 Darrell Pilling
Darrell Pilling Richard H. Gomer
Richard H. Gomer- Department of Biology, Texas A&M University, College Station, TX, United States
Pentraxins such as serum amyloid P (SAP; also known as PTX2) regulate several aspects of the innate immune system. SAP inhibits the differentiation of monocyte-derived fibroblast-like cells called fibrocytes, promotes the formation of immuno-regulatory macrophages, and inhibits neutrophil adhesion to extracellular matrix proteins. In this minireview, we describe how these effects of SAP have led to its possible use as a therapeutic, and how modulating SAP effects might be used for other therapeutics. Fibrosing diseases such as pulmonary fibrosis, cardiac fibrosis, liver fibrosis, and renal fibrosis are associated with 30–45% of deaths in the US. Fibrosis involves both fibrocyte differentiation and profibrotic macrophage differentiation, and possibly because SAP inhibits both of these processes, in 9 different animal models, SAP inhibited fibrosis. In Phase 1B and Phase 2 clinical trials, SAP injections reduced the decline in lung function in pulmonary fibrosis patients, and in a small Phase 2 trial SAP injections reduced fibrosis in myelofibrosis patients. Acute respiratory distress syndrome/ acute lung injury (ARDS/ALI) involves the accumulation of neutrophils in the lungs, and possibly because SAP inhibits neutrophil adhesion, SAP injections reduced the severity of ARDS in an animal model. Conversely, depleting SAP is a potential therapeutic for amyloidosis, topically removing SAP from wound fluid speeds wound healing in animal models, and blocking SAP binding to one of its receptors makes cultured macrophages more aggressive toward tuberculosis bacteria. These results suggest that modulating pentraxin signaling might be useful for a variety of diseases.
Introduction: SAP and Debris Clearance
SAP (PTX2) is a member of the pentraxin family of proteins that includes C-reactive protein (CRP; PTX1) and pentraxin-3 (PTX3). SAP is made by hepatocytes and secreted into the blood (1, 2). Searches of proteomics and RNA-seq databases suggests that the liver is the major source of SAP. In humans and most mammals, the levels of SAP in the plasma are maintained at relatively constant levels, between 20 and 50 μg/ml (3–5). There is little evidence for sequence variation of SAP at the genomic or amino acid level. In mice, SAP acts as an acute phase protein, with levels rising up to 20-fold following an inflammatory insult (6, 7). SAP is a pentameric protein with sequence and structural similarity to CRP (8–10). The structure of SAP (and CRP) pentamers is a flat disk with a hole in the middle (11, 12). The crystal structure of PTX3 has yet to be determined, but models based on site-directed mutagenesis, electron microscopy, and small-angle X-ray scattering data suggests that PTX3 is an octamer of two tetramers (13). Each SAP molecule has two Ca++ atoms bound to it, and the pentamer thus has 10 Ca++ atoms on one side of the disk. With the help of the bound Ca++, this side of the disk binds to a variety of molecules including apoptotic debris, bacterial polysaccharides, amyloid deposits, and bacterial toxins (1, 14, 15). Phagocytic cells such as monocytes and macrophages then bind the SAP, CRP, or PTX3, and engulf the debris or other material the pentraxin has bound (16). CRP and PTX3 can similarly bind a variety of debris molecules (17, 18). Proteins with strong similarity to SAP (and CRP and PTX3) are present in the hemolymph of horseshoe crabs (19, 17), so this debris clearance mechanism appears to have evolved during the early evolution of animals (Figure 1).
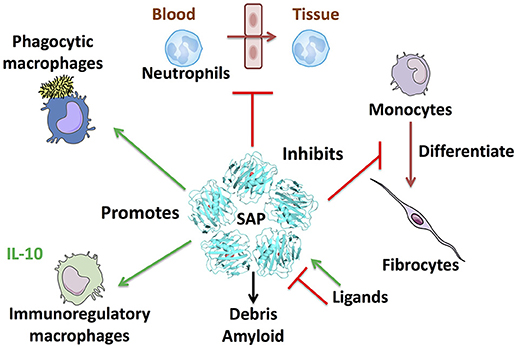
Figure 1. SAP regulates multiple aspects of immune responses. Some of the known effects of SAP are shown clockwise from top: SAP inhibits neutrophil adhesion to extracellular matrix and inhibits neutrophil movement into tissues. SAP binds to FcγR and DC-SIGN to inhibit monocyte to fibrocyte differentiation. SAP also binds multiple plasma proteins such as the complement component C1q and mannose-binding lectin (MBL) to promote phagocytosis of bacteria and regulate macrophage differentiation. SAP opsonizes bacteria and cell debris to promote removal by macrophages, and binds amyloid deposits. Finally, SAP promotes immuno-regulatory, and M1 phagocytic macrophages.
Removing Stuck SAP as a Possible Therapeutic for Amyloidosis
Amyloidosis is a disease where misfolded proteins aggregate and form large deposits in a tissue, leading to organ dysfunction (20, 21). SAP was originally isolated as a serum-derived protein found in all types of amyloid deposits (hence the serum amyloid part of its name) (22, 23). SAP was found to be a pentameric protein, hence the P part of its name (24–27). SAP is also easily purified by incubating serum with certain types of agarose in the presence of calcium, washing unbound protein off, and then eluting fairly pure bound SAP with a calcium chelator (28, 29). One possibility is that the SAP in the amyloid deposits binds to the misfolded proteins in an attempt to opsonize them for phagocytosis, but cannot pull proteins out of the deposit, and the SAP then gets stuck in the deposits. SAP knockout mice have reduced severity of experimentally-induced amyloidosis, suggesting that the stuck SAP exacerbates the amyloid deposit formation and/or hinders the ability of other opsins to pull the amyloid complexes apart (30). The Pepys group found a small molecule compound that causes two human SAP pentamers to stick to each other, and this complex is then quickly cleared from the circulation (31). In SAP knockout mice expressing human SAP, the compound decreased serum SAP levels but did not reduce the severity of experimentally-induced amyloidosis (31). Adding anti-SAP antibodies to this treatment however did reduce experimentally-induced amyloidosis, suggesting that reducing SAP levels is a possible therapeutic for amyloidosis (32–35).
Intermezzo 1: Wound Healing and Fibrosis
Most plant and animal tissues have a remarkable ability to heal mechanical wounds, indicating a strong evolutionary pressure for wound healing (36). In vertebrates, a typical dermal wound fills with scar tissue consisting of fibroblasts, connective tissue, and a capillary bed, and then is covered with an epithelium (37). Unfortunately, inappropriate wound healing responses to perceived wounds cause fibrosing diseases, where scar tissue forms in an internal organ, leading to organ dysfunction. There are at least 62 different fibrosing diseases, and these are associated with 30–45% of deaths in the US (38, 39). Examples of fibrosing diseases include cardiac fibrosis, probably triggered by reduced blood flow to part of the heart, and this fibrosis accounts for a significant fraction of the 450,000 deaths per year from cardiovascular disease in the US (40, 41). Other fibrosing diseases are cirrhosis of the liver, triggered by damage from viral infections, alcohol, or other chemical insults (39), end-stage kidney disease in diabetics, where the scar tissue formation is probably triggered by damage from high glucose levels (42), and pulmonary fibrosis, where particulate matter such as coal dust, and other unknown factors, triggers the progressive formation of scar tissue in the lungs (43). The only FDA-approved therapeutics for fibrosis are two drugs which slow, but do not stop, the progression of pulmonary fibrosis (44).
Intermezzo 2: Fibrocytes
In the 1850's, James Paget examined healing wounds and observed that cells from the blood enter the wound and then differentiate into elongated cells with an oval nucleus [see Figure 14, page 127 in (45)]. Bucala et al. found that these cells originate from bone marrow derived circulating CD14+ monocytes and express markers such as such as CD34 and CD45 that identify them as bone marrow-derived cells, as well as markers such as collagen that identify them as fibroblast-like cells (46–49). They named the cells fibrocytes. Although fibrocytes are rarely observed in normal tissues, they are present in high numbers in healing wounds (46, 50, 51) and fibrotic lesions in pulmonary fibrosis (51–58), keloid scars (59, 60), asthma (52, 61, 62), chronic kidney disease (63, 64, 65), and nephrogenic systemic fibrosis (66). Fibrocytes are also present in the fibrotic lesions in animal models of pulmonary fibrosis (53, 67–75), liver fibrosis (71) and renal fibrosis (73, 76). In addition to contributing to the mass of fibrotic lesions, fibrocytes promote angiogenesis (77), which can then promote the growth of the scar tissue, and secrete TGF-β (78), which causes resident fibroblasts to proliferate and increase their collagen production. Fibroblasts thus have a multiplicative effect on scar tissue formation. At the time we started working on fibrocytes, nothing was known about extracellular factors that regulate their differentiation, and thus why fibrocytes are present in wounds but not normal tissues, and how to control them.
