 Perrine Bortolotti
Perrine Bortolotti Emmanuel Faure
Emmanuel Faure Eric Kipnis
Eric Kipnis- 1Meakins-Christie Laboratories, Department of Medicine, Research Institute of the McGill University Health Center, Montreal, QC, Canada
- 2Surgical Critical Care Unit, Department of Anesthesiology and Critical Care, Centre Hospitalier Regional et Universitaire de Lille, Lille, France
- 3Host-Pathogen Translational Research, Faculté de Médecine, Université Lille 2 Droit et Santé, Lille, France
Trauma remains a leading cause of death worldwide. Hemorrhagic shock and direct injury to vital organs are responsible for early mortality whereas most delayed deaths are secondary to complex pathophysiological processes. These processes result from imbalanced systemic reactions to the multiple aggressions associated with trauma. Trauma results in the uncontrolled local and systemic release of endogenous mediators acting as danger signals [damage-associated molecular patterns (DAMPs)]. Their recognition by the innate immune system triggers a pro-inflammatory immune response paradoxically associated with concomitant immunosuppression. These responses, ranging in intensity from inappropriate to overwhelming, promote the propagation of injuries to remote organs, leading to multiple organ failure and death. Some of the numerous DAMPs released after trauma trigger the assembly of intracellular multiprotein complexes named inflammasomes. Once activated by a ligand, inflammasomes lead to the activation of a caspase. Activated caspases allow the release of mature forms of interleukin-1β and interleukin-18 and trigger a specific pro-inflammatory cell death termed pyroptosis. Accumulating data suggest that inflammasomes, mainly NLRP3, NLRP1, and AIM2, are involved in the generation of tissue damage and immune dysfunction after trauma. Following trauma-induced DAMP(s) recognition, inflammasomes participate in multiple ways in the development of exaggerated systemic and organ-specific inflammatory response, contributing to organ damage. Inflammasomes are involved in the innate responses to traumatic brain injury and contribute to the development of acute respiratory distress syndrome. Inflammasomes may also play a role in post-trauma immunosuppression mediated by dysregulated monocyte functions. Characterizing the involvement of inflammasomes in the pathogenesis of post-trauma syndrome is a key issue as they may be potential therapeutic targets. This review summarizes the current knowledge on the roles of inflammasomes in trauma.
Introduction
According to the World Health Organization and the Global Burden of Disease study, injuries are responsible for five million deaths per year worldwide (1, 2). Trauma-related mortality is usually described as trimodal: immediate (minutes), early (hours), and late (days to weeks) (3–5). Immediate and early mortalities, mainly due to overwhelming brain injuries, massive bleeding, and critical injuries to vital organs, have remained stable over the past three decades, accounting for approximatively 60 and 25% of overall trauma-related mortality, respectively (2–4). Interestingly, a decline in late deaths has been observed in high-income countries, suggesting that improvement in care is successful to reduce mortality. However, post-trauma care remains challenging, as late mortality still accounts for 10–30% of all trauma-related deaths (3, 5). Trauma patients often develop a “post-injury syndrome,” resulting from an imbalanced systemic reaction to the multiple insults occurring after trauma (6). Initial tissue damage, blood loss and subsequent secondary tissue injuries, lead to the local and systemic release of endogenous mediators acting as danger signals [damage-associated molecular patterns (DAMPs)] (7, 8). Recognition of DAMPs by the innate immune system triggers both an intense pro-inflammatory immune response and a concomitant anti-inflammatory response (9–11). While excessive inflammation promotes the development and propagation of secondary tissue injuries beyond the initial traumatic foci, the anti-inflammatory response leads to host defense impairment and sepsis, contributing altogether to multiple organ dysfunctions, and ultimately, death (12).
To date, treatment of the “post-injury syndrome” is limited and mainly supportive. However, a greater understanding of the underlying pathophysiology has led to the development of adjuvant therapeutic strategies. Treatments aiming to control excessive inflammation, monitor immune cell functions, and restore immune responses have emerged, with sometimes encouraging results (9, 11, 13). Among the potential mechanisms involved in the “post-injury syndrome,” inflammasomes could become promising targets. Indeed, some of the trauma-induced DAMPs are recognized by NOD-like receptors (NLRs) leading to the assembly of intracellular multiprotein complexes named inflammasomes (Figure 1). Accumulating data suggest that inflammasomes are involved in the generation of tissue damage as they promote an exaggerated systemic and organ-specific inflammatory response (14). Inflammasome activation in immune cells, endothelial cells, and platelets contribute to microcirculatory dysfunctions and play a critical role in tissue injuries after ischemia and reperfusion (15). Inflammasomes are involved in the development of traumatic brain injury (TBI) (16) and acute respiratory distress syndrome (ARDS) (17). Finally, inflammasomes may also play a role in monocyte dysfunctions involved in post-trauma immunosuppression (18).
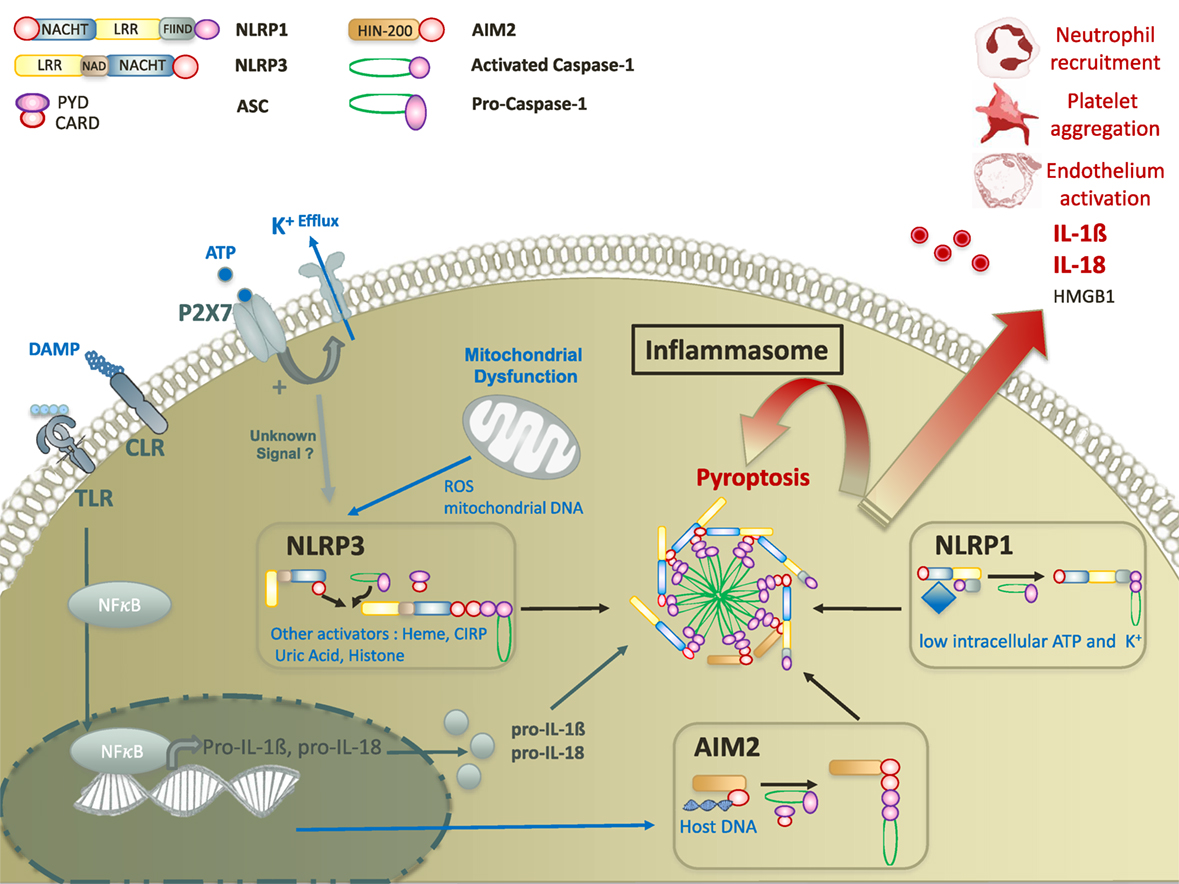
Figure 1. Molecular mechanisms of damage-associated molecular pattern mediated inflammasome activation. Inflammasome activation requires two signals. The first signal ultimately leads to the translocation of NFκB transcription factor to promote the expression of pro IL-18 and pro-IL-1β. The second signal is mediated by a nod-like receptor (NLRP3, AIM2, or NLRP1). Upon activation, NLR activates caspase-1 with or without recruitment of adaptor protein ASC. Activated caspase-1 leads to inflammasome formation and to the secretion of mature forms of IL-18 and IL-1β, and the release of HMGB1. In addition, inflammasomes promote a specific pro-inflammatory cell death, called pyroptosis that contributes to tissue damage in various cellular subtypes. Abbreviations: CIRP, cold-inducible RNA-binding protein; CLR, C-type lectin receptor; ROS, reactive oxygen species; ATP, adenosine triphosphate; TLR, toll-like receptor; IL, interleukine; AIM, absent in melanoma; NFκB, nuclear factor-kappa B; HMGB1, high mobility group box 1.
In this review, we put into perspective how inflammasomes contribute to the pathophysiology of early post-trauma organ dysfunction, systemically and at specific organ levels. We also discuss the involvement of inflammasomes in the development of post-trauma immunosuppression. Finally, based on existing data from therapeutic strategies targeting inflammasomes in inflammatory diseases, we propose therapeutic perspectives highlighting the challenges of research on inflammasomes in trauma.
Description and Functions of Inflammasomes
General Description
Inflammasomes were initially described in 2002 as caspase-1 activating multiprotein complexes (19). Since this seminal report, multiple distinct inflammasomes have been identified. Both pathogen-associated molecular patterns (PAMPs) and endogenous damage-associated molecular patterns (DAMPs) can trigger inflammasome activation. In the context of trauma, we will focus on the inflammasome-mediated response to DAMPs. Inflammasomes are highly expressed in immune cells, especially monocytes and macrophages where they were first discovered. However, inflammasomes are also expressed in a wide range of parenchymal, endothelial (20, 21), and epithelial cell (22, 23) types. Beyond modulation of inflammation, inflammasomes are involved in many physiological processes such as metabolism, cell survival, or tumorigenesis. Consequently, disturbances of inflammasome activity have been involved in a wide range of diseases associated with inflammatory disorders (24). In depth, recent reviews describe inflammasome structures, functions, and regulatory mechanisms providing a translational overview of their role in health and disease, and their potential in development of new immunomodulatory therapeutics (25–30). Here, we provide a brief description of inflammasome biology to introduce their role in post-trauma immune disorders.
Structure and Functions of Inflammasomes
Inflammasome assembly results from the oligomerization of sensor complexes in response to an intracytoplasmic trigger, allowing procaspase 1 recruitment and activation through interactions of homotypic caspase recruitment domains (CARDs). Depending on the sensor, an adaptor protein known as ASC (apoptosis-associated speck-like protein containing a CARD) is required for caspase recruitment (19). Inflammasomes are named after their intracellular receptor, including NLRs, AIM2-like receptors, RIG-I-like receptors, or pyrin (28). NLRs, the most studied receptors to date, are highly conserved throughout evolution, suggesting an important role in host defense to aggression. NLRs contain an N-terminal signaling domain supporting effector functions, and a C-terminal domain containing leucine rich repeat (LRR) sequences which is usually involved in the interaction with ligands (Table 1). N-terminal and C-terminal domains flank a nucleotide-binding domain that defined NLRs (28) (Figure 1). NLRP3 and NLRP1 inflammasomes may be activated by a wide range of exogenous and endogenous compounds, making them the perfect candidates for inflammasome-mediated inflammatory response in trauma (Table 1) (31, 32). Among non-NLR inflammasomes, absent in melanoma (AIM)2 is one of the most studied. Specifically triggered by cytosolic deoxiribonucleic acid (DNA), AIM2 activation has been reported in trauma. Like NLRP3, activated AIM2 receptor nucleates ASC through PYD–PYD interactions to form AIM2 inflammasome. The structure of NLRP1 slightly differs as it contains both PYD and CARD domains, resulting in the ability to recruit procaspase 1 with or without ASC (33, 34) (Figure 1). Other non-NLR receptors have been shown to assemble inflammasomes, but their roles in critical injury have rarely, if ever, been studied. Of note, some differences in the structural characteristics or mechanisms of assembly have been observed between mouse and human. Thus, the interpretation of results from in vivo animal models should always take into account such possible discrepancies (35).
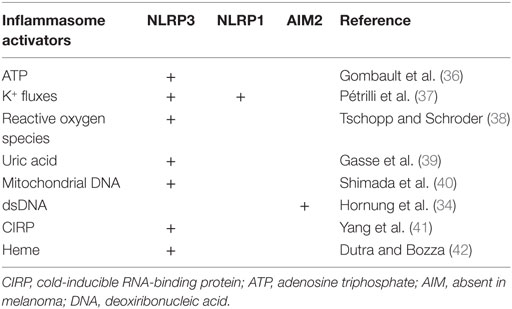
Table 1. Inflammasome activators and activated inflammasomes in trauma.
