
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 September 2018
Sec. Mucosal Immunity
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01956
 Valentina P. Sebastián1
Valentina P. Sebastián1 Geraldyne A. Salazar1
Geraldyne A. Salazar1 Irenice Coronado-Arrázola1
Irenice Coronado-Arrázola1 Bárbara M. Schultz1
Bárbara M. Schultz1 Omar P. Vallejos1
Omar P. Vallejos1 Loni Berkowitz1,2
Loni Berkowitz1,2 Manuel M. Álvarez-Lobos1,2
Manuel M. Álvarez-Lobos1,2 Claudia A. Riedel3
Claudia A. Riedel3 Alexis M. Kalergis1,4
Alexis M. Kalergis1,4 Susan M. Bueno1*
Susan M. Bueno1*Heme Oxygenase 1 (HMOX1) is an enzyme that catalyzes the reaction that degrades the heme group contained in several important proteins, such as hemoglobin, myoglobin, and cytochrome p450. The enzymatic reaction catalyzed by HMOX1 generates Fe2+, biliverdin and CO. It has been shown that HMOX1 activity and the by-product CO can downmodulate the damaging immune response in several models of intestinal inflammation as a result of pharmacological induction of HMOX1 expression and the administration of non-toxic amounts of CO. Inflammatory Bowel Diseases, which includes Crohn's Disease (CD) and Ulcerative Colitis (UC), are one of the most studied ailments associated to HMOX1 effects. However, microbiota imbalances and infections are also important factors influencing the occurrence of acute and chronic intestinal inflammation, where HMOX1 activity may play a major role. As part of this article we discuss the immune modulatory capacity of HMOX1 during IBD, as well during the infections and interactions with the microbiota that contribute to this inflammatory disease.
Heme oxygenase 1 (HMOX1) is an enzyme that catalyzes the first step of the oxidative degradation of the heme group, which is a rate-limiting reaction that releases the following molecules as by-products: carbon monoxide (CO), free iron, and biliverdin (which is lately reduced to bilirubin) (1). HMOX1 is composed of 288 amino acid residues with an active site located between the first two alpha-helixes (2). The exact reactions leading to the conversion of hemoglobin, hemin, and myoglobin into bilirubin were identified in 1969, when Tenhunen, Marver, and Schmid described that this reaction was catalyzed by a heme oxygenase enzyme, based on two observations. First, the reaction required a metal chelate and the formation of only one isomer of bile pigment, suggesting the participation of an enzyme in this reaction. Second, the insertion of two hydroxyl groups that indicated that the cleavage of the porphyrin ring was an oxidative reaction. Based on these observations, authors described that heme oxygenase stoichiometrically required NADPH and molecular oxygen to generate the same amount of carbon monoxide and bilirubin (3, 4). Few years later, Maines and Kappas described that, not only heme is a substrate for heme oxygenase, but also Cobalt protoporphyrin IX (CoPP). Using microsomal fractions, a NADPH-generating system and biliverdin reductase, the authors measured CoPP oxidation. These results suggested that CoPP can be bound to and oxidized by heme oxygenase, as well as is heme. On the contrary, although nonreactive metalloporphyrins, such as Ni-heme, bind to the enzyme these molecules are not oxidized by HMOX1 and inhibit its enzymatic activity (5). Another important metalloporphyrin that inhibits HMOX1 activity is tin-protoporphyrin (SnPP), a molecule that was early evaluated as a potential therapy for hyperbilirubinemia in newborns, due to its capacity of inhibit the formation of bile pigments (6). SnPP has the ability to strongly inhibit heme oxygenase activity by binding to the same site than does heme and CoPP, competing with these substrate molecules. At the same time, SnPP treatment increases the amount of heme oxygenase protein in rat liver cells, probably, as a compensatory response to the reduced activity of the enzyme (7).
Although HMOX1 is expressed in all mammalian tissues at basal levels, expression levels of this enzyme can significantly increase in cells or tissues where red blood cells or hemoglobin are degraded by macrophages, such as the spleen, liver, bone marrow, and kidney. In these tissues HMOX1 expression can be induced either by substrate (heme) or by other physical and chemical stimuli, including metalloporphyrins (8), oxidative stress, pathogen-associated molecular patterns (PAMPs), and cytokines. The enzyme localization was first described in the microsomal fraction of spleen and liver cells of Sprague-Dawley rats (3). The association with the endoplasmic reticulum (ER) membrane under physiological conditions was later confirmed for this enzyme (9). However, under stress conditions, HMOX1 is capable of translocating to other cellular membranous compartments. Under lipopolysaccharide (LPS) and heme treatments, HMOX1 is found associated with cytochrome c-containing fractions, suggesting a possible localization in mitochondria. Stimulation with LPS, heme and hypoxia resulted in localization in low-density fractions, with high expression of caveolin-1, the principal structural component of caveolae or lipid rafts. Furthermore, HMOX1 also colocalized with NADPH:cytochrome P-450 reductase and biliverdin reductase, suggesting a functional role of HMOX1 in lipid raft activity (10). HMOX 1 nuclear localization has also been described in astroglial cells during differentiation. Confocal microscopy showed that HMOX1 at day 7 is localized in cytosol and perinuclear region, moving to the nucleus and nucleolus after 2 and 3 weeks of differentiation (11). Furthermore, biliverdin reductase has been localized in nuclear fraction of rat kidney, in response to HMOX1 inducers, LPS and bromobenzene. These data suggest a dynamic localization for HMOX1 and that this molecule can display enzymatic activity at the various sites (12).
Because of the wide range of molecules that can trigger the induction of HMOX1 expression, the complete network of gene regulation leading to HMOX1 expression is not yet fully understood. Nevertheless, there are some signaling pathways and transcription factors that have been well associated with the HMOX1 response (Figure 1B). The main regulators of HMOX1 transcription described so far is the transcription factor Nrf2 [nuclear factor (erythroid-derived 2)-like 2] (13) and the inducible repressor Bach1 (BTB and CNC homology 1) (14). Indeed, the effect of CoPP over HMOX1 induction involves the participation of these two regulators (15). Nrf2 is a basic Leucine zipper protein that regulates the expression of antioxidant proteins, as the response to oxidative stress, including HMOX1 (16). Several are the stimuli and signaling pathways leading Nrf2 to induce HMOX1 expression. For instance, exposure to the flavonoid Orientin (Ori) can alleviate hydrogen peroxide-induced oxidative impairment in RAW264.7 cells, by induction of Nrf2/HMOX1 axis, through c-Jun N-terminal kinases (JNK) and phosphoinositide-3 kinase (PI3K)/protein kinase B (AKT) activation (17). Cytokines are another stimuli for HMOX1 expression (18) and it has been demonstrated that in human macrophages, IL-10 and IL-6-induced expression of HMOX1 required the activation of the signal transducer and activator of transcription 3 (STAT3) (19). On the contrary, Bach1 is a repressor of HMOX1 in physiological conditions, when Bach1 is absent, HMOX1 is constitutively expressed (14). Moreover, a deficiency of Bach1 protects against osteoarthritis and from oxidative stress-induced injury through the upregulation of HMOX1 (20, 21).
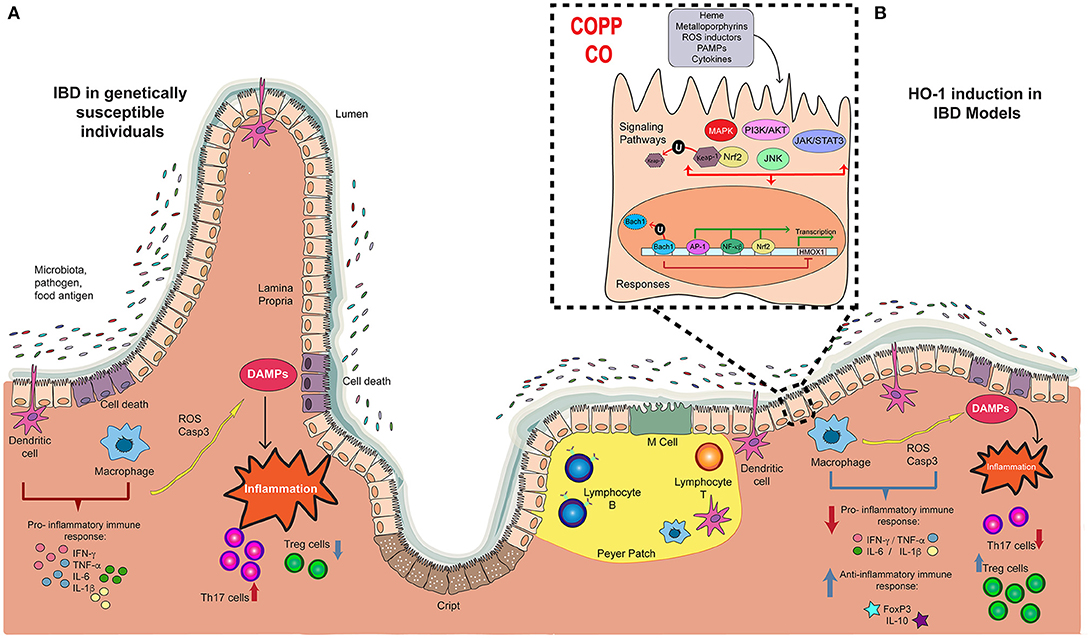
Figure 1. Players in the development and progression of IBD and the potential therapeutic effect of Hemoxigenase-1. (A) The pathology of IBD in genetically susceptible patients is characterized by the recognition of microbiota, pathogens and food antigens developing a pro-inflammatory immune response, with an augmented production of pro-inflammatory cytokines such as IFN-g, TNF- α, IL-6, and IL-1b. This response triggers tissue damage by the production of reactive oxygen species (ROS) and Caspase-3, which releases damage-associated molecular patterns (DAMPs) that induces the intestinal inflammation, with high Th17 profile T cell number, and reduced Treg cells. (B) In some IBD models, treatment with CoPP or CO reduces the pro-inflammatory immune response described above, and induces the production of anti-inflammatory molecules, such as FoxP3 and IL-10. Consequently, cell death is reduced, diminishing Th17 response. Also, CoPP or CO increase number of Treg cells. This anti-inflammatory effect result in reduced colitis, suggesting that HMOX1 activity is an important target in the development of new therapies for IBD.
