 Zhidong Hu
Zhidong Hu Ling Gu
Ling Gu Chun-Ling Li
Chun-Ling Li Tsugumine Shu
Tsugumine Shu Douglas B. Lowrie
Douglas B. Lowrie Xiao-Yong Fan
Xiao-Yong Fan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 03 August 2018
Sec. Vaccines and Molecular Therapeutics
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01796
This article is part of the Research Topic Innovative Therapeutic and Vaccine Approaches Against Respiratory Pathogens View all 20 articles
The kinds of vaccine-induced T cell responses that are beneficial for protection against Mycobacterium tuberculosis (Mtb) infection are not adequately defined. We had shown that a novel Sendai virus vectored vaccine, SeV85AB, was able to enhance immune protection induced by bacille Calmette–Guérin (BCG) in a prime-boost model. However, the profile of T cell responses boosted by SeV85AB was not determined. Herein, we show that the antigen-specific CD4+ and CD8+ T cell responses were both enhanced by the SeV85AB boost after BCG. Different profiles of antigen-specific po T cell subsets were induced in the local (lung) and systemic (spleen) sites. In the spleen, the CD4+ T cell responses that were enhanced by the SeV85AB boost were predominately IL-2 responses, whereas in the lung the greater increases were in IFN-γ- and TNF-α-producing CD4+ T cells; in CD8+ T cells, although IFN-γ was enhanced in both the spleen and lung, only IL-2+TNF-α+CD8+ T subset was boosted in the latter. After a challenge Mtb infection, there were significantly higher levels of recall IL-2 responses in T cells. In contrast, IFN-γ-producing cells were barely boosted by SeV85AB. After Mtb challenge a central memory phenotype of responding CD4+ T cells was a prominent feature in SeV85AB-boosted mice. Thus, our data strongly suggest that the enhanced immune protection induced by SeV85AB boosting was associated with establishment of an increased capacity to recall antigen-specific IL-2-mediated T cell responses and confirms this Sendai virus vector system as a promising candidate to be used in a heterologous prime-boost immunization regimen against TB.
Tuberculosis (TB) remains among the most deadly health challenge to humankind. Bacille Calmette–Guérin (BCG), the attenuated form of Mycobacterium bovis, has been used for over 80 years to protect children against severe forms of TB (1). However, its protective efficacy against pulmonary TB was found to vary from 0 to 80% in adults (2), hence a more effective vaccine is needed.
T cell responses are regarded as a critical factor in containment of Mycobacterium tuberculosis (Mtb) infection. After phagocytosis, Mtb preferentially resides in phagosomes, where its antigens are processed and assembled onto MHC-II molecules for presentation to CD4+ T cells (3). During the course of Mtb-driven differentiation, the T cells can gain the capacity to simultaneously produce two or more key cytokines and are called poly-functional T cells, which are considered to be superior effectors of protective immunity as compared to cells that produce only one cytokine (4). Specially, IFN-γ, IL-2, and TNF-α secreting CD4+ T cells, which are known as Th1 cells, are regarded as crucial for activation of effector functions to control intracellular Mtb and are correlated with protection (5, 6).
Although the role of CD8+ T cell-mediated immune responses against TB infection is less well defined than that of Th1 CD4+ T cells, these cells are also considered to play a crucial role in optimal immunity and protection. It was shown that CD8+ T cells were essential against Mtb infection in the models of mice (7, 8), cattle (9), and macaques (10). Furthermore, vaccine-induced antigen-specific CD8+ T cell responses were found to contribute to strong or modest immune protection in several studies (11–14). Recently, we reported that a novel Sendai virus vectored vaccine, SeV85AB, induced robust T cell responses and substantial protection against Mtb infection, which was mainly mediated by CD8+ T cells (15).
