
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 11 September 2018
Sec. Molecular Innate Immunity
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02072
 Mohamad N. Ali1,2
Mohamad N. Ali1,2 Gopinath Kasetty2
Gopinath Kasetty2 Malin Elvén1
Malin Elvén1 Saud Alyafei3
Saud Alyafei3 Sandra Jovic2
Sandra Jovic2 Arne Egesten2
Arne Egesten2 Heiko Herwald3
Heiko Herwald3 Artur Schmidtchen1,4,5
Artur Schmidtchen1,4,5 Praveen Papareddy1,3*
Praveen Papareddy1,3*Tissue factor pathway inhibitor-2 (TFPI-2) has previously been characterized as an endogenous anticoagulant. TFPI-2 is expressed in the vast majority of cells, mainly secreted into the extracellular matrix. Recently we reported that EDC34, a C-terminal peptide derived from TFPI-2, exerts a broad antimicrobial activity. In the present study, we describe a previously unknown antimicrobial mode of action for the human TFPI-2 C-terminal peptide EDC34, mediated via binding to immunoglobulins of the classes IgG, IgA, IgE, and IgM. In particular the interaction of EDC34 with the Fc part of IgG is of importance since this boosts interaction between the immunoglobulin and complement factor C1q. Moreover, we find that the binding increases the C1q engagement of the antigen-antibody interaction, leading to enhanced activation of the classical complement pathway during bacterial infection. In experimental murine models of infection and endotoxin challenge, we show that TFPI-2 is up-regulated in several organs, including the lung. Correspondingly, TFPI-2−/− mice are more susceptible to pulmonary Pseudomonas aeruginosa bacterial infection. No anti-coagulant role of TFPI-2 was observed in these models in vivo. Furthermore, in vivo, the mouse TFPI-2-derived C-terminal peptide VKG24, a homolog to human EDC34 is protective against systemic Escherichia coli bacterial infection. Moreover, in sputum from cystic fibrosis patients TFPI-2 C-terminal fragments are generated and found associated with immunoglobulins. Together our data describe a previously unknown host defense mechanism and therapeutic importance of TFPI-2 against invading Gram-negative bacterial pathogens.
The exploration of immuno-modulating proteins and peptides has attracted a significant interest within the past years (1, 2). To this end, techniques such as phage display (3, 4) and peptide array technologies (5) had been developed that allow the detection of potential interactions between these peptides and their potential interaction partners. Notably, the mammalian immune system is in the focus of such analyses as there are many reports showing that it can by modulated by a variety of exogenous peptides. During million years of evolution the mammalian immune system has evolved into a complex and self-regulating network comprised by many components that orchestrate with each other to ascertain maximum protection for the host (6). However, some microorganisms have succeeded to manipulate the host response by deceiving the immune defense system and crossing the firewall of protection. Though bacteria can very easily adapt to new hostile environments, there have been only be a few examples showing that they can also counteract the activity of antimicrobial peptides (AMPs). Thus, AMPs demonstrate a significant potential as novel therapeutic agents. Many researchers have therefore extensively investigated different classes of endogenous peptides derived for instance from mammalian neutrophils, i.e., cathelicidins and defensins (7). AMPs are found in all life forms including bacteria, fungi, plants, invertebrates, and vertebrates (8). They are essential components of the innate immune system (9), as they help to ensure the maximum protection for the host against pathogens and invading microbes (10, 11). In addition to their involvement in direct extermination of various microorganisms, AMPs can mediate diverse immunomodulatory responses (12, 13). These include stimulation of macrophages, lymphocytes and neutrophils (9), induction of angiogenesis (14), triggering in wound healing (15), and neutralizing lipopolysaccharide endotoxins derived from Gram-negative bacteria (16).
During the past decade there have been several findings on HDPs generated from proteins involved in coagulation including thrombin (17–19), HCAII (20), TFPI-1 (21) among others. There are also host derived peptides generated through proteolytic cleavage by bacterial proteases, as recently discovered in the case of Pseudomonas aeruginosa (22). P. aeruginosa infect by several additional strategies, targeting matrix remodeling, impressively proven by the TIMP-1−/− mice and their resistance to infection by this particular pathogen. Importantly, the protective role in the case of TIMP1−/− mice was accompanied by increased inflammatory and complement-dependent immune regulation (23). Structurally related, to an extent of approximately 50% on both nucleotide and amino acid level to TFPI-1, is it's sister protein TFPI-2 also called Matrix Serine Proteinase Inhibitor (MSPI) and Placental Protein 5 (PP5) (24–26). TFPI-2 exerts not only antimicrobial properties against Gram-negative infection (27)—the protein is also present in skin and the C-terminal region have shown to be up-regulated at the edges of both acute and chronic wounds (28). Importantly, TFPI-2 has previously been shown to inhibit several MMPs, both indirectly and through protein-protein interactions. The importance of the MMP inhibitory capabilities of TFPI-2 during dysfunctional matrix turnover as in arthritis, atherosclerosis and cancer are getting increased attention (29–31). The possibility to modify local concentrations of MMPs and their inhibitors has a large therapeutic potential in many immunopathologies, including those seen during infections (32).
Previous studies have shown that complement activation can be amplified by targeting certain proteins such as complement factor C3 (33). Elvington et al. for instance described in 2012 a method showing that complement activation can be used as cancer therapy. To this end the authors employed a fusion protein of a complement receptor and an IgG Fc fragment to improve the outcome of mAb therapy in a murine metastatic cancer model (33). In another study, we found that EDC34, a peptide derived from human TFPI-2, enhances the binding of C1q to bacterial surfaces. Moreover EDC34 is able to up regulate the antimicrobial activity of C3a, which in turn can cause amplification of the classical complement system pathway (27). These findings further reveal that EDC34 has a significant capacity to eradicate in particular Gram-negative bacteria in vivo (27, 28). Based on these results, we therefore decided to investigate the mode of action of C-terminal TFPI-2 region under infectious disease conditions. Our finding demonstrates that EDC34 is capable of binding to immunoglobulins (IgA, IgG, IgE, and IgM), by specifically engaging the Fc region of IgG. These finding were confirmed in in vivo infection studies using a peptide derived from the C-terminal part of murine TFPI-2 and were further underlined by employing TFPI-2−/− mice experiments. Together our results suggest an important role of the C-terminal TFPI-2 region in boosting the host defense against invading pathogens.
The peptides listed in Table 1 were synthesized by Biopeptide Co., San Diego, CA. The purity (>95%) of these peptides was confirmed by mass spectral analysis (MALDI-ToF Voyager). The bacterial isolate Escherichia coli ATCC 25922 was obtained from the American Type Culture Collection and Xen41 was obtained form PerkinElmer. Mouse VKG24 peptide was synthesized by Ontores (Shanghai, China).

Table 1. C-terminal human TFPI-2 derived peptides.
Escherichia coli was grown to mid-logarithmic phase in 10 ml (3% w/v) of trypticase soy broth (TSB). Bacteria were washed and re-suspended in 10 mM Tris, pH 7.4 to obtain a dilution of 1% of bacterial suspension (1–2 × 109 colony forming units cfu/ml). Subsequently, 6.6 μl of bacteria were added to 15 ml of melted underlay agarose gel consisting of 0.03% (w/v) TSB, 1% (w/v) low electroendosmosis type (EEO) agarose and 0.02% (v/v) Tween 20 (Sigma) with or without 150 mM NaCl. The mixture was poured to a 144 mm sterile petri dish and left to solidify. After that, 4 mm diameter wells were punctured and 100 μM peptides were added to each well. Plates were incubated for 3 h at 37°C and then covered by adding 15 ml of over-lay gel consisting of 6% TSB and 1% Low-EEO agarose in Millipore water. After 16–24 h of incubation at 37°C, the peptides diffusion around each well formed the zone of inhibition, which corresponded to the peptide's antimicrobial activity.
