 Carl Weidinger
Carl Weidinger Jörn F. Ziegler
Jörn F. Ziegler Marilena Letizia
Marilena Letizia Franziska Schmidt
Franziska Schmidt Britta Siegmund
Britta Siegmund
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 12 October 2018
Sec. Cytokines and Soluble Mediators in Immunity
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01974
This article is part of the Research Topic Cytokines and Intestinal Mucosal Immunity View all 12 articles
Fat tissue was initially described for its endocrine and metabolic function. Over the last two decades increasing evidence indicated a close interaction with the immune system. Partly responsible for this immune modulatory function are soluble factors released by the fat tissue, most prominently the so-called adipokines. These discoveries led to the question how adipokines influence inflammatory diseases. Linking inflammation and adipose tissue, Crohn's disease, a chronic inflammatory bowel disease, is of particular interest for studying the immune modulatory properties of adipokines since it is characterized by a hyperplasia of the mesenteric fat that subsequently is creeping around the inflamed segments of the small intestine. Thus, the role of several adipokines in the creeping fat as well as in intestinal inflammation was recently explored. The present review selected the four adipokines adiponectin, apelin, chemerin, and leptin and provides a working model based on the available literature how these factors participate in the maintenance of intestinal immune homeostasis.
Adipokines represent a group of mediators primarily released by adipocytes that modulate a variety of metabolic functions within the fat tissue and additional organs such as liver, brain and muscle. In addition to the metabolic functions, a regulatory role within the immune system was identified early on for a number of adipokines. This includes adiponectin which in first reports was shown to suppress mature macrophage function (1) as well as leptin that was identified as a T cell stimulatory factor (2). These initial publications induced a plethora of studies exploring the impact of various adipokines in several inflammatory diseases. Why is intestinal inflammation of particular interest for the regulatory function of adipokines?
In Crohn's disease, one subtype of inflammatory bowel disease (IBD), characteristic changes of the mesenteric fat suggest that the mesenteric fat might play a central immune modulatory role in the pathogenesis of Crohn's diseases (3). In this disease, the mesenteric fat becomes hyperplastic and is creeping around the inflamed segments of the small intestine, suggesting a direct role of this fat tissue compartment in Crohn's disease. In order to provide a concise overview how adipokines regulate intestinal inflammation, we here selected adipokines for which experimental data in intestinal inflammation were available. Thus, we chose apelin, adiponectin, chemerin, and leptin. In order to include the relevant literature, we chose the names of the respective adipokine in combination with either colitis, IBD, Crohn's disease, or ulcerative colitis as search terms in PubMed. This review will first focus on the selected adipokines (summary provided in Table 1), namely apelin, adiponectin, chemerin and leptin, and then provide a disease-relevant working model.

Table 1. Adipokines and the regulatory effect on intestinal homeostasis.
Early on, apelin has been shown to be elevated in the colonic tissue of mice suffering from colitis as well as in humans with IBD. Apelin was mainly expressed by epithelial cells and addition of synthetic apelin in cell culture increased epithelial cell proliferation (12). More recently, apelin was described to play a significant role in the development as well as the stabilization of lymphatic vessels (13, 14). Remarkably, defects in the lymphatic transport of the mesentery have previously been suggested to be involved in the pathogenesis of Crohn's disease. This is reflected by an increased density of lymphatic vessels as well as the presence of tertiary lymphoid organs in the mesentery of Crohn's disease patients (30, 31). Accordingly, apelin was shown to be highly expressed in the mesenteric adipose tissue of Crohn's disease patients. In addition, the administration of apelin to Il10−/− mice with established colitis resulted not only in an amelioration of disease by lowering the production of pro-inflammatory cytokines such as TNFα, IL-6, and IL-1β but furthermore enhanced intestinal lymphatic function. This was shown by an increased lymphatic vessel density. In addition, lymphangiography indicated an augmented lymphatic drainage (15). Based on these findings, one can propose a regenerative function of apelin on intestinal epithelial cells as well as a supportive role with regard to intestinal lymphatic drainage.