Specific Features of Inflammasomes as Innate Immune Receptors
Like toll-like receptors (TLRs), inflammasomes were recently demonstrated to orchestrate innate immune responses to aggression (25). However, the localizations, mechanisms of activation, and signaling pathways differ between these two pattern recognition receptors (PRRs) (43). TLRs are transmembrane proteins localized either to the cell surface or within endosomes. Thereby, TLRs mainly recognize extracellular compounds via their extracellular or intra-endosomal LRR motif (44). Conversely, inflammasomes are intra-cytosolic sensors detecting mostly intracellular stimuli (25). Whereas TLRs activation results from binding to well described specific ligands, the triggers for inflammasome activation are more heterogeneous, ranging from the recognition of specific ligands to the sensing of disturbances in the intracellular environment (29). Activation of TLRs mainly leads to a transcriptional immune response via either the MyD88/nuclear factor-kappa B, mitogen-activated protein kinase, or TRIF pathways, resulting in the synthesis of pro-inflammatory proteins, cytokines, and type I Interferon (IFN) (45). Conversely, inflammasome activation does not support any direct transcriptional activity but allows the caspase-1 dependent cleavage of pro-interleukine (IL)-1β and pro-IL-18 into mature forms (27). Importantly, inflammasome activation generally requires a priming step allowing the transcription of the inflammasome components genes (46, 47), in which TLRs are critically involved (47). The second signal comes from the detection of an intracellular “danger” by the cytoplasmic sensor (31). Finally, TLR-signaling was demonstrated to promote various forms of programmed cell death such as autophagy, apoptosis, or necrosis, whereas inflammasomes trigger specific caspase-1-dependent pyroptotic cell death (45, 48).
Inflammasomes: A Double-Edged Sword in Host Defense
Inflammation is an evolutionarily conserved, protective response to harmful stimuli mounted to preserve or restore the integrity of the body. However, the intensity, duration, and compartmentalization of the inflammatory response needs to be tightly regulated. Excessive, extensive, or prolonged inflammation is responsible for secondary damage, as observed during ARDS or trauma (12). Beyond their critical role in detection and control of intracellular pathogens (49, 50), studies suggest that inflammasomes contribute to tissue regeneration through inflammasome-dependent cytokines which promote effective clearance of damaged cells and tissue repair (22, 51). Alongside these beneficial roles, there is evidence that inflammasome activation is also responsible for unbalanced, excessive inflammation (52). Some of the numerous DAMPs released in trauma are known activators of inflammasomes, suggesting the potential role of inflammasome activation in the pathogenesis of trauma-associated immune disorders (10).
Damp-Mediated Inflammasome Activation in Trauma
The emergence of the concept of danger rather than “non-self” as the trigger of the innate immune response was a turning point in the history of immunology (53). From this perspective, it is not surprising that various pathologic conditions associated with inflammatory disorders, such as trauma, sepsis, or ARDS, share common pathophysiologic features (54, 55). Through the activation of identical PRRs, PAMPs initiate the immune response to infection whereas DAMPs trigger “sterile inflammation” (10). Conversely to PAMPs that are exogenous compounds of infectious origin, DAMPs are mostly endogenous self-molecules reflecting the alteration of cellular integrity. This explains why some authors proposed the term “alarmin” to specifically designate damage-associated endogenous compounds and broaden the DAMP acronym to “danger-associated molecular pattern” including both alarmins and PAMPs (56, 57). In this review, we chose to use the term DAMP as “damage-associated molecular pattern,” equivalent to alarmin.
The definition and classification of DAMPs are still debated, as any intracellular compound could potentially be a DAMP. DAMPs should be released after cell stress or damage and reflect damage intensity. In addition, DAMPs should trigger an inflammatory response through identified receptors, measurable at physiological concentrations. However, all the endogenous molecules supporting a pro-inflammatory role cannot always be strictly classified as DAMPs based on these criteria (10, 56, 58). Because the nature of DAMPs is extremely heterogenous, and the receptors involved in their recognition are often redundant with other DAMPs or PAMPs, the conceptual framework of DAMPs is not constrained to specific molecular groups or unique signaling pathways. Pragmatically however, the concept of DAMPs provides the opportunity to distinguish exogenous danger signals such as microbial patterns, from endogenous danger signals that can be recognized through innate immune sensors and/or trigger an immune response. Consistently, it has been proposed to take into account the clinical relevance of the compounds in the pathogenesis of the inflammatory response to injury to define DAMPs (10, 56, 58). Indeed, some of these molecules can trigger inflammation through direct interaction with host cells, but also indirectly via complement activation for example. Direct interactions with host cells occurs either via cell surface and extracellular milieu receptors, such as TLRs or receptor for advanced glycation endproducts (RAGE), or via intracellular receptors such as NLRs (10, 59). The intra- versus extracellular compartmentalization of DAMPs determines the nature of activated PRRs (56). In trauma, mechanical tissue injuries and blood loss, associated with secondary events including ischemia/reperfusion (I/R), hypothermia, hypoxia, coagulopathy, or neuroendocrine disorders, lead to cell stress and cell death (9–11). Both cell stress and cell death lead to the active or passive intra- and extracellular release of intracellular compounds that are the major source of DAMPs (56). Several classes of compounds have been identified ranging from the small uric acid or adenosine triphosphate (ATP) molecules to large proteins over 50 kDa. This tremendous structural diversity associated with the extensive variety of PRRs and their mechanisms of interaction highlights the complexity of DAMP signaling, which was previously reviewed (56).
Here, we focus on characterized DAMPs involved in the trauma-related, inflammasome-dependent inflammatory response.
DAMPs are classified depending on their origin, mitochondrial, cytosolic, or nucleic. Mitochondrial DAMPs identified in trauma-induced response are: mitochondrial DNA, reactive oxygen species (ROS), and ATP, which promote NLRP3 inflammasome activation, and NLRP1 inflammasome activation for ATP (38). Cytosolic DAMPs are mostly represented by uric acid, and cold-inducible RNA-binding protein (CIRP), a highly conserved chaperone protein that belongs to the family of cold shock proteins, both of them activating the NLRP3 inflammasome pathway (39, 41). Heme is also reported to activate NLRP3 inflammasome, participating in hemolysis-induced lethality in trauma (42).
Two main nucleic DAMPs are demonstrated to participate in trauma-induced immune response: histones (60) and host DNA (61). Host DNA interacts directly with AIM2 inflammasome, through its HIN domain (46). Histones activate the NLRP3 inflammasome, but the precise pathways are not characterized (60).
The diversity of stimuli described as NLRP3 and NLRP1 activators makes the hypothesis of direct interaction unlikely, but rather supports the existence of common intracellular pathway(s) leading to inflammasome assembly. DAMP-mediated NLRP3 and NLRP1 activation remains poorly understood despite years of research. However, the reduction of intracellular potassium levels through K+ efflux channel is recognized as a downstream convergence point (37) (Figure 1).
Interestingly, inflammasome activation could result from extracellular DAMPs released by surrounding stressed or dying cells, or via intracellular nucleic and mitochondrial DAMPs translocating into the cytosol in response to cell stress (56).
The nature of these DAMPs and the inflammasomes to which they are related are summarized in Table 1. Importantly, one should keep in mind that other uncharacterized ligands could also be potential activators of the inflammasomes after trauma.
Priming and activation of inflammasomes through DAMPs have been proven in trauma-like models. In proof-of-concept experiments, Iyer et al. show that murine macrophages exposed to necrotic cells produced by pressure disruption, hypoxic injury, or complement-mediated damage, elicit NLRP3 inflammasome priming and activation (62). Likewise, injection of necrotic cells in the peritoneum of wild-type mice leads to NLRP3 activation (62). Consistently, recent studies show the priming and activation of the NLRP3 inflammasome in the injured tissues of rodents in response to mechanical stress (63, 64). Similar observations have been made in the brain, heart, kidney, and testis of mice exposed to hypoxia–ischemia (15). Here, we describe how inflammasome signaling participates in the pathogenesis of remote tissue and organ damage in this context.
Inflammasomes in the Pathogenesis of Remote Tissue and Organ Damage after Trauma
The pathogenesis of remote organ injury after trauma is a multifactorial process that has already been expertly reviewed (9, 11, 12, 55, 65). The post-trauma period is often associated with an intense pro-inflammatory response termed “systemic inflammatory response syndrome” or SIRS. SIRS often exceeds its functions of clearing and repair, promoting tissue damage independently of the initial injury (12, 55). The concomitant dysfunction of endothelium, coagulation, and immune system promotes the onset and the self-perpetuation of secondary damages to tissues (Figure 2). The extreme expression of this immune deregulation is multiorgan dysfunction syndrome (MODS) which occurs in approximately in one to four trauma patients, and represents the leading cause of “late” death in trauma (66). Because DAMPs both drive and result from tissue damage, they play a central role in the pathogenesis of trauma-related critical illness, supporting a vicious cycle of injury (55). Multiple studies suggest that inflammasome activation, notably NLRP3, is involved in the pathogenesis of SIRS and MODS after trauma.
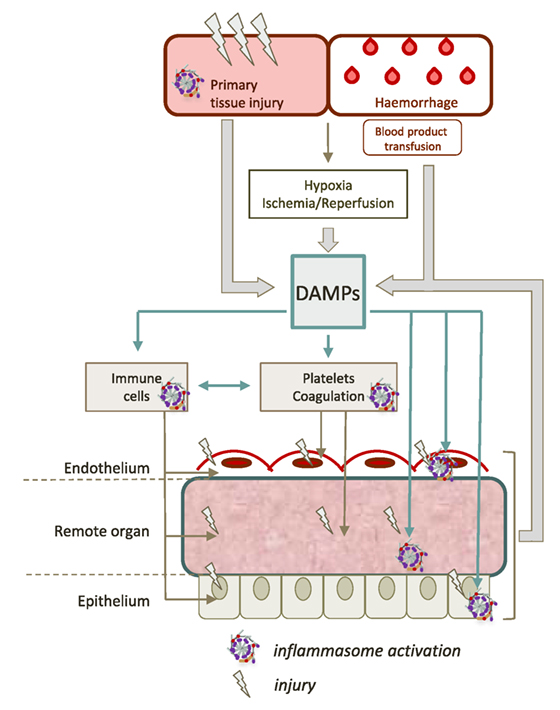
Figure 2. Contribution of inflammasomes to remote tissue damage after trauma. The initial trauma injury induces the release of a large amount of different damage-associated molecular patterns (DAMPs). DAMPs can activate inflammasomes in various cell types such as hematopoietic cells (platelets, neutrophils, monocytes, and macrophages), epithelial cells, or endothelial cells. This dramatic inflammasome activation leads to increase the release of new DAMPs, maintaining a vicious circle, that contributes to worsen organ damage and propagate the pro-inflammatory response.
Limits to the Study of Host Response Pathophysiology in Trauma
In adult trauma studies, the variability in the timeframes of biological sampling, in patients’ underlying conditions, and in patient management, particularly in the late phase of trauma in which patients are variably addressed to critical care units, intensive care units, step down units, or surgical wards leads to excessively challenging interpretation of results. To address these problems, strictly controlled animal models greatly simplify addressing physiopathological questions, albeit with inherent limitations. The advantages and limitations of animal models of trauma were reviewed by Tsukamoto and Pape (67) and Deitch (68). Regardless of the limitations of animal models, some can provide information even on late phase of trauma and may corroborate clinical observations. For instance, while in adult patients with trauma brain injury, a significant reduction in phagocytosis capacity has been observed more than 3 weeks after the first insult, in a murine brain injury model, a reduced amount of phagocytic cells and a decline of the thymic mass were observed after 2 months post-injury (69, 70). Considering this example, promising data from animal models may be useful even in studying the later phases of trauma and trauma-induced long-term immune disorders.
Inflammasomes in Uncontrolled Inflammation After Trauma
Both inflammasome-mediated pro-inflammatory mediator release and pyroptotic cell death contribute to initiating, enhancing, and propagating inflammation after trauma (30, 71). In a rat model of acute severe stress, Maslanik et al. demonstrate that caspase-1 activity promotes the production of IL-1β and IL-18 in the circulation and in peripheral tissues (72). In a mouse model of tissue contusion, Starzl et al. show that NLRP3 inflammasome activation is associated with the severity of the inflammatory response through IL-1β and IL-18 secretion (64). Through multiple systemic and local effects, IL-1β is an essential actor of the inflammatory and immune response (73). IL-1β induces the expression of multiple pro-inflammatory genes such as IL-6, IL-8, MCP-1, IL-1α and β, platelet activating factor, and eicosanoids. IL-1β is responsible for the production of acute phase proteins such as CRP and coagulation factors. By enhancing the production of cell surface adhesion molecules and chemo-attractants, IL-1β promotes leukocyte recruitment and activation to the site of inflammation (74, 75). Although less studied than IL-1β, IL-18 also promotes the expression of adhesion proteins, pro-inflammatory cytokines, and chemokines beside its major role as an IFNγ inducer (76). Interestingly, it has been shown that inflammasome activation also plays a critical role in the passive and active extracellular release of high mobility group box 1 (HMGB1), a well-characterized DAMP in trauma (77). HMGB1, a ubiquitously expressed DNA-binding protein located in the nucleus, increases in patients serum and CSF after trauma (78, 79), triggering both pro-inflammatory response and leukocyte recruitment via its main receptors RAGE and TLR2/4 (80, 81). HMGB1 is also been involved in post-trauma immunosuppression (82). Therefore, HMGB1 has been proposed as a prognostic biomarker and as a potential therapeutic target in trauma (82, 83).