Our Entry Into the SAP, Fibrocytes, Wound Healing, and Fibrosis Fields
Our lab had been studying the ability of diffusible secreted factors to indicate the local density and composition of cells in a tissue, using the eukaryotic amoeba Dictyostelium discoideum as a model system (79, 80). We decided to try to look for cell density sensing factors secreted by human white blood cells. To simplify the purification of any such factors from the extracellular medium, human peripheral blood mononuclear cells (PBMCs) were cultured in serum-free medium. Some of the cells became long, spindle-shaped cells after 3–5 days (81, 82). Videomicroscopy indicated that these spindle shaped cells were quite motile, and they stained for fibrocyte markers (81). Fibrocytes did not appear during this timeframe when serum was present, indicating that something in serum inhibits fibrocyte differentiation.
Since removing something that inhibits fibrocyte differentiation might potentiate wound healing, and conversely adding something that inhibits fibrocyte differentiation might inhibit fibrosis, we abandoned the search for human density sensing factors and purified the fibrocyte differentiation inhibitor from human serum. It turned out to be SAP (81). SAP also inhibits the differentiation of mouse, rat, and dog PBMCs into fibrocytes (83–86). CRP has no significant effect on, and PTX3 potentiates, fibrocyte differentiation, indicating that the three pentraxins differentially affect fibrocytes (81, 87). When PBMCs were cultured in serum that was depleted of SAP, fibrocytes rapidly appeared, indicating that SAP is the main endogenous inhibitor of fibrocyte differentiation in the blood (81).
Pentraxins also regulate macrophages (17, 39, 88–95). In addition to inhibiting fibrocyte differentiation, SAP inhibits pro-fibrotic macrophages, and promotes the formation of immuno-regulatory macrophages (84, 95–105). Although SAP can bind complement component C1q and mannose-binding lectin, these proteins have very modest effects on the ability of SAP to affect macrophage phenotypes (95, 106–108). We refer the reader to the above references and reviews for information on the complexity of pentraxin (including SAP) regulation of macrophages that is beyond the simplicity of this minireview (Figure 1).
Removing SAP as a Possible Therapeutic for Wound Healing
Since after blood clots, a wound is covered with serum, and serum contains SAP, and SAP inhibits fibrocyte differentiation and thus wound healing, an intriguing possibility is that removing SAP from wound fluid might potentiate fibrocyte differentiation and wound healing. Wound dressings with Ca++ and the type of agarose originally used to purify SAP from serum were tested on full thickness dermal wounds in rats. These dressings speeded healing of these wounds, as well as partial thickness dermal wounds in pigs (109). In the pig wounds, the agarose/ Ca++ dressings caused wound to heal faster than wounds treated with commercial dressings such as Tielle, Intrasite, and Xeroform. Although SAP levels in humans are unaffected by inflammation, serum SAP levels in the general population range from 20 to 60 μg/ml (3, 5, 110). Compared to controls, patients with low levels of SAP have better survival of skin grafts, supporting the idea that reducing SAP levels might help wound healing (111). In part because the wound dressing market is basically saturated, efforts to fund clinical tests of this SAP-depleting dressing have been unsuccessful.
Adding SAP as a Possible Therapeutic for Fibrosing Diseases
A simple non-surgical animal model of a fibrosing disease is pulmonary fibrosis in mice and rats, where a drug called bleomycin can be pipetted through the mouth into the airway, and within 14 days causes pulmonary fibrosis (112, 113). In the bleomycin model, SAP injections led to reduced numbers of fibrocytes in the lungs and reduced fibrosis in rats and mice, and delaying SAP injections until inflammation and fibrosis was already apparent (therapeutic dosing) also reduced symptoms (84). SAP injections have now been shown to inhibit inflammation and fibrosis in other models of pulmonary fibrosis (102, 103), cardiac fibrosis (96, 97), radiation-induced oral mucositis (101), allergic airway disease (100), autoimmune encephalomyelitis (114), corneal wound healing (75), and two models of renal fibrosis/ end stage kidney disease (98). An obvious question about using SAP as a therapeutic for fibrosis is that this might block wound healing. We found that SAP injections slow, but do not stop, dermal wound healing in mice (115).
SAP Efficacy as an Antifibrotic in Clinical Trials
Compared to control mice, mice lacking SAP have strongly increased pulmonary fibrosis in response to bleomycin, indicating that an endogenous function of SAP is to reduce fibrosis (116). Compared to controls, patients with renal fibrosis, pulmonary fibrosis, scleroderma, myelofibrosis, rheumatoid arthritis, and mixed connective tissue disease tend to have low levels of SAP, supporting the idea that fibrosis might in part involve a SAP deficiency (81, 98, 103, 117). One initial problem obtaining NIH funding to study SAP and fibrosis was that people confused SAP with serum amyloid A (SAA; a completely different, and probably not beneficial protein). We encountered this with a grant application where a reviewer denounced our efforts to inject animals with SAA. After politely explaining that SAP was not SAA, the grant was funded. An early worker in the SAP field encountered this too, and published a letter in Nature entitled “Serum Amyloid P component (not Serum Amyloid Protein)” (118). For this and other reasons, colleagues used the alternative SAP nomenclature Pentraxin 2 (PTX2) (26, 119), and called the recombinant SAP used for clinical trials PRM-151 (120). Compared to standard of care, injections of recombinant human SAP/PTX2 improved lung function in a Phase 1b trial and a Phase 2 trial in pulmonary fibrosis patients (5, 121). In the 28-weeks Phase 2 trial, SAP injections slowed the decline in forced exhalation volume (FEV), and essentially stopped the decline in the distance patients could walk in 6 min. 61 of the patients receiving SAP in this trial were also taking either pirfenidone or nintedanib, FDA-approved drugs that slow the decline in lung function in pulmonary fibrosis, while 16 other patients treated with SAP were not taking these drugs. Intriguingly, the 16 patients not taking these drugs who were treated with SAP appeared to show on average a very slight improvement in forced exhalation volume and an improvement in how far they could walk in 6 min, suggesting the exciting possibility that SAP might be able to partially reverse pulmonary fibrosis. Myelofibrosis is a fibrosis of the bone marrow (122). SAP injections also reduced fibrosis and improved bone marrow function in a 27-patient Phase 2 trial on myelofibrosis patients (123).
SAP Pharmacology
The plasma clearance rates for patients treated with SAP is ~24 to 30 h (5, 124). In the Phase 2 clinical trial, and in earlier trials, efficacy was observed with monthly dosing. The apparent paradox of how something with a short plasma half-life could show efficacy with monthly dosing can be answered by looking at the tissue half-life, which for healthy volunteers is 7.2 days (124). SAP, as well as CRP and PTX3, have long been known as opsonins that helps phagocytic cells ingest debris (16, 125–129). In fibrosis, debris and other tissue insults are thought to both initiate as well as potentiate fibrosis (39). Clearing debris from the vicinity of a fibrotic lesion is very likely one mechanism whereby SAP inhibits fibrosis. The debris is not detectable in the circulation, rather it is localized to the vicinity of the fibrotic lesion. Thus this beneficial effect of SAP occurs in the tissue rather than in the circulation. Although SAP has some modest effects on macrophage differentiation from monocytes (changes in the expression of a small number of surface markers in some but not all of the macrophages) (99, 95), macrophage polarization from one macrophage phenotype to another macrophage phenotype (again, even more subtle changes in the expression of a small number of surface markers) (95), and neutrophil adhesion to tissue extracellular matrix components (see below), the most obvious effect of SAP on innate immune cells is its ability to completely inhibit the differentiation of monocytes into fibrocytes. All of these effects on innate immune cells affect what the cells do after they have entered a tissue. A reasonable assumption is thus that the SAP effects occur in the tissue, specifically in the vicinity of the fibrotic lesion, rather than in the circulation, and thus that the key half-life is the tissue rather than plasma half-life.