The adipokine adiponectin plays a key role in regulating insulin sensitivity and had previously been shown to play an anti-inflammatory role in atherosclerosis (4). The study by Yamamoto et al. analyzed the adiponectin expression in the creeping fat of Crohn's disease patients, that revealed an upregulation of adiponectin expression in creeping fat of Crohn's disease patients when compared to non-creeping fat of Crohn's disease patients as well as fat from ulcerative colitis patients or controls (11).
A study by Fayad et al. explored the function of adiponectin in intestinal inflammation by subjecting adiponectin-deficient animals to two models of experimental colitis, the model of dextran sulfate sodium (DSS)- and the model of trinitrobenzene sulphonic acid (TNBS)-induced colitis. Adiponectin was shown to rather induce the production of pro-inflammatory cytokines in the colon as adiponectin stimulation of colon organ cultures of DSS-treated mice resulted in increased production of IL-6 and MIP-2, whereas adiponectin-deficient APN knock out mice were protected from experimental colitis. Thus, these data were somewhat contrary to the findings described for atherosclerosis (8). A second study by Nishihara et al. explored again adiponectin-deficient mice in the DSS and TNBS model of colitis. In contrast to the previous study, adiponectin protected from inflammation, possibly mediated by a direct anti-inflammatory effect on the colonic epithelial cells (7). These discrepancies between the two studies as well as in several in vitro studies are most likely due to different types of knockout mice or adiponectin used (9).
In line with the first study, Ogunwobi and colleagues provide data analyzing the effect of adiponectin on colon epithelial cells by using the colonic epithelial cell line HT-29 and by carefully distinguishing globular adiponectin from full-length adiponectin. Remarkably, in particular globular adiponectin mediated pro-proliferative as well as pro-inflammatory effects through activation of extracellular-signal regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK) NF-κB signaling on colonic epithelial cells (5). When the human NCM60 epithelial cell line was exposed to fat-conditioned media obtained from IBD patients, cells showed a reduced expression of adiponectin receptor 1 (AdipoR1). Silencing of AdipoR1 in mice resulted in an exacerbation of TNBS-induced colitis (10).
In a recent study, adiponectin-deficient mice treated with DSS exhibited more severe colitis accompanied by an increased presence of activated B cells, pro-inflammatory cytokines such as IL-1β, IL-4, and IL-6 and increased STAT3 signaling in the colon. The epithelium of the knockout animals revealed a decrease in cell proliferation as well as increased apoptosis and cellular stress. In in vitro experiments these effects could be reversed by adiponectin. These data are supporting the concept that adiponectin maintains intestinal homeostasis (6).
Chemerin has been shown to serve as chemo-attractant for cells of the innate immune system (16). Serum from Crohn's disease (n = 230), ulcerative colitis patients (n = 80), and healthy controls (n = 80) was recently compared for the expression of chemerin and adiponectin. Chemerin was elevated in IBD patients, whereas adiponectin was decreased (19). An in vivo study furthermore indicated that the administration of chemerin resulted in aggravation of DSS-induced colitis in mice by augmenting TNFα and IL-6 production, whereas a decrease in IL-10—producing anti-inflammatory macrophages could be detected. This could be confirmed in vitro, where the presence of chemerin prevented macrophages from polarizing into an anti-inflammatory phenotype resulting in impaired expression of Arginase-1 and IL-10 (17). The chemokine-like receptor 1 (CMKLR1) is the receptor for chemerin. Mice deficient in CMKLR1 developed DSS-induced colitis in a delayed time course, albeit ultimately presenting with a similar disease activity (18).
Leptin was initially identified as a satiety factor (20, 32). Work by Lord and colleagues revealed that the adipokine leptin supports T cell proliferation and results in increased T helper cell type 1 (Th1) and in suppressed Th2 cytokine production. In addition, administration of leptin to starved mice resulted in an abrogation of starvation-induced immunosuppression (2). The additional characterization of three children with congenital leptin deficiency indicated that leptin substitution not only reversed their metabolic dysfunction including insulin resistance and fatty liver degeneration, but furthermore increased the number of circulating CD4+ T cells as well as their proliferative capacity (33). These findings raised the question whether leptin exerts a modulating effect on autoimmune diseases. First evidence was provided from the model of experimental autoimmune encephalomyelitis. Here, leptin-deficient (ob/ob) mice were protected from T cell-mediated neuronal damage and leptin substitution augmented the disease susceptibility in mice. Additional work on the same model revealed that CNS-infiltrating T cells themselves are able to produce leptin and that this effect could be blocked by an anti-leptin receptor antibody (34, 35).