Together with inflammasome-dependent pro-inflammatory mediators, pyroptosis contributes to the overwhelming inflammatory response. Pyroptosis is characterized by cytoplasm swelling and plasma membrane rupture (84). Pyroptosis allows the destruction of disturbed cells (85), but also leads to the release of the intracellular content into the extracellular space, contributing to “sterile” inflammation. Further inflammasome-dependent mechanisms allow the amplification of the inflammatory burst (27). First, the inflammasome apparatus per se, by promoting a large amount of active caspase 1 in response to a single trigger, contributes to amplify the signal (30, 86). Second, inflammasome components themselves, by releasing activated ASC into the extracellular environment, induce further inflammasome activation in surrounding cells (30, 71).
Altogether, inflammasome-mediated inflammation also promotes the recruitment and activation of immune cells through a concentration gradient of DAMPs, cytokines, and chemokines (87). Inflammatory leukocytes are major effectors of post-injury secondary damage (88). When exposed to inflammatory mediators, immune cells start activating. This “priming” step promotes leukocytes adhesion to the microvascular endothelium and their extravasation into tissue through the vascular wall (89). The release by activated leukocytes of a wide array of mediators such as ROS, proteases, cytokines, chemokines, and lipid mediators into ischemic tissues promotes further endothelial barrier and parenchymal tissue damage (90).
Inflammasomes and Endothelial Dysfunction
Systemic release of DAMPs triggers the diffuse activation of endothelial cells in an organ-specific manner. Endothelial cells do not just form a mechanical barrier but rather constitute an active regulatory organ which plays an essential role in vascular homeostasis and host defense (91). Under physiological conditions, the endothelium continuously maintains an antithrombotic environment by regulating platelet activation, and balancing inhibitors and activators of coagulation and fibrinolysis (91). Under pathological conditions such as trauma, endothelial functions are critical to control hemorrhage by promoting a procoagulant environment while preventing massive thrombosis (92). Vasoconstriction triggered by the decrease in endothelial nitric oxide (NO) production, platelet adhesion, and expression of procoagulant proteins such as endothelial tissue factor, allow the formation of adherent thrombi (Figure 2) (92). The endothelium also regulates the homing and recruitment of leukocytes. Activated endothelium overexpresses cell surface adhesion molecules, chemotactic and activating factors, setting the stage for immune cell recruitment even in non-injured, non-infected remote organs (92). Both clot formation and leukocyte infiltration are mandatory for the control of bleeding and the secondary tissue repair. However, these mechanisms also contribute to worsen, disseminate, and perpetuate injury as well (Figure 2) (92).
There is evidence for the involvement of inflammasomes in endothelial dysfunction, even though specific studies in the context of trauma remain scarce. It has been shown in vitro that several DAMPs such as ATP or CIRP activate the NLRP3 inflammasome in endothelial cells (41, 93). The most striking data on the topic arise from studies performed in lung endothelium. Three recent studies using in vivo mouse models show that hemorrhagic shock (HS) induces NLRP3 priming and activation in lung endothelial cells, enhancing the pro-inflammatory response through pyroptosis and IL-1β secretion (20, 94, 95). Yang et al. demonstrate that endothelial cell pyroptosis leads to increased inflammation and injury in the lung of mice subjected to a “two-hit” model of HS followed by lipopolysaccharide (LPS). In this model, major neutrophil alveolar recruitment and interstitial edema are attenuated in caspase-1-null mice (94). Xiang et al. find a role for hemorrhage-related ROS production in the activation of endothelial NLRP3 inflammasome, leading to increased levels of IL-1β in BAL (Figure 3) (20). A similar result is found by Xu et al., showing an increase in NLRP3 expression in endothelial cells responsible for higher IL-1β level in bronchoalveolar lavage fluid (20, 94, 95). These inflammasome-mediated responses contribute to endothelial damage associated with vascular leakage, edema, increased leukocyte infiltration, and cytokine release in the lungs (84, 87). Besides their effects on endothelial cells, inflammasomes also modulate other actors of vascular dysfunction and secondary damages, including platelets and coagulation.
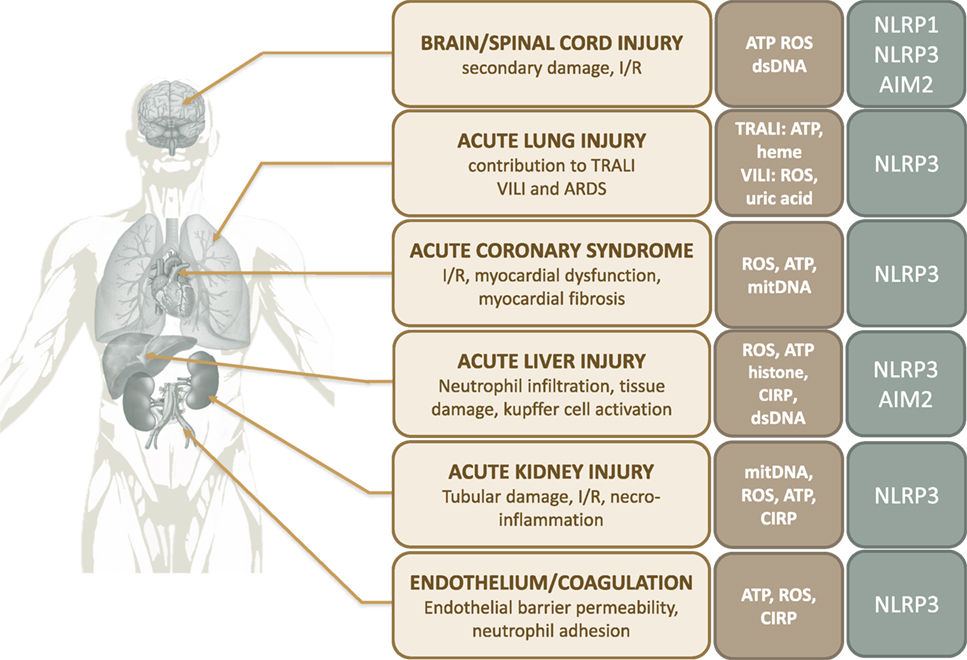
Figure 3. Contribution of inflammasomes to organ damage after trauma. Abbreviations: ARDS, acute respiratory distress syndrome; ATP, adenosine triphosphate; CIRP, cold-inducible RNA-binding protein; I/R, ischemia/reperfusion; ROS, reactive oxygen species; TRALI, transfusion-related acute lung injury; VILI, ventilator-induced lung injury.
Inflammasomes, Platelet Functions, and Coagulation
Because they are anucleate, platelets were initially considered as simple procoagulant effectors. However, recent research established that platelets also support critical pro-inflammatory and anti-infectious properties beside their role in hemostasis (96, 97). Indeed, activated platelets play a critical role in the inflammatory response, especially in ischemia-reperfusion. Platelets possess various immune receptors, cell surface adhesion molecules, and a broad spectrum of immunomodulatory mediators contained in preformed granules (98). When activated by circulating DAMPs, platelets adhere to endothelial cells and leukocytes to form aggregates. Adherent platelets can either release the content of their granules or synthetize pro-inflammatory mediators (99). Platelets modulate endothelial and immune cell functions. For instance, platelets enhance adhesion molecule expression and chemokine secretion of endothelial cells (98). Most importantly, platelets strongly promote leukocyte activation and adhesion to post-ischemic microvessels, enhancing their recruitment to sites of tissue damage (100). Platelets also directly modulate neutrophil functions such as phagocytosis and degranulation. Thus, platelets play a critical role in reinforcing the pro-inflammatory cycle leading to and perpetuating organ injury (97, 100).
Inflammasomes have been recently shown to be involved in the pro-inflammatory roles of platelets in both infectious and non-infectious diseases (97, 98). The effects of inflammasome activation on platelets, reviewed by Hottz et al., are essentially mediated by the production of IL-1β (97). Briefly, it has been recently discovered that platelets are capable of assembling functional NLRP3 inflammasomes leading to the synthesis of IL-1β (101). Although anucleate, platelets possess a fully functional translational apparatus associated with constitutively present mRNA transcripts, including IL-1β (99, 102). These properties allow rapid synthesis of mature IL-1β after platelet activation (99). Although no study exists in the specific field of trauma, the role of inflammasomes on platelet functions has been studied in vitro in models of vascular injury or thrombin-induced platelet activation. These studies show that inflammasome-mediated IL-1β secretion, either in a paracrine fashion or through the release of IL-1β-rich microparticles, activate endothelial cells, with subsequent increase in endothelial permeability, neutrophil adhesion, and endothelial transmigration (97). Aside from their effects on platelet functions, inflammasomes are involved in driving a procoagulant response. Rothmeier et al. show that ATP-driven inflammasome activation in murine bone marrow-derived macrophages promotes the intracellular trafficking of tissue-factor onto the surface of MPs and thus facilitates the release of highly procoagulant MPs (103). These results suggest a deleterious role of DAMP-induced inflammasome activation in the onset of coagulopathy.
Inflammasome in Ischemia-Reperfusion
Endothelial, platelet, and coagulation dysfunction together with leukocyte activation actively contribute to worsen initial tissue ischemia. While changes in cell physiology under short-acting ischemic conditions represent an appropriate protective process, prolonged or intense ischemia unconditionally leads to cell damage and death (100). In addition, reperfusion of either primary injured or distant ischemic organs is also a critical actor of tissue damage and DAMP production through “ischemia-reperfusion syndrome” (104). At the cellular level, reperfusion triggers massive production of ROS, calcium overload, and severe mitochondrial dysfunction, which are all critical events in the progression of cell death (100). The active contribution of inflammasomes to I/R-induced injuries has been demonstrated in vivo using experimental models of mice deficient for inflammasome components or IL-1β and IL-18 signaling pathways. The inflammasomes have been involved in the pathogenesis of injury in the heart, liver, kidneys, central nervous system (CNS), and lungs exposed to I/R.
In the heart, I/R-related phenomenon are responsible for cardiac cell death, enhancing the size and severity of myocardial infarction, and promoting fibrosis after ischemic events (105). The NLRP3 inflammasome has been reported to play a major role in this process via IL-1β and IL-18 secretion (95–98) (Figure 3). It has been shown in mouse models of myocardial I/R that inflammasome activation in cardiac fibroblasts (106), cardiomyocytes (107), and cardiac endothelial cells (108) are essential to the inflammatory response driving myocardial I/R injury. Inflammasome activation promotes myocardial inflammatory cell infiltration and increased pro-inflammatory cytokine expression in cardiac tissue. Consistently, myocardial infarct size, contractile dysfunction, and fibrosis are decreased in ASC and caspase-1 deficient mice (106, 109). Likewise, pharmacological inhibition of NLRP3 or IL-1β/IL-18 signaling protects the heart from I/R injury in a rodent model of cardiac ischemia (110–114) (Figure 3). In this context, the role of ROS as activators of the NLRP3 inflammasomes seems predominant (15).
As for the heart, the critical role of inflammasomes is well documented in several acute and chronic human kidney diseases (115). As a result, inflammasome signaling is already an effective therapeutic target for some selected patients with chronic renal failure (115). Inflammasomes are involved in the pathogenesis of acute kidney injury (AKI) related to I/R (15) (Figure 3). With crucial functions in maintaining water and sodium homeostasis, overall plasma electrolyte regulation and detoxification, the kidneys receive more than 20% of the cardiac output. To support their functions, the kidneys and particularly renal tubular epithelial cells have a high mitochondrial density which requires a high continuous energy supply. Thus, they are vulnerable to oxygen and energy deprivation. The release of mitochondrial components, ROS, and other DAMPs from damaged tubular epithelial cells promotes acute cell death (Figure 3). CIRP is also highlighted to participate in I/R-induced damage during AKI, in a murine model using CIRP-KO mice (116) (Figure 3). The subsequent pro-inflammatory burst promotes fluid leakage and parenchymal infiltration of inflammatory cells, worsening renal dysfunction and injury (117). Subsequent tubular necrosis and loss of functional nephrons lead to the clinical syndrome of AKI, explaining the high frequency of acute renal failure in critical illness (118). Once again, DAMPs are the initiators of the inflammatory response. There is substantial evidence that the inflammasome–IL-1β axis plays a key role in the primary mechanisms leading to renal necroinflammation. Macrophages and dendritic cells, in addition to most renal parenchymal cells, express inflammasomes (115). DAMPs released from necrotic cells have been shown to activate the NLRP3 inflammasome in renal tissue. Indeed, Iyer et al. observe an upregulation of Nlrp3 and Asc gene expression in a mouse model of nonlethal renal I/R injury, associated with intense acute tubular necrosis (62). Nlrp3, Asc, and Casp1/11 deficient animals are consistently protected from renal necroinflammation (115, 119). Nlrp3-deficiency dramatically improves the survival of animals in lethal renal ischemic injury and provides functional protection against renal failure as suggested by significantly lower plasma urea and creatinine levels. This protection is associated with reduced neutrophil infiltration and IL-1β levels in the renal interstitium of Nlrp3-deficient mice (62, 120, 121). The role of IL-1β and IL-18 in acute renal failure is still debated as the results of blockade experiments are controversial (122). Shigeoka et al. find that blocking these cytokines fails to improve creatinine levels and histopathological damage in murine models (120). However, Wu et al. demonstrate that IL-18-null mice show better kidney function, less tubular damage, and reduced necroinflammation. The same observations have been made when mice are pretreated with the IL-18 antagonist IL-18BP (123). Last, NLRP3 might impair tissue repair during the reperfusion phase, as Nlrp3-null mice show reduced tubular necrosis and apoptosis together with increased proliferation of tubular epithelial cells after I/R injury (124).
