 Jingyun Luan
Jingyun Luan Dianwen Ju
Dianwen Ju
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 25 September 2018
Sec. Inflammation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02201
This article is part of the Research Topic Patho- and Physiological Roles of Inflammasomes View all 7 articles
Inflammasomes have emerged as critical innate sensors of host immune that defense against pathogen infection, metabolism syndrome, cellular stress and cancer metastasis in the liver. The assembly of inflammasome activates caspase-1, which promotes the maturation of interleukin-1β (IL-1β) and interleukin-18 (IL-18), and initiates pyroptotic cell death (pyroptosis). IL-18 exerts pleiotropic effects on hepatic NK cells, priming FasL-mediated cytotoxicity, and interferon-γ (IFN-γ)-dependent responses to prevent the development of liver diseases. However, considerable attention has been attracted to the pathogenic role of inflammasomes in various acute and chronic liver diseases, including viral hepatitis, nanoparticle-induced liver injury, alcoholic and non-alcoholic steatohepatitis. In this review, we summarize the latest advances on the physiological and pathological roles of inflammasomes for further development of inflammasome-based therapeutic strategies for human liver diseases.
Innate immune system is well-known as the first line defense against pathogen associated molecular patterns (PAMPs) derived from microbial pathogens (e.g., bacteria, parasites, viruses) and damage associated molecular patterns (DAMPs) produced by host cells (e.g., cellular stress, cytosolic DNA, damage) (1, 2). Inflammasomes are critical innate immune sensors involved in maintaining the cellular health in response to cytosolic pathogens or stress signals (3). Inflammasomes are cytoplasmic multiprotein complexes typically composed of three components: (i) a sensor molecule consisting of NOD-like receptors (NLRs), absent in melanoma 2 (AIM2) or pyrin, (ii) an adaptor protein, and (iii) an effector molecule procaspase-1 (4). Upon stimulation, inflammasome complexes are assembled to process the cleavage of caspase-1, which activates proinflammatory cytokines interleukin-1β (IL-1β) and interleukin-18 (IL-18), and a cytosolic protein gasdermin D (GSDMD) (5). Cleaved GSDMD forms pores on the plasma membrane, which induces pyroptotic cell death and permits the release of IL-1β and IL-18 into the extracellular space (6, 7). These two proinflammatory cytokines and pyroptosis exert both beneficial and deleterious effects in the liver, which will be discussed in this review.
The liver is a “first past” organ that continually challenged with diverse microbial particles derived from intestine through the portal circulation. During this process, a large number of cytosolic pathogens can be sensed by inflammasomes, which are pivotal in evoking adaptive immunity for complete clearance (8–10). In addition, hepatocytes are susceptible to the infection of various liver-tropic viruses that leads to the pathogenesis of virus-related liver diseases. The emerging importance of inflammasomes in response to viral infection points to another area for the involvement of inflammasomes (11).
Paradoxically, inflammasomes are essential for liver defense against pathogens and danger signals, but excessive activation of inflammasomes promotes the pathogenesis of various liver diseases (Table 1). Thus, a definitive understanding of the roles of inflammasomes in the liver is essential for the development of inflammasome-based therapies.
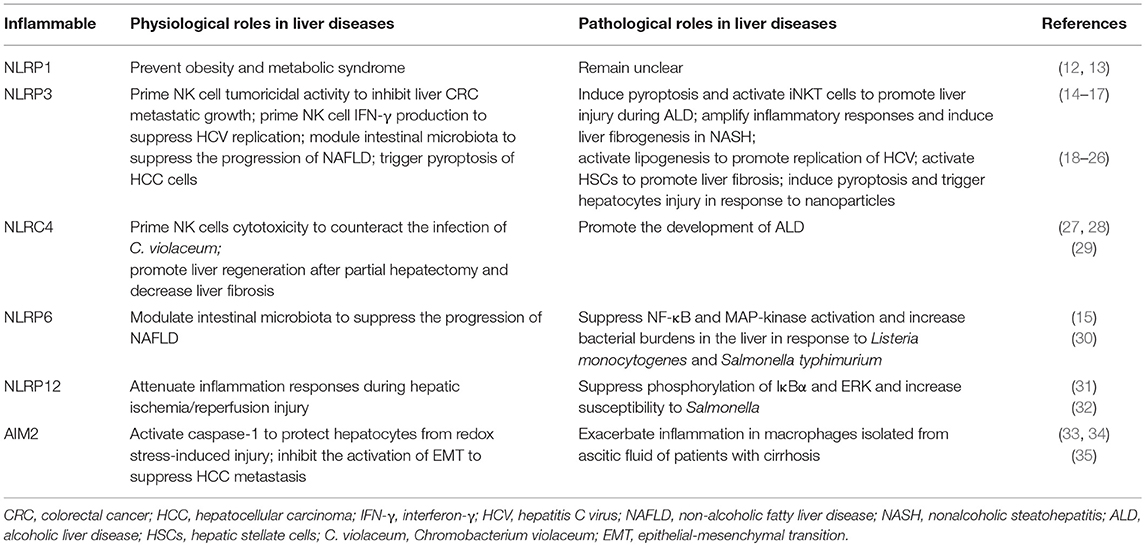
Table 1. Roles of inflammasomes in the liver diseases.
