BMCs + 3 × 107 splenocytes + 5 × 103 32D-FLT3ITD without MAX.16H5 IgG1 pre-treatment did not significantly increase until the end of the experiment (4.6 ± 4.0%, n = 5, day 9–20) whereas the human CD135+ events in animals receiving a MAX.16H5 IgG1 pre-treated graft increased until the end of the experiment (17.9 ± 17.2%, n = 5, day 27–56; day −2 vs. day 20, P < 0.001; day −2 vs. endpoint, P = 0.008; Nemenyi tests) (Figure 3D and Table S2). However, additional rank correlation tests (Kendall's tau) against the precise day after transplantation indicated that CD135+ event ratios increased in both groups over time (rτ = 0.842, 95% confidence interval 0.716–0.968 without MAX.16H5 IgG1 pre-incubation; rτ = 0.665, 95% confidence interval 0.380–0.950 with MAX.16H5 IgG1 pre-incubation).
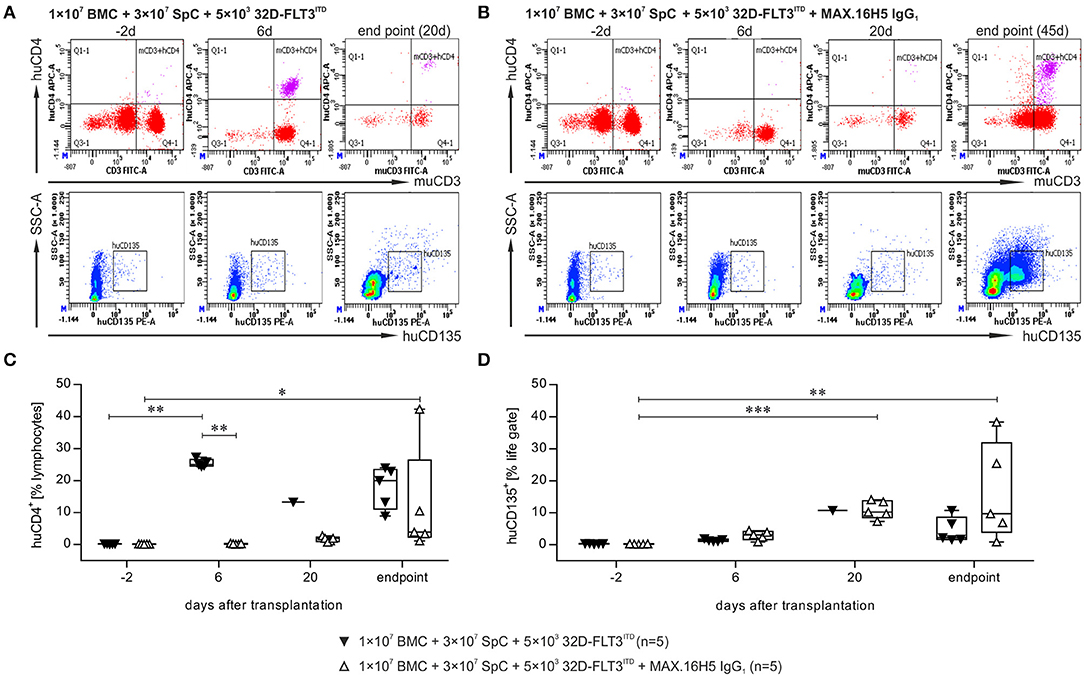
Figure 3. Flow cytometric analysis of the immune reconstitution (murine CD3+/human CD4+) as well as tumor growth (human CD135+) after transplantation of bone marrow and spleen cell grafts pre-incubated with MAX.16H5 IgG1 or without antibody pre-treatment in co-transplantation experiments with 5 × 103 32D-FLT3ITD cells. All recipient animals received 1 × 107 bone marrow cells (BMC) and 3 × 107 splenocytes (SpC) in these experiments. (A,B) Representative plots of flow cytometric analyses after transplantation of (A) one animal with untreated or (B) one animal with MAX.16H5 IgG1 pre-treated grafts at different time points [in (A): day −2, day 6, endpoint; in (B): day −2, day 6, day 20, endpoint] using specific antibodies for murine CD3+/human CD4+ or human CD135+. In the lower rows, human CD135+ signals are depicted together with side-scattered light signals. (C) Human CD4+ events as percentages of lymphocyte gates in the blood samples of recipient mice. Every symbol indicates one animal at a defined time point. Engraftment and proliferation of human CD4+ cells were detected in five out of five animals receiving an antibody untreated graft and in two out of five animals receiving a MAX.16H5 IgG1 incubated graft. The immune cell engraftment was delayed in animals receiving the MAX.16H5 IgG1 pre-incubated grafts in comparison to the control group. (D) Engraftment of the 32D-FLT3ITD cells was analyzed by immunofluorescent staining of blood samples (marker: human CD135). The human CD135+ counts in the blood samples from mice transplanted with 1 × 107 BMCs + 3 × 107 splenocytes + 5 × 103 32D-FLT3ITD without MAX.16H5 IgG1 pre-treatment did not increase significantly until the end of the experiment (day 9–20) whereas the human CD135+ events in animals receiving a MAX.16H5 IgG1 pre-treated graft increased until they reached termination criteria (day 27–56). (C,D) Nemenyi tests with Bonferroni correction computed the comparative statistics. *P < 0.05, **P < 0.01, ***P < 0.001.
Tumor Growth in Different Organs After Allogeneic Transplantation
Bone marrow (BM), spleen, and liver samples were examined for tumor growth by using flow cytometric analysis of human CD135 at the endpoint of each animal. The presented data is a collection of endpoint measurements combining samples from mice, which were analyzed as soon as they had to be taken out of the experiment or as soon as they died. These endpoints ranged from day 8 (e.g., mice receiving an untreated graft) to day 56 (e.g., when mice received MAX.16H5 antibody incubated grafts). For mice receiving a treated or untreated graft (1 × 107 BMCs + 3 × 107 splenocytes) without co-transplantation of 32D-FLT3ITD AML cells, human CD135+ events were not detectable neither in BM and liver nor in spleen preparations (Figures 4A,B and Table S3).