In an early study, systemic leptin concentrations were determined during the course of acute experimental colitis. In this study, the concentration of leptin in the plasma increased in TNBS-induced colitis and indomethacin-induced ileitis and correlated with disease severity. However, this observed increase was only transient and returned to control concentrations over time (36). The association of a distinct disease to plasma levels has proven difficult for several disease entities in the past. However, independent studies revealed a strong up-regulation of leptin expression in the mesenteric fat of Crohn's disease patients (28, 37).
In our own work, we were able to demonstrate that leptin-deficient ob/ob mice are protected from DSS-induced colitis and that leptin administration reverses disease susceptibility in mice (24). To confirm the previously established concept that leptin mediates pro-inflammatory effects, at least partly, via T cells, we performed a T cell transfer model of colitis and transferred naive CD4+ T cells lacking the signaling Ob-Rb-isoform of the leptin receptor. In fact, the development of colitis was significantly delayed using this approach, indicating that the stimulatory effect of leptin plays a crucial role in this model (25). Additional data showing that ob/ob mice are protected in models driven by either Th1 (TNBS) or Th2 (oxazolone) cells underline that the T cell-stimulating capacity of leptin is important for the observed effects (27). However, this enhancing factor does not apply for all T cell subpopulations, since leptin has been shown to inhibit the proliferation of regulatory T (Treg, FoxP3+CD4+CD25+) cells. Accordingly, the absence of leptin, as demonstrated for ob/ob and db/db mice (mice deficient for the signaling Ob-Rb isoform of the leptin receptor), resulted in an increased proliferation of functional Treg cells (38). Likewise, Reis and colleagues could demonstrate that Leprfl/fl−CD4-Cre mice harboring a conditional knock out of the leptin receptor in their CD4+ T cell compartment also showed higher frequencies of FOXP3+ Treg cells under steady state conditions. Importantly, Lepr-deficient T cells displayed a severe defect in Th17 differentiation due to a decreased activation of the STAT3 signaling cascade, resulting in impaired cytokine production of IL-17 and IFNγ, which protected recipient Rag-−/− mice receiving Lepr-deficient T cells from transfer colitis (23).
Data from the experimental autoimmune encephalomyelitis model have previously indicated that leptin produced by T cells might play a key role in mediating disease severity (23, 35). Thus, we performed again the T cell transfer model of colitis and chose this time naive T cells isolated from ob/ob mice as disease-inducing cell population in comparison to wild-type cells. In contrast to the data cited above, no differences were observed with regard to disease severity or cytokine production, implying that leptin produced by T cells is irrelevant for intestinal inflammation (26).
Recent data indicate that the administration of a pegylated leptin antagonist (PG-MLA) was sufficient to protect from chronic experimental colitis. Amelioration of colitis was here associated with a decrease in the expression of mucosal pro-inflammatory cytokines and an increase of mucosal Treg cells (39).
Having shown that leptin modulates intestinal inflammation via the T cell compartment, the question arose whether there is an additional effect on epithelial cells. The leptin receptor (Ob-Rb) is in fact expressed in human colonic tissue as well as on the HT-29 colon cancer cell line. Stimulation of HT-29 cells with leptin was followed by activation of p42/44 MAPK as well as increased proliferation in vitro and in vivo (21). An interesting aspect was added by the study of Sitaraman and colleagues that were able to demonstrate that inflamed colonic epithelial cells produce and release leptin into the intestinal lumen. The presence of luminal leptin resulted in the activation of the NF- κB pathway in intestinal epithelial cells. Rectal application of leptin was followed by intestinal inflammation and epithelial wall damage (22). Following up on these luminal effects, Hoda et al. analyzed the effect of luminal leptin on ion transport capacities under inflammatory conditions. Human intestinal epithelial cells (T84) as well as intestinal tissue from a rat model of chemotherapy-induced enterocolitis were analyzed in Ussing chambers. These served to determine the transepithelial short-circuit current I(sc). The presence of leptin resulted in an increase of basal I(sc) of T84 cells, which was mediated by activation of the MAPK pathway. In line, in the enterocolits model luminal leptin equally induced an increase in I(sc), that was more prominent in the proximal colon (40).