The enzymatic activity of HMOX1 was initially associated with the immune response during inflammatory processes involved in organs rejection. Early on was observed that the mechanisms protecting xenografts from being rejected involved a rapid increase of HMOX1 expression by the endothelial and smooth muscle cells from mouse cardiac xenografts transplanted into rats (22). Two years later, experiments performed in a mouse-to-rat cardiac transplant model showed that the inhibition of HMOX1 activity by tin-protoporphyrin resulted in an earlier organ rejection, but when these same rats were additionally treated with exogenous CO, the long-term graft survival was restored (23). These studies demonstrated the important role that HMOX1 can play in the immune modulation involved in transplants, and that this immunomodulatory effect was driven by CO (23).
Tardif and collaborators demonstrated that Dendritic Cells (DCs) treated with lipopolysaccharide (LPS) and CO showed a diminished capacity to present antigens to T cells and this effect was due to a reduced fusion of endosomes and lysosomes, that is required for antigen presentation (24). Such reduced fusion of endosomes and lysosomes is probably caused by ATP reduction in the cell, as a result of impaired mitochondrial function caused by CO (25–27). The activation of adaptive immunity requires antigen presentation resulting in cytokine secretion, which finally derives in cell infiltration and inflammation. Therefore, impaired antigen presentation will result in reduced inflammation. Another of the HMOX1 effects is the induction of anti-inflammatory cytokines production, such as IL-10. However, the interplay between IL-10 and HMOX1 seems to be much more complex. For instance, in murine macrophages it has been shown that treatment with recombinant IL-10 induces, in a dose-dependent manner, HMOX1 production measured by Western Blot and that such an induction is mediated by the MAPK p38 activity (28), which is modulated by CO (29). In addition, HMOX1 induction has been reported to enhance macrophages polarization into an IL-10-producing anti-inflammatory (M2) phenotype (30).
The anti-inflammatory properties of HMOX1 have also conferred it with a protective capacity on different tissues that have suffered injury. For instance, hypoxia can induce the secretion of several pro-inflammatory cytokines in lungs, producing severe inflammation and structural changes of vessels. Consistently, transgenic mice that constitutively express HMOX1 in the lungs submitted to hypoxia showed that an increased HMOX1 expression resulted in less inflammation and reduced vessel hypertrophy, as compared with wild type mice (31). Furthermore, the pro-inflammatory cytokines and chemokines expressed in normal conditions were significantly suppressed in HMOX1 transgenic mice (31). Similarly, the anti-inflammatory effects of HMOX1 induction were observed in FcγRIIb−/− mice, which are a model of systemic lupus erythematosus (SLE). These FcγRIIb−/− mice spontaneously develop proteinuria and renal inflammation, which are delayed by the induction of HMOX1 or carbon monoxide treatment. Indeed, carbon monoxide administration decreased the expansion of CD11b+ cells, prevented the decline of CD4+ FoxP3+ regulatory T (Treg) cells and reduced the titers of anti-histone antibodies, as compared to untreated mice (32). It has been also reported that patients with SLE show reduced expression of HMOX1 in circulating monocytes, suggesting that in myeloid cells, the expression of HMOX1 could contribute to modulate the host inflammatory response (33). Moreover, several studies support the notion that HMOX1 overexpression in the brain and spinal cord of multiple sclerosis patients could play an important role in multiple sclerosis patients, which was also observed in the experimental autoimmune encephalomyelitis (EAE) model (34). It has been observed that multiple sclerosis patients show a decreased expression of HMOX1 in mononuclear cells from the peripheral blood during disease exacerbation, similar to SLE patients (33, 35). A recent study has suggested an association between HMOX1 and HMOX2 polymorphisms and the risk of developing multiple sclerosis in Spanish Caucasian men (36). Additionally, possible associations between variants in HMOX-1 genes and the risk of Parkinson's disease have been also reported (37, 38). Furthermore, it has been also described that HMOX1 is up-regulated during essential tremor and restless legs syndrome, suggesting a possible link between these ailments and the activity of HMOX1 (39).
Several studies have also revealed the relationship between HMOX1 and the pathophysiology of both acute and chronic intestinal inflammation (40). In the following chapter, we will discuss the studies performed up to date that support this notion.
Chronic intestinal inflammation can be triggered by several conditions, such as infections and autoimmune diseases. It is characterized by immune cell infiltration, edema and loss of the epithelium structure (41). The most studied chronic intestinal inflammations are denominated Inflammatory Bowel Diseases (IBD). These are chronic inflammatory pathologies of unknown etiology, which includes Crohn's Disease (CD) and Ulcerative Colitis (UC). IBD usually begins in early adulthood, and the symptoms include recurrent diarrhea, abdominal pain and the presence of blood in the stool (42). As part of Crohn's disease, inflammation can occur in any section of the gastrointestinal tract, whereas ulcerative colitis is typically confined to the colon (42). Although in recent years there has been a significant improvement in understanding the pathophysiology of these pathologies, their incidence has significantly increased worldwide and their etiology remains unclear (41). Unfortunately, there is still no cure for IBD, and the benefits of surgery are usually only temporary (43). Currently, the treatment of IBD is based on drugs to reduce symptoms and inflammation, such as 5-aminosalicylic acid, corticosteroids, immunomodulators, and biological therapy (44). However, these treatments are only palliative and most of them can have significant side effects. For this reason, there is a broad interest to understand in more detail the pathophysiology of these diseases in order to identify new therapeutic targets.
Even though the specific cause of IBD is not clear, it is known that infections, genetic predisposition, and environmental factors can contribute to disease progression (45–52). Importantly, several studies on this field suggest that IBD is caused by an inappropriate immune response to enteric commensal bacteria in genetically susceptible hosts (53–56). Such an unbalanced immune response leads to a persistent and relapsing inflammation of different portions of the gastrointestinal tract (41). Supporting this hypothesis are genome-wide association studies that have identified approximately 200 inflammatory bowel disease-associated loci (57). Most of the genes that have been linked to IBD are related to host defense against infection, particularly to the interaction between the host mucosal immune system and microbes at the epithelial cell surface and inside the gut lumen (58). The best-known example is the association of CD with nod2 variants (59). nod2 encodes the primary receptor for muramyl dipeptide (MDP) and is essential for bacterial recognition. Defective nod2 prevents the clearance of intracellular bacteria, due to a deficient antimicrobial response, which favors the activation of the immune response against the microbiota (60).
Dysregulation of immune response in the pathogenesis of IBD is also associated with an imbalance between pro- and anti-inflammatory molecules involved in both innate and adaptive immunity, leading to the development of excessive and chronic inflammation (61). In physiological conditions, the intestinal mucosa represents a mechanical barrier between immune system and microorganisms (62) and the abundant immunogenic stimuli induce infiltration of lymphocytes, monocytes, mast cells, and eosinophils (63). This physiological response is a tightly regulated phenomenon, which is important to keep the integrity of the gastrointestinal barrier (62). However, under certain pathological conditions this mechanism of control fails, leading to a persistent detrimental inflammation of the digestive tract. Along these lines, it has been reported that patients with IBD display an activated mucosal immune system, with high infiltration of plasma cells, neutrophils and macrophages, accompanied by overexpression of pro-inflammatory cytokines and various chemokines that promote the recruitment of inflammatory cells (64). Together, cytokines, chemokines, reactive oxygen species and other mediators result in exacerbated local inflammation and tissue damage (42) (Figure 1A).
Chronic inflammatory response in CD is characterized by an abnormal Th1 and Th17 immune responses, which are triggered by increased mucosal levels of cytokines, including tumor necrosis factor-alpha (TNF-α), interferon-gamma (IFN-γ), IL-12, IL-17, and IL-18 (Figure 1A). In contrast, the production of IL-4, IL-5, and IL-13 is increased in UC, which leads to a Th2-polarized immune response. Although in both disorders the immune response is different, genetically susceptible individuals have an inappropriate mucosal immune response against their gut microbiota, mediated by an excessive activation of effector T cell subsets and/or a deficiency of Treg cells (65) (Figure 1A). Indeed, Treg cells play a crucial role in the maintenance of intestinal homeostasis through TGF-β- and IL-10-dependent mechanisms. For instance, IL-10 is a potent anti-inflammatory cytokine produced by both Treg and non-Treg cells, capable of suppressing the production of pro-inflammatory mediators to limit the inflammatory response. In fact, IL-10-deficient mice are highly susceptible to colitis, due to aberrant immune response to commensal bacteria. This abnormal response is more severe when combined with a deficiency of TGF-β signaling (66). Previous studies have indicated that IL-10 production is suppressed in the intestine during IBD. However, the molecular and cellular mechanisms behind the reduction of IL-10 secretion observed during IBD have not yet been fully understood. In addition, it has been suggested that IL-10 could induce other molecules with anti-inflammatory activity by a positive feedback circuit, which leads to the amplification of this response and the inhibition of intestinal inflammation. Abnormal low levels of IL-10 during these diseases could account for the inflammatory imbalance in the intestinal tissue (67).