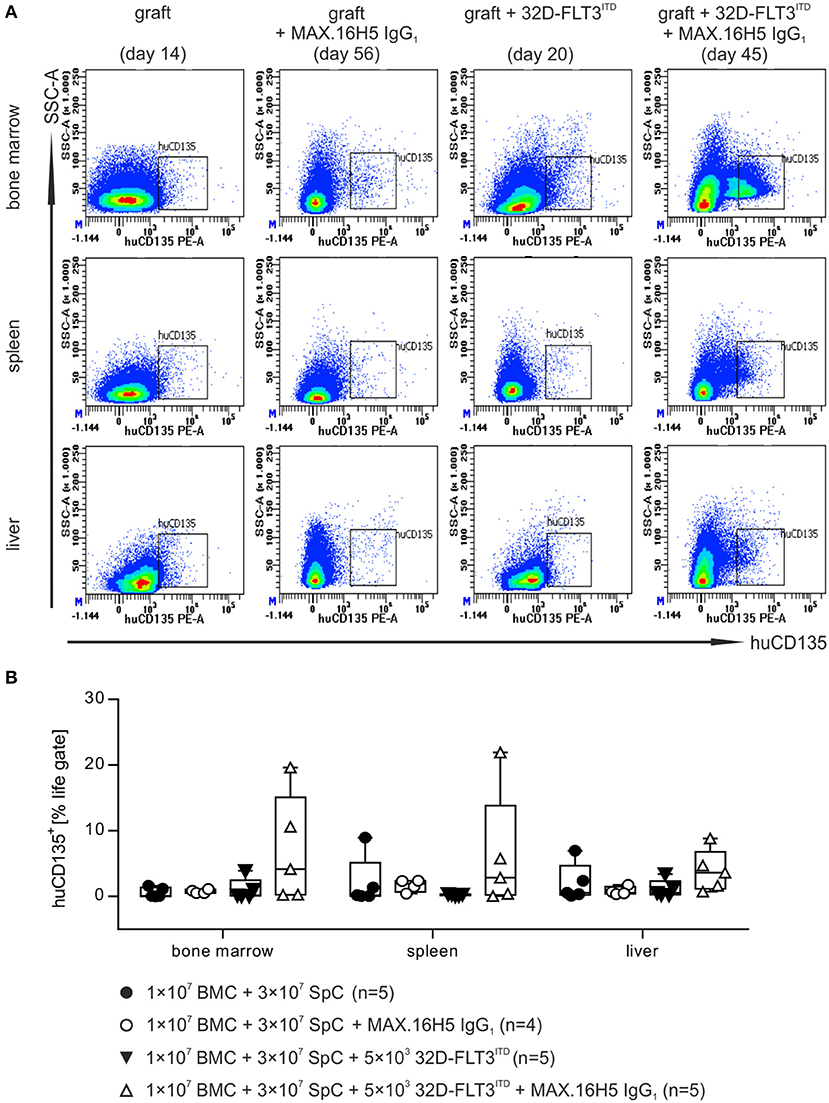
Figure 4. Flow cytometric analysis of human CD135+ events in different organs after transplantation of bone marrow and spleen cell grafts pre-incubated either with or without MAX.16H5 IgG1 in combination with or without co-transplantation of 32D-FLT3ITD cells. The manifestation of human CD135+ cells in different organs (bone marrow, spleen and liver) from animals receiving 1 × 107 bone marrow cells (BMC) and 3 × 107 splenocytes (SpC) together with AML cells was analyzed by flow cytometry. (A) Representative plots from flow cytometric analyses of human CD135+ events after transplantation of untreated or MAX.16H5 IgG1-treated grafts with or without 5 × 103 32D-FLT3ITD cells. At the endpoint, bone marrow, spleen, and liver samples from the transplanted mice were examined regarding human CD135 expression. (B) Human CD135+ events as percentage of the viable cell parent gate in organ preparations of recipient mice. Every symbol indicates one animal at a defined time point. The presented data is a collection of endpoint measurements combining samples from mice, which were analyzed as soon as they had to be taken out of the experiment or as soon as they died. Engraftment and proliferation of human CD135+ cells were detected in two out of five animals receiving MAX.16H5 IgG1 pre-incubated grafts in the co-transplantation experiments with 5 × 103 32D-FLT3ITD cells. Kruskal-Wallis test indicated no significant differences (P = 0.171).
Mice receiving 32D-FLT3ITD AML cells and an untreated graft showed no increased human CD135+ counts in the BM (1.1 ± 1.6%, n = 5), spleen (0.3 ± 0.1%, n = 5), or liver preparations (1.2 ± 1.3%, n = 5) (Figures 4A,B and Table S3). In comparison, human CD135+ signals were detected in BM, spleen, and liver preparations in two out of five mice receiving antibody pre-treated grafts and 32D-FLT3ITD cells (BM: 7.0 ± 8.2%, spleen: 6.2 ± 9.1%, liver: 3.9 ± 3.2%, n = 5) (Figure 4 and Table S3).
MAX.16H5 IgG1 Pre-Incubation Reduces Apoptotic Cells in the Gut (ileum) of Transplanted Mice
Apoptotic cells in the gut of mice were analyzed by TUNEL staining. Mice receiving an antibody untreated graft without co-transplantation of 32D-FLT3ITD cells showed slightly higher apoptotic cell counts (median: 0.54 counts/cm2, 25% percentile: 0.07, 75% percentile: 0.83, n = 5, Figures 5A,F) compared to corresponding mice transplanted with a MAX.16H5 IgG1 antibody pre-incubated graft (median: 0.10 counts/cm2, 25% percentile: 0.06, 75% percentile: 0.25, n = 4) (Figures 5B,F). This difference was not significant (Wilcoxon rank sum test).