Continual exposure to orally-ingested antigens and intestinally-released microbial products sensitizes the liver to various bacterial and parasitic infections. Inflammasomes assembled by NLRs are known as innate immune sensors that detect cytosolic contaminations or perturbations (36, 37). NLRC4 is demonstrated to defense against bacterial infections through detecting bacterial flagellin or two bacterial type III secretion systems (T3SSs) (38–42). Studies on the liver-tropic pathogen Chromobacterium violaceum have shown that NLRC4-deficient mice are more susceptible to infection (27). Activated NLRC4 inflammasome processes the production of IL-18, which primes NK cell cytotoxicity to clear hepatocyte replication niches (27). Additionally, exogenous administration of IL-18 is protective against the infection of C. violaceum and another liver-tropic bacteria Listeria monocytogenes (27). Although IL-18 therapy does not fully reduce bacterial burdens to the normal level, considerable potential still exists in activating NLRC4-IL-18 pathway as a combination therapy to counteract bacteria with T3SSs or flagellin.
In addition to participating in liver defense against bacterial pathogens, inflammasomes are also identified to control parasitic infections. Canonical inflammasomes process the cleavage of caspase-1, while noncanonical inflammasomes are implicated in the activation of caspase-11 (43, 44). Loss of all inflammasome signalings by knocking out caspase-1/11 leads to higher hepatic parasitic load and lower survival of mice in response to Trypanosoma cruzi (T. cruzi) (45, 46). The activation of caspase-1 is highly dependent on lysosomal cathepsin B, as pharmacological inhibition of cathepsin B significantly reduces the production of IL-1β during T. cruzi infection (45). Furthermore, the lack of IL-1β in caspase-1/11-deficient mice is accompanied by downregulated hepatic interleukin-17+CD8+ and interferon-γ (IFN-γ)+CD8+ T cells, implying that inflammasome-mediated IL-1β is involved in promoting liver adaptive immunity to control this parasitic infection (46). This finding is in line with previous studies showing the importance of IL-1β for the differentiation of Th17 cells and antigen-driven T cells (47–49). Thus, the interplay between inflammasomes and adaptive immune system in the defense against pathogen infection is an interesting aspect for future investigations.
Viral infection typically initiates a cascade of innate immune responses, which restrict viral spread and provoke adaptive immunity for complete removal of virus. As innate immune sensors, inflammasomes have a prominent role in defense against hepatitis virus infections (14, 50, 51). Mice deficient in caspase-1/11 are more susceptible to mouse hepatitis virus (MHV) infection, suggesting that inflammasomes as a whole are protective (51). Further explorations have identified that inflammasome-dependent cytokine, IL-18, is required for host defense against MHV, as IL-18 receptor (IL-18R) deficiency reduces the production of IFN-γ by activated T cells, causing elevated viral replication and poor survival of MHV-infected mice (51). Despite evidence for the involvement of IL-18 in MHV inhibition, many unanswered questions remain, including which inflammasome mediates the maturation of IL-18 and how this inflammasome is activated during MHV infection.
Beyond T cells, IL-18 is also involved in promoting the production of IFN-γ by NK cells (14). Studies performed on an in vitro model of hepatitis C virus (HCV) replication have shown that monocytes can detect HCV-infected hepatocytes and respond by secreting IL-18 in an NLRP3 inflammasome-dependent manner, which subsequently stimulates NK cell-derived IFN-γ, causing suppression on HCV (14). This finding is supported by the observation of a higher expression of IL-18R on NK cells than other cell populations (27). However, this protective effect seems to be specific for monocytes, because macrophage-derived IL-1β amplifies inflammatory responses during HCV infection (52). Thus, special consideration is required for inflammasome-related inflammatory responses when applying inflammasome-based therapies for hepatitis virus infection.
Oxidative stress induced by excessive reactive oxygen species (ROS) has emerged as a hallmark of liver injury (53). During this process, nuclear DNA and mitochondrial DNA (mtDNA) are released into the cytosol which act as DAMPs to activate innate immunity (54). Recent progress has been made in identifying that inflammasomes have a broader role in protecting hepatocytes from oxidative stress-induced injury by responding to mtDNA (55, 56). For example, activation of caspase-1 increases resistance to oxidative stress-induced liver inflammation and damage during hemorrhagic shock with resuscitation (HS/R) (55). Follow-up studies have shown that activation of caspase-1 is mediated by AIM2 inflammasome, as AIM2 deficiency reduces the production of caspase-1 and aggravates hepatocellular cell death (33). Considering that AIM2 inflammasome is an intracellular receptor that recognizes dysfunctional DNA, further investigations have demonstrated that AIM2 interacts with the immunogenic DNA sensor, high mobility group box 1 (HMGB1), to facilitate hepatoprotective effects (33). Another interesting finding is that AIM2 inflammasome-mediated caspase-1 upregulates the expression of beclin-1, which initiates autophagy to clear damaged mitochondria in hepatocytes, thus reducing the generation of ROS and degrading damaged mtDNA (33, 55). Together, these findings suggest that AIM2 inflammasome is on the crossroad of innate immune sensor and beneficial autophagy to protect against oxidative stress-induced liver injury. In addition, the emerging importance of hepatocytes (nonimmune cell type) in regulation of the immune response is another important area for further investigations.
One additional organ-specific feature of liver that makes it sensitive to inflammasome is that liver serves as the second largest storehouse of lipid next to adipose tissues (57). Emerging evidences have indicated that inflammasomes are critical regulators in suppressing hepatic lipid deposition (58, 59). Mice deficient in NLRP1 spontaneously develop hepatic steatosis, and the syndrome is aggravated on high-fat diet (HFD), while NLRP1MUT (an activating mutation in NLRP1a) mice are devoid of lipid vacuoles in the liver (12). The anti-obesity ability of NLRP1 inflammasome appears to be dependent on IL-18, as knockout of IL-18 reverses its protective effects (12, 13). In addition, exogenous administration of IL-18 counteracts steatohepatitis in mice upon HFD, further highlighting the importance of NLRP1-IL-18 signaling in controlling metabolic syndromes (12). From the mechanistic perspective, these studies also raise an interesting question about what is the trigger that leads to the activation of NLRP1 inflammasome in metabolic liver diseases, which needs to be more clearly elucidated.