A recent study was able to link leptin, cell metabolism and intestinal epithelial function. The authors provided evidence indicating that leptin is able to induce lipid droplet formation in intestinal epithelial cells (IEC-6). This was accompanied by an increased production of CXCL1/CINC-1, CCL2/MCP-1, and TGFβ. In line with previous data, leptin induced cell proliferation. The process of lipid droplet induction and the associated effects depended on the mammalian target of rapamycin (mTOR) pathway, since it was completely abrogated in the presence of rapamycin (41). Thus, one can conclude that leptin, very similar to adiponectin, exerts direct effects on intestinal epithelial cells and consequently plays a role in intestinal homeostasis.
The question arises how the mesenteric fat gets involved in the first place. Courageous proposals suggest that changes in the mesenteric fat tissue might in fact be the initiating event of Crohn's disease. However, this is not only difficult to proof but it also seems to be more likely that the transmural inflammation and consecutive chronic bacterial translocation is the initiating factor for these changes. Crohn's disease, in contrast to ulcerative colitis, presents with a transmural inflammation and can also affect the small intestine. Several experimental models as well as studies in humans provided evidence of an increased translocation of bacteria into the mesenteric fat (42, 43). Previous data had already proven that adipocytes as well as preadipocytes express functional receptors of the innate immune system including toll-like receptors (Tlr) and nucleotide oligomerization domains 1 (NOD1) and 2 (NOD2) (44, 45). Accordingly, while wild type mice featured an up-regulation of pro-inflammatory mediators in chronic DSS colitis, Myd88−/− mice, thus mice with a dysfunctional innate immune system, failed to respond to these translocalizing bacteria and showed a significantly increased mortality, suggesting that the mesenteric fat, serves as a potential second barrier (42, 46). In line, wild-type mice revealed an up-regulation of leptin in the mesenteric fat whereas this was not observed in Tlr9−/− mice. However, IL-6 production was unaffected by the absence of Tlr9 (47), indicating that other innate receptors contribute to the effects described before (42).
What is the subsequent effect of adipokines produced within the mesenteric adipose tissue? Leptin has been shown to not only modulate the polarization of the T helper cell compartment but to equally affect together with adiponectin the myeloid compartment. Leptin as well as adiponectin favored the polarization of anti-inflammatory macrophages, the dominant macrophage population detectable in creeping fat of Crohn's disease patients (29). In addition, as described above for mice (42) and patients with Crohn's disease (43), the mesenteric fat develops an effector response to the translocalizing antigens. Remarkably, a dysfunction of this effector response, as discussed above for the Myd88−/− mice, results in an increased mortality, thus suggesting a primarily protective function. Together this fits into the concept that the mesenteric fat represents a second barrier and thereby prevents the Crohn's disease patient from continuous bacteremia (48, 49).
A summary of the effects mediated by adipokines is provided in Figure 1.