Two observations suggest that the half-life of SAP in a fibrotic lesion may be considerably longer than 7.2 days. First, amyloid deposits resemble in many ways fibrotic lesions, and the half-life of SAP in amyloid deposits is 24–27 days (124, 130, 131). Second, in mice where fibrosis was induced in one kidney by obstructing the ureter, injected SAP localized to the fibrotic kidney, with much less localization to the non-injured contralateral kidney (98). Together, these arguments and results support the idea that even with a short plasma half-life, monthly injections of SAP can be efficacious.
SAP Inhibition of Fibrocytes Allows an Assessment of the Possible Effect of Factors Such as Dietary Salt on Fibrosis
Human PBMC cultured in serum-free medium differentiate into easily identifiable (by microscopy) fibrocytes, and adding different concentrations of SAP to inhibit this generates a standard curve of SAP effects. This allows a simple assay to look at the effects of various conditions or compounds on this process. For instance, ELISA assays of sera from keloid patients (these patients form greatly exaggerated dermal scars) showed normal levels of SAP, but the fibrocyte assay on keloid patient PBMC showed that these cells are relatively insensitive to SAP (132). A variety of compounds affect fibrocyte differentiation and/or the ability of SAP to inhibit fibrocyte differentiation (57, 63, 82, 87, 117, 133–142). One compound that may be clinically relevant is NaCl, which when added to increase the medium NaCl concentration by 25 mM (this level of increase can be seen in the serum after a very salty meal) potentiates fibrocyte differentiation and inhibits the SAP effect, possibly explaining why high salt intake is associated with a propensity for cardiac fibrosis (137). Peritoneal dialysis can lead to peritoneal fibrosis, and we found that peritoneal dialysis fluid and dialysis fluid components such as NaCl also promote fibrocyte differentiation and impede SAP (139). In support of this connection between salt and fibrocytes, low dietary salt reduces the severity of bleomycin-induced pulmonary fibrosis in mice, suggesting that low salt diets may be beneficial for fibrosis patients (143).
SAP Can Override Other Profibrotic Factors
A variety of signals promote wound healing and fibrosis. For instance, TGF-β1 is an extracellular signal that drives fibrosis (144, 145), and in mice, conditional expression of TGF-β1 in the lungs causes pulmonary fibrosis (103, 146). In this model, SAP injections stopped and reversed fibrosis (103). We found that although quiescent fibroblasts secrete the protein Slit2 to inhibit fibrocyte differentiation (essentially telling incoming monocytes that no more fibroblast-cells are needed) (138), fibroblasts activated by the pro-fibrotic signal TNF-α secrete the protein lumican, which promotes fibrocyte differentiation (142). Thankfully, SAP can override the effect of lumican on fibrocytes (142). Other signals that promote fibrocyte differentiation and profibrotic macrophage differentiation include thrombin activated during blood clotting (this may thus initiate the fibrocyte component of wound healing), and tryptase released from mast cells (147, 148). SAP also competes with these signals to inhibit fibrocytes and macrophages (149, 148). In addition, SAP inhibits fibrocyte differentiation induced by IL-4, IL-13, high molecular weight hyaluronic acid, and PTX3 (82, 87, 136, 141). Together, these results suggest that one reason SAP appears to be effective in the clinic as an anti-fibrotic is a fortunate dominance of SAP over these signals.
Elucidating SAP Receptors Led to Small-Molecule SAP Mimetics
Fcγ receptors (FcγRs) bind the Fc domain of IgG immunoglobulins (150). Once aggregated IgG cross-links multiple FcγRs (this prevents monomeric IgGs from activating FcγRs), a signaling cascade is activated through tyrosine kinases to initiate an immune response (151). Phagocytic cells, such as monocytes, bind SAP, CRP, and PTX3 using different combinations of FcγRs (16, 86, 98, 152–156), and the structure of SAP bound to FcγRIIa and modeling of SAP binding to other Fc receptors has been published (157, 158). In support of the hypothesis that SAP inhibits fibrocyte differentiation by binding to FcγRs, we found that cross-linked but not monomeric IgG also inhibits fibrocyte differentiation (159). Mouse monocytes lacking FcγRI, or human monocytes with siRNA-reduced FcγRI, had a reduced sensitivity to SAP, while mouse cells lacking other FcγRs had normal or enhances sensitivity to SAP, indicating that FcγRI mediates SAP signaling (86). Surprisingly, monocytes from cells lacking all four known FcγRs still responded to SAP, indicating that a different receptor also mediates SAP signaling (156, 160).
To help elucidate SAP signaling, we mutated SAP protein surface amino acids that were different from CRP, and the mutant SAPs were assayed for their ability to inhibit fibrocyte differentiation (SAP and CRP are have highly similar amino acid sequences and structures, but CRP does not inhibit fibrocyte differentiation) (81, 87)). None of the mutant SAPs completely abrogated SAP activity (86, 156). One amino acid initially overlooked was a glycosylated asparagine on SAP that is a non-glycosylated alanine on CRP, and when SAP was desialylated, the SAP largely lost its ability to inhibit fibrocyte differentiation; conversely when the CRP alanine was mutated to an asparagine, the asparagine became glycosylated and the glycosylated CRP inhibited fibrocyte differentiation (160). This suggested that a polysaccharide receptor might help to sense SAP, and we found that the C-type lectin DC-SIGN mediated SAP effects on monocytes (160). Other workers found a variety of compounds that block the ability of polysaccharides to bind DC-SIGN, and three of these potently inhibited fibrocyte differentiation. One of the DC-SIGN-binding molecules showed efficacy in a mouse pulmonary fibrosis model at 0.001 mg/kg (160). These results suggest that small molecules that mimic SAP might be useful as therapeutics for fibrosing diseases.
Adding SAP as a Possible Therapeutic for Neutrophil-Driven Diseases
Inflammatory lesions recruit neutrophils to the site of damage (161, 162). This however can sometimes be counterproductive; for instance some patients with damaged lungs develop acute respiratory distress syndrome/ acute lung injury (ARDS/ ALI), where neutrophils enter the lungs and release proteases and reactive oxygen species. This causes further damage and further neutrophil recruitment and subsequent damage, and this vicious cycle results in the ~40% mortality seen in the ~200,000 ARDS patients each year in the US (163). SAP decreases neutrophil binding to extracellular matrix components (164–166), and in a mouse model of ARDS, SAP injections starting 24 h after injury reduced the number of neutrophils in the lungs (166). The small-molecule SAP mimetic discussed above also showed efficacy in this ARDS model (160). These results suggest that SAP and SAP mimetics might be useful as therapeutics for neutrophil-driven diseases such as ARDS/ ALI.
Blocking SAP Signaling as a Possible Therapeutic for Diseases Such as Tuberculosis
M1 macrophages are highly aggressive against bacteria and other pathogens, but SAP, which is a constitutive component of the blood, pushes macrophages toward an anti-inflammatory/anti-fibrotic phenotype (90, 95, 99, 100, 103, 156, 167, 168). Tuberculosis bacteria can live inside macrophages, where they also push the host macrophage away from a M1 phenotype to help the survival of the parasitic bacteria (169). To test the hypothesis that blocking SAP signaling to macrophages would reduce regulatory macrophages and increase M1 macrophages, we screened 3,000 compounds for the ability to inhibit the binding of SAP to FcγRI, and found 12 that reduced this binding (170). In support of the hypothesis, SAP potentiated the proliferation of Mycobacterium smegmatis and Mycobacterium tuberculosis in human macrophages, and in the presence of SAP, 2 of the compounds reduced the intra-macrophage proliferation of these bacteria (170).
Conclusion
Pentraxins are ancient and fascinating molecules. Increasing levels of SAP either locally or systemically is showing promise as a therapeutic for a variety of diseases where the ability of SAP to help clear debris and calm the innate immune system is beneficial. Conversely, decreasing levels of SAP, or decreasing SAP effects, shows promise as potential therapeutics where unleashing the innate immune system is beneficial. An intriguing possibility is that altering levels of other pentraxins might similarly be useful as stand-alone therapeutics or in combination with manipulations of SAP levels for even more diseases.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was funded by NIH R01 HL132919 and NIH R01 HL118507.