Another two inflammasomes, NLRP3 and NLRP6, have also been reported to negatively regulate the progression of non-alcoholic fatty liver disease (NAFLD) (15). NLRP3- or NLRP6-deficient mice develop exacerbated hepatic steatosis with a microbiome dysbiosis on either HFD or methionine-choline-deficient diet (MCDD) (15). Interestingly, the microbiome dysbiosis in NLRP3−/− or NLRP6−/− mice can be transferred to co-housed wide type (WT) mice and is strongly correlated with the severity of hepatic disorders (15). A possible explanation is that exposure to HFD or MCDD alters the gut microbiota composition and function, which increase the translocation of bacterial products into the liver, thus aggravating hepatic steatosis and inflammation, termed “gut-liver axis” (60–62). NLRP6 inflammasome is recognized as a potent modulator for maintaining gut homeostasis (63). One group of microbial-derived metabolites triggers the activation of NLRP6-caspase-1 axis and subsequently leads to the proteolytic processing of IL-18 (63). IL-18 not only elicits anti-microbial peptides synthesis to control the composition of the gut microbiota, but also upregulates interleukin-22 signaling to promote wound healing (63, 64). The uncovered interactions between inflammasomes and hepatic lipid metabolism provide novel therapeutic targets for the treatment of hepatic steatosis.
NLRP3 inflammasome has been recognized to be important for tumor control by directly activating pyroptotic cell death or secreting death-inducing cytokines (65). Evidence of NLRP3 inflammasome in suppressing hepatic tumor growth comes from the study showing that colorectal cancer (CRC) metastatic liver tumor burden is exacerbated in NLRP3-deficient mice (16). The tumor-suppressive effect of NLRP3 inflammasome on liver CRC metastasis is highly dependent on IL-18, which promotes the maturation of hepatic NK cells and primes FasL-mediated cytotoxicity (16, 17). This study provides insight into the innate immunity circulation between CRC-induced activation of NLRP3 inflammasome in kupffer cell- and NK cell-mediated cytotoxicity. In addition to priming tumoricidal activity of hepatic NK cell, NLRP3 inflammasome also induces caspase-1-mediated pyroptosis to control the proliferation of hepatocellular carcinoma (HCC) cells (66). Downregulated expression of NLRP3 inflammasome in HCC tissues correlates with the aggravation of carcinoma, while reconstitution of NLRP3 inflammasome dramatically reverses the malignant phenotype of HCC (67). These findings highlight the significance of NLRP3 inflammasome in preventing hepatic tumor growth, but it also exerts pro-carcinogenic effects for gastric and prostate cancers, indicting the protective role of NLRP3 inflammasome in cancer development may be organ or cell specific (65).
ALD is triggered by excessive alcohol consumption that can progress from fatty liver to severe cirrhosis, liver failure and HCC (68). The involvement of NLRP3 inflammasome in ALD has been demonstrated by a robust expression of NLRP3, caspase-1 and IL-1β in alcohol-fed mice, while liver inflammation and steatosis are dramatically attenuated in NLRP3−/− or caspase-1−/− mice (18). The proinflammatory cytokine IL-1β leads to the recruitment of invariant natural killer T (iNKT) cells, which promotes the influx of neutrophils for exacerbated hepatitis (19).
Beyond regulating IL-1β activation, NLRP3 inflammasome also facilitates the occurrence of pyroptosis (20). It has been confirmed that the pyroptosis determinant protein GSDMD is activated in the liver of mice suffering ALD (69). Adenoviral expression of cleaved GSDMD in hepatocytes aggravates the severity of liver inflammation and damage. Interestingly, one type of miRNA, miR-148a, has recently been demonstrated to suppress pyroptosis in ALD (70). The hepatocyte specific expression of miR-148a by lentivirus delivery directly inhibits the interaction between thioredoxin-interacting protein and NLRP3, leading to attenuated pyroptosis and decreased incidence of ALD (70). These reports provide evidence that NLRP3 inflammasome is pathogenic in the development of ALD, however, the trigger that initiates the activation of NLRP3 remains to be determined.
NASH is a progressive type of NAFLD with chronic hepatic damage and inflammation (71). The hepatocellular damage is associated with toxic effects induced by accumulated lipids, such as saturated fatty acid (ceramide and palmitate) and cholesterol crystals (72). Emerging evidence has accumulated that NLRP3 inflammasome is activated by these toxic lipids as a pathogenic mechanism for the development of NASH in murine models (21, 73–76). Increased levels of NLRP3, GSDMD and IL-1β are observed in the liver of patients with NASH (77). Beyond amplifying inflammatory responses, activation of NLRP3 inflammasome also promotes liver fibrogenesis during NASH, as blockade of NLRP3 improves NASH pathology by simultaneously suppressing liver inflammation and fibrosis (78). The latest research has demonstrated the importance of GSDMD-mediated pyroptosis in the process of NASH, as GSDMD−/− mice develop remarkably attenuated steatohepatitis compared to WT mice, further confirming the detrimental role of NLRP3 inflammasome signaling pathway in NASH (77).