Insufficient induction of T cell responses by BCG immunization might underlie the vaccine’s inadequacies and boosting these responses by novel vaccines might be an appropriate vaccine strategy (16, 17). However, which kind of T cell responses are beneficial for the anti-TB immune protection remains controversial (18, 19); notably, the classical marker, IFN-γ, was found to play a minor role in, or even be detrimental to, the anti-TB immunity (20–22). Although we had shown that intra-nasal delivery of the SeV85AB vaccine was able to enhance immune protection induced by BCG in a prime-boost model (15), the profile of T cell responses boosted by SeV85AB was not determined. Herein, we show that SeV85AB boosting established substantial T cell responses in the lung that differed from systemic immunity; there were different profiles of antigen-specific poly-functional T cell subsets in the lung compared with the spleen. After challenge by Mtb infection, SeV85AB-boosted mice had significantly higher levels of recall CD4+ and CD8+ T cell responses, which were mainly mediated by IL-2. In contrast, the IFN-γ-producing cells were barely boosted by SeV85AB. The proportion of cells with central memory phenotype of peptides-responding CD4+ T cells was elevated in SeV85AB-boosted mice after Mtb challenge. Our study, therefore, lends strong support to the adoption of Sendai virus as a promising vector system to be used in a heterologous prime-boost immunization regimen against TB.
This study was approved by the Institutional Animal Care and Use Committee and was performed according to the guidelines of the Laboratory Animal Ethical Board of Shanghai Public Health Clinical Center. Specific pathogen-free female BALB/c mice aged 6–8 weeks were immunized with BCG subcutaneously [s.c., 5 × 106 CFU (colony forming units), in 100 µl PBS] in each hind leg and boosted intra-nasally (i.n.) with SeV85AB [1 × 107 cell infectious units (CIU), in 20 µl PBS]. BCG, SeV85AB single immunizations, and PBS were used as controls. For the evaluation of primary cellular immune responses, 4 weeks after vaccination, animals were sacrificed, then, the lungs and spleens were aseptically removed for antigen-specific T cell immune response assessments. For the evaluation of recall immune responses after infection, the mice were challenged through a respiratory route by the Mtb virulent strain H37Rv 4 weeks after immunization and maintained in a level 3 bio-containment animal facility. Five weeks later, the mice were sacrificed and lungs sampled to assess recall responses by intracellular staining (ICS) as described below.
Spleen was mechanically disrupted and single splenocytes were filtered, and then subjected to red blood cell lysis. Lung was gently minced by scissors and then incubated with DNase I (10 U, Thermo) and collagenase IV (1 mg/ml, Invitrogen) in 10 ml R10 medium (RPMI-1640 medium containing 10% fetal bovine serum and 1% Penicillin and Streptomycin) for 30 min at 37°C. The collagenase-digested tissue pieces were filtered through a 70 µm cell strainer (Fisher Scientific) by gently squashing with the plunger of a syringe. The cell suspension was centrifuged and red blood cells were lysed. The single splenocytes and lung lymphocytes were washed and re-suspended in R10 medium.
Enzyme-linked immunospot assays were performed according to IFN-γ ELISPOT kit protocols (BD Biosciences). Briefly, 96-well plates were coated with anti-mouse IFN-γ antibody at 4°C overnight. Then, they were washed and blocked with R10 medium at room temperature for 2 h. Isolated lung cells or splenocytes were added at 2 × 105 cells per well with peptide pools (5 µg/ml) or PPD (10 µg/ml). PMA (50 ng/ml, Sigma) and ionomycin (1 µg/ml, Sigma) stimulation were used as positive controls. The cells were stimulated at 37°C for 20 h. The cell suspension was aspirated and washed with PBST (PBS containing 0.5% Tween-20), and then incubated with anti-mouse IFN-γ biotinylated detection antibodies for 2 h. The cell suspension was aspirated and washed with PBST and then incubated with streptavidin-horseradish superoxidase conjugated anti-biotin antibodies for an additional 1 h. After washing with PBST and PBS, AEC substrate solution was added and incubated for 30 min before rinsing away with water. The plates were air-dried and analyzed with Immunospot Reader (Champspot III, Beijing Sage Creation Science).
The peptides were synthesized by GL Biochem (Shanghai, China) with 95% purity. MPVGGQSSF, 70-78aa; TFLTSELPGWLQANRHVKPT, 99-118aa; GLSMAASSALTL, 124-125aa and YAGAMSGL, 145-152aa were used as an Ag85A peptide pool. IYAGSLSAL, 144-152aa; ALLDPSQGMGPSLIG, 151-165aa; GPSSDPAWERNDPTQQIP, 181-198aa and HSWEYWGAQLNAMKGDLQ, 262-279aa were used as an Ag85B peptide pool.