A similar role for NLRP3 and AIM2 inflammasomes has been recently reported in hepatic I/R injury (87, 125, 126) (Figure 3). DAMPs locally released during I/R, namely ROS, ATP, or extracellular histones, have been shown to trigger inflammasomes activation (Figure 3) in Kupffer cells, which critically contribute to the exacerbation of the local inflammatory response (87, 125, 126). Inflammasome-mediated liver injury in I/R arises from mechanisms involving pro-inflammatory cytokines and excessive neutrophil infiltration (127). McDonald et al. and others demonstrate that the I/R-related pro-inflammatory environment leads to the adhesion of circulating neutrophils within liver sinusoids, and facilitates their migration through healthy tissue toward damaged tissue (87). Consistently, both the inhibition of NLRP3 inflammasome activity in mice exposed to liver I/R is associated with decreased levels of cytokines and neutrophil infiltration in hepatic tissue, with a dramatic reduction in histological damage and transaminase release (87, 125, 128, 129). Similar results are found when inhibiting IL-1β signaling via blocking antibodies or IL-1-receptor antagonist (87, 127, 128). Finally, blocking inflammasome activators such as CIRP increases the overall survival in a mouse model of hepatic I/R, by decreasing neutrophils recruitment and nitrosative stress level (130).
In addition to their roles in I/R-related injury, inflammasomes are involved in brain and lung dysfunction following trauma.
Contribution of Inflammasomes to Specific Organ Dysfunctions after Trauma
Brain Injury
Damage to CNS structures is a major determinant of vital and functional prognosis in trauma patients. Thus, studying the mechanisms leading to CNS injuries following trauma is crucial. While the primary traumatic insult directly leads to immediate tissue damage, the pro-inflammatory innate immune response to injury, termed “neuroinflammation,” is responsible for additional secondary cellular damage and extension of the lesions (131, 132). Among the multiple actors involved in neuroinflammation following trauma, the inflammasomes, specifically NLRP1 and NLRP3, play a major role (133, 134) (Figure 3). Although it has been shown that inflammasome-mediated IL-1β production contributes to CNS tissue repair after trauma (135), the pro-inflammatory response resulting from inflammasome activation, together with glial and neuronal pyroptotic cell death, promote the secondary insult that contributes to worsen and extend initial damage (131). Many cell types, including endothelial cells (21), microglia (136), astrocytes, and neurons (133) are capable of assembling inflammasomes. The role of inflammasomes in TBI and spinal cord injury (SCI) has been recently reviewed (133, 134).
NLRP1 retains a special interest in the context of neurotrauma as a major actor of induction and diffusion of inflammation (16). Importantly, the NLRP1 inflammasome is already preassembled before any stimulation in neurons and other CNS cells (133). It has been proposed that this preassembly of the NLRP1 inflammasome in the CNS may facilitate rapid innate immune response after CNS trauma, or may serve to maintain a constant low level of IL-1β in these cells (133) (Figure 3). CNS NLRP1 activation is triggered by multiple stimuli including the activation of pannexin-1 channels, triggered by high extracellular potassium concentrations (137). Likewise, P2X4 purinergic receptors activated by extracellular ATP induce NLRP1 inflammasome activation (133) (Figure 3).
de Rivero Vaccari et al. report the increased expression of NLRP1 inflammasome, ASC, caspase-1, and subsequent IL-1β in the brain and spinal cord motor neurons following trauma (138, 139). Consistently, Satchell et al. observe that caspase 1 and IL-1β proteins are increased in cerebrospinal fluid in infants and children after severe TBI (140). Moreover, de Rivero Vaccari et al. find that NLRP1 inflammasome proteins are present in exosomes derived from cerebral spinal fluid of patients with SCI and/or TBI (141). These exosomes, by exposing neighboring cells to their cargo of proteins such as IL-1β as well as inflammasome components, contribute to the diffusion of inflammation in the CNS (142).
NLRP3 expression and activation also increase in the brain after direct trauma or following ischemia in rodent models, and NLRP3 knockdown leads to a reduction of brain damage and inflammatory mediators in animal models (143, 144). Consistently, Wallisch et al. report increased NLRP3 levels in CSF from children with severe TBI, which is independently associated with poor outcome (145).
Considering the role of inflammasomes in the particular case of cerebral ischemia, it has been shown that activated inflammasomes, namely NLRP3 and AIM2, increase inflammation, infarct size, and neurovascular damage (144, 146). IL-1β-triggered inflammation is a major contributor to cell death in the ischemic brain, but some studies report inflammasome-mediated effects that are independent of IL-1β production. Consistently, inflammasome inhibition or blockade of IL-1β both significantly decrease neuronal cell death in the brain or spinal cord of ischemic animals (147). The link between inflammasome activity, CNS inflammation, and functional outcome leads some authors to suggest the use of inflammasome protein levels in cerebrospinal fluid of brain-injured patients as biomarkers of functional prognosis (148). Interestingly, regulatory mechanisms that downregulate NLRP1 or NLRP3 activity exhibit a protective role against CNS injury (133, 134). For example, Lin et al. show that the downregulation of NLRP1 activity by heme oxygenase-1 decreases NLRP1 inflammasome-induced neuronal death and improves functional recovery in a rat model of spinal cord compression (149). Experimental inhibition of inflammasomes in rodent models of TBI or SCI, targeting either the inflammasome components ASC or NLR, caspase-1 or IL-1β, shows promising results in term of histopathological improvement and improved functional outcome (132, 138, 149–153) (Figure 3).
However, in TBI, inflammasome activation is not the only driver of organ dysfunction. It has indeed been shown that the CNS, particularly through the Hypothalamic–Pituitary–Adrenal axis, drives control of the peripheral immune response (154). This topic has been previously extensively reviewed (155, 156). In brief, the studies suggest that CNS injury impacts both circulating immune cells populations and function, potentially contributing to secondary damage to remote organs and susceptibility to infection (88, 157).
Acute Lung Injury
The lung is a vital organ supporting blood oxygenation and decarboxylation necessary to aerobic life. Gas exchange between blood and air is highly dependent on the integrity of the alveolar-capillary membrane. Any event leading to tissue damage may impair oxygenation and compromise survival. Thus, lung immune responses and inflammatory processes have to be tightly regulated to deal with aggression while maintaining lung structure and homeostasis compatible with respiratory function. The lungs are exposed through the pulmonary vessels to central venous blood conveying systemic DAMPs from injured tissues. Thus, the lungs are highly susceptible to developing innate responses leading to acute lung injury in response to systemic inflammation encountered in critical illness. Accumulating evidence in the literature suggests that inflammasome-dependent excessive inflammation is involved in the pathogenesis of acute lung injury (158, 159).
It has been shown that trauma-related acute respiratory conditions lead to systemic and local NLRP3 inflammasome activation. Dolinay et al. report the increase of caspase 1, IL-1β, and IL-18 mRNA and subsequent IL-18 and caspase 1 protein levels in peripheral blood of patients with trauma-related ARDS (160). Other teams report NLRP3 inflammasome expression and activation in the lungs of mice undergoing I/R acute lung injury (20, 95, 159) (Figure 3). Various conditions encountered in trauma, such as HS, lung contusion (150, 151), burns (161), ventilator-induced lung injury (VILI), or transfusion-related acute lung injury (TRALI), have been shown to activate the NLRP3 inflammasome in the lung. Animal studies report NLRP3 inflammasome activation in lung endothelial cells and alveolar macrophages (AMs) after HS, resulting in the amplification of local inflammation and IL-1β secretion (20, 94, 95). Consistently, inhibition of the NLRP3 inflammasome attenuates acute lung injury, attested by the decrease in histopathologic damages, reduction in myeloperoxidase activity and inflammatory cytokines in lung tissue (161) (Figure 3).
Inflammasomes have also been implicated in the development of VILI. VILI is caused by lung overinflation during mechanical ventilation (MV) responsible for baro- and volo-trauma that induce overwhelming inflammation (162) (Figure 3). Recent studies demonstrate that sterile inflammation in response to MV is NLRP3-dependent. In mice, MV increases inflammasome gene expression in lung tissue and AMs (160, 163–165). Concomitantly, MV enhances IL-Iβ and L-18 protein levels in the lung and bronchoalveolar lavage. Genetic deletion of Nlrp3, Caspase-1, or Il-18 is associated with reduced MV-induced lung injury, assessed by alveolar neutrophil infiltration, alveolar-interstitial edema, BAL fluid protein content, and pulmonary cell apoptosis (160). Likewise, IL-18 or IL-1β neutralizing antibodies significantly reduce MV-induced inflammatory lung injury. The authors show that AMs are the main cell type involved in IL-1β and IL-18 production (160). Though the exact mechanisms remain to be completely elucidated, it is hypothesized that biomechanical cell trauma related to MV may lead to the release of intracellular DAMPs inducing inflammasome activation (163). Indeed, Wu et al. show that NLRP3 inflammasomes are activated in mouse AMs exposed to cyclic stretch in vitro. They report that mitochondrial generation of ROS, together with uric acid release, are responsible for stretch-induced NLRP3 inflammasome activation (164). Consistently, Kuipers et al. find increased uric acid levels in BAL fluid of ventilated mice (163). Finally, patients suffering from ARDS also show increased IL-18 concentration in the serum, whose levels were correlated with lactate concentration, APACHE II score, and mortality (160).
Last, inflammasomes may play a role in the pathogenesis of TRALI (Figure 3). Patients with severe trauma often require blood transfusions. TRALI is a rare but severe complication occurring within 6 h of blood transfusion (166). The pathogenesis of TRALI remains unclear, but might arise from the injury of the pulmonary microvasculature in a “two-hit” model (167). The “first hit” or priming step is an initial injury to the pulmonary endothelium that promotes endothelial cell activation. Mechanisms of this initial pulmonary endothelial injury may be as diverse as MV, direct traumatic injury, and/or sepsis. Endothelial activation leads to adherence, activation, and sequestration of neutrophils in the pulmonary capillary beds. The “second hit” is mediated by the blood transfusion itself. Indeed, blood storage is associated with a varying degree of hemolysis, which releases DAMPs into the stored blood units. The second exposure of the primed lung circulation to DAMPs is thought to further activate neutrophils, triggering the intense pro-inflammatory response that leads to acute lung injury (167). Some authors propose that the NLRP3 inflammasome plays a role in this inflammatory process, linking the two-hit pathogenesis of TRALI with the two-step activation of inflammasomes (167). Indeed, the priming step of TRALI may coincide with the expression of NLRP3 inflammasome in the different types of immune and endothelial cells in the lung, while the second step involving DAMPs such as heme or extracellular ATP, may trigger inflammasome activation and subsequent inflammation (168). However, this hypothesis remains to be confirmed (167).
Impairment of Inflammasome Functions as a Component of Trauma-Induced Immunosuppression
After severe injury, an anti-inflammatory response named compensatory anti-inflammatory response syndrome (CARS), occurs concomitantly to the pro-inflammatory response. When excessive or persistent, CARS leads to the severe systemic anti-inflammatory response syndrome, which promotes immunosuppression, secondary infections, and late or persisting organ dysfunctions (12, 169). The deactivation of monocytes/macrophages is an important component of immunosuppression following trauma (157–160). Interestingly, inflammasomes are major effectors of monocyte/macrophage immune functions. Although no direct proof exists, two studies suggest that inflammasome function impairment in immune cells may be involved in post-trauma immunosuppression.
Relja et al. show that NLRP1 gene expression after LPS stimulation is reduced in monocytes isolated from trauma patients compared to healthy volunteers (18). The decrease in mRNA levels of NLRP1 exists upon admission to the emergency department and persists over 10 days of immune monitoring. Although NLRP1 protein levels are not assessed in this study, the release of IL-1β from monocytes of trauma patients decreases but is restored with NLRP1 transfection. The authors hypothesize that restoring NLRP1 activity may improve the immune response to PAMPs/DAMPs in this context. However, the role of the other critical inflammasomes such as NLRP3 is not concomitantly assessed (18). In a non-trauma study that enrolled 51 patients who had undergone cardiopulmonary resuscitation after cardiac arrest, the levels of AIM2 gene expression and activity are downregulated in monocytes of resuscitated patients compared to control patients (170). Conversely, NLRP3, ASC, and IL-1β are upregulated at 12, 24, and 48 h following cardiac arrest. Interestingly, a time-dependent decrease in monocyte NLRP1, NLRP3, ASC, and IL-1β mRNA expression levels is observed in patients who die during the first 30 days after CPR, whereas survivors have stable expression of these transcripts over time. Non-survivors at 30 days have significantly lower mRNA levels of NLRP3 and CASP1 48 h after return of spontaneous circulation following resuscitation, with the same trend observed for NLRP1. The authors also find a reduced ability to release IL-1β in whole blood and monocytes isolated from patients after CPR (170). The impaired systemic IL-1β response of leukocytes from trauma patients was already observed prior to the discovery of inflammasomes although they were yet to be supported by a physiological explanation. Indeed, a decrease in IL-1β protein levels in a group of 14 trauma patients compared to healthy controls was observed until day 5 post-trauma (171). Likewise, other studies on the early inflammatory response in multiple-trauma patients found that blood IL-1β levels in post-trauma patients weakly increased compared to other pro-inflammatory cytokines like tumor necrosis factor α or IL-6 (172). At the time, it was therefore concluded that IL-1β was not a good candidate in the search of potential early predictive markers for systemic inflammatory response after trauma (172).