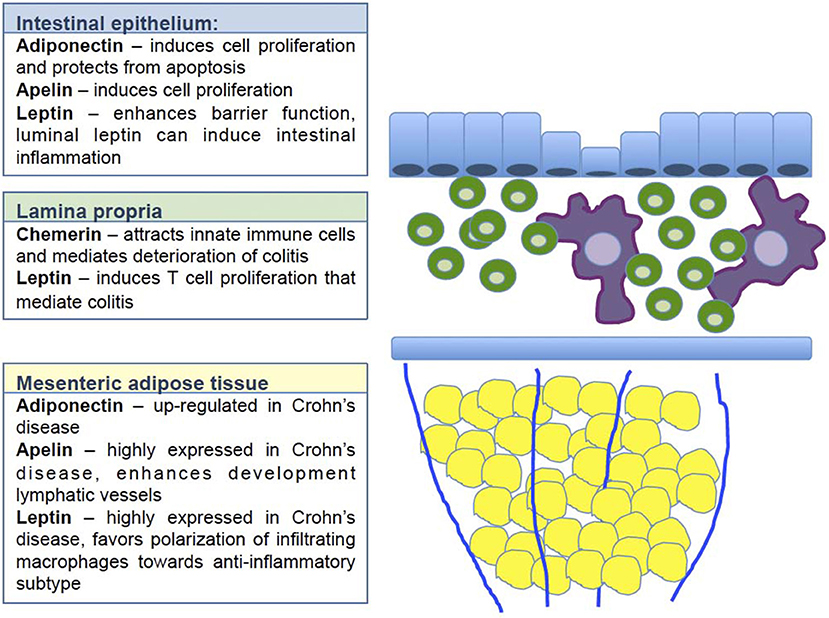
Figure 1. Regulatory effect of selected adipokines on intestinal homeostasis. The figure illustrates at which layer of the intestinal wall the selected adipokines exert their respective regulatory function. Blue indicates the intestinal epithelial layer. Adiponectin, apelin and leptin have been shown to enhance cell proliferation and enhance barrier function. The majority of these data derive from in vitro data, thus it remains open how these adipokines contribute to the complex crosstalk between epithelial cells and immune cells in vivo. Green indicates the lamina propria with all cell populations included. Chemerin has been shown here to attract innate immune cells resulting in a deterioration of colitis. Leptin induces T cell proliferation thus enhancing inflammation. Yellow indicates the mesenteric fat tissue. Adiponectin, apelin and leptin are all up-regulated in this compartment. Apelin enhances the function of the lymphatic vessels, that are known to be dysfunctional in Crohn's disease. Leptin strongly influences the polarization of infiltrating monocytes toward rather anti-inflammatory macrophages.
Continuous work over the last two decades has substantially provided insights into the regulation of adipokines and intestinal inflammation. The data discussed for adiponectin, apelin, chemerin, and leptin within this review indicate a complex specific function for each individual adipokine in the regulation of intestinal inflammation. However, the summary also reveals the shortcomings of our current understanding. Thus, the complex interplay between the adipose tissue and the immune system presents a hot topic for current research. In our view, it is rather unlikely that adipokines will serve as therapeutic option for patients with IBD. Nevertheless, the overall metabolic condition of a patient, e.g., body mass index, presence of diabetes, might strongly influence the adipokine response on one hand, on the other hand, anti-inflammatory treatment in IBD patients might result in direct changes of the adipokine and thus metabolic profile. Therefore, this particular cross-regulation will become even more important in the foreseeable future.
JZ, ML, and FS contributed distinct parts of the manuscript. CW participated in writing and finalizing the manuscript. BS wrote the first draft and finalized the manuscript.
BS served as consultant for Abbvie, Falk, Janssen, Hospira, MSD and Takeda and received speaker's fees from Abbvie, Falk, Ferring, Hospira, Janssen MSD, Takeda. BS received a research grant from Pfizer. All money went to the Charité.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
CW acknowledges funding by the Berlin Institute of Health and the German Research Foundation (DFG, We-5303/3-1); BS acknowledges funding by the DFG, the Deutsche Krebshilfe, the Berlin Institute of Health. CW and BS are PIs in the TRR 241 IEC in IBD funded by the DFG, JZ is supported by a student grant of the German Society of Internal Medicine (DGIM). We acknowledge support from the German Research Foundation (DFG) and the Open Access Publication Fund of Charité–Universitätsmedizin Berlin.
1. Yokota T, Oritani K, Takahashi I, Ishikawa J, Matsuyama A, Ouchi N, et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood (2000) 96:1723–32.
2. Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature (1998) 394:897–901. doi: 10.1038/29795
3. Crohn BB, Ginzburg L, Oppenheimer GD. Regional ileitis, a pathologic and clinical entity. J Am Med Assoc. (1932) 99:1323–9. doi: 10.1001/jama.1932.02740680019005
4. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature (2003) 423:762–9. doi: 10.1038/nature01705
5. Ogunwobi OO, Beales IL. Adiponectin stimulates proliferation and cytokine secretion in colonic epithelial cells. Regul Pept. (2006) 134:105–13. doi: 10.1016/j.regpep.2006.02.001
6. Obeid S, Wankell M, Charrez B, Sternberg J, Kreuter R, Esmaili S, et al. Adiponectin confers protection from acute colitis and restricts a B cell immune response. J Biol Chem. (2017) 292:6569–82. doi: 10.1074/jbc.M115.712646
7. Nishihara T, Matsuda M, Araki H, Oshima K, Kihara S, Funahashi T, et al. Effect of adiponectin on murine colitis induced by dextran sulfate sodium. Gastroenterology (2006) 131:853–61. doi: 10.1053/j.gastro.2006.06.015
8. Fayad R, Pini M, Sennello JA, Cabay RJ, Chan L, Xu A, et al. Adiponectin deficiency protects mice from chemically induced colonic inflammation. Gastroenterology (2007) 132:601–14. doi: 10.1053/j.gastro.2006.11.026
9. Fantuzzi G. Adiponectin in inflammatory and immune-mediated diseases. Cytokine (2013) 64:1–10. doi: 10.1016/j.cyto.2013.06.317
10. Sideri A, Stavrakis D, Bowe C, Shih DQ, Fleshner P, Arsenescu V, et al. Effects of obesity on severity of colitis and cytokine expression in mouse mesenteric fat. Potential role of adiponectin receptor 1. Am J Physiol Gastrointest Liver Physiol. (2015) 308:G591–604. doi: 10.1152/ajpgi.00269.2014
11. Yamamoto K, Kiyohara T, Murayama Y, Kihara S, Okamoto Y, Funahashi T, et al. Production of adiponectin, an anti-inflammatory protein, in mesenteric adipose tissue in Crohn's disease. Gut (2005) 54:789–96. doi: 10.1136/gut.2004.046516
12. Han S, Wang G, Qiu S, De La Motte C, Wang HQ, Gomez G, et al. Increased colonic apelin production in rodents with experimental colitis and in humans with IBD. Regul Pept. (2007) 142:131–7. doi: 10.1016/j.regpep.2007.02.002
13. Sawane M, Kajiya K, Kidoya H, Takagi M, Muramatsu F, Takakura N. Apelin inhibits diet-induced obesity by enhancing lymphatic and blood vessel integrity. Diabetes (2013) 62:1970–80. doi: 10.2337/db12-0604
14. Kim JD, Kang Y, Kim J, Papangeli I, Kang H, Wu J, et al. Essential role of Apelin signaling during lymphatic development in zebrafish. Arterioscler Thromb Vasc Biol. (2014) 34:338–45. doi: 10.1161/ATVBAHA.113.302785
15. Ge Y, Li Y, Chen Q, Zhu W, Zuo L, Guo Z, et al. Adipokine apelin ameliorates chronic colitis in Il-10(-/-) mice by promoting intestinal lymphatic functions. Biochem Pharmacol. (2018) 148:202–12. doi: 10.1016/j.bcp.2018.01.011
16. Wittamer V, Franssen JD, Vulcano M, Mirjolet JF, Le Poul E, Migeotte I, et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J Exp Med. (2003) 198:977–85. doi: 10.1084/jem.20030382
17. Lin Y, Yang X, Yue W, Xu X, Li B, Zou L, et al. Chemerin aggravates DSS-induced colitis by suppressing M2 macrophage polarization. Cell Mol Immunol. (2014) 11:355–66. doi: 10.1038/cmi.2014.15
18. Dranse HJ, Rourke JL, Stadnyk AW, Sinal CJ. Local chemerin levels are positively associated with DSS-induced colitis but constitutive loss of CMKLR1 does not protect against development of colitis. Physiol Rep. (2015) 3:e12497. doi: 10.14814/phy2.12497