The splenocytes or lung cells were stimulated with Ag85AB peptide pool (5 µg/ml) or PPD (10 µg/ml) in 96-well plates at 37°C for 1 or 14 h, respectively. Then, cells were incubated for an additional 5 h after the addition of 1 µl/ml Monensin and Brefeldin A (BD Biosciences), PMA (50 ng/ml), and inonomycin (1 µg/ml) stimulation were used as positive controls. After stimulation, cells were washed with wash PBS containing 2% fetal bovine serum and then incubated on ice with the mixture of antibodies for 30 min, then washed and subjected to fixation and permeation with fix/perm buffer (BD Bioscience). After washing, the cells were incubated with antibodies against intracellular cytokines on ice for another 30 min. Then, the cells were re-suspended for flow cytometry analysis (LSRFortessa, BD Biosciences). At least 50,000 T cells were harvested.
The following antibodies were used in this study: CD3-Pacific Blue (clone 17A2) from BioLegend, and CD44-FITC (clone IM7), CD62L-Percp-Cyanine 5.5 (clone MEL-14), TNF-α-PE-Cyanine 7 (clone MP6-XT22) from eBioscience, and CD4-Alexa Fluor 647 (clone RM4-5), IFN-γ-APC-Cyanine 7 (clone XMG1.2), IL-2-PE (clone JES6-5H4) from BD Biosciences.
The statistical analysis was performed using GraphPad Prism 6 software. One-way ANOVA was used to determine the statistical significance for comparison of multiple groups, and two-way ANOVA was used for the grouped analysis.
The experiment protocol is shown diagrammatically in Figure 1A. Typical IFN-γ ELISPOT results are shown in Figure 1B. As expected, SeV85AB vaccination induced Ag85AB-specific immune responses, and this effect was significantly greater in BCG-primed mice, both in the spleen (Figure 1C) and in the lung (Figure 1D). Although PPD-specific cell responses were only slightly induced by SeV85AB, mice receiving SeV85AB boost secreted higher levels of IFN-γ compared with SeV85AB or BCG single immunization (Figures 1C,D).