The impairment of inflammasome activation found in trauma patients is consistent with the data observed in the post-septic immunosuppression syndrome. Alterations in NLRP1 mRNA expression are detectable in human monocytes isolated from patients with septic shock and seem associated with monocyte deactivation. Monocytes from patients in the early phase of septic shock show alterations of caspase-1 gene expression. Also, relative mRNA copy numbers for ASC, caspase-1, and NLRP1 are significantly lower. Finally, NLRP1 mRNA levels are linked to survival in patients with sepsis and correlated with SAPS II scores. In accordance with the observations made in trauma patients, these data suggest that impairment of inflammasome functions in immune cells occurs during the earliest stages of the illness and are involved in monocyte deactivation (173).
The dampening of inflammasome signaling in circulating immune cells is the opposite of what appears to happen in tissues and organs. This is similar to discrepancies reported in the cytokine profiles between blood and injured organs in post-injury patients. Most studies show that IL-1β level increases in damaged tissue while remaining stable or decreasing in blood (156). This illustrates the challenging nature of dynamic host response monitoring, beginning with the selection of the best clinical samples in which to assess the immune response. Whole blood samples are readily and repeatedly accessible and allow characterization of circulating immune cell populations and activation through increasingly clinically available flow cytometry. However, due to the compartmentalization of inflammation in severe injury (156, 174), the information obtained from whole blood may not be extrapolated to injured tissues/organs.
Whether the impairment of inflammasome signaling in monocytes of severely injured patients plays a role in the post-trauma immunosuppression phenotype or only represents a biomarker for severity or immune impairment is uncertain. Data focusing on the involvement of inflammasomes in preserving an effective immune response against secondary infection have yet to be provided.
Some of the treatments that have been evaluated in the post-trauma immunosuppression syndrome interfere with the inflammasome pathway. Glucocorticoids (GC) have long been known as anti-inflammatory compounds. However, an increasing amount of studies suggests that GC also support a pro-inflammatory role through inflammasome priming. GC have been proposed as an important trigger for neuroinflammation through NLRP3/NLRP1 inflammasome priming in microglial cells (175). Moreover, GC induce NLRP3 expression in human macrophages in vitro, sensitizing cells to inflammasome triggers, and facilitating inflammasome-mediated release of pro-inflammatory molecules (176). Although no evidence supports this hypothesis to date, the effect of GC-induced inflammasome priming may be a potential mechanism for the anti-VAP effect of GC in severe trauma patients (177, 178), as the role of macrophages and IL-1β in host defense against respiratory infections is well known (179).
Likewise, IFNγ and GM-CSF therapies have been proposed to restore the functions of immune cells after critical injury through HLA-DR induction, especially in a selected population of patients with slow mHLA-DR recovery (180, 181). There is evidence in the literature suggesting that both IFNγ and GM-CSF modulate inflammasome signaling. The role of IFNγ in inflammasome activation has been reviewed recently and remains ambiguous (182). On one hand, it has been shown that IFNγ could upregulate NLRP3 components expression (182). On the other hand, IFNγ has been shown to indirectly inhibit NLRP3 assembly and activity via iNOS induction and NO production. Consistently, iNOS−/− mice show enhanced NLRP3 activity and higher mortality in a model of LPS-induced sepsis (183). A few studies report that GM-CSF enhances NLRP3 activity and IL-1β production by macrophages. Shaw et al. show that monocyte/macrophages from GM-CSF-neutralized mice produce less IL-1β in vivo and ex vivo, together with a decreased expression of NLRP3, pro/active IL-1β, and pro/active caspase 1. Consistently, in vitro-derived GM-CSF-differentiated macrophages express higher levels of NLRP3, caspase 1, and IL-1β compared to M-CSF-differentiated macrophages (184). Similar results have been found when studying the inflammasome activity in human monocyte-derived macrophages differentiated in the presence of GM-CSF in vitro (185, 186).
Taken together, these data may suggest some potential inflammasome-mediated mechanisms of GC, IFNγ, and GM-CSF to prevent secondary infections in post-injury immunosuppression and highlight the need for in-depth understanding of the underlying pathophysiology to guide the development of targeted, personalized therapies.
Future Perspectives and Concluding Remarks
When considering another deregulated immune response in trauma, trauma-induced coagulopathy, one can realize how deeply deciphering its pathophysiology has changed the management of trauma care (24). Trauma-induced coagulopathy is now routinely modulated as early as prehospital care through the prophylactic administration of tranexamic acid to counter early hyperfibrinolysis, is monitored throughout patient care using routine laboratory exams or point-of-care thromboelastography/metry, and is treated with a better understanding of transfusion thresholds and beneficial blood product ratios (187). These improvements in the early management of trauma care stem from decades of deciphering a complex innate immune response that goes far beyond clot formation and clearance (188).
Likewise, we have yet to fully understand the later phase of some trauma patients who develop organ failures and/or secondary infections leading to sepsis, both responsible for late trauma deaths. The emerging characterization of trauma-induced immunosuppression seems to be a major underlying mechanism of such late phase morbidity and mortality (189).
Given the roles of the inflammasomes we have reviewed above in generating the initial inflammatory burst and/or amplifying inflammatory responses, the inflammasomes are potentially key actors involved in late trauma organ failure and trauma-induced immunosuppression. Early post-trauma inhibition of inflammasome activation, by preventing pyroptosis-related DAMP release and IL-1β production in damaged tissues, may help to break the vicious cycle of injury propagation and may be useful as an early biological damage-control therapy (9, 156). Likewise, such strategies may be interesting in preventing secondary, DAMP-mediated tissue damage and remote organ injury (156). These hypotheses remain to be proven clinically as the majority the studies are based on animal or in vitro experimental models.
Inhibitors of inflammasome signaling already exist; some of which are commercially available and successfully applied to other diseases. Most of these treatments target IL-1β or IL-18: recombinant IL-1R antagonist (anakinra), IL-1β blocking antibody (canakinumab), IL-1 receptor soluble decoy (rilonacept), and IL-18-binding protein soluble receptors or blocking antibodies. Such treatments have transformed patient management, prognosis and quality of life in (auto)inflammatory diseases such as gout, type I diabetes or hereditary disorders like cryopyrin-associated autoinflammatory syndrome (CAPS) (190). The recent discovery of small molecule inhibitors selectively targeting the NLRP3 inflammasome rather than IL-1β might be promising for future research (191, 192). As pointed out by the authors, the potential benefits of these molecules would be increased therapeutic potential through the simultaneous blockade of IL-1β, IL-18, and pyroptosis, and a better safety profile, by only inhibiting NLRP3-dependant IL-1β and IL-18 production while preserving their secretion by other inflammasomes or redundant pathways against infection (191).
Indeed, the essential role of inflammasomes in antimicrobial defense raises the issue of potentially increasing the risk of secondary sepsis when interfering with inflammasomes. The prospective follow-up of CAPS patients treated with anakinra shows a median yearly rate of 7.7 adverse events per patient, among which most of the severe adverse events are infections (193). Likewise, a systematic review finds that 129 (5.1%) severe infections were reported in 2,896 patients treated with anakinra (194). Conversely to its effect in such chronic diseases, the role of anakinra in severe acute infections may be less clear due to concomitant potential beneficial effects in sepsis. Indeed, although anakinra in sepsis yields no benefit on primary outcomes in a historic phase III trial (195), a post hoc data reanalysis shows that it significantly improves in survival of a subgroup of patients with sepsis and concurrent features of hepatobiliary dysfunction/disseminated intravascular coagulation similar to macrophage activation syndrome (196). Considering that both SIRS and CARS severity are related to the intensity of the initial trigger as suggested by an increasing number of studies (181, 197, 198), early inhibition of the inflammasome activity may also prevent/decrease the intensity of the post-trauma immunosuppression by attenuating initial biological damage (199).
In their recent review on the immune response to trauma, Huber-Lang et al. provide an excellent overview of the promising strategies that are currently evaluated to treat the post-trauma syndrome (9). As debated above, the constant improvement in the comprehension of the post-trauma syndrome pathophysiology enables the development of pioneering strategies which belong to the field of precision medicine. In a break with past treatments that aimed to dampen or enhance the immune response (GC, prostaglandin, aprotinin, pro immunonutrition, etc.) (180), current approaches are directed toward the main actors of the innate immune response involved in the early “biological damage,” including DAMPs, complement, coagulation, glycocalyx, macrophages, and neutrophils. Such strategies could be considered as “preventive” rather than “curative” for the treatment of SIRS, CARS, and PICS. In addition, personalized evaluation of the immune profile combined with “boosting” immunotherapies are currently assessed for the management of the post-trauma immunosuppression. Because they might contribute to both post-trauma biological damage and immunosuppression, inflammasome represents a relevant pathway to explore in these perspectives.
This review is in favor of a trauma research framework at the interface with a relatively recent field of immunology that may lead to the characterization of a trauma-induced inflammasomopathy and perspectives for modulation with an impact on patient outcomes that remain to be determined.
Author Contributions
PB, EF, and EK conceived and designed the review. PB wrote the first draft of the manuscript. PB, EF, and EK wrote sections of the manuscript. EF conceived and drafted the figures. EK, PB, and EF provided critical revisions to the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AIM, absent in melanoma; AKI, acute kidney injury; ALR, AIM2-like receptor; ASC, apoptosis-associated speck-like protein containing CARD; ARDS, acute respiratory distress syndrome; ATP, adenosine triphosphate; BALF, bronchoalveolar lavage fluid; CARD, caspase recruitment domain; CARS, compensatory anti-inflammatory response syndrome; CIRP, cold-inducible RNA-binding protein; CLR, C-type lectin receptor; CNS, central nervous system; DAMP, damage-associated molecular pattern; DNA, deoxiribonucleic acid; HMGB1, high mobility group box 1; HS, hemorrhagic shock; IFN, interferon; IL, interleukine; I/R, ischemia/reperfusion; LPS, lipopolysaccharide; MODS, multiorgan dysfunction syndrome; MV, mechanical ventilation; NFκB, nuclear factor-kappa B; NLR, nod-like receptor; NLRC, nucleotide-binding oligomerization domain, Leucine rich repeat and caspase domain containing; NLRP, nucleotide-binding oligomerization domain, Leucine rich repeat and pyrin domain containing; NOD, nucleotide oligomerization binding domain; PAMP, pathogen-associated molecular pattern; PRR, pattern recognition receptor; PYD, pyrin domain; RAGE, receptor for advanced glycation endproducts; RLR, RIG-I-like receptor; RNA, ribonucleic acid; ROS, reactive oxygen species; SARS, systemic anti-inflammatory response syndrome; SCI, spinal cord injury; SIRS, systemic inflammatory response syndrome; TBI, traumatic brain injury; TLR, toll-like receptor; TNF, tumor necrosis factor; TRALI, transfusion-related acute lung injury; VILI, ventilator-induced lung injury.