Escherichia coli bacteria were grown to mid-logarithmic phase in 10 ml of (3% w/v) Todd-Hewitt (TH), bacteria were washed and diluted in buffer 10 mM Tris, pH 7.4, containing 150 mM NaCl, either alone or with the presence of human 20% citrate plasma, lepirudin, heparin, EDTA plasma, or serum. 1–2 × 106 cfu/ml bacteria were incubated in a total volume of 50 μl with the indicated concentrations of C-terminal TFPI-2 derived peptides for 2 h at 37°C. Alternatively, 1–2 × 106 cfu/ml of E. coli bacteria in the presence or absence of EDC34 were incubated either in PBS alone or PBS supplemented with 25% of human citrated plasma, immunoglobulins depleted citrated plasma, or immunoglobulins depleted citrated plasma supplemented with 10 μg of IgG/μl, for 2 h at 37°C. Serial of dilutions of the reaction mixture were plated on TH agar plates, followed by the incubation at 37°C overnight. The number of colony forming units was determined as a percentage to control sample.
Three μg of IgG or IgA or IgE or IgM were incubated in 20 μl of buffer (10 mM Tris, 150 mM NaCl, pH 7.4), either alone or with 0.5 μg (6 μM) of EDC34 for 1 h at 37°C. Samples were diluted in SDS buffer and analyzed under non-reducing conditions by SDS-PAGE on Criterion TGX Any kD Gel (BioRad). Alternatively, EDC34 (6 μM) was incubated either alone or in PBS containing 20% serum, citrate, lepirudin, EDTA plasma at different time points (15, 30, 60, and 120 min). Samples were analyzed under non-reducing conditions.
Six μM of EDC34 was either incubated alone or with IgGs in 10 mM Tris, 150 mM NaCl, pH 7.4 buffer, under similar concentrations for 15, 30, 60, 120 min or 24 h at 37°C. In another experiment, 6 μM of EDC34 was pre-incubated with 50 μM of TCEP (tris(2-carboxyethyl)phosphine) for 30 min at room temperature before IgG (6 μM) was added. The mixture was further incubated for 24 h at 37°C. In another experiment, 6 μM of IgG or Fab-IgG or Fc-IgG were incubated with or without 6 μM of EDC34 in 10 mM Tris, 150 mM NaCl, pH 7.4 buffer for 30 min at 37°C. Proteins and peptides were transferred to nitrocellulose membranes (Hybond-C), blocked by 5% (w/v) skimmed milk, washed and incubated with rabbit polyclonal antibodies to CAK27 or rabbit polyclonal antibodies to IgG (or 1:1,000) (Dako) or rabbit polyclonal antibodies to C1q (or 1:1,000) (Dako). Polyclonal swine anti-rabbit immunoglobulin secondary HRP antibody (1:1,000) was used for detection and an enhanced chemiluminescent Substrate (LumiGLO) developing system (Upstate cell signaling solutions).
In a total volume of 500 μl PBS buffer, 2 μM of EDC34, and 8 μM of IgG were incubated for 24 h at 37°C. Subsequently, E. coli bacteria (1–2 × 106 cfu/ml) were added, and then the mixture was incubated for additional 1 h. In a parallel experiment, the bacteria in the presence of absence of EDC34 were incubated for 30 min at 37°C before IgG was added for 1 h at 37°C. Samples were then centrifuged, bacterial pellets were washed twice with PBS and then re-suspended in 360 μl of 0.1 M glycine, pH 2.0 to elute the bound proteins. The pH of the eluted material was raised to 7.5 with 1 M Tris. Thereafter, 100 μl of 100% TCA were added to precipitate the eluted proteins followed by incubation for 10 min at −20° C followed by a centrifugation step for 20 min at 15,000 × g (4°C). The pellets were washed with acetone and left to air dry before they were dissolved in loading dye and analyzed under non-reducing conditions by SDS-PAGE, followed by Western blot analysis.
The experiments were performed according to the manufacture's instructions. Briefly, total albumins and (>98%) and immunoglobulins (IgG, IgA, IgM, IgE, and IgD >99%) were depleted in human citrated plasma using PureProteomeTM Human Albumin/Immunoglobulin Depletion Kit (Millipore).
IgG was immobilized (15 μg/mL) on a CM5 sensor chip via amine coupling in a 10 mM sodium acetate buffer (pH 4). A blank immobilization was performed in flow cell one. EDC34 was injected at different concentrations (25–400 nM) at a flow rate of 30 μl/min and a temperature of 25°C over the flow cells using a running buffer containing 10 mm Hepes, 150 mm NaCl, 0.005% surfactant P20, and 3.4 mM EDTA (pH 7.5). Regeneration of the sensor surface was conducted in 0.5 M sodium chloride. The analysis was performed using a Biacore X100 instrument (GE Healthcare, Uppsala, Sweden) and the data was fitted using a 1:1 binding model.
TFPI-2 conditional knockout mice were generated by Deltagen (San Mateo, CA, USA). Male chimeric mice were mated with C57BL/6J female mice, and F1 heterozygous mice TFPI-2+/−, were mated with each other to yield complete knockout mice TFPI-2−/− (Data sheet 1). All experiments were performed facilitating female and male mice at the age between of 6 and 8 weeks according to protocols approved by the Animal Ethics Committee, Lund, Sweden. Animals were housed under standard conditions of light and temperature and had free access to standard laboratory chow and water.
Escherichia coli ATCC 25922 bacteria were grown to reach mid-exponential phase (OD620~0.4), washed twice in PBS and then diluted in PBS. Balb/c mice (7 weeks, n = 6 per group) were intraperitoneally (i. p) injected with 100 μl (8–8.5 × 108 cfu/ml) of E. coli. One hour after bacterial injection, VKG24 (500 μg per mouse) or PBS buffer alone were injected (i. p). Mice were sacrificed after 8 h post-infection and cfu were evaluated in blood, lung, spleen, kidney and liver.
AIN-93M-Purified diet (ENVIGO) was provided for 1 week to mice, after 8 h post-infection mice were anesthetized and L-021 dye 25 mg per kg (Wako) was injected subcutaneously (s. c.), after 10–12 min of incubation. Reactive oxygen species (ROS) production was detected by measuring fluorescence in radiance (p/sec) for control and treated mice using IVIS® Spectrum (version 4.4, Caliper Life Sciences). Imaging was performed in groups of 2 and 3 animals at a time.
Escherichia coli ATCC 25922 bacteria were grown to mid-exponential phase (OD620~0.5), harvested, washed in PBS and diluted in the same buffer to 3–4 × 108 cfu/ml. Two hundred microliter of the bacterial suspension was injected intraperitoneally (i. p) into C57BL/6 mice. In another experiment, C57BL/6 mice were i. p. injected with 8 mg/kg of E. coli O111:B4 LPS (Sigma). In both cases, for evaluation of TFPI-2 expression, mice were sacrificed 0, 1, 2, 4, 6, and 12 h post-infection, and lung, brain, spleen, liver, kidney, small, and large intestine were harvested in trizol reagent. Before isolating RNA all organs were stored at −80°C.
Animals were housed under standard conditions of light and temperature, with access to chow and water ad libitum. Pseudomonas aeruginosa Xen41 bacteria were grown to logarithmic phase (OD620~0.5), harvested, washed in PBS, pH 7.4 and diluted in PBS to either 3–4 × 108 cfu/ml, and kept on ice until injection. Fifty microliter of the bacterial suspension was administered intra nasally into mice. The survival data were obtained by following the animals daily up to 7 days monitoring. Mice reaching the pre-defined endpoint criteria were sacrificed and counted as non-survivors. For bacterial dissemination, mice were sacrificed after 12 h and cfu were determined in BALF and lung.
The cytokines IL-6, IL-10, MCP-1, IFN-γ, TNF-α were measured in plasma and BAL fluid from mice intra-nasally infected with P. aeruginosa using the Cytometric bead array (CBA) Mouse Inflammation Kit # 552364 (Becton Dickinson AB) according to the manufacturer's instructions on a FACSCalibur flow cytometer (Becton Dickinson AB). All plasma samples were stored at −80°C before the analysis.
Values are shown as mean with SEM. For statistical evaluation of two experimental groups, one-way with Tukey's multiple comparisons post-test was used and for comparison of survival curves the Mantel-Cox's test. Viable count and radial diffusion assay data are presented as mean with SD. All statistical evaluations were performed using the GraphPad Prism software 7.0 with *p < 0.05, **p < 0.01, *** < 0.001, ****p < 0.0001, and ns, not significant.