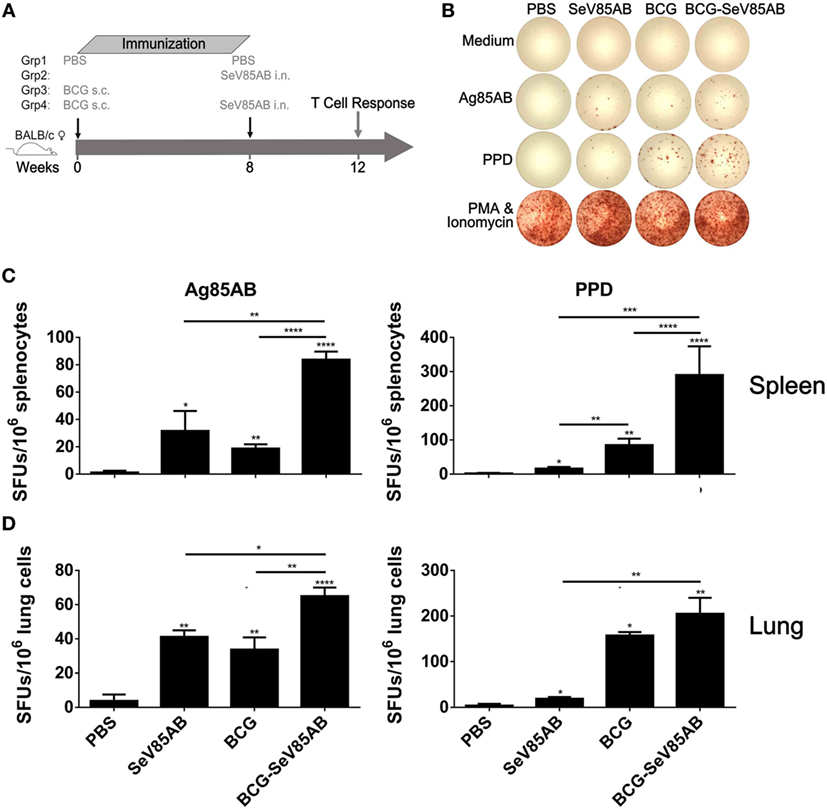
Figure 1. Boosting with SeV85AB increased the IFN-γ responses primed by bacille Calmette–Guérin (BCG) vaccination. (A) Immunization and detection schedule. Mice primed by BCG were i.n. boosted with SeV85AB or not, and 4 weeks later, they were sacrificed for assays of cellular immune responses. Controls received PBS, SeV85AB, or BCG only, at the indicated time points. (B–D) The IFN-γ responses determined by enzyme linked immunospot assay. Representative dot plots are shown in (B). Cells from spleen (C) and lung (D) were stimulated for 20 h with Ag85AB peptide pool (5 µg/ml) or PPD (10 µg/ml) in 96-well plates and then the IFN-γ-secreting cells were detected and counted. PMA plus Ionomycin stimulation was used as positive control. Data are representative of two independent experiments with at least four mice per group. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
At 4 weeks after vaccination, splenocytes were obtained from the different vaccination groups and stimulated with the Ag85AB peptide pool or incubated with medium as control (The gating strategy is shown in Figure S1 in Supplementary Material, CD4+ T cells were gated as CD3+CD4+ cells and CD8+ T cells were defined as CD3+CD4− cells) and the results of flow cytometric ICS detection of IFN-γ, IL-2, and TNF-α positive T cells were compared between groups (representative mono-positive dot plots are shown in Figure 2A and dual-positive plots are shown in Figure S2 in Supplementary Material). Consistent with the ELISPOT assay, the production of all three cytokines in CD4+ T cells was significantly enhanced by SeV85AB boost (Figure 2B), whereas only IFN-γ was