One potential negative regulator of NLRP3 inflammasome during NASH is autophagy (79). Defective autophagy causes the accumulation of dysfunctional mitochondria and increased production of ROS, which is required for the activation of NLRP3 inflammasome (80, 81). In contrast, induction of autophagy by ezetimibe dampens NLRP3 inflammasome activity and ameliorates hepatic lipid accumulation and inflammation on MCD (82). All these findings imply the beneficial role of autophagy in NASH via the suppression of NLRP3 inflammasome.
On the contrary to the above studies suggesting that NLRP3 promotes NASH, an experimental research has shown that NLRP3 deficiency leads to increased bacteremia and aggravated NASH (15). These diverse functions of NLRP3 have been explained by the evidence of different activities of NLRP3 inflammasome in different organs during NASH (83). In the liver, expression of NLRP3 inflammasome is upregulated and responsible for the pathogenesis of NASH, but downregulated in the gut that protects against alteration of intestinal bacteria (83, 84). These observations suggest that liver-specific blockade of NLRP3 inflammasome is necessary to afford improvement in liver inflammation and steatosis but devoid of gut microbial dysbiosis.
Hepatitis viruses preferentially infect hepatocytes and cause liver inflammation with high mortality, among which approximately 90% are attributable to chronic infection induced by hepatitis B virus (HBV) and HCV (85). Several studies suggest that NLRP3 inflammasome is the central player in the pathophysiology of viral hepatitis (22, 86, 87). The hepatic expression of NLRP3, caspase-1 and IL-1β are significantly higher in patients with active untreated chronic HBV than those in chronic remission (86). In addition, a strong correlation is demonstrated between levels of IL-1β and severity of liver inflammation in HBV patients, implying that NLRP3-mediated IL-1β is the potential driving force of HBV-induced viral hepatitis.
NLRP3 inflammasome is also identified in HCV infection by the observation of an upregulated expression of NLRP3 signaling pathway in monocytes and macrophages with HCV (87). During HCV infection, NLRP3 inflammasome can serve as either a beneficial or a detrimental role. On one hand, kupffer cell has been identified as the primary cell source of IL-1β in chronic HCV patients, and the production of IL-1β by kupffer cell is associated with amplified inflammatory responses (52). In addition, NLRP3 inflammasome stimulates lipid droplet formation in hepatocytes, which promotes the morphogenesis and replication of HCV, thus contributing to the pathogenesis of liver diseases (22). On the other hand, NLRP3-mediated activation of IL-18 in monocytes stimulates the production of IFN-γ to prime resistance to HCV infection (14). These observations suggest that NLRP3 inflammasome derived from macrophages, hepatocytes and monocytes exerts different functions, which requires in-depth investigations for the potential link of NLRP3 inflammasome activation between these cells in HCV infection.
The optimal physicochemical properties of nanoparticles make them widely used for disease diagnosis, imaging, and treatment, but their clinical applications are greatly hampered by fulminant hepatitis and liver injury (88). Tremendous efforts are being made to clarify the underling mechanisms, and recent studies have identified that NLRP3-mediated pyroptosis serves as a critical pathogenic factor in the liver injury induced by multiple nanoparticles, including rare-earth oxide (REO), quantum dots and mesoporous silica (23–26). For example, REO treatment activates NLRP3 inflammasome, leading to the occurrence of pyroptosis and secretion of IL-1β in kupffer cells, both of which are suppressed by GSDMD knockdown (24). Coating REO with a small peptide RE-1 inhibits NLRP3 activation via reducing ROS generation and calcium influx, thus attenuating REO-elicited inflammation, further confirming the pathogenic role of NLRP3 in REO-induced toxicity (23). Detailed insights into the mechanism have shown that the sustained activation of NLRP3 in response to REO is mediated by lysosomal damage and massive release of cathepsin B (24). Collectively, these studies implicate the involvement of NLRP3 inflammasome-mediated pyroptosis in nanoparticle-induced liver injury, which may provide novel strategies for controlling nanoparticles-mediated adverse effects.
Liver fibrosis is a pathogenic result of chronic liver diseases, such as ASH and NASH, and characterized by deposition of extracellular matrix (ECM) (89). Hepatic stellate cells (HSCs) are the primary cells for the storage of ECM, and multiple functional changes of HSCs can be caused by NLRP3 inflammasome, including suppression of chemotaxis, upregulation of collagen and transforming growth factor-β (90, 91). These functions are confirmed by the study that knocking in NLRP3 induces the activation of HSCs and subsequent accumulation of ECM proteins, while fibrogenesis is not reversed by IL-1Ra therapy, indicating that some other regulators of NLRP3 inflammasome pathway instead of IL-1β promote fibrogenesis (20).
In addition to the direct effect on innate immunity, NLRP3 inflammasome has an essential role in shaping adaptive immune responses in liver fibrosis. In particular, IL-1β promotes the differentiation of Th17 cells to secret interleukin-17 (IL-17), which is a critical proinflammatory cytokine in amplifying inflammation responses and perpetuating liver fibrosis driven by NLRP3 inflammasome activation (92). Collectively, NLRP3 inflammasome pathway appears to be central to the pathogenesis of liver fibrosis, and this uncovered link may open avenues for novel therapeutics for liver fibrosis.
In summary, inflammasomes are central components of innate immune system that protect liver from pathogen infection, metabolism syndrome, oxidative stress and tumor growth, however, excessive immune responses mediated by inflammasomes may promote the pathogenesis of various liver diseases. The dual functions of inflammasomes pose a challenge in designing inflammasome-based therapies. Therefore, it is important to better understand the precise mechanisms underlying the activation of inflammasomes. To date, the pathological role of NLRP3 inflammasome in liver diseases have been extensively studied (Figure 1). Future investigations need to elucidate the hepatic importance of other inflammasomes, which may hopefully provide new targets for the treatment of liver diseases.