Many factors including pH, salt concentrations, or interacting proteins, have been described to alter the antimicrobial activity of a number of AMPs (30). To test whether these parameters have an effect on EDC34, the antimicrobial activity of the peptide was analyzed under five different plasma conditions (including human serum, citrate plasma, lepirudin-, heparin-, and EDTA-plasma, respectively). As shown in Figure 1A, EDC34 is able to completely eradicate the bacteria in citrate plasma (CP), while its antimicrobial activity is decreased in lepirudin or EDTA plasma as well as in serum. When the experiment was performed with heparin plasma the bactericidal effect of EDC34 was significantly blocked resulting in the survival rates rate exceeding 80%. The latter findings were expected since also many other heparin-binding AMPs lose their antimicrobial activity in the presence of heparin (34). Peptide stability can be influenced by many different conditions including proteolytic degradation. To test whether EDC34 is resistant to proteolysis, the peptide was incubated with three different anticoagulant plasmas as well as with serum. Samples were collected after different time points and subjected to western blot analysis probing with an antibody against the C-terminal part of TFPI-2. When incubated with serum, EDC34 was completely degraded after an incubation time of 15 min. However, when the experiment was performed in citrate, lepirudin, or EDTA-plasma, we noticed that EDC34 immediately formed a complex with a protein with an apparent molecular weight of about 180 kDa (Figure 1B). In this complex EDC34 seemed to be more stable as it started to degrade in EDTA plasma after 60 min and remained intact for more than 2 h in citrate and lepirudin plasma.
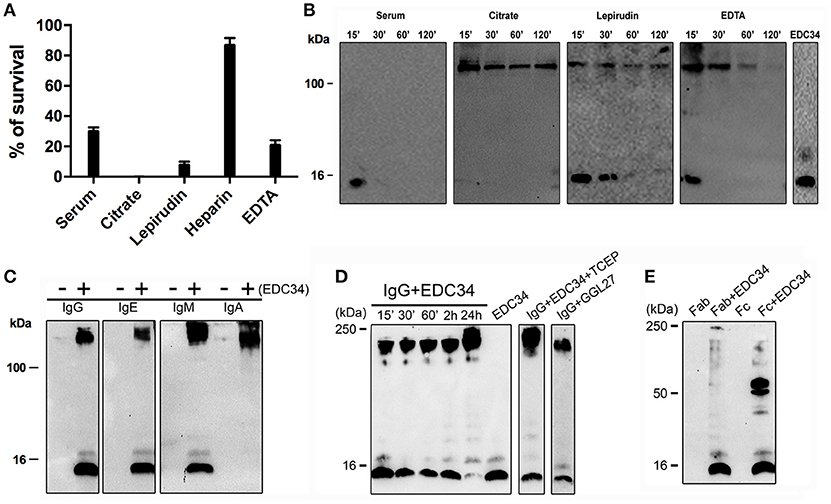
Figure 1. EDC34 binds to immunoglobulins. (A) The antimicrobial activity of EDC34 (6 μM) was tested against E. coli bacteria in 10 mM Tris, 150 mM NaCl, pH 7.4 buffer, containing 20% of serum or citrate or lepirudin or heparin or EDTA plasma. Mean with SD are shown (n = 3). (B) 6 μM of EDC34 were incubated in PBS alone (control) or a buffer containing 20% of serum, citrate, lepirudin, or EDTA plasma for 15 or 30 or 60 or 120 min at 37°C followed by electrophoretic analysis. EDC34 (1 μg) in 10 mM Tris was served control. (C) 0.5 μg of EDC34 were incubated with 3 μg of IgG, IgA, IgE, or IgM dissolved in 10 mM Tris, 150 mM NaCl, pH 7.4 buffer, for 1 h at 37°C. (D) EDC34 was incubated with IgG in 1:1 ratio (6 μM) at the indicated time points, or pre-incubated for 30 min with 50 μM of TCEP followed by an additional 24-h incubation step at 37°C. GGL27 in the absence of plasma served a control. (E) 6 μM of EDC34 were incubated with IgG Fab or Fc fragments (6 μM) for 30 min at 37°C. Samples were separated on SDS-PAGE and probed with an antibody against CAK27 a C-terminal TFPI-2 peptide region.
IgG is one of most abundant proteins in plasma and has a molecular weight in the range of 180 kDa. Thus, we hypothesized that EDC34 can interact with IgG or even with other immunoglobulins. In order to test this hypothesis, EDC34 was incubated with different classes of human purified immunoglobulins, i.e., IgA, IgG, IgE, and IgM and binding was tested by immunoblotting. Our results show that EDC34 avidly binds to all immunoglobulins tested (Figure 1C). When the interaction of the peptide with IgG was studied over time, we found that it was entirely absorbed after a 24 h incubation (Figure 1D). EDC34 contains two cysteine amino acids and to exclude that the interaction between EDC34 and IgG is the result of unspecifically newly formed disulphide bridges, the interaction was further studied under reducing conditions. Moreover, we used a control peptide GGL27 from TFPI-1 (Figure 1D) (35). We next explored whether EDC34 interacts with the Fab or Fc part of IgG. To this end the peptide was incubated with the two fragments. Binding was then assessed by western blot analysis, showing that only the Fc fragment is able to bind to EDC34, further underlining the specificity of this interaction (Figure 1E).
To investigate whether EDC34 can alter the antigen-antibody interactions during infection, human IgG was pre-incubated with EDC34 and mixed with E. coli bacteria. Figure 2A shows that the interaction of EDC34 with human IgG triggered increased opsonization of immunglobulin on the bacterial surface. This effect was less pronounced when EDC34 was first preincubated with E. coli bacteria and then later mixed with human IgG (Figure 2A). We next tested whether the binding of EDC34 to IgG can promote activation of the classical pathway of complement. To this end we focused on C1q, as the protein is part of the first subcomponent of the classical complement pathway (36). When EDC34 was added to citrate plasma for 1 h we observed a recruitment of C1q to the surface of E. coli bacteria, which was not seen when plasma alone was incubated with the bacteria (Figure 2B). This effect was not dependent on the conditions (reducing or non-reducing) that were chosen. Together the results show that EDC34 is needed for the assembly of C1q at the bacterial surface, which is in line with our previous findings (27). Having shown that EDC34 promotes IgG-mediated opsonization of C1q at the surface of E. coli bacteria, we next tested whether this interaction can trigger bactericidal activity. Figure 3C shows that the incubation of EDC34 with bacteria in PBS exerts a moderate killing activity, while almost 100% killing was noted in citrate plasma. Bacterial survival was increased when immunoglobulin-depleted plasma was used and upon reconstitution with IgG the depleted plasma retained its full antimicrobial activity in the presence of peptide (Figures 2C,D). These results demonstrated the important role of EDC34 in boosting complement-mediated bacterial killing.