increased in CD8+ T cells (Figure 2C). The SeV85AB boost induced higher levels of antigen-specific poly-functional CD4+ T cell responses, in which the increased percentage of dual-positive IFN-γ+IL-2+ and IL-2+TNF-α+ cells were statistically significant (Figures 3A,C). Additionally, the mono-positive IFN-γ+CD8+ subset was significantly increased (Figures 3B,C).
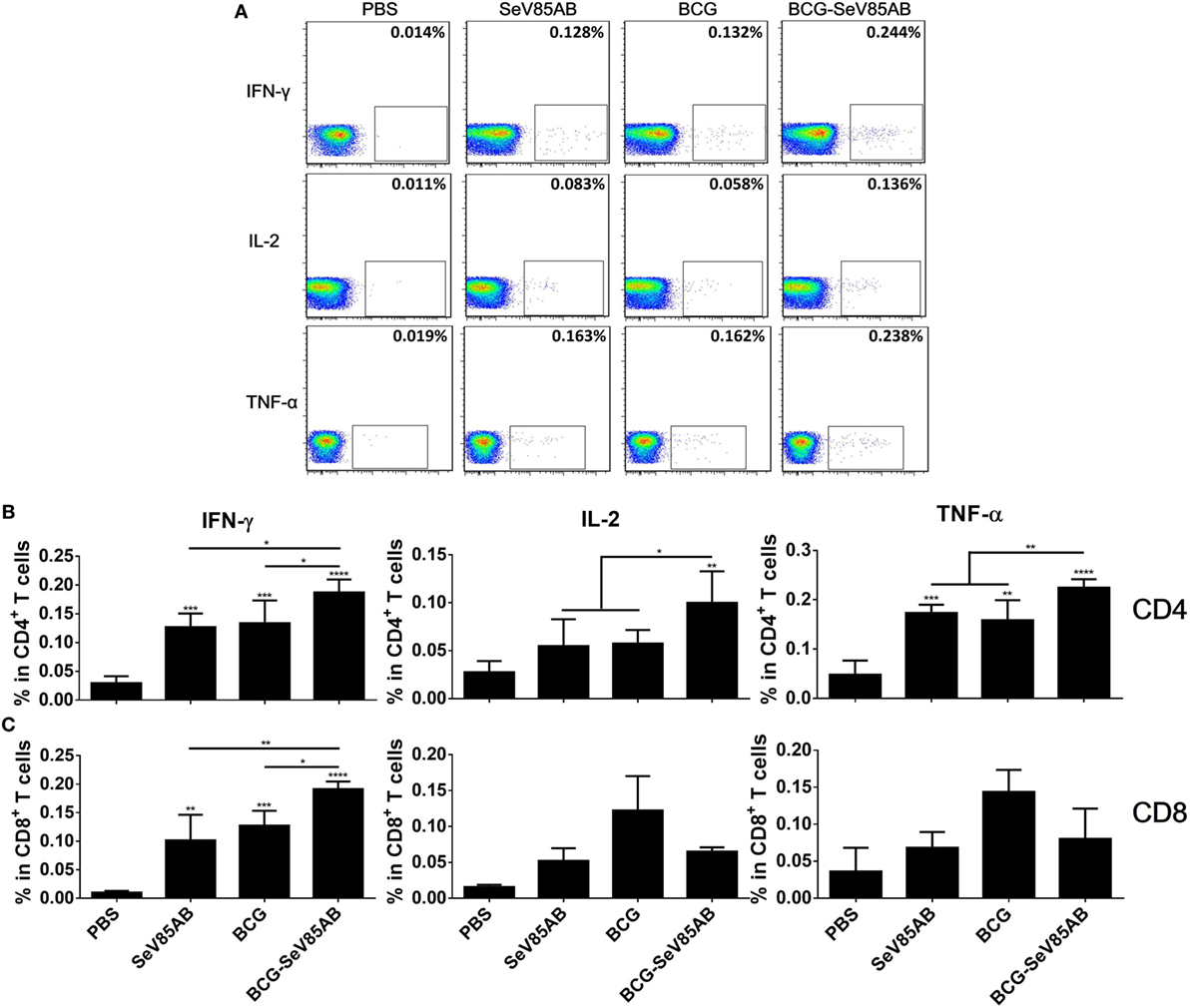
Figure 2. Flow cytometric analysis of ICS in the splenocytes of vaccinated mice. Splenocytes were collected 4 weeks after the last inoculation, incubated with Ag85AB peptides (5 µg/ml) in the presence of monensin and brefeldin A, and analyzed for cytokine production by ICS assay. CD3+CD4+ cells (CD4+ T cells) and CD3+CD4− cells (CD8+ T cells) producing IFN-γ, IL-2, and TNF-α were analyzed. Representative flow cytometric plots of intracellular staining in CD4+ T cells are shown in (A), and summary data of single cytokine producing CD4+ T cells (B) and CD8+ T cells (C) are shown with significant differences indicated. Data are representative of two independent experiments with at least four mice per group. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
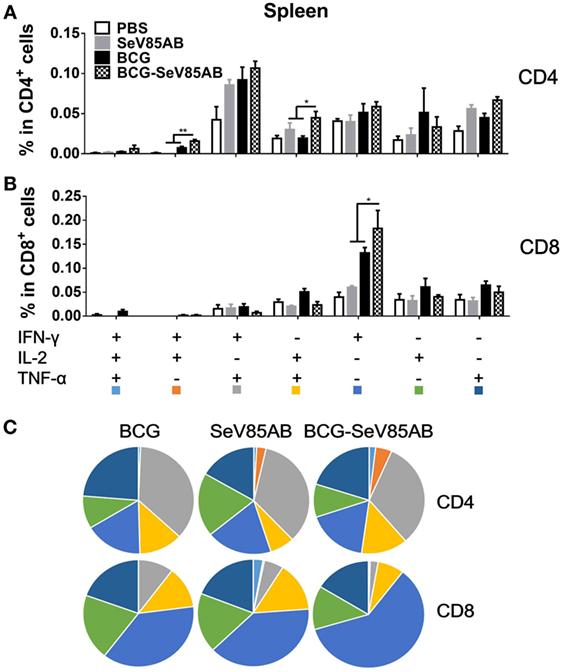
Figure 3. Characterization of poly-functional Ag85AB-specific T cell responses in the spleen. T cells producing IFN-γ, IL-2, and TNF-α were distinguished by ICS assay as seven subpopulations based on the production of these three cytokines in any combination. The percentage of subpopulations as components of the total CD4+ (A) or CD8+ (B) T cell population and the pie chart analysis (C) are shown. Significant differences in frequency of poly-functional T cell subsets are indicated. Data are representative of two independent experiments with at least four mice per group. *P < 0.05 and **P < 0.01.
