 Zlatko Kopecki
Zlatko Kopecki Natalie E. Stevens
Natalie E. Stevens Heng T. Chong
Heng T. Chong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 10 August 2018
Sec. Cytokines and Soluble Mediators in Immunity
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01833
This article is part of the Research Topic Autoantibodies View all 89 articles
Atopic dermatitis (AD) is a chronic pruritic inflammatory skin disease characterized by excessive inflammation and disrupted skin barrier function. Although the etiology of AD is not completely understood, clinical and basic studies suggest increasing involvement of autoantibodies against intracellular proteins. An actin remodeling protein, Flightless I (Flii), has been shown to promote development of inflammatory mediated skin conditions and impairment of skin barrier development and function. Here, we sought to determine the effect of altering Flii expression on the development of AD and its contribution to autoimmune aspects of inflammatory skin conditions. Ovalbumin (OVA)-induced AD skin-like disease was induced in Flii heterozygous (Flii+/−), wild-type (Flii+/+), and Flii transgenic (FliiTg/Tg) mice by epicutaneous exposure to OVA for 3 weeks; each week was separated by 2-week resting period. Reduced Flii expression resulted in decreased disease severity and tissue inflammation as determined by histology, lymphocytic, and mast cell infiltrate and increased anti-inflammatory IL-10 cytokine levels and a marked IFN-γ Th1 response. In contrast, Flii over-expression lead to a Th2 skewed response characterized by increased pro-inflammatory TNF-α cytokine production, Th2 chemokine levels, and Th2 cell numbers. Sera from OVA-induced AD skin-like disease Flii+/− mice showed a decreased level of autoreactivity while sera from FliiTg/Tg mice counterparts showed an altered autoantibody profile with strong nuclear localization favoring development of a more severe disease. These findings demonstrate autoimmune responses in this model of OVA-induced AD-like skin disease and suggest that Flii is a novel target, whose manipulation could be a potential approach for the treatment of patients with AD.
Atopic dermatitis (AD) is one of the most common heterogeneous inflammatory skin diseases affecting 20% of children and 1–3% of adults worldwide (1). The disease is associated with impairments in the skin barrier and variable clinical indicators including occurrence of eczematous lesions, pruritus, and cheilitis (1). The etiology of AD is complex and is often characterized by abnormal immunological pathways that manifest in an imbalance of T-helper (Th)1 and Th2 responses (2). Typically, AD is described as having a biphasic course consisting of an acute inflammatory Th2-dominated phase associated with IgE production and a chronic phase distinguished by reappearance of Th1 responses, tissue remodeling, and dermal thickening (3). Histopathologically, AD is characterized by an inflammatory infiltrate consisting of CD4+ memory T cells, mast cells, and eosinophils and a controlled temporal-spatial expression of pro-inflammatory cytokines and chemokines driving atopic inflammation of the skin (4).
Research within the last decade has found an association between AD and autoantibody development, suggesting the contribution of autoantibodies to the pathogenesis of AD (3, 5–8). It is proposed that tissue damage induced by AD allows exposure of intracellular antigens that are normally inaccessible by antibodies to the extracellular space, where they can interact with B cells and antibodies (9). A broad spectrum of IgE targeting self-antigens have been identified in above 90% of severe AD patients and high-avidity IgG autoantibodies have been proposed as potential diagnostic markers for severe AD (5, 10–12). The fact that these autoantibodies are associated with disease severity implicates their role in both humoral and cellular immunity in AD pathogenesis (5, 12). Antinuclear antibodies have been shown to have both biological and clinical significance acting as sensors of cellular stress and inflammation associated with environmental factors (13). While the presence of autoantibodies can have a protective role as natural autoantibodies (14), the presence of nuclear autoantibodies in AD has been suggested to lead to the continual provocation of the immune system hence contributing to the severity and chronicity of disease.
Flightless I (Flii) is a highly conserved and unique member of the gelsolin family of actin remodeling proteins and a nuclear receptor activator affecting the transcriptional activity of many modulators of tissue remodeling and inflammation (15, 16). Flii has been demonstrated to regulate cytokine secretion and cellular inflammatory responses via its intracellular and extracellular effects on toll-like receptors (17–20). Flii expression increases in skin during development and in response to inflammation, injury, skin cancer development, wound healing, and skin blistering (21–23). Over-expression of Flii delays the development of an intact skin barrier in the embryonic skin of mice and impairs the recovery of the epidermal barrier post injury via its effects on tight-junction formation (22). A recent study has shown that reducing Flii levels either genetically or using Flii neutralizing antibodies decreases erythema, inflammatory cell infiltrate, and pro-inflammatory cytokine secretion in a mouse model of psoriasiform dermatitis (24). Taken together, these findings suggest a possible role for Flii in inflammatory responses mediating AD. Using an OVA-induced AD-like skin mouse model (25), this study aimed to investigate the effect of differential Flii gene expression on development of AD via characterization of inflammatory and autoimmune responses responsible for AD pathogenesis.
Mice were maintained according to the Australian Code for the Care and Use of Animals for Scientific Purposes under protocols approved by the Child Youth and Women’s Health Service Animal Ethics Committee (AEC916/06/2015). Mice with the BALB/c background and wild-type controls were obtained from inbred litters. Flii-deficient heterozygous null mice (Flii+/−) and mice carrying the complete human Flii gene on a cosmid transgene were maintained as described previously (26, 27). Heterozygous transgenic mice FliiTg/+ were made by crossing Flii+/+ with cosmid transgene Flii+/−. These transgenic mice were intercrossed to obtain animals homozygous for the transgene FliiTg/Tg which carry two copies of the mouse Flii gene and two copies of the human Flii transgene.
Atopic dermatitis was induced via epicutaneous exposure of mice to OVA as previously described (25). Briefly, 10- to 12-week-old female wild-type (Flii+/+), Flii heterozygous (Flii+/−), and Flii transgenic (FliiTg/Tg) mice (n = 8/genotype) were anesthetized using isoflurane inhalation, the skin on the back of mice was shaved and then tape stripped four times by a transparent adhesive tape (Tegaderm) to introduce a standardized skin injury. A gauze patch (1 × 1 cm2) soaked with 100 µl of 0.1% OVA (OVA group) in saline or 0.9% saline (control group) was placed on the back skin and secured with Tegaderm dressing. The experiment comprised three 1-week exposures with a 2-week interval between each exposure week (Figure 1A). Clinical images of affected skin, transepidermal water loss (TEWL), and erythema measurements were recorded at the end of the third sensitization week. The level of skin erythema was measured daily using a handheld DermaLab Unit (Cortex Technology) following manufacturer’s instructions. This instrument uses skin reflectance spectroscopy to determine the redness of inflamed skin. The instrument was blanked prior to placing a probe directly onto the inflamed AD-like lesions on dorsal skin and a reading obtained as previously described (24). Measurements of TEWL were obtained with a calibrated Vapometer evaporimeter (Delfin Technologies, Finland). The probe was allowed to equilibrate for approximately 30 s before a brief measurement period (8 s) as indicated in manufacturer’s guidelines. The Vapometer was placed directly onto the inflamed AD-like lesions on dorsal skin (using an 11-mm adaptor) and held there securely for the duration of the measurement. For both erythema and TEWL analysis, three separate measurements were taken per mouse to ensure the entire region of AD-like inflamed back skin was assessed as previously described (24). Mice were then euthanized and skin biopsies collected for histology analysis, immunohistochemistry, and mRNA extraction. Blood was collected for autoimmunity experiments.