As described above, several studies have associated IL-10 production and HMOX1 activity with the development of IBD (67). Therefore, it has been important to determine the contribution of the HMOX1 enzyme to the advancement of intestinal inflammation. Under healthy conditions, HMOX1 expresses in the intestine at low levels, but it is significantly induced upon triggering of an inflammatory state (68). HMOX1 mRNA expression and HMOX1 protein production are increased in inflamed colonic mucosa of UC patients. In addition, HMOX1 expression is also decreased in mononuclear cells from colonic submucosa from UC patients, as compared to normal mucosa from healthy subjects (69). Similar data were obtained during the occurrence of three different inflammatory conditions: Helicobacter pylori-positive gastritis patients, active UC and active CD. Immunohistochemical staining showed a higher expression of HMOX1 in gastric mucosa of Helicobacter pylori-positive gastritis patients and in colonic mucosa of active UC and active CD patients, as compared to healthy controls (70). In the next section, studies performed to evaluate the effect of HMOX1 modulation in IBD will be discussed.
The contribution of HMOX1 to the prevention or reduction of intestinal inflammation has been shown in different models of IBD, which are summarized in Table 1. It has been reported that in the model of colitis induction by the administration of Dextran Sulfate Sodium (DSS) an increase in the expression of HMOX1 led to an amelioration of the intestinal inflammation. Induction of HMOX1 by intraperitoneal injection of CoPP at day 1 and 3 after DSS treatment significantly reduced the intestinal histological damage as compared to control animals (72). Another study also described the cytoprotective role of HMOX1 in the model of colitis induced by DSS in mice. In this study, a treatment with hydrogen-rich water was performed in mice treated with DSS, resulting in decreased inflammation and disease score, increased level of anti-oxidative markers and reduced production of inflammatory molecules. Importantly, it was shown that this treatment up-regulated HMOX1 expression (78). In support of this observation, another study showed that mice treated with the HMOX1 inducer hemin previous to the induction of DSS colitis displayed reduced histological damage, which was associated to an increased expression of HMOX1. Furthermore, a decreased proportion of Th17 cells and increased number of Treg cells were found in mesenteric lymph nodes (MLN) and spleen of hemin-pretreated mice that were exposed to DSS (77).
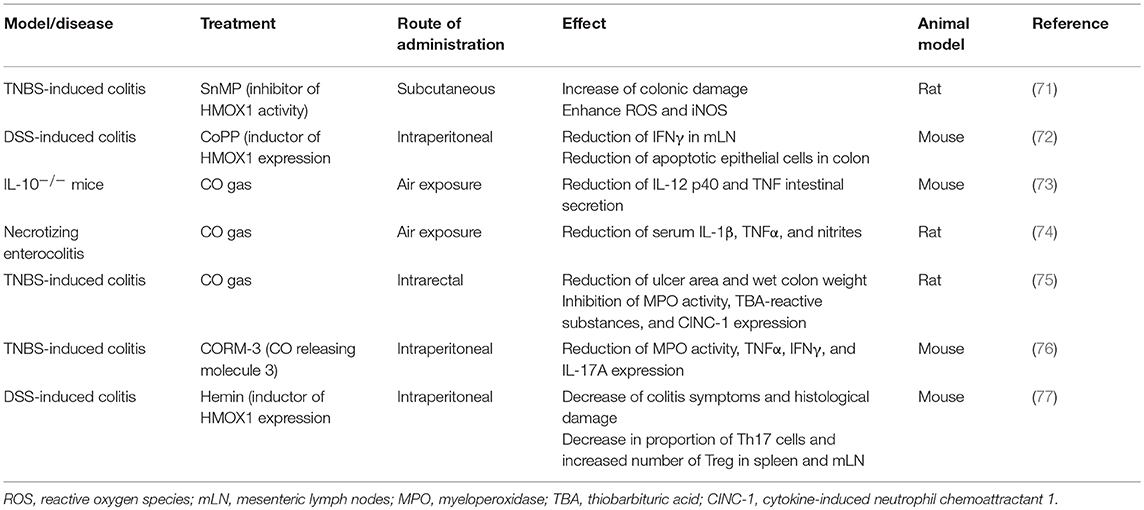
Table 1. Protective effect of HMOX1/CO in inflammatory intestinal models.
Another mouse model of colitis induction, in which the effect of HMOX-1 was evaluated, is the treatment with 2,4,6-trinitrobenzene sulfonic acid (TNBS). Both HMOX1 mRNA expression and enzymatic activity were induced after TNBS treatment. Further, when the HMOX1 inhibitor tin mesoporphyrin (SnMP) was used in these assays, HMOX1 activity was reduced and colonic damage increased (71).
An analysis of the role of HMOX1 and IBD has been also performed using patient samples, including both in vitro and in vivo experiments. In these studies, IBD patients showed a heterogeneous distribution of HMOX1 expression in the colonic mucosa, as compared to patients with intestinal damage produced by conditions different from IBD, such as diverticulitis. During these ailments the expression of HMOX1 was shown to be highly increased in epithelial cells of colonic crypts and in macrophages (72). Consistently, some drugs used in patients with IBD have shown to have a beneficial effect through the induction of HMOX1. For example, it was demonstrated that 5-aminosalicylic acid (5-ASA), an anti-inflammatory drug often used to treat IBD, reduces the TNBS-provoked colonic inflammatory injury in rats, by increasing colonic HMOX1 activity (79). Another example is Tranilast, a mastocyte stabilizer used in some countries as IBD treatment. The simultaneous intrarectal administration of DSS and Tranilast to C57BL/6 mice resulted in an attenuated colitis, produced by low pro-inflammatory cytokines, increased anti-inflammatory cytokines and promoted HMOX1 expression (80). In another field, in vitro studies have demonstrated that treatment with plant extracts of Atractylodes macrocephala (AM) or Taraxacum herba (TH) increased HMOX1 production. Oral pre-administration of AM and TH can rescue mice from DSS-induced colitis by inhibiting inflammatory mediators via inactivated extracellular signal regulated kinase (ERKs) and repressed NFκB and signal transducer and activator of transcription 3 (81).
Given that HMOX1 might be involved in the immune processes leading to intestinal inflammation, CO has been considered as a therapeutic tool to reduce the symptoms of this disease (82). The first evidence supporting that CO could work as a therapy for IBD is the observation that smoking can work as a protective factor in UC patients (83, 84). It was shown that exposure to CO ameliorates chronic intestinal inflammation in IL-10−/− mice by interfering with the IFN-γ signaling that mediates inflammation in this model (73). Similar results were found in another murine model of IBD. In this case, mice were treated with TNBS to induce intestinal inflammation, and CO gas was administrated intrarectally at 200 ppm per day. TNBS- and CO-treated mice showed significantly smaller ulcers in colon, less colon weight and reduced myeloperoxidase (MPO) activity. Further, CO-treated mice showed reduced thiobarbituric acid (TBA)-reactive substances and cytokine-induced neutrophil chemoattractant 1 (CINC-1) expression in colonic mucosa, as compared with TNBS-air treated mice (75). In a necrotizing enterocolitis rat model, inhaled CO gas also protected against intestinal inflammation and decreased levels of TNF-α (74). However, exposition to CO is not the only way to administrate low doses of CO. Carbon monoxide-releasing molecules (CORM) have been widely used to corroborate the effectiveness of CO. A study showed that intraperitoneal CORM-3 injection in mice for 3 days prior to TNBS treatment attenuated distal colon damage and histological score. In an in vitro approximation, CORM-3 treatment reduced IFN-γ and TNF-α in CD4+ T cells stimulated with anti-CD3/CD28 antibodies (76).
The evidences listed in Tables 1, 2 support the important role of HMOX1 during the pathology of IBD in animal models and in humans. However, to elucidate the molecular mechanism of HMOX1 activity would help to understand those effects and to the development of new potential therapies for IBD (Figure 1B).