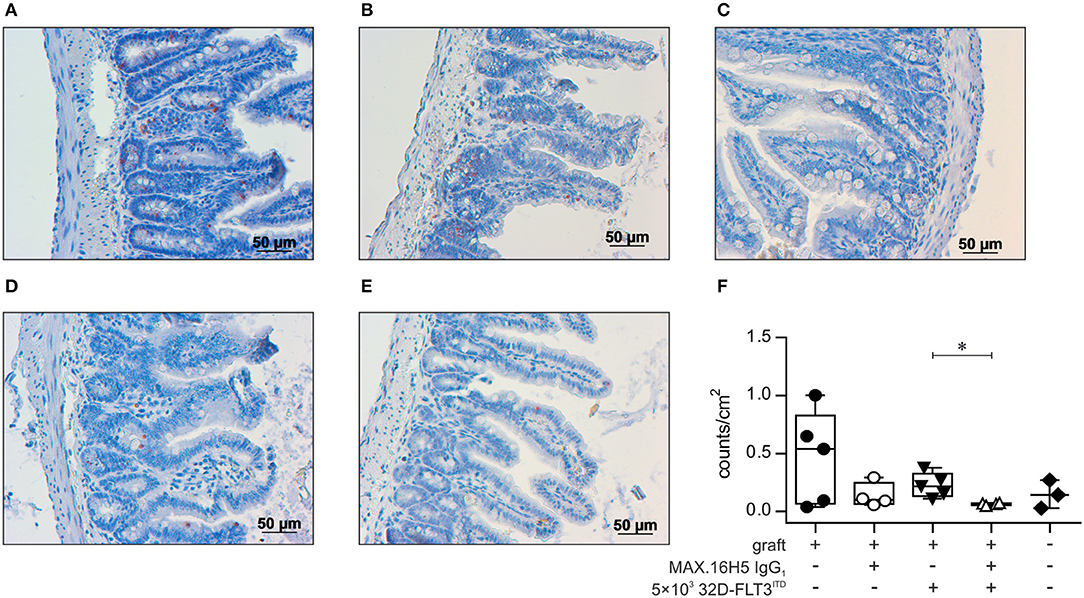
Figure 5. Histological analysis of gut tissue (ileum) from transplanted animals by TUNEL assay. Histological sections of gut tissues from all C3H/HeN mice receiving 1 × 107 bone marrow cells (BMC) and 3 × 107 splenocytes (SpC) after pre-incubation with MAX.16H5 IgG1 or an untreated graft with or without co-transplantation of 5 × 103 32D-FLT3ITD cells were examined regarding apoptotic cell count. NaCl treated mice served as the control group. Five individual visual fields from one histological slide of the gut of each mouse were randomly chosen and microscopically recorded in 10 × magnification, and apoptotic cell counts were analyzed with ImageJ. (A–E) Representative histological ileum images of the transplanted animals (magnification 20 ×) after TUNEL staining. TUNEL stained gut sections of animals receiving 1 × 107 bone marrow and 3 × 107 spleen cells (A) without antibody pre-incubation and (B) with MAX.16H5 IgG1 pre-incubation are shown. (C) Ileum TUNEL staining from one mouse receiving a non-incubated graft together with 5 × 103 32D-FLT3ITD cells. (D) Histological section from one mouse receiving a MAX.16H5 IgG1 pre-incubated graft together with 32D-FLT3ITD AML cells. (E) TUNEL staining of a gut section from a NaCl control animal. Red areas indicate apoptotic nuclei in the basal crypts of the mucosa, which are a diagnostic criterion in acute GVHD (80). Apoptotic nuclei in the apical mucosal layer were excluded from the analysis. (F) Group-separated boxplots indicating the group medians and quartiles together with each symbol representing the mean of the examined apoptotic nuclei count per cm2 (based on the image in 10 × magnification) of all counted visual fields per mouse. Wilcoxon rank sum tests on unprocessed data were used for comparative statistics. *P < 0.05.
A significantly increased apoptosis rate (P = 0.016, Wilcoxon rank sum test) was detected in the gut from mice which were co-transplanted with 32D-FLT3ITD cells and 1 × 107 BMCs + 3 × 107 splenocytes without prior MAX.16H5 IgG1 incubation (median: 0.22 counts/cm2, 25% percentile: 0.13, 75% percentile: 0.33, n = 5, Figures 5C,F) in comparison to mice receiving an antibody incubated graft and 32D-FLT3ITD AML cells (median: 0.07 counts/cm2, 25% percentile: 0.05, 75% percentile: 0.07, n = 4, Figures 5D,F). Nevertheless, the apoptotic cell counts in the guts from the irradiated control group (median: 0.14 counts/cm2, 25% percentile: 0.03, 75% percentile: 0.27, n = 3) were comparable to those of the mice transplanted with 1 × 107 BMCs, 3 × 107 splenocytes and 5 × 103 32D-FLT3ITD cells (Figures 5E,F). As only two comparisons were drawn, Wilcoxon rank sum tests on unprocessed data were performed without correction.
In the multifactorial analysis, MAX.16H5 IgG1 pre-treatment of the transplants was a significant (P = 0.002) as well as relevant factor indicating reduced clinical scores (interaction between the individual and time factor as cause of error variance, partial eta-squared = 0.450). On the other hand, neither the tumor transplantation nor the interaction between MAX.16H5 IgG1 pre-treatment and tumor transplantation exerted a significant influence. This indicates a worsened general health state of mice after the co-transplantation with AML cells carrying the FLT3-ITD mutation independently of the MAX.16H5 pre-incubation.
Impact of Graft Transplantation on Toxicity
For toxicity analysis, creatinine and uric acid concentrations in plasma samples of mice which received a graft (1 × 107 bone marrow and 3 × 107 spleen cells) after a short-term pre-incubation with MAX.16H5 IgG1 or without pre-incubation in combination with 5 × 103 32D-FLT3ITD cells were examined (For detailed description of the performed assays see also Supplementary Materials and Methods). A strong and significant increase in creatinine levels was detected in both groups starting on day 6 after transplantation [without antibody: day−2: 179.60 ± 11.08 μmol/L, day 6: 566.40 ± 46.85 μmol/L, P < 0.001 (n = 5); with anti-human CD4 antibody MAX.16H5 IgG1: day−2: 226.8 ± 154.37 μmol/L, day 6: 639.60 ± 45.59 μmol/L, P < 0.001 (n = 5)] until the end of the experiment (Figure S6A, with anti-human CD4 antibody MAX.16H5 IgG1: day −2 vs. day 14, vs. day 20, and vs. day 28, P < 0.001, pre-defined contrasts only taking minimum sample sizes of 4 into account in simultaneous tests for general linear hypothesis, Bonferroni adjustment). The plasma samples of mice in the group receiving an untreated graft showed significantly higher uric acid concentration on day 6 (305.94 ± 39.38 μmol/L, n = 5) than on day −2 (137.48 ± 30.81 μmol/L, P < 0.001, pre-defined contrasts only taking minimum sample sizes of 4 into account in simultaneous tests for general linear hypothesis, Bonferroni adjustment). In comparison, no difference in the uric acid concentration was observed between day −2 and day 6 in the plasma of those mice receiving MAX.16H5 IgG1 pre-treated grafts (Figure S6B). The uric acid concentrations measured on day 6 were significantly different between the animals receiving grafts either without (305.94 ± 39.38 μmol/L, n = 5) or with MAX.16H5 IgG1 pre-incubation (156.84 ± 41.12 μmol/L, n = 5, P = 0.003) (Figure S6B). Nevertheless, starting on day 14 increased uric acid concentration were observed within the group receiving an antibody pre-incubated graft (day−2: 120.93 ± 17.61 μmol/L, n = 5, day 14: 215.84 ± 97.52 mol/L, n = 5). Uric acid concentrations in plasma samples of these mice receiving an antibody pre-incubated graft were significantly different between day−2 (120.93 ± 17.61 μmol/L, n = 5) and day 28 (254.30 ± 59.14 μmol/L, n = 5, P = 0.019) and were maintained until the end of the experiment (day 56: 390.70 μmol/L, n = 1). Calcium and phosphate plasma levels were not significantly different between mice receiving grafts with or without MAX.16H5 IgG1 antibody pre-incubation (data not shown).