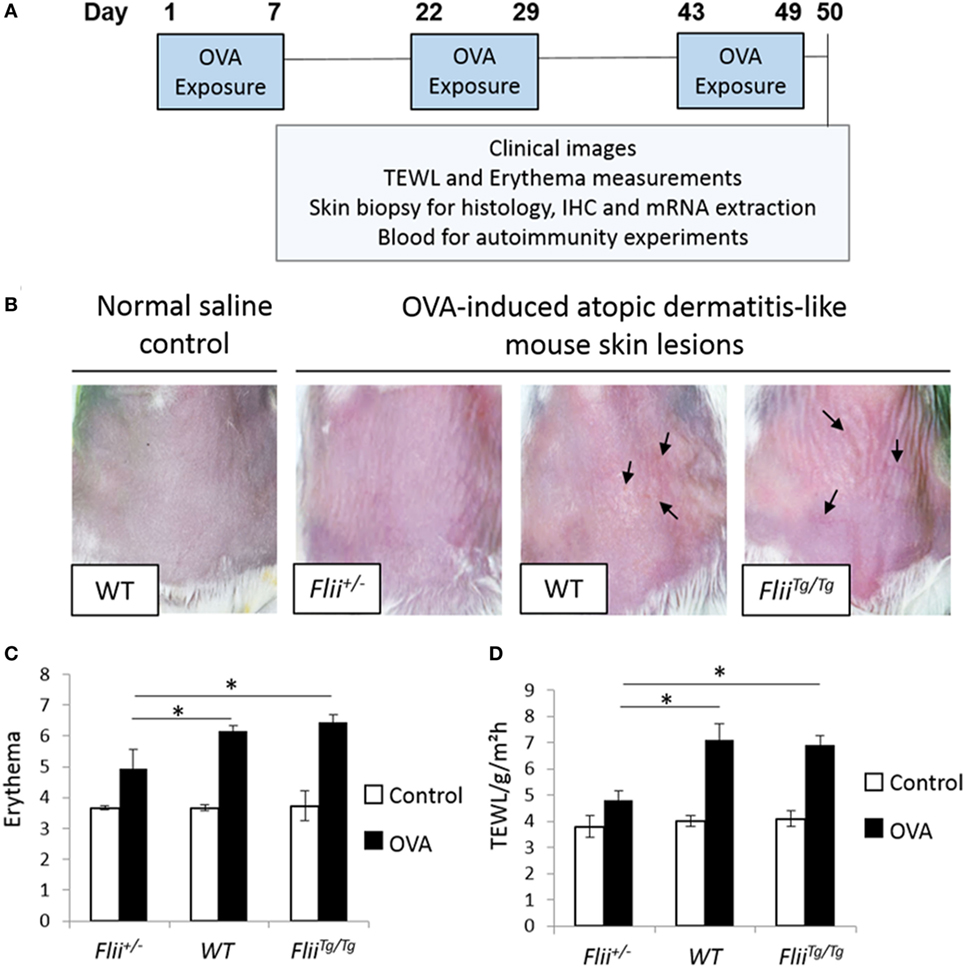
Figure 1. Reduced Flii expression leads to decreased development of OVA-induced atopic dermatitis (AD)-like lesions. (A) Ovalbumin (OVA) exposure protocol, including a total of three 1-week exposures to 100 µl 0.1% OVA or saline control on patches separated from each other by 2-week resting interval. (B) Representative clinical images of OVA-induced AD-like skin lesions in mice differential Flii expression illustrating high degree of erythema, skin thickening, and scaling (black arrowhead) in WT and FliiTg/Tg mice exposed to OVA. (C,D) OVA exposure leads to development of AD-like lesions demonstrated by increased erythema and transepidermal water loss (TEWL). Decreasing Flii expression reduces development of AD-like lesions with significantly decreased erythema and TEWL measurements compared to OVA exposed skin of WT and FliiTg/Tg animals. n = 8. Mean ± SEM. *<0.05.
Paraffin-embedded fixed tissue samples were stained with hematoxylin and eosin or toluidine blue following established protocols (28). Level of skin inflammation was assessed using skin thickness measurements, and analysis of inflammatory and mast cell numbers in lesional skin of OVA-induced AD-like skin lesions of Flii+/−, wild-type, and FliiTg/Tg mice using high magnification images and light microscopy using optimized protocols using Image Pro-Plus 5.1 program (MediaCybernetics Inc.) as previously described (24). Skin thickness included epidermal and dermal measurements of OVA-induced AD-like lesions; inflammatory cell number was assessed by counting total inflammatory cells in 10 different HPF of view in OVA-induced AD-like lesions, and mast cell analysis included counting toludine blue positive mast cells in entire sections of OVA-induced AD-like skin lesions per mm2. Immunohistochemistry was also performed on paraffin-embedded fixed AD-like skin lesions following antigen retrieval according to the manufacturer’s protocols (DAKO Corporation, Glostrup, Denmark). Following blocking in 3% normal goat serum, primary antibodies against CD4 [rat monoclonal, #14-9766-82 (Thermo Scientific, Australia) (1:200)], T-bet [rabbit polyclonal, #PA5-40573 (Thermo Scientific, Australia) (1:100)], GATA-3 [rabbit polyclonal, #ab106625 (Abcam Australia) (1:100)], and ROR-γ [rabbit monoclonal, #ab207082 (Abcam Australia) (1:3,000)] were applied and slides were incubated at 4°C overnight in a humidified chamber before application of species-specific, Alexa Flour-488 or Alexa Flour-594 secondary antibodies (Invitrogen, Australia) for 1 h at room temperature. Finally, slides were washed and mounted in Fluorescence Mounting Medium (Dako, Australia). Images were captured on Olympus microscope and CellSense Live Science Imaging Software program (Olympus, Germany) used for counting the positive cells in the AD-like lesions of Flii+/−, wild-type, and FliiTg/Tg mice. Negative controls included replacing primary antibodies with normal rabbit IgG, or normal mouse IgG. For verification of staining, non-specific binding was determined by omitting primary or secondary antibodies. All control sections had negligible immunofluorescence.
Harvested tissue was snap-frozen in liquid nitrogen and total RNA was isolated using Ultraclean Tissue and Cell RNA Isolation Kit (MoBio Laboratories, Carlsbad, CA, USA) according to the manufacturer’s protocol. Total cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA, USA) according to manufacturer’s protocol. Quantitative PCR was performed using iQ SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA, USA) in triplicate reactions. The plates were placed in a CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA). Reactions underwent 30 s at 95°C, then 40 cycles of 5 s at 95°C, 20 s at 60°C, and 10 s at 95°C before determination of melt curve between 65 and 95°C. GAPDH and CyPA were used as reference genes and inter-reaction calculator method was applied for all plates. For relative comparison, the cycle threshold value (Ct) was analyzed using the ΔΔCt method and data reported as Ct normalized to reference genes. Sequences for PCR primers are listed in Table S1 in Supplementary Material.