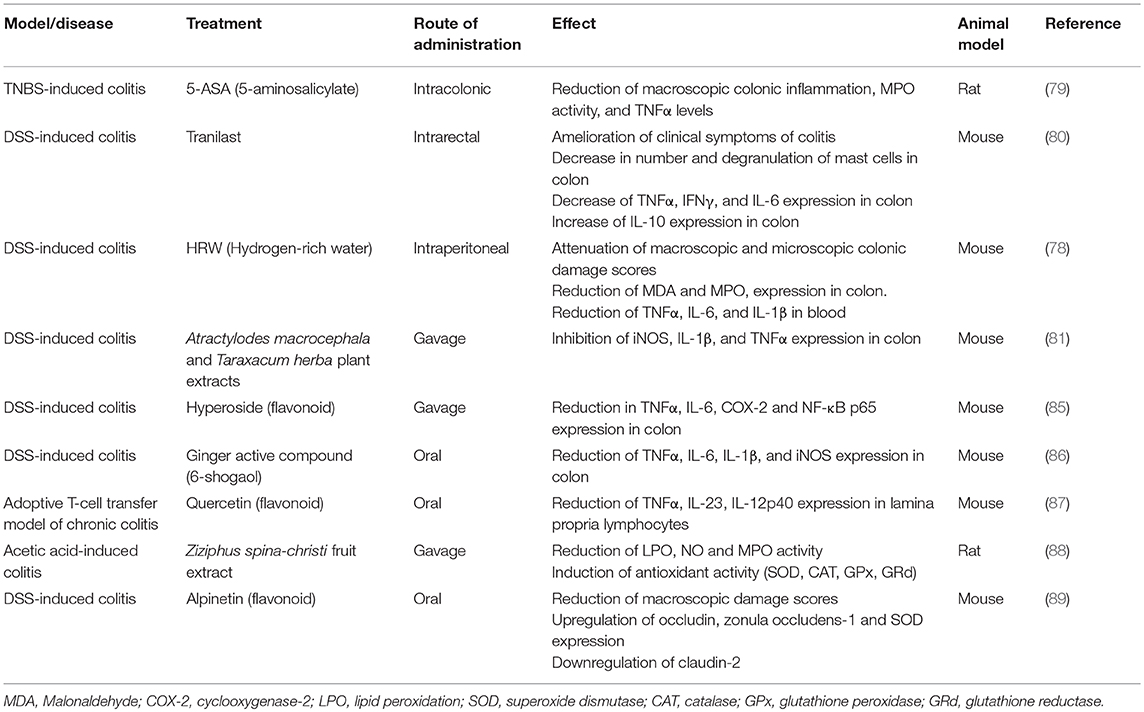
Table 2. Protective effect of different compounds in inflammatory intestinal models that involves HMOX1 pathways.
An important cause of intestinal inflammation is the infection by pathogenic bacteria, such as Salmonella enterica. Although little is known about the exact effect of HMOX1 on bacterial infections, it has been described that HMOX1 plays a role in the clearance of Salmonella by the immune system. For instance, in an acute inflammation model, where C57BL/6 mice were treated with streptomycin to eliminate the microbiota, prior to Salmonella enterica serovar Typhimurium (S. Typhimurium) oral infection, treatment with CoPP reduced the presence of Salmonella DNA in the mesenteric lymph nodes, lamina propria, liver and spleen (90). Furthermore, in vitro depletion of HMOX1 in murine macrophages reduced these cells bactericidal activity against E. coli, Enterococcus faecalis and S. Typhimurium. These data suggest that the observed CO effect is due to the capacity to promote bacterial clearance in phagocytic cells associated to the intestine (90). A recent in vitro study supports these findings, in which the bacterial clearance capacity of murine and human macrophages was tested. Specifically, it was observed that in HMOX1-deficient mice, macrophages have bacterial killing defects that were restored with the administration of CO. Further, CO treatment results in an enhanced bacterial clearance capacity in mouse and human macrophages (91). In contrast, CO has no effect on bactericidal activity of macrophages isolated from NALP3-deficient and caspase3-deficient mice. Consequently, the capacity of macrophages of killing bacteria depends on CO-mediated inflammasome activation (91). However, apparently contradictory results were observed in murine macrophages transfected with hmox1 shRNA. In these macrophages, intracellular S. Typhimurium survival was reduced upon hmox1 knockdown, an effect attributed to iron availability inside the cell (92).
Other bacteria that can be affected by the action of HMOX1 in the intestine are members of the intestinal microbiota. Recent research has revealed that alterations in the intestinal microbiota play a key role in the development of colitis and other inflammatory conditions of the gut (55). For example, there are studies that support an interaction between the host and molecules produced by commensal bacteria. Some of these are Short Chain Fatty Acid (SCFA), such as acetate, butyrate and propionate, which can promote an anti-inflammatory response in the intestine (93).
In addition, when C57BL/6 mice are exposed to specific pathogen free (SPF) enteric microbiota after been raised under germ free conditions, colonic HMOX1 expression is increased almost 6 times, and this induction depends on IL-10 and Nrf2 (90). These data support the notion that HMOX1 not only works by inducing an anti-inflammatory cytokine profile, but also by interacting with intestinal microbiota.
As well as mammals, other organisms have enzymes with functions equivalent to HMOX1. It has been described that some bacteria express heme-degrading proteins that act as a complex group of molecules that work together to catalyze heme degradation (94, 95). Some enteric pathogens and commensal bacteria express this HMOX1 homolog, which may affect intestinal immune response. Corynebacterium diphteriae has a heme-oxygenase (HmuO) that has 33% sequence identity and 70% of homology with human HMOX1 (96). Most of the known bacterial HO are described for pathogens, such as Yersinia pestis (Hmu), Neisseriae spp. (HemO), Pseudomonas aeruginosa (PigA), Mycobacterium tuberculosis (MhuD), and Staphylococcus aureus (IsdG, IsdI). All of these bacteria harbor genes for HO that encode functional proteins (94, 97–101).
Some strains of E. coli have a heme uptake gene cluster that is required for harnessing available iron in the environment, presented as heme group. Bacterial analog of HMOX1 called ChuS is encoded by chuS. Metabolic products are the same as human HMOX1. For instance, HO enzyme of E. coli O157:H7 produces CO efficiently, despite having a different structure from HMOX1. Additionally, enteric bacteria as Enterobacter and Shigella may have similar proteins (95, 102). Other proteins that are involved in heme uptake machinery are heme receptor called ChuA (103) present in the outer membrane, periplasmic protein ChuT which binds to heme, inner membrane protein ChuU and the ATPase ChuV (104) (Figure 2B).
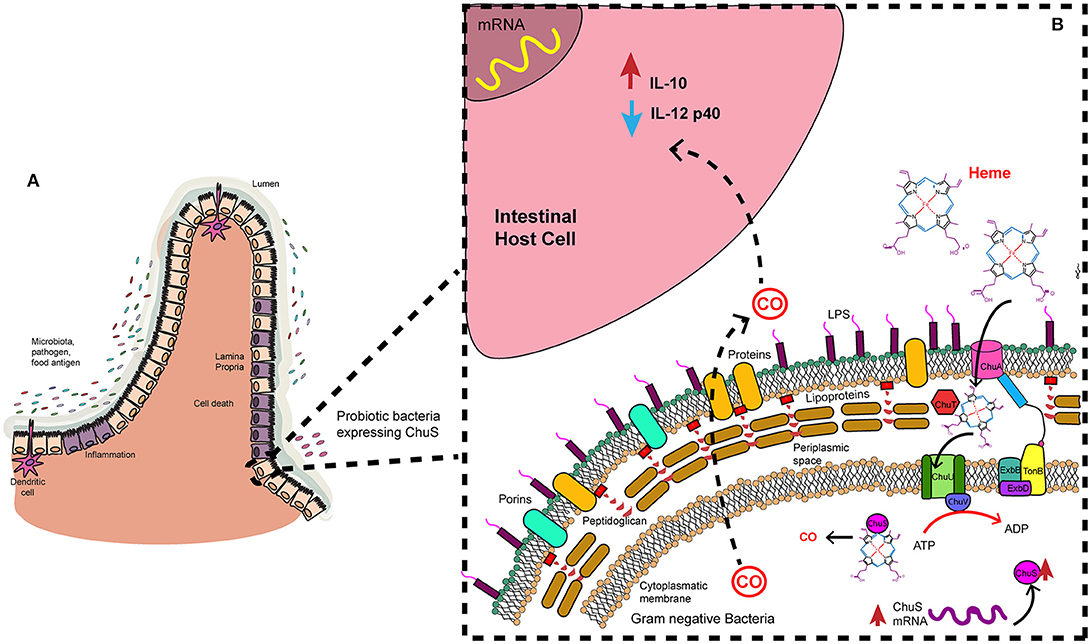
Figure 2. Heme uptake and degradation by gram negative bacteria. Microbiota is in constant interaction with the epithelial cells. (A) In an inflammation context in the intestine some bacteria may modulate immune response. (B) That is the case of a probiotic strain with ChuS. These bacteria have an outer membrane receptor (ChuA) catches specifically heme group. Periplasmic protein (ChuT) binds substrate and carry it to inner membrane protein (ChuU), where ATPase direct the transport of the substrate to the cytoplasm. There, HMOX1 analog (ChuS) binds heme group and catalyzes its degradation. CO is a product of this reaction and may be released to the environment where it exerts its anti-inflammatory effect on host cells.
A recent study described the anti-inflammatory action of an E. coli strain isolated from mice gut, named NC101, which has chuS gene. In mono-association studies, germ-free SvEV/129 IL-10 deficient mice were treated with this bacterial strain. A correlation was found between chuS expression on feces and the progression of inflammation. In addition, composition of intestinal bacteria was altered when C57BL/6 wild type mice were exposed to carbon monoxide for 14 days. Also, chuS expression was increased in that point. Interestingly these changes returned to baseline after 14 days without CO exposure. Additionally, when BMDMs were cultured with an E. coli NC101 strain that overexpresses chuS pro-inflammatory cytokine IL-12p40 production was decreased. On the other hand, IL-10 production increased, promoting an anti-inflammatory scenario. These changes in cytokines expression were promoted by a soluble factor released by bacteria. Finally, CO abundance found in liver of mice infected with the strain that overexpresses chuS was higher than in those infected with a strain that lacks chuS. Moreover, in the first group of mice mentioned before expression of HMOX1 and Nqo1 were significantly increased same as IL-10 production (105).