Conflict of Interest Statement
DP and RG are inventors on patents for the use of SAP as a therapeutic for fibrosing diseases, and patents for the use of SAP-depleting materials to enhance wound healing. DP and RG are members of the Science Advisory Board of, and have stock options from, Promedior, a start-up company that is developing SAP as a therapeutic for fibrosing diseases, and receive a share of milestone payments made by Promedior to Rice University.
The reviewer AM and handling Editor declared their shared affiliation.
Acknowledgments
We thank Tejas Karhadkar and Kristen Consalvo for comments on the manuscript. Some components of the figure were adjusted from originals generated by Servier Medical Art, under a Creative Commons License.
References
1. Pepys MB, Booth DR, Hutchinson WL, Gallimore JR, Collins PM, Hohenester E. Amyloid P component. A critical review. Amyloid (1997) 4:274–95. doi: 10.3109/13506129709003838
2. Hutchinson WL, Hohenester E, Pepys MB. Human serum amyloid P component is a single uncomplexed pentamer in whole serum. Mol Med. (2000) 6:482–93. doi: 10.1007/BF03401789
3. Nelson SR, Tennent GA, Sethi D, Gower PE, Ballardie FW, Amatayakul-Chantler S, et al. Serum amyloid P component in chronic renal failure and dialysis. Clin Chim Acta (1991) 200:191–9. doi: 10.1016/0009-8981(91)90090-Y
4. Steel DM, Whitehead AS. The major acute phase reactants: C-reactive protein, serum amyloid P component and serum amyloid A protein. Immunol Today (1994) 15:81–8. doi: 10.1016/0167-5699(94)90138-4
5. Dillingh MR, van den Blink B, Moerland M, van Dongen MGJ, Levi M, Kleinjan A, et al. Recombinant human serum amyloid P in healthy volunteers and patients with pulmonary fibrosis. Pulmonary Pharmacol Therapeut. (2013) 26:672–6. doi: 10.1016/j.pupt.2013.01.008
6. Pepys MB, Baltz M, Gomer K, Davies AJ, Doenhoff M. Serum amyloid P-component is an acute-phase reactant in the mouse. Nature (1979) 278:259–61. doi: 10.1038/278259a0
7. Le PT, Muller MT, Mortensen RF. Acute phase reactants of mice. I Isolation of serum amyloid P- component (SAP) and its induction by a monokine. J Immunol. (1982) 129:665–72.
8. Du Clos TW. The interaction of C-reactive protein and serum amyloid P component with nuclear antigens. Mol Biol Rep. (1996) 23:253–60. doi: 10.1007/BF00351177
9. Kravitz M, Pitashny M, Shoenfeld Y. Protective Molecules–C-Reactive Protein (CRP), Serum Amyloid P (SAP), Pentraxin3 (PTX3), Mannose-Binding Lectin (MBL), and Apolipoprotein A1 (Apo A1), and Their Autoantibodies: Prevalence and Clinical Significance in Autoimmunity. J Clin Immunol. (2005) 25:582–91. doi: 10.1007/s10875-005-7828-2
10. Agrawal A, Singh PP, Bottazzi B, Garlanda C, Mantovani A. Pattern recognition by pentraxins. Adv Exp Med Biol. (2009) 653:98–116. doi: 10.1007/978-1-4419-0901-5_7
11. Emsley J, White HE, O'Hara BP, Oliva G, Srinivasan N, Tickle IJ, et al. Structure of pentameric human serum amyloid P component. Nature (1994) 367:338–45. doi: 10.1038/367338a0
12. Shrive AK, Cheetham GM, Holden D, Myles DA, Turnell WG, Volanakis JE, et al. Three dimensional structure of human C-reactive protein. Nat Struct Biol. (1996) 3:346–54. doi: 10.1038/nsb0496-346
13. Garlanda C, Bottazzi B, Magrini E, Inforzato A, Mantovani A. PTX3, a Humoral Pattern Recognition Molecule, in Innate Immunity, Tissue Repair, and Cancer. Physiol Rev. (2018) 98:623–39. doi: 10.1152/physrev.00016.2017
14. Pepys MB, Dyck RF, de Beer FC, Skinner M, Cohen AS. Binding of serum amyloid P-component (SAP) by amyloid fibrils. Clin ExpImmunol. (1979) 38:284–93.
15. Hamazaki H. Ca(2+)-dependent binding of human serum amyloid P component to Alzheimer's beta-amyloid peptide. J Biol Chem. (1995) 270:10392–4. doi: 10.1074/jbc.270.18.10392
16. Bharadwaj D, Mold C, Markham E, Du Clos TW. Serum amyloid P component binds to Fc gamma receptors and opsonizes particles for phagocytosis. J Immunol. (2001) 166:6735–41. doi: 10.4049/jimmunol.166.11.6735
17. Du Clos TW. Pentraxins: structure, function, and role in inflammation. ISRN Inflamm. (2013) 2013:379040. doi: 10.1155/2013/379040
18. Bottazzi B, Inforzato A, Messa M, Barbagallo M, Magrini E, Garlanda C, et al. The pentraxins PTX3 and SAP in innate immunity, regulation of inflammation and tissue remodelling. J Hepatol. (2016) 64:1416–27. doi: 10.1016/j.jhep.2016.02.029
19. Iwanaga S. The molecular basis of innate immunity in the horseshoe crab. Curr Opin Immunol. (2002) 14:87–95. doi: 10.1016/S0952-7915(01)00302-8
20. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Eng J Med. (2003) 349:583–96. doi: 10.1056/NEJMra023144
21. Sipe JD, Benson MD, Buxbaum JN, Ikeda SI, Merlini G, Saraiva MJM, et al. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid (2014) 21:221–4. doi: 10.3109/13506129.2014.964858
22. Cathcart ES, Comerford FR, Cohen AS. Immunologic studies on a protein extracted from human secondary amyloid. N Engl J Med. (1965) 273:143–6. doi: 10.1056/NEJM196507152730306
23. Cathcart ES, Wollheim FA, Cohen AS. Plasma protein constituents of amyloid fibrils. J Immunol. (1967) 99:376–85.
24. Bladen HA, Nylen MU, Glenner GG. The ultrastructure of human amyloid as revealed by the negative staining technique. J Ultrastruct Res. (1966) 14:449–59. doi: 10.1016/S0022-5320(66)80075-8
25. Benson Merrill D, Skinner M, Shirahama T, Cohen Alan S. P-component of amyloid. Isolation from human serum by affinity chromatography. Arthr Rheumat. (1976) 19:749–54. doi: 10.1002/1529-0131(197607/08)19:4<749::AID-ART1780190415>3.0.CO;2-C
26. Osmand AP, Friedenson B, Gewurz H, Painter RH, Hofmann T, Shelton E. Characterization of C-reactive protein and the complement subcomponent C1t as homologous proteins displaying cyclic pentameric symmetry (pentraxins). Proc Natl Acad Sci USA. (1977) 74:739–43. doi: 10.1073/pnas.74.2.739
27. Pepys MB. Amyloidosis. Annu Rev Med. (2006) 57:223–41. doi: 10.1146/annurev.med.57.121304.131243
28. Pepys MB, Dash AC, Munn EA, Feinstein A, Skinner M, Cohen AS, et al. Isolation of amyloid p component (protein ap) from normal serum as a calcium-dependent binding protein. Lancet (1977) 309:1029–31. doi: 10.1016/S0140-6736(77)91260-0
29. Painter RH, De Escallon I, Massey A, Pinteric L, Stern SB. The structure and binding characteristics of serum amyloid protein (9.5S alpha 1-glycoprotein). Ann N Y Acad Sci. (1982) 389:199–215. doi: 10.1111/j.1749-6632.1982.tb22138.x
30. Botto M, Hawkins PN, Bickerstaff MC, Herbert J, Bygrave AE, McBride A, et al. Amyloid deposition is delayed in mice with targeted deletion of the serum amyloid P component gene. Nat Med. (1997) 3:855–9. doi: 10.1038/nm0897-855
31. Pepys MB, Herbert J, Hutchinson WL, Tennent GA, Lachmann HJ, Gallimore JR, et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature (2002) 417:254–9. doi: 10.1038/417254a
32. Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature (2010) 468:93–7. doi: 10.1038/nature09494
33. Richards DB, Cookson LM, Berges AC, Barton SV, Lane T, Ritter JM, et al. Therapeutic clearance of amyloid by antibodies to serum amyloid P component. N Engl J Med. (2015) 373:1106–14. doi: 10.1056/NEJMoa1504942
34. Nuvolone M, Merlini G. Systemic amyloidosis: novel therapies and role of biomarkers. Nephrol Dialysis Trans. (2017) 32:770–80. doi: 10.1093/ndt/gfw305
35. Richards DB, Cookson LM, Barton SV, Liefaard L, Lane T, Hutt DF, et al. Repeat doses of antibody to serum amyloid P component clear amyloid deposits in patients with systemic amyloidosis. Sci Transl Med. (2018) 10:eaan3128. doi: 10.1126/scitranslmed.aan3128
36. Eming SA, Wynn TA, Martin P. Inflammation and metabolism in tissue repair and regeneration. Science (2017) 356:1026–30. doi: 10.1126/science.aam7928
37. Eming SA, Martin P, Tomic-Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Trans Med. (2014) 6:265sr266. doi: 10.1126/scitranslmed.3009337
38. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. (2012) 18:1028–40. doi: 10.1038/nm.2807
39. Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host responses in tissue repair and fibrosis. Ann Rev Pathol. (2013) 8:241–76. doi: 10.1146/annurev-pathol-020712-163930
40. Assayag P, Carre F, Chevalier B, Delcayre C, Mansier P, Swynghedauw B. Compensated cardiac hypertrophy: arrhythmogenicity and the new myocardial phenotype. I Fibr Cardiovas Res. (1997) 34:439–44. doi: 10.1016/S0008-6363(97)00073-4
41. Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol. (1998) 32:1454–9. doi: 10.1016/S0735-1097(98)00407-0
42. Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. (2014) 124:2333–40. doi: 10.1172/JCI72271
43. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Res Crit Care Med. (2011) 183:788–824. doi: 10.1164/rccm.2009-040GL
44. Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Eng J Med. (2018) 378:1811–23. doi: 10.1056/NEJMra1705751
45. Paget J. Lectures on surgical pathology: delivered at the Royal College of Surgeons of England. Philadelphia: Lindsay & Blakiston (1854).
46. Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. (1994) 1:71–81.
47. Chesney J, Bacher M, Bender A, Bucala R. The peripheral blood fibrocyte is a potent antigen-presenting cell capable of priming naive T cells in situ. Proc Natl Acad Sci USA. (1997) 94:6307–12. doi: 10.1073/pnas.94.12.6307
48. Chesney J, Metz C, Stavitsky AB, Bacher M, Bucala R. Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J Immunol. (1998) 160:419–25.
49. Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. (2001) 166:7556–62. doi: 10.4049/jimmunol.166.12.7556
50. Yang L, Scott PG, Giuffre J, Shankowsky HA, Ghahary A, Tredget EE. Peripheral blood fibrocytes from burn patients: identification and quantification of fibrocytes in adherent cells cultured from peripheral blood mononuclear cells. Lab Invest. (2002) 82:1183–92. doi: 10.1097/01.LAB.0000027841.50269.61
51. Mori L, Bellini A, Stacey MA, Schmidt M, Mattoli S. Fibrocytes contribute to the myofibroblast population in wounded skin and originate from the bone marrow. Exp Cell Res. (2005) 304:81–90. doi: 10.1016/j.yexcr.2004.11.011
52. Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol. (2003) 171:380–9. doi: 10.4049/jimmunol.171.1.380
53. Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R. Circulating fibrocytes: collagen-secreting cells of the peripheral blood. Int J Biochem Cell Biol. (2004) 36:598–606. doi: 10.1016/j.biocel.2003.10.005
54. Yang L, Scott PG, Dodd C, Medina A, Jiao H, Shankowsky HA, et al. Identification of fibrocytes in postburn hypertrophic scar. Wound Repair Regener. (2005) 13:398–404. doi: 10.1111/j.1067-1927.2005.130407.x
55. Mehrad B, Burdick MD, Zisman DA, Keane MP, Belperio JA, Strieter RM. Circulating peripheral blood fibrocytes in human fibrotic interstitial lung disease. Biochem Biophys Res Commun. (2007) 353:104–8. doi: 10.1016/j.bbrc.2006.11.149
56. Andersson-Sjöland A, de Alba CG, Nihlberg K, Becerril C, Ramírez R, Pardo A, et al. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol. (2008) 40:2129–40. doi: 10.1016/j.biocel.2008.02.012
57. Reilkoff RA, Bucala R, Herzog EL. Fibrocytes: emerging effector cells in chronic inflammation. Nat Rev Immunol. (2011) 11:427–35. doi: 10.1038/nri2990
58. Just SA, Lindegaard H, Hejbøl EK, Davidsen JR, Bjerring N, Hansen SWK, et al. Fibrocyte measurement in peripheral blood correlates with number of cultured mature fibrocytes in vitro and is a potential biomarker for interstitial lung disease in Rheumatoid Arthritis. Respirat Res. (2017) 18:141. doi: 10.1186/s12931-017-0623-9
59. Aiba S, Tagami H. Inverse correlation between CD34 expression and proline-4-hydroxylase immunoreactivity on spindle cells noted in hypertrophic scars and keloids. J Cutan Pathol. (1997) 24:65–9. doi: 10.1111/j.1600-0560.1997.tb01098.x
60. Shin JU, Kim SH, Kim H, Noh JY, Jin S, Park CO, et al. TSLP is a potential initiator of collagen synthesis and an activator of CXCR4/SDF-1 axis in keloid pathogenesis. J Invest Dermatol. (2016) 136:507–15. doi: 10.1016/j.jid.2015.11.008
61. Isgr òM, Bianchetti L, Marini MA, Mattoli S. Involvement of fibrocytes in allergen-induced T cell responses and rhinovirus infections in asthma. Biochem Biophys Res Commun. (2013) 437:446–51. doi: 10.1016/j.bbrc.2013.06.099
62. Mattoli S. Involvement of fibrocytes in asthma and clinical implications. Clin Exp Allergy (2015) 45:1497–509. doi: 10.1111/cea.12525
63. Niedermeier M, Reich B, Rodriguez Gomez M, Denzel A, Schmidbauer K, Gobel N, et al. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci USA. (2009) 106:17892–7. doi: 10.1073/pnas.0906070106
64. Sakai N, Furuichi K, Shinozaki Y, Yamauchi H, Toyama T, Kitajima S, et al. Fibrocytes are involved in the pathogenesis of human chronic kidney disease. Hum Pathol. (2010) 41:672–8. doi: 10.1016/j.humpath.2009.10.008
65. Wada T, Sakai N, Sakai Y, Matsushima K, Kaneko S, Furuichi K. Involvement of bone-marrow-derived cells in kidney fibrosis. Clin Exp Nephrol. (2011) 15:8–13. doi: 10.1007/s10157-010-0372-2
66. Cowper SE. Nephrogenic fibrosing dermopathy: the first 6 years. Curr Opin Rheumatol. (2003) 15:785–90. doi: 10.1097/00002281-200311000-00017
67. Direkze NC, Forbes SJ, Brittan M, Hunt T, Jeffery R, Preston SL, et al. Multiple organ engraftment by bone-marrow-derived myofibroblasts and fibroblasts in bone-marrow-transplanted mice. Stem Cells (2003) 21:514–20. doi: 10.1634/stemcells.21-5-514
68. Epperly MW, Guo H, Gretton JE, Greenberger JS. Bone marrow origin of myofibroblasts in irradiation pulmonary fibrosis. Am J Res Cell Mol Biol. (2003) 29:213–24. doi: 10.1165/rcmb.2002-0069OC
69. Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest. (2004) 113:243–52. doi: 10.1172/JCI200418847
70. Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. (2004) 114:438–46. doi: 10.1172/JCI200420997
71. Kisseleva T, Uchinami H, Feirt N, Quintana-Bustamante O, Segovia JC, Schwabe RF, et al. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. (2006) 45:429–38. doi: 10.1016/j.jhep.2006.04.014
72. Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB. The Role of CCL12 in the Recruitment of Fibrocytes and Lung Fibrosis. Am J Respirat Cell Mol Biol. (2006) 35:175–81. doi: 10.1165/rcmb.2005-0239OC
73. Sakai N, Wada T, Yokoyama H, Lipp M, Ueha S, Matsushima K, et al. Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc Natl Acad Sci. (2006) 103:14098–103. doi: 10.1073/pnas.0511200103
74. Varcoe RL, Mikhail M, Guiffre AK, Pennings G, Vicaretti M, Hawthorne WJ, et al. The role of the fibrocyte in intimal hyperplasia. J Thromb Haemost. (2006) 4:1125–33. doi: 10.1111/j.1538-7836.2006.01924.x
75. Santhiago MR, Singh V, Barbosa FL, Agrawal V, Wilson SE. Monocyte development inhibitor PRM-151 decreases corneal myofibroblast generation in rabbits. Exp Eye Res. (2011) 93:810–7. doi: 10.1016/j.exer.2011.08.015
76. Sakai N, Wada T, Matsushima K, Bucala R, Iwai M, Horiuchi M, et al. The renin-angiotensin system contributes to renal fibrosis through regulation of fibrocytes. J Hypert. (2008) 26:780–90. doi: 10.1097/HJH.0b013e3282f3e9e6
77. Hartlapp I, Abe R, Saeed RW, Peng T, Voelter W, Bucala R, et al. Fibrocytes induce an angiogenic phenotype in cultured endothelial cells and promote angiogenesis in vivo. FASEB J. (2001) 15:2215–24. doi: 10.1096/fj.01-0049com