References
1. Haagsma JA, Graetz N, Bolliger I, Naghavi M, Higashi H, Mullany EC, et al. The global burden of injury: incidence, mortality, disability-adjusted life years and time trends from the Global Burden of Disease study 2013. Inj Prev (2016) 22:3–18. doi:10.1136/injuryprev-2015-041616
2. Lashoher A, Schneider EB, Juillard C, Stevens K, Colantuoni E, Berry WR, et al. Implementation of the World Health Organization Trauma Care Checklist Program in 11 centers across multiple economic strata: effect on care process measures. World J Surg (2017) 41:954–62. doi:10.1007/s00268-016-3759-8
3. Gunst M, Ghaemmaghami V, Gruszecki A, Urban J, Frankel H, Shafi S. Changing epidemiology of trauma deaths leads to a bimodal distribution. Proc (Bayl Univ Med Cent) (2010) 23:349–54. doi:10.1080/08998280.2010.11928649
4. Abbasi H, Bolandparvaz S, Yadollahi M, Anvar M, Farahgol Z. Time distribution of injury-related in-hospital mortality in a trauma referral center in South of Iran (2010-2015). Medicine (Baltimore) (2017) 96:e6871. doi:10.1097/MD.0000000000006871
5. Sobrino J, Shafi S. Timing and causes of death after injuries. Proc (Bayl Univ Med Cent) (2013) 26:120–3. doi:10.1080/08998280.2013.11928934
6. Lenz A, Franklin GA, Cheadle WG. Systemic inflammation after trauma. Injury (2007) 38:1336–45. doi:10.1016/j.injury.2007.10.003
7. Hirsiger S, Simmen H-P, Werner CML, Wanner GA, Rittirsch D. Danger signals activating the immune response after trauma. Mediators Inflamm (2012) 2012:315941. doi:10.1155/2012/315941
8. Zedler S, Faist E. The impact of endogenous triggers on trauma-associated inflammation. Curr Opin Crit Care (2006) 12:595–601. doi:10.1097/MCC.0b013e3280106806
9. Huber-Lang M, Lambris JD, Ward PA. Innate immune responses to trauma. Nat Immunol (2018) 19:327–41. doi:10.1038/s41590-018-0064-8
10. Manson J, Thiemermann C, Brohi K. Trauma alarmins as activators of damage-induced inflammation. Br J Surg (2012) 99(Suppl 1):12–20. doi:10.1002/bjs.7717
11. Lord JM, Midwinter MJ, Chen Y-F, Belli A, Brohi K, Kovacs EJ, et al. The systemic immune response to trauma: an overview of pathophysiology and treatment. Lancet (2014) 384:1455–65. doi:10.1016/S0140-6736(14)60687-5
12. Sauaia A, Moore FA, Moore EE. Postinjury inflammation and organ dysfunction. Crit Care Clin (2017) 33:167–91. doi:10.1016/j.ccc.2016.08.006
13. Marik PE, Flemmer M. The immune response to surgery and trauma: implications for treatment. J Trauma Acute Care Surg (2012) 73:801–8. doi:10.1097/TA.0b013e318265cf87
14. Ojcius D, Saïd-Sadier N. Alarmins, inflammasomes and immunity. Biomed J (2012) 35:437. doi:10.4103/2319-4170.104408
15. Minutoli L, Puzzolo D, Rinaldi M, Irrera N, Marini H, Arcoraci V, et al. ROS-mediated NLRP3 inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid Med Cell Longev (2016) 2016:2183026. doi:10.1155/2016/2183026
16. McKee CA, Lukens JR. Emerging roles for the immune system in traumatic brain injury. Front Immunol (2016) 7:556. doi:10.3389/fimmu.2016.00556
17. Han S, Mallampalli RK. The acute respiratory distress syndrome: from mechanism to translation. J Immunol (2015) 194:855–60. doi:10.4049/jimmunol.1402513
18. Relja B, Horstmann JP, Kontradowitz K, Jurida K, Schaible A, Neunaber C, et al. Nlrp1 inflammasome is downregulated in trauma patients. J Mol Med (2015) 93:1391–400. doi:10.1007/s00109-015-1320-0
19. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi:10.1016/S1097-2765(02)00599-3
20. Xiang M, Shi X, Li Y, Xu J, Yin L, Xiao G, et al. Hemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cells. J Immunol (2011) 187:4809–17. doi:10.4049/jimmunol.1102093
21. Nagyőszi P, Nyúl-Tóth Á, Fazakas C, Wilhelm I, Kozma M, Molnár J, et al. Regulation of NOD-like receptors and inflammasome activation in cerebral endothelial cells. J Neurochem (2015) 135:551–64. doi:10.1111/jnc.13197
22. Santana PT, Martel J, Lai H-C, Perfettini J-L, Kanellopoulos JM, Young JD, et al. Is the inflammasome relevant for epithelial cell function? Microbes Infect (2016) 18:93–101. doi:10.1016/j.micinf.2015.10.007
23. Sellin ME, Maslowski KM, Maloy KJ, Hardt W-D. Inflammasomes of the intestinal epithelium. Trends Immunol (2015) 36:442–50. doi:10.1016/j.it.2015.06.002
24. Lamkanfi M, Vande Walle L, Kanneganti T-D. Deregulated inflammasome signaling in disease. Immunol Rev (2011) 243:163–73. doi:10.1111/j.1600-065X.2011.01042.x
25. Horvath GL, Schrum JE, De Nardo CM, Latz E. Intracellular sensing of microbes and danger signals by the inflammasomes. Immunol Rev (2011) 243:119–35. doi:10.1111/j.1600-065X.2011.01050.x
26. Maltez VI, Miao EA. NAIP inflammasomes give the NOD to bacterial ligands. Trends Immunol (2014) 35:503–4. doi:10.1016/j.it.2014.10.002
27. Guo H, Callaway JB, Ting JP-Y. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21:677–87. doi:10.1038/nm.3893
28. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi:10.1038/nri.2016.58
29. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi:10.1038/nri3452
30. Sharma D, Kanneganti T-D. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol (2016) 213:617–29. doi:10.1083/jcb.201602089
31. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci (2016) 41:1012–21. doi:10.1016/j.tibs.2016.09.002
32. Chavarria-Smith J, Vance RE. The NLRP1 inflammasomes. Immunol Rev (2015) 265:22–34. doi:10.1111/imr.12283
33. Proell M, Riedl SJ, Fritz JH, Rojas AM, Schwarzenbacher R. The nod-like receptor (NLR) family: a tale of similarities and differences. PLoS One (2008) 3:e2119. doi:10.1371/journal.pone.0002119
34. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458:514–8. doi:10.1038/nature07725
35. Grandjean T, Boucher A, Thepaut M, Monlezun L, Guery B, Faudry E, et al. The human NAIP-NLRC4-inflammasome senses the Pseudomonas aeruginosa T3SS inner-rod protein. Int Immunol (2017) 29:377–84. doi:10.1093/intimm/dxx047
36. Gombault A, Baron L, Couillin I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol (2013) 3:414. doi:10.3389/fimmu.2012.00414
37. Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ (2007) 14:1583–9. doi:10.1038/sj.cdd.4402195
38. Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol (2010) 10:210–5. doi:10.1038/nri2725
39. Gasse P, Riteau N, Charron S, Girre S, Fick L, Pétrilli V, et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med (2009) 179:903–13. doi:10.1164/rccm.200808-1274OC
40. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36(3):401–14. doi:10.1016/j.immuni.2012.01.009
41. Yang W-L, Sharma A, Wang Z, Li Z, Fan J, Wang P. Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Sci Rep (2016) 6:26571. doi:10.1038/srep26571
42. Dutra FF, Bozza MT. Heme on innate immunity and inflammation. Front Pharmacol (2014) 5:115. doi:10.3389/fphar.2014.00115
43. Fukata M, Vamadevan AS, Abreu MT. Toll-like receptors (TLRs) and nod-like receptors (NLRs) in inflammatory disorders. Semin Immunol (2009) 21:242–53. doi:10.1016/j.smim.2009.06.005
44. Botos I, Segal DM, Davies DR. The structural biology of toll-like receptors. Structure (2011) 19:447–59. doi:10.1016/j.str.2011.02.004
45. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol (2014) 5:1–8. doi:10.3389/fimmu.2014.00461
46. Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458:509–13. doi:10.1038/nature07710
47. Patel MN, Carroll RG, Galván-Peña S, Mills EL, Olden R, Triantafilou M, et al. Inflammasome priming in sterile inflammatory disease. Trends Mol Med (2017) 23:165–80. doi:10.1016/j.molmed.2016.12.007
48. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol (2009) 7:99–109. doi:10.1038/nrmicro2070
49. Vande Walle L, Lamkanfi M. Inflammasomes: caspase-1-activating platforms with critical roles in host defense. Front Microbiol (2011) 2:3. doi:10.3389/fmicb.2011.00003
50. Vladimer GI, Marty-Roix R, Ghosh S, Weng D, Lien E. Inflammasomes and host defenses against bacterial infections. Curr Opin Microbiol (2013) 16:23–31. doi:10.1016/j.mib.2012.11.008
51. Artlett CM. Inflammasomes in wound healing and fibrosis. J Pathol (2013) 229:157–67. doi:10.1002/path.4116
52. de Torre-Minguela C, Mesa Del Castillo P, Pelegrín P. The NLRP3 and Pyrin inflammasomes: implications in the pathophysiology of autoinflammatory diseases. Front Immunol (2017) 8:43. doi:10.3389/fimmu.2017.00043
53. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol (1994) 12:991–1045. doi:10.1146/annurev.iy.12.040194.005015
54. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J Exp Med (2011) 208:2581–90. doi:10.1084/jem.20111354
55. Ma KC, Schenck EJ, Pabon MA, Choi AMK. The role of danger signals in the pathogenesis and perpetuation of critical illness. Am J Respir Crit Care Med (2017) 197(3):300–9. doi:10.1164/rccm.201612-2460PP
56. Schaefer L. Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem (2014) 289:35237–45. doi:10.1074/jbc.R114.619304
57. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol (2007) 81:1–5. doi:10.1189/jlb.0306164
58. Harris HE, Raucci A. Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep (2006) 7:774–8. doi:10.1038/sj.embor.7400759
59. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol (2008) 8:279–89. doi:10.1038/nri2215
60. Silk E, Zhao H, Weng H, Ma D. The role of extracellular histone in organ injury. Cell Death Dis (2017) 8:e2812. doi:10.1038/cddis.2017.52
61. Gögenur M, Burcharth J, Gögenur I. The role of total cell-free DNA in predicting outcomes among trauma patients in the intensive care unit: a systematic review. Crit Care (2017) 21(1):14. doi:10.1186/s13054-016-1578-9
62. Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A (2009) 106:20388–93. doi:10.1073/pnas.0908698106
63. Albalawi F, Lu W, Beckel JM, Lim JC, McCaughey SA, Mitchell CH. The P2X7 receptor primes IL-1β and the NLRP3 inflammasome in astrocytes exposed to mechanical strain. Front Cell Neurosci (2017) 11:227. doi:10.3389/fncel.2017.00227
64. Starzl R, Wolfram D, Zamora R, Jefferson B, Barclay D, Ho C, et al. Cardiac arrest disrupts caspase-1 and patterns of inflammatory mediators differently in skin and muscle following localized tissue injury in rats: insights from data-driven modeling. Front Immunol (2015) 6:587. doi:10.3389/fimmu.2015.00587
65. Osuka A, Ogura H, Ueyama M, Shimazu T, Lederer JA. Immune response to traumatic injury: harmony and discordance of immune system homeostasis. Acute Med Surg (2014) 1:63–9. doi:10.1002/ams2.17
66. Ciesla DJ, Moore EE, Johnson JL, Burch JM, Cothren CC, Sauaia A. A 12-year prospective study of postinjury multiple organ failure: has anything changed? Arch Surg (2005) 140:432–8; discussion 438–40. doi:10.1001/archsurg.140.5.432
67. Tsukamoto T, Pape H-C. Animal models for trauma research: what are the options? Shock (2009) 31:3–10. doi:10.1097/SHK.0b013e31817fdabf
68. Deitch EA. Animal models of sepsis and shock: a review and lessons learned. Shock (1998) 9:1–11. doi:10.1097/00024382-199801000-00001
69. Liao Y, Liu P, Guo F, Zhang Z-Y, Zhang Z. Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLoS One (2013) 8:e68963. doi:10.1371/journal.pone.0068963
70. Schwulst SJ, Trahanas DM, Saber R, Perlman H. Traumatic brain injury-induced alterations in peripheral immunity. J Trauma Acute Care Surg (2013) 75:780–8. doi:10.1097/TA.0b013e318299616a
71. Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol (2014) 15(8):738–48. doi:10.1038/ni.2919
72. Maslanik T, Mahaffey L, Tannura K, Beninson L, Greenwood BN, Fleshner M. The inflammasome and danger associated molecular patterns (DAMPs) are implicated in cytokine and chemokine responses following stressor exposure. Brain Behav Immun (2013) 28:54–62. doi:10.1016/j.bbi.2012.10.014
73. Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity (2013) 39:1003–18. doi:10.1016/j.immuni.2013.11.010
74. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol (2009) 27:519–50. doi:10.1146/annurev.immunol.021908.132612
75. Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol (2010) 10:89–102. doi:10.1038/nri2691
76. Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol (2013) 4:289. doi:10.3389/fimmu.2013.00289
77. Vande Walle L, Kanneganti T-D, Lamkanfi M. HMGB1 release by inflammasomes. Virulence (2014) 2:162–5. doi:10.4161/viru.2.2.15480
78. Peltz ED, Moore EE, Eckels PC, Damle SS, Tsuruta Y, Johnson JL, et al. HMGB1 is markedly elevated within 6 hours of mechanical trauma in humans. Shock (2009) 32:17–22. doi:10.1097/SHK.0b013e3181997173