These in vitro and in vivo findings contribute to the hypothesis that ChuS may be used to modulate immune response in an inflammation context (Figure 2A). A probiotic strain with ChuS enzyme may be able to decrease inflammation in the gut by secreting CO as a product of heme uptake process. CO would act in host cells (gut epithelium) modulating IL-12p40 and IL-10 expression and production. Consequently, inflammation will be ameliorated, leading to an anti-inflammatory scenario (Figure 2B).
Another aspect of intestinal inflammation where HMOX1 is involved is cancer. HMOX1 has been associated with different types of cancer (106), including prostate cancer (107), bladder cancer (108), skin cancer (109), and colorectal cancer (CRC). The latter is an important issue due to all the evidences that support the hypothesis that HMOX1 is a key immune modulator in intestine. It has been demonstrated that HMOX1 is highly expressed in CRC (110) and high expression of HMOX1 in small-intestine adenocarcinomas is associated with lower pT (primary tumor) category, less pancreatic invasion and longer overall survival than those with low HMOX1 expression (111). Another study also showed that HMOX1 was over-expressed in human invasive CRC, associated with longer overall survival time. Using 1,2-dimethylhydrazine (DMH)-induced CRC animal model, the authors showed that HMOX1 expression is enhanced as tumor progress, and that this effect is due to the induction of cell cycle arrest and apoptosis (112). In vitro studies also showed augmented expression of HMOX1 in primary colon cancer tissues and HCT116 colon cancer cells compared with normal surrounding tissue and with normal epithelial cell line, respectively. Moreover, treatment with CORM-3 inhibited cell proliferation of HCT116 cells (113). An important feature of intestinal immune response are macrophages. Particularly, CX3CR1+ macrophages contribute to intestinal homeostasis, controlling T cell response to commensal bacteria (114, 115). CX3CR1 deficient mice could not resolve intestinal inflammation in azoxymethane (AOM)/DSS-induced colitis-associated cancer and have higher colitis score and polyps, compared with wildtype mice. These authors showed that CX3CR1 deficient mice has lower HMOX1 expression in adenomatous colon tissues, and treatment with CoPP restored the recovery reducing intestinal inflammation and preventing carcinogenesis (116).
Based on all these evidence, HMOX1 has been proposed as a target for cancer treatment (106), including its inducible repressor BACH1 (117), or its positive regulator Nrf2 (118).
Since HMOX1 was originally described, strong evidence has supported the notion that this enzyme is a major modulator of immune response in several pathologies. Given that the number of IBD cases is increasing worldwide, it is very important to bring to light the etiology of these chronic diseases and new therapeutic alternatives. The evidences discussed in this review support the notion that HMOX1 activation reduces inflammation in the gut in several animal models of IBD, through the induction of anti-inflammatory cytokines pathways. Importantly, it has been demonstrated an association between IBD development and genes involved in the proper immune response to infecting microbes. Although the exact relation between HMOX1 and microorganisms colonizing—or infecting—the gastrointestinal tract is not totally elucidated yet, it is interesting that enzymes with similar functions than HMOX1 have been described in bacteria, particularly ChuS. Importantly, this bacterial enzyme has been associated with the induction of an anti-inflammatory environment in a mouse colitis model (105). Therefore, intestinal bacterial might be contributing to prevent intestinal inflammation through the byproducts of HMOX1 enzymatic activity. Furthermore, intestinal infections are an important factor in the development of IBD (51) and most of the evidence available to date supports the notion that HMOX1 promotes bacterial clearance, diminishing intestinal inflammation (90). Moreover, it has been demonstrated that CO can interact directly with heme groups of the bacterial electron transport chain, suggesting that HMOX1 has an effect in bacterial infections not only through the modulation of the immune response, but by the direct interaction with the pathogen (119). In another field, HMOX1 has also been associated with colorectal cancer, being suggested as a novel target for cancer therapy (106).
Because of the existing evidence of HMOX1 role in intestinal inflammation, many therapy targets can be proposed. Given that most of the anti-inflammatory and cytoprotective effects of HMOX1 have been attributed to CO, this could be an effective treatment. However, it is well known that CO in mammals is toxic from determined concentrations. For these reasons, it is very important to elucidate the exact mechanisms by which HMOX1 and CO are exerting all the effects described here, in order to design novel therapeutic strategies to deliver CO in a safe manner.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by the Fondo Nacional de Desarrollo Científico y Tecnológico FONDECYT #1131012, #1161525 and #1170964, and #1161525; Millennium Institute on Immunology and Immunotherapy P09/016-F, Iniciativa Científica Milenio from the Ministry of Economy and Tourism; the Puente Proyect P1715/2017, Dirección de Investigación, Vicerrectoría de Investigación from the Pontificia Universidad Católica de Chile; the National Council of Scientific and Technological Research (CONICYT) (National PhD grants #21140273, #21140169, #21171014, and #63140215); the PhD grant provided to VS by the Vicerrectoría de Investigación from the Pontificia Universidad Católica de Chile.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ryter SW, Alam J, Choi AMK. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. (2006) 86:583–650. doi: 10.1152/physrev.00011.2005
2. Schuller DJ, Wilks A, Ortiz de Montellano PR, Poulos TL. Crystal structure of human heme oxygenase-1. Nat Struct Biol. (1999) 6:860–67. doi: 10.1038/12319
3. Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxgenase. Proc Natl Acad Sci USA. (1968) 61:748–55.
4. Tenhunen R, Marver HS, Schmid R. Microsomal heme oxygenase: characterization of the enzyme. J Biol Chem. (1969) 244:6388–94.
5. Maines MD, Kappas A. Enzymatic oxidation of cobalt protoporphyrin IX: observations on the mechanism of heme oxygenase action. Biochemistry (1977) 16:419–23. doi: 10.1021/bi00622a012
6. Drummond GS, Kappas A. Prevention of neonatal hyperbilirubinemia by tin protoporphyrin IX, a potent competitive inhibitor of heme oxidation. Proc Natl Acad Sci USA. (1981) 78:6466–70. doi: 10.1073/pnas.78.10.6466
7. Sardana MK, Kappas A. Dual control mechanism for heme oxygenase: tin(IV)-protoporphyrin potently inhibits enzyme activity while markedly increasing content of enzyme protein in liver. Proc Natl Acad Sci USA. (1987) 84:2464–8. doi: 10.1073/pnas.84.8.2464
8. Shan Y, Pepe J, Lu TH, Elbirt KK, Lambrecht RW, Bonkovsky HL. Induction of the heme oxygenase-1 gene by metalloporphyrins. Arch Biochem Biophys. (2000) 380:219–27. doi: 10.1006/abbi.2000.1921
9. Ishikawa K, Sato M, Yoshida T. Expression of rat heme oxygenase in Escherichia coli as a catalytically active, full-length form that binds to bacterial membranes. Eur J Biochem. (1991) 202:161–5. doi: 10.1111/j.1432-1033.1991.tb16357.x
10. Kim HP, Wang X, Galbiati F, Ryter SW, Choi AMK. Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. FASEB J. (2004) 18:1080–9. doi: 10.1096/fj.03-1391com
11. Li Volti G, Ientile R, Abraham NG, Vanella A, Cannavò G, Mazza F, et al. Immunocytochemical localization and expression of heme oxygenase-1 in primary astroglial cell cultures during differentiation: effect of glutamate. Biochem Biophys Res Commun. (2004) 315:517–24. doi: 10.1016/j.bbrc.2004.01.090
12. Maines MD, Ewing JF, Huang TJ, Panahian N. Nuclear localization of biliverdin reductase in the rat kidney: response to nephrotoxins that induce heme oxygenase-1. J Pharmacol Exp Ther. (2001) 296:1091–7. Available online at: http://jpet.aspetjournals.org/content/296/3/1091?ijkey=278873cf695dba36df7ce40befa6d7a6b5a6d476&keytype2=tf_ipsecsha
13. Alam J, Stewart D, Touchard C, Boinapally S, Choi AMK, Cook JL. Nrf2, a cap'n'collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. (1999) 274:26071–8.
14. Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, et al. Hemoprotein Bach1 regulates enhancer availbility of home oxygenase-1 gene. EMBO J. (2002) 21:5216–24. Available online at: http://emboj.embopress.org/content/embojnl/21/19/5216.full.pdf
15. Shan Y, Lambrecht RW, Donohue SE, Bonkovsky HL. Role of Bach1 and Nrf2 in up-regulation of the heme oxygenase-1 gene by cobalt protoporphyrin. FASEB J. (2006) 20:2651–3. doi: 10.1096/fj.06-6346fje
16. Chen HH, Chen YT, Huang YW, Tsai HJ, Kuo CC. 4-Ketopinoresinol, a novel naturally occurring ARE activator, induces the Nrf2/HO-1 axis and protects against oxidative stress-induced cell injury via activation of PI3K/AKT signaling. Free Radic Biol Med. (2012) 52:1054–66. doi: 10.1016/j.freeradbiomed.2011.12.012
17. Xiao Q, Piao R, Wang H, Li C, Song L. Orientin-mediated Nrf2/HO-1 signal alleviates H2O2-induced oxidative damage via induction of JNK and PI3K/AKT activation. Int J Biol Macromol. (2018) 118:747–55. doi: 10.1016/j.ijbiomac.2018.06.130
18. Gómez-Hurtado I, Zapater P, Bellot P, Pascual S, Pérez-Mateo M, Such J, Francés R. Interleukin-10-mediated heme oxygenase 1-induced underlying mechanism in inflammatory down-regulation by norfloxacin in cirrhosis. Hepatology (2011) 53:935–44. doi: 10.1002/hep.24102
19. Ricchetti GA, Williams LM, Foxwell BMJ. Heme oxygenase 1 expression induced by IL-10 requires STAT-3 and phosphoinositol-3 kinase and is inhibited by lipopolysaccharide. J Leukoc Biol. (2004) 76:719–26. doi: 10.1189/jlb.0104046.Journal
20. Takada T, Miyaki S, Ishitobi H, Hirai Y, Nakasa T, Igarashi K, Lotz MK, Ochi M. Bach1 deficiency reduces severity of osteoarthritis through upregulation of heme oxygenase-1. Arthritis Res Ther. (2015) 17:1–11. doi: 10.1186/s13075-015-0792-1
21. Kondo K, Ishigaki Y, Gao J, Yamada T, Imai J, Sawada S, Muto A, Oka Y, Igarashi K, Katagiri H. Bach1 deficiency protects pancreatic β-cells from oxidative stress injury. AJP Endocrinol Metab. (2013) 305:E641–8. doi: 10.1152/ajpendo.00120.2013
22. Soares MP, Lin Y, Anrather J, Csizmadia E, Takigami K, Sato K, Grey ST, Colvin RB, Choi AM, Poss KD, et al. Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat Med. (1998) 4:1073–7. doi: 10.1038/2063
23. Sato K, Balla J, Otterbein L, Smith RN, Brouard S, Lin Y, et al. Carbon monoxide generated by heme oxygenase-1 suppresses the rejection of mouse-to-rat cardiac transplants. J Immunol. (2001) 166:4185–94. doi: 10.4049/jimmunol.166.6.4185
24. Tardif V, Riquelme SA, Remy S, Carreño LJ, Cortés CM, Simon T, et al. Carbon monoxide decreases endosome-lysosome fusion and inhibits soluble antigen presentation by dendritic cells to T cells. Eur J Immunol. (2013) 43:2832–44. doi: 10.1002/eji.201343600
25. Riquelme SA, Pogu J, Anegon I, Bueno SM, Kalergis AM. Carbon monoxide impairs mitochondria-dependent endosomal maturation and antigen presentation in dendritic cells. Eur J Immunol. (2015) 45:3269–88. doi: 10.1002/eji.201545671
26. Riquelme SA, Carreño LJ, Espinoza JA, Mackern-Oberti JP, Alvarez-Lobos MM, Riedel CA, et al. Modulation of antigen processing by haem-oxygenase 1. Implications on inflammation and tolerance. Immunology (2016) 149:1–12. doi: 10.1111/imm.12605
27. Blancou P, Tardif V, Simon T, Rémy S, Carreño LJ, Kalergis AM, et al. Immunoregulatory properties of heme oxygenase-1. Methods Mol Biol. (2011) 677:431–47. doi: 10.1007/978-1-60761-869-0
28. Lee T-S, Chau L-Y. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. (2002) 8:240–6. doi: 10.1038/nm0302-240
29. Sheikh SZ, Hegazi RA, Kobayashi T, Onyiah JC, Russo SM, Matsuoka K, et al. An anti-inflammatory role for carbon monoxide and heme oxygenase-1 in chronic Th2-mediated murine colitis. J Immunol. (2011) 186:5506–13. doi: 10.4049/jimmunol.1002433
30. Sierra-Filardi E, Vega MA, Sánchez-Mateos P, Corbí AL, Puig-Kröger A. Heme oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology (2010) 215:788–95. doi: 10.1016/j.imbio.2010.05.020
31. Minamino T, Christou H, Hsieh CM, Liu Y, Dhawan V, Abraham NG, et al. Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc Natl Acad Sci USA. (2001) 98:8798–803. doi: 10.1073/pnas.161272598
32. Mackern-Oberti JP, Llanos C, Carreño LJ, Riquelme SA, Jacobelli SH, Anegon I, et al. Carbon monoxide exposure improves immune function in lupus-prone mice. Immunology (2013) 140:123–32. doi: 10.1111/imm.12124
33. Herrada AA, Llanos C, Mackern-Oberti JP, Carreño LJ, Henriquez C, Gómez RS, et al. Haem oxygenase 1 expression is altered in monocytes from patients with systemic lupus erythematosus. Immunology (2012) 136:414–24. doi: 10.1111/j.1365-2567.2012.03598.x
34. Stahnke T, Stadelmann C, Netzler A, Brück W, Richter-Landsberg C. Differential upregulation of heme oxygenase-1 (HSP32) in glial cells after oxidative stress and in demyelinating disorders. J Mol Neurosci. (2007) 32:25–37. doi: 10.1007/s12031-007-0005-8
35. Fagone P, Patti F, Mangano K, Mammana S, Coco M, Touil-Boukoffa C, Chikovani T, Di Marco R, Nicoletti F. Heme oxygenase-1 expression in peripheral blood mononuclear cells correlates with disease activity in multiple sclerosis. J Neuroimmunol. (2013) 261:82–6. doi: 10.1016/j.jneuroim.2013.04.013
36. Agúndez JAG, García-Martín E, Martínez C, Benito-León J, Millán-Pascual J, Díaz-Sánchez M, et al. Heme oxygenase-1 and 2 common genetic variants and risk for multiple sclerosis. Sci Rep. (2016) 6:20830. doi: 10.1038/srep20830
37. Ayuso P, Martinez C, Pastor P, Lorenzo-Betancor O, Luengo A, Jimenez-Jimenez FJ, et al. An association study between Heme oxygenase-1 genetic variants and Parkinson's disease. Front Cell Neurosci. (2014) 8:298. doi: 10.3389/fncel.2014.00298
38. Tian S, Yang X, Zhao Q, Zheng J, Huang H, Chen Y, An R, Xu Y. Association between a heme oxygenase-2 genetic variant and risk of Parkinson's disease in Han Chinese. Neurosci Lett. (2017) 642:119–22. doi: 10.1016/j.neulet.2017.02.008
39. García-Martín E, Jiménez-Jiménez FJ, Alonso-Navarro H, Martínez C, Zurdo M, Turpín-Fenoll L, et al. Heme oxygenase-1 and 2 common genetic variants and risk for restless legs syndrome. Medicine (2015) 94:e1448. doi: 10.1097/MD.0000000000001448
40. Naito YNA, Takagi T, Yoshikawa T. Heme oxygenase-1 : a new therapeutic target for inflammatory bowel disease. Aliment Pharmacol Ther. (2004) 20:177–184. doi: 10.1111/j.1365-2036.2004.01992.x
41. De Souza HSP, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. (2016) 13:13–27. doi: 10.1038/nrgastro.2015.186
42. Baumgart DC, Sandborn WJ. Crohn's disease. Lancet (2012) 380:1590–605. doi: 10.1016/S0140-6736(12)60026-9
43. Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology (2011) 140:1785–94. doi: 10.1053/j.gastro.2011.01.055
44. Fakhoury M, Negrulj R, Mooranian A, Al-Salami H. Inflammatory bowel disease: clinical aspects and treatments. J Inflamm Res. (2014) 7:113–20. doi: 10.2147/JIR.S65979
45. Schultz BM, Paduro CA, Salazar GA, Salazar-Echegarai FJ, Sebastián VP, Riedel CA, et al. A potential role of Salmonella infection in the onset of inflammatory bowel diseases. Front Immunol. (2017) 8:191. doi: 10.3389/fimmu.2017.00191
46. Ishihara S, Aziz MM, Yuki T, Kazumori H, Kinoshita Y. Inflammatory bowel disease: review from the aspect of genetics. J Gastroenterol. (2009) 44:1097–108. doi: 10.1007/s00535-009-0141-8
47. Hou JK, Lee D, Lewis J. Diet and inflammatory bowel disease: review of patient-targeted recommendations. Clin Gastroenterol Hepatol. (2014) 12:1592–600. doi: 10.1016/j.cgh.2013.09.063
