 Robin Maximilian Awad
Robin Maximilian Awad Yannick De Vlaeminck
Yannick De Vlaeminck Johannes Maebe†
Johannes Maebe† Cleo Goyvaerts
Cleo Goyvaerts Karine Breckpot
Karine Breckpot- Laboratory of Molecular and Cellular Therapy, Department of Biomedical Sciences, Vrije Universiteit Brussel, Brussels, Belgium
Tumor cells frequently produce soluble factors that favor myelopoiesis and recruitment of myeloid cells to the tumor microenvironment (TME). Consequently, the TME of many cancer types is characterized by high infiltration of monocytes, macrophages, dendritic cells and granulocytes. Experimental and clinical studies show that most myeloid cells are kept in an immature state in the TME. These studies further show that tumor-derived factors mold these myeloid cells into cells that support cancer initiation and progression, amongst others by enabling immune evasion, tumor cell survival, proliferation, migration and metastasis. The key role of myeloid cells in cancer is further evidenced by the fact that they negatively impact on virtually all types of cancer therapy. Therefore, tumor-associated myeloid cells have been designated as the culprits in cancer. We review myeloid cells in the TME with a focus on the mechanisms they exploit to support cancer cells. In addition, we provide an overview of approaches that are under investigation to deplete myeloid cells or redirect their function, as these hold promise to overcome resistance to current cancer therapies.
Introduction to the Role of Myeloid Cells in Cancer
Until the beginning of the Twenty-first century, cancer was considered a disease afflicting a single cell. This oversimplified view has made way for a new perspective. As tumors develop, a tireless cross-talk takes place between heterogeneous malignant cells and neighboring parenchymal, stromal, and recruited immune cells. These cells, along with the extracellular matrix, and soluble mediators, form the tumor microenvironment (TME). The composition of the immune infiltrate in the TME largely determines cancer progression and the cancer's sensitivity to various therapies. Tumor-infiltrating CD8+ T lymphocytes are key in controlling cancer cells. Consequently, activating de novo or existing CD8+ T cells using strategies like cancer vaccines and immune checkpoint therapy has attracted considerable attention (1). The durable responses obtained with these therapies have prompted the use of cancer immunotherapy in the standard of care of various cancer types. Despite these vast improvements in cancer treatments, a significant number of patients does not benefit from current cancer immunotherapy. There is ample evidence that myeloid cells, present within the TME, are at the basis of this therapy failure. Therefore, tumor-infiltrating myeloid cells (TIMs) are considered relevant therapeutic targets (2).
Myeloid cells are a heterogeneous group of immune cells that belong to the innate immune system. Among the myeloid cells, monocytes, macrophages, dendritic cells (DCs), and granulocytes have received much attention. These cells, each in their own way, play an essential role in tissue homeostasis. Moreover, monocytes, macrophages and DCs are well known for their ability to regulate T cell responses, thereby bridging innate and adaptive immunity. Tumor cells take advantage of myeloid cells to maintain tissue homeostasis by exploiting the myeloid cells' capacity to produce inflammatory mediators [e.g., interleukin-6 [IL-6] and tumor necrosis factor-α [TNF-α]], growth factors that affect tumor proliferation and vessel formation [e.g., transforming growth factor-β [TGF-β] and vascular endothelial growth factor [VEGF]], and enzymes that degrade matrix proteins [e.g., matrix metallo-proteinases [MMPs]] (3, 4). Moreover, tumor cells take advantage of the myeloid cells' ability to keep T cell responses in check. Thus instructed by tumor cells, myeloid cells aid in creating a TME that is characterized by chronic inflammation, immunosuppression, and continuously proliferating tumor cells that can disseminate. These are important hallmarks of cancer (5). Paradoxically, myeloid cells have also been implicated in the resolution of cancer (4). Myeloid cells can exert profound antitumor functions such as direct tumor cell killing next to indirect tumor cell killing through activation of among others CD8+ T cells. In the remainder of the introduction, we discuss the pro- and antitumor properties of different TIM subsets in the context of the cancer immunoediting paradigm (6). We moreover discuss TIMs and how they influence various cancer therapies. Finally we provide an overview of approaches that have been studied to target TIMs and as such enhance the efficacy of current cancer therapies.
The Development and Phenotype of Tumor-Infiltrating Myeloid Cells in a Nutshell
Tumor-infiltrating myeloid cells (TIMs) constitute a heterogeneous population of cells that are characterized by diversity and plasticity. Many TIMs originate from circulating monocytes and granulocytes, which in turn stem from bone marrow-derived hematopoietic stem cells (HSCs; Figure 1). In the absence of activation signals, persistent stimulation by tumor-derived factors incites monocyte and granulocyte progenitors to divert from their intrinsic pathway of terminal differentiation into mature macrophages, DCs or granulocytes. Instead differentiation into pathological, alternatively activated immature myeloid cells is favored. These immature myeloid cells include tumor-associated DCs (TADCs), tumor-associated neutrophils (TANs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs). Alternative to this “emergency myelopoiesis,” TAMs can originate from tissue-resident macrophages, which in turn can be of embryonic or monocytic origin (7–9). These tissue-resident macrophages undergo changes in phenotype and function during carcinogenesis, and proliferation seems key to maintain TAMs derived from tissue-resident macrophages.
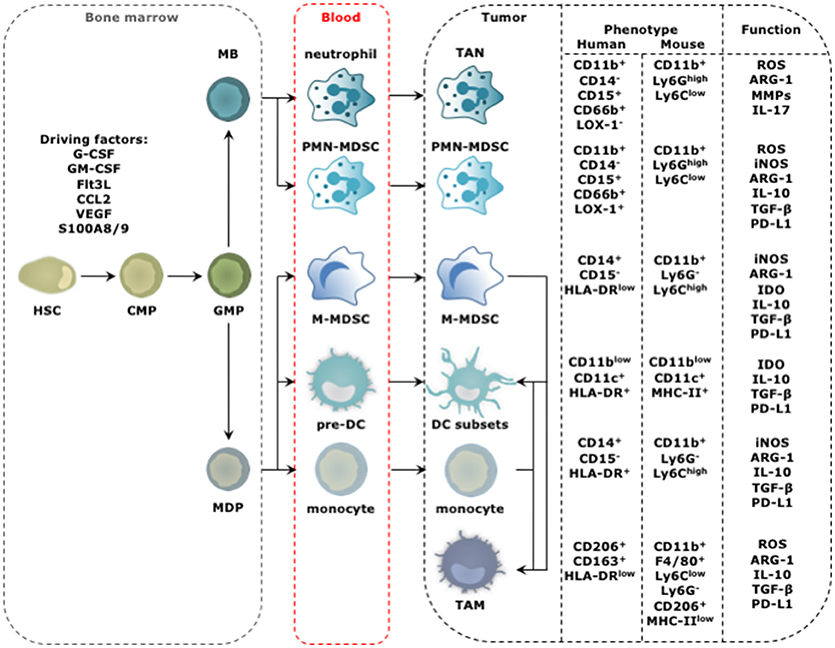
Figure 1. Progression from HSC to tumor-promoting TIM. The distinct steps in the progression from HSC to TIM occur at different locations and start with amplification and differentiation of the HSC and its progenitors, including the common myeloid progenitor (CMP), granulocyte-monocyte progenitor (GMP), myeloblast (MB), and monocyte-dendritic cell progenitor (MDP) in the bone marrow. New myeloid cells are released into the blood stream ready to migrate to the tumor bed. This process is regulated by molecular signals produced by cancer cells and is further amplified by molecular signals produced by among others TIMs. These factors include granulocyte (G) and granulocyte macrophage (GM) colony stimulating factor (CSF), Fms-like tyrosine kinase 3-ligand (Flt3-L), chemokine (C-C motif) ligand 2 (CCL2), VEGF and S100A8/9. The phenotype of TIMs of human and mouse origin, and their functional hallmarks are shown. For TADCs, TANs, and TAMs the figure is simplified as different subsets with either anti- (stimulatory) or protumor (regulatory) functions are discriminated for these cell types. The figure focuses on the subsets with protumor activity.
Among the TIMs, MDSCs have attracted considerable attention. This TIM population has been divided in polymorphonuclear (PMN) and monocytic (M) MDSCs, which morphologically and phenotypically resemble TANs and monocytes, respectively (10, 11). The most important criterion defining MDSCs is their ability to suppress T cells. Since the tumor is the soil for tumor-promoting myeloid cell types, the term MDSCs and its narrow division into PMN- and M-MDSCs is debatable (3). Two observations argue for a more broad term that gathers immature myeloid cells under the same umbrella. First, several conditions have been described to promote the rapid differentiation of M-MDSCs to either PMN-MDSCs, TAMs or TADCs, highlighting the plasticity of myeloid cells and their ability to shift shape in function of the encountered signals (12–14). Second, the hallmark feature, suppressing T cells, is not unique to MDSCs. Also monocytes, DCs and macrophages can acquire a T cell suppressive phenotype in the TME (3, 15). The above findings hamper pigeonholing of the plastic TIM compartment into discrete subsets but force us to acknowledge the fact that the tumor reigns its environment according to its own rules and molds TIMs that functionally engage in a cross-talk, to serve the tumor. Nonetheless, several TIM subsets can display functions that oppose tumor progression, e.g., activation of tumor-rejecting CD8+ T cells. Therefore, we previously proposed to divide TIMs according to their function into myeloid stimulatory cells associated with immune activation, and myeloid regulatory cells associated with immune suppression and wound healing (3).
The functional polarization of TIMs toward aiding or opposing tumor growth is often explained using the T helper (TH) 1/2 paradigm of CD4+ T cells. In particular TAMs and TANs have been subdivided in so-called type 1 and type 2 TIMs, as such categorizing them into TIMs that exert anti- or protumor activities, respectively (16–21). Although the division in type 1 and type 2 is certainly of interest, it should be emphasized that any form of strict classification does not reflect the complexity of the in vivo situation. In vivo TIMs are exposed to a multitude of environmental stimuli, which can give opposing cues, resulting in mixed profiles, rather than a strict type 1 or type 2 profile. This has been well documented for TAMs (22–25).
Mature TADCs and type 1 TAMs (TAM1) are considered immunostimulatory TIMs. Both are characterized by high expression of antigen presenting molecules (e.g., MHC-I & -II), co-stimulatory molecules (e.g., CD80 and CD86), next to secretion of high levels of stimulatory cytokines (e.g., IL-6, IL-12, IL-23, and TNF-α) (24). In contrast, MDSCs, type 2 TAMs (TAM2), and tolerogenic TADCs (tolDCs) are considered immunoregulatory TIMs. These share many functional features that enable strong immunosuppression, among which expression of various enzymes like inducible nitric oxide synthase (iNOS), arginase-1 (ARG-1), and indoleamine 2,3 deoxygenase (IDO), secretion of cytokines such as IL-10 and TGF-β, and expression of co-inhibitory molecules like programmed death ligand 1 (PD-L1; Figure 1) (3). Next to immunosuppression, these immunoregulatory TIMs are responsible for tissue remodeling, promotion of tumor cell proliferation and angiogenesis, thereby favoring cancer progression in various ways.
The protumor traits of TIMs are exploited by most tumors through cultivation of regulatory TIMs till large collections of TAM2, MDSCs, TAN2, and small amounts of immature tolDCs are amongst others established. This installs feed forward loops, as many factors produced by regulatory TIMs enforce the generation, recruitment, and function of new regulatory TIMs. These factors include VEGF, TGF-β, S100A8/9 proteins, et cetera. Because the protumor pathways and mechanisms used by regulatory TIMs show redundancy, tackling only one subset or pathway will likely be insufficient to revert cancer progression. In the following sections we discuss the activities of TIMs in more detail in function of the immunoediting paradigm (6).
Myeloid Cells Have a Cat and Mouse Relationship With Cancer Cells
Immunoediting is a result of a dynamic process of intricate interactions between immune cells and cancer cells. In this process nascent transformed cells are initially recognized and eliminated by various cells of the innate and adaptive immune system. This continuous immune attack results in selection of transformed cells with non-immunogenic traits, which can evade an immune attack. This heralds the equilibrium phase in which malignant cells co-exist with immune cells. Some of these exert antitumor activities, while others exert tumor-promoting activities. Finally, cancer cells are no longer effectively recognized and killed by the immune system, and thus escape to progress in a full-blown malignant tumor. Myeloid cells have been implicated in each phase of this process (Figures 2, 3).
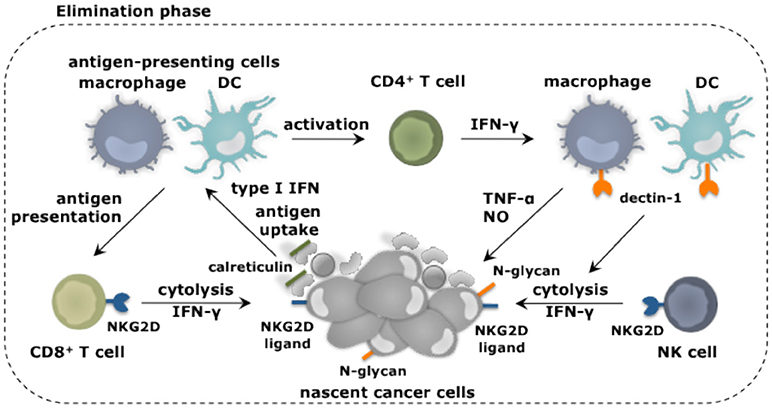
Figure 2. Myeloid cells and their role in elimination of cancer cells. Myeloid cells in addition to other immune cell types fulfill unique as well as redundant functions to achieve tumor cell elimination. Nascent cancer cells are detected by NK cells through the expression of specific ligands, e.g., ligands for the receptor NKG2D. This results in NK cell-mediated killing of the cancer cells, which can be further enhanced through the activation of macrophages and DCs through binding of their receptor dectin-1 to N-glycan structures expressed on certain tumor cells. Cancer cell fragments as well as cancer cells expressing surface calreticulin can be ingested by macrophages (provided that SIRPα is not activated) and DCs. As such these antigen-presenting cells acquire TAAs and can activate CD4+ and CD8+ T cells. IFN-γ produced by these T cells as well as NK cells is one of the mechanisms exploited to kill cancer cells. This cancer cell killing can be further amplified by activation of the tumoricidal program (NO and TNF-α release) of macrophages via IFN-γ.
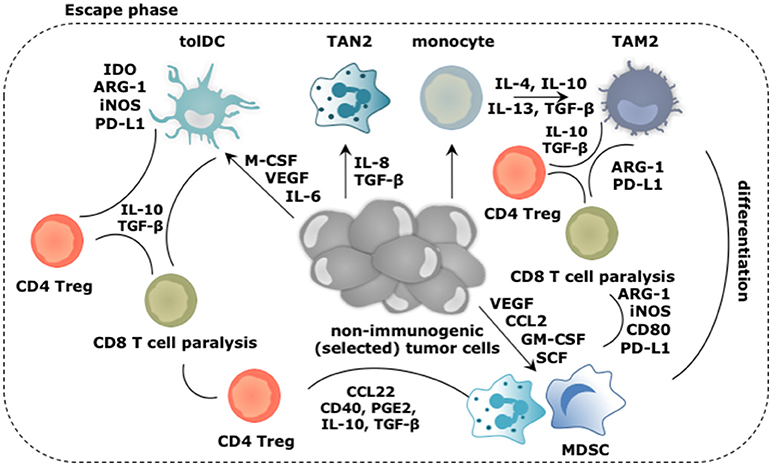
Figure 3. Main roles played by the myeloid cells during the escape of cancer cells from immune mediated destruction. Soluble factors secreted by tumor and immune cells, like CCL2, IL-4, IL-6, IL-8, IL-10, IL-13, GM-CSF, M-CSF, VEGF, and SCF create a TME in which arriving and local myeloid cells are molded into immunosuppressive TIMs. Of these, TAM2, TAN2, and MDSCs are the most abundant, while tolDCs are less frequent in the TME. Cytokines like IL-10 and TGF-β extensively contribute in creating the immunosuppressive TME. These TIMs moreover express a multitude of enzymes (e.g., IDO, ARG-1, iNOS) and surface markers (e.g., CD40, CD80, PD-L1) that support them to dampen antitumor immunity. Moreover, TIMs are not only instructed to suppress antitumor responses under influence of growth factors and cytokines in the TME, they are further instructed to perform activities that are in tune with the needs of the tumor cells. These activities include sustaining chronic inflammation, promoting neo-angiogenesis, tumor growth, invasion and metastasis. These activities are mediated by secretion of soluble factors (e.g., cytokines, growth factors, and proteases). These are not shown in the figure.