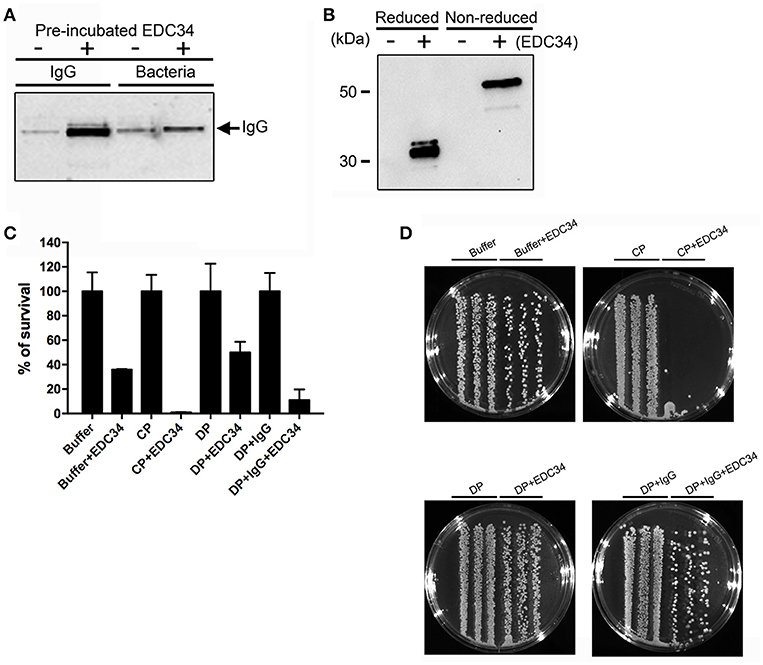
Figure 2. Interaction of EDC34 with IgG enhances C1q deposition on bacterial surfaces. (A) EDC34 (2 μM) was incubated with IgG (8 μM) for 24 h in PBS buffer. E. coli bacteria were for 1 h at 37°C. Alternatively, EDC34 was incubated with E. coli bacteria and then supplemented with IgG (see also Materials and Methods). Samples were separated on SDS-PAGE followed by immunoblotting, and probed with anti-IgG antibodies. (B) E. coli bacteria were incubated with citrate plasma supplemented with 10 μM of EDC34 for 1 h at 37°C. Proteins at the bacterial surface were eluted, concentrated by a pull down assay, and subjected to SDS-PAGE followed by immunoblotting. Recovered C1q was immuno-detected with anti-C1q antibodies. (C) Using viable count assays, antimicrobial activity of EDC34 against E. coli was studied. Bacteria and EDC34 were incubated in a PBS buffer, a PBS buffer supplemented with 25 % citrate plasma (CP), a PBS buffer supplemented with 25% immunoglobulin-depleted citrate plasma (DP), or a PBS buffer supplemented with 25% immunoglobulin-depleted citrate plasma reconstituted with IgGs. Mean with SD are shown (n = 3). (D) Colony cultures of E. coli bacteria grown in the indicated conditions shown in petri dishes.
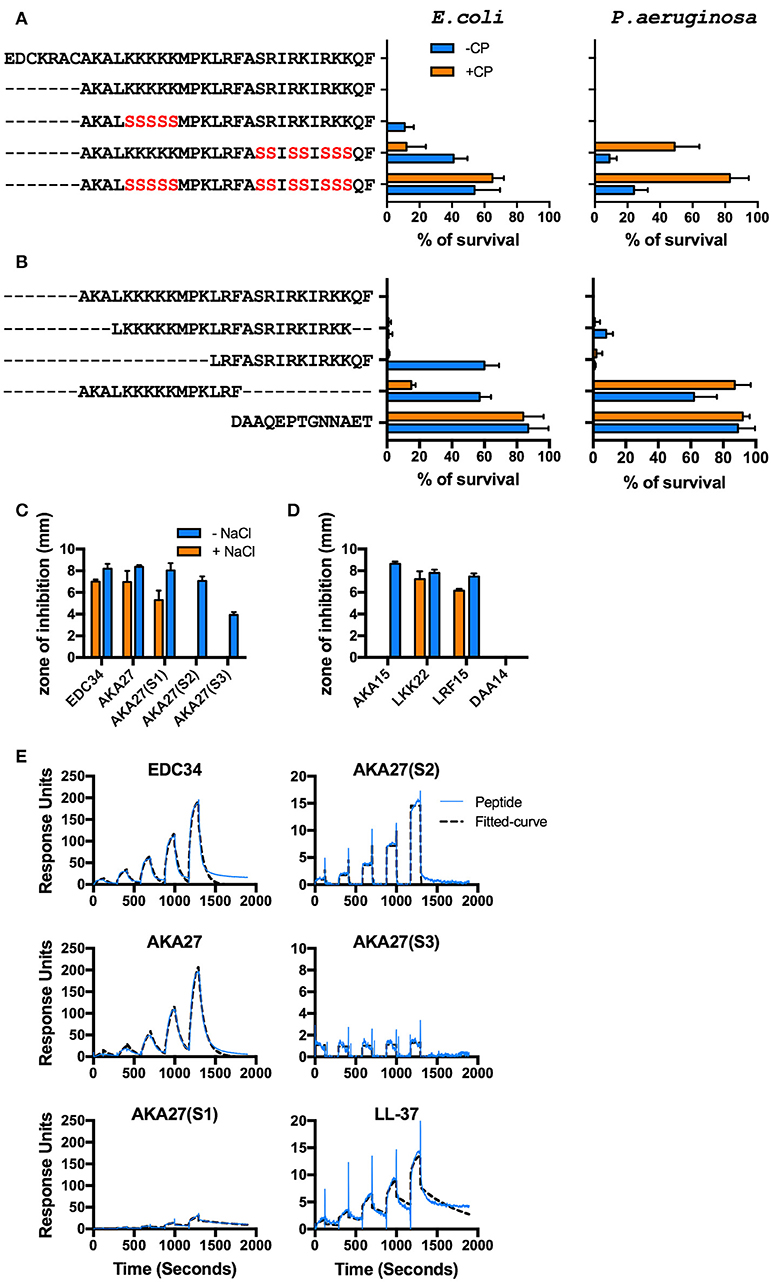
Figure 3. Mapping of EDC34 epitope. (A,B) C-terminal TFPI-2 derived peptides at indicated concentrations were incubated with E. coli and P. aeruginosa bacteria in the presence or absence of 20% citrate plasma, and their antimicrobial effects were measured in viable count assays. (C,D) The zone of inhibition of each peptide (X-axis) against E. coli bacteria was measured in the presence or absence of 150 mM NaCl using radial diffusion assays (RDAs). DAA14 was used as a negative control. Mean with SD is shown (n = 3). (E) Surface plasmon resonance analysis of the interaction between IgG and the indicated peptides in the concentration range of 25–400 nM is shown.
In order to map the epitope responsible for antimicrobial activity, the bactericidal effect of EDC34 and shortened versions of the peptide (Table 1) were tested using E. coli and P. aeruginosa. To exclude a possible influence of disulphide bonding, a truncated peptide (AKA27) was employed lacking the first seven amino acids, including two cysteine residues. Notably, AKA27 consists of several patches with clustered positively charged amino acids. Earlier studies have demonstrated these amino acids play an important role for the antimicrobial activity of many AMPs (37). To test whether this also applies to AKA27, arginine and lysine residues of the peptide were sequentially replaced by a neutral amino acid (serine) as depicted in Table 1. While AKA27(S1) remained its full antimicrobial potential in 20% human plasma, the bactericidal activity was decreased when AKA27(S2) or AKA27(S3) were analyzed under the same experimental conditions (Figure 3A). To further characterize the active site, truncated versions of AKA27 were synthesized. The sequences of these peptides (LKK22, AKA15, and LRF15) as well as the sequence of a control peptide (DAA14), derived from the N-terminal region of TFPI-2, are listed in Table 1. As shown in Figure 2B, LKK22, spanning the central core region of AKA27, and LRF15, lacking the N-terminal portion of AKA27, retained their antimicrobial activity, when incubated with bacteria in the presence of plasma. However, AKA15, lacking the C-terminal part of AKA27, and the control peptide (DAA14) had no antimicrobial activity under these conditions (Figure 3B). The same activity pattern was recorded using radial diffusion assays (RDAs). Here the antimicrobial activity of the peptides was tested under low and high salt conditions. While in the absence of salt all peptides with the exception DAA14 (negative control) had full or some antimicrobial activity, only EDC34, AKA27, AKA27(S1), LKK22, and LRF15 were able impair bacterial growth under high salt conditions (Figures 3C,D). These findings suggest that the salt concentration might be in part responsible that AKA27(S2), AKA27(S3), and AKA15 have lost their antimicrobial activity in plasma. Finally, we determined the affinity of EDC34, AKA27, AKA27(S1), AKA27(S2), and AKA27(S3) for human IgG. LL-37 was used as control. SPR analysis were performed by running the peptides over an IgG coted chip. As depicted in Figure 3E, EDC34 and AKA27 have highest affinity for IgG, while the affinity of other peptides showed was significantly lower (Figure 3E). Together these findings support the notion that the C-terminal region of AKA27 is important for the interaction with IgG and subsequent activation of the classical complement pathway.