78. Wang JF, Jiao H, Stewart TL, Shankowsky HA, Scott PG, Tredget EE. Fibrocytes from burn patients regulate the activities of fibroblasts. Wound Repair Regen. (2007) 15:113–21. doi: 10.1111/j.1524-475X.2006.00192.x
79. Yuen IS, Jain R, Bishop JD, Lindsey DF, Deery WJ, Van Haastert PJ, et al. A density-sensing factor regulates signal transduction in Dictyostelium. J Cell Biol. (1995) 129:1251–62. doi: 10.1083/jcb.129.5.1251
80. Brock DA, Gomer RH. A cell-counting factor regulating structure size in Dictyostelium. Genes Dev. (1999) 13:1960–9. doi: 10.1101/gad.13.15.1960
81. Pilling D, Buckley CD, Salmon M, Gomer RH. Inhibition of fibrocyte differentiation by serum amyloid P. J Immunol. (2003) 17:5537–46. doi: 10.4049/jimmunol.171.10.5537
82. Shao DD, Suresh R, Vakil V, Gomer RH, Pilling D. Pivotal Advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. J Leukocyte Biol. (2008) 83:1323–33. doi: 10.1189/jlb.1107782
83. Pilling D, and Gomer RH. Regulatory pathways for fibrocyte differentiation. In: Bucala, R, editor. Fibrocytes-New Insights into Tissue Repair and Systemic Fibroses (2007). p. 37–60. doi: 10.1142/9789812706690_0003
84. Pilling D, Roife D, Wang M, Ronkainen SD, Crawford JR, Travis EL, et al. Reduction of bleomycin-induced pulmonary fibrosis by serum amyloid P. J Immunol. (2007) 179:4035–44. doi: 10.4049/jimmunol.179.6.4035
85. Crawford JR, Pilling D, Gomer RH. Improved serum-free culture conditions for spleen-derived murine fibrocytes. J Immunol Methods (2010) 363:9–20. doi: 10.1016/j.jim.2010.09.025
86. Crawford JR, Pilling D, Gomer RH. FcγRI mediates serum amyloid P inhibition of fibrocyte differentiation. J Leukocyte Biol. (2012) 92:699–711. doi: 10.1189/jlb.0112033
87. Pilling D, Cox N, Vakil V, Verbeek JS, Gomer RH. The long pentraxin PTX3 promotes fibrocyte differentiation. PLoS ONE (2015) 10:e0119709. doi: 10.1371/journal.pone.0119709
88. Bottazzi B, Doni A, Garlanda C, Mantovani A. An integrated view of humoral innate immunity: pentraxins as a paradigm. Ann Rev Immunol. (2010) 28:157–83. doi: 10.1146/annurev-immunol-030409-101305
89. Devaraj S, Jialal I. C-reactive protein polarizes human macrophages to an M1 phenotype and inhibits transformation to the M2 phenotype. Arterioscler Thromb Vas Biol. (2011) 31:1397–402. doi: 10.1161/ATVBAHA.111.225508
90. Zhang W, Xu W, Xiong S. Macrophage differentiation and polarization via phosphatidylinositol 3-Kinase/Akt–ERK signaling pathway conferred by serum amyloid P component. J Immunol. (2011a) 187:1764–77. doi: 10.4049/jimmunol.1002315
91. Zhao XQ, Zhang MW, Wang F, Zhao YX, Li JJ, Wang XP, et al. CRP enhances soluble LOX-1 release from macrophages by activating TNF-α converting enzyme. J Lipid Res. (2011) 52:923–33. doi: 10.1194/jlr.M015156
92. Lu J, Marjon KD, Mold C, Du Clos TW, Sun PD. Pentraxins and Fc receptors. Immunol Rev. (2012) 250:230–8. doi: 10.1111/j.1600-065X.2012.01162.x
93. Cox N, Pilling D, Gomer RH. Serum amyloid P: a systemic regulator of the innate immune response. J Leukocyte Biol. (2014) 96:739–43. doi: 10.1189/jlb.1MR0114-068R
94. Pegues MA, McWilliams IL, Szalai AJ. C-reactive protein exacerbates renal ischemia-reperfusion injury: are myeloid derived suppressor cells to blame? Am J Physiol Renal Physiol. (2016) 311:F176–81. doi: 10.1152/ajprenal.00107.2016
95. Pilling D, Galvis-Carvajal E, Karhadkar TR, Cox N, Gomer RH. Monocyte differentiation and macrophage priming are regulated differentially by pentraxins and their ligands. BMC Immunol. (2017) 18:30. doi: 10.1186/s12865-017-0214-z
96. Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci USA. (2006) 103:18284–9. doi: 10.1073/pnas.0608799103
97. Haudek SB, Trial J, Xia Y, Gupta D, Pilling D, Entman ML. Fc receptor engagement mediates differentiation of cardiac fibroblast precursor cells. Proc Natl Acad Sci USA. (2008) 105:10179–84. doi: 10.1073/pnas.0804910105
98. Castano AP, Lin SL, Surowy T, Nowlin BT, Turlapati SA, Patel T, et al. Serum amyloid P inhibits fibrosis through Fc gamma R-dependent monocyte-macrophage regulation in vivo. Sci Trans Med. (2009) 1:5ra13. doi: 10.1126/scitranslmed.3000111
99. Pilling D, Fan T, Huang D, Kaul B, Gomer RH. Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PLoS ONE (2009) 4:e7475. doi: 10.1371/journal.pone.0007475
100. Moreira AP, Cavassani KA, Hullinger R, Rosada RS, Fong DJ, Murray L, et al. Serum amyloid P attenuates M2 macrophage activation and protects against fungal spore-induced allergic airway disease. J Allergy Clin Immunol. (2010) 126:712–721 e717. doi: 10.1016/j.jaci.2010.06.010
101. Murray L, Kramer M, Hesson D, Watkins B, Fey E, Argentieri R, et al. Serum amyloid P ameliorates radiation-induced oral mucositis and fibrosis. Fibrogen Tissue Repair (2010) 3:11. doi: 10.1186/1755-1536-3-11
102. Murray LA, Rosada R, Moreira AP, Joshi A, Kramer MS, Hesson DP, et al. Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS ONE (2010) 5:e9683. doi: 10.1371/journal.pone.0009683
103. Murray LA, Chen Q, Kramer MS, Hesson DP, Argentieri RL, Peng X, et al. TGF-beta driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int J Biochem Cell Biol. (2011) 43:154–62. doi: 10.1016/j.biocel.2010.10.013
104. Nakagawa N, Barron L, Gomez IG, Johnson BG, Roach AM, Kameoka S, et al. Pentraxin-2 suppresses c-Jun/AP-1 signaling to inhibit progressive fibrotic disease. JCI Insight. (2016) 1:e87446. doi: 10.1172/jci.insight.87446
105. Chen W, Pilling D, Gomer RH. C-reactive protein (CRP) but not the related pentraxins serum amyloid P and PTX3 inhibits the proliferation and induces apoptosis of the leukemia cell line Mono Mac 6. BMC Immunol. (2017) 18:47. doi: 10.1186/s12865-017-0230-z
106. Sorensen IJ, Nielsen EH, Andersen O, Danielsen B, Svehag SE. Binding of complement proteins C1q and C4bp to serum amyloid P component (SAP) in solid contra liquid phase. Scand J Immunol. (1996) 44:401–7. doi: 10.1046/j.1365-3083.1996.d01-326.x
107. Bottazzi B, Vouret-Craviari V, Bastone A, De Gioia L, Matteucci C, Peri G, et al. Multimer formation and ligand recognition by the long pentraxin PTX3. Similarities and differences with the short pentraxins C-reactive protein and serum amyloid P component. J Biol Chem. (1997) 272:32817–23. doi: 10.1074/jbc.272.52.32817
108. Ma YJ, Doni A, Skjoedt MO, Honore C, Arendrup M, Mantovani A, et al. Heterocomplexes of mannose-binding lectin and the pentraxins PTX3 or serum amyloid P component trigger cross-activation of the complement system. J Biol Chem. (2011) 286:3405–17. doi: 10.1074/jbc.M110.190637