The Elimination Phase
Our knowledge on the events occurring during the elimination phase of the immunoediting process is largely inferred from mouse cancer model studies. These show that an intact lymphoid immune cell compartment is central to the principle of cancer cell elimination. The role of myeloid cells in elimination of nascent transformed cells is less frequently studied with only a limited number of publications on the subject. Nonetheless, several cutting edge studies show that myeloid cells contribute significantly to this stage of the immunoediting process.
Transformed cells can express ligands for the receptor natural killer (NK) group 2 member D (NKG2D), which in mice is expressed on NK cells, CD8+ T cells and activated macrophages (26, 27), while in humans its expression is limited to NK cells and CD8+ T cells (28). Binding of NKG2D on NK cells sets of its cytolytic functions (29). Other receptors expressed on NK cells that have been implicated in tumor cell recognition and eradication are NKp30, NKp44, and NKp46 (30), and CD226 (DNAM-1) (31). Also loss of MHC-I, which frequently occurs on cancers cells (32), triggers the cytolytic properties of NK cells, as receptors such as Ly49 that provides inhibitory signals to NK cells, are no longer engaged. Studies show that NK cells with reduced Ly49 expression are unable to reject transplanted tumors, suggesting that proper functioning of NK cells is key to tumor rejection in vivo (33). Macrophages and DCs can enhance NK cell-mediated cytolytic activity against tumor cells (34, 35). It was established through co-culture experiments that certain tumor cells express N-glycan structures, which can bind dectin-1 on macrophages and DCs, resulting in downstream activation of interferon regulatory factor 5 (IRF5) (35). The activity of IRF5 was critical to induce expression of INAM, a membrane bound protein known to activate NK cells via homophilic interaction (36).
It is further well established that transformed cells express antigens generated due to mutations, epigenetic alterations or aberrant processing of proteins. These tumor-associated antigens (TAAs) are displayed on MHC-I molecules and can be recognized by CD8+ T cells (37). These CD8+ T cells can receive co-stimulation via triggering of their receptor NKG2D by ligands expressed on the transformed cells (38). It has been further proposed that tumor-rejecting CD8+ T cells are stimulated by DCs and macrophages that have taken up fragments of NK cell targeted cancer cells (39). Moreover, cancer cells can express surface calreticulin in response to stress (40). The surface expression of calreticulin serves as an “eat me” signal, allowing DCs to acquire TAAs, which can be cross-presented to CD8+ T cells. It was shown that endogenous type I IFN is required to initiate such an antitumor CD8+ T cell response, since mice of which the CD8α+ DCs lack IFNAR are unable to induce lymphocyte-mediated tumor rejection (41). This endogenous type I IFN could be a result of a DNA damage response, resulting in sensing of cytosolic DNA by the cGAS/STING pathway (42). Next to DCs, macrophages can acquire TAAs by ingesting calreticulin positive cells. As such they could further assist in the activation of CD8+ T cells. However, this function of macrophages is counteracted by the expression of SIRPα. This receptor binds to CD47, which is constitutively expressed in various cancer types and serves as a “don't eat me” signal (43).
Another way in which macrophages could contribute to the elimination of nascent cells is through release of nitric oxide (NO) and TNF-α, which in experimental settings can be induced through NK2GD triggering (27). Also other stimuli, which are more relevant to the human setting, such as IFN-γ, anti-CD40 and toll-like receptor (TLR) ligands, can induce the release of NO and TNF-α, thereby stimulating the tumoricidal activity of macrophages (44). At least for IFN-γ, there are studies that suggest this might occur during the early stages of cancer formation. It was shown in a model of MHC-II negative multiple myeloma that macrophages are rapidly recruited and present TAAs to CD4+ T cells, which in turn activate the tumoricidal program in macrophages through secretion of IFN-γ (45). Also NK cells and CD8+ T cells produce high levels of IFN-γ, thereby provoking the hypothesis that also these lymphocytes can stimulate the tumoricidal properties of macrophages.
The Equilibrium Phase
The coordinated immune attack, executed during the elimination phase, can annihilate transformed cells. However, continued pressure of the immune system can result in selection of cancer cells that can prevail. Cancer cells can avoid immune recognition in several ways, among which downregulation of antigen processing machinery, or expression of TAAs or MHC-I molecules. Consequently, cancer cells with reduced immunogenicity are selected for. Several preclinical studies have provided evidence for a role of CD8+ T cells in this process (46). The clinical relevance hereof was suggested in several cancer trials, showing selective downregulation of the targeted TAA or MHC-I in recurring tumors (47–49). Also myeloid cells were shown to play a role in shaping the immunogenicity of tumors. By educating CD8+ T cells, they contribute indirectly to the tumorigenicity. A more direct action was suggested for TAM1 (50). It was shown that in the in absence of T cells, NK cells can activate TAM1 through the production of IFN-γ, and that these TAM1 served as modulators of the tumor's immunogenicity. In addition to camouflaging themselves, cancer cells co-opt regulatory circuits to dampen immune responses. These include but are not limited to expression of PD-L1, recruitment of regulatory T cells (Tregs), and regulatory myeloid cells. The many mechanisms exploited by regulatory cells to paralyze tumor-specific effector cells and as such create a tolerant environment for cancer cells has been reviewed elsewhere (51, 52). Consequently, tumor clones survive the elimination checkpoint and form a clinically occult tumor that co-exists with host immune cells. During this dynamic equilibrium, immune cells with anti- and protumor activities can be found in the vicinity of the tumor cells (53).
Cell death coinciding with the presence of surface calreticulin and alarm signals such as type I IFN is key to antitumor immune activation by DCs (41). This type of cell death, when further associated with release of adenosine triphosphate (ATP), high mobility group box 1 (HMGB1) proteins, and annexin A1 (ANXA1), has been coined immunogenic cell death (ICD). While cell death due to ROS in combination with endoplasmic reticulum (ER) stress leads to ICD in tumor cells, apoptosis induced by exposure of tumor cells to IFN-γ and TNF-α is less immunogenic (54). This can induce quiescence, implicated in tumor dormancy (55, 56). Also, the pro-inflammatory signals that HMGB1 normally triggers on myeloid cells are strongly diminished when this protein gets oxidized (57). Moreover prolonged production of ROS and RNS by myeloid cells can increase the events of DNA mutation in tumor cells, and as such be a cause of cancer progression in itself (58). Several mechanisms are built into the inflammatory cascade that occurs during the elimination phase, to prevent excessive collateral tissue damage and auto-immunity. For example, debris clearance through phagocytosis and autophagy results in secretion of TGF-β, which functions as a strong immunosuppressant and activates type 2 polarized TIMs, resulting in CXCL5-mediated inflammation and tumor growth (25, 59). Even TH1-linked cytokines can help in the accumulation of TIMs with regulatory functions. Both IFN-γ and IL-1β were shown to participate in the mobilization, recruitment, and inhibitory activity of MDSCs (60). Transcription factors that govern these events shift from pro-inflammatory IRF8 and signal transducer and activator of transcription (STAT) 1, 2, 3, and 6 to CCAAT-enhancer-binding protein β (C/EBPβ) and PI3Kγ, generating immature TIMs. Moreover glycolysis, the metabolic pathway preferred by TIMs and tumor cells during the elimination phase, mediates production of proteins, nucleic acids, lipids, lactate, adenosine, and consequently immunosuppressive acidification of the TME. As a result TIMs undergo peroxisome proliferator-activated receptor gamma (PPARγ) and STAT-mediated metabolic reprogramming (61). They switch from glycolysis to oxidative phosphorylation and lipid catabolism (fatty acid oxidation). Inhibition of the glycolysis pathway in TAN1, TAM1, and mature DCs shortcuts their activities, thereby favoring the activity of tumor-promoting TIMs, eventually leading to the escape phase, where immunologically sculpted tumors grow progressively, become clinically apparent and have an established immunosuppressive TME (62).
The Escape Phase
The early immune response to cancer cells is purposed to deal with the danger at hand and is characterized by acute inflammation. However, persistence of cancer cells coinciding with the co-existence of immune cells with pro- and antitumor properties results in smoldering inflammation. This chronic inflammation is mainly sustained by TAM2, TAN2, and MDSCs, which are the main TIM subsets found within many established cancers. This is not surprising as tumor cells produce factors such as G-CSF, GM-CSF, and VEGF to ensure continuous recruitment of myeloid progenitors into the circulation. Under physiological conditions, GM-CSF drives myelopoiesis as well as DC differentiation, while G-CSF and M-CSF establish the differentiation, proliferation and survival of granulocytes and macrophages respectively (10). In chronic inflammation however, GM-CSF downregulates IRF8 in DC progenitors, and thus results in reduced DC development next to recruitment of more MDSCs. Furthermore, M-CSF suppresses the differentiation of DCs while enhancing TAM2 polarization (63). Therefore, this factor has been attributed as a predictor of poor prognosis (64). G-CSF can mobilize granulocytic myeloid cells from the bone marrow to promote angiogenesis. Moreover, immature TIMs will drive the recruitment of more regulatory TIMs resulting in a “never-healing-wound.” Factors that aid in the recruitment of myeloid cells, such as monocytes, DCs, MDSCs, and TAN2 to the TME, are among others IL-4, IL-6, IL-8, IL-10, S100A8/A9, VEGF, and TGF-β (65).
The tumor promoting functions of TIMs are typically activated by TLR triggering and binding of cytokines and growth factors such as IFN-γ, IL-4, IL-6, IL-10, IL-13, and TGF-β to their responding receptor. The signaling cascade following receptor engagement results in upregulation of various transcription factors, including STAT1, 3 and 6, mitogen activated protein kinase-extracellular regulated kinase (MAPK-ERK) and nuclear factor-κB (NF-κB), with STAT3 being a master regulator in most myeloid cell subsets (66–68). Once activated, TIMs perform overlapping functions. TAM2 and MDSCs are well known for their ability to suppress T cell-mediated tumor killing (Figure 3) since both can exert direct immunosuppressive effects using cell-cell contact (e.g., CD80 and PD-L1), secreted factors (e.g., IL-10 and TGF-β) and expression of enzymes (e.g., ARG-1, iNOS, IDO) (69–75). In addition, TAM2, and MDSCs engage Tregs to aid them in suppressing antitumor immune responses. To that end, these TIMs express and produce several factors that stimulate the differentiation (e.g., IDO, CD40, prostaglandin E2 [PGE2], IL-10, TGF-β), or recruitment of Tregs (e.g., CCL22) (76–78). TAM2 can further interfere with the activity of TAM1, suppressing the expression of IFN-γ and IL-12, thereby impacting on direct tumor cell killing as well as the activation of killer T cells (65). Although TAN2 are not notorious for their T cell suppressive activity, it has been shown that eliminating TAN2 results in increased cytotoxic T cell activity, a phenomenon that was linked to TGF-β, which favors TAN2 accumulation and induces ARG-1 expression (16, 79). Although DCs are not abundantly present in the established TME, it is essential to discuss their role in facilitating the escape of tumor cells from immune killing (Figure 3). Conditions in the TME like accumulation of extracellular adenosine and lactate, VEGF, M-CSF, TGF-β, IL-6, and hypoxia, act in concert to inhibit antigen presentation by DCs, therefore inhibit activation of adaptive antitumor immunity (80–82). Moreover during tumor progression, subsets of DCs are able to reach tumor-draining lymph nodes and stimulate proliferation of Tregs in a TGF-β dependent way, turning immature DCs from a victim to a partner in crime (83). It has even been described that mature DCs within the TME can exert protumor properties through expression of IDO, ARG-1, and iNOS. These mature but tolDCs block the proliferation of T cells and in the presence of TGF-β further stimulate Treg differentiation (84–87).
Promoting angiogenesis by secreting VEGF, angiopoietin 1, and 2 and GM-CSF, and tumor cell dissemination by secreting MMPs and cathepsins are two other features shared between TAN2, tolDCs, TAM2, and MDSCs (88). Moreover, it has been proposed that TIMs like MDSCs and TAN2 are responsible for creating a pre-metastatic niche (89–93). In these pre-metastatic niches, factors are produced to attract and later on arrest tumor cells such as IL-1β, prokineticin 2, and MMPs while neighboring cells are stimulated to secrete VEGF to facilitate the arrival of tumor cells.
In conclusion, the TME instructs TIMs to perform functions in tune with the tumor's needs. These functions are related to chronic inflammation, tissue remodeling and immunosuppression, and promote tumor growth and metastasis, as these functions enable tumor cells to acquire nutrients, proliferate, survive, migrate, and escape immune elimination.
Cancer Therapies and Myeloid Cells: Mutual Influence and Implications
Experimental studies have provided ample evidence that myeloid cells influence virtually every type of cancer therapy, ranging from chemotherapy (CT), radiotherapy (RT), tumor targeted therapy to immunotherapy (Figure 4). Therefore, the following sections will briefly discuss the interplay with various cancer therapies.
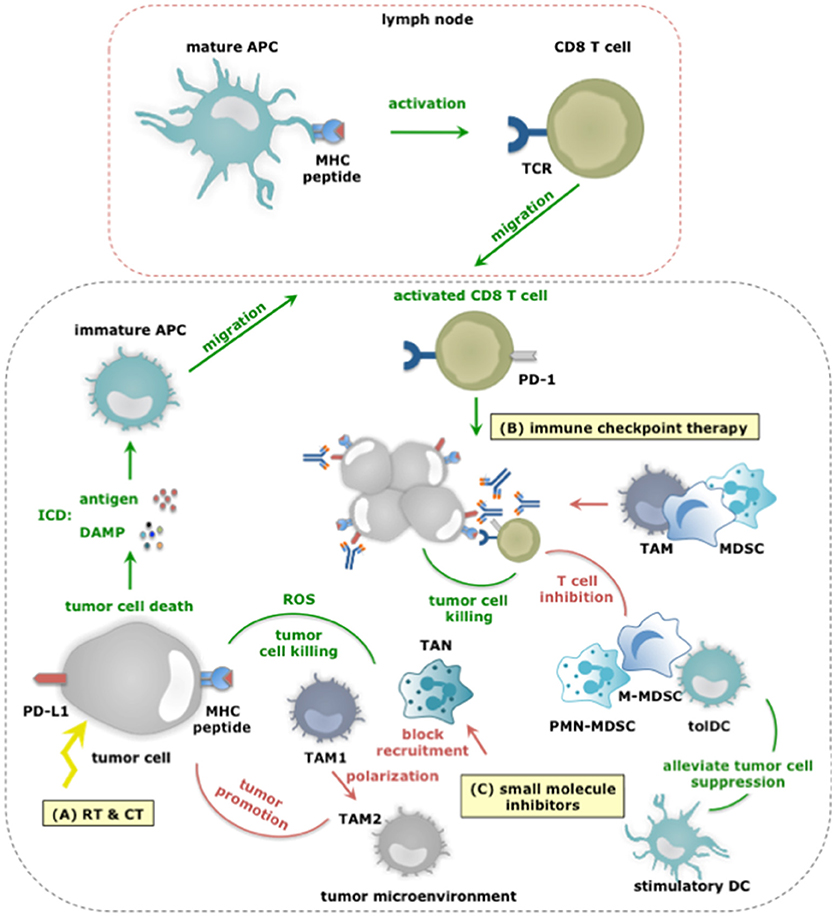
Figure 4. The dual role of myeloid cells in anticancer therapy responses. (A) RT and CT regimens can induce ICD, thereby alarming antigen-presenting cells (APCs) of the danger that is posed by cancer cells. This alarm is given through the release of various DAMPs (e.g., HMGB1, ATP, et cetera) and facilitates cross priming of tumor-specific CD8+ T cells in tumor draining lymph nodes. Activated CD8+ T cells subsequently infiltrate the tumor in search of TAA-expressing cancer cells, and upon recognition exploit various mechanisms to kill cancer cells. While, TAM1 and TANs can further enhance the efficacy of CT by releasing ROS, thereby enhancing tumor cell death, other TIMs including MDSCs can counteract the effects or RT and CT not in the least by inhibiting CD8+ T cells. (B) Within the TME, CD8+ T cells are often rendered tolerant via immunosuppressive factors expressed by tolDCs, TAM2, MDSCs, tumor cells (and Tregs). One strategy is expression of PD-L1 that binds to PD-1 on activated T cells. Consequently, immune checkpoint inhibition has been studied to alleviate PD-L1:PD-1 mediated immunosuppression. Monoclonal antibodies are actively used for this purpose, however, were shown to be captured by TAMs using their FcγR and were shown to be counteracted by MDSCs. Nonetheless, the success of immune checkpoint inhibition is correlated to the presence of immunostimulatory TIMs and CD8+ T cells within the TME. (C) Small molecule inhibitors targeting protein kinases implicated in tumor cell progression have been shown to exert effects on TIMs. Some of these effects are beneficial for therapy outcome. For instance BRAF inhibitors revert the suppression exerted by melanoma cells on TADCs. However the majority of these effects potentiate TIMs to exert tumor-promoting functions or prevent TIMs from entering the TME to oppose tumor growth. Examples hereof are Imatinib that stimulates TAM1 to TAM2 polarization, BRAF inhibitors that restore the MDSC compartment through induction of CCL2 and MET inhibitors that avoid recruitment of neutrophils with cytotoxic capactity (iNOS mediated NO release).