Discussion
GVHD is one of the main complications leading to a high mortality rate among patients after allogenic HSCT (57–59). AML patients presenting with an aberrant FLT genotype are regularly transplanted when their prognosis is intermediate or poor (60). These patients receive extensive chemotherapy and midostaurin as induction therapy prior to HSCT (60). To date, HSCT is often the only curative therapy option for patients suffering from leukemia (61, 62). New therapy strategies need to be designed ensuring the prevention of GVHD while supporting or maintaining the GVL effect (63, 64). In the past, it was shown that the graft treatment with the anti-human CD4 antibodies MAX.16H5 IgG1 (murine) or IgG4 (chimeric) was able to prevent GVHD in a xenogeneic NSG HSCT model (27) and also to preserve the GVL effect in a murine mastocytoma model (MAX.16H5 IgG1) (26). Due to these promising results, a murine full MHC mismatch transplantation model for AML was established to investigate whether MAX.16H5 IgG1 also exerts a beneficial effect regarding GVHD prevention in the background of leukemia. A highly aggressive AML subtype carrying the ITD mutation in the FLT3 receptor was used in this tumor model (65). The oncogenic potential of 32D cells transfected with FLT3ITD or FLT3wt has already been described in a syngeneic setting elsewhere (45). It is important to note that wild-type FLT3 is regularly expressed on CD34+ hematopoietic stem cells and plays a role in the development of different immune cells (e.g., NK cells, dendritic cells, B cells) [reviewed in (66)]. For co-transplantation experiments, 32D-FLT3ITD cells were administered together with the allogeneic hematopoietic graft of CD4/DR3 mice, which was shortly pre-incubated with MAX.16H5 IgG1 before transplantation in C3H/HeN mice as previously described (26). In this work it could be shown that lethally irradiated mice, which received BMC and splenocyte transplants from CD4/DR3 mice without MAX.16H5 IgG1 pre-treatment died within 20 days due to the development of GVHD. In comparison, the groups transplanted with MAX.16H5 IgG1 pre-treated grafts survived significantly longer in this model. We assume that the development of severe GVHD was hampered due to the antibody treatment. Nevertheless, the appearance of severe acute GVHD was not prevented in all mice receiving antibody pre-treated grafts, but the GVHD development was decelerated and effectively reduced in this transplantation setting. It should be pointed out that the development of GVHD in mice models is influenced by different parameters such as the applied irradiation dose, a variable proportion of T cell subsets (e.g., CD4+) residing in the graft, and genetic disparity in MHC and minor histocompatibility antigens between donor and recipient [reviewed in (67)].
The engraftment of the transplanted immune cell grafts was confirmed by flow cytometric detection of circulating human CD4+ events in the blood of recipient animals. Within a week following transplantation, no T cells positive for human CD4 were detected in those groups receiving grafts pre-incubated with MAX.16H5 IgG1 antibodies (with or without co-transplanted AML cells). In an earlier study it was shown that the engraftment of MAX.16H5 antibody incubated grafts was delayed in comparison to mice receiving a graft treated with an isotype control antibody (27). Eventually, by the end of the experiments, 5 out of 6 mice showed a stable engraftment of human immune cells (27). The appearance of murine CD3+/murine CD8+ events in the blood samples was comparable between the groups receiving grafts with or without antibody pre-treatment and with or without AML co-administration. It is known that the clinical outcome after HSCT is strongly dependent on the cellular composition of the donor grafts (68). In several studies, different researchers showed that the depletion of T cells and a non-myeloablative conditioning regimen led to a higher risk of graft failure and decreased graft function in transplanted patients (69). We already showed in previous in vitro experiments that in comparison to the depleting antibody OKT3 the MAX.16H5 IgG1 antibody was not able to deplete T cells by activating the complement system (complement-dependent cytotoxicity) (27). We also discussed a potential mode of action for the induced tolerance in regard to transplanted stem cell grafts after MAX.16H5 IgG1 antibody incubation elsewhere (27, 29). The binding of the antibody to CD4+ T cells may shift their development toward an anergic, tolerogenic state resulting in a decreased GVHD activation after HSCT (27, 29). Tolerance induction by a CD4 directed antibody was also described by Helling et al. where the humanized CD4-specific monoclonal antibody tregalizumab was examined regarding its immunomodulatory potential (70). Tregalizumab modulated regulatory T cells in a T cell receptor-independent manner inducing an unique phosphorylation pattern of signaling molecules in CD4+ cells (70). The antibody recognizes the second domain of the CD4 molecule on T cells (70). In contrast, the binding of CD4-directed antibodies to the first domain D1 of CD4 was described to be possibly connected to the induction of T cell anergy due to a steric hindrance of the CD4/MHC class II interaction after antibody binding (70). However, the mechanisms underlying the immunomodulatory features of the MAX.16H5 IgG1 antibody still remain elusive and have to be addressed in further studies.
According to data obtained in a former study (29), we showed in this work that a single dose treatment with MAX.16H5 IgG1 did not cause graft failure and was sufficient to prevent GVHD in allogeneic transplanted mice. Nevertheless, in the present study two out of five mice, which received antibody pre-incubated grafts, showed increased counts of human CD135+ events toward the end of the experiment, thus indicating a higher tumor burden and engraftment of the FLT3ITD AML cells. It has to be noted that mice receiving an antibody-incubated graft survived significantly longer than mice, which were transplanted with control grafts, leading to different experimental “endpoints” in this setting. The prolonged life span of these particular mice may cause re-proliferation of 32D-FLT3ITD AML explaining the increased CD135+ events. However, at comparable time points (until day 20), both groups (with and without MAX.16H5 antibody incubation) showed similar levels of CD135+ events in the blood. The growth and kinetics of tumor cells (e.g., AML) in mouse models are of special importance for dataset analysis and interpretation. Therefore, extensive literature search was implemented beforehand regarding AML tumorigenesis in mouse models. In these studies, AML cells were mainly transplanted in a syngeneic setting without irradiation of the animals. Mizuki et al. investigated 32D-FLT3ITD and 32D-FLT3wt cells regarding their tumorigenic potential and the development of leukemia-resembling disease in a syngeneic setting using non-irradiated C3H mice (45). The group showed that the injection of 1 × 106 32D-FLT3ITD cells led to a rapid development of a leukemia-like disease and that the mice died within 4–5 weeks after tumor cell application. The animals also showed signs of splenomegaly. In contrast, mice receiving 32D-FLT3wt cells did not develop a leukemic disease until up to 3 months after tumor cell injection (45). According to these data, we showed in a previous work that non-irradiated C3H mice transplanted with 1 × 106 32D-FLT3ITD cells (syngeneic setting) died within 21 days after tumor cell application (71). Moreover, an increased CD135+ cell count was detected by flow cytometry in peripheral blood samples of mice which died a few days after blood collection. In preparation for the immune cell graft transplantation used in this current study, irradiation was performed in all animals to eradicate their immune system. The comparison between models described in literature and the allogeneic transplantation setting we describe in this manuscript is hampered due to the lethal irradiation of mice. Irradiated mice will die when hematopoiesis is not rescued by the transplantation of an immune cell graft. Although we impressively showed that GVHD was significantly reduced in our co-transplantation experiments and the antibody did not cause graft failure, our FLT3ITD positive AML C3H mouse model revealed some limitations, especially regarding the AML kinetics due to the lethal irradiation protocol. Further investigations are required to obtain data regarding GVHD and GVL activity of MAX.16H5 antibody-incubated immune cell grafts using an NSG xenograft transplantation model in the background of AML. Using this approach, mice do not have to be lethally irradiated in preparation for graft transplantation and the kinetics of the tumor cell growth can be implemented additionally.