In order to explore the function of TFPI-2 during systemic inflammation we analyzed the expression levels of TFPI-2 in two relevant animal models, a murine endotoxin-induced model and an E. coli infection model. TFPI-2 expression was investigated in spleen, lung, brain, small, and large intestine, liver and kidney in challenged/infected or untreated wild type mice using real-time quantitative PCR (Figure 4A). The results showed that TFPI-2 mRNA expression was induced by 10-fold in the spleen already 1 h post-injection of LPS and peaked at 2 h followed by a transient decrease until 12 h post-injection. The second most effected organ in terms of TFPI-2 expression was the brain showing a peak of expression 4 h post-injection. The same was found for lungs. In addition, we examined kidney, large and small intestine and liver which all showed an additional increase in TFPI-2 expression upon systemic inflammation induced by LPS from E. coli. The expression was normalized to time-point 0 h and where expression was 1 time fold change in relation to house- keeping gene β-actin.
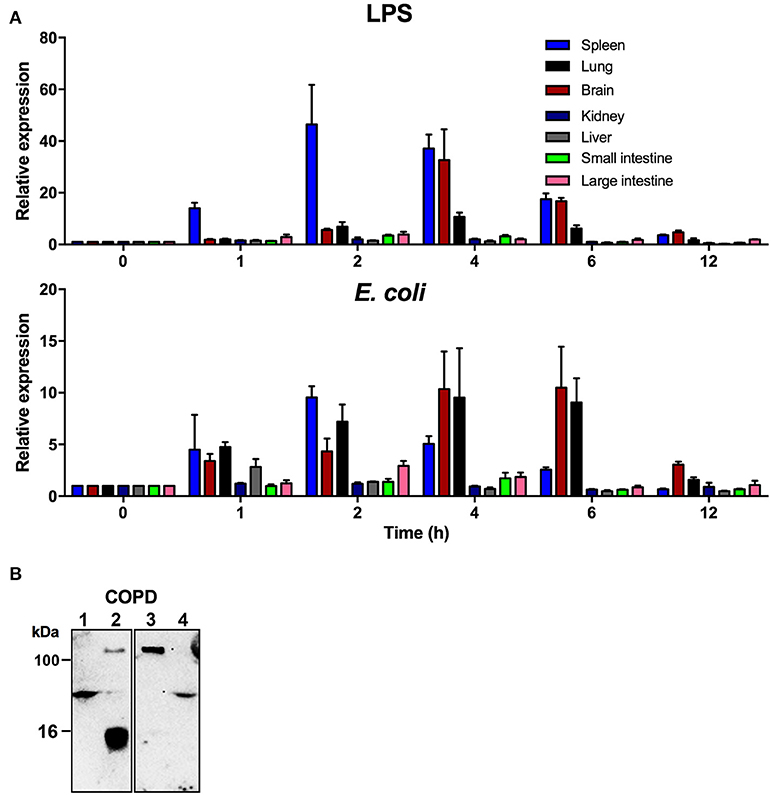
Figure 4. TFPI-2 is highly expressed during endotoxin-induced inflammation and sepsis. (A) To measure TFPI-2 expression levels in various organs during inflammation and systemic bacterial infection TFPI-2+/+ mice were intraperitoneally injected with either 8 mg/kg LPS or E. coli. TFPI-2 mRNA expression levels was measured at indicated hours post-injection in spleen, lung, brain, kidney, liver, small intestine and large intestine (n = 3). Expression is presented as relative gene expression levels in relation to housekeeping gene β-actin. The data shown mean ± SEM. (B) Identification of TFPI-2 in COPD patient sputum samples (Lane: 1-4). Cleavage fragments of TFPI-2 were analyzed by immunoblotting using polyclonal antibodies against C-terminal EDC34 peptide.
We have previously showed that TFPI-2 is highly expressed and that C-terminal fragments are present in chronic skin wounds. In addition to the previous human and current TFPI-2 expression mouse data, we explored whether C-terminal fragments of TFPI-2 are present in sputum samples from COPD patients. Western blot analysis using polyclonal antibodies against the C-terminal part of TFPI-2 shows that TFPI-2 is highly expressed and that C-terminal fragments are indeed associated with immunoglobulins in sputum from a COPD patient (Figure 4B).
In the last series of experiments, we employed in vivo models to explore both therapeutic and physiological aspects of our findings. To this end, we infected mice intraperitoneally with E. coli bacteria. One hour after the bacterial challenge, mice were treated with buffer or the mouse-derived TFPI-2 C-terminal peptide VKG24, a mouse analog of human EDC34. After an 8-h incubation, IVIS® Spectrum images were taken to detect ROS production. Mice were then sacrificed and blood, kidney, lung, liver and spleen were collected to determine bacterial dissemination. As shown in Figure 5A, ROS production in VKG24-treated mice was noticeably decreased when compared with buffer-treated mice. In accordance with these results, we also noticed that bacterial spreading to the different organs was reduced in peptide-treated mice when compared to the controls (Figure 5B).
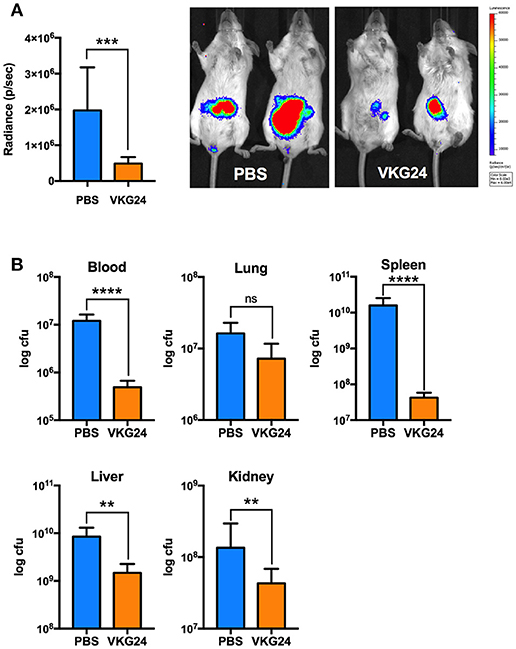
Figure 5. Mouse TFPI-2 peptide VKG24 protects mice from E. coli-induced sepsis. Mice were infected with 100 μl (8–8.5 × 108 cfu/ml) E. coli bacteria. One hour after infection animals were treated (i. p.) with PBS and VKG24 (500 μg per mouse) for 8 h. Mice (n = 5) were then anesthetized and injected (s. c.) with L-012 dye to detect ROS production. IVIS® Spectrum with Life Image® version 4.4 was used for imaging. (A) ROS production in radiance (p/sec) for control mice (PBS) and mice treated with VKG24 (PBS & VKG24 n = 5). Representative IVIS® Spectrum images indicating ROS production from two control (PBS) and treated (VKG24) mice are included. (B) After the IVIS examination, mice were scarified and organs were harvested for cfu count measurements (n = 5; liver **p < 0.0048, kidney **p < 0.0036, spleen ****p < 0.0001, ***p < 0.0007, blood ****p < 0.0001).
Given the high expression of TFPI-2 in the lungs during inflammation, we next wanted to pinpoint the role of TFPI-2 in the respiratory tract. We performed intranasal P. aeruginosa infection using TFPI-2−/− and TFPI-2+/+ mice. TFPI-2−/− mice exhibited a significantly lower survival rate compared to wild type mice (p = 0.003) (Figure 6A). In addition, bacterial load was measured in both bronchioalveolar lavage fluid (BALF) and lung tissue was measured after 12 h post-infection (Figure 6B). Indeed TFPI-2−/− mice exhibited larger amounts of bacteria in lung tissue compared to wild type mice. Hence, TFPI-2−/− mice are more prone to P. aeruginosa induced lung infection. To evaluate if the full-length protein of TFPI-2, in addition to limit bacterial infiltration, exerts host defense properties by keeping inflammation at bay during infection we measured inflammatory cytokine levels in both BALF and citrate plasma after 12 h post-infection. On a systemic level (plasma) 12 h post-infection IL-6, IL-10, and MCP-1 was higher in TFPI-2−/− mice and no significant changes were observed in INF-γ, TNFα, and IL-12p70 (Figure 6C), whereas in BALF fluid, IL-10, and MCP-1 was higher in TFPI-2−/− mice. To rule out the possible anticoagulant activity role of TFPI-2 in TFPI-2−/− mice, we induced LPS mediated septic shock in TFPI-2+/+ and TFPI-2−/− mice and determined clotting times, platelet counts and cytokines in whole blood after 6 h post-LPS challenge. Measurements of clotting times upon endotoxemia did not reveal any difference between TFPI-2+/+ and TFPI-2−/− mice (Figure 7A). No significant differences were observed in platelet counts (Figure 7B), cytokine levels between TFPI-2+/+ and TFPI-2−/− animals (Figure 7C), indicating that TFPI-2 is not involved in anticoagulation and inflammation. Together our results demonstrate a previously undisclosed mechanism based on boosting of complement-dependent immune response to infection. A protective role of TFPI-2 was demonstrated during lung infection by employing TFPI-2−/− and wild type mice. By using an animal model of infection and treatment with a C-terminal mouse homolog of EDC34, we found that our findings can be used for the development of novel antimicrobial therapies.