Myeloid Cells and Radiotherapy
Radiotherapy (RT) uses ionizing radiation to kill malignant cells and can induce full-blown ICD in a dose-dependent manner (94). ICD allows TADCs to acquire TAAs as well as stimuli that enforce their maturation, therefore enable them to activate tumor-specific CD8+ T cells (95, 96). Moreover, low doses of RT can reprogram TAMs into TAM1, which can contribute to the recruitment and activation of tumor-specific T cells (97). However, RT-mediated immunogenic effects can be masked (98, 99). In an established TME, TIMs are instructed to counteract antitumor immune responses. Depending on the dose, its fractionation and also the tumor model, RT can recruit myeloid cells and even polarize them into tumor-promoting immunosuppressive cells (100). The latter finding suggests that depletion of TIMs could be a strategy to enhance the outcome of RT. It was indeed shown in several experimental studies that depletion of TAMs, MDSCs and neutrophils enhances the efficacy of RT (101–103). Caution must however be taken to ensure that RT-mediated effects on TADCs and TAM1 polarization are not lost, as both contribute to the abscopal effects of RT.
Myeloid Cells and Chemotherapy
Chemotherapy (CT) comes in many colors and shapes. Similar to RT certain CT regimens, like use of anthracyclines, can induce full-blown ICD, thereby acting on TADCs, which in turn activate tumor-specific CD8+ T cells. The efficacy of anthracyclines was shown to depend on TADCs (104, 105). Also TAMs can play an advantageous role during CT treatment. These cells can act as drug depots when CT compounds are delivered in nanoparticles, thereby ensuring local and prolonged delivery of the therapy (106). Moreover, ROS production by TAMs and TANs is observed upon treatment with oxaliplatin, and aids in stimulating tumor cell death (107). Chemotherapies can further act on TIM numbers, either depleting or amplifying them. Gemcitabine and 5-fluorouracil are examples of chemotherapies that reduce MDSC numbers. By relieving MDSC-mediated immunosuppression these treatments enable CD8+ T cell-dependent anticancer immune responses (108). Other chemotherapeutics, like paclitaxel show a more complicated profile. While paclitaxel depletes MDSCs (which express P450 reductase), it increases TAMs (12, 109). In case of the latter, inhibiting TAM accumulation enhanced therapy outcome (110). This study suggests that TAMs can hamper the efficacy of CT. This is confirmed by several other studies, and unfortunately is not limited to TAMs. Also MDSCs have been linked to CT failure. TAMs and MDSCs can induce drug resistance for instance by producing cysteine cathepsins that protect tumor cells from being killed by chemotherapeutic agents like taxol, and even promote tumor growth by enforcing chronic inflammation (111, 112). Moreover, TAMs can secrete IL-10 in response to CT and as such enforce immunosuppression (113). Thus, TIMs are both friend and foe when treating cancer patients with chemotherapies (and RT), posing the challenge to identify and selectively act on the enemy, while leaving friendly TIMs unharmed.
Myeloid Cells and Tumor Targeting Small-Molecule Cancer Therapy
Our growing knowledge on driver mutations that provide cancer cells with a growth and survival advantage has allowed the development of small-molecule drugs that target the molecular alterations arising from these mutations. Proteins with kinase activity such as stem cell factor receptor (c-kit), BRAF, MEK, and MET are examples of targets for which small-molecule inhibitors were clinically tested. In clinical trials these small-molecule inhibitors initially reduce the tumor size (114, 115). However, they have little effect on the long haul, which might at least in part be due to activities of myeloid cells. For instance Imatinib, a tyrosine kinase inhibitor that works via activating mutations of c-kit, has been evaluated in gastrointestinal cancer. Imatinib was shown to induce tumor cell apoptosis. While this is the envisaged outcome, it was further observed that TAM1, which mainly populated the gastrointestinal stroma were polarized to TAM2 due to activation of C/EBPβ upon interaction with dying tumor cells (116). BRAF and MEK are protein kinases of the MAPK-ERK pathway, which in solid tumors such as melanoma enable proliferation and cell survival. Consequently, mutations of BRAF and MEK, which continuously activate MAPK-ERK are linked to oncogenesis. Small-drug inhibitors were developed to counteract the uncontrolled growth and survival of cancer cells through BRAF and MEK mutations (117–120). Development of resistance to BRAF inhibitors in a mouse melanoma model was linked to restoration of the MDSC compartment. This depended on reactivation of the MAPK pathway followed by production of CCL2, a myeloid cell attractant. Modulating MDSCs using depleting antibodies (anti-Gr-1) or blocking their recruitment (CCR2 antagonist) prohibited the outgrowth of these BRAF resistant melanoma tumors (121). Also TAMs and TAM-derived TNF-α have been implicated in failure of BRAF inhibitors in mouse melanoma models (122). Blocking TAM development (inhibition M-CSFR) improved the efficacy of BRAF inhibitors (123). On the other hand, MEK, and BRAF inhibitors revert TADC suppression enforced by mutated melanoma cells in vitro (124, 125). Finally MET (hepatocyte growth factor receptor) is considered a molecular target in several cancers, including non-small cell lung cancer, gastrointestinal cancer, and hepatocellular carcinoma (114). Inhibition of MET also impacts on neutrophils, which rely on MET activity to home to tumors and exert cytotoxic responses such as iNOS-mediated release of NO (126). The above studies show that further studies are required to understand how inhibiting kinases impact on myeloid cells and how myeloid cells can be manipulated to allow more durable responses in patients.
Myeloid Cells and Immune Checkpoint Therapy
Immune activation and activity are tightly regulated by several mechanisms to avoid immunopathology (e.g., auto-immunity). Cancer cells hijack these mechanisms to escape immunosurveillance. They express ligands, such as PD-L1, which triggers PD-1, an inhibitory receptor expressed on tumor-reactive immune cells, such as CD8+ T cells. Consequently, the tumor-reactive immune cells become paralyzed. Monoclonal antibodies (mAbs) that bind PD-1 or PD-L1 and block this inhibitory immune checkpoint have ushered a new era for cancer immunotherapy (127–129). To date, five mAbs that target the PD-1:PD-L1 pathway have been approved for treatment of patients with solid tumors, including melanoma and non-small cell lung cancer. These mAbs are pembrolizumab and nivolumab, which antagonize PD-1, and avelumab, darvulumab and atezolizumab, which antagonize PD-L1. These mAbs have signified a turning point in the treatment of a significant number of patients (130). Regrettably the full potential of these mAbs has not yet been achieved.
A considerable number of patients and cancers are refractory at the start of treatment or become resistant during the course of treatment and therefore fail the therapy. Several hypotheses exist to explain this therapy failure, one of them being that TIMs are responsible for the resistance to immune checkpoint blocking therapy. This hypothesis resulted from the observation that high infiltration of tumors with immunosuppressive myeloid cells correlates with poor prognosis and immune checkpoint therapy resistance (131, 132). Mainly MDSCs and TAMs have been implicated in resistance to immune checkpoint blocking mAbs. Therefore, merely blocking PD-1:PD-L1 interactions might be insufficient to overcome TIM-mediated inhibition of CD8+ T cells. A line of thought that is reinforced by the observation that MDSC depletion in experimental models was shown to enhance antitumor immune responses and overcome resistance (133). Moreover, functional modulation of MDSCs by epigenetic drugs, sensitized several experimental cancer models that were previously resistant to immune checkpoint therapy (134, 135). Further TAMs have been described to inhibit PD-1:PD-L1 therapy by removal of anti-PD-1 mAbs from PD-1+ CD8+ T cells using their FcγR (136). Additionally emerging data report that both metabolic and inflammatory pathways can enhance the expression of PD-L1 on myeloid cells (71, 137, 138). These studies provide the first proof that immunosuppessive TIMs can hamper immune checkpoint therapy via several redundant strategies. However, there is also proof that immunostimulatory TIMs are a pre-requisite for immune checkpoint success. It was shown that binding of monocytes and DCs by anti-PD-L1 mAbs is at least in part responsible for the subsequent control of tumor growth (139, 140). The need for DCs to guarantee successful checkpoint therapy has further been evidenced in experiments, in which delivery of bone marrow-derived DCs to tumors resulted in T cell recruitment and sensitization to anti-PD-1 therapy (141). Similar to the conclusion on myeloid cells in RT, CT and targeted therapies, we can conclude that immune checkpoint therapy can be supported by TIM allies, i.e. TIMs that can activate CD8+ T cells, while immunosuppressive TIMs that exert several means to paralyze CD8+ T cells, can be regarded as the saboteurs of this therapy. Besides the PD-1:PD-L1 pathway, other checkpoint inhibiting receptors or their ligands have been reported to be expressed by one or more TIM subsets. For instance, the expression of T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) has been described in TADCs while ‘V-domain Ig suppressor of T cell activation’ (VISTA) is predominantly expressed on hematopoietic cells, and in multiple murine cancer models found at particularly high levels on TIMs. Further the “T cell immunoreceptor with Ig and ITIM domains” (TIGIT) has been reported to bind CD155 on DCs and macrophages while the main ligand of “Lymphocyte-activation gene 3” (LAG-3) is represented by MHC-II, expressed in various degrees on almost all TIMs. Future investigation is therefore warranted to find out which role TIMs play in other checkpoint inhibiting mechanisms.
Strategies Developed to Target Myeloid Cells in Order to Resolve Cancer
An increasing number of experimental and clinical studies have been published on targeting of myeloid cells in cancer with close to 400 studies published in the last 5 years. Overall the approaches to target myeloid cells with tumor-promoting characteristics like TAM2, TAN2, and MDSCs can be divided into two main strategies; reducing their numbers by depleting them or by blocking their recruitment, and repolarization of their function. Conversely, attempts have been made to enhance the numbers and function of myeloid cells with antitumor properties. In the following sections we highlight several promising strategies (Figure 5).
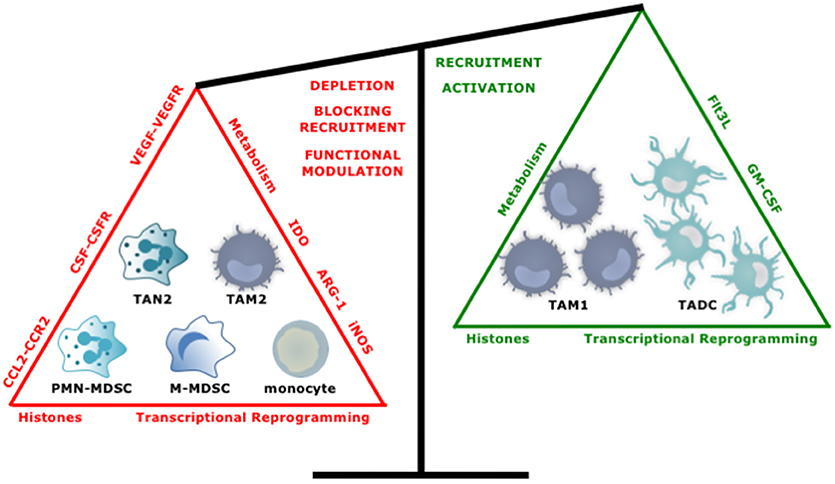
Figure 5. Tipping the balance toward myeloid cells with an antitumor phenotype. Several approaches have been studied to increase the ratio of anti- over protumor TIMs. These include, depleting or repolarizing tumor-promoting TIMs, and attracting and activating antitumor TIMs.
Manipulating Myeloid Cell Numbers
Depleting myeloid cells and even specific subsets, as a strategy to reduce myeloid cell numbers, can be performed using antibodies that target myeloid cell specific surface makers such as CD11b, Gr-1 or Ly6G. Furthermore also transgenic mouse strains can be used with permanent or conditional myeloid cell ablation. Conditional deletion circumvents embryonic lethality while precluding analysis of gene function on a well-defined time point in the adult tissues. The two most commonly used models are based on the addition of diphtheria toxin and/or Cre/LoxP recombinase. The former model is based on a knock-in of the diphtheria toxin receptor behind the promotor of a cell type characterizing marker such as CD11b or CD11c-diphteria toxin receptor mice. In contrast, the generation of conditional Cre/LoxP knockout mice is a multiple-step process, which involves mating flox mutant with tissue specific Cre-expressing mouse lines. As such the latter allows conditional myeloid cell subtype specific ablation in a particular tissue of interest.
Such experimental approaches have delivered a proof-of-concept that depletion of myeloid cells with immunosuppressive functions can delay tumor growth, while depleting stimulatory myeloid cells has the opposite effect (142, 143). As these approaches do not limit myeloid cell depletion to the TME, it is however not certain that the observed effects can in part be ascribed to depletion of myeloid cells in the periphery. Moreover, complete depletion strategies are not attractive for translation to a human setting. Therefore, other pharmacological and gene-based approaches have been developed to interfere with the accumulation of suppressive myeloid cells. These are based on our current knowledge on how TAM, MDSC and TAN myelopoiesis and recruitment are driven. Accumulation of TAMs in the TME depend strongly on CCL2:CCR2 and M-CSF:M-CSFR signaling, therefore different approaches using mAbs (144–146), small-molecule inhibitors (145, 147, 148) and RNA interference (149) were devised to block these pathways. Overall these studies showed reduced TAM numbers and correlating herewith, delayed tumor outgrowth in various mouse cancer models. These studies further provided insight into the effect of CCL2:CCR2 and M-CSF:M-CSFR blockade, and how treatment regimens might evoke unwanted effects.
When blockade of the CCL2:CCR2 axis was halted in mice, a sudden burst of monocytes released from the bone marrow was observed, and an increased frequency of metastasis and linked herewith decreased survival of animals was the consequence (110). Use of CCL2 versus CCR2 knockout mice further revealed that deficiency in CCL2 resulted in delayed outgrowth of mammary carcinoma, while deficiency in CCR2 enhanced tumor outgrowth (150). Differences in transcriptional programs in monocytes deficient in CCR2 vs. CCL2 might explain these disparate effects of CCR2 vs. CCL2 disruption on progression of mammary carcinoma. While this study did not show differences in monocyte or TAM infiltration, it is conceivable that other TIM subsets might play a role. For instance, also MDSCs can act on CCL2 through their expression of CXCR2 (151). Similar to interfering with the CCL2:CCR2 axis and TAM accumulation, interfering with both the CCR2+ TAMs and CXCR2+ TANs has profound effects at least when used in combination with CT (152, 153). It is therefore of utmost importance to identify all relevant players when blocking a certain molecule in order to fully understand the potential strengths and weaknesses of the depletion approach.