In order to assess the degree of autoimmunity in OVA-induced AD-like skin of Flii+/−, wild-type, and FliiTg/Tg mice, sub-confluent primary wild-type mouse keratinocytes were stained with mouse sera of AD-induced mice following established protocols (13, 29). Briefly, primary keratinocytes were isolated from murine epidermis as previously described (30), grown on glass coverslips and washed in 1× phosphate-buffered saline before paraformaldehyde (4%) fixation (10 min at room temperature). Fixed cells were subsequently permeabilized using 0.2% Triton-X-100 and 0.5% BSA in 1× phosphate-buffered saline (5 min at room temperature) before incubation with pooled murine serum (n = 3) diluted (1 in 10) in 0.5% BSA in 1× phosphate-buffered saline for 1 h. Bound murine IgG was detected with Alexa Flour 633 goat anti-mouse IgG (1 in 1,000; #A21050; Invitrogen, Mulgrave, VIC, Australia) in 1× phosphate-buffered saline for 1 h at room temperature. Following repeated 2 min washes with 1× phosphate-buffered saline, cells were stained with DAPI nucleic acid stain (Sigma-Aldrich) for 5 min at room temperature before washing and mounting for imaging. Negative controls included omitting the incubation with mouse serum. All control sections had negligible immunofluorescence. Staining pattern of autoantibodies was assessed using the Olympus microscope and CellSense Live Science Imaging Software program (Olympus, Germany).
Protein was extracted from WT murine keratinocytes using standard protein extraction protocols (31). Samples of extracted protein (20 µg) were run on 10% SDS-PAGE gels (30 min; 200V) and transferred to nitrocellulose by wet transfer (1 h; 100V). Membranes were cut into strips, blocked in 3% bovine serum albumin (Sigma) for 30 min, and probed with murine serum diluted (1 in 10) in tris-buffered saline containing 3% BSA and 0.1% Tween overnight. After washing, horseradish peroxidase-conjugated goat anti-murine immunoglobulin secondary antibody was added for a further 1 h at room temperature. Stringent washes were performed before detection of horseradish peroxidase and exposure using GeneSnap analysis program (SynGene, Frederick, MD, USA).
Statistical differences were determined using the Student’s t-test or one-way ANOVA. For data not following a normal distribution, the Mann–Whitney U test was performed. A P value of less than 0.05 was considered significant.
The repeated epicutaneous exposure of OVA (Figure 1A) on mouse skin results in an AD-like skin disease with histological and phenotypic features of human AD, including erythema, skin thickening, and a localized immune response (25). Flii homozygous (Flii−/−) mice are embryonic lethal (26); therefore, using the OVA-induced AD-like skin mouse model, the severity of OVA-induced AD-like lesions was determined in response to different Flii gene levels in Flii heterozygous (Flii+/−), normal (Flii+/+), and Flii transgenic (FliiTg/Tg) mice. All three mice genotypes developed localized inflammation which increased following each week of OVA exposure. Flii-deficient mice showed reduced levels of inflammation and scaling (Figure 1B). Spectrophotometric measurement of the redness of the OVA exposed skin showed that Flii-deficient mice had significantly less erythema (Figure 1B) than wild-type and FliiTg/Tg counterparts at day 50 of the experiment. Similarly, the degree of TEWL was also significantly reduced in Flii+/− mice and very similar to that observed in control animals (Figure 1C). Control mice administered saline only showed no evidence of AD-like inflammation macroscopically or any development of erythema or TEWL (Figures 1B–D).
A hallmark of AD is skin thickening with a marked influx of leukocyte and mast cell inflammatory infiltrate. All OVA-induced AD-like skin sections showed evidence of AD compared to control saline exposed skin including a degree of epidermal hyperplasia and edema coupled with inflammatory lymphocytic and mast cell dermal infiltrate (Figures 2A–C). Examining the skin thickness in OVA-induced AD-like skin lesions revealed that deficient Flii mice had significantly thinner skin than OVA-induced AD-like skin lesions of wild-type and FliiTg/Tg mice (Figure 2A). Assessment of lymphocytic and mast cell dermal infiltrate showed that Flii deficiency consistently resulted in significantly decreased inflammatory cell numbers (Figures 2B,C). In contrast, both normal and increased Flii gene expression resulted in thickened epidermis, increased numbers of inflammatory cells (Figure 2B) and mast cells (Figure 2C). Control mice skin exposed to saline only gauze patch showed no evidence of AD-like dermatitis features microscopically and had low levels of inflammatory infiltrate in the dermis (Figures 2A–C).
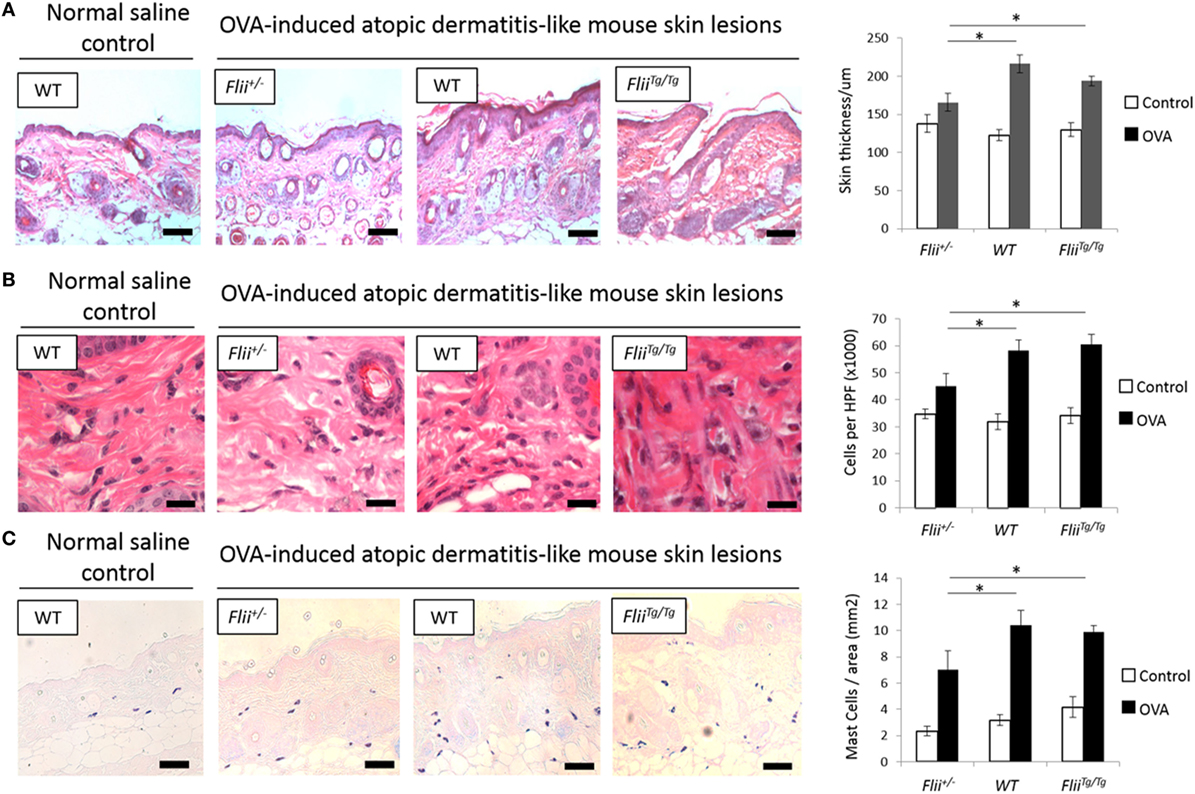
Figure 2. Mice with reduced Flii expression develop less severe OVA-induced atopic dermatitis (AD) skin-like lesions and tissue inflammation. (A) Representative images and graphical analysis of skin thickness in Flii+/−, WT, and FliiTg/Tg mice with OVA-induced AD skin-like lesions illustrating evidence of epidermal hyperplasia, edema, and lymphocytic infiltrate. Magnification 4× scale bar 200 µm. (B) Representative images and graphical analysis of lymphocytic inflammatory infiltrate in in Flii+/−, WT, and FliiTg/Tg mice with OVA-induced AD skin-like lesions illustrating decreased inflammation in Flii+/− mice skin. Magnification 1,000× scale bar 25 µm. (C) Representative images and graphical analysis of mast cells numbers in Flii+/−, WT, and FliiTg/Tg mice with OVA-induced AD skin-like lesions illustrating decreased inflammation in Flii+/− mice skin. Magnification 4× scale bar 200 µm. n = 8. Mean ± SEM. *<0.05.