One of the examined mice receiving 32D-FLT3ITD AML cells and the pre-incubated graft showed a lower amount of CD135+ events but an increased count for CD8+ events, whereas the results were reversed in a second mouse. Since it is known that CD8+ cells play a major role in the immune defense against tumor cells [reviewed in 72)], there may also be a correlation between tumor cell counts and CD8+ cell counts in this experimental setting. A plausible deduction of a specific mechanism and a conclusion for a correlation was not made since the effects were observed in single animals only. For a better understanding of the underlying mechanism further studies have to focus specifically on this topic. Regarding our used assay we cannot exclude a cross-reactivity of the used mouse anti-human CD135 antibody since low levels of human CD135+ events were also detected in mice that were not co-transplanted with AML cells.
However, one mouse of the group receiving the antibody pre-incubated graft and FLT3ITD AML cells survived without developing AML and showed a successful engraftment without any signs of GVHD. We assume that mice receiving an antibody treated graft together with AML cells exhibited a prevention of GVHD by the graft incubation with the anti-human CD4 antibody MAX.16H5 IgG1. Unfortunately, the GVL effect mediated by the graft was not strong enough and HSCT alone was not sufficient to cure AML carrying a FLT3ITD mutation in this model. In general, co-transplantation of 32D-FLT3ITD (mutated) AML cells with an immune cell graft in lethally irradiated mice revealed that MAX.16H5 IgG1 antibody pre-incubation of the graft cells prolonged their survival significantly. Nevertheless, when 32D-FLT3wt AML cells were used for co-transplantation, lower splenocyte counts were sufficient to reduce AML cells with or without antibody pre-incubation of the graft reflecting the aggressive proliferation properties of FLT3ITD AML cells. Moreover, in comparison to the control group (without MAX.16H5 IgG1 pre-incubation), no tumor cells were detected in the blood of the transplanted mice on day 54 after transplantation if mice received an antibody treated graft together with 32D-FLT3wt AML cells. When ITD-mutated AML cells were investigated under the same experimental conditions, the control group receiving an untreated graft died within 18 days, whereas the mice receiving a MAX.16H5 IgG1 treated graft died within 35 days after allogeneic transplantation. A slight increase in human CD135+ counts was observed in both groups on day 5, possibly due to a subsequent manifestation of the tumor cells in the organs. Because of this tissue manifestation, no AML cells were detectable in the periphery. Even though a long-term anti-tumor effect was not mediated by graft transplantation, the incubation with the anti-human CD4 antibody MAX.16H5 IgG1 improved the survival of the mice significantly. The efficient prevention of GVHD after immune cell transplantation mediated by the MAX.16H5 antibody graft incubation prolonged the survival of the respective animals paving the way for 32D-FLT3ITD AML cell proliferation later on thereby causing the death of all mice examined. Unfortunately, the transplantation of higher graft cell counts in combination with MAX.16H5 antibody pre-incubation only further prolonged the survival of the mice but showed no long lasting anti-tumor effect toward the ITD mutated AML cells in this model. These findings are supported by the work of other groups, showing that constitutively activated STAT5 in FLT3ITD mutated cells leads to uncontrolled proliferation, factor independent growth and radiation resistance in humans and mice (45).
AML patients carrying cytogenetically mutated FLT3ITD are affected by high post-HSCT relapse rates and diminished overall survival (73–75). A study from Whiteman et al. showed that 87% of patients carrying a FLT3ITD/− AML died within 12 months. In comparison, 74% of patients with a wild type (FLT3wt/wt) AML and 65% of patients with a heterozygous (FLT3ITD/wt) AML were alive 12 months after autologous transplantation (36). Another study by Bienz et al. revealed a three-year overall survival of one patient with FLT3ITD AML (out of 19 patients) compared to 11 patients with FLT3wt AML (out of 48 patients) after intensive chemotherapy and allogeneic transplantation (76). Comparable results were reviewed by Beitinjaneh et al. in an article where 11 different studies were examined (77). This indicates that the cure of FLT3ITD mutated AML by allogeneic HSCT can be challenging, since this mutation is involved in the activation of proliferative signaling cascades inhibiting hematopoietic differentiation and apoptosis in these cells [reviewed in (78)]. However, HSCT and the graft-mediated GVL effect are key players in the successful treatment of many AML patients (79).
The results of the present study indicate that the anti-human CD4 antibody MAX.16H5 IgG1 successfully impairs GVHD development following allogeneic HSCT which is still the main complication arising from HSCT. Although the successful allogeneic transplantation did not induce long-term anti-tumor effects in the mice, the prevention of GVHD by incubating graft cells with the anti-CD4 antibody prolonged their survival significantly thereby opening a time frame for additional treatment options. In further experiments, the results regarding FLT3ITD mutated AML will be investigated in NSG mice in order to verify our findings in a more patient-related setting.
Author Contributions
NH, SF, and GB designed the research; NH, CM, AM, CH, CW, and SF ran the experiments; NH, CM, AK, SF, LS, SD, DG, AW, and CH analyzed results; NH, CM, AK, BZ, LS, SF, and CH created figures and wrote the paper.
Funding
This study was supported by grants from Dr. Werner Jackstädt Stiftung and German society for hematology and medical oncology (DGHO, CH), German Federal Ministry of Education and Research (BMBF, C4107).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the animal attendants of the Medical Experimental Center (Faculty of Medicine, University of Leipzig) and our colleagues from the Translational Centre for Regenerative Medicine, University of Leipzig for providing and breeding the CD4/DR3 mice, Katrin Eisenbruch and Ina Patties for preparing the irradiation of recipient mice. Study design, collection of data, analysis, decision to publish and preparation of the manuscript were not influenced.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02408/full#supplementary-material
Abbreviations
AML, acute myeloid leukemia; BM, bone marrow; BMC, bone marrow cells; CD, cluster of differentiation; CTL, cytotoxic T lymphocyte; FLT3, FMS like tyrosine-protein kinase 3; GVHD, graft-vs. -host disease; GVL, graft-vs.-leukemia; HLA-DR3, human leukocyte antigen isotype DR3; HSCT, hematopoietic stem cell transplantation; hu, human; ITD, internal tandem duplication; MHC, major histocompatibility complex; mu, murine; NK, natural killer; NSG, NOD.Cg-Prkdcscid IL-2rgtm1Wjl/SzJ; PBS, phosphate buffered saline; SD, standard deviation; SpC, splenocytes; SSC-A, side scatter area; TUNEL, TdT-mediated dUTP-biotin nick end labeling; WBC, white blood cell; wt, wild type.