Targeting the M-CSF:M-CSFR pathway is also interfere with TAM accumulation. This approach delays tumor growth in all experimental models tested, either when used as a stand-alone therapy (146, 147), or when used in combination with other therapies (101, 102, 109, 113). Certain studies in which the M-CSF:M-CSFR pathway was targeted reported on tumor regression, which was not linked to numerical changes in TAMs in the TME. However, changes in the TAM phenotype, particularly downregulation of TAM2 associated features, were reported in these studies (145, 149). Because interrupting CCL2:CCR2 and M-CSF:M-CSFR signaling has been beneficial in many experimental models, these pathway are being evaluated in patients both as mono- (146) and combination therapies (CT and immune checkpoint blocking therapy) (154).
Another strategy that is explored in the context of reducing tumor-promoting myeloid cell numbers in the TME is interfering with VEGF:VEGFR signaling. VEGFR is highly expressed by TIMs and interaction with VEGF contributes to their migration and differentiation into immunosuppressive cells (155). Thus, interfering with VEGF:VEGFR signaling might have a dual role on TIMs, reducing their numbers and repolarizing their function (in addition to affecting angiogenesis). In view of attracting TIMs, VEGF:VEGFR signaling was shown to attract macrophages (156) and monocytes in a CCR2-dependent way (157). It was further shown that also recruitment of MDSCs to the TME of non-small cell lung carcinoma is reduced upon blockade of VEGF:VEGFR signaling. However, recruitment of monocytes increased upon treatment with anti-VEGF antibodies in this study (158). Several anti-VEGF-based drugs, including bevacizumab, avastin and axitinib, targeting VEGFR are used for treatment of various human cancers (159–161). While the anti-VEGFR targeting drugs used in these studies are mainly studied in view of angiogenesis, it was shown at least for the drug axitinib that it impacts on myeloid cells. A dose-dependent reduced activation of STAT3 was observed in axitinib-exposed DCs. Despite this favorable change, these DCs were impaired in their T cell stimulatory activity (162). In contrast, it was shown in a model of melanoma brain metastasis that axitinib increases the number of M-MDSCs, and reverts their function from T cell suppressive to stimulating cells with a subsequent delay in disease progression (163). Therefore, this study suggests that enriching myeloid cells with antitumor properties might be an attractive approach to treat cancer. This idea is supported by experimental and clinical studies in which ex vivo DCs (often engineered to secrete stimulatory cytokines), delivered to the tumor, mediate disease control (164, 165).
The use of Flt3-L to enhance DC numbers has been extensively studied, since Flt3:Flt3-L mediated signaling is involved in the generation of various DC subsets from bone marrow progenitor cells (166, 167). In experimental models, delivery of Flt3-L (alone or in combination with CT) increased DC numbers and resulted in delayed tumor outgrowth (168–171). Evidence supporting the use of Flt3-L in a clinical setting came from a study in which recombinant human Flt3-L was administered to expand DCs in vivo. These DCs were collected by leukapheresis, pulsed with tumor antigen-derived peptides and used for vaccination, eliciting tumor regression in 2 out of 12 patients (172).
Also GM-CSF has been studied as a means to enhance DC numbers in vivo. This might seem controversial given the role of GM-CSF in myelopoiesis. It is certainly a strategy that has to be approached with caution, as blocking GM-CSF has been shown to reduce accumulation of monocytes and myeloid precursor cells, and was shown to delay tumor progression, while delivering GM-CSF to tumors, for instance using the oncolytic virus T-VEC, was shown to induce tumor-specific T cells, most likely as a consequence of DC activation. In this clinical study, delivery of GM-CSF by T-VEC also resulted in a reduced number of MDSCs (173, 174). The latter is surprising as engineering of tumors to express high levels of GM-CSF has been linked to MDSC accumulation in various experimental models (175, 176).
Overall these studies support the idea that decreasing the number of TIMs with tumor-promoting activities, while increasing TIMs with antitumor activities, is a valuable approach in the fight against cancer. These studies however also point out that tipping the balance toward increased numbers of antitumor TIMs by acting on pathways involved in myelopoiesis is a challenging endeavor.
Manipulating the Function of Myeloid Cells
Myeloid cells are characterized by high plasticity. Their ability to shift shape according to the cues they receive, has instigated research into strategies that promote their anti- and/or inhibit their protumor activities.
Manipulating Genetic Programs in Myeloid Cells
Genetic programs, controlled by transcriptional networks, and epigenetic mechanisms, dictate the phenotype, and function of myeloid cells. Therefore, reprogramming them has been a focus of interest. Triggering TLRs on TIMs, in particular TLR3 and TLR9, has been extensively studied in this context, as these jumpstart transcriptional networks. It was shown that the T cell suppressive activity of MDSCs is reduced when exposed to agonists of TLR3 or TLR9 (177–180). Exposing MDSCs to the TLR9 ligand CpG stimulated them to produce TH1 activating cytokines and even differentiate into TAM1, a switch that was driven by IFN-α, produced by plasmacytoid DCs after CpG stimulation (178, 181). Besides its effect on MDSCs, TLR3 triggering also converts TAM2 to TAM1 and facilitates cross-priming of antigen-specific CD8+ T cells by TADCs, thereby promoting tumor regression (182, 183).
The use of TLR4 to functionally reprogram DCs was achieved by a DC unique genetic approach, which is likely explanatory for its success, as it was previously shown that inflammation primed MDSCs are activated by lipopolysaccharide (LPS), a well-known TLR4 ligand, to produce increased levels of IL-10 and to downregulate production of IL-12 by macrophages (184).
The CD40:CD40 ligand (CD40L) interaction may represent another interesting target to dampen the protumor transcriptional program of MDSCs, while stimulating the antitumor transcriptional program of TAMs and TADCs. Agonistic CD40 mAbs were shown to abrogate MDSC:Treg communication and to mature MDSCs (185, 186). Macrophages are activated upon treatment with these CD40 mAbs, enabling them to infiltrate tumors and exert tumoricidal activities (187). The observation that anti-CD40 mAbs enable CD8+ T cell independent tumor cell killing, prompted the use of a fully humanized CD40 agonist mAb (CP-870.893) in a phase I clinical trial, showing that its combination with gemcitabine activates antitumor immune responses (188). Activation of DCs by CD40 licenses them to perform their CD8+ T cell stimulatory function (189). Combining CD40 and TLR3 ligands was shown to endow human and mouse tolDCs with stimulatory properties (190). Also TLR4 in addition to CD70 delivery was studied in combination with CD40 activation of TADCs (191). It was shown that TADCs modified with CD70 and simultaneously activated via CD40 and TLR4, were able to migrate to tumor draining lymph nodes and stimulate tumor-specific CD8+ T cells.
Cytokines represent another means of functionally reprogramming myeloid cells. In this context, IL-12 and type I IFNs have been shown to stimulate antitumor properties, while IL-10 and TGF-β have been linked to tumor-promoting properties. Consequently, exposing TIMs to IL-12, IFN-α or IFN-β, and/or shielding them from IL-10 and TGF-β has been studied. Delivery of IL-12 to the TME was shown to have substantial antitumor effects, which were correlated to reprogramming of MDSCs, TADCs and TAMs into antigen presenting cells with CD8+ T cell activating capacity (175, 192–196). The antitumor stimulating properties of IL-12 were confirmed by the clinical responses observed in patients with renal cell carcinoma, melanoma and peritoneal metastasis from ovarian cancer upon IL-12 treatment (197–202). Type I IFNs, in particular IFN-α and IFN-β, have long been shown to exert tumoricidal effects and act as strong inducers of myeloid cell polarization, mainly for MDSCs and TADCs (203, 204). Although IFN-α has shown promising clinical results in hematopoietic cancers, little success was achieved in solid tumors (205). This might be related to its systemic administration or its use as a single agent in these trials. It was shown in preclinical studies that combining IFN-β stimulation of myeloid cells with simultaneous blockade of TGF-β signaling, is a viable strategy to reprogram MDSCs and DCs, thereby facilitating CD8+ T cell responses (206). Also IL-10 switches on immunosuppressive transcriptional programs in myeloid cells. IL-10 receptor blocking antibodies, used alone or in combination with CpG, restored IL-12 expression through activation of DCs, and stimulated antitumor responses (113). However, in some studies ablating IL-10 was associated with an increased level of MDSCs and Tregs in TME and in the tumor-draining lymph nodes (207).
Strategies to manipulate the transcriptional programs in myeloid cells more directly have been developed as well, including approaches to down- or upregulate transcription factors like STAT3 and NF-κB (208, 209). Transcription factor STAT3 has been designated the master regulator of the immunosuppressive activity of myeloid cells (66, 67). Studies with small-molecule inhibitors, acting on JAK2:STAT3 signaling, showed delayed tumor growth, which in some cases coincided with a TAM2 to TAM1 switch, an increase of MDSCs or was independent of TIM modulation (210, 211). In clinical trials, small molecular inhibitors showed limited efficacy and substantial side effects, prompting the development of more targeted and more specific strategies based on RNA interference and decoy oligonucleotides. Some of these entered clinical testing (212–214). More selective delivery of STAT3 inhibiting small interfering RNA (siRNA) was achieved by coupling them to CpG. Therefore, this strategy takes advantage of the positive effects of CpG on TLR9 positive myeloid cells as well as the potential effects of STAT3 inhibition. Upon treatment, TLR9 expressing myeloid cells like PMN-MDSCs, showed reduced immunosuppressive capacity. Also TLR9 expressing tumor cells lost the resistance to apoptosis by interfering with STAT3 signaling (212, 213, 215, 216). The less activated status of TADCs and their overall reduced ability to respond to TLR stimulation has been related with STAT3 hyperactivity (215). Therefore, STAT3 inhibition has been studied in the context of TADC modulation. Herein, advantage was taken of the TADCs' ability to take up nanoparticles to deliver STAT3 siRNA and polyinosinic:polycytidylic acid (polyI:C), a well-known agonist of TLR3. This strategy enabled TADCs to upregulate the expression of co-stimulatory molecules and IL-12, and to induce potent antitumor immune responses (215). Next to STAT3, NF-κB is known to regulate the expression of many tumor-promoting genes, including VEGF, IL-6, TNF-α, and cyclooxygenase 2 (COX2), which support its crucial role in the activation of TAM2 in the TME (217, 218). Inhibition of NF-κB allows TAMs to acquire a tumoricidal phenotype, resulting in vivo in regression of advanced tumors (219). In TADCs, NF-κB activity is inhibited by FOXO3, which has been related to the TADCs' immunosuppressive activity (220). Activation of NF-κB in DCs has been extensively studied albeit mainly in the context of cancer vaccination. Overall these studies showed that selective activation of NF-κB in DCs of human or mouse origin enhances their ability to activate CD8+ T cells (209, 221). Another transcription factor that recently gained attention is PI3Kγ, as this transcription factor was shown to govern expression of ARG-1 and TGF-β in TAMs, therefore represents another important regulator of the immune suppressive phenotype of TAMs (222). Furthermore, PI3Kγ in myeloid cells has been directly linked to the success of immune checkpoint therapy, pinpointing PI3Kγ as an attractive target in cancer immunotherapy (222, 223).
The genetic programs active in myeloid cells are also regulated at the epigenetic level (138). Alternatively activated bone marrow-derived macrophages have enhanced levels of acetylated histones. This was linked to expression of TAM2 genes (224). Bromodomain and extra-terminal motif proteins (225) or histone deacetylase inhibitors (HDACi) (226–228), which interfere with chromatin remodeling, skew toward a TAM1 phenotype and were shown to mediate tumor regression in response to immune checkpoint therapy in a model in which monotherapy with anti-PD1 antibodies failed (228). Also MDSCs were shown to be numerically and functionally affected by HDACi (134, 135). Inhibition of HDAC reduced the number of MDSCs as well as the expression of hallmark enzymes, including ARG-1 and iNOS, making several experimental cancer models sensitive to immune checkpoint therapy (134, 135). HDACi are currently studied in combination with checkpoint blockers in patients with metastatic and unresectable HER2/neu negative breast cancer (138).
Manipulating the Metabolism of Myeloid Cells
Because the metabolism of TIMs is linked to their function, changing metabolic pathways has been studied as a strategy to target myeloid cells (229). MDSCs have an enhanced expression of CD36, a fatty acid translocase that enables uptake and oxidation of fatty acids, and as such an increase in their immunosuppressive capacity (230). Inhibiting fatty acid oxidation decreases the immunosuppressive capacity of MDSCs, and moreover mediates antitumor effects in experimental models when combined with low-dose CT and adoptive cell therapy (231). Metabolic pathways were also studied in TADCs. Fatty acid metabolism in DCs has been linked to their ability to cross-present antigens, a critical process to stimulate tumor-specific CD8+ T cells (232). Nonetheless, enhanced levels of lipids as observed in several preclinical mouse cancer models and cancer patients has a negative impact on DC cross-presentation. Normalization of lipid levels in these cancer models restored the functional activity of lipid rich DCs and enabled them to become more potent when used in a cancer vaccine (233). It was further shown that anaerobic glycolysis after TLR stimulation is essential to upregulate among others co-stimulatory molecules and IL-12 (234, 235). Also the metabolic signature of TAMs is correlated to their function (236, 237). The direct link between TAM metabolism, tumor vasculature and metastasis, was recently shown using a genetic approach. Herein genetic deletion of regulated in development and DNA damage responses 1 (REDD1), an inhibitor of mTOR, which is highly expressed by TAM2 was shown to increase glucose uptake and direct TAM2 toward glycolysis (238), a hallmark of TAM1 (239). Consequently, TAMs competed with tumor endothelial cells for glucose consumption, which resulted in stabilization of the tumor vasculature, thereby preventingmetastasis (238).
Manipulating Distinct Tumor Promoting Functions
Several enzymes expressed by TIMs have been inextricably connected to their immunosuppressive function, among which IDO, ARG-1, iNOS, and COX2, and have therefore been a focus of research. IDO is a tryptophan degrading enzyme that can be induced in MDSCs, TADCs, TAMs, and to some extent TANs in response to stimuli such as pro-inflammatory cytokines, TLR ligands, hormones, PGE2, and contact dependent stimuli such as B7:cytotoxic T lymphocyte antigen 4 (CTLA-4) interactions (10, 240–242). The net effect of IDO mediated conversion of tryptophan to kynurenine is induction of T cell anergy, apoptosis, and commitment of CD4+ T cells toward immunosuppressive Tregs (243). These Tregs can further amplify the immunosuppressive TME by endowing TADCs with tolerogenic properties (e.g., upregulation of IDO), and by recruiting and activating MDSCs, which exploit expression of IDO, ARG-1, and iNOS in addition to immunosuppressive cytokines and co-inhibitory molecules to suppress T cells (244). The tryptophan catabolism is therefore an important mechanism of cancer immune escape. Expression of IDO has been linked to poor clinical prognosis in several cancers, as it correlates with decreased survival as well as increased risk of metastasis (245). Therefore, strategies to disrupt IDO's enzymatic activity or expression have been studied extensively. In animal studies, genetic inhibition of IDO expression resulted in infiltration, and activation of granulocytes that established a TME favoring tumor growth (246). The tyrosine kinase inhibitor imatinib, which inhibits COX2 activity, therefore blocks PGE2 production a potent stimulus for IDO upregulation, has been successfully used in the context of IDO inhibition (247, 248). Also small-molecule inhibitors of IDO exhibit anticancer activity and cooperate with immunotherapy, RT or CT to trigger rapid regression of aggressive tumors otherwise resistant to treatment (249). Several IDO inhibitors are under clinical evaluation either as a mono- or combination therapy (e.g., checkpoint blockade and CT) (250). The IDO inhibitors most intensively studied in clinical trials are epacadostat and indoximod. In a phase I trial, epacadostat showed to normalize kynurenine levels in patients with advanced solid malignancies (251). In phase I-III trials, epacadostat showed promising results in combination with various checkpoint inhibitors, including durvalumab (anti-PD-L1), pembrolizumab (anti-PD-1), and ipilimumab (anti-CTLA-4), in different cancer types (252). Indoximod was shown to counteract the immunosuppressive effects of kynurenine, to activate multiple immune cells and to stimulate Treg to TH1 conversion (253–255). Ongoing phase II-III clinical studies investigate the effect of indoximod in combination with the checkpoint inhibitor nivolumab (anti-PD-1) in patients with metastatic melanoma. Further development resulted in a compound, NLG802, the prodrug of indoximod, which in animal studies induced antitumor responses at lower doses than indoximod. Recently, NLG802 entered phase I clinical testing (256).