OVA-induced AD-like skin lesions of Flii+/−, wild-type, and FliiTg/Tg mice were assessed for Flii and levels of cytokines and chemokines mediating the Th1 and Th2 inflammatory responses during AD pathogenesis. Flii-deficient mice showed approximately 25% decrease in Flii levels, while Flii over-expressing mice had a twofold increase in Flii levels compared to wild-type counterparts (Figure 3). Flii gene levels were found to affect number of key cytokines and chemokines responsible for development of AD-like lesions. Notably, main Flii deficiency favored a Th1 immune response and decreased inflammation with significant threefold increase in IFN-γ mRNA levels compared to FliiTg/Tg mice, decreased pro-inflammatory IL-4, and increased anti-inflammatory IL-10 mRNA levels compared to both wild-type and FliiTg/Tg mice (Figure 3). Interestingly, Flii+/− OVA-induced AD-like skin lesions also showed increased IL-5 and IL-6 mRNA levels when compared to FliiTg/Tg mice but not wild-type counterparts (Figure 3). In contrast, over-expression of Flii resulted in similar cytokine levels to wild-type counterparts, except significantly increased TNF-α mRNA expression and significantly reduced IFN-γ mRNA expression when compared to both Flii-deficient and wild-type mice, which would favor more severe AD manifestation. Cytokine mRNA levels of IL-13, IL-23, and IL-17A did not differ between OVA-induced AD-like skin lesions of three genotypes (Figure 3). Expression of CCL22 mRNA was significantly increased in OVA-induced AD-like skin lesions of FliiTg/Tg mice compared to wild-type mice, while OVA-induced AD-like skin lesions of Flii+/− mice showed elevated CCL17 chemokine mRNA expression compared to FliiTg/Tg mice (Figure 4). CXCL9 chemokine mRNA levels were found to be similar in all three genotypes (Figure 4). The unbalanced skewed ratio of Th1/Th2 response in FliiTg/Tg mice was confirmed by immunohistochemical co-staining of T cell subset markers, namely Th1 (CD4 and T-bet), Th2 (CD4 and GATA-3), and Th17 (CD4 and ROR-γ) showing increased Th2 cell numbers in dermal area of FliiTg/Tg mice (Figures 5A–D).

Figure 3. OVA-induced atopic dermatitis (AD) skin-like lesions of Flii-deficient mice exhibit increased anti-inflammatory cytokine levels. mRNA levels of Flii, pro-inflammatory, and anti-inflammatory cytokines responsible for manifestation of AD were analyzed in the OVA-induced AD skin-like lesions of Flii+/−, wild-type, and FliiTg/Tg animals. OVA-induced AD skin-like lesions of Flii-deficient mice have significantly higher IFN-γ, reduced IL-4 signaling, and significantly higher levels of anti-inflammatory IL-10 while the Flii transgenic counterparts show significantly increased TNF-α and significantly reduced IFN-γ compared to wild-type animals. n = 6. Mean ± SEM. *<0.05.
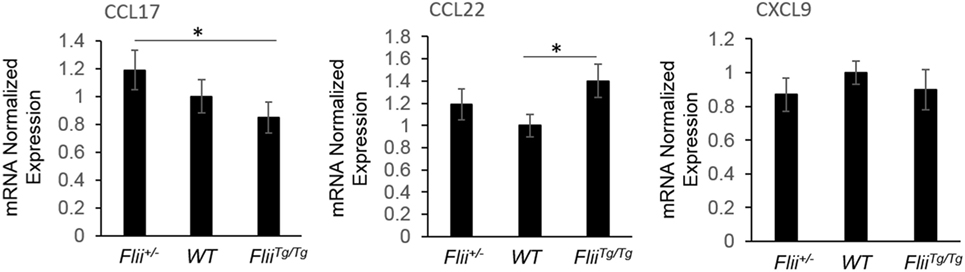
Figure 4. Flii expression affects Th2 chemokine levels in OVA-induced atopic dermatitis (AD) skin-like lesions. mRNA levels of CCL17, CCL22, and CXCL9 chemokines responsible for manifestation of AD were analyzed in the OVA-induced AD skin-like lesions of Flii+/−, wild-type, and FliiTg/Tg animals. OVA-induced AD skin-like lesions of Flii+/− mice have significantly higher levels of CCL17 compared to FliiTg/Tg counterparts, while CCL22 was significantly increased in OVA-induced AD skin-like lesions of FliiTg/Tg mice compared to wild-type controls. n = 6. Mean ± SEM. *<0.05.