19. Weigert J, Obermeier F, Neumeier M, Wanninger J, Filarsky M, Bauer S, et al. Circulating levels of chemerin and adiponectin are higher in ulcerative colitis and chemerin is elevated in Crohn's disease. Inflamm Bowel Dis. (2010) 16:630–7. doi: 10.1002/ibd.21091
20. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372:425–32. doi: 10.1038/372425a0
21. Hardwick JC, Van Den Brink GR, Offerhaus GJ, Van Deventer SJ, Peppelenbosch MP. Leptin is a growth factor for colonic epithelial cells. Gastroenterology (2001) 121:79–90. doi: 10.1053/gast.2001.25490
22. Sitaraman S, Liu X, Charrier L, Gu LH, Ziegler TR, Gewirtz A, et al. Colonic leptin: source of a novel proinflammatory cytokine involved in IBD. FASEB J. (2004) 18:696–8. doi: 10.1096/fj.03-0422fje
23. Reis BS, Lee K, Fanok MH, Mascaraque C, Amoury M, Cohn LB, et al. Leptin receptor signaling in T cells is required for Th17 differentiation. J Immunol. (2015) 194:5253–60. doi: 10.4049/jimmunol.1402996
24. Siegmund B, Lehr HA, Fantuzzi G. Leptin: a pivotal mediator of intestinal inflammation in mice. Gastroenterology (2002) 122:2011–25. doi: 10.1053/gast.2002.33631
25. Siegmund B, Sennello JA, Jones-Carson J, Gamboni-Robertson F, Lehr HA, Batra A, et al. Leptin receptor expression on T lymphocytes modulates chronic intestinal inflammation in mice. Gut (2004) 53:965–72. doi: 10.1136/gut.2003.027136
26. Fantuzzi G, Sennello JA, Batra A, Fedke I, Lehr HA, Zeitz M, et al. Defining the role of T cell-derived leptin in the modulation of hepatic or intestinal inflammation in mice. Clin Exp Immunol. (2005) 142:31–8. doi: 10.1111/j.1365-2249.2005.02898.x
27. Batra A, Okur B, Glauben R, Erben U, Ihbe J, Stroh T, et al. Leptin: a critical regulator of CD4+ T-cell polarization in vitro and in vivo. Endocrinology (2010) 151:56–62. doi: 10.1210/en.2009-0565
28. Zulian A, Cancello R, Micheletto G, Gentilini D, Gilardini L, Danelli P, et al. Visceral adipocytes: old actors in obesity and new protagonists in Crohn's disease? Gut (2012) 61:86–94. doi: 10.1136/gutjnl-2011-300391
29. Kredel LI, Batra A, Stroh T, Kuhl AA, Zeitz M, Erben U, et al. Adipokines from local fat cells shape the macrophage compartment of the creeping fat in Crohn's disease. Gut (2013) 62:852–62. doi: 10.1136/gutjnl-2011-301424
30. Rahier JF, De Beauce S, Dubuquoy L, Erdual E, Colombel JF, Jouret-Mourin A, et al. Increased lymphatic vessel density and lymphangiogenesis in inflammatory bowel disease. Aliment Pharmacol Ther. (2011) 34:533–43. doi: 10.1111/j.1365-2036.2011.04759.x
31. Randolph GJ, Bala S, Rahier JF, Johnson MW, Wang PL, Nalbantoglu I, et al. Lymphoid aggregates remodel lymphatic collecting vessels that serve mesenteric lymph nodes in Crohn disease. Am J Pathol. (2016) 186:3066–73. doi: 10.1016/j.ajpath.2016.07.026
32. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature (1998) 395:763–70. doi: 10.1038/27376
33. Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. (2002) 110:1093–103. doi: 10.1172/JCI0215693
34. Matarese G, Di Giacomo A, Sanna V, Lord GM, Howard JK, Di Tuoro A, et al. Requirement for leptin in the induction and progression of autoimmune encephalomyelitis. J Immunol. (2001) 166:5909–16. doi: 10.4049/jimmunol.166.10.5909
35. Sanna V, Di Giacomo A, La Cava A, Lechler RI, Fontana S, Zappacosta S, et al. Leptin surge precedes onset of autoimmune encephalomyelitis and correlates with development of pathogenic T cell responses. J Clin Invest. (2003) 111:241–50. doi: 10.1172/JCI200316721
36. Barbier M, Cherbut C, Aube AC, Blottiere HM, Galmiche JP. Elevated plasma leptin concentrations in early stages of experimental intestinal inflammation in rats. Gut (1998) 43:783–90. doi: 10.1136/gut.43.6.783