ARG-1 is another amino acid degrading enzyme. It converts L-arginine into L-ornithine and urea, and can be upregulated in MDSCs, TAM2 and tolDCs by multiple factors, including PGE2 (produced by COX2), GM-CSF, TGF-β, IL-6, and IL-10, where the latter two both act via STAT3 (257–260). Also hypoxia has been linked to ARG-1 expression (261). Expression of ARG-1 by myeloid cells has been unambiguously linked to tumor promoting activities, and L-ornithine was shown to further stimulate alternative activation of myeloid cells (262–264). iNOS is also an L-arginine converting enzyme, which produces NO and citrulline as a result. Expression of iNOS has been shown in TAM1, inflammatory DCs and MDSCs in response to IL-1β, IL-6, IFN-γ, TNF-α, and TLR4 agonists, and has been linked to both anti- and protumor activities (265, 266). This dual role of iNOS and NO is likely depending on the levels, source and timing of NO production, and is further influenced by the TME. In early events of tumor development some myeloid cells like macrophages can generate high concentrations of NO, thereby inducing tumor cell apoptosis. However, in an established TME, myeloid cells are reprogrammed to support tumor progression. At that time low amounts of NO act pro-angiogenic and enhance tumor growth and metastasis by inducing growth factors such as VEGF and MMPs. Moreover, a temporal relationship between the expression of ARG-1 and iNOS in TIMs such as macrophages has been linked to immunosuppression. It was proposed that ARG-1 occurs prior to iNOS expression to stop T cell proliferation and that subsequent iNOS expression and as such NO release, stimulates T cell apoptosis, thereby ending the anticancer immune responses (267). Therefore, strategies to block ARG-1 and iNOS have been developed. Inhibitors like nor-N-hydroxy-L-arginine and aminoguanidin (or 1400W) downregulate ARG-1 and iNOS activity in MDSCs, restore T cell antitumor immunity and reduce tumor progression (176, 268). Moreover, recent studies showed that combining ARG-1 inhibition with immune checkpoint therapy or RT is a promising strategy (244, 269). Several reports recommend targeting both ARG-1 and iNOS to augment the therapeutic effect (270). Phosphodiesterase-5 (PDE5) inhibitors, like sildenafil, tadalafil, and vardenafil, were shown to decrease ARG-1 and iNOS expression. Consequently the suppressive activity of MDSCs was blocked which ultimately boosted tumor-specific T cell responses (271–273). Similarly, NO-releasing aspirin was shown to reduce ARG-1 and iNOS, and enhance the number and function of tumor-specific T cells (274). The prior use of PDE5 inhibitors and NO-releasing aspirin in other indications prompted their use in clinical trials, often in combination with other cancer therapies including RT, CT, and immunotherapy (vaccination, immunomodulatory drugs and immune checkpoint blockade) (275). While many trials are ongoing, pilot trials in metastatic melanoma patients report on its safety and further show a decreased frequency and function of MDSCs, correlating with an increased T cell reactivity and improved clinical outcome (276–278). While these studies mainly focused on the role of MDSCs, it is conceivable that these agents could also modulate other ARG-1 and iNOS expressing myeloid cells like TAMs and TADCs.
COX2 is the enzyme responsible for the metabolic conversion of arachidonic acid into various prostaglandins including PGE2, which has an immunosuppressive function as evidenced by its ability to induce IDO and ARG-1 expression in various TIMs. Another link to immunosuppressive amino acid degrading enzymes and COX2 in myeloid cells is the observation that NO is involved in regulating COX2 expression (279). Inhibition of COX2 in mouse and human myeloid cell progenitors was shown to inhibit MDSC and TAM2 generation, and favor differentiation toward mature APCs (280–283). Genetic deletion of COX2 in myeloid cells in vivo was shown to reduce macrophage infiltration, increase T cell numbers in the tumor and linked herewith reduce tumor progression (284). Pharmacological inhibition of COX2 was also shown to delay tumor growth in various studies and even synergize with immunotherapy. In these studies, COX2 inhibition was linked to reduced MDSC accumulation, and reduced IDO or ARG-1 expression (248, 285–288).
Overall, inhibition of key immunosuppressive enzymes was shown to (partially) revert the immunosuppressive TME and as such facilitate cancer specific immunity. Future preclinical and clinical studies will have to substantiate if targeting one or more of the suppressive mechanisms exerted by TIMs suffices to control cancer progression.
Conclusion
In this review we emphasize the multifaceted functions that TIMs possess to support cancer manifestation, progression, and metastasis next to the roadblocks they form to successful therapeutic interventions. This is evidenced by the abundance of publications on the subject of TAMs, TADCs, TANs, MDSCs, and the commonalities they share such as plasticity and immunosuppressive functions. So far, vast efforts has been invested in the development of pharmacological compounds targeting TIMs to alter their cell number or repolarize their functions. These strategies resulted in significant therapeutic benefits in several preclinical models and are currently clinically evaluated as additional modalities in cancer treatment.
Nonetheless, caution is warranted when neutralizing immune factors that are not unique to the tumor and its TME (e.g., PGE2, ROS, iNOS, or TGF-β), since they are indispensable in maintaining homeostasis. Therefore, TIM depleting or function altering therapies targeting one of these factors, must be transient or local in nature. For future therapies, it will be essential to expand our knowledge on mechanisms that drive the immunosuppressive barricades herded by TIMs. Also, the type of cancer should be considered since some myeloid compositions can be beneficial in one type while unfavorable in another. Since migration patterns and metabolic aberrations occur during the several stages of tumor progression, it is important to push current methodology toward a combination of functional imaging and advanced in silico analysis. This will be essential to understand and predict the dynamics of the TME as well as why some predicted therapies underperform in vivo.
Conventional therapies are often focused on eliminating a particular subset among the abundance of myeloid cells neglecting those that are desirable. Tumor lyses, clearance and antigen presentation are essential modalities that are especially designated to myeloid cells, however they are still undervalued. Instead of eradicating a complete TIM population we rather suggest to mold the myeloid cell composition and polarization state, by combining several myeloidal immunotherapies, based on the redundancy and commonalities in TIM phenotype and function. In conclusion, we believe that reorganizing the myeloid landscape can clear away the roadblock to a successful cancer therapy.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Laboratory of Molecular and Cellular Therapy is supported by the Scientific Fund Willy Gepts of the University Hospital Brussels; the Strategic Research Program of the Vrije Universiteit Brussel; the National Cancer Plan of the Federal Ministry of Health, the Stichting tegen Kanker, Kom op tegen Kanker (Stand up to Cancer), the Flemish cancer society, the Institute for Science and Innovation (IWT), the Research Foundation Flanders (FWO-V), the European Union's FP7 Research and Innovation funding program, the ERA-NET TRANSCAN funding program and the Melanoma Research Alliance. YDV has received an Emmanuel van der Schueren grant from Kom op tegen Kanker and a doctoral grant strategic basic research from FWO-V. CG is a fellow of Kom op tegen Kanker (Stand up to Cancer), the Flemish cancer society.
References
1. Durgeau A, Virk Y, Corgnac S, Mami-Chouaib F. Recent advances in targeting CD8 T-Cell immunity for more effective cancer immunotherapy. Front Immunol. (2018) 9:14. doi: 10.3389/fimmu.2018.00014
2. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
3. De Vlaeminck Y, Gonzalez-Rascon A, Goyvaerts C, Breckpot K. Cancer-associated myeloid regulatory cells. Front Immunol. (2016) 7:113. doi: 10.3389/fimmu.2016.00113
4. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454:436–44. doi: 10.1038/nature07205
5. Hanahan D, Weinberg Robert A. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
6. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
7. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science (2014) 344:921–5. doi: 10.1126/science.1252510
8. Tymoszuk P, Evens H, Marzola V, Wachowicz K, Wasmer MH, Datta S, et al. In situ proliferation contributes to accumulation of tumor-associated macrophages in spontaneous mammary tumors. Eur J Immunol. (2014) 44:2247–62. doi: 10.1002/eji.201344304
9. Campbell MJ, Tonlaar NY, Garwood ER, Huo D, Moore DH, Khramtsov AI, et al. Proliferating macrophages associated with high grade, hormone receptor negative breast cancer and poor clinical outcome. Breast Cancer Res Treat. (2011) 128:703–11. doi: 10.1007/s10549-010-1154-y
10. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
11. Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN-MDSC: Their biological role and interaction with stromal cells. Semin Immunol. (2018) 35:19–28. doi: 10.1016/j.smim.2017.12.004
12. Liechtenstein T, Perez-Janices N, Gato M, Caliendo F, Kochan G, Blanco-Luquin I, et al. A highly efficient tumor-infiltrating MDSC differentiation system for discovery of anti-neoplastic targets, which circumvents the need for tumor establishment in mice. Oncotarget (2014) 5:7843–57. doi: 10.18632/oncotarget.2279
13. Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity (2016) 44:303–15. doi: 10.1016/j.immuni.2016.01.014
14. Youn JI, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol. (2013) 14:211–20. doi: 10.1038/ni.2526
15. Singel KL, Segal BH. Neutrophils in the tumor microenvironment: trying to heal the wound that cannot heal. Immunol Rev. (2016) 273:329–43. doi: 10.1111/imr.12459
16. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
17. Keskinov AA, Shurin MR. Myeloid regulatory cells in tumor spreading and metastasis. Immunobiology (2015) 220:236–42. doi: 10.1016/j.imbio.2014.07.017
18. Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer (2016) 16:431–46. doi: 10.1038/nrc.2016.52
19. Dehne N, Mora J, Namgaladze D, Weigert A, Brune B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr Opin Pharmacol. (2017) 35:12–9. doi: 10.1016/j.coph.2017.04.007
20. Mills CD, Shearer J, Evans R, Caldwell MD. Macrophage arginine metabolism and the inhibition or stimulation of cancer. J Immunol. (1992) 149:2709–14.
21. Mills CD. Anatomy of a Discovery: M1 and M2 Macrophages. Front Immunol. (2015) 6:212. doi: 10.3389/fimmu.2015.00212
22. Van den Bossche J, Bogaert P, van Hengel J, Guerin CJ, Berx G, Movahedi K, et al. Alternatively activated macrophages engage in homotypic and heterotypic interactions through IL-4 and polyamine-induced E-cadherin/catenin complexes. Blood (2009) 114:4664–74. doi: 10.1182/blood-2009-05-221598
23. Ghassabeh GH, De Baetselier P, Brys L, Noel W, Van Ginderachter JA, Meerschaut S, et al. Identification of a common gene signature for type II cytokine-associated myeloid cells elicited in vivo in different pathologic conditions. Blood (2006) 108:575–83. doi: 10.1182/blood-2005-04-1485
24. Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer (2017) 117:1583–91. doi: 10.1038/bjc.2017.356
25. Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H, et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci USA. (2011) 108:12425–30. doi: 10.1073/pnas.1106645108
26. Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity (2002) 17:19–29. doi: 10.1016/S1074-7613(02)00333-3
27. Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol. (2000) 1:119–26. doi: 10.1038/77793
28. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999) 285:727–9. doi: 10.1126/science.285.5428.727
29. Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med. (2013) 210:2057–69. doi: 10.1084/jem.20130783
30. Kruse PH, Matta J, Ugolini S, Vivier E. Natural cytotoxicity receptors and their ligands. Immunol Cell Biol. (2014) 92:221–9. doi: 10.1038/icb.2013.98
31. Bryceson YT, March ME, Barber DF, Ljunggren HG, Long EO. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J Exp Med. (2005) 202:1001–12. doi: 10.1084/jem.20051143
32. Garrido F, Algarra I. MHC antigens and tumor escape from immune surveillance. Adv Cancer Res. (2001) 83:117–58. doi: 10.1016/S0065-230X(01)83005-0
33. Tu MM, Mahmoud AB, Wight A, Mottashed A, Belanger S, Rahim MM, et al. Ly49 family receptors are required for cancer immunosurveillance mediated by natural killer cells. Cancer Res. (2014) 74:3684–94. doi: 10.1158/0008-5472.CAN-13-3021
34. Fernandez NC, Lozier A, Flament C, Ricciardi-Castagnoli P, Bellet D, Suter M, et al. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat Med. (1999) 5:405–11. doi: 10.1038/7403
35. Chiba S, Ikushima H, Ueki H, Yanai H, Kimura Y, Hangai S, et al. Recognition of tumor cells by Dectin-1 orchestrates innate immune cells for anti-tumor responses. Elife (2014) 3:e04177. doi: 10.7554/eLife.04177
36. Ebihara T, Azuma M, Oshiumi H, Kasamatsu J, Iwabuchi K, Matsumoto K, et al. Identification of a polyI:C-inducible membrane protein that participates in dendritic cell–mediated natural killer cell activation. J Exp Med. (2010) 207:2675–87. doi: 10.1084/jem.20091573
37. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer (2014) 14:135–46. doi: 10.1038/nrc3670
38. Markiewicz MA, Carayannopoulos LN, Naidenko OV, Matsui K, Burack WR, Wise EL, et al. Costimulation through NKG2D enhances murine CD8+ CTL function: similarities and differences between NKG2D and CD28 costimulation. J Immunol. (2005) 175:2825–33. doi: 10.4049/jimmunol.175.5.2825
39. Finn OJ. Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann Oncol. (2012) 23 (Suppl. 8):viii6–9. doi: 10.1093/annonc/mds256
40. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell (2005) 123:321–34. doi: 10.1016/j.cell.2005.08.032
41. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. (2011) 208:1989–2003. doi: 10.1084/jem.20101158
42. Shen YJ, Le Bert N, Chitre AA, Koo CX, Nga XH, Ho SS, et al. Genome-derived cytosolic DNA mediates type I interferon-dependent rejection of B cell lymphoma cells. Cell Rep. (2015) 11:460–73. doi: 10.1016/j.celrep.2015.03.041
43. Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med. (2010) 2:63ra94. doi: 10.1126/scitranslmed.3001375
44. Vicetti Miguel RD, Cherpes TL, Watson LJ, McKenna KC. CTL induction of tumoricidal nitric oxide production by intratumoral macrophages is critical for tumor elimination. J Immunol. (2010) 185:6706–18. doi: 10.4049/jimmunol.0903411
45. Corthay A, Skovseth DK, Lundin KU, Rosjo E, Omholt H, Hofgaard PO, et al. Primary antitumor immune response mediated by CD4+ T cells. Immunity (2005) 22:371–83. doi: 10.1016/j.immuni.2005.02.003
46. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature (2012) 482:400–4. doi: 10.1038/nature10755
47. von Boehmer L, Mattle M, Bode P, Landshammer A, Schafer C, Nuber N, et al. NY-ESO-1-specific immunological pressure and escape in a patient with metastatic melanoma. Cancer Immun. (2013) 13:12. doi: 10.5167/uzh-79657
48. Nicholaou T, Chen W, Davis ID, Jackson HM, Dimopoulos N, Barrow C, et al. Immunoediting and persistence of antigen-specific immunity in patients who have previously been vaccinated with NY-ESO-1 protein formulated in ISCOMATRIX. Cancer Immunol Immunother. (2011) 60:1625–37. doi: 10.1007/s00262-011-1041-3
49. Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol Res. (2017) 5:1152–61. doi: 10.1158/2326-6066.CIR-17-0189
50. O'Sullivan T, Saddawi-Konefka R, Vermi W, Koebel CM, Arthur C, White JM, et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J Exp Med. (2012) 209:1869–82. doi: 10.1084/jem.20112738
51. Ahrends T, Borst J. The opposing roles of CD4(+) T cells in anti-tumour immunity. Immunology (2018) 154. doi: 10.1111/imm.12941
52. Ben-Meir K, Twaik N, Baniyash M. Plasticity and biological diversity of myeloid derived suppressor cells. Curr Opin Immunol. (2018) 51:154–61. doi: 10.1016/j.coi.2018.03.015
53. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol. (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
54. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer (2012) 12:860–75. doi: 10.1038/nrc3380
55. Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature (2013) 494:361–5. doi: 10.1038/nature11824
56. Kursunel MA, Esendagli G. The untold story of IFN-gamma in cancer biology. Cytokine Growth Factor Rev. (2016) 31:73–81. doi: 10.1016/j.cytogfr.2016.07.005
57. Yang H, Lundback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med. (2012) 18:250–9. doi: 10.2119/molmed.2011.00389
58. El-Kenawi A, Ruffell B. Inflammation, ROS, and Mutagenesis. Cancer Cell. (2017) 32:727–9. doi: 10.1016/j.ccell.2017.11.015
59. Roca H, Jones JD, Purica MC, Weidner S, Koh AJ, Kuo R, et al. Apoptosis-induced CXCL5 accelerates inflammation and growth of prostate tumor metastases in bone. J Clin Invest. (2017) 128:248–66. doi: 10.1172/JCI92466
60. Tamadaho RSE, Hoerauf A, Layland LE. Immunomodulatory effects of myeloid-derived suppressor cells in diseases: Role in cancer and infections. Immunobiology (2018) 223:432–42. doi: 10.1016/j.imbio.2017.07.001