References
1. Ottinger HD, Ferencik S, Beelen DW, Lindemann M, Peceny R, Elmaagacli AH, et al. Hematopoietic stem cell transplantation: contrasting the outcome of transplantations from HLA-identical siblings, partially HLA-mismatched related donors, and HLA-matched unrelated donors. Blood (2003) 102:1131–7. doi: 10.1182/blood-2002-09-2866
2. Dickinson AM, Norden J, Li S, Hromadnikova I, Schmid C, Schmetzer H, et al. Graft-versus-Leukemia effect following hematopoietic stem cell transplantation for leukemia. Front Immunol. (2017) 8:496. doi: 10.3389/fimmu.2017.00496
3. Jamani K, Russell JA, Daly A, Stewart D, Savoie L, Duggan P, et al. Prognosis of grade 3-4 acute GVHD continues to be dismal. Bone Marrow Transplant. (2013) 48(10):1359–61. doi: 10.1038/bmt.2013.59
4. Martin PJ, Inamoto Y, Flowers ME, Carpenter PA. Secondary treatment of acute graft-versus-host disease: a critical review. Biol.Blood Marrow Transplant. (2012) 18:982–8. doi: 10.1016/j.bbmt.2012.04.006
5. Magenau J, Runaas L, Reddy P. Advances in understanding the pathogenesis of graft-versus-host disease. Br J Haematol. (2016) 173:190–205. doi: 10.1111/bjh.13959
6. Matthews K, Lim Z, Afzali B, Pearce L, Abdallah A, Kordasti S, et al. Imbalance of effector and regulatory CD4 T cells is associated with graft-versus-host disease after hematopoietic stem cell transplantation using a reduced intensity conditioning regimen and alemtuzumab. Haematologica (2009) 94:956–66. doi: 10.3324/haematol.2008.003103
7. Simonetta F, Alvarez M, Negrin RS. Natural killer cells in graft-versus-host-disease after allogeneic hematopoietic cell transplantation. Front Immunol. (2017) 8:465. doi: 10.3389/fimmu.2017.00465
8. Cohen JL, Trenado A, Vasey D, Klatzmann D, Salomon BL. CD4+ CD25+ immunoregulatory T cells. J Exp Med. (2002) 196:401–6. doi: 10.1084/jem.20020090
9. Li PG, Di CS, Montanari M, Moretta L, Manca F. Specific removal of alloreactive T-cells to prevent GvHD in hemopoietic stem cell transplantation: rationale, strategies and perspectives. Blood Rev. (2016) 30:297–307. doi: 10.1016/j.blre.2016.03.001
10. Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood (1990) 75:555–62.
11. Nash RA, Storb R. Graft-versus-host effect after allogeneic hematopoietic stem cell transplantation: GVHD and GVL. Curr Opin Immunol. (1996) 8:674–80.
12. de Aquino MT, Malhotra A, Mishra MK, Shanker A. Challenges and future perspectives of T cell immunotherapy in cancer. Immunol Lett. (2015) 166:117–33. doi: 10.1016/j.imlet.2015.05.018
13. Apetoh L, Smyth MJ, Drake CG, Abastado J-P, Apte RN, Ayyoub M, et al. Consensus nomenclature for CD8+ T cell phenotypes in cancer. Oncoimmunology (2015) 4:e998538. doi: 10.1080/2162402X.2014.998538
14. Glimcher LH, Kara CJ. Sequences and factors: a guide to MHC class-II transcription. Annu Rev Immunol. (1992) 10:13–49. doi: 10.1146/annurev.iy.10.040192.000305
15. Zhou Y, Zha J, Lin Z, Fang Z, Zeng H, Zhao J, et al. CD4+ T cell-mediated cytotoxicity is associated with MHC class II expression on malignant CD19+ B cells in diffuse large B cell lymphoma. Exp Cell Res. (2018) 362:287–92. doi: 10.1016/j.yexcr.2017.11.029
16. Thibodeau J, Bourgeois-Daigneault M-C, Lapointe R. Targeting the MHC Class II antigen presentation pathway in cancer immunotherapy. Oncoimmunology (2012) 1:908–16. doi: 10.4161/onci.21205
17. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol. (2015) 35(Suppl.):S185–98. doi: 10.1016/j.semcancer.2015.03.004
18. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity (2004) 21:137–48. doi: 10.1016/j.immuni.2004.07.017
19. Hadrup S, Donia M, Thor Straten P. Effector CD4 and CD8 T cells and their role in the tumor microenvironment. Cancer Microenviron. (2013) 6:123–33. doi: 10.1007/s12307-012-0127-6
20. Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature (1998) 393:480–3. doi: 10.1038/31002
21. Abe BT, Shin DS, Mocholi E, Macian F. NFAT1 supports tumor-induced anergy of CD4+ T cells. Cancer Res. (2012) 72:4642–51. doi: 10.1158/0008-5472.CAN-11-3775
22. Staveley-O'Carroll K, Sotomayor E, Montgomery J, Borrello I, Hwang L, Fein S, et al. Induction of antigen-specific T cell anergy: an early event in the course of tumor progression. Proc Natl Acad Sci USA. (1998) 95:1178–83. doi: 10.1073/pnas.95.3.1178
23. Kern DE, Klarnet JP, Jensen MC, Greenberg PD. Requirement for recognition of class II molecules and processed tumor antigen for optimal generation of syngeneic tumor-specific class I-restricted CTL. J Immunol. (1986) 136:4303.
24. Greaves P, Gribben JG. The role of B7 family molecules in hematologic malignancy. Blood (2013) 121:734–44. doi: 10.1182/blood-2012-10-385591
25. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. (2009) 229:152–72. doi: 10.1111/j.1600-065X.2009.00782.x
26. Schmidt F, Hilger N, Oelkrug C, Svanidze E, Ruschpler P, Eichler W, et al. Flow cytometric analysis of the graft-versus-Leukemia-Effect after hematopoietic stem cell transplantation in mice. Cytometry A (2015) 87:334–45 doi: 10.1002/cyto.a.22619
27. Hilger N, Glaser J, Müller C, Halbich C, Müller A-M, Schwertassek, U, et al. Attenuation of graft-versus-host-disease in NOD scid IL-2Rgamma(-/-) (NSG) mice by ex vivo modulation of human CD4+ T cells. Cytometry A (2016) 89:803–15. doi: 10.1002/cyto.a.22930
28. Repke H, Gabuzda D, Palù G, Emmrich F, Sodroski J. Effects of CD4 synthetic peptides on HIV type I envelope glycoprotein function. J Immunol. (1992) 149:1809–16.