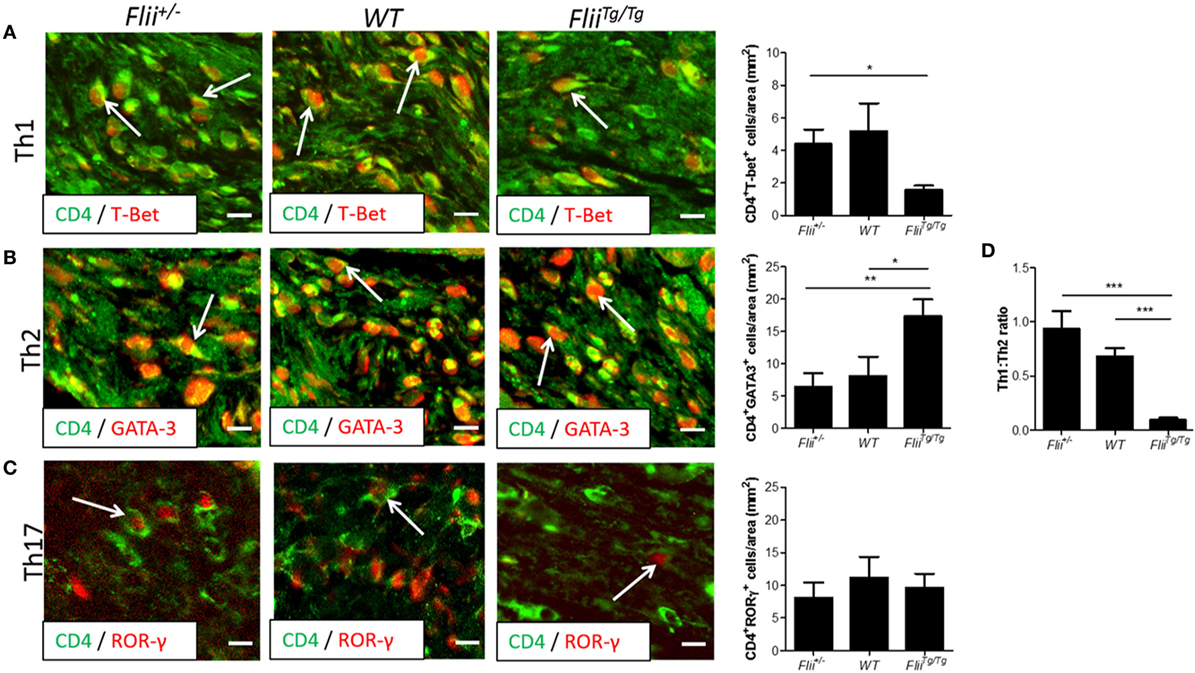
Figure 5. Mice with increased Flii expression exhibit increased Th2 cell numbers within OVA-induced atopic dermatitis (AD) skin-like lesions. (A) Representative images and graphical analysis of Th1 (CD4+T-bet+) cell numbers in AD skin-like lesions within Flii+/−, WT, and FliiTg/Tg mice. (B) Representative images and graphical analysis of Th2 (CD4+GATA-3+) cell numbers in AD skin-like lesions within Flii+/−, WT, and FliiTg/Tg mice. (C) Representative images and graphical analysis of Th17 (CD4+RORγ+) cell numbers in AD skin-like lesions within Flii+/−, WT, and FliiTg/Tg mice. (D) Ratio of Th1 to Th2 cells within AD skin-like lesions of Flii+/−, WT, and FliiTg/Tg mice. Magnification 4× scale bar 200 µm. n = 6. Mean ± SEM. *<0.05.
To compare autoantibody formation in the OVA-induced AD-like disease model between Flii-deficient, wild-type, and Flii-overexpressing mice, serum samples collected at day 50 were assessed by immunofluorescence and immunoblot analysis. Primary keratinocytes from wild-type mice were fixed and probed with pooled sera from experimental mice and staining patterns analyzed using fluorescence microscopy. Cytoplasmic and perinuclear staining within keratinocytes was apparent after incubation with sera from all three genotypes, however, only sera from OVA-induced AD-like disease FliiTg/Tg mice produced strong nuclear staining patterns (Figure 6A). To further elucidate autoreactivity in these samples, keratinocyte whole-cell lysates were separated via SDS-PAGE, transferred to nitrocellulose membrane, and probed with pooled sera (Figure 6B). Additionally, autoreactivity patterns of individual mouse sera are shown in Figure S1 in Supplementary Material. Regions of differential autoreactivity were detected at 145, 70, 60, and 43 kDa band sizes (black arrows). Immunoblot analysis of sera (Figure 6C) from individual mice found that autoreactivity was highest in wild-type mice (50%) and FliiTg/Tg mice (43%), while Flii-deficient (Flii+/−) mice showed the lowest degree of autoreactivity (25%). Positive autoreactive bands at 70 kDa were also different in FliiTg/Tg mice compared to the two other genotypes, which may be responsible for the nuclear staining pattern observed in immunofluorescence analysis. Immunoblot analysis of sera from normal non-dermatitis mice from each genotype were also analyzed; no autoreactivity was detected (Figure S2 in Supplementary Material).
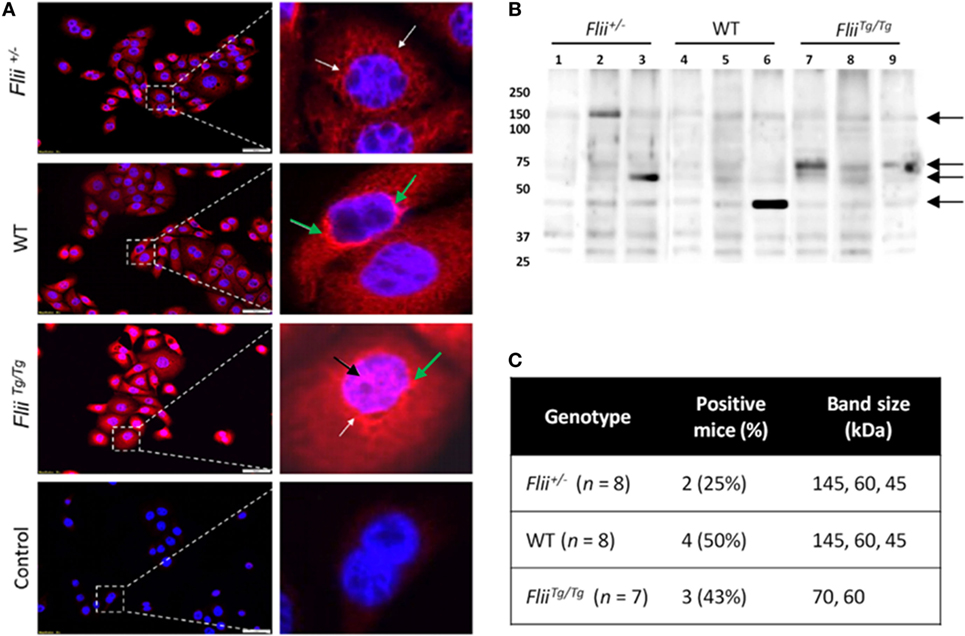
Figure 6. Over-expression of Flii produces an altered autoantibody profile in murine OVA-induced atopic dermatitis (AD) skin-like disease. (A) Representative images of IF microscopy staining patterns produced by mouse autoreactive IgG (red) and DAPI nuclear counterstaining (blue) in primary mouse keratinocytes. Magnification 20×; scale bar 50 µm. Antibodies from OVA-induced AD skin-like lesions of Flii+/− and WT mice producing a predominantly cytoplasmic (white arrow) and perinuclear (green arrow) staining patterns, while antibodies from OVA-induced AD skin-like lesions of FliiTg/Tg mice produce a strong cytoplasmic, perinuclear (green arrow), and nuclear (black arrow) staining patterns. (B) Western blot analysis of murine keratinocyte proteins probed with pooled sera from OVA-induced AD-like mice (2–3 mice per lane); black arrows represent regions of autoreactivity. (C) Autoreactivity summary of Western blot results from murine keratinocyte lysate probed with sera from individual mice.
Flii is a cytoskeletal protein with important roles in skin development, tight junction function, skin barrier establishment, and recovery post injury which is upregulated in response to tissue inflammation and wounding (22, 31). The secreted form of Flii has been shown to affect innate immune signaling pathways and modulate cell activity and cytokine secretion from fibroblasts and macrophages in vitro (17, 20). Recent studies have described the contribution of Flii over-expression in exacerbation of inflammatory conditions, including psoriasiform dermatitis (24) and an autoimmune inflammation-mediated epidermolysis bullosa acquisita (23, 32).