61. Mohamed E, Al-Khami AA, Rodriguez PC. The cellular metabolic landscape in the tumor milieu regulates the activity of myeloid infiltrates. Cell Mol Immunol. (2018) 15:421–7. doi: 10.1038/s41423-018-0001-7
62. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science (2011) 331:1565–70. doi: 10.1126/science.1203486
63. Lin EY, Gouon-Evans V, Nguyen AV, Pollard JW. The macrophage growth factor CSF-1 in mammary gland development and tumor progression. J Mammary Gland Biol Neoplasia (2002) 7:147–62. doi: 10.1023/A:1020399802795
64. Zhang X, Xue D, Hao F, Xie L, He J, Gai J, et al. Remodeling and spacing factor 1 overexpression is associated with poor prognosis in renal cell carcinoma. Oncol Lett. (2018) 15:3852–7. doi: 10.3892/ol.2018.7797
65. Chimal-Ramirez GK, Espinoza-Sanchez NA, Fuentes-Panana EM. Protumor activities of the immune response: insights in the mechanisms of immunological shift, oncotraining, and oncopromotion. J Oncol. (2013) 2013:835956. doi: 10.1155/2013/835956
66. Dufait I, Van Valckenborgh E, Menu E, Escors D, De Ridder M, Breckpot K. Signal transducer and activator of transcription 3 in myeloid-derived suppressor cells: an opportunity for cancer therapy. Oncotarget (2016) 7:42698–715. doi: 10.18632/oncotarget.8311
67. Kortylewski M, Yu H. Role of Stat3 in suppressing anti-tumor immunity. Curr Opin Immunol. (2008) 20:228–33. doi: 10.1016/j.coi.2008.03.010
68. Arce F, Breckpot K, Stephenson H, Karwacz K, Ehrenstein MR, Collins M, et al. Selective ERK activation differentiates mouse and human tolerogenic dendritic cells, expands antigen-specific regulatory T cells, and suppresses experimental inflammatory arthritis. Arthritis Rheum. (2011) 63:84–95. doi: 10.1002/art.30099
69. Maenhout SK, Van Lint S, Emeagi PU, Thielemans K, Aerts JL. Enhanced suppressive capacity of tumor-infiltrating myeloid-derived suppressor cells compared with their peripheral counterparts. Int J Cancer (2014) 134:1077–90. doi: 10.1002/ijc.28449
70. Ballbach M, Dannert A, Singh A, Siegmund DM, Handgretinger R, Piali L, et al. Expression of checkpoint molecules on myeloid-derived suppressor cells. Immunol Lett. (2017) 192:1–6. doi: 10.1016/j.imlet.2017.10.001
71. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci USA. (2017) 114:1117–22. doi: 10.1073/pnas.1612920114
72. Takahashi H, Sakakura K, Kudo T, Toyoda M, Kaira K, Oyama T, et al. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget (2017) 8:8633–47. doi: 10.18632/oncotarget.14374
73. Chen J, Ye Y, Liu P, Yu W, Wei F, Li H, et al. Suppression of T cells by myeloid-derived suppressor cells in cancer. Hum Immunol. (2017) 78:113–9. doi: 10.1016/j.humimm.2016.12.001
74. Mougiakakos D, Jitschin R, von Bahr L, Poschke I, Gary R, Sundberg B, et al. Immunosuppressive CD14+HLA-DRlow/neg IDO+ myeloid cells in patients following allogeneic hematopoietic stem cell transplantation. Leukemia (2013) 27:377–88. doi: 10.1038/leu.2012.215
75. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. (2013) 190:3783–97. doi: 10.4049/jimmunol.1390024
76. Baratelli F, Lin Y, Zhu L, Yang SC, Heuze-Vourc'h N, Zeng G, et al. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol. (2005) 175:1483–90. doi: 10.4049/jimmunol.175.3.1483
77. Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. (2010) 70:99–108. doi: 10.1158/0008-5472.CAN-09-1882
78. Zoso A, Mazza EM, Bicciato S, Mandruzzato S, Bronte V, Serafini P, et al. Human fibrocytic myeloid-derived suppressor cells express IDO and promote tolerance via Treg-cell expansion. Eur J Immunol. (2014) 44:3307–19. doi: 10.1002/eji.201444522
79. Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C, et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest. (2014) 124:5466–80. doi: 10.1172/JCI77053
80. Mancino A, Schioppa T, Larghi P, Pasqualini F, Nebuloni M, Chen IH, et al. Divergent effects of hypoxia on dendritic cell functions. Blood (2008) 112:3723–34. doi: 10.1182/blood-2008-02-142091
81. Elia AR, Cappello P, Puppo M, Fraone T, Vanni C, Eva A, et al. Human dendritic cells differentiated in hypoxia down-modulate antigen uptake and change their chemokine expression profile. J Leukoc Biol. (2008) 84:1472–82. doi: 10.1189/jlb.0208082
82. Yang M, Ma C, Liu S, Shao Q, Gao W, Song B, et al. HIF-dependent induction of adenosine receptor A2b skews human dendritic cells to a Th2-stimulating phenotype under hypoxia. Immunol Cell Biol. (2010) 88:165–71. doi: 10.1038/icb.2009.77
83. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med. (2005) 202:919–29. doi: 10.1084/jem.20050463
84. Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science (2002) 297:1867–70. doi: 10.1126/science.1073514
85. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. (2004) 4:762–74. doi: 10.1038/nri1457
86. Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood (2008) 112:1822–31. doi: 10.1182/blood-2008-02-136325
87. Liu Q, Zhang C, Sun A, Zheng Y, Wang L, Cao X. Tumor-educated CD11bhighIalow regulatory dendritic cells suppress T cell response through arginase I. J Immunol. (2009) 182:6207–16. doi: 10.4049/jimmunol.0803926
88. Albini A, Bruno A, Noonan DM, Mortara L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: implications for immunotherapy. Front Immunol. (2018) 9:527. doi: 10.3389/fimmu.2018.00527
89. Gordon-Weeks AN, Lim SY, Yuzhalin AE, Jones K, Markelc B, Kim KJ, et al. Neutrophils promote hepatic metastasis growth through fibroblast growth factor 2-dependent angiogenesis in mice. Hepatology (2017) 65:1920–35. doi: 10.1002/hep.29088
90. Tabaries S, Ouellet V, Hsu BE, Annis MG, Rose AA, Meunier L, et al. Granulocytic immune infiltrates are essential for the efficient formation of breast cancer liver metastases. Breast Cancer Res. (2015) 17:45. doi: 10.1186/s13058-015-0558-3
91. Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci USA. (2010) 107:21248–55. doi: 10.1073/pnas.1015855107
92. Shi H, Zhang J, Han X, Li H, Xie M, Sun Y, et al. Recruited monocytic myeloid-derived suppressor cells promote the arrest of tumor cells in the premetastatic niche through an IL-1beta-mediated increase in E-selectin expression. Int J Cancer. (2017) 140:1370–83. doi: 10.1002/ijc.30538
93. Safarzadeh E, Orangi M, Mohammadi H, Babaie F, Baradaran B. Myeloid-derived suppressor cells: Important contributors to tumor progression and metastasis. J Cell Physiol. (2018) 233:3024–36. doi: 10.1002/jcp.26075
94. Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology (2014) 3:e28518. doi: 10.4161/onci.28518
95. Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, et al. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. (2011) 71:2488–96. doi: 10.1158/0008-5472.CAN-10-2820
96. Gupta A, Probst HC, Vuong V, Landshammer A, Muth S, Yagita H, et al. Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J Immunol. (2012) 189:558–66. doi: 10.4049/jimmunol.1200563
97. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. (2013) 24:589–602. doi: 10.1016/j.ccr.2013.09.014
98. Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. (2013) 105:256–65. doi: 10.1093/jnci/djs629
99. Russell JS, Brown JM. The irradiated tumor microenvironment: role of tumor-associated macrophages in vascular recovery. Front Physiol. (2013) 4:157. doi: 10.3389/fphys.2013.00157
100. Vatner RE, Formenti SC. Myeloid-derived cells in tumors: effects of radiation. Semin Radiat Oncol. (2015) 25:18–27. doi: 10.1016/j.semradonc.2014.07.008
101. Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. (2013) 73:2782–94. doi: 10.1158/0008-5472.CAN-12-3981
102. Shiao SL, Ruffell B, DeNardo DG, Faddegon BA, Park CC, Coussens LM. TH2-Polarized CD4(+) T cells and macrophages limit efficacy of radiotherapy. Cancer Immunol Res. (2015) 3:518–25. doi: 10.1158/2326-6066.CIR-14-0232
103. Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. (2014) 124:687–95. doi: 10.1172/JCI67313
104. Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity (2013) 39:211–27. doi: 10.1016/j.immuni.2013.07.017
105. Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity (2016) 44:343–54. doi: 10.1016/j.immuni.2015.11.024
106. Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, et al. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat Commun. (2015) 6:8692. doi: 10.1038/ncomms9692
107. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science (2013) 342:967–70. doi: 10.1126/science.1240527
108. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. (2010) 70:3052–61. doi: 10.1158/0008-5472.CAN-09-3690
109. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. (2011) 1:54–67. doi: 10.1158/2159-8274.CD-10-0028
110. Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature (2014) 515:130–3. doi: 10.1038/nature13862
111. Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. (2011) 25:2465–79. doi: 10.1101/gad.180331.111
112. Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. (2013) 19:57–64. doi: 10.1038/nm.2999
113. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell (2014) 26:623–37. doi: 10.1016/j.ccell.2014.09.006
114. Mo HN, Liu P. Targeting MET in cancer therapy. Chronic Dis Transl Med. (2017) 3:148–53. doi: 10.1016/j.cdtm.2017.06.002
115. Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, et al. Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther. (2015) 147:22–31. doi: 10.1016/j.pharmthera.2014.11.001
116. Cavnar MJ, Zeng S, Kim TS, Sorenson EC, Ocuin LM, Balachandran VP, et al. KIT oncogene inhibition drives intratumoral macrophage M2 polarization. J Exp Med. (2013) 210:2873–86. doi: 10.1084/jem.20130875
117. Hoeflich KP, O'Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. (2009) 15:4649–64. doi: 10.1158/1078-0432.CCR-09-0317
118. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta (2007) 1773:1263–84. doi: 10.1016/j.bbamcr.2006.10.001
119. Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell (2004) 116:855–67. doi: 10.1016/S0092-8674(04)00215-6
120. Zhang BH, Guan KL. Activation of B-Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. Embo J. (2000) 19:5429–39. doi: 10.1093/emboj/19.20.5429
121. Steinberg SM, Shabaneh TB, Zhang P, Martyanov V, Li Z, Malik BT, et al. Myeloid cells that impair immunotherapy are restored in melanomas with acquired resistance to BRAF inhibitors. Cancer Res. (2017) 77:1599–610. doi: 10.1158/0008-5472.CAN-16-1755
122. Smith MP, Sanchez-Laorden B, O'Brien K, Brunton H, Ferguson J, Young H, et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFalpha. Cancer Discov. (2014) 4:1214–29. doi: 10.1158/2159-8290.CD-13-1007
123. Mok S, Tsoi J, Koya RC, Hu-Lieskovan S, West BL, Bollag G, et al. Inhibition of colony stimulating factor-1 receptor improves antitumor efficacy of BRAF inhibition. BMC Cancer (2015) 15:356. doi: 10.1186/s12885-015-1377-8
124. Ott PA, Henry T, Baranda SJ, Frleta D, Manches O, Bogunovic D, et al. Inhibition of both BRAF and MEK in BRAF(V600E) mutant melanoma restores compromised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol Immunother. (2013) 62:811–22. doi: 10.1007/s00262-012-1389-z
125. Ott PA, Bhardwaj N. Impact of MAPK pathway activation in BRAF(V600) melanoma on T cell and dendritic cell function. Front Immunol. (2013) 4:346. doi: 10.3389/fimmu.2013.00346
126. Finisguerra V, Di Conza G, Di Matteo M, Serneels J, Costa S, Thompson AA, et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature (2015) 522:349–53. doi: 10.1038/nature14407
127. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
128. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
129. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
130. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer (2018) 118:9–16. doi: 10.1038/bjc.2017.434
131. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. (2014) 63:247–57. doi: 10.1007/s00262-013-1508-5
132. Bjoern J, Juul Nitschke N, Zeeberg Iversen T, Schmidt H, Fode K, Svane IM. Immunological correlates of treatment and response in stage IV malignant melanoma patients treated with Ipilimumab. Oncoimmunology (2016) 5:e1100788. doi: 10.1080/2162402X.2015.1100788
133. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. (2014) 6:237ra67. doi: 10.1126/scitranslmed.3007974
134. Orillion A, Hashimoto A, Damayanti NP, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat neutralizes myeloid derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res. (2017) 23:5187–201. doi: 10.1158/1078-0432.CCR-17-0741
135. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci. (2014) 111:11774. doi: 10.1073/pnas.1410626111
136. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In vivo imaging reveals a tumor-associated macrophage–mediated resistance pathway in anti–PD-1 therapy. Sci Trans Med. (2017) 9 :eaal3604. doi: 10.1126/scitranslmed.aal3604
137. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
138. Porta C, Sica A, Riboldi E. Tumor-associated myeloid cells: new understandings on their metabolic regulation and their influence in cancer immunotherapy. Febs J. (2018) 285:717–33. doi: 10.1111/febs.14288
139. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. (2009) 206:1327–37. doi: 10.1084/jem.20082173
140. Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. (2003) 9:562–7. doi: 10.1038/nm863
141. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature (2015) 523:231–5. doi: 10.1038/nature14404
142. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature (2015) 522:345–8. doi: 10.1038/nature14282
143. Srivastava MK, Zhu L, Harris-White M, Kar UK, Huang M, Johnson MF, et al. Myeloid suppressor cell depletion augments antitumor activity in lung cancer. PLoS ONE (2012) 7:e40677. doi: 10.1371/annotation/5c756e7d-6e97-416f-836a-dced97cf46af
144. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature (2011) 475:222–5. doi: 10.1038/nature10138
145. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. (2014) 74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723
146. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell (2014) 25:846–59. doi: 10.1016/j.ccr.2014.05.016
147. Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology (2013) 2:e26968. doi: 10.4161/onci.26968
148. Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. (2013) 73:1128–41. doi: 10.1158/0008-5472.CAN-12-2731
149. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. (2013) 19:1264–72. doi: 10.1038/nm.3337
150. Chen X, Wang Y, Nelson D, Tian S, Mulvey E, Patel B, et al. CCL2/CCR2 regulates the tumor microenvironment in HER-2/neu-driven mammary carcinomas in mice. PLoS ONE (2016) 11:e0165595. doi: 10.1371/journal.pone.0165595
151. Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. (2012) 72:876–86. doi: 10.1158/0008-5472.CAN-11-1792
152. Izhak L, Wildbaum G, Weinberg U, Shaked Y, Alami J, Dumont D, et al. Predominant expression of CCL2 at the tumor site of prostate cancer patients directs a selective loss of immunological tolerance to CCL2 that could be amplified in a beneficial manner. J Immunol. (2010) 184:1092–101. doi: 10.4049/jimmunol.1090025
153. Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, et al. Targeting both tumour-associated CXCR2 <sup>+ </sup> neutrophils and CCR2 <sup>+ </sup> macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut (2017) 67:1112–3. doi: 10.1136/gutjnl-2017-313738
154. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. (2015) 27:462–72. doi: 10.1016/j.ccell.2015.02.015
155. Horikawa N, Abiko K, Matsumura N, Hamanishi J, Baba T, Yamaguchi K, et al. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin Cancer Res. (2017) 23:587–99. doi: 10.1158/1078-0432.CCR-16-0387
156. Linde N, Lederle W, Depner S, van Rooijen N, Gutschalk CM, Mueller MM. Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J Pathol. (2012) 227:17–28. doi: 10.1002/path.3989
157. Avraham-Davidi I, Yona S, Grunewald M, Landsman L, Cochain C, Silvestre JS, et al. On-site education of VEGF-recruited monocytes improves their performance as angiogenic and arteriogenic accessory cells. J Exp Med. (2013) 210:2611–25. doi: 10.1084/jem.20120690
158. Chen XW, Sun JG, Zhang LP, Liao XY, Liao RX. Recruitment of CD11b(+)Ly6C(+) monocytes in non-small cell lung cancer xenografts challenged by anti-VEGF antibody. Oncol Lett. (2017) 14:615–22. doi: 10.3892/ol.2017.6236
159. D'Agostino RB Sr. Changing end points in breast-cancer drug approval–the Avastin story. N Engl J Med. (2011) 365:e2. doi: 10.1056/NEJMp1106984
160. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly Diagnosed Glioblastoma. N Engl J Med. (2014) 370:699–708. doi: 10.1056/NEJMoa1308573
161. Du Four S, Maenhout SK, Benteyn D, De Keersmaecker B, Duerinck J, Thielemans K, et al. Disease progression in recurrent glioblastoma patients treated with the VEGFR inhibitor axitinib is associated with increased regulatory T cell numbers and T cell exhaustion. Cancer Immunol Immunother. (2016) 65:727–40. doi: 10.1007/s00262-016-1836-3
162. Heine A, Held SA, Daecke SN, Riethausen K, Kotthoff P, Flores C, et al. The VEGF-Receptor inhibitor axitinib impairs dendritic cell phenotype and function. PLoS ONE (2015) 10:e0128897. doi: 10.1371/journal.pone.0128897
163. Du Four S, Maenhout SK, De Pierre K, Renmans D, Niclou SP, Thielemans K, et al. Axitinib increases the infiltration of immune cells and reduces the suppressive capacity of monocytic MDSCs in an intracranial mouse melanoma model. Oncoimmunology (2015) 4:e998107. doi: 10.1080/2162402X.2014.998107
164. Nishioka Y, Hirao M, Robbins PD, Lotze MT, Tahara H. Induction of systemic and therapeutic antitumor immunity using intratumoral injection of dendritic cells genetically modified to express interleukin 12. Cancer Res. (1999) 59:4035–41.