48. Ananthakrishnan AN, Khalili H, Konijeti GG, Higuchi LM, de Silva P, Fuchs CS, et al. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn's disease. Gut (2014) 63:776–84. doi: 10.1136/gutjnl-2013-305304
49. Berkowitz L, Schultz BM, Salazar GA, Pardo-Roa C, Sebastián VP, Álvarez-Lobos MM, et al. Impact of cigarette smoking on the gastrointestinal tract inflammation: opposing effects in Crohn's disease and ulcerative colitis. Front Immunol. (2018) 9:74. doi: 10.3389/fimmu.2018.00074
50. Parkes GC, Whelan K, Lindsay JO. Smoking in inflammatory bowel disease: impact on disease course and insights into the aetiology of its effect. J Crohns Colitis (2014) 8:717–25. doi: 10.1016/j.crohns.2014.02.002
51. Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology (2008) 134:577–94. doi: 10.1053/j.gastro.2007.11.059
52. Hokari R, Matsunaga H, Miura S. Effect of dietary fat on intestinal inflammatory diseases. J Gastroenterol Hepatol. (2013) 28:33–36. doi: 10.1111/jgh.12252
53. Emge JR, Huynh K, Miller EN, Kaur M, Reardon C, Barrett KE, et al. Modulation of the microbiota-gut-brain axis by probiotics in a Murine model of inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. (2016) 310:G989–98. doi: 10.1152/ajpgi.00086.2016
54. Bassett S, Young W, Barnett M, Cookson A, McNabb W, Roy N. Changes in composition of caecal microbiota associated with increased colon inflammation in interleukin-10 gene-deficient mice inoculated with Enterococcus species. Nutrients (2015) 7:1798–816. doi: 10.3390/nu7031798
55. Maharshak N, Packey CD, Ellermann M, Manick S, Siddle JP, Young Huh E, et al. Altered enteric microbiota ecology in interleukin 10-deficient mice during development and progression of intestinal inflammation. Gut Microbes (2013) 4:316–324. doi: 10.4161/gmic.25486
56. Nagalingam NA, Lynch SV. Role of the microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. (2012) 18:968–80. doi: 10.1002/ibd.21866
57. Huang H, Fang M, Jostins L, Umićević Mirkov M, Boucher G, Anderson CA, et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature (2017) 547:173–78. doi: 10.1038/nature22969
58. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491:119–24. doi: 10.1038/nature11582
59. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature (2001) 411:599–603. doi: 10.1038/35079107
60. Wehkamp J, Harder J, Weichenthal M, Schwab M, Schäffeler E, Schlee M, et al. NOD2 (CARD15) mutations in Crohn's disease are associated with diminished mucosal alpha-defensin expression. Gut (2004) 53:1658–64. doi: 10.1136/gut.2003.032805
61. Xu X-R. Dysregulation of mucosal immune response in pathogenesis of inflammatory bowel disease. World J Gastroenterol. (2014) 20:3255. doi: 10.3748/wjg.v20.i12.3255
62. Harrison OJ, Maloy KJ. Innate immune activation in intestinal homeostasis. J Innate Immun. (2011) 3:585–93. doi: 10.1159/000330913
63. Carpenter HA, Talley NJ. The importance of clinicopathological correlation in the diagnosis of inflammatory conditions of the colon: histological patterns with clinical implications. Am J Gastroenterol. (2000) 95:878–96. doi: 10.1111/j.1572-0241.2000.01924.x
64. Ajuebor MN, Swain MG. Role of chemokines and chemokine receptors in the gastrointestinal tract. Immunology (2002) 105:137–43. doi: 10.1046/j.1365-2567.2002.01309.x
65. Strober W, Fuss IJ. Pro-inflammatory cytokines in the pathogenesis of IBD. Gastroenterology (2011) 140:1756–67. doi: 10.1002/nbm.3066.Non-invasive
66. Ruiz PA, Shkoda A, Kim SC, Sartor RB, Haller D. IL-10 gene-deficient mice lack TGF-beta/Smad-mediated TLR2 degradation and fail to inhibit proinflammatory gene expression in intestinal epithelial cells under conditions of chronic inflammation. Ann N Y Acad Sci. (2006) 1072:389–94. doi: 10.1196/annals.1326.023
67. Shah N, Kammermeier J, Elawad M, Glocker EO. Interleukin-10 and interleukin-10-receptor defects in inflammatory bowel disease. Curr Allergy Asthma Rep. (2012) 12:373–9. doi: 10.1007/s11882-012-0286-z
68. Takagi T, Naito Y, Uchiyama K, Yoshikawa T. The role of heme oxygenase and carbon monoxide in inflammatory bowel disease. Redox Rep. (2010) 15:193–201. doi: 10.1179/174329210X12650506623889
69. Takagi T, Naito Y, Mizushima K, Nukigi Y, Okada H, Suzuki T. Increased intestinal expression of heme oxygenase-1 and its localization in patients with ulcerative colitis. J Gastroenterol Hepatol. (2008) 23:S229–33. doi: 10.1111/j.1440-1746.2008.05443.x
70. Barton SGRG, Rampton DS, Winrow VR, Domizio P, Feakins RM. Expression of heat shock protein 32 (hemoxygenase-1) in the normal and inflamed human stomach and colon: an immunohistochemical study. Cell Stress Chaperones (2003) 8:329. doi: 10.1379/1466-1268(2003)008 < 0329:EOHSPH>2.0.CO;2
71. Wang WP, Guo X, Koo MWL, Wong BCY, Lam SK, Ye YN, et al. Protective CHC. Protective role of heme oxygenase-1 on trinitrobenzene sulfonic acid-induced colitis in rats. Am J Physiol Gastrointest Liver Physiol. (2001) 281:G586–94. doi: 10.1152/ajpgi.2001.281.2.G586
72. Paul G, Bataille F, Obermeier F, Bock J, Klebl F, Strauch U, et al. Analysis of intestinal haem-oxygenase-1 (HO-1) in clinical and experimental colitis. Clin Exp Immunol. (2005) 140:547–55. doi: 10.1111/j.1365-2249.2005.02775.x
73. Hegazi RAF, Rao KN, Mayle A, Sepulveda AR, Otterbein LE, Plevy SE. Carbon monoxide ameliorates chronic murine colitis through a heme oxygenase 1-dependent pathway. J Exp Med. (2005) 202:1703–13. doi: 10.1084/jem.20051047
74. Zuckerbraun BS, Otterbein LE, Boyle P, Jaffe R, Upperman J, Zamora R, et al. Carbon monoxide protects against the development of experimental necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol. (2005) 289:G607–13. doi: 10.1152/ajpgi.00055.2005
75. Takagi T, Naito Y, Uchiyama K, Okuda T, Suzuki T, Tsuboi H, et al. Colonic insufflation with carbon monoxide gas inhibits the development of intestinal inflammation in rats. Med Gas Res. (2012) 2:23. doi: 10.1186/2045-9912-2-23
76. Fukuda W, Takagi T, Katada K, Mizushima K, Okayama T, Yoshida N, et al. Anti-inflammatory effects of carbon monoxide-releasing molecule on trinitrobenzene sulfonic acid-induced colitis in mice. Dig Dis Sci. (2014) 59:1142–51. doi: 10.1007/s10620-013-3014-1
77. Zhang L, Zhang Y, Zhong W, Di C, Lin X, Xia Z. Heme oxygenase-1 ameliorates dextran sulfate sodium-induced acute murine colitis by regulating Th17/Treg cell balance. J Biol Chem. (2014) 289:26847–58. doi: 10.1074/jbc.M114.590554
78. Shen N-Y, Bi J-B, Zhang J-Y, Zhang S-M, Gu J-X, Qu K, Liu C. Hydrogen-rich water protects against inflammatory bowel disease in mice by inhibiting endoplasmic reticulum stress and promoting heme oxygenase-1 expression. World J Gastroenterol. (2017) 23:1375. doi: 10.3748/wjg.v23.i8.1375
79. Horváth K, Varga C, Berkó A, Pósa A, László F, Whittle BJR. The involvement of heme oxygenase-1 activity in the therapeutic actions of 5-aminosalicylic acid in rat colitis. Eur J Pharmacol. (2008) 581:315–23. doi: 10.1016/j.ejphar.2007.12.004
80. Sun X, Suzuki K, Nagata M, Kawauchi Y, Yano M, Ohkoshi S, et al. Rectal administration of tranilast ameliorated acute colitis in mice through increased expression of heme oxygenase-1. (2010) 60:93–101. doi: 10.1111/j.1440-1827.2009.02490.x
81. Han K, Park J, Jeong M, Han Y, Go E, Park J, et al. Heme oxygenase-1 induction and anti-inflammatory actions of atractylodes macrocephala and taraxacum herba extracts prevented colitis and was more effective than sulfasalazine in preventing relapse. Gut Liver (2017) 11:655–66. doi: 10.5009/gnl16496
82. Takagi T, Uchiyama K, Naito Y. The therapeutic potential of carbon monoxide for inflammatory bowel disease. Digestion (2015) 91:13–8. doi: 10.1159/000368765