To determine if Flii affects AD development and severity, mice with low (Flii+/−), normal (Flii+/+), and high (FliiTg/Tg) expression of the Flii gene were exposed to repeated epicutaneous exposure to OVA and induced to form AD-like skin lesions. In contrast to our original hypothesis, Flii over-expression did not result in a more exacerbated development of erythema and AD-like skin lesions. However, reduced Flii levels in Flii-deficient mice led to significantly decreased development of AD-like skin lesions as marked by decreased erythema, TEWL, epidermal hyperplasia, and lymphocytic and mast cell tissue infiltrate compared to both wild-type and FliiTg/Tg mice. Despite increased IFN-γ levels, the AD lesions of Flii+/− mice had reduced IL-4, which may contribute significantly to the reduced skin thickening in this model of AD (33). These findings are in agreement with studies which demonstrated that reducing Flii expression, either genetically or using Flii neutralizing antibodies, decreased tissue inflammation, and disease severity in mouse models of psoriasiform dermatitis and epidermolysis bullosa acquisita (23, 24, 32).
Genetically modified mice engineered to over-express Th2 cytokines develop skin barrier defects and AD spontaneously (34). The formation of AD lesions is known to be triggered by production of Th2 cytokines by mast cells and CD4+ T cells which also promote IgE production by B cells, while Th1 cells secrete IFN-γ to suppress proliferation of Th2 cells and IgE synthesis (35, 36). The dominance of Th2 cytokines in AD cause decreased expression of fillagrin and other barrier promoting molecules found in the skin (3). Examining the levels of cytokines and chemokines produced in AD-like lesional skin of Flii+/−, wild-type, and FliiTg/Tg mice revealed decreased IFN-γ levels in FliiTg/Tg mice suggestive of Th2 immune responses, while Flii+/− had increased IFN-γ levels and decreased clinical AD severity suggestive of potential Flii effect on IgE synthesis, however, this is yet to be investigated. Analysis of T-helper subsets within lesional skin showed a significantly Th2 skewed response in OVA-induced AD-like lesions of FliiTg/Tg mice with higher numbers of CD4+GATA3+ cells compared to AD-like lesions of Flii+/− or wild-type counterparts who showed significantly higher Th1:Th2 ratio. Additionally, AD-like lesions of FliiTg/Tg mice showed increased pro-inflammatory TNF-α cytokine levels and while TNF-α has been demonstrated to promote the AD development (37) we did not observe increased AD severity in these mice in this model of OVA-induced AD. As mast cell numbers were not significantly increased in FliiTg/Tg mice, and T-helper cells were heavily skewed toward a Th2 phenotype within FliiTg/Tg lesions, excess TNF-α was likely produced by other cell types such as macrophages, which is further supported by increased CCL22 chemokine levels observed in FliiTg/Tg mice. Interestingly, previous studies have shown that secreted Flii reduces macrophage secretion of TNF-α in vitro (20) while in vivo studies using a mouse model of psoriasiform dermatitis showed reduced TNF-α levels in response to reduced Flii (24) which was not observed in this model of OVA-induced AD. These findings may reflect inherent differences between in vivo and in vitro studies, for example differences in complex in vivo, multicellular environments, as well as differences in different models of inflammatory skin diseases.
In agreement with previous studies demonstrating decreased tissue inflammation and inflammatory cytokine secretion in Flii-deficient mice (24, 32), OVA-induced AD-like skin lesions of Flii+/− mice showed a reduced inflammatory response marked by significantly increased anti-inflammatory IL-10 secretion as well as significantly increased IFN-γ and significantly reduced IL-4 levels. Indeed, IL-4 has been shown to be essential for eosinophil recruitment, Th2 cell differentiation, and IgE production (38). The increased CCL17 expression observed in AD lesions of Flii+/− mice compared to FliiTg/Tg mice may be a potential mechanism to restore Th1/Th2 balance, as CCL17 has previously been shown to induce a Th2-dominated inflammatory reaction (39). Reduction of Flii levels did not alter IL-23 and IL-17A cytokine levels as previously observed in psoriasiform dermatitis (24) suggesting that Th17 responses are not involved in this model of AD. Despite significant differences in IFN-γ levels between three genotypes, CXCL9 chemokine levels were not altered between genotypes in this model of AD. IL-13 levels were also not significantly different between three genotypes, however, there was a trend to higher levels in FliiTg/Tg mice OVA-induced AD-like skin lesions and IL-13 cytokine has previously been linked to autoantibody production in early rheumatoid arthritis (40).
Autoimmunity has been increasingly recognized to play part in exacerbating the severity of AD (6, 9, 12) as a consequence of both humoral and cellular and immunity (6). It is postulated that IFN-γ signaling during AD pathogenesis may promote the development of autoimmunity as IFN-γ overexpressing mice spontaneously develop autoantibodies (41, 42) and deletion of the IFN-γ receptor inhibits autoantibody production in lupus-prone mice (43). On the basis of our findings showing altered AD severity and altered Th1/Th2 responses including significantly altered IFN-γ expression with different Flii genotypes, we examined the effect of Flii levels on autoimmunity in OVA-induced AD-like skin lesions. The decreased severity of AD observed in Flii+/− mice also correlated with the reduced degree of autoreactivity (50% reduction vs WT), and further studies are required to investigate whether the autoantibodies from OVA induced AD-like skin mice contribute to or are a product of the observed AD-like symptoms. Immunoblot analysis of sera from normal non-dermatitis mice showed no autoreactivity suggesting that autoantibody development was disease-specific. Interestingly, both Flii+/− and wild-type mice showed similar autoantibody staining patterns and regions of differential autoreactivity. FliiTg/Tg mice showed similar levels of autoreactivity compared to wild-type mice, however, in addition to the cytoplasmic and perinuclear staining pattern observed in all genotypes, the sera of these mice had a strong nuclear staining pattern. This pattern may indicate the presence of disease-mediating antinuclear antibodies which have been clinically associated with AD and other inflammatory skin disorders (44–46). Flii has previously been shown to affect TLR signaling pathways, both intracellularly and extracellularly, hence modulating innate inflammatory responses and directly impacting immune signaling, however, the potential role of Flii in autoimmunity has not been explored to date. Further studies are required to determine if autoreactivity in Flii genetic mice is a direct result of the impact of Flii on immune signaling or if it is secondary to increased pathology observed in these animals. In addition, determining the subtype of autoreactive immunoglobulins developed in the OVA-challenged murine model of AD would allow more comparison to be drawn to findings of previous clinical cohorts (10, 47).
While major differences between human AD and murine models have been demonstrated (47, 48), models of AD promote our understanding of the complex pathogenesis of human AD, and identify potential novel targets for design of targeted biologics (49). Here, we have demonstrated that Flii is a novel target in AD and that reducing its levels decreased the severity of AD in the ovalbumin-challenged murine model of AD. Additionally, we have examined the role of autoimmunity in this model of AD and while the exact mechanisms are yet to be identified, our results suggest that the effects of Flii upon Th1/Th2 balance and autoimmunity are important during AD pathogenesis.
Mice were maintained according to the Australian Code for the Care and Use of Animals for Scientific Purposes under protocols approved by the Child Youth and Women’s Health Service Animal Ethics Committee (AEC916/06/2015).
AC and ZK conceived all the experiments with assistance from NS. ZK and NS carried out experiments and analysis with the assistance of HC and GY. ZK, NS, and AC wrote the manuscript and all authors contributed to the manuscript preparation and approved the final submitted and published versions.