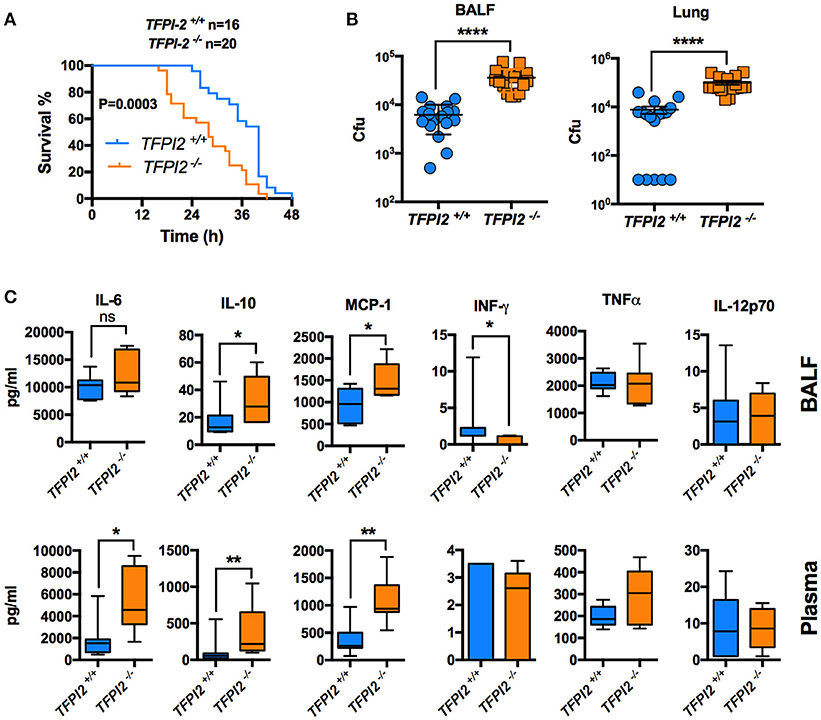
Figure 6. TFPI-2−/− mice are susceptible to acute Pseudomonas aeruginosa lung infection. (A) Survival. Fifty microliter of P. aeruginosa Xen41 bacteria (3–4 × 108 cfu/ml) were administered intra-nasally (i.n.) into TFPI-2+/+ and TFPI-2−/− mice. Survival of the mice was monitored every 2 h. The data is pooled from three different experiments with a total number of 16 wild type mice and 20 TFPI-2−/− mice. (B) BALF and lung tissue was harvested, placed on ice, homogenized (lung tissue), and colony-forming units (cfu) was determined 12 h after infection (n = 17). (C) Cytokines were analyzed in BALF and plasma (n = 7).
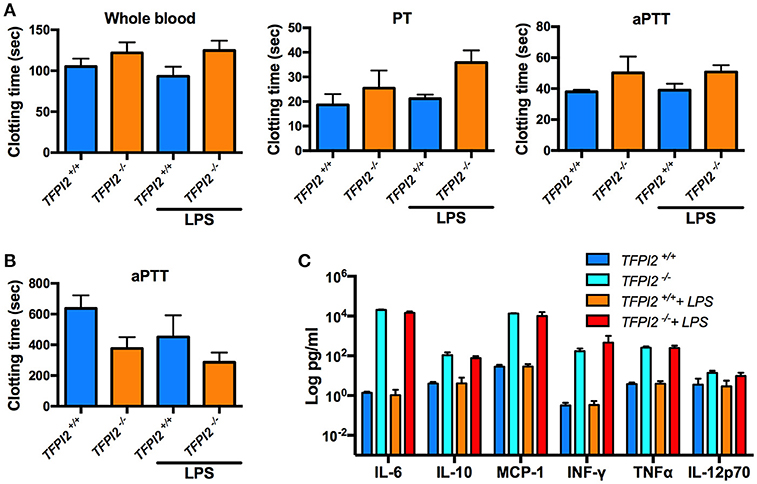
Figure 7. In vivo TFPI-2 has no significant impact on coagulation. Mice were intraperitoneally injected with either 8 mg/kg LPS, clotting time, platelets and cytokines were measured after 6 h post-LPS challenge. (A) Clotting times was measured in whole blood. Extrinsic pathway, prothrombin time (PT) and the intrinsic pathway activated partial thrombin time (aPTT) was determined in TFPI-2+/+ and TFPI-2−/− citrate plasma. (B,C) Total number of platelets counts and indicated cytokine levels were analyzed in blood from TFPI-2+/+ and TFPI-2−/− mice. No significant differences were observed in between the groups.
In this work we present a novel mechanism mediated by TFPI-2 C-terminal peptides to promote complement mediated bacterial killing. In addition, using TFPI-2−/− mice we for the first time show a protective role of TFPI-2 during Gram-negative infection. Studies using artificial membranes and live organisms have illustrated that the vast majority of AMPs exert their antimicrobial activity by permeabilizing the membrane of the pathogen. Thus a mechanism, leading to a burst of the bacterial cell wall, has been described for instance for defensins (38, 39), bactenecins (40), and magainins (41, 42). However, there are also other mechanisms, such as the breakage of single-strand DNA by defensins (43), production of hydrogen peroxide by magainins (44), or induction of apoptosis by lactoferricin (45) that can in addition contribute to an elimination of the pathogen. Based on these findings we hypothesized that EDC34 utilizes different strategies to exert its antimicrobial activities. This assumption was based on previous studies showing the peptide has multifunctional properties, as it not only displays antimicrobial activity, but it can also activate the classical complement system and the intrinsic pathway of coagulation (27, 28).
The complement system is activated by three distinctive routes, known as the classical, alternative, and lectin pathway, respectively. Though all three cascades lead to the generation of the same effector functions, only the classical pathway requires the opsonization of immunoglobulins at the bacterial surfaces (46). Recent work has shown that EDC34 can amplify the classical pathway (28). However, the underlying mechanisms were not addressed in previous studies. Here we show that EDC34 exerts its immunomodulatory effect by binding to immunoglobulins which then helps to augment complement activation. Though the interaction of C1q with antibodies is normally needed during this process, it can also occur in an antibody-independent mechanism. To this end C1q binds directly to the surface of the pathogen where it triggers structural changes in the C1s/C1r complex. This then leads to production of C3a and C3b molecules that play a critical role in inflammation, bacterial killing, and phagocytosis (46). To initiate classical pathway C1q has to bind with its CH2 domain to the Fc region of IgG (47, 48). Our results show that the interaction of EDC34 with the Fc region of IgG enhances the binding of immunoglobulin to C1q which then in turn leads to an increased activation of the classical complement pathway. Notably, the interaction of EDC34 with the complement system appears crucial since the endogenous antimicrobial activity of the peptide was not sufficient for a complete eradication of the invading pathogen.
TFPI-2 is mainly expressed in brain, spleen and lung during sepsis and LPS-induced systemic inflammation in mice (C57BL/6). Surprisingly, TFPI-2−/− mice did not show any significant impact on coagulation during endotoxin-induced sepsis. This is contradictory to the previously claimed functional relevance as an endogenous anti-coagulant, similar to the highly homologous protein TFPI-1 (49). Platelets are well established as a cell type that maintains hemostasis and thrombus growth. Rather than an anti-coagulant, TFPI-2 display characteristics of a host-defense protein with a possible wide variety of functions based on it's expression in the different organs. Based on the reported data we have for the first time shown that the TFPI-2 plays a prominent role in host defense against Gram-negative lung infection in mice. TFPI-2 could therefore constitute an important acute phase reactant in lung tissue with bactericidal and immune-regulatory functions. Based on the detection of proteolytically digested fragments of TFPI-2 found in sputum from patients with COPD, our data further implies that proteases can generate TFPI-2 C-terminal fragments with host defense activities. Such investigations, along side of monitoring of correlations to enzymatic activity and clinical outcome, would likely generate implications for novel treatment routes for COPD patients.