109. Gomer RH, Pilling D, Kauvar L, Ellsworth S, Pissani S, Real L, et al. A Serum Amyloid P-binding hydrogel speeds healing of partial thickness wounds in pigs. Wound Repair Regen. (2009) 17:397–404. doi: 10.1111/j.1524-475X.2009.00482.x
110. Sen JW, Recke C, Rahbek L, Skogstrand K, Heegaard NH. Structural, quantitative and functional comparison of amyloid P component in sera from patients with systemic lupus erythematosus and healthy donors. Scand J Immunol. (2002) 56:645–51. doi: 10.1046/j.1365-3083.2002.01178.x
111. Zhang YM, Fang YD, Wang YC, Wang SL, Lei ZY, Liu XW, et al. Role of serum amyloid P in skin graft survival and wound healing in burned patients receiving skin grafts. Clin Chim Acta (2011) 412:227–9. doi: 10.1016/j.cca.2010.09.036
112. Moore BB, Lawson WE, Oury TD, Sisson TH, Raghavendran K, Hogaboam CM. Animal models of fibrotic lung disease. Am J Respirat Cell Mol Biol. (2013) 49:167–79. doi: 10.1165/rcmb.2013-0094TR
113. Tashiro J, Rubio GA, Limper AH, Williams K, Elliot SJ, Ninou I, et al. Exploring animal models that resemble idiopathic pulmonary fibrosis. Front Med. (2017) 4:118. doi: 10.3389/fmed.2017.00118
114. Ji Z, Ke ZJ, Geng JG. SAP suppresses the development of experimental autoimmune encephalomyelitis in C57BL/6 mice. Immunol Cell Biol. (2011) 90:388–95. doi: 10.1038/icb.2011.51
115. Naik-Mathuria B, Pilling D, Crawford JR, Gay AN, Smith CW, Gomer RH, et al. Serum amyloid P inhibits dermal wound healing. Wound Repair Regen. (2008) 16:266–73. doi: 10.1111/j.1524-475X.2008.00366.x
116. Pilling D, Gomer RH. Persistent lung inflammation and fibrosis in serum amyloid P component (Apcs -/-) knockout mice. PLoS ONE (2014) 9:e93730. doi: 10.1371/journal.pone.0093730
117. Verstovsek S, Manshouri T, Pilling D, Bueso-Ramos CE, Newberry KJ, Prijic S, et al. Role of neoplastic monocyte-derived fibrocytes in primary myelofibrosis. J Exp Med. (2016) 213:1723–40. doi: 10.1084/jem.20160283
118. Pepys MB. Serum amyloid P component (not serum amyloid protein). Nat Med. (1999) 5:852–3. doi: 10.1038/11272
119. Pontet M, D'Asnieres M, Gache D, Escaig J, Engler R. A new pentraxin (serum amyloid P-component) in the rat: evidence for two quaternary structures and effect of ligands on self-association. Biochim Biophys Acta (1981) 671:202–10. doi: 10.1016/0005-2795(81)90135-5
120. Duffield JS, Lupher ML Jr. PRM-151 (recombinant human serum amyloid P/pentraxin 2) for the treatment of fibrosis. Drug News Perspect. (2010) 23:305–15. doi: 10.1358/dnp.2010.23.5.1444206
121. Raghu G, van den Blink B, Hamblin MJ, Brown AW, Golden JA, Ho LA, et al. Effect of recombinant human pentraxin 2 vs placebo on change in forced vital capacity in patients with idiopathic pulmonary fibrosis: a randomized clinical trial. JAMA (2018) 319:2299–307. doi: 10.1001/jama.2018.6129
122. Barbui T, Thiele J, Gisslinger H, Kvasnicka HM, Vannucchi AM, Guglielmelli P, et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. (2018) 8:15. doi: 10.1038/s41408-018-0054-y
123. Verstovsek S, Mesa RA, Foltz LM, Gupta V, Mascarenhas JO, Ritchie EK, et al. PRM-151 in myelofibrosis: durable efficacy and safety at 72 Weeks. Blood (2015) 126:56.
124. Hawkins PN, Wootton R, Pepys MB. Metabolic studies of radioiodinated serum amyloid P component in normal subjects and patients with systemic amyloidosis. J Clin Invest. (1990) 86:1862–9. doi: 10.1172/JCI114917
125. Mortensen RF, Osmand AP, Lint TF, Gewurz H. Interaction of C-reactive protein with lymphocytes and monocytes: complement-dependent adherence and phagocytosis. J Immunol. (1976) 117:774–81.
126. Singh PP, Gervais F, Skamene E, Mortensen RF. Serum amyloid P-component-induced enhancement of macrophage listericidal activity. InfectImmun (1986) 52:688–94.
127. de Haas CJ, van Leeuwen EM, van Bommel T, Verhoef J, van Kessel KP, van Strijp JA. Serum amyloid P component bound to gram-negative bacteria prevents lipopolysaccharide-mediated classical pathway complement activation. InfectImmun (2000) 68:1753–9. doi: 10.1128/IAI.68.4.1753-1759.2000
128. Noursadeghi M, Bickerstaff MC, Gallimore JR, Herbert J, Cohen J, Pepys MB. Role of serum amyloid P component in bacterial infection: protection of the host or protection of the pathogen. Proc Natl Acad Sci USA. (2000) 97:14584–9. doi: 10.1073/pnas.97.26.14584
129. Diniz SN, Nomizo R, Cisalpino PS, Teixeira MM, Brown GD, Mantovani A, et al. PTX3 function as an opsonin for the dectin-1-dependent internalization of zymosan by macrophages. J Leukocyte Biol. (2004) 75:649–56. doi: 10.1189/jlb.0803371
130. Hawkins PN, Myers MJ, Lavender JP, Pepys MB. Diagnostic radionuclide imaging of amyloid: biological targeting by circulating human serum amyloid P component. Lancet (1988) 1:1413–8. doi: 10.1016/S0140-6736(88)92235-0
131. Hawkins PN, Aprile C, Capri G, Vigano L, Munzone E, Gianni L, et al. Scintigraphic imaging and turnover studies with iodine-131 labelled serum amyloid P component in systemic amyloidosis. Eur J Nucl Med. (1998) 25:701–8. doi: 10.1007/s002590050272
132. Naylor MC, Lazar DA, Zamora IJ, Mushin OP, Yu L, Brissett AE, et al. Increased in vitro differentiation of fibrocytes from keloid patients is inhibited by serum amyloid P. Wound Repair Regen. (2012) 20:277–83. doi: 10.1111/j.1524-475X.2012.00782.x
133. Wang J, Jiao H, Stewart TL, Shankowsky HA, Scott PG, Tredget EE. Improvement in postburn hypertrophic scar after treatment with IFN-alpha2b is associated with decreased fibrocytes. J Interferon Cytokine Res. (2007) 27:921–30. doi: 10.1089/jir.2007.0008
134. Maharjan AS, Pilling D, Gomer RH. Toll-like receptor 2 agonists inhibit human fibrocyte differentiation. Fibrogenesis Tissue Repair (2010) 3:23. doi: 10.1186/1755-1536-3-23
135. Gan Y, Reilkoff R, Peng X, Russell T, Chen Q, Mathai SK, et al. Role of semaphorin 7a signaling in transforming growth factor β1–induced lung fibrosis and scleroderma-related interstitial lung disease. Arthrit Rheumat. (2011) 63:2484–94. doi: 10.1002/art.30386
136. Maharjan AS, Pilling D, Gomer RH. High and low molecular weight hyaluronic acid differentially regulate human fibrocyte differentiation. PLoS ONE (2011) 6:e26078. doi: 10.1371/journal.pone.0026078
137. Cox N, Pilling D, Gomer RH. NaCl potentiates human fibrocyte differentiation. PLoS ONE (2012) 7:e45674. doi: 10.1371/journal.pone.0045674