79. Parker TM, Nguyen AH, Rabang JR, Patil A-A, Agrawal DK. The danger zone: systematic review of the role of HMGB1 danger signalling in traumatic brain injury. Brain Inj (2016) 31:2–8. doi:10.1080/02699052.2016.1217045
80. Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A. HMGB1: endogenous danger signaling. Mol Med (2008) 14:476–84. doi:10.2119/2008-00034.Klune
81. Venereau E, Schiraldi M, Uguccioni M, Bianchi ME. HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol Immunol (2012) 55(1):76–82. doi:10.1016/j.molimm.2012.10.037
82. Ruan X, Darwiche SS, Cai C, Scott MJ, Pape H-C, Billiar TR. Anti-HMGB1 monoclonal antibody ameliorates immunosuppression after peripheral tissue trauma: attenuated T-lymphocyte response and increased splenic CD11b (+) Gr-1 (+) myeloid-derived suppressor cells require HMGB1. Mediators Inflamm (2015) 2015:458626. doi:10.1155/2015/458626
83. Wang X-W, Karki A, Zhao X-J, Xiang X-Y, Lu Z-Q. High plasma levels of high mobility group box 1 is associated with the risk of sepsis in severe blunt chest trauma patients: a prospective cohort study. J Cardiothorac Surg (2014) 9:874–7. doi:10.1186/s13019-014-0133-5
84. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci (2017) 42:245–54. doi:10.1016/j.tibs.2016.10.004
85. Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol (2012) 28:137–61. doi:10.1146/annurev-cellbio-101011-155745
86. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell (2014) 156:1193–206. doi:10.1016/j.cell.2014.02.008
87. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CCM, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science (2010) 330:362–6. doi:10.1126/science.1195491
88. Hazeldine J, Hampson P, Lord JM. The impact of trauma on neutrophil function. Injury (2014) 45:1824–33. doi:10.1016/j.injury.2014.06.021
89. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13:159–75. doi:10.1038/nri3399
90. Kruger P, Saffarzadeh M, Weber ANR, Rieber N, Radsak M, von Bernuth H, et al. Neutrophils: between host defence, immune modulation, and tissue injury. PLoS Pathog (2015) 11:e1004651. doi:10.1371/journal.ppat.1004651
91. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol (2007) 7:803–15. doi:10.1038/nri2171
92. Greven J, Pfeifer R, Zhi Q, Pape HC. Update on the role of endothelial cells in trauma. Eur J Trauma Emerg Surg (2017) 43:1–11. doi:10.1007/s00068-017-0812-8
93. Li Y, Yang X, He Y, Wang W, Zhang J, Zhang W, et al. Negative regulation of NLRP3 inflammasome by SIRT1 in vascular endothelial cells. Immunobiology (2017) 222:552–61. doi:10.1016/j.imbio.2016.11.002
94. Yang J, Zhao Y, Zhang P, Li Y, Yang Y, Yang Y, et al. Hemorrhagic shock primes for lung vascular endothelial cell pyroptosis: role in pulmonary inflammation following LPS. Cell Death Dis (2016) 7:e2363. doi:10.1038/cddis.2016.274
95. Xu P, Wen Z, Shi X, Li Y, Fan L, Xiang M, et al. Hemorrhagic shock augments Nlrp3 inflammasome activation in the lung through impaired pyrin induction. J Immunol (2013) 190:5247–55. doi:10.4049/jimmunol.1203182
96. Chatterjee M, Geisler T. Inflammatory contribution of platelets revisited: new players in the arena of inflammation. Semin Thromb Hemost (2016) 42:205–14. doi:10.1055/s-0035-1570081
97. Hottz ED, Monteiro APT, Bozza FA, Bozza PT. Inflammasome in platelets: allying coagulation and inflammation in infectious and sterile diseases? Mediators Inflamm (2015) 2015:435783. doi:10.1155/2015/435783
98. Jenne CN, Urrutia R, Kubes P. Platelets: bridging hemostasis, inflammation, and immunity. Int J Lab Hematol (2013) 35:254–61. doi:10.1111/ijlh.12084
99. Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, et al. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell (2005) 122:379–91. doi:10.1016/j.cell.2005.06.015
100. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol (2012) 298:229–317. doi:10.1016/B978-0-12-394309-5.00006-7
101. Hottz ED, Lopes JF, Freitas C, Valls-de-Souza R, Oliveira MF, Bozza MT, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood (2013) 122:3405–14. doi:10.1182/blood-2013-05-504449
102. Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, et al. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol (2001) 154:485–90. doi:10.1083/jcb.200105058
103. Rothmeier AS, Marchese P, Petrich BG, Furlan-Freguia C, Ginsberg MH, Ruggeri ZM, et al. Caspase-1-mediated pathway promotes generation of thromboinflammatory microparticles. J Clin Invest (2015) 125:1471–84. doi:10.1172/JCI79329
104. Eltzschig HK, Eckle T. Ischemia and reperfusion – from mechanism to translation. Nat Med (2011) 17:1391–401. doi:10.1038/nm.2507
105. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med (2007) 357:1121–35. doi:10.1056/NEJMra071667
106. Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation (2011) 123:594–604. doi:10.1161/CIRCULATIONAHA.110.982777
107. Qiu Z, Lei S, Zhao B, Wu Y, Su W, Liu M, et al. NLRP3 inflammasome activation-mediated pyroptosis aggravates myocardial ischemia/reperfusion injury in diabetic rats. Oxid Med Cell Longev (2017) 2017:9743280. doi:10.1155/2017/9743280
108. Liu Y, Lian K, Zhang L, Wang R, Yi F, Gao C, et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res Cardiol (2014) 109:415. doi:10.1007/s00395-014-0415-z
109. Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc Natl Acad Sci U S A (2001) 98:2871–6. doi:10.1073/pnas.041611398
110. Marchetti C, Chojnacki J, Toldo S, Mezzaroma E, Tranchida N, Rose SW, et al. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol (2014) 63:316–22. doi:10.1097/FJC.0000000000000053
111. Dong W, Yang R, Yang J, Yang J, Ding J, Wu H, et al. Resveratrol pretreatment protects rat hearts from ischemia/reperfusion injury partly via a NALP3 inflammasome pathway. Int J Clin Exp Pathol (2015) 8:8731–41.
112. Mastrocola R, Penna C, Tullio F, Femminò S, Nigro D, Chiazza F, et al. Pharmacological inhibition of NLRP3 inflammasome attenuates myocardial ischemia/reperfusion injury by activation of RISK and mitochondrial pathways. Oxid Med Cell Longev (2016) 2016:5271251. doi:10.1155/2016/5271251
113. Venkatachalam K, Prabhu SD, Reddy VS, Boylston WH, Valente AJ, Chandrasekar B. Neutralization of interleukin-18 ameliorates ischemia/reperfusion-induced myocardial injury. J Biol Chem (2009) 284:7853–65. doi:10.1074/jbc.M808824200
114. Gu H, Xie M, Xu L, Zheng X, Yang Y, Lv X. The protective role of interleukin-18 binding protein in a murine model of cardiac ischemia/reperfusion injury. Transpl Int (2015) 28:1436–44. doi:10.1111/tri.12683
115. Anders H-J. Of inflammasomes and alarmins: IL-1β and IL-1α in kidney disease. J Am Soc Nephrol (2016) 27:2564–75. doi:10.1681/ASN.2016020177
116. Cen C, Yang W-L, Yen H-T, Nicastro JM, Coppa GF, Wang P. Deficiency of cold-inducible ribonucleic acid-binding protein reduces renal injury after ischemia-reperfusion. Surgery (2016) 160:473–83. doi:10.1016/j.surg.2016.04.014
117. Rosin DL, Okusa MD. Dangers within: DAMP responses to damage and cell death in kidney disease. J Am Soc Nephrol (2011) 22:416–25. doi:10.1681/ASN.2010040430
118. Duann P, Lianos EA, Ma J, Lin P-H. Autophagy, innate immunity and tissue repair in acute kidney injury. Int J Mol Sci (2016) 17:E662. doi:10.3390/ijms17050662
119. Yang J-R, Yao F-H, Zhang J-G, Ji Z-Y, Li K-L, Zhan J, et al. Ischemia-reperfusion induces renal tubule pyroptosis via the CHOP-caspase-11 pathway. Am J Physiol Renal Physiol (2014) 306:F75–84. doi:10.1152/ajprenal.00117.2013
120. Shigeoka AA, Mueller JL, Kambo A, Mathison JC, King AJ, Hall WF, et al. An Inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J Immunol (2010) 185:6277–85. doi:10.4049/jimmunol.1002330
121. Kim H-J, Lee DW, Ravichandran K, Keys OD, Akcay A, Nguyen Q, et al. NLRP3 inflammasome knockout mice are protected against ischemic but not cisplatin-induced acute kidney injury. J Pharmacol Exp Ther (2013) 346:465–72. doi:10.1124/jpet.113.205732
122. Furuichi K, Wada T, Iwata Y, Kokubo S, Hara A, Yamahana J, et al. Interleukin-1-dependent sequential chemokine expression and inflammatory cell infiltration in ischemia-reperfusion injury. Crit Care Med (2006) 34:2447–55. doi:10.1097/01.CCM.0000233878.36340.10
123. Wu H, Craft ML, Wang P, Wyburn KR, Chen G, Ma J, et al. IL-18 contributes to renal damage after ischemia-reperfusion. J Am Soc Nephrol (2008) 19:2331–41. doi:10.1681/ASN.2008020170
124. Bakker PJ, Butter LM, Claessen N, Teske GJD, Sutterwala FS, Florquin S, et al. A tissue-specific role for Nlrp3 in tubular epithelial repair after renal ischemia/reperfusion. Am J Pathol (2014) 184:2013–22. doi:10.1016/j.ajpath.2014.04.005
125. Huang H, Chen HW, Evankovich J, Yan W, Rosborough BR, Nace GW, et al. Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J Immunol (2013) 191:2665–79. doi:10.4049/jimmunol.1202733
126. Kim H-Y, Kim S-J, Lee S-M. Activation of NLRP3 and AIM2 inflammasomes in Kupffer cells in hepatic ischemia/reperfusion. FEBS J (2015) 282:259–70. doi:10.1111/febs.13123
127. Sadatomo A, Inoue Y, Ito H, Karasawa T, Kimura H, Watanabe S, et al. Interaction of neutrophils with macrophages promotes IL-1β maturation and contributes to hepatic ischemia-reperfusion injury. J Immunol (2017) 199(9):3306–15. doi:10.4049/jimmunol.1700717
128. Kamo N, Ke B, Ghaffari AA, Shen X-D, Busuttil RW, Cheng G, et al. ASC/caspase-1/IL-1β signaling triggers inflammatory responses by promoting HMGB1 induction in liver ischemia/reperfusion injury. Hepatology (2013) 58:351–62. doi:10.1002/hep.26320
129. Zhu P, Duan L, Chen J, Xiong A, Xu Q, Zhang H, et al. Gene silencing of NALP3 protects against liver ischemia-reperfusion injury in mice. Hum Gene Ther (2011) 22:853–64. doi:10.1089/hum.2010.145
130. Godwin A, Yang W-L, Sharma A, Khader A, Wang Z, Zhang F, et al. Blocking cold-inducible RNA-binding protein protects liver from ischemia-reperfusion injury. Shock (2015) 43:24–30. doi:10.1097/SHK.0000000000000251
131. Hinson HE, Rowell S, Schreiber M. Clinical evidence of inflammation driving secondary brain injury: a systematic review. J Trauma Acute Care Surg (2015) 78:184–91. doi:10.1097/TA.0000000000000468
132. Simon DW, McGeachy MJ, Bayır H, Clark RSB, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol (2017) 13:572. doi:10.1038/nrneurol.2017.116
133. de Rivero Vaccari JP, Dietrich WD, Keane RW. Activation and regulation of cellular inflammasomes: gaps in our knowledge for central nervous system injury. J Cereb Blood Flow Metab (2014) 34:369–75. doi:10.1038/jcbfm.2013.227
134. Mortezaee K, Khanlarkhani N, Beyer C, Zendedel A. Inflammasome: its role in traumatic brain and spinal cord injury. J Cell Physiol (2017) 233:5160–5169. doi:10.1002/jcp.26287
135. Herx LM, Rivest S, Yong VW. Central nervous system-initiated inflammation and neurotrophism in trauma: IL-1 beta is required for the production of ciliary neurotrophic factor. J Immunol (2000) 165:2232–9. doi:10.4049/jimmunol.165.4.2232
136. Gustin A, Kirchmeyer M, Koncina E, Felten P, Losciuto S, Heurtaux T, et al. NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One (2015) 10:e0130624. doi:10.1371/journal.pone.0130624
137. Silverman WR, de Rivero Vaccari JP, Locovei S, Qiu F, Carlsson SK, Scemes E, et al. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J Biol Chem (2009) 284:18143–51. doi:10.1074/jbc.M109.004804
138. Tomura S, de Rivero Vaccari JP, Keane RW, Bramlett HM, Dietrich WD. Effects of therapeutic hypothermia on inflammasome signaling after traumatic brain injury. J Cereb Blood Flow Metab (2012) 32:1939–47. doi:10.1038/jcbfm.2012.99
139. de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW. A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci (2008) 28:3404–14. doi:10.1523/JNEUROSCI.0157-08.2008
140. Satchell MA, Lai Y, Kochanek PM, Wisniewski SR, Fink EL, Siedberg NA, et al. Cytochrome c, a biomarker of apoptosis, is increased in cerebrospinal fluid from infants with inflicted brain injury from child abuse. J Cereb Blood Flow Metab (2005) 25:919–27. doi:10.1038/sj.jcbfm.9600088
141. de Rivero Vaccari JP, Brand F, Adamczak S, Lee SW, Perez-Barcena J, Wang MY, et al. Exosome-mediated inflammasome signaling after central nervous system injury. J Neurochem (2016) 136(Suppl 1):39–48. doi:10.1111/jnc.13036
142. Qu Y, Franchi L, Núñez G, Dubyak GR. Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol (2007) 179:1913–25. doi:10.4049/jimmunol.179.3.1913