The representative staining results of flow cytometric ICS detection of IFN-γ, IL-2, and TNF-α mono-producing lung T cells after Ag85AB stimulation are shown in Figure 4A and dual-positive plots are shown in Figure S3 in Supplementary Material. The secretion of all three cytokines was significantly boosted, both in CD4+ T and CD8+ T cells (Figures 4B,C). The poly-functional T cell subset analysis further showed that in CD4+ T cells the dual-positive IFN-γ+TNF-α+ and mono-positive IFN-γ+ subsets were significantly boosted (Figures 5A,C), whereas in the CD8+ T cells, the IFN-γ+ and IL-2+TNF-α+ subsets were boosted (Figures 5B,C). In the spleen, SeV85AB boost dominantly increased IL-2 responses in CD4+ T cells (Figure 3A), whereas in the lung, the greater increases were in IFN-γ positive CD4+ T cells (Figure 5B). In the spleen, the CD8+ T cell responses had been only modestly enhanced (Figure 3B), whereas in the lung, IL-2+TNF-α+ and IFN-γ+ cells had been significantly increased by SeV85AB (Figure 5B). In addition, PPD-specific CD4+ T cell responses in the spleen and CD8+ T cell responses in the lung were also boosted by SeV85AB (Figure S4 in Supplementary Material). These divergences confirmed that the mucosal immunization with SeV85AB had a different capacity to establish immune memory in the systemic immune system compared with local site in the lung, confirming our previous observation (15). Taken together, these data demonstrated that SeV85AB boost induced higher levels of poly-functional antigen-specific CD4+ and CD8+ T cell responses primed by BCG vaccine.

Figure 4. Flow cytometric analysis of ICS in the lung cells of vaccinated mice. Lung cells were collected, stimulated, and analyzed for cytokine production by ICS assay as described for splenocytes in Figure 2. Representative flow cytometric data plots from CD4+ T cells (A) and summarized data of single cytokine producing CD4+ T cells (B) and CD8+ T cells (C) are shown. The percentages of responding cells were compared as indicated. Data are representative of two independent experiments with at least four mice per group. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
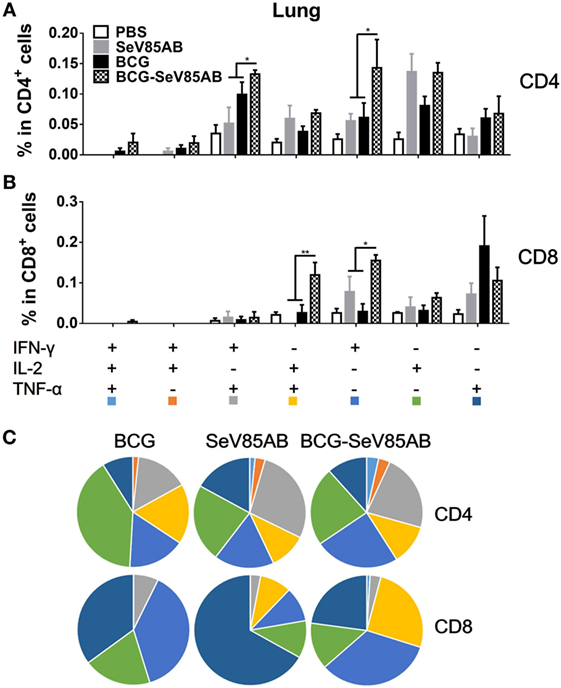
Figure 5. Characterizations of poly-functional Ag85AB-specific T cell responses in the lung. T cells secreting IFN-γ, IL-2, and TNF-α were distinguished as seven distinct subpopulations as described in Figure 3. The percentage of subpopulation as components of the total CD4+ (A) and CD8+ (B) T cell populations and the pie chart analysis (C) are shown. Significant differences in frequency of poly-functional T cell subsets are indicated. Data are representative of two independent experiments with at least four mice per group. *P < 0.05 and **P < 0.01.