165. Mazzolini G, Alfaro C, Sangro B, Feijoo E, Ruiz J, Benito A, et al. Intratumoral injection of dendritic cells engineered to secrete interleukin-12 by recombinant adenovirus in patients with metastatic gastrointestinal carcinomas. J Clin Oncol. (2005) 23:999–1010. doi: 10.1200/JCO.2005.00.463
166. Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. (2003) 3:650–65. doi: 10.1038/nrc1169
167. Watowich SS, Liu YJ. Mechanisms regulating dendritic cell specification and development. Immunol Rev. (2010) 238:76–92. doi: 10.1111/j.1600-065X.2010.00949.x
168. Lynch DH, Andreasen A, Maraskovsky E, Whitmore J, Miller RE, Schuh JC. Flt3 ligand induces tumor regression and antitumor immune responses in vivo. Nat Med. (1997) 3:625–31. doi: 10.1038/nm0697-625
169. Esche C, Subbotin VM, Maliszewski C, Lotze MT, Shurin MR. FLT3 ligand administration inhibits tumor growth in murine melanoma and lymphoma. Cancer Res. (1998) 58:380–3.
170. Hou S, Kou G, Fan X, Wang H, Qian W, Zhang D, et al. Eradication of hepatoma and colon cancer in mice with Flt3L gene therapy in combination with 5-FU. Cancer Immunol Immunother. (2007) 56:1605–13. doi: 10.1007/s00262-007-0306-3
171. Tandon M, Vemula SV, Sharma A, Ahi YS, Mittal S, Bangari DS, et al. EphrinA1-EphA2 interaction-mediated apoptosis and Flt3L-induced immunotherapy inhibits tumor growth in a breast cancer mouse model. J Gene Med. (2012) 14:77–89. doi: 10.1002/jgm.1649
172. Fong L, Hou Y, Rivas A, Benike C, Yuen A, Fisher GA, et al. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci USA. (2001) 98:8809–14. doi: 10.1073/pnas.141226398
173. Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol. (2009) 27:5763–71. doi: 10.1200/JCO.2009.24.3675
174. Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. (2010) 17:718–30. doi: 10.1245/s10434-009-0809-6
175. Liechtenstein T, Perez-Janices N, Blanco-Luquin I, Goyvaerts C, Schwarze J, Dufait I, et al. Anti-melanoma vaccines engineered to simultaneously modulate cytokine priming and silence PD-L1 characterized using ex vivo myeloid-derived suppressor cells as a readout of therapeutic efficacy. Oncoimmunology (2014) 3:e945378. doi: 10.4161/21624011.2014.945378
176. Dufait I, Schwarze JK, Liechtenstein T, Leonard W, Jiang H, Escors D, et al. Ex vivo generation of myeloid-derived suppressor cells that model the tumor immunosuppressive environment in colorectal cancer. Oncotarget. (2015) 6:12369–82. doi: 10.18632/oncotarget.3682
177. De Santo C, Salio M, Masri SH, Lee LY-H, Dong T, Speak AO, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus–induced myeloid-derived suppressor cells in mice and humans. J Clin Invest. (2008) 118:4036–48. doi: 10.1172/JCI36264
178. Shirota Y, Shirota H, Klinman DM. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J Immunol. (2012) 188:1592–9. doi: 10.4049/jimmunol.1101304
179. Takeda Y, Kataoka K, Yamagishi J, Ogawa S, Seya T, Matsumoto M. A TLR3-specific adjuvant relieves innate resistance to PD-L1 blockade without cytokine toxicity in tumor vaccine immunotherapy. Cell Rep. (2017) 19:1874–87. doi: 10.1016/j.celrep.2017.05.015
180. Shime H, Matsumoto M, Seya T. Double-stranded RNA promotes CTL-independent tumor cytolysis mediated by CD11b(+)Ly6G(+) intratumor myeloid cells through the TICAM-1 signaling pathway. Cell Death Differ. (2017) 24:385–96. doi: 10.1038/cdd.2016.131
181. Zoglmeier C, Bauer H, Norenberg D, Wedekind G, Bittner P, Sandholzer N, et al. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin Cancer Res. (2011) 17:1765–75. doi: 10.1158/1078-0432.CCR-10-2672
182. Shime H, Matsumoto M, Oshiumi H, Tanaka S, Nakane A, Iwakura Y, et al. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proc Natl Acad Sci USA. (2012) 109:2066–71. doi: 10.1073/pnas.1113099109
183. Matsumoto M, Tatematsu M, Nishikawa F, Azuma M, Ishii N, Morii-Sakai A, et al. Defined TLR3-specific adjuvant that induces NK and CTL activation without significant cytokine production in vivo. Nat Commun. (2015) 6:6280. doi: 10.1038/ncomms7280
184. Bunt SK, Clements VK, Hanson EM, Sinha P, Ostrand-Rosenberg S. Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J Leukoc Biol. (2009) 85:996–1004. doi: 10.1189/jlb.0708446
185. Ko H-J, Lee J-M, Kim Y-J, Kim Y-S, Lee K-A, Kang C-Y. Immunosuppressive myeloid-derived suppressor cells can be converted into immunogenic APCs with the help of activated NKT cells: an alternative cell-based antitumor vaccine. J Immunol. (2009) 182:1818–28. doi: 10.4049/jimmunol.0802430
186. De Santo C, Arscott R, Booth S, Karydis I, Jones M, Asher R, et al. Invariant NKT cells modulate the suppressive activity of Serum Amyloid A-differentiated IL-10-secreting neutrophils. Nat Immunol. (2010) 11:1039–46. doi: 10.1038/ni.1942
187. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (2011) 331:1612–6. doi: 10.1126/science.1198443
188. Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. (2013) 19:6286–95. doi: 10.1158/1078-0432.CCR-13-1320
189. Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. (1996) 184:747–52. doi: 10.1084/jem.184.2.747
190. Scarlett UK, Cubillos-Ruiz JR, Nesbeth YC, Martinez DG, Engle X, Gewirtz AT, et al. In situ stimulation of CD40 and Toll-like receptor 3 transforms ovarian cancer-infiltrating dendritic cells from immunosuppressive to immunostimulatory cells. Cancer Res. (2009) 69:7329–37. doi: 10.1158/0008-5472.CAN-09-0835
191. Van Lint S, Renmans D, Broos K, Goethals L, Maenhout S, Benteyn D, et al. Intratumoral delivery of TriMix mRNA results in T-cell activation by cross-presenting dendritic cells. Cancer Immunol Res. (2016) 4:146–56. doi: 10.1158/2326-6066.CIR-15-0163
192. Cavallo F, Giovarelli M, Forni G, Signorelli P, Musiani P, Modesti A, et al. Antitumor efficacy of adenocarcinoma cells engineered to produce interleukin 12 (IL-12) or other cytokines compared with exogenous IL-12. JNCI. J Natl Cancer Inst. (1997) 89:1049–58. doi: 10.1093/jnci/89.14.1049
193. Tanriover N, Ulu MO, Sanus GZ, Bilir A, Canbeyli R, Oz B, et al. The effects of systemic and intratumoral interleukin-12 treatment in C6 rat glioma model. Neurol Res. (2008) 30:511–7. doi: 10.1179/174313208X289516
194. Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. (2011) 121:4746–57. doi: 10.1172/JCI58814
195. Kerkar SP, Restifo NP. The power and pitfalls of IL-12. Blood (2012) 119:4096. doi: 10.1182/blood-2012-03-415018
196. Watkins SK, Egilmez NK, Suttles J, Stout RD. IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophages in vitro and in vivo. J Immunol. (2007) 178:1357–62. doi: 10.4049/jimmunol.178.3.1357
197. Haicheur N, Escudier B, Dorval T, Negrier S, De Mulder PH, Dupuy JM, et al. Cytokines and soluble cytokine receptor induction after IL-12 administration in cancer patients. Clin Exp Immunol. (2000) 119:28–37. doi: 10.1046/j.1365-2249.2000.01112.x
198. Portielje JE, Lamers CH, Kruit WH, Sparreboom A, Bolhuis RL, Stoter G, et al. Repeated administrations of interleukin (IL)-12 are associated with persistently elevated plasma levels of IL-10 and declining IFN-gamma, tumor necrosis factor-alpha, IL-6, and IL-8 responses. Clin Cancer Res. (2003) 9:76–83.
199. Yin XL, Wang N, Wei X, Xie GF, Li JJ, Liang HJ. Interleukin-12 inhibits the survival of human colon cancer stem cells in vitro and their tumor initiating capacity in mice. Cancer Lett. (2012) 322:92–7. doi: 10.1016/j.canlet.2012.02.015
200. Gollob JA, Mier JW, Veenstra K, McDermott DF, Clancy D, Clancy M, et al. Phase I trial of twice-weekly intravenous interleukin 12 in patients with metastatic renal cell cancer or malignant melanoma: ability to maintain IFN-gamma induction is associated with clinical response. Clin Cancer Res. (2000) 6:1678–92.
201. Motzer RJ, Rakhit A, Schwartz LH, Olencki T, Malone TM, Sandstrom K, et al. Phase I trial of subcutaneous recombinant human interleukin-12 in patients with advanced renal cell carcinoma. Clin Cancer Res. (1998) 4:1183–91.
202. Cebon J, Jager E, Shackleton MJ, Gibbs P, Davis ID, Hopkins W, et al. Two phase I studies of low dose recombinant human IL-12 with Melan-A and influenza peptides in subjects with advanced malignant melanoma. Cancer Immun. (2003) 3:7.