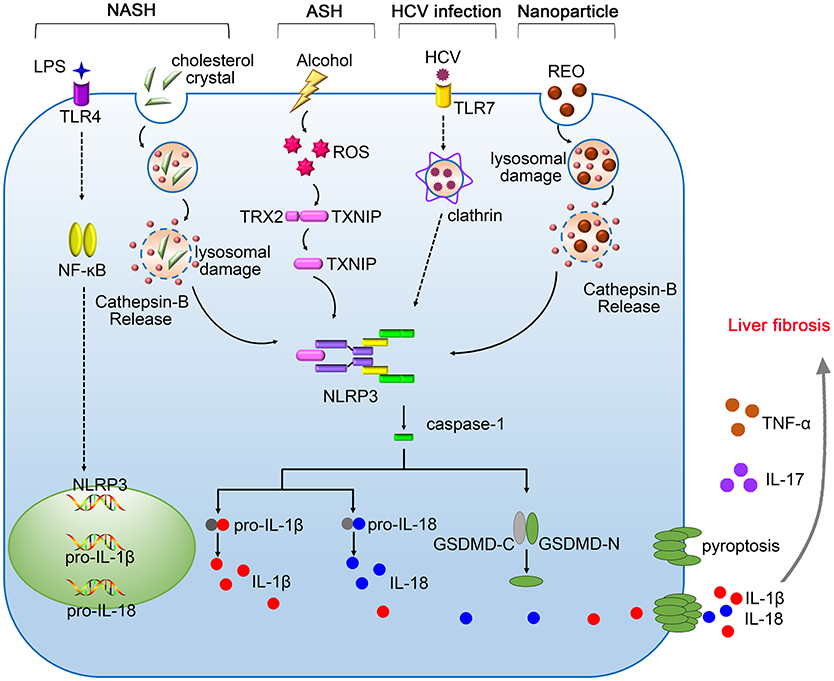
Figure 1. The pathogenic roles of NLRP3 inflammasome in liver diseases. Gut-derived PAMPs, such as lipopolysaccharide (LPS), activate nuclear factor kappa B (NF-κB) signaling pathway, promoting the expression of pro-IL-1β, and pro-IL-18. The NLRP3 inflammasome in the liver is activated by serious danger signals, such as cholesterol crystals, ethanol, and REO nanoparticles. Excessive alcohol consumption stimulates the generation of ROS, which facilitates the cleavage of TXNIP and contributes to assembly of NLRP3 inflammasome. The activation of NLRP3 inflammasome in response to HCV infection requires the recognition by Toll-like receptor-7 (TLR-7) and clathrin-mediated endocytosis. Pyroptosis features GSDMD pores on the membrane, allowing the release of IL-1β and IL-18 into the extracellular space. NLRP3 inflammasome cooperates with TNF-α and IL-17 contributing to the pathogenesis of liver fibrosis.
JL and DJ conceived the review. JL wrote the manuscript. DJ revised the manuscript. All authors have read and approved the final version of the manuscript.
This work is funded by National Natural Science Foundation of China (No. 81573332, No.81773620), National Key Basic Research Program of China (2015CB931800), China Postdoctoral Science Foundation (No. 2017M611462, 2018T110352).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. (2010) 10:826–37. doi: 10.1038/nri2873
2. Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. (2011) 32:157–64. doi: 10.1016/j.it.2011.01.005
3. Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. (2012) 28:137–61. doi: 10.1146/annurev-cellbio-101011-155745
4. Sharma D, Kanneganti TD. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol. (2016) 213:617–29. doi: 10.1083/jcb.201602089
5. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. (2017) 277:61–75. doi: 10.1111/imr.12534
6. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
7. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity (2018) 48:35–44. doi: 10.1016/j.immuni.2017.11.013
8. Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology (2012) 143:1158–72. doi: 10.1053/j.gastro.2012.09.008
9. Ayres JS. Inflammasome-microbiota interplay in host physiologies. Cell Host Microbe (2013) 14:491–7. doi: 10.1016/j.chom.2013.10.013
10. Próchnicki T, Latz E. Inflammasomes on the crossroads of innate immune recognition and metabolic control. Cell Metab. (2017) 26:71–93. doi: 10.1016/j.cmet.2017.06.018
11. Chen IY, Ichinohe T. Response of host inflammasomes to viral infection. Trends Microbiol. (2015) 23:55–63. doi: 10.1016/j.tim.2014.09.007
12. Murphy AJ, Kraakman MJ, Kammoun HL, Dragoljevic D, Lee MK, Lawlor KE, et al. IL-18 production from the NLRP1 inflammasome prevents obesity and metabolic syndrome. Cell Metab. (2016) 23:155–64. doi: 10.1016/j.cmet.2015.09.024
13. Netea MG, Joosten LA. The NLRP1-IL18 connection: a stab in the back of obesity-induced inflammation. Cell Metab. (2016) 23:6–7. doi: 10.1016/j.cmet.2015.12.014
14. Serti E, Werner JM, Chattergoon M, Cox AL, Lohmann V, Rehermann B, et al. Monocytes activate natural killer cells via inflammasome induced interleukin 18 in response to hepatitis C virus replication. Gastroenterology (2014) 147:209–20. doi: 10.1053/j.gastro.2014.03.046
15. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature (2012) 482:179–85. doi: 10.1038/nature10809
16. Dupaul-Chicoine J, Arabzadeh A, Dagenais M, Douglas T, Champagne C, Morizot A, et al. The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity (2015) 43:751–63. doi: 10.1016/j.immuni.2015.08.013
17. Dagenais M, Saleh M. Linking cancer-induced Nlrp3 inflammasome activation to efficient NK cell-mediated immunosurveillance. Oncoimmunology (2016) 5:e1129484. doi: 10.1080/2162402X.2015.1129484
18. Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. (2012) 122:3476–89. doi: 10.1172/JCI60777
19. Cui K, Yan G, Xu C, Chen Y, Wang J, Zhou R, et al. Invariant NKT cells promote alcohol-induced steatohepatitis through interleukin-1β in mice. J Hepatol. (2015) 62:1311–8. doi: 10.1016/j.jhep.2014.12.027
20. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology (2014) 59:898–910. doi: 10.1002/hep.26592
21. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