We had shown that mucosal boosting of SeV85AB improved the protection of BCG vaccination against Mtb challenge in a prime-boost model (15). In this study, the protective efficacy afforded by SeV85AB boost was confirmed (data not shown), and the recall T cell responses that occurred upon the Mtb infection were especially assayed (Figure 6A). IFN-γ ELISPOT results showed that, at 5 weeks postinfection, mice that had received SeV85AB boosting before the infection had developed only slight, if any, overall increase of Ag85AB- (Figure 6B) and PPD- (Figure 6C) specific responses in the lung compared with the response to infection after SeV85AB or BCG single vaccination. However, SeV85AB boosting resulted in a response to infection that contained a higher percentage of Ag85AB-specific poly-functional lung T cells, notably CD4+ T cells with the phenotypes IL-2+TNF-α+/IL-2+, and CD8+ T cells of phenotypes IFN-γ+IL-2+TNF-α+/IL-2+ (Figure 7).
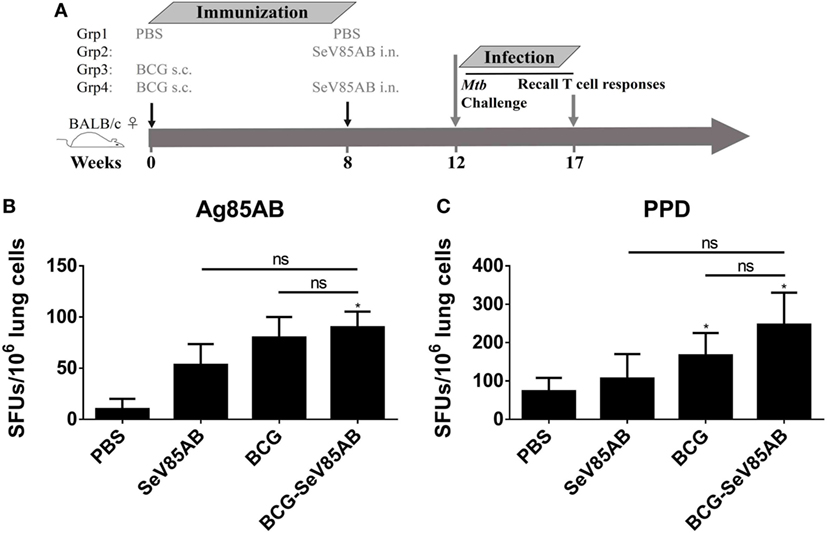
Figure 6. Recall T cell responses against specific stimulation post Mycobacterium tuberculosis (Mtb) challenge determined by IFN-γ Enzyme Linked Immunospot (ELISPOT) assay. (A) Immunization and infection schedule. Vaccinated mice were aerosol challenged 4 weeks later with virulent Mtb H37Rv. Five weeks postinfection, recall lung T cell responses were determined. (B,C) The IFN-γ responses by ELISPOT assay. Cells from the infected lung were stimulated for 20 h with Ag85AB peptide pool (B) or PPD (C) in 96-well plates, respectively. Significant differences in frequency between T cell subsets are indicated. Data are representative of two independent experiments with three mice per group. ns, no significant difference.