143. Liu H-D, Li W, Chen Z-R, Hu Y-C, Zhang D-D, Shen W, et al. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res (2013) 38:2072–83. doi:10.1007/s11064-013-1115-z
144. Thakkar R, Wang R, Sareddy G, Wang J, Thiruvaiyaru D, Vadlamudi R, et al. NLRP3 Inflammasome activation in the brain after global cerebral ischemia and regulation by 17β-estradiol. Oxid Med Cell Longev (2016) 2016:8309031. doi:10.1155/2016/8309031
145. Wallisch JS, Simon DW, Bayır H, Bell MJ, Kochanek PM, Clark RSB. Cerebrospinal fluid NLRP3 is increased after severe traumatic brain injury in infants and children. Neurocrit Care (2017) 27:44–50. doi:10.1007/s12028-017-0378-7
146. Denes A, Coutts G, Lénárt N, Cruickshank SM, Pelegrín P, Skinner J, et al. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc Natl Acad Sci U S A (2015) 112:4050–5. doi:10.1073/pnas.1419090112
147. Nesic O, Xu GY, McAdoo D, High KW, Hulsebosch C, Perez-Pol R. IL-1 receptor antagonist prevents apoptosis and caspase-3 activation after spinal cord injury. J Neurotrauma (2001) 18:947–56. doi:10.1089/089771501750451857
148. Adamczak S, Dale G, de Rivero Vaccari JP, Bullock MR, Dietrich WD, Keane RW. Inflammasome proteins in cerebrospinal fluid of brain-injured patients as biomarkers of functional outcome: clinical article. J Neurosurg (2012) 117:1119–25. doi:10.3171/2012.9.JNS12815
149. Lin W-P, Xiong G-P, Lin Q, Chen X-W, Zhang L-Q, Shi J-X, et al. Heme oxygenase-1 promotes neuron survival through down-regulation of neuronal NLRP1 expression after spinal cord injury. J Neuroinflammation (2016) 13:52. doi:10.1186/s12974-016-0521-y
150. Ma J, Xiao W, Wang J, Wu J, Ren J, Hou J, et al. Propofol inhibits NLRP3 inflammasome and attenuates blast-induced traumatic brain injury in rats. Inflammation (2016) 39:2094–103. doi:10.1007/s10753-016-0446-8
151. Jiang W, Huang Y, He F, Liu J, Li M, Sun T, et al. Dopamine D1 receptor agonist A-68930 inhibits NLRP3 inflammasome activation, controls inflammation, and alleviates histopathology in a rat model of spinal cord injury. Spine (2016) 41:E330–4. doi:10.1097/BRS.0000000000001287
152. Jiang W, Huang Y, Han N, He F, Li M, Bian Z, et al. Quercetin suppresses NLRP3 inflammasome activation and attenuates histopathology in a rat model of spinal cord injury. Spinal Cord (2016) 54:592–6. doi:10.1038/sc.2015.227
153. de Rivero Vaccari JP, Dietrich WD, Keane RW. Therapeutics targeting the inflammasome after central nervous system injury – ScienceDirect. Transl Res (2016) 167:35–45. doi:10.1016/j.trsl.2015.05.003
154. Webster JI, Tonelli L, Sternberg EM. Neuroendocrine regulation of immunity. Annu Rev Immunol (2002) 20:125–63. doi:10.1146/annurev.immunol.20.082401.104914
155. Hazeldine J, Lord JM, Belli A. Traumatic brain injury and peripheral immune suppression: primer and prospectus. Front Neurol (2015) 6:235. doi:10.3389/fneur.2015.00235
156. Islam MN, Bradley BA, Ceredig R. Sterile post-traumatic immunosuppression. Clin Transl Immunology (2016) 5:e77. doi:10.1038/cti.2016.13
157. Kong X-D, Bai S, Chen X, Wei H-J, Jin W-N, Li M-S, et al. Alterations of natural killer cells in traumatic brain injury. Neurosci Bull (2014) 30:903–12. doi:10.1007/s12264-014-1481-9
158. Hosseinian N, Cho Y, Lockey RF, Kolliputi N. The role of the NLRP3 inflammasome in pulmonary diseases. Ther Adv Respir Dis (2015) 9:188–97. doi:10.1177/1753465815586335
159. Grailer JJ, Canning BA, Kalbitz M, Haggadone MD, Dhond RM, Andjelkovic AV, et al. Critical role for the NLRP3 inflammasome during acute lung injury. J Immunol (2014) 192:5974–83. doi:10.4049/jimmunol.1400368
160. Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med (2012) 185:1225–34. doi:10.1164/rccm.201201-0003OC
161. Han S, Cai W, Yang X, Jia Y, Zheng Z, Wang H, et al. ROS-mediated NLRP3 inflammasome activity is essential for burn-induced acute lung injury. Mediators Inflamm (2015) 2015:1–16. doi:10.1155/2015/720457
162. Santos dos CC, Slutsky AS. The contribution of biophysical lung injury to the development of biotrauma. Annu Rev Physiol (2006) 68:585–618. doi:10.1146/annurev.physiol.68.072304.113443
163. Kuipers MT, Aslami H, Janczy JR, van der Sluijs KF, Vlaar APJ, Wolthuis EK, et al. Ventilator-induced lung injury is mediated by the NLRP3 inflammasome. Anesthesiology (2012) 116:1104–15. doi:10.1097/ALN.0b013e3182518bc0
164. Wu J, Yan Z, Schwartz DE, Yu J, Malik AB, Hu G. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J Immunol (2013) 190:3590–9. doi:10.4049/jimmunol.1200860
165. Santos dos CC. The role of the inflammasome in ventilator-induced lung injury. Am J Respir Crit Care Med (2012) 185:1141–4. doi:10.1164/rccm.201204-0649ED
166. Barrett NA, Kam PCA. Transfusion-related acute lung injury: a literature review. Anaesthesia (2006) 61:777–85. doi:10.1111/j.1365-2044.2006.04742.x
167. Land WG. Transfusion-related acute lung injury: the work of DAMPs. Transfus Med Hemother (2013) 40:3–13. doi:10.1159/000345688
168. Mendonça R, Silveira AAA, Conran N. Red cell DAMPs and inflammation. Inflamm Res (2016) 65:665–78. doi:10.1007/s00011-016-0955-9
169. Vanzant EL, Lopez CM, Ozrazgat-Baslanti T, Ungaro R, Davis R, Cuenca AG, et al. Persistent inflammation, immunosuppression, and catabolism syndrome after severe blunt trauma. J Trauma Acute Care Surg (2014) 76:21–9; discussion 29–30. doi:10.1097/TA.0b013e3182ab1ab5
170. Asmussen A, Fink K, Busch H-J, Helbing T, Bourgeois N, Bode C, et al. Inflammasome and toll-like receptor signaling in human monocytes after successful cardiopulmonary resuscitation. Crit Care (2016) 20:170. doi:10.1186/s13054-016-1340-3
171. Schinkel C, Zimmer S, Kremer JP, Walz A, Rordorf-Adam C, Henckel von Donnersmarck G, et al. Comparative analysis of transcription and protein release of the inflammatory cytokines interleukin-1 beta (IL-1 beta) and interleukin-8 (IL-8) following major burn and mechanical trauma. Shock (1995) 4:241–6. doi:10.1097/00024382-199510000-00002
172. Guisasola MC, Ortiz A, Chana F, Alonso B, Vaquero J. Early inflammatory response in polytraumatized patients: cytokines and heat shock proteins. A pilot study. Orthop Traumatol Surg Res (2015) 101:607–11. doi:10.1016/j.otsr.2015.03.014
173. Fahy RJ, Exline MC, Gavrilin MA, Bhatt NY, Besecker BY, Sarkar A, et al. Inflammasome mRNA expression in human monocytes during early septic shock. Am J Respir Crit Care Med (2008) 177:983–8. doi:10.1164/rccm.200703-418OC
174. Cavaillon J-M, Annane D. Compartmentalization of the inflammatory response in sepsis and SIRS. J Endotoxin Res (2006) 12:151–70. doi:10.1177/09680519060120030301
175. Frank MG, Watkins LR, Maier SF. The permissive role of glucocorticoids in neuroinflammatory priming: mechanisms and insights. Curr Opin Endocrinol Diabetes Obes (2015) 22:300–5. doi:10.1097/MED.0000000000000168
176. Busillo JM, Azzam KM, Cidlowski JA. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J Biol Chem (2011) 286:38703–13. doi:10.1074/jbc.M111.275370
177. Roquilly A, Broquet A, Jacqueline C, Masson D, Segain JP, Braudeau C, et al. Hydrocortisone prevents immunosuppression by interleukin-10+ natural killer cells after trauma-hemorrhage. Crit Care Med (2014) 42:e752–61. doi:10.1097/CCM.0000000000000658
178. Asehnoune K, Seguin P, Allary J, Feuillet F, Lasocki S, Cook F, et al. Hydrocortisone and fludrocortisone for prevention of hospital-acquired pneumonia in patients with severe traumatic brain injury (Corti-TC): a double-blind, multicentre phase 3, randomised placebo-controlled trial. Lancet Respir Med (2014) 2:706–16. doi:10.1016/S2213-2600(14)70144-4
179. Mayer-Barber KD, Yan B. Clash of the cytokine titans: counter-regulation of interleukin-1 and type I interferon-mediated inflammatory responses. Cell Mol Immunol (2017) 14:22–35. doi:10.1038/cmi.2016.25
180. Spruijt NE, Visser T, Leenen LP. A systematic review of randomized controlled trials exploring the effect of immunomodulative interventions on infection, organ failure, and mortality in trauma patients. Crit Care (2010) 14:R150. doi:10.1186/cc9218
181. Lukaszewicz A-C, Grienay M, Resche-Rigon M, Pirracchio R, Faivre V, Boval B, et al. Monocytic HLA-DR expression in intensive care patients: interest for prognosis and secondary infection prediction*. Crit Care Med (2009) 37:2746–52. doi:10.1097/CCM.0b013e3181ab858a
182. Kopitar-Jerala N. The role of interferons in inflammation and inflammasome activation. Front Immunol (2017) 8:873. doi:10.3389/fimmu.2017.00873
183. Yang Q, Yu C, Yang Z, Wei Q, Mu K, Zhang Y, et al. Deregulated NLRP3 and NLRP1 inflammasomes and their correlations with disease activity in systemic lupus erythematosus. J Rheumatol (2014) 41:444–52. doi:10.3899/jrheum.130310
184. Shaw OM, Steiger S, Liu X, Hamilton JA, Harper JL. Brief Report: granulocyte-macrophage colony-stimulating factor drives monosodium urate monohydrate crystal-induced inflammatory macrophage differentiation and NLRP3 inflammasome up-regulation in an in vivo mouse model. Arthritis Rheumatol (2014) 66:2423–8. doi:10.1002/art.38730
185. Ahn S, Jeong D, Oh SJ, Ahn J, Lee SH, Chung DH. GM-CSF and IL-4 produced by NKT cells inversely regulate IL-1β production by macrophages. Immunol Lett (2017) 182:50–6. doi:10.1016/j.imlet.2017.01.003
186. Budai MM, Tőzsér J, Benkő S. Different dynamics of NLRP3 inflammasome-mediated IL-1β production in GM-CSF- and M-CSF-differentiated human macrophages. J Leukoc Biol (2017) 101:1335–47. doi:10.1189/jlb.3A0716-300RR
187. Simmons JW, Pittet J-F, Pierce B. Trauma-induced coagulopathy. Curr Anesthesiol Rep (2014) 4:189–99. doi:10.1007/s40140-014-0063-8
188. Cohen MJ. Translational approaches to coagulopathy after trauma: towards targeted treatment. PLoS Med (2017) 14:e1002359. doi:10.1371/journal.pmed.1002359
189. Ward NS, Casserly B, Ayala A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med (2008) 29:617–25. doi:10.1016/j.ccm.2008.06.010
190. López-Castejón G, Pelegrín P. Current status of inflammasome blockers as anti-inflammatory drugs. Expert Opin Investig Drugs (2012) 21:995–1007. doi:10.1517/13543784.2012.690032
191. Lamkanfi M, Dixit VM. A new lead to NLRP3 inhibition. J Exp Med (2017) 214:3147–9. doi:10.1084/jem.20171848
192. Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21:248–55. doi:10.1038/nm.3806
193. Kullenberg T, Löfqvist M, Leinonen M, Goldbach-Mansky R, Olivecrona H. Long-term safety profile of anakinra in patients with severe cryopyrin-associated periodic syndromes. Rheumatology (Oxford) (2016) 55:1499–506. doi:10.1093/rheumatology/kew208
194. Cabral VP, de Andrade CAF, Passos SRL, de Martins M, Hökerberg YH. Severe infection in patients with rheumatoid arthritis taking anakinra, rituximab, or abatacept: a systematic review of observational studies. Rev Bras Reumatol Engl Ed (2016) 56(6):543–50. doi:10.1016/j.rbr.2016.07.008
195. Opal SM, Fisher CJ, Dhainaut JF, Vincent JL, Brase R, Lowry SF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The interleukin-1 receptor antagonist sepsis investigator group. Crit Care Med (1997) 25:1115–24. doi:10.1097/00003246-199707000-00010
196. Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med (2016) 44:275–81. doi:10.1097/CCM.0000000000001402
197. Binkowska AM, Michalak G, Słotwiński R. Current views on the mechanisms of immune responses to trauma and infection. Cent Eur J Immunol (2015) 40:206–16. doi:10.5114/ceji.2015.52835
198. Leijte GP, Custers H, Gerretsen J, Heijne A, Roth J, Vogl T, et al. Increased Plasma levels of danger-associated molecular patterns are associated with immune suppression and postoperative infections in patients undergoing cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. Front Immunol (2018) 9:663. doi:10.3389/fimmu.2018.00663
Keywords: inflammasome, trauma, DAMP, inflammation, immunosuppression
Citation: Bortolotti P, Faure E and Kipnis E (2018) Inflammasomes in Tissue Damages and Immune Disorders After Trauma. Front. Immunol. 9:1900. doi: 10.3389/fimmu.2018.01900
Received: 12 December 2017; Accepted: 31 July 2018;
Published: 16 August 2018
Edited by:
Julien Pottecher, Hôpitaux Universitaires de Strasbourg, FranceReviewed by:
Antoine Roquilly, University of Nantes, FranceDidier Payen, Université Sorbonne Paris Cité, France
Copyright: © 2018 Bortolotti, Faure and Kipnis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric Kipnis, ZWtpcG5pc0BnbWFpbC5jb20=