83. Calkins BM. A meta-analysis of the role of smoking in inflammatory bowel disease. Dig Dis Sci. (1989) 34:1841–54.
84. Beaugerie L, Massot N, Carbonnel F, Cattan S, Gendre JP, Cosnes J. Impact of cessation of smoking on the course of ulcerative colitis. Am J Gastroenterol. (2001) 96:2113–6. doi: 10.1111/j.1572-0241.2001.03944.x
85. Yang L, Shen L, Li Y, Li Y, Yu S, Wang S. Hyperoside attenuates dextran sulfate sodium-induced colitis in mice possibly via activation of the Nrf2 signalling pathway. J Inflamm. (2017) 14:25. doi: 10.1186/s12950-017-0172-5
86. Zhang M, Xu C, Liu D, Han MK, Wang L, Merlina D. Oral delivery of nanoparticles loaded with ginger active compound, 6-Shogaol, attenuates ulcerative colitis and promotes wound healing in a murine model of ulcerative colitis. J Crohn's Colitis (2018) 12:217–29. doi: 10.1093/ecco-jcc/jjx115
87. Ju S, Ge Y, Li P, Tian X, Wang H, Zheng X, et al. Dietary quercetin ameliorates experimental colitis in mouse by remodeling the function of colonic macrophages via a heme oxygenase-1-dependent pathway. Cell Cycle (2018) 17:53–63. doi: 10.1080/15384101.2017.1387701
88. Almeer RS, Mahmoud SM, Amin HK, Abdel Moneim AE. Ziziphus spina-christi fruit extract suppresses oxidative stress and p38 MAPK expression in ulcerative colitis in rats via induction of Nrf2 and HO-1 expression. Food Chem Toxicol. (2018) 115:49–62. doi: 10.1016/j.fct.2018.03.002
89. Tan Y, Zheng C. Effects of alpinetin on intestinal barrier function, inflammation and oxidative stress in dextran sulfate sodium-induced ulcerative colitis mice. Am J Med Sci. (2018) 355:377–86. doi: 10.1016/j.amjms.2018.01.002
90. Onyiah JC, Sheikh SZ, Maharshak N, Steinbach EC, Russo SM, Kobayashi T, et al. Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance. Gastroenterology (2013) 144:789–98. doi: 10.1053/j.gastro.2012.12.025
91. Wegiel B, Larsen R, Gallo D, Chin BY, Harris C, Mannam P, et al. Macrophages sense and kill bacteria through carbon monoxide – dependent inflammasome activation. J Clin Invest. (2014) 124:4926–40. doi: 10.1172/JCI72853DS1
92. Mitterstiller A-M, Haschka D, Dichtl S, Nairz M, Demetz E, Talasz H, et al. Heme oxygenase 1 controls early innate immune response of macrophages to Salmonella typhimurium infection. Cell Microbiol. (2016) 18:1374–89. doi: 10.1111/cmi.12578
93. Sun M, Wu W, Liu Z, Cong Y. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J Gastroenterol. (2017) 52:1–8. doi: 10.1007/s00535-016-1242-9
94. Skaar EP, Gaspar AH, Schneewind O. IsdG and IsdI, heme-degrading enzymes in the cytoplasm of Staphylococcus aureus. J Biol Chem. (2004) 279:436–43. doi: 10.1074/jbc.M307952200
95. Suits MDL, Pal GP, Nakatsu K, Matte A, Cygler M, Jia Z. Identification of an Escherichia coli O157:H7 heme oxygenase with tandem functional repeats. Proc Natl Acad Sci USA. (2005) 102:16955–60. doi: 10.1073/pnas.0504289102
96. Wilks A, Schmitt MP. Expression and characterization of a heme oxygenase (Hmu O) from Corynebacterium diphtheriae. J Biol Chem. (1998) 273:837–41. Available online at: http://www.jbc.org/content/273/2/837.abstract
97. Engel RR, Matsen JM, Chapman SS, Schwartz S. Carbon monoxide production from heme compounds by bacteria. J Bacteriol. (1972) 112:1310–15.
98. Schmitt MP. Transcription of the Corynebacterium diphtheriae hmuO gene is regulated by iron and heme. Infect Immun. (1997) 65:4634–41. Available online at: http://www.ncbi.nlm.nih.gov/pubmed/9353044
99. Thompson JM, Jones HA, Perry RD. Molecular characterization of the hemin uptake locus (hmu) from Yersinia pestis and analysis of hmu mutants for hemin and hemoprotein utilization. Infect Immun. (1999) 67:3879–92.
100. Zhu W, Wilks A, Stojiljkovic I. Degradation of heme in gram-negative bacteria: the product of the hemO gene of neisseriae is a heme oxygenase. J Bacteriol. (2000) 182:6783–90. doi: 10.1128/JB.182.23.6783-6790.2000
101. Ratliff M, Zhu W, Deshmukh R, Wilks A, Stojiljkovic I. Homologues of neisserial heme oxygenase in gram-negative bacteria: degradation of heme by the product of the pigA gene of Pseudomonas aeruginosa. J Bacteriol. (2001) 183:6394–403. doi: 10.1128/JB.183.21.6394-6403.2001
102. Suits MDL, Jaffer N, Jia Z. Structure of the Escherichia coli O157:H7 heme oxygenase ChuS in complex with heme and enzymatic inactivation by mutation of the heme coordinating residue His-193. J Biol Chem. (2006) 281:36776–782. doi: 10.1074/jbc.M607684200
103. Torres AG, Payne SM. Haem iron-transport system in enterohaemorrhagic Escherichia coli O157:H7. Mol Microbiol. (1997) 23:825–33. doi: 10.1046/j.1365-2958.1997.2641628.x
104. Stojiljkovic I, Hantke K. Transport of hemin across the cytoplasmic membrane through a hemin-specific periplasmic binding-protein-dependent transport-system in Yersinia-enterocolitica. Mol Microbiol. (1994) 13:719–32. doi: 10.1111/j.1365-2958.1994.tb00465.x
105. Maharshak N, Ryu HS, Fan T, Onyiah JC, Otterbein SL, Wong R, et al. Escherichia coli heme oxygenase modulates host innate immune responses. Microbiol Immunol. (2015) 59:452–65. doi: 10.1111/1348-0421.12282.Escherichia
106. Chau L-Y. Heme oxygenase-1: emerging target of cancer therapy. J Biomed Sci. (2015) 22:22. doi: 10.1186/s12929-015-0128-0
107. Nemeth Z, Li M, Csizmadia E, Döme B, Johansson M, Liao Persson J, et al. Heme oxygenase-1 in macrophages controls prostate cancer progression. Oncotarget (2015) 6:33675–88. doi: 10.18632/oncotarget.5284
108. Yim MS, Ha YS, Kim IY, Yun SJ, Choi YH, Kim WJ. HMOX1 is an important prognostic indicator of nonmuscle invasive bladder cancer recurrence and progression. J Urol. (2011) 185:701–5. doi: 10.1016/j.juro.2010.09.081
109. Lee J, Mailar K, Yoo OK, Choi WJ, Keum YS. Marliolide inhibits skin carcinogenesis by activating NRF2/ARE to induce heme oxygenase-1. Eur J Med Chem. (2018) 150:113–26. doi: 10.1016/j.ejmech.2018.02.068
110. Yin H, Fang J, Liao L, Maeda H, Su Q. Upregulation of heme oxygenase-1 in colorectal cancer patients with increased circulation carbon monoxide levels, potentially affects chemotherapeutic sensitivity. BMC Cancer (2014) 14:436. doi: 10.1186/1471-2407-14-436
111. Jun S-Y, Hong S-M, Bae YK, Kim HK, Jang KY, Eom DW. Clinicopathological and prognostic significance of heme oxygenase-1 expression in small intestinal adenocarcinomas. Pathol Int. (2018) 68:294–300. doi: 10.1111/pin.12657
112. Andrés NC, Fermento ME, Gandini NA, Romero AL, Ferro A, Donna LG, et al. Heme oxygenase-1 has antitumoral effects in colorectal cancer: involvement of p53. Exp Mol Pathol. (2014) 97:321–31. doi: 10.1016/j.yexmp.2014.09.012
113. Oláh G, Módis K, Törö G, Hellmich MR, Szczesny B, Szabo C. Role of endogenous and exogenous nitric oxide, carbon monoxide and hydrogen sulfide in HCT116 colon cancer cell proliferation. Biochem Pharmacol. (2018) 149:186–204. doi: 10.1016/j.bcp.2017.10.011
114. Panea C, Farkas AM, Goto Y, Abdollahi-Roodsaz S, Lee C, Koscsó B, et al. Intestinal monocyte-derived macrophages control commensal- specific Th17 responses. Cell Rep. (2016) 12:1314–24. doi: 10.1016/j.celrep.2015.07.040.Intestinal
115. Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science (2005) 307:254–8. doi: 10.1126/science.1102901
116. Marelli G, Erreni M, Anselmo A, Taverniti V, Guglielmetti S, Mantovani A, et al. Heme-oxygenase-1 production by intestinal CX3CR1+macrophages helps to resolve inflammation and prevents carcinogenesis. Cancer Res. (2017) 77:4472–85. doi: 10.1158/0008-5472.CAN-16-2501
117. Davudian S, Mansoori B, Shajari N, Mohammadi A, Baradaran B. BACH1, the master regulator gene: a novel candidate target for cancer therapy. Gene (2016) 588:30–7. doi: 10.1016/j.gene.2016.04.040
118. Chang LC, Fan CW, Tseng WK, Chein HP, Hsieh TY, Chen JR, et al. The ratio of Hmox1/Nrf2 mRNA level in the tumor tissue is a predictor of distant metastasis in colorectal cancer. Dis Markers (2016) 2016:2–7. doi: 10.1155/2016/8143465
119. Rana N, McLean S, Mann BE, Poole RK. Interaction of the carbon monoxide-releasing molecule Ru(CO)3Cl(glycinate) (CORM-3) with Salmonella enterica serovar Typhimurium: in situ measurements of CO binding by integrating cavity dual beam spectrophotometry. Microbiology (2014) 160:2771–9. doi: 10.1099/mic.0.081042-0
Keywords: heme oxygenase-1, inflammatory bowel disease, carbon monoxide, infection, inflammation, nuclear factor erythroid 2-related factor 2, colorectal cancer, microbiota
Citation: Sebastián VP, Salazar GA, Coronado-Arrázola I, Schultz BM, Vallejos OP, Berkowitz L, Álvarez-Lobos MM, Riedel CA, Kalergis AM and Bueno SM (2018) Heme Oxygenase-1 as a Modulator of Intestinal Inflammation Development and Progression. Front. Immunol. 9:1956. doi: 10.3389/fimmu.2018.01956
Received: 09 May 2018; Accepted: 08 August 2018;
Published: 12 September 2018.
Edited by:
Marcello Chieppa, Ospedale Specializzato in Gastroenterologia Saverio de Bellis (IRCCS), ItalyReviewed by:
Paola Allavena, Humanitas Clinical and Research Center, ItalyCopyright © 2018 Sebastián, Salazar, Coronado-Arrázola, Schultz, Vallejos, Berkowitz, Álvarez-Lobos, Riedel, Kalergis and Bueno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan M. Bueno, c2J1ZW5vQGJpby5wdWMuY2w=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.