37. Siegmund B. Mesenteric fat in Crohn's disease: the hot spot of inflammation? Gut (2012) 61:3–5. doi: 10.1136/gutjnl-2011-301354
38. De Rosa V, Procaccini C, Cali G, Pirozzi G, Fontana S, Zappacosta S, et al. A key role of leptin in the control of regulatory T cell proliferation. Immunity (2007) 26:241–55. doi: 10.1016/j.immuni.2007.01.011
39. Singh UP, Singh NP, Guan H, Busbee B, Price RL, Taub DD, et al. Leptin antagonist ameliorates chronic colitis in IL-10(-)/(-) mice. Immunobiology (2013) 218:1439–51. doi: 10.1016/j.imbio.2013.04.020
40. Hoda MR, Scharl M, Keely SJ, Mccole DF, Barrett KE. Apical leptin induces chloride secretion by intestinal epithelial cells and in a rat model of acute chemotherapy-induced colitis. Am J Physiol Gastrointest Liver Physiol. (2010) 298:G714–721. doi: 10.1152/ajpgi.00320.2009
41. Fazolini NP, Cruz AL, Werneck MB, Viola JP, Maya-Monteiro CM, Bozza PT. Leptin activation of mTOR pathway in intestinal epithelial cell triggers lipid droplet formation, cytokine production and increased cell proliferation. Cell Cycle (2015) 14:2667–76. doi: 10.1080/15384101.2015.1041684
42. Batra A, Heimesaat MM, Bereswill S, Fischer A, Glauben R, Kunkel D, et al. Mesenteric fat - control site for bacterial translocation in colitis? Mucosal Immunol. (2012) 5:580–91. doi: 10.1038/mi.2012.33
43. Peyrin-Biroulet L, Gonzalez F, Dubuquoy L, Rousseaux C, Dubuquoy C, Decourcelle C, et al. Mesenteric fat as a source of C reactive protein and as a target for bacterial translocation in Crohn's disease. Gut (2012) 61:78–85. doi: 10.1136/gutjnl-2011-300370
44. Batra A, Pietsch J, Fedke I, Glauben R, Okur B, Stroh T, et al. Leptin-dependent toll-like receptor expression and responsiveness in preadipocytes and adipocytes. Am J Pathol. (2007) 170:1931–41. doi: 10.2353/ajpath.2007.060699
45. Stroh T, Batra A, Glauben R, Fedke I, Erben U, Kroesen A, et al. Nucleotide oligomerization domains 1 and 2: regulation of expression and function in preadipocytes. J Immunol. (2008) 181:3620–7. doi: 10.4049/jimmunol.181.5.3620
46. Kredel L, Batra A, Siegmund B. Role of fat and adipokines in intestinal inflammation. Curr Opin Gastroenterol. (2014) 30:559–65. doi: 10.1097/MOG.0000000000000116
47. Karrasch T, Schmid A, Kopp A, Obermeier F, Hofmann C, Schaffler A. Impact of toll-like-receptor-9 (TLR9) deficiency on visceral adipose tissue adipokine expression during chronic DSS-induced colitis in mice. Exp Clin Endocrinol Diabetes (2015) 123:80–7. doi: 10.1055/s-0034-1398502
48. Kredel LI, Siegmund B. Adipose-tissue and intestinal inflammation - visceral obesity and creeping fat. Front Immunol. (2014) 5:462. doi: 10.3389/fimmu.2014.00462
Keywords: adipokines, inflammatory bowel disease, Crohn's disease, creeping fat, immune modulation
Citation: Weidinger C, Ziegler JF, Letizia M, Schmidt F and Siegmund B (2018) Adipokines and Their Role in Intestinal Inflammation. Front. Immunol. 9:1974. doi: 10.3389/fimmu.2018.01974
Received: 27 May 2018; Accepted: 10 August 2018;
Published: 12 October 2018.
Edited by:
Fabio Cominelli, Case Western Reserve University, United StatesReviewed by:
Angela Bonura, Consiglio Nazionale Delle Ricerche (CNR), ItalyCopyright © 2018 Weidinger, Ziegler, Letizia, Schmidt and Siegmund. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Britta Siegmund, YnJpdHRhLnNpZWdtdW5kQGNoYXJpdGUuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.