138. Pilling D, Zheng Z, Vakil V, Gomer RH. Fibroblasts secrete Slit2 to inhibit fibrocyte differentiation and fibrosis. Proc Natl Acad Sci USA. (2014) 111:18291–6. doi: 10.1073/pnas.1417426112
139. Herlihy SE, Starke HE, Lopez-Anton M, Cox N, Keyhanian K, Fraser DJ, et al. Peritoneal dialysis fluid and some of its components potentiate fibrocyte differentiation. Perit Dial Int. (2016) 36:367–73. doi: 10.3747/pdi.2014.00284
140. Karhadkar TR, Pilling D, Cox N, Gomer RH. Sialidase inhibitors attenuate pulmonary fibrosis in a mouse model. Sci Rep. (2017) 7:15069. doi: 10.1038/s41598-017-15198-8
141. Zhong JN, Lan L, Chen YF, Huang G, He GZ, Yang J, et al. IL-4 and serum amyloid P inversely regulate fibrocyte differentiation by targeting store-operated Ca2+ channels. Pharmacol Rep. (2018) 70:22–8. doi: 10.1016/j.pharep.2017.07.005
142. Pilling D, Vakil V, Cox N, Gomer RH. TNF-α-stimulated fibroblasts secrete lumican to promote fibrocyte differentiation. Proc Natl Acad Sci USA. (2015) 112:11929–34. doi: 10.1073/pnas.1507387112
143. Chen W, Pilling D, Gomer RH. Dietary NaCl affects bleomycin-induced lung fibrosis in mice. Exp Lung Res. (2017) 43:395–406. doi: 10.1080/01902148.2017.1385110
144. Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Ann Rev Pathol. (2014) 9:157–79. doi: 10.1146/annurev-pathol-012513-104706
145. Kim KK, Sheppard D, Chapman HA. TGF-beta1 Signaling and Tissue Fibrosis. Cold Spring Harb Perspect Biol. (2017) 10:a022293. doi: 10.1101/cshperspect.a022293
146. Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J Exp Med. (2004) 200:377–89. doi: 10.1084/jem.20040104
147. White MJV, Glenn M, Gomer RH. Trypsin potentiates human fibrocyte differentiation. PLoS ONE (2013) 8:e70795. doi: 10.1371/journal.pone.0070795
148. White MJ, Roife D, Gomer RH. Galectin-3 binding protein secreted by breast cancer cells inhibits monocyte-derived fibrocyte differentiation. J Immunol. (2015) 195:1858–67. doi: 10.4049/jimmunol.1500365
149. White MJ, Gomer RH. Trypsin, tryptase, and thrombin polarize macrophages towards a pro-fibrotic M2a phenotype. PLoS ONE (2015) 10:e0138748. doi: 10.1371/journal.pone.0138748
150. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
151. Daeron M. Fc Receptor Biology. Ann Rev Immunol. (1997) 15:203–34. doi: 10.1146/annurev.immunol.15.1.203
152. Mortensen RF, Zhong W. Regulation of phagocytic leukocyte activities by C-reactive protein. J Leukoc Biol. (2000) 67:495–500. doi: 10.1002/jlb.67.4.495
153. Mold C, Gresham HD, Du Clos TW. Serum amyloid P component and C-reactive protein mediate phagocytosis through murine Fc gamma Rs. J Immunol. (2001) 166:1200–5. doi: 10.4049/jimmunol.166.2.1200
154. Bodman-Smith KB, Gregory RE, Harrison PT, Raynes JG. FcgammaRIIa expression with FcgammaRI results in C-reactive protein- and IgG-mediated phagocytosis. J Leukocyte Biol. (2004) 75:1029–35. doi: 10.1189/jlb.0703306
155. Moalli F, Doni A, Deban L, Zelante T, Zagarella S, Bottazzi B, et al. Role of complement and Fc{gamma} receptors in the protective activity of the long pentraxin PTX3 against Aspergillus fumigatus. Blood (2010) 116:5170–80. doi: 10.1182/blood-2009-12-258376
156. Cox N, Pilling D, Gomer RH. Distinct Fcγ receptors mediate the effect of serum amyloid p on neutrophil adhesion and fibrocyte differentiation. J Immunol. (2014) 193:1701–8. doi: 10.4049/jimmunol.1400281
157. Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, Sun PD. Structural recognition and functional activation of FcgR by innate pentraxins. Nature (2008) 456:989–92. doi: 10.1038/nature07468
158. Lu J, Marjon KD, Mold C, Marnell L, Du Clos TW, Sun P. Pentraxins and IgA share a binding hot-spot on FcαRI. Prot Sci. (2014) 23:378–86. doi: 10.1002/pro.2419
159. Pilling D, Tucker NM, Gomer RH. Aggregated IgG inhibits the differentiation of human fibrocytes. J Leukocyte Biol. (2006) 79:1242–51. doi: 10.1189/jlb.0805456
160. Cox N, Pilling D, Gomer RH. DC-SIGN activation mediates the differential effects of SAP and CRP on the innate immune system and inhibits fibrosis in mice Proc Natl Acad Sci USA. (2015) 112:8385–90. doi: 10.1073/pnas.1500956112
161. Nourshargh S, Hordijk PL, Sixt M. Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol. (2010) 11:366–78. doi: 10.1038/nrm2889
162. Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. (2011) 17:1381–90. doi: 10.1038/nm.2514
163. Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. (2011) 17:293–307. doi: 10.2119/molmed.2010.00138
164. Galkina EV, Nazarov PG, Polevschikov AV, Berestovaya LK, Galkin VE, Bychkova NV. Interactions of C-reactive protein and serum amyloid P component with interleukin-8 and their role in regulation of neutrophil functions. Russ J Immunol. (2000) 5:363–74.
165. Stibenz D, Grafe M, Debus N, Hasbach M, Bahr I, Graf K, et al. Binding of human serum amyloid P component to L-selectin. Eur J Immunol. (2006) 36:446–56. doi: 10.1002/eji.200425360
166. Maharjan AS, Roife D, Brazill D, Gomer RH. Serum amyloid P inhibits granulocyte adhesion. Fibrogenesis Tissue Repair (2013) 6:2. doi: 10.1186/1755-1536-6-2
167. Wang Y, Guo Y, Wang X, Huang J, Shang J, Sun S. Human serum amyloid P functions as a negative regulator of the innate and adaptive immune responses to DNA vaccines. The J Immunol. (2011) 186:2860–70. doi: 10.4049/jimmunol.1003641
168. Sato T, Kameyama T, Noto T, Ueno H, Inoue H. Enhanced expression of hemoglobin scavenger receptor CD163 in accumulated macrophages within filtered debris between acute coronary syndromes and stable angina pectoris. Int Heart J. (2015) 56:150–6. doi: 10.1536/ihj.14-224
169. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
Keywords: pentraxin, serum amyloid P component (SAP), fibrosis, macrophage, fibrocyte, pulmonary fbrosis
Citation: Pilling D and Gomer RH (2018) The Development of Serum Amyloid P as a Possible Therapeutic. Front. Immunol. 9:2328. doi: 10.3389/fimmu.2018.02328
Received: 27 July 2018; Accepted: 19 September 2018;
Published: 16 October 2018.
Edited by:
Barbara Bottazzi, Humanitas Clinical and Research Center, ItalyReviewed by:
Amrita Dosanjh, Pediatric Pulmonologist, San Diego, CA, United StatesAlberto Mantovani, Humanitas Research Hospital, Italy
Copyright © 2018 Pilling and Gomer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Darrell Pilling, ZHBpbGxpbmdAYmlvLnRhbXUuZWR1
Richard H. Gomer, cmdvbWVyQHRhbXUuZWR1