IP associated with this project has been filed by AbRegen Pty Ltd., of which AC is a shareholder and both AC and ZK are named inventors on associated patents.
The handling Editor declared a past co-authorship with one of the authors AC.
AC is supported by NHMRC Senior Research Fellowship (GNT#1102617) and ZK is supported by a Future Industries Institute Foundation Fellowship.
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01833/full#supplementary-material.
Table S1. Primer sequences used in real-time qPCR.
1. Plotz SG, Wiesender M, Todorova A, Ring J. What is new in atopic dermatitis/eczema? Expert Opin Emerg Drugs (2014) 19(4):441–58. doi:10.1517/14728214.2014.953927
2. D’Auria E, Banderali G, Barberi S, Gualandri L, Pietra B, Riva E, et al. Atopic dermatitis: recent insight on pathogenesis and novel therapeutic target. Asian Pac J Allergy Immunol (2016) 34(2):98–108. doi:10.12932/AP0732.34.2.2016
4. Homey B, Steinhoff M, Ruzicka T, Leung DY. Cytokines and chemokines orchestrate atopic skin inflammation. J Allergy Clin Immunol (2006) 118(1):178–89. doi:10.1016/j.jaci.2006.03.047
5. Cipriani F, Ricci G, Leoni MC, Capra L, Baviera G, Longo G, et al. Autoimmunity in atopic dermatitis: biomarker or simply epiphenomenon? J Dermatol (2014) 41(7):569–76. doi:10.1111/1346-8138.12464
6. Navarrete-Dechent C, Perez-Mateluna G, Silva-Valenzuela S, Vera-Kellet C, Borzutzky A. Humoral and cellular autoreactivity to epidermal proteins in atopic dermatitis. Arch Immunol Ther Exp (Warsz) (2016) 64(6):435–42. doi:10.1007/s00005-016-0400-3
7. Tang TS, Bieber T, Williams HC. Does "autoreactivity" play a role in atopic dermatitis? J Allergy Clin Immunol (2012) 129(5):1209–15e2. doi:10.1016/j.jaci.2012.02.002
8. Watanabe K, Muro Y, Sugiura K, Tomita Y. IgE and IgG(4) autoantibodies against DFS70/LEDGF in atopic dermatitis. Autoimmunity (2011) 44(6):511–9. doi:10.3109/08916934.2010.549157
9. Muro Y. Autoantibodies in atopic dermatitis. J Dermatol Sci (2001) 25(3):171–8. doi:10.1016/S0923-1811(01)00084-6
10. Watanabe K, Muro Y, Sugiura K, Akiyama M. High-avidity IgG autoantibodies against DFS70/LEDGF in atopic dermatitis. J Clin Cell Immunol (2013) 4:170. doi:10.4172/2155-9899.1000170
11. Ohkouchi K, Mizutani H, Tanaka M, Takahashi M, Nakashima K, Shimizu M. Anti-elongation factor-1alpha autoantibody in adult atopic dermatitis patients. Int Immunol (1999) 11(10):1635–40. doi:10.1093/intimm/11.10.1635
12. Mittermann I, Aichberger KJ, Bunder R, Mothes N, Renz H, Valenta R. Autoimmunity and atopic dermatitis. Curr Opin Allergy Clin Immunol (2004) 4(5):367–71. doi:10.1097/00130832-200410000-00007
13. Ochs RL, Mahler M, Basu A, Rios-Colon L, Sanchez TW, Andrade LE, et al. The significance of autoantibodies to DFS70/LEDGFp75 in health and disease: integrating basic science with clinical understanding. Clin Exp Med (2016) 16(3):273–93. doi:10.1007/s10238-015-0367-0
14. Silosi I, Silosi CA, Boldeanu MV, Cojocaru M, Biciusca V, Avramescu CS, et al. The role of autoantibodies in health and disease. Rom J Morphol Embryol (2016) 57(2 Suppl):633–8.
15. Chan H, Kopecki Z, Waters J, Powell BC, Arkell R, Cowin AJ. Cytoskeletal protein Flightless I differentially affects TGF-β isoform expression in both in vitro and in vivo wound models. Wound Pract Res (2014) 22(3):169–81.
16. Choi JS, Choi SS, Kim ES, Seo YK, Seo JK, Kim EK, et al. Flightless-1, a novel transcriptional modulator of PPARgamma through competing with RXRalpha. Cell Signal (2015) 27(3):614–20. doi:10.1016/j.cellsig.2014.11.035
17. Cowin AJ, Lei N, Franken L, Ruzehaji N, Offenhauser C, Kopecki Z, et al. Lysosmal secretion of Flightless I upon injury has the potential to alter inflammation. Commun Integr Biol (2012) 5(6):546–9. doi:10.4161/cib.21928
18. Ruzehaji N, Mills SJ, Melville E, Arkell R, Fitridge R, Cowin AJ. The influence of Flightless I on toll-like-receptor-mediated inflammation in a murine model of diabetic wound healing. Biomed Res Int (2013) 2013:389792. doi:10.1155/2013/389792
19. Wang T, Chuang TH, Ronni T, Gu S, Du YC, Cai H, et al. Flightless I homolog negatively modulates the TLR pathway. J Immunol (2006) 176(3):1355–62. doi:10.4049/jimmunol.176.3.1355
20. Lei N, Franken L, Ruzehaji N, Offenhauser C, Cowin AJ, Murray RZ. Flightless, secreted through a late endosome/lysosome pathway, binds LPS and dampens cytokine secretion. J Cell Sci (2012) 125(Pt 18):4288–96. doi:10.1242/jcs.099507
21. Kopecki Z, Yang GN, Jackson JE, Melville EL, Calley MP, Murrell DF, et al. Cytoskeletal protein Flightless I inhibits apoptosis, enhances tumor cell invasion and promotes cutaneous squamous cell carcinoma progression. Oncotarget (2015) 6(34):36426–40. doi:10.18632/oncotarget.5536
22. Kopecki Z, Yang GN, Arkell RM, Jackson JE, Melville E, Iwata H, et al. Flightless I over-expression impairs skin barrier development, function and recovery following skin blistering. J Pathol (2014) 232(5):541–52. doi:10.1002/path.4323
23. Kopecki Z, Arkell RM, Strudwick XL, Hirose M, Ludwig RJ, Kern JS, et al. Overexpression of the Flii gene increases dermal-epidermal blistering in an autoimmune ColVII mouse model of epidermolysis bullosa acquisita. J Pathol (2011) 225(3):401–13. doi:10.1002/path.2973
24. Chong HT, Yang GN, Sidhu S, Ibbetson J, Kopecki Z, Cowin AJ. Reducing Flightless I expression decreases severity of psoriasis in an imiquimod-induced murine model of psoriasiform dermatitis. Br J Dermatol (2017) 176(3):705–12. doi:10.1111/bjd.14842
25. Wang G, Savinko T, Wolff H, Dieu-Nosjean MC, Kemeny L, Homey B, et al. Repeated epicutaneous exposures to ovalbumin progressively induce atopic dermatitis-like skin lesions in mice. Clin Exp Allergy (2007) 37(1):151–61. doi:10.1111/j.1365-2222.2006.02621.x