29. Fricke S, Hilger N, Fricke C, Schönfelder U, Behre G, Ruschpler P, et al. Prevention of graft-versus-host-disease with preserved graft-versus-leukemia-effect by ex vivo and in vivo modulation of CD4+ T-cells. Cell Mol Life Sci. (2014) 71:2135–48. doi: 10.1007/s00018-013-1476-0
30. Nasilowska-Adamska B, Czyz A, Markiewicz M, Rzepecki P, Piatkowska-Jakubas B, Paluszewska M, et al. Mild chronic graft-versus-host disease may alleviate poor prognosis associated with FLT3 internal tandem duplication for adult acute myeloid leukemia following allogeneic stem cell transplantation with myeloablative conditioning in first complete remission: a retrospective study. Eur J Haematol. (2016) 96:236–44. doi: 10.1111/ejh.12575
31. Tschan-Plessl A, Halter JP, Heim D, Medinger M, Gerull S. Synergistic effect of sorafenib and cGvHD in patients with high-risk FLT3-ITD+AML allows long-term disease control after allogeneic transplantation. Ann Hematol. (2015) 94:1899–905. doi: 10.1007/s00277-015-2461-5
32. Grafone T, Palmisano M, Nicci C, Storti S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: biology and treatment. Oncol Rev. (2012) 6:e8. doi: 10.4081/oncol.2012.e8
33. Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer (2003) 3:650–65. doi: 10.1038/nrc1169
34. Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. (2012) 366:1079–89. doi: 10.1056/NEJMoa1112304
35. Rockova V, Abbas S, Wouters BJ, Erpelinck CA, Beverloo HB, Delwel R, et al. Risk stratification of intermediate-risk acute myeloid leukemia: integrative analysis of a multitude of gene mutation and gene expression markers. Blood (2011) 118:1069–76. doi: 10.1182/blood-2011-02-334748
36. Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res. (2001) 61:7233–9.
37. Pratz KW, Levis M. How I treat FLT3-mutated AML. Blood (2017) 129:565–71. doi: 10.1182/blood-2016-09-693648
38. Chen F, Ishikawa Y, Akashi A, Naoe T, Kiyoi H. Co-expression of wild-type FLT3 attenuates the inhibitory effect of FLT3 inhibitor on FLT3 mutated leukemia cells. Oncotarget. (2016) 7:47018–32. doi: 10.18632/oncotarget.10147
39. Kadia TM, Ravandi F, Cortes J, Kantarjian H. New drugs in acute myeloid leukemia. Ann Oncol. (2016) 27:770–8. doi: 10.1093/annonc/mdw015
40. Gallogly MM, Lazarus HM. Midostaurin: an emerging treatment for acute myeloid leukemia patients. J Blood Med. (2016) 7:73–83. doi: 10.2147/JBM.S100283
41. European Medicines Agency. Rydapt: Midostaurin (2017). Available online at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004095/human_med_002155.jsp&mid=WC0b01ac058001d124.
42. U.S. Food and Drug Administration. Midostaurin (2017). Available online at: 5756.htm
43. Fricke S, Rothe K, Hilger N, Ackermann M, Oelkrug C, Fricke C, et al. Allogeneic bone marrow grafts with high levels of CD4+ CD25+ FoxP3+ T cells can lead to engraftment failure. Cytometry A (2012) 81:476–88. doi: 10.1002/cyto.a.22061
44. Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J, Crawford JM, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood (1996) 88:3230–9.
45. Mizuki M, Fenski R, Halfter H, Matsumura I, Schmidt R, Müller C, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. (2000) 96:3907–14.
46. Gerloff D, Grundler R, Wurm AA, Brauer-Hartmann D, Katzerke C, Hartmann JU, et al. NF-κB/STAT5/miR-155 network targets PU.1 in FLT3-ITD-driven acute myeloid leukemia. Leukemia (2015) 29:535–47. doi: 10.1038/leu.2014.231
47. Oelkrug C, Hilger N, Schönfelder U, Boltze J, Sack U, Fricke C, et al. Modelling hematological parameters after total body irradiation. Int J Radiat Biol. (2014) 90:538–46. doi: 10.3109/09553002.2014.899443
48. Loo DT. In situ detection of apoptosis by the TUNEL assay: an overview of techniques. Methods Mol Biol. (2011) 682:3–13. doi: 10.1007/978-1-60327-409-8_1
50. R Core Team. R: A Language Environment for Statistical Computing. Vienna, Wien (2017). Available online at: https://www.R-project.org/.
51. Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biomet J. (2008) 50:346–63. doi: 10.1002/bimj.200810425
52. Peters G-JY. Diamond plots: a tutorial to introduce a visualisation tool that facilitates interpretation and comparison of multiple sample estimates while respecting their inaccuracy. PsyArXiv (2017). doi: 10.31234/osf.io/fzh6c
53. Andri Signorell et mult. al. (2017). DescTools: Tools for Descriptive Statistics. R package version 0.99.20. Available online at: https://CRAN.R-project.org/package=DescTools
54. Hothorn T, Hornik K, van de Wiel MA, Zeileis A. A lego system for conditional inference. Am Stat. (2006) 60:257–63. doi: 10.1198/000313006X118430
55. Hothorn T, Hornik K, van de Wiel MA, Zeileis A. Implementing a class of permutation tests: the coin package. J Stat Softw. (2008) 28:1–23. doi: 10.18637/jss.v028.i08
57. Fricke S. Entwicklung präklinischer Transplantationsmodelle und Innovativer Immunologischer Therapien zur Prävention der Transplantat-Gegen-Wirt-Erkrankung (GvHD) mit Erhaltenem Anti-Tumor-Effekt Nach Hämatopoetischen Stammzelltransplantationen. Leipzig: Habilitationsschrift (2015).
58. del Campo L, León NG, Palacios DC, Lagana C, Tagarro D. Abdominal complications following hematopoietic stem cell transplantation. Radiographics (2014) 34:396–412. doi: 10.1148/rg.342135046
59. Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet (2009) 373:1550–61. doi: 10.1016/S0140-6736(09)60237-3
60. Röllig C, Beelen DW, Braess J, Greil R, Niederwieser D, Passweg J, et al. Akute Myeloische Leukämie (AML): Leitlinie ICD-10 C92.-, ICD-10 C93.-Empfehlungen der Fachgesellschaft zur Diagnostik und Therapie hämatologischer und onkologischer Erkrankungen (2018). Available online at: https://www.onkopedia.com/de/onkopedia/guidelines/akute-myeloische-leukaemie-aml/@@view/pdf/index.pdf.