As a proof of concept from a therapeutic perspective, we employed an animal model of infection using VKG24, the murine TFPI-2-derived homolog to EDC34 (27). Previous work has shown that VKG24 exerts, like EDC34, anti-inflammatory activity and protects mice from fatal LPS shock (50). It was further reported that VKG24 exhibits antimicrobial killing activity against E. coli under in vitro conditions (50). Based on these findings we decided to test the effect of VKG24 in an E. coli sepsis model. Our results show that bacterial growth was reduced in blood, liver, kidney, spleen, and lungs when infected mice were treated with VKG24. Along these lines we also noted that ROS production was notably lower in these mice, which indicates that VKG24 exerts an antimicrobial activity by lowering inflammation at the site of infection. Our current study describes increased cytokine levels detected in TFPI-2−/− bacterial challenged mice. This is in line with previously published study where we showed reduced cytokine levels in LPS challenged mice treated with VKG24 peptide (50), indicating potent immunomodulatory activity of C-terminal TFPI-2 region against Gram-negative infections. In conclusion, our data show that EDC34 possesses therapeutic potential, as application of the peptide or its murine homolog not only can kill bacteria by permeabilizing their membranes, but also by boosting activation of the classical complement pathway (27). In vivo experiments further reveal the peptide motif exerts a systemic bacterial killing activity combined with a non-pathological inflammatory response, which in combination provides a promising approach for future therapeutic applications. Taken together, the evidence presented in this work demonstrates that TFPI-2 is mainly a host-defense protein, while the anti-coagulative functions likely play a minor role in normal hemostasis.
PP and AS designed the research. MA, GK, ME, SA, SJ, and PP performed the experiments. AE, HH, AS, and PP contributed analytic tools,reagents,materials,analysis tools. PP analyzed the data and wrote the paper.
This work was supported by grants from the Alfred Österlund Foundation, Crafoord Foundation, Welander-Finsen, the Swedish Foundation for Strategic Research, the Swedish Research Council (projects 2012-1883), the Knut and Alice Wallenberg Foundation, The Swedish Government Funds for Clinical Research (ALF), and the Royal Physiographic Society in Lund. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AT and handling Editor declared their shared affiliation.
We would like to thank Pia Andersson and Ann-Charlotte Strömdahl for expert technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02072/full#supplementary-material
1. Neduva V, Linding R, Su-Angrand I, Stark A, de Masi F, Gibson TJ, et al. Systematic discovery of new recognition peptides mediating protein interaction networks. PLoS Biol. (2005) 3:e405. doi: 10.1371/journal.pbio.0030405
2. Tan SH, Hugo W, Sung WK, Ng SK. A correlated motif approach for finding short linear motifs from protein interaction networks. BMC Bioinformatics (2006) 7:502. doi: 10.1186/1471-2105-7-502
3. Dalby PA, Hoess RH, DeGrado WF. Evolution of binding affinity in a WW domain probed by phage display. Protein Sci. (2000) 9:2366–76. doi: 10.1110/ps.9.12.2366
4. Tong AH, Drees B, Nardelli G, Bader GD, Brannetti B, Castagnoli L, et al. A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science (2002) 295:321–4. doi: 10.1126/science.1064987
5. Wiedemann U, Boisguerin P, Leben R, Leitner D, Krause G, Moelling K, et al. Quantification of PDZ domain specificity, prediction of ligand affinity and rational design of super-binding peptides. J Mol Biol. (2004) 343:703–18. doi: 10.1016/j.jmb.2004.08.064
6. Bistrian B. Systemic response to inflammation. Nutr Rev. (2007) 65(12 Pt 2):S170–2. doi: 10.1301/nr.2007.dec.S170-S172
7. Ganz T, Selsted ME, Lehrer RI. Defensins. Eur J Haematol. (1990) 44:1–8. doi: 10.1111/j.1600-0609.1990.tb00339.x
9. Bowdish DM, Davidson DJ, Lau YE, Lee K, Scott MG, Hancock RE. Impact of LL-37 on anti-infective immunity. J Leukoc Biol. (2005) 77:451–9. doi: 10.1189/jlb.0704380
10. Hancock RE, Lehrer R. Cationic peptides: a new source of antibiotics. Trends Biotechnol. (1998) 16:82–8. doi: 10.1016/S0167-7799(97)01156-6
11. Hancock RE, Diamond G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. (2000) 8:402–10. doi: 10.1016/S0966-842X(00)01823-0
12. Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. (2006) 24:1551–7. doi: 10.1038/nbt1267
13. Schuerholz T, Brandenburg K, Marx G. Antimicrobial peptides and their potential application in inflammation and sepsis. Crit Care (2012) 16:207. doi: 10.1186/cc11220
14. Elsbach P. What is the real role of antimicrobial polypeptides that can mediate several other inflammatory responses? J Clin Invest. (2003) 111:1643–1645. doi: 10.1172/JCI18761
15. Nijnik A, Madera L, Ma S, Waldbrook M, Elliott MR, Easton DM, et al. Synthetic cationic peptide IDR-1002 provides protection against bacterial infections through chemokine induction and enhanced leukocyte recruitment. J Immunol. (2010) 184:2539–50. doi: 10.4049/jimmunol.0901813
16. Rosenfeld Y, Papo N, Shai Y. Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides. Peptide properties and plausible modes of action. J Biol Chem. (2006) 281:1636–43. doi: 10.1074/jbc.M504327200
17. Kasetty G, Papareddy P, Kalle M, Rydengard V, Morgelin M, Albiger B, et al. Structure-activity studies and therapeutic potential of host defense peptides of human thrombin. Antimicrob Agents Chemother. (2011) 55:2880–90. doi: 10.1128/AAC.01515-10
18. Kalle M, Papareddy P, Kasetty G, Morgelin M, van der Plas MJ, Rydengard V, et al. Host defense peptides of thrombin modulate inflammation and coagulation in endotoxin-mediated shock and Pseudomonas aeruginosa sepsis. PLoS ONE (2012) 7:e51313. doi: 10.1371/journal.pone.0051313
19. Hansen FC, Kalle-Brune M, van der Plas MJ, Stromdahl AC, Malmsten M, Morgelin M, et al. The thrombin-derived host defense peptide GKY25 inhibits endotoxin-induced responses through interactions with lipopolysaccharide and macrophages/monocytes. J Immunol. (2015) 194:5397–406. doi: 10.4049/jimmunol.1403009
20. Kalle M, Papareddy P, Kasetty G, van der Plas MJ, Morgelin M, Malmsten M, et al. A peptide of heparin cofactor II inhibits endotoxin-mediated shock and invasive Pseudomonas aeruginosa infection. PLoS ONE (2014) 9:e102577. doi: 10.1371/journal.pone.0102577
21. Papareddy P, Kalle M, Kasetty G, Morgelin M, Rydengard V, Albiger B, et al. C-terminal peptides of tissue factor pathway inhibitor are novel host defense molecules. J Biol Chem. (2010) 285:28387–98. doi: 10.1074/jbc.M110.127019
22. van der Plas MJ, Bhongir RK, Kjellstrom S, Siller H, Kasetty G, Morgelin M, et al. Pseudomonas aeruginosa elastase cleaves a C-terminal peptide from human thrombin that inhibits host inflammatory responses. Nat Commun. (2016) 7:11567. doi: 10.1038/ncomms11567