22. McRae S, Iqbal J, Sarkar-Dutta M, Lane S, Nagaraj A, Ali N, et al. The hepatitis C virus-induced NLRP3 inflammasome activates the sterol regulatory element-binding protein (SREBP) and regulates lipid metabolism. J Biol Chem. (2016) 291:3254–67. doi: 10.1074/jbc.M115.694059
23. Yao H, Zhang Y, Liu L, Xu Y, Liu X, Lin J, et al. Inhibition of lanthanide nanocrystal-induced inflammasome activation in macrophages by a surface coating peptide through abrogation of ROS production and TRPM2-mediated Ca(2+) influx. Biomaterials (2016) 108:143–56. doi: 10.1016/j.biomaterials.2016.08.036
24. Mirshafiee V, Sun B, Chang CH, Liao YP, Jiang W, Jiang J, et al. Toxicological profiling of metal oxide nanoparticles in liver context reveals pyroptosis in kupffer cells and macrophages versus apoptosis in hepatocytes. ACS Nano (2018) 12:3836–52. doi: 10.1021/acsnano.8b01086
25. Zhang X, Luan J, Chen W, Fan J, Nan Y, Wang Y, et al. Mesoporous silica nanoparticles induced hepatotoxicity via NLRP3 inflammasome activation and caspase-1-dependent pyroptosis. Nanoscale (2018) 10:9141–52. doi: 10.1039/c8nr00554k
26. Lu Y, Xu S, Chen H, He M, Deng Y, Cao Z, et al. CdSe/ZnS quantum dots induce hepatocyte pyroptosis and liver inflammation via NLRP3 inflammasome activation. Biomaterials (2016) 90:27–39. doi: 10.1016/j.biomaterials.2016.03.003
27. Maltez VI, Tubbs AL, Cook KD, Aachoui Y, Falcone EL, Holland SM. Inflammasomes coordinate pyroptosis and natural killer cell cytotoxicity to clear infection by a ubiquitous environmental bacterium. Immunity (2015) 43:987–97. doi: 10.1016/j.immuni.2015.10.010
28. DeSantis DA, Ko CW, Wang L, Lee P, Croniger CM. Constitutive activation of the Nlrc4 inflammasome prevents hepatic fibrosis and promotes hepatic regeneration after partial hepatectomy. Mediators Inflamm. (2015) 2015:909827. doi: 10.1155/2015/909827
29. DeSantis DA, Ko CW, Liu Y, Liu X, Hise AG, Nunez G, et al. Alcohol-induced liver injury is modulated by Nlrp3 and Nlrc4 inflammasomes in mice. Mediators Inflamm. (2013) 2013:751374. doi: 10.1155/2013/751374
30. Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature (2012) 488:389–93. doi: 10.1038/nature11250
31. Haga H, Yan IK, Borrelli DA, Matsuda A, Parasramka M, Shukla N, et al. Extracellular vesicles from bone marrow-derived mesenchymal stem cells protect against murine hepatic ischemia/reperfusion injury. Liver Transpl. (2017) 23:791–803. doi: 10.1002/lt.24770
32. Zaki MH, Man SM, Vogel P, Lamkanfi M, Kanneganti TD. Salmonella exploits NLRP12-dependent innate immune signaling to suppress host defenses during infection. Proc Natl Acad Sci USA. (2014) 111:385–90. doi: 10.1073/pnas.1317643111
33. Sun Q, Loughran P, Shapiro R, Shrivastava IH, Antoine DJ, Li T, et al. Redox-dependent regulation of hepatocyte absent in melanoma 2 inflammasome activation in sterile liver injury in mice. Hepatology (2017) 65:253–68. doi: 10.1002/hep.28893
34. Chen SL, Liu LL, Lu SX, Luo RZ, Wang CH, Wang H, et al. HBx-mediated decrease of AIM2 contributes to hepatocellular carcinoma metastasis. Mol Oncol. (2017) 11:1225–40. doi: 10.1002/1878-0261.12090
35. Lozano-Ruiz B, Bachiller V, García-Martínez I, Zapater P, Gómez-Hurtado I, Moratalla A, et al. Absent in melanoma 2 triggers a heightened inflammasome response in ascitic fluid macrophages of patients with cirrhosis. J Hepatol. (2015) 62:64–71. doi: 10.1016/j.jhep.2014.08.027
36. Ulland TK, Ferguson PJ, Sutterwala FS. Evasion of inflammasome activation by microbial pathogens. J Clin Invest. (2015) 125:469–77. doi: 10.1172/JCI75254
37. Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. (2012) 13:325–32. doi: 10.1038/ni.2231
38. Cerqueira DM, Pereira MS, Silva AL, Cunha LD, Zamboni DS. Caspase-1 but not caspase-11 is required for NLRC4-mediated pyroptosis and restriction of infection by flagellated legionella species in mouse macrophages and in vivo. J Immunol. (2015) 195:2303–11. doi: 10.4049/jimmunol.1501223
39. Sellin ME, Müller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe (2014) 16:237–48. doi: 10.1016/j.chom.2014.07.001
40. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477:596–600. doi: 10.1038/nature10510
41. Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci USA. (2013) 110:14408–13. doi: 10.1073/pnas.1306376110