203. Lorenzi S, Mattei F, Sistigu A, Bracci L, Spadaro F, Sanchez M, et al. Type I IFNs control antigen retention and survival of CD8alpha(+) dendritic cells after uptake of tumor apoptotic cells leading to cross-priming. J Immunol. (2011) 186:5142–50. doi: 10.4049/jimmunol.1004163
204. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. (2011) 208:2005–16. doi: 10.1084/jem.20101159
205. Picozzi VJ, Abrams RA, Decker PA, Traverso W, O'Reilly EM, Greeno E, et al. Multicenter phase II trial of adjuvant therapy for resected pancreatic cancer using cisplatin, 5-fluorouracil, and interferon-alfa-2b-based chemoradiation: ACOSOG Trial Z05031. Ann Oncol. (2011) 22:348–54. doi: 10.1093/annonc/mdq384
206. Van der Jeught K, Joe PT, Bialkowski L, Heirman C, Daszkiewicz L, Liechtenstein T, et al. Intratumoral administration of mRNA encoding a fusokine consisting of IFN-beta and the ectodomain of the TGF-beta receptor II potentiates antitumor immunity. Oncotarget (2014) 5:10100–13. doi: 10.18632/oncotarget.2463
207. Tanikawa T, Wilke CM, Kryczek I, Chen GY, Kao J, Nunez G, et al. Interleukin-10 ablation promotes tumor development, growth, and metastasis. Cancer Res. (2012) 72:420–9. doi: 10.1158/0008-5472.CAN-10-4627
208. Breckpot K, Escors D. Dendritic cells for active anti-cancer immunotherapy: targeting activation pathways through genetic modification. Endocr Metab Immune Disord Drug Targets. (2009) 9:328–43. doi: 10.2174/187153009789839156
209. Arce F, Kochan G, Breckpot K, Stephenson H, Escors D. Selective activation of intracellular signalling pathways in dendritic cells for cancer immunotherapy. Anticancer Agents Med Chem. (2012) 12:29–39. doi: 10.2174/187152012798764679
210. Maenhout SK, Du Four S, Corthals J, Neyns B, Thielemans K, Aerts JL. AZD1480 delays tumor growth in a melanoma model while enhancing the suppressive activity of myeloid-derived suppressor cells. Oncotarget (2014) 5:6801–15. doi: 10.18632/oncotarget.2254
211. He W, Zhu Y, Mu R, Xu J, Zhang X, Wang C, et al. A Jak2-selective inhibitor potently reverses the immune suppression by modulating the tumor microenvironment for cancer immunotherapy. Biochem Pharmacol. (2017) 145:132–46. doi: 10.1016/j.bcp.2017.08.019
212. Spinetti T, Spagnuolo L, Mottas I, Secondini C, Treinies M, Rüegg C, et al. TLR7-based cancer immunotherapy decreases intratumoral myeloid–derived suppressor cells and blocks their immunosuppressive function. Oncoimmunology (2016) 5:e1230578. doi: 10.1080/2162402X.2016.1230578
213. Kortylewski M, Moreira D. Myeloid cells as a target for oligonucleotide therapeutics: turning obstacles into opportunities. Cancer Immunol Immunother. (2017) 66:979–88. doi: 10.1007/s00262-017-1966-2
214. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. (2018) 9:398. doi: 10.3389/fimmu.2018.00398
215. Luo Z, Wang C, Yi H, Li P, Pan H, Liu L, et al. Nanovaccine loaded with poly I:C and STAT3 siRNA robustly elicits anti-tumor immune responses through modulating tumor-associated dendritic cells in vivo. Biomaterials (2015) 38:50–60. doi: 10.1016/j.biomaterials.2014.10.050
216. Zhang Q, Hossain DM, Duttagupta P, Moreira D, Zhao X, Won H, et al. Serum-resistant CpG-STAT3 decoy for targeting survival and immune checkpoint signaling in acute myeloid leukemia. Blood (2016) 127:1687–700. doi: 10.1182/blood-2015-08-665604
217. Biswas SK, Tergaonkar V. Myeloid differentiation factor 88-independent Toll-like receptor pathway: Sustaining inflammation or promoting tolerance? Int J Biochem Cell Biol. (2007) 39:1582–92. doi: 10.1016/j.biocel.2007.04.021
218. Oshima H, Oshima M, Inaba K, Taketo MM. Hyperplastic gastric tumors induced by activated macrophages in COX-2/mPGES-1 transgenic mice. Embo J. (2004) 23:1669–78. doi: 10.1038/sj.emboj.7600170
219. Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. (2008) 205:1261–8. doi: 10.1084/jem.20080108
220. Thompson MG, Larson M, Vidrine A, Barrios K, Navarro F, Meyers K, et al. FOXO3-NF-kappaB RelA protein complexes reduce proinflammatory cell signaling and function. J Immunol. (2015) 195:5637–47. doi: 10.4049/jimmunol.1501758
221. Breckpot K, Aerts-Toegaert C, Heirman C, Peeters U, Beyaert R, Aerts JL, et al. Attenuated expression of A20 markedly increases the efficacy of double-stranded RNA-activated dendritic cells as an anti-cancer vaccine. J Immunol. (2009) 182:860–70. doi: 10.4049/jimmunol.182.2.860
222. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature (2016) 539:437–42. doi: 10.1038/nature19834
223. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature (2016) 539:443–7. doi: 10.1038/nature20554
224. Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife (2016) 5:e11612. doi: 10.7554/eLife.11612
225. Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, et al. Suppression of inflammation by a synthetic histone mimic. Nature (2010) 468:1119–23. doi: 10.1038/nature09589
226. Wang H, Cheng F, Woan K, Sahakian E, Merino O, Rock-Klotz J, et al. Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J Immunol. (2011) 186:3986–96. doi: 10.4049/jimmunol.1001101
227. Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, et al. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci USA. (2012) 109:E2865–74. doi: 10.1073/pnas.1121131109
228. Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature (2017) 543:428–32. doi: 10.1038/nature21409
229. Al-Khami AA, Rodriguez PC, Ochoa AC. Energy metabolic pathways control the fate and function of myeloid immune cells. J Leukoc Biol. (2017) 102:369–80. doi: 10.1189/jlb.1VMR1216-535R
230. Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology (2017) 6:e1344804. doi: 10.1080/2162402X.2017.1344804
231. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. (2015) 3:1236–47. doi: 10.1158/2326-6066.CIR-15-0036
232. Bougneres L, Helft J, Tiwari S, Vargas P, Chang BH, Chan L, et al. A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity (2009) 31:232–44. doi: 10.1016/j.immuni.2009.06.022
233. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. (2010) 16:880–6. doi: 10.1038/nm.2172
234. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood (2010) 115:4742–9. doi: 10.1182/blood-2009-10-249540
235. Dong H, Bullock TN. Metabolic influences that regulate dendritic cell function in tumors. Front Immunol. (2014) 5:24. doi: 10.3389/fimmu.2014.00024
236. Netea-Maier RT, Smit JWA, Netea MG. Metabolic changes in tumor cells and tumor-associated macrophages: a mutual relationship. Cancer Lett. (2018) 413:102–9. doi: 10.1016/j.canlet.2017.10.037
237. Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol. (2017) 8:289. doi: 10.3389/fimmu.2017.00289
238. Wenes M, Shang M, Di Matteo M, Goveia J, Martin-Perez R, Serneels J, et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab. (2016) 24:701–15. doi: 10.1016/j.cmet.2016.09.008
239. Mills EL, O'Neill LA. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur J Immunol. (2016) 46:13–21. doi: 10.1002/eji.201445427
240. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer (2004) 4:71–8. doi: 10.1038/nrc1256
241. Rotondo R, Barisione G, Mastracci L, Grossi F, Orengo AM, Costa R, et al. IL-8 induces exocytosis of arginase 1 by neutrophil polymorphonuclears in nonsmall cell lung cancer. Int J Cancer (2009) 125:887–93. doi: 10.1002/ijc.24448
242. Veglia F, Gabrilovich DI. Dendritic cells in cancer: the role revisited. Curr Opin Immunol. (2017) 45:43–51. doi: 10.1016/j.coi.2017.01.002
243. Mbongue JC, Nicholas DA, Torrez TW, Kim NS, Firek AF, Langridge WH. The role of indoleamine 2, 3-Dioxygenase in immune suppression and autoimmunity. Vaccines (2015) 3:703–29. doi: 10.3390/vaccines3030703
244. Timosenko E, Hadjinicolaou AV, Cerundolo V. Modulation of cancer-specific immune responses by amino acid degrading enzymes. Immunotherapy (2017) 9:83–97. doi: 10.2217/imt-2016-0118
245. Godin-Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Cancer Res. (2011) 17:6985–91. doi: 10.1158/1078-0432.CCR-11-1331
246. Manuel ER, Diamond DJ. A road less traveled paved by IDO silencing: Harnessing the antitumor activity of neutrophils. Oncoimmunology (2013) 2. doi: 10.4161/onci.23322
247. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. (2011) 17:1094–100. doi: 10.1038/nm.2438
248. von Bergwelt-Baildon MS, Popov A, Saric T, Chemnitz J, Classen S, Stoffel MS, et al. CD25 and indoleamine 2,3-dioxygenase are up-regulated by prostaglandin E2 and expressed by tumor-associated dendritic cells in vivo: additional mechanisms of T-cell inhibition. Blood (2006) 108:228–37. doi: 10.1182/blood-2005-08-3507
249. Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. (2014) 63:721–35. doi: 10.1007/s00262-014-1549-4
250. Hornyák L, Dobos N, Koncz G, Karányi Z, Páll D, Szabó Z, et al. The Role of Indoleamine-2,3-dioxygenase in cancer development, diagnostics, and therapy. Front Immunol. (2018) 9:151. doi: 10.3389/fimmu.2018.00151
251. Beatty GL, O'Dwyer PJ, Clark J, Shi JG, Bowman KJ, Scherle PA, et al. First-in-human phase I study of the oral inhibitor of indoleamine 2,3-Dioxygenase-1 epacadostat (INCB024360) in patients with advanced solid malignancies. Clin Cancer Res. (2017) 23:3269–76. doi: 10.1158/1078-0432.CCR-16-2272
252. Gangadhar TC, Hamid O, Smith DC, Bauer TM, Wasser JS, Olszanski AJ, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. (2016) 27(Suppl. 6):1110PD. doi: 10.1093/annonc/mdw379.06
253. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-expressed IDO recruits and activates MDSCs in a Treg-dependent manner. Cell Rep. (2015) 13:412–24. doi: 10.1016/j.celrep.2015.08.077
254. Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology (2012) 1:1460–8. doi: 10.4161/onci.21716
255. Sharma MD, Hou DY, Baban B, Koni PA, He Y, Chandler PR, et al. Reprogrammed Foxp3(+) regulatory T cells provide essential help to support cross-presentation and CD8(+) T cell priming in naive mice. Immunity (2010) 33:942–54. doi: 10.1016/j.immuni.2010.11.022
256. Mautino MR, Kumar S, Zhuang H, Waldo J, Jaipuri F, Potturi H, et al. Abstract 4076: a novel prodrug of indoximod with enhanced pharmacokinetic properties. Cancer Research. (2017) 77(13 Suppl.):4076. doi: 10.1158/1538-7445.AM2017-4076
257. Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest. (2013) 123:1580–9. doi: 10.1172/JCI60083
258. Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. (2002) 169:2253–63. doi: 10.4049/jimmunol.169.5.2253
259. Boutard V, Havouis R, Fouqueray B, Philippe C, Moulinoux JP, Baud L. Transforming growth factor-beta stimulates arginase activity in macrophages. Implications for the regulation of macrophage cytotoxicity. J Immunol. (1995) 155:2077–84.
260. Jost MM, Ninci E, Meder B, Kempf C, Van Royen N, Hua J, et al. Divergent effects of GM-CSF and TGFbeta1 on bone marrow-derived macrophage arginase-1 activity, MCP-1 expression, and matrix metalloproteinase-12: a potential role during arteriogenesis. Faseb J. (2003) 17:2281–3. doi: 10.1096/fj.03-0071fje
261. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. (2010) 207:2439–53. doi: 10.1084/jem.20100587
262. de Boniface J, Mao Y, Schmidt-Mende J, Kiessling R, Poschke I. Expression patterns of the immunomodulatory enzyme arginase 1 in blood, lymph nodes and tumor tissue of early-stage breast cancer patients. Oncoimmunology (2012) 1:1305–12. doi: 10.4161/onci.21678
263. Yang Z, Ming XF. Functions of arginase isoforms in macrophage inflammatory responses: impact on cardiovascular diseases and metabolic disorders. Front Immunol. (2014) 5:533. doi: 10.3389/fimmu.2014.00533
264. Toor SM, Syed Khaja AS, El Salhat H, Bekdache O, Kanbar J, Jaloudi M, et al. Increased levels of circulating and tumor-infiltrating granulocytic myeloid cells in colorectal cancer patients. Front Immunol. (2016) 7:560. doi: 10.3389/fimmu.2016.00560
265. Vannini F, Kashfi K, Nath N. The dual role of iNOS in cancer. Redox Biol. (2015) 6:334–43. doi: 10.1016/j.redox.2015.08.009
266. Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front Immunol. (2017) 8:93. doi: 10.3389/fimmu.2017.00093
267. Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res. (2007) 13(2 Pt 2):721s−6s. doi: 10.1158/1078-0432.CCR-06-2197
268. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. (2004) 64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465
269. Crittenden MR, Savage T, Cottam B, Baird J, Rodriguez PC, Newell P, et al. Expression of arginase I in myeloid cells limits control of residual disease after radiation therapy of tumors in mice. Radiat Res. (2014) 182:182–90. doi: 10.1667/RR13493.1
270. Bronte V, Kasic T, Gri G, Gallana K, Borsellino G, Marigo I, et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med. (2005) 201:1257–68. doi: 10.1084/jem.20042028
271. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. (2006) 203:2691–702. doi: 10.1084/jem.20061104
272. Meyer C, Sevko A, Ramacher M, Bazhin AV, Falk CS, Osen W, et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc Natl Acad Sci USA. (2011) 108:17111–6. doi: 10.1073/pnas.1108121108
273. Lin S, Wang J, Wang L, Wen J, Guo Y, Qiao W, et al. Phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. Am J Cancer Res. (2017) 7:41–52.
274. De Santo C, Serafini P, Marigo I, Dolcetti L, Bolla M, Del Soldato P, et al. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc Natl Acad Sci USA. (2005) 102:4185–90. doi: 10.1073/pnas.0409783102
275. Wesolowski R, Markowitz J, Carson WE III. Myeloid derived suppressor cells - a new therapeutic target in the treatment of cancer. J Immunother Cancer. (2013) 1:10. doi: 10.1186/2051-1426-1-10
276. Califano JA, Khan Z, Noonan KA, Rudraraju L, Zhang Z, Wang H, et al. Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res. (2015) 21:30–8. doi: 10.1158/1078-0432.CCR-14-1716
277. Weed DT, Vella JL, Reis IM, De la fuente AC, Gomez C, Sargi Z, et al. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell Carcinoma. Clin Cancer Res. (2015) 21:39–48. doi: 10.1158/1078-0432.CCR-14-1711
278. Hassel JC, Jiang H, Bender C, Winkler J, Sevko A, Shevchenko I, et al. Tadalafil has biologic activity in human melanoma. Results of a pilot trial with Tadalafil in patients with metastatic Melanoma (TaMe). Oncoimmunology (2017) 6:e1326440. doi: 10.1080/2162402X.2017.1326440
279. Kim SH, Roszik J, Grimm EA, Ekmekcioglu S. Impact of l-arginine metabolism on immune response and anticancer immunotherapy. Front Oncol. (2018) 8:67. doi: 10.3389/fonc.2018.00067
280. Talmadge JE, Hood KC, Zobel LC, Shafer LR, Coles M, Toth B. Chemoprevention by cyclooxygenase-2 inhibition reduces immature myeloid suppressor cell expansion. Int Immunopharmacol. (2007) 7:140–51. doi: 10.1016/j.intimp.2006.09.021
281. Eruslanov E, Daurkin I, Ortiz J, Vieweg J, Kusmartsev S. Pivotal Advance: Tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages by altering intracellular PGE(2) catabolism in myeloid cells. J Leukoc Biol. (2010) 88:839–48. doi: 10.1189/jlb.1209821
282. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood (2011) 118:5498–505. doi: 10.1182/blood-2011-07-365825
283. Na YR, Yoon YN, Son DI, Seok SH. Cyclooxygenase-2 inhibition blocks M2 macrophage differentiation and suppresses metastasis in murine breast cancer model. PLoS ONE. (2013) 8:e63451. doi: 10.1371/journal.pone.0063451
284. Chen EP, Markosyan N, Connolly E, Lawson JA, Li X, Grant GR, et al. Myeloid Cell COX-2 deletion reduces mammary tumor growth through enhanced cytotoxic T-lymphocyte function. Carcinogenesis (2014) 35:1788–97. doi: 10.1093/carcin/bgu053
285. Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. (2005) 65:3044–8. doi: 10.1158/0008-5472.CAN-04-4505
286. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. (2007) 67:4507–13. doi: 10.1158/0008-5472.CAN-06-4174
287. Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer (2010) 10:464. doi: 10.1186/1471-2407-10-464
Keywords: cancer, tumor microenvironment, immature myeloid cell, macrophage, dendritic cell, myeloid-derived suppressor cell
Citation: Awad RM, De Vlaeminck Y, Maebe J, Goyvaerts C and Breckpot K (2018) Turn Back the TIMe: Targeting Tumor Infiltrating Myeloid Cells to Revert Cancer Progression. Front. Immunol. 9:1977. doi: 10.3389/fimmu.2018.01977
Received: 18 June 2018; Accepted: 13 August 2018;
Published: 31 August 2018.
Edited by:
Fabrizio Mattei, Istituto Superiore di Sanità (ISS), ItalyReviewed by:
Abhishek D. Garg, KU Leuven, BelgiumNadege Bercovici, Centre National de la Recherche Scientifique (CNRS), France
Copyright © 2018 Awad, De Vlaeminck, Maebe, Goyvaerts and Breckpot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karine Breckpot, a2FyaW5lLmJyZWNrcG90QHZ1Yi5iZQ==
†These authors have contributed equally to this work
‡These authors share senior authorship