26. Campbell HD, Fountain S, McLennan IS, Berven LA, Crouch MF, Davy DA, et al. Fliih, a gelsolin-related cytoskeletal regulator essential for early mammalian embryonic development. Mol Cell Biol (2002) 22(10):3518–26. doi:10.1128/MCB.22.10.3518-3526.2002
27. Cowin AJ, Adams DH, Strudwick XL, Chan H, Hooper JA, Sander GR, et al. Flightless I deficiency enhances wound repair by increasing cell migration and proliferation. J Pathol (2007) 211(5):572–81. doi:10.1002/path.2143
28. Kajisa T, Yamaguchi T, Hu A, Suetake N, Kobayashi H. Hydrogen water ameliorates the severity of atopic dermatitis-like lesions and decreases interleukin-1beta, interleukin-33, and mast cell infiltration in NC/Nga mice. Saudi Med J (2017) 38(9):928–33. doi:10.15537/smj.2017.9.20807
29. Ochs RL, Muro Y, Si Y, Ge H, Chan EK, Tan EM. Autoantibodies to DFS 70 kd/transcription coactivator p75 in atopic dermatitis and other conditions. J Allergy Clin Immunol (2000) 105(6 Pt 1):1211–20. doi:10.1067/mai.2000.107039
30. Kopecki Z, Arkell R, Powell BC, Cowin AJ. Flightless I regulates hemidesmosome formation and integrin-mediated cellular adhesion and migration during wound repair. J Invest Dermatol (2009) 129(8):2031–45. doi:10.1038/jid.2008.461
31. Kopecki Z, Cowin AJ. Flightless I: an actin-remodelling protein and an important negative regulator of wound repair. Int J Biochem Cell Biol (2008) 40(8):1415–9. doi:10.1016/j.biocel.2007.04.011
32. Kopecki Z, Ruzehaji N, Turner C, Iwata H, Ludwig RJ, Zillikens D, et al. Topically applied Flightless I neutralizing antibodies improve healing of blistered skin in a murine model of epidermolysis bullosa acquisita. J Invest Dermatol (2012) 133(4):1008–16. doi:10.1038/jid.2012.457
33. Matsunaga MC, Yamauchi PS. IL-4 and IL-13 inhibition in atopic dermatitis. J Drugs Dermatol (2016) 15(8):925–9.
34. Vestergaard C, Yoneyama H, Murai M, Nakamura K, Tamaki K, Terashima Y, et al. Overproduction of Th2-specific chemokines in NC/Nga mice exhibiting atopic dermatitis-like lesions. J Clin Invest (1999) 104(8):1097–105. doi:10.1172/JCI7613
35. Matsuda H, Watanabe N, Geba GP, Sperl J, Tsudzuki M, Hiroi J, et al. Development of atopic dermatitis-like skin lesion with IgE hyperproduction in NC/Nga mice. Int Immunol (1997) 9(3):461–6. doi:10.1093/intimm/9.3.461
36. Yamanaka K, Mizutani H. The role of cytokines/chemokines in the pathogenesis of atopic dermatitis. Curr Probl Dermatol (2011) 41:80–92. doi:10.1159/000323299
37. Danso MO, van Drongelen V, Mulder A, van Esch J, Scott H, van Smeden J, et al. TNF-alpha and Th2 cytokines induce atopic dermatitis-like features on epidermal differentiation proteins and stratum corneum lipids in human skin equivalents. J Invest Dermatol (2014) 134(7):1941–50. doi:10.1038/jid.2014.83
38. Nedoszytko B, Sokolowska-Wojdylo M, Ruckemann-Dziurdzinska K, Roszkiewicz J, Nowicki RJ. Chemokines and cytokines network in the pathogenesis of the inflammatory skin diseases: atopic dermatitis, psoriasis and skin mastocytosis. Postepy Dermatol Alergol (2014) 31(2):84–91. doi:10.5114/pdia.2014.40920
39. Vestergaard C, Deleuran M, Gesser B, Larsen CG. Thymus- and activation-regulated chemokine (TARC/CCL17) induces a Th2-dominated inflammatory reaction on intradermal injection in mice. Exp Dermatol (2004) 13(4):265–71. doi:10.1111/j.0906-6705.2004.00149.x
40. Silosi I, Boldeanu MV, Cojocaru M, Biciusca V, Padureanu V, Bogdan M, et al. The relationship of cytokines IL-13 and IL-17 with autoantibodies profile in early rheumatoid arthritis. J Immunol Res (2016) 2016:3109135. doi:10.1155/2016/3109135
41. Seery JP. IFN-gamma transgenic mice: clues to the pathogenesis of systemic lupus erythematosus? Arthritis Res (2000) 2(6):437–40. doi:10.1186/ar124
42. Watt FM. Transgenic mice expressing IFN-gamma in the epidermis are a model of inflammatory skin disease and systemic lupus erythematosus. Ernst Schering Res Found Workshop (2005) (50):277–91. doi:10.1007/3-540-26811-1_16
43. Haas C, Ryffel B, Le Hir M. IFN-gamma receptor deletion prevents autoantibody production and glomerulonephritis in lupus-prone (NZB x NZW)F1 mice. J Immunol (1998) 160(8):3713–8.
44. Higashi N, Niimi Y, Aoki M, Kawana S. Clinical features of antinuclear antibody-positive patients with atopic dermatitis. J Nippon Med Sch (2009) 76(6):300–7. doi:10.1272/jnms.76.300
45. Wozniacka A, Salamon M, McCauliffe D, Sysa-Jedrzejowska A. Antinuclear antibodies in rosacea patients. Postepy Dermatol Alergol (2013) 30(1):1–5. doi:10.5114/pdia.2013.33372
46. Silvy F, Bertin D, Bardin N, Auger I, Guzian MC, Mattei JP, et al. Antinuclear antibodies in patients with psoriatic arthritis treated or not with biologics. PLoS One (2015) 10(7):e0134218. doi:10.1371/journal.pone.0134218
47. Ewald DA, Noda S, Oliva M, Litman T, Nakajima S, Li X, et al. Major differences between human atopic dermatitis and murine models, as determined by using global transcriptomic profiling. J Allergy Clin Immunol (2017) 139(2):562–71. doi:10.1016/j.jaci.2016.08.029
48. Martel BC, Lovato P, Baumer W, Olivry T. Translational animal models of atopic dermatitis for preclinical studies. Yale J Biol Med (2017) 90(3):389–402.
Keywords: atopic dermatitis, flightless I, autoantibody, inflammation, skin barrier
Citation: Kopecki Z, Stevens NE, Chong HT, Yang GN and Cowin AJ (2018) Flightless I Alters the Inflammatory Response and Autoantibody Profile in an OVA-Induced Atopic Dermatitis Skin-Like Disease. Front. Immunol. 9:1833. doi: 10.3389/fimmu.2018.01833
Received: 21 December 2017; Accepted: 25 July 2018;
Published: 10 August 2018
Edited by:
Ralf J. Ludwig, Universität zu Lübeck, GermanyReviewed by:
Luciana D’Apice, Consiglio Nazionale Delle Ricerche (CNR), ItalyCopyright: © 2018 Kopecki, Stevens, Chong, Yang and Cowin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zlatko Kopecki, emxhdGtvLmtvcGVja2lAdW5pc2EuZWR1LmF1
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.