23. Fanjul-Fernández M, Folgueras AR, Cabrera S, López-Otín C. Matrix metalloproteinases: evolution, gene regulation and functional analysis in mouse models. Biochim Biophys Acta (2010) 1803:3–19. doi: 10.1016/j.bbamcr.2009.07.004
24. Sprecher CA, Grant FJ, Grimm G, O'Hara PJ, Norris F, Norris K, et al. Molecular cloning of the cDNA for a human amyloid precursor protein homolog: evidence for a multigene family. Biochemistry (1993) 32:4481–6. doi: 10.1021/bi00068a002
25. Sprecher CA, Kisiel W, Mathewes S, Foster DC. Molecular cloning, expression, and partial characterization of a second human tissue-factor-pathway inhibitor. Proc Natl Acad Sci USA. (1994) 91:3353–7. doi: 10.1073/pnas.91.8.3353
26. Liu Y, Stack SM, Lakka SS, Khan AJ, Woodley DT, Rao JS, et al. Matrix localization of tissue factor pathway inhibitor-2/matrix-associated serine protease inhibitor (TFPI-2/MSPI) involves arginine-mediated ionic interactions with heparin and dermatan sulfate: heparin accelerates the activity of TFPI-2/MSPI toward plasmin. Arch Biochem Biophys. (1999) 370:112–8. doi: 10.1006/abbi.1999.1371
27. Papareddy P, Kalle M, Sorensen OE, Malmsten M, Morgelin M, Schmidtchen A. The TFPI-2 derived peptide EDC34 improves outcome of gram-negative sepsis. PLoS Pathog. (2013) 9:e1003803. doi: 10.1371/journal.ppat.1003803
28. Papareddy P, Kalle M, Sorensen OE, Lundqvist K, Morgelin M, Malmsten M, et al. Tissue factor pathway inhibitor 2 is found in skin and its C-terminal region encodes for antibacterial activity. PLoS ONE (2012) 7:e52772. doi: 10.1371/journal.pone.0052772
29. Herman MP, Sukhova GK, Kisiel W, Foster D, Kehry MR, Libby P, et al. Tissue factor pathway inhibitor-2 is a novel inhibitor of matrix metalloproteinases with implications for atherosclerosis. J Clin Invest. (2001) 107:1117–26. doi: 10.1172/JCI10403
30. Crawley JT, Goulding DA, Ferreira V, Severs NJ, Lupu F. Expression and localization of tissue factor pathway inhibitor-2 in normal and atherosclerotic human vessels. Arterioscler Thromb Vasc Biol. (2002) 22:218–24. doi: 10.1161/hq0102.101842
31. Hong J, Liu R, Chen L, Wu B, Yu J, Gao W, et al. Conditional knockout of tissue factor pathway inhibitor 2 in vascular endothelial cells accelerates atherosclerotic plaque development in mice. Thromb Res. (2016) 137:148–56. doi: 10.1016/j.thromres.2015.11.010
32. Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci. (2002) 115:3719–27. doi: 10.1242/jcs.00063
33. Elvington M, Huang Y, Morgan BP, Qiao F, van Rooijen N, Atkinson C, et al. A targeted complement-dependent strategy to improve the outcome of mAb therapy, and characterization in a murine model of metastatic cancer. Blood (2012) 119:6043–51. doi: 10.1182/blood-2011-10-383232
34. Andersson E, Rydengård V, Sonesson A, Mörgelin M, Björck L, Schmidtchen A. Antimicrobial activities of heparin-binding peptides. Eur J Biochem. (2004) 271:1219–26. doi: 10.1111/j.1432-1033.2004.04035.x
35. Papareddy P, Kasetty G, Alyafei S, Smeds E, Salo-Ahen OMH, Hansson SR, et al. An ecoimmunological approach to study evolutionary and ancient links between coagulation, complement and Innate immunity. Virulence (2018) 9:724–37. doi: 10.1080/21505594.2018.1441589
36. Perkins SJ, Kerckaert JP, Loucheux-Lefebvre MH. The shapes of biantennary and tri/tetraantennary alpha 1 acid glycoprotein by small-angle neutron and X-ray scattering. Eur J Biochem. (1985) 147:525–31. doi: 10.1111/j.0014-2956.1985.00525.x
37. Jiang Z, Vasil AI, Hale JD, Hancock RE, Vasil ML, Hodges RS. Effects of net charge and the number of positively charged residues on the biological activity of amphipathic alpha-helical cationic antimicrobial peptides. Biopolymers (2008) 90:369–83. doi: 10.1002/bip.20911
38. Cociancich S, Ghazi A, Hetru C, Hoffmann JA, Letellier L. Insect defensin, an inducible antibacterial peptide, forms voltage-dependent channels in Micrococcus luteus. J Biol Chem (1993) 268:19239–45.
39. Wimley WC, Selsted ME, White SH. Interactions between human defensins and lipid bilayers: evidence for formation of multimeric pores. Protein Sci. (1994) 3:1362–73. doi: 10.1002/pro.5560030902
40. Gennaro R, Skerlavaj B, Romeo D. Purification, composition, and activity of two bactenecins, antibacterial peptides of bovine neutrophils. Infect Immun. (1989) 57:3142–6.
41. Tytler EM, Anantharamaiah GM, Walker DE, Mishra VK, Palgunachari MN, Segrest JP. Molecular basis for prokaryotic specificity of magainin-induced lysis. Biochemistry (1995) 34:4393–401. doi: 10.1021/bi00013a031
42. Ludtke SJ, He K, Heller WT, Harroun TA, Yang L, Huang HW. Membrane pores induced by magainin. Biochemistry (1996) 35:13723–8. doi: 10.1021/bi9620621
43. Gera JF, Lichtenstein A. Human neutrophil peptide defensins induce single strand DNA breaks in target cells. Cell Immunol. (1991) 138:108–20. doi: 10.1016/0008-8749(91)90136-Y
44. Leem JY, Nishimura C, Kurata S, Shimada I, Kobayashi A, Natori S. Purification and characterization of N-β-alanyl-5-S-glutathionyl-3,4-dihydroxyphenylalanine, a novel antibacterial substance of Sarcophaga peregrina (flesh fly). J Biol Chem. (1996) 271:13573–77. doi: 10.1074/jbc.271.23.13573
45. Andres MT, Viejo-Diaz M, Fierro JF. Human lactoferrin induces apoptosis-like cell death in Candida albicans: critical role of K+-channel-mediated K+ efflux. Antimicrob Agents Chemother. (2008) 52:4081–8. doi: 10.1128/AAC.01597-07
46. Janeway CA Jr, Travers P, Walport M, Shlomchik MJ. The distribution and functions of immunoglobulin isotypes. In: Immunobiology, the Immune System in Health and Disease. New York, NY: Garland Science (2001).
47. Yasmeen D, Ellerson JR, Dorrington KJ, Painter RH. The structure and function of immunoglobulin domains. IV. The distribution of some effector functions among the Cgamma2 and Cgamma3 homology regions of human immunoglobulin G1. J Immunol. (1976) 116:518–26.
48. Duncan AR, Winter G. The binding site for C1q on IgG. Nature (1988) 332:738–40. doi: 10.1038/332738a0
49. Udagawa K, Yasumitsu H, Esaki M, Sawada H, Nagashima Y, Aoki I, et al. Subcellular localization of PP5/TFPI-2 in human placenta: a possible role of PP5/TFPI-2 as an anti-coagulant on the surface of syncytiotrophoblasts. Placenta (2002) 23:145–53. doi: 10.1053/plac.2001.0774
Keywords: TFPI-2, antimicrobial peptide, bacteria, complement, immunoglobulins, sepsis
Citation: Ali MN, Kasetty G, Elvén M, Alyafei S, Jovic S, Egesten A, Herwald H, Schmidtchen A and Papareddy P (2018) TFPI-2 Protects Against Gram-Negative Bacterial Infection. Front. Immunol. 9:2072. doi: 10.3389/fimmu.2018.02072
Received: 31 May 2018; Accepted: 21 August 2018;
Published: 11 September 2018.
Edited by:
Uday Kishore, Brunel University London, United KingdomReviewed by:
Anthony George Tsolaki, Brunel University London, United KingdomCopyright © 2018 Ali, Kasetty, Elvén, Alyafei, Jovic, Egesten, Herwald, Schmidtchen and Papareddy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Praveen Papareddy, cHJhdmVlbi5wYXBhcmVkZHlAbWVkLmx1LnNl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.