61. Appelbaum FR. Haematopoietic cell transplantation as immunotherapy. Nature (2001) 411:385–9. doi: 10.1038/35077251
62. Levine JE, Logan B, Wu J, Alousi AM, Ho V, Bolaños-Meade J, et al. Graft-versus-host disease treatment: predictors of survival. Biol Blood Marrow Transplant. (2010) 16:1693–9. doi: 10.1016/j.bbmt.2010.05.019
63. Heinrichs J, Li J, Nguyen H, Wu Y, Bastian D, Daethanasanmak A, et al. CD8+ Tregs promote GVHD prevention and overcome the impaired GVL effect mediated by CD4+ Tregs in mice. Oncoimmunology (2016) 5:e1146842. doi: 10.1080/2162402X.2016.1146842
64. Auletta JJ, Eid SK, Wuttisarnwattana P, Silva I, Metheny L, Keller MD, et al. Human mesenchymal stromal cells attenuate graft-versus-host disease and maintain graft-versus-leukemia activity following experimental allogeneic bone marrow transplantation. Stem cells (2015) 33:601–14. doi: 10.1002/stem.1867
65. Müller-Tidow C, Steur C, Mizuki M, Schwable J, Brandts C, Berdel WE, et al. Mutationen des Wachstumsfaktor-Rezeptors Flt3 bei Akuter Myeloischer Leukamie - Transformation myeloischer Zellen durch Ras-abhangige und Ras-unabhangige Mechanismen. Dtsch Med Wochenschr. (2002) 127:2195–200. doi: 10.1055/s-2002-34942
66. Chen Y, Pan Y, Guo Y, Zhao W, Ho WT, Wang J, et al. Tyrosine kinase inhibitors targeting FLT3 in the treatment of acute myeloid leukemia. Stem Cell Investig. (2017) 4:48. doi: 10.21037/sci.2017.05.04
67. Schroeder MA, DiPersio JF. Mouse models of graft-versus-host disease: advances and limitations. Dis Model Mech. (2011) 4:318–33. doi: 10.1242/dmm.006668
68. Impola U, Larjo A, Salmenniemi U, Putkonen M, Itälä-Remes M, Partanen J. Graft immune cell composition associates with clinical outcome of allogeneic hematopoietic stem cell transplantation in patients with AML. Front Immunol. (2016) 7:523. doi: 10.3389/fimmu.2016.00523
69. Marmont AM, Horowitz MM, Gale RP, Sobocinski K, Ash RC, van Bekkum DW, et al. T-cell depletion of HLA-identical transplants in leukemia. Blood (1991) 78:2120–30.
70. Helling B, König M, Dälken B, Engling A, Krömer W, Heim K, et al. A specific CD4 epitope bound by tregalizumab mediates activation of regulatory T cells by a unique signaling pathway. Immunol Cell Biol. (2015) 93:396–405. doi: 10.1038/icb.2014.102
71. Gerloff D, Wurm AA, Hartmann JU, Hilger N, Müller A-M, Katzerke, C, et al. Next generation sequencing and functional analysis of Mirna expression in acute myeloid leukemia patients with different FLT3 mutations: block of MiR-155 in FLT3-ITD driven AML leads to downregulation of myeloid blasts in vivo. Blood (2015) 126:2438. Retrieved from http://www.bloodjournal.org/content/126/23/2438
72. Barnes TA, Amir E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br J Cancer (2017) 117:451–60. doi: 10.1038/bjc.2017.220
73. Schmid C, Labopin M, Socie G, Daguindau E, Volin L, Huynh A, et al. Outcome of patients with distinct molecular genotypes and cytogenetically normal AML after allogeneic transplantation. Blood (2015) 126:2062–9. doi: 10.1182/blood-2015-06-651562
74. Sengsayadeth SM, Jagasia M, Engelhardt BG, Kassim A, Strickland SA, Goodman S, et al. Allo-SCT for high-risk AML-CR1 in the molecular era: impact of FLT3/ITD outweighs the conventional markers. Bone Marrow Transplant. (2012) 47:1535–7. doi: 10.1038/bmt.2012.88
75. Song Y, Magenau J, Li Y, Braun T, Chang L, Bixby D, et al. FLT3 mutational status is an independent risk factor for adverse outcomes after allogeneic transplantation in AML. Bone Marrow Transplant. (2016) 51:511–20. doi: 10.1038/bmt.2015.170
76. Bienz M, Ludwig M, Leibundgut EO, Mueller BU, Ratschiller D, Solenthaler M, et al. Risk assessment in patients with acute myeloid leukemia and a normal karyotype. Clin Cancer Res. (2005) 11:1416–24. doi: 10.1158/1078-0432.CCR-04-1552
77. Beitinjaneh A, Jang S, Roukoz H, Majhail NS. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations in acute promyelocytic leukemia: a systematic review. Leuk Res. (2010) 34:831–6. doi: 10.1016/j.leukres.2010.01.001
78. de Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. (2016) 6:e441. doi: 10.1038/bcj.2016.50
79. Kanate AS, Pasquini MC, Hari PN, Hamadani M. Allogeneic hematopoietic cell transplant for acute myeloid leukemia: current state in 2013 and future directions. World J Stem Cells (2014) 6:69–81. doi: 10.4252/wjsc.v6.i2.69
80. Shulman HM, Cardona DM, Greenson JK, Hingorani S, Horn T, Huber E, et al. NIH consensus development project on criteria for clinical trials in chronic graft-versus-host disease: II. The 2014 Pathology Working Group Report. Biol Blood Marrow Transplant. (2015) 21:589–603. doi: 10.1016/j.bbmt.2014.12.031
Keywords: anti-human CD4 antibody MAX.16H5, hematopoietic stem cell transplantation, graft-vs.-host disease, acute myeloid leukemia, 32D-FLT3ITD, graft-vs.-leukemia, C3H/HeN
Citation: Hilger N, Mueller C, Stahl L, Mueller AM, Zoennchen B, Dluczek S, Halbich C, Wickenhauser C, Gerloff D, Wurm AA, Behre G, Kretschmer A and Fricke S (2018) Incubation of Immune Cell Grafts With MAX.16H5 IgG1 Anti-Human CD4 Antibody Prolonged Survival After Hematopoietic Stem Cell Transplantation in a Mouse Model for Fms Like Tyrosine Kinase 3 Positive Acute Myeloid Leukemia. Front. Immunol. 9:2408. doi: 10.3389/fimmu.2018.02408
Received: 23 April 2018; Accepted: 28 September 2018;
Published: 22 October 2018.
Edited by:
Aurore Saudemont, GlaxoSmithKline, United KingdomReviewed by:
Nirmalya Chatterjee, Harvard Medical School, United StatesAlix Eden Seif, Children's Hospital of Philadelphia, United States
Copyright © 2018 Hilger, Mueller, Stahl, Mueller, Zoennchen, Dluczek, Halbich, Wickenhauser, Gerloff, Wurm, Behre, Kretschmer and Fricke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nadja Hilger, bmFkamEuaGlsZ2VyQGl6aS5mcmF1bmhvZmVyLmRl
† These authors have contributed equally to this work
‡ These authors share authorship