42. Batista JH, da Silva Neto JF. Chromobacterium violaceum pathogenicity: updates and insights from genome sequencing of novel chromobacterium species. Front Microbiol. (2017) 8:2213. doi: 10.3389/fmicb.2017.02213
43. Lamkanfi M, Dixit V. Mechanisms and functions of inflammasomes. Cell (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
44. Yoon L, Sharad K. A single cut to pyroptosis. Oncotarget (2015) 6:36926–7. doi: 10.18632/oncotarget.6142
45. Goncalves VM, Matteucci KC, Buzzo CL, Miollo BH, Ferrante D, Torrecilhas AC, et al. NLRP3 controls Trypanosoma cruzi infection through a caspase-1-dependent IL-1R-independent NO production. PLoS Negl Trop Dis. (2013) 7:e2469. doi: 10.1371/journal.pntd.0002469
46. Paroli AF, Gonzalez PV, Díaz-Luján C, Onofrio LI, Arocena A, Cano RC, et al. NLRP3 Inflammasome and caspase-1/11 pathway orchestrate different outcomes in the host protection against Trypanosoma cruzi acute infection. Front Immunol. (2018) 9:913. doi: 10.3389/fimmu.2018.00913
47. Ben-Sasson SZ, Wang K, Cohen J, Paul WE. IL-1beta strikingly enhances antigen-driven CD4 and CD8 T-cell responses. Cold Spring Harb Symp Quant Biol. (2013) 78:117–24. doi: 10.1101/sqb.2013.78.021246
48. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. (2014) 14:585–600. doi: 10.1038/nri3707
49. Luan J, Zhang X, Wang S, Li Y, Fan J, Chen W, et al. NOD-Like receptor protein 3 inflammasome-dependent IL-1β accelerated ConA-induced hepatitis. Front Immunol. (2018) 9:758. doi: 10.3389/fimmu.2018.00758
50. Watashi K, Liang G, Iwamoto M, Marusawa H, Uchida N, Daito T, et al. Interleukin-1 and tumor necrosis factor-α trigger restriction of hepatitis B virus infection via a cytidine deaminase activation-induced cytidine deaminase (AID). J Biol Chem. (2013) 288:31715–27. doi: 10.1074/jbc.M113.501122
51. Zalinger ZB, Elliott R, Weiss SR. Role of the inflammasome-related cytokines IL-1 and IL-18 during infection with murine coronavirus. J Neurovirol. (2017) 23:845–54. doi: 10.1007/s13365-017-0574-4
52. Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, et al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. (2013) 9:e1003330. doi: 10.1371/journal.ppat.1003330
53. Gill R, Tsung A, Billiar T. Linking oxidative stress to inflammation: Toll-like receptors. Free Radic Biol Med. (2010) 48:1121–32. doi: 10.1016/j.freeradbiomed
54. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464:104–7. doi: 10.1038/nature08780
55. Sun Q, Gao W, Loughran P, Shapiro R, Fan J, Billiar TR, et al. Caspase 1 activation is protective against hepatocyte cell death by up-regulating beclin 1 protein and mitochondrial autophagy in the setting of redox stress. J Biol Chem. (2013) 288:15947–58. doi: 10.1074/jbc.M112.426791
56. Menzel CL, Sun Q, Loughran PA, Pape HC, Billiar TR Scott MJ, et al. Caspase-1 is hepatoprotective during trauma and hemorrhagic shock by reducing liver injury and inflammation. Mol Med. (2011) 17:1031–8. doi: 10.2119/molmed.2011.00015
57. Böttcher JP, Knolle PA, Stabenow D. Mechanisms balancing tolerance and immunity in the liver. Dig Dis. (2011) 29:384–90. doi: 10.1159/000329801
58. Febbraio MA. Role of interleukins in obesity: implications for metabolic disease. Trends Endocrinol Metab. (2014) 25:312–9. doi: 10.1016/j.tem.2014.02.004
59. Netea MG, Joosten LA, Lewis E, Jensen DR, Voshol PJ, Kullberg BJ, et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat Med. (2006) 12:650–6. doi: 10.1038/nm1415
60. Murphy EF, Cotter D, Healy S, Marques TM, O'Sullivan O, Fouhy F, et al. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut (2010) 59:1635–42. doi: 10.1136/gut.2010.215665
61. de Wit N, Derrien M, Bosch-Vermeulen H, Oosterink E, Keshtkar S, Duval C, et al. Saturated fat stimulates obesity and hepatic steatosis and affects gut microbiota composition by an enhanced overflow of dietary fat to the distal intestine. Am J Physiol Gastrointest Liver Physiol. (2012) 303:G589–99. doi: 10.1152/ajpgi.00488.2011
62. Anitha M, Reichardt F, Tabatabavakili S, Nezami BG, Chassaing B, Mwangi S, et al. Intestinal dysbiosis contributes to the delayed gastrointestinal transit in high-fat diet fed mice. Cell Mol Gastroenterol Hepatol. (2016) 2:328–39. doi: 10.1016/j.jcmgh.2015.12.008
63. Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell (2015) 163:1428–43. doi: 10.1016/j.cell.2015.10.048.
64. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature (2012) 491:259–63. doi: 10.1038/nature11535
65. Zhiyu W, Wang N, Wang Q, Peng C, Zhang J, Liu P, et al. The inflammasome: an emerging therapeutic oncotarget for cancer prevention. Oncotarget (2016) 7:50766–80. doi: 10.18632/oncotarget.9391
66. Wei Q, Mu K, Li T, Zhang Y, Yang Z, Jia X, et al. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Invest. (2014) 94:52–62. doi: 10.1038/labinvest.2013.126
67. Wei Q, Guo P, Mu K, Zhang Y, Zhao W, Huai W, et al. Estrogen suppresses hepatocellular carcinoma cells through ERβ-mediated upregulation of the NLRP3 inflammasome. Lab Invest. (2015) 95:804–16. doi: 10.1038/labinvest.2015.63
68. Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology (2011) 141:1572–85. doi: 10.1053/j.gastro.2011.09.002
69. Khanova E, Wu R, Wang W, Yan R, Chen Y, French SW, et al. Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology (2018) 67:1737–53. doi: 10.1002/hep.29645
70. Heo MJ, Kim TH, You JS, Blaya D, Sancho-Bru P, Kim SG. Alcohol dysregulates miR-148a in hepatocytes through FoxO1, facilitating pyroptosis via TXNIP overexpression. Gut (2018). doi: 10.1136/gutjnl-2017-315123. [Epub ahead of print].
71. Suzuki A, Diehl AM. Nonalcoholic Steatohepatitis. Annu Rev Med. (2017) 68:85–98. doi: 10.1146/annurev-med-051215-031109
72. Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol. (2018) 68:280–295. doi: 10.1016/j.jhep.2017.11.014
73. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. (2011) 17:179–88. doi: 10.1038/nm.2279
74. Camell CD, Nguyen KY, Jurczak MJ, Christian BE, Shulman GI, Shadel GS, et al. Macrophage-specific de novo synthesis of ceramide is dispensable for inflammasome-driven inflammation and insulin resistance in obesity. J Biol Chem. (2015) 290:29402–13. doi: 10.1074/jbc.M115.680199
75. Kanneganti TD, Dixit VD. Immunological complications of obesity. Nat Immunol. (2012) 13:707–12. doi: 10.1038/ni.2343
76. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology (2011) 54:133–44. doi: 10.1002/hep.24341
77. Xu B, Jiang M, Chu Y, Wang W, Chen D, Li X, et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J Hepatol. (2018) 68:773–82. doi: 10.1016/j.jhep.2017.11.040
78. Wree A, McGeough MD, Peña CA, Schlattjan M, Li H, Inzaugarat ME, et al. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med. (2014) 92:1069–82. doi: 10.1007/s00109-014-1170-1
79. Abdelaziz DH, Khalil H, Cormet-Boyaka E, Amer AO. The cooperation between the autophagy machinery and the inflammasome to implement an appropriate innate immune response: do they regulate each other? Immunol Rev. (2015) 265:194–204. doi: 10.1111/imr.12288
80. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. (2011) 12:408–15. doi: 10.1038/ni.2022
81. Wen H, Ting JP, O'Neill LA. A role for the NLRP3 inflammasome in metabolic diseases—did Warburg miss inflammation? Nat Immunol. (2012) 13:352–7. doi: 10.1038/ni.2228
82. Kim SH, Kim G, Han DH, Lee M, Kim I, Kim B, et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy (2017) 13:1767–81. doi: 10.1080/15548627.2017.1356977
83. De Minicis S, Rychlicki C, Agostinelli L, Saccomanno S, Candelaresi C, Trozzi L. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology (2014) 59:1738–49. doi: 10.1002/hep.26695
84. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity (2010) 32:379–91. doi: 10.1016/j.immuni.2010.03.003
85. Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet (2015) 386:743–800. doi: 10.1016/S0140-6736(15)60692-4
86. Molyvdas A, Georgopoulou U, Lazaridis N, Hytiroglou P, Dimitriadis A, Foka P, et al. The role of the NLRP3 inflammasome and the activation of IL-1β in the pathogenesis of chronic viral hepatic inflammation. Cytokine (2018) 110:389–96. doi: 10.1016/j.cyto.2018.04.032
87. Chattergoon MA, Latanich R, Quinn J, Winter ME, Buckheit RW, Blankson JN, et al. HIV and HCV activate the inflammasome in monocytes and macrophages via endosomal Toll-like receptors without induction of type 1 interferon. Plos Pathog. (2014) 10:e1004082. doi: 10.1371/journal.ppat.1004082
88. Wang J, Zhou G, Chen C, Yu H, Wang T, Ma Y, et al. Acute toxicity and biodistribution of different sized titanium dioxide particles in mice after oral administration. Toxicol Lett. (2007) 168:176–85 doi: 10.1016/j.toxlet.2006.12.001
90. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. (2013) 4:2823. doi: 10.1038/ncomms3823
91. Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, et al. Inflammasome-mediated regulation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. (2009) 296:G1248–57. doi: 10.1152/ajpgi.90223.2008
Keywords: inflammasome, liver, pyroptosis, innate immune, adaptive immune, metabolism
Citation: Luan J and Ju D (2018) Inflammasome: A Double-Edged Sword in Liver Diseases. Front. Immunol. 9:2201. doi: 10.3389/fimmu.2018.02201
Received: 30 June 2018; Accepted: 05 September 2018;
Published: 25 September 2018.
Edited by:
Rongbin Zhou, University of Science and Technology of China, ChinaReviewed by:
John R. Lukens, University of Virginia, United StatesCopyright © 2018 Luan and Ju. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dianwen Ju, ZGlhbndlbmp1QGZ1ZGFuLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.