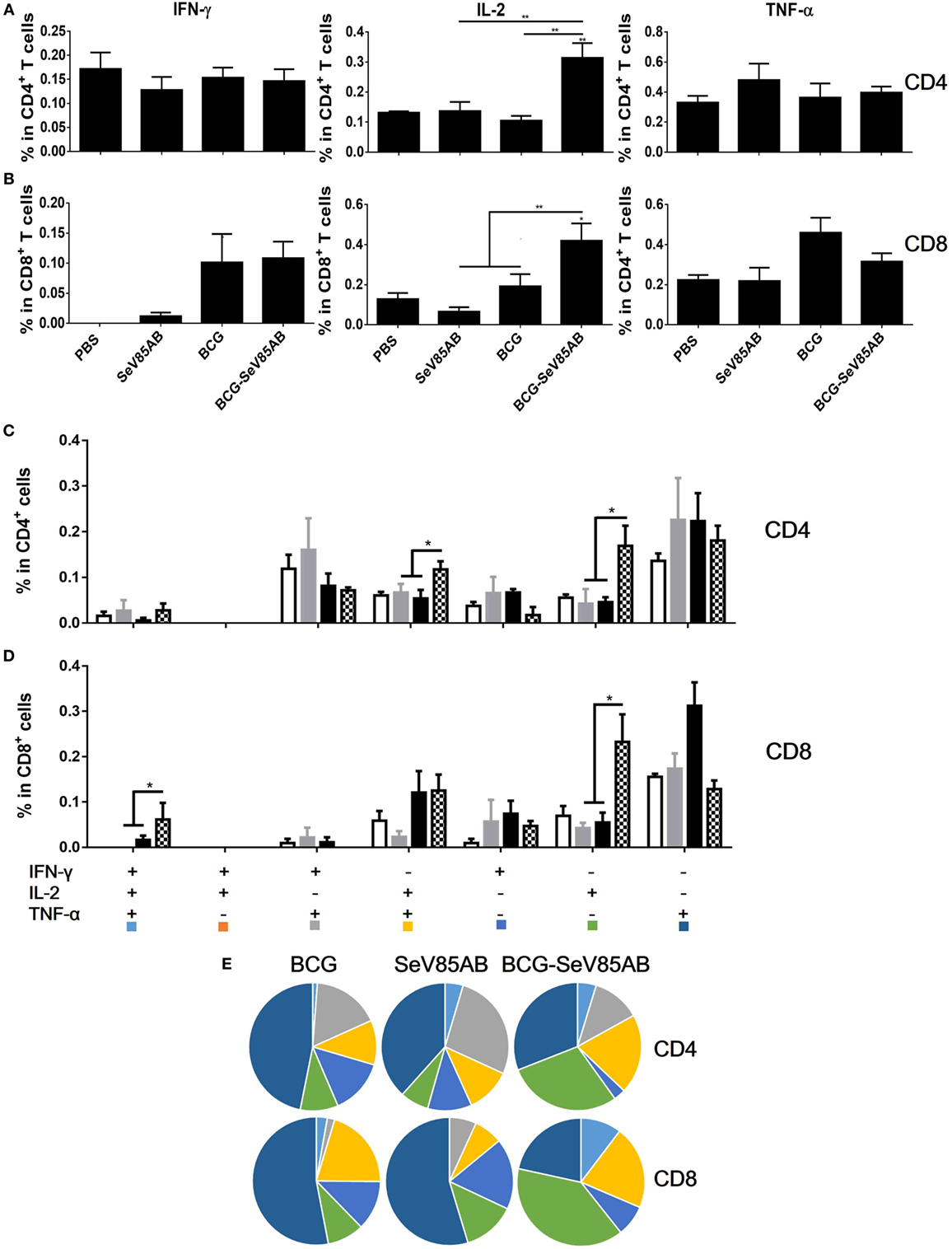
Figure 7. Recall T cell responses against specific stimulation post Mycobacterium tuberculosis challenge determined by ICS assay. The immunization and infection schedule were described in Figure 6A. Lung cells were stimulated ex vivo with Ag85AB peptides in the presence of monensin and brefeldin A and analyzed for cytokine production by ICS assay. T cells producing IFN-γ, IL-2, and TNF-α were analyzed and their percentage in CD4+ T cells (A) and CD8+ T cells (B) are shown. The percentage of seven subpopulations based on the production of three cytokines in any combination in the total CD4+ (C) and CD8+ (D) T cells and the pie chart analysis (E) are shown. Significant differences in frequency of T cell subsets are indicated. Data are representative of two independent experiments with three mice per group. *P < 0.05 and **P < 0.01.
The memory phenotypes of antigen-responding CD4+ T cells that secreted at least one of the cytokines IFN-γ, IL-2, or TNF-α were further determined (The gating strategy is shown in Figure S5 in Supplementary Material). As a result, SeV85AB boost led to significantly higher levels CD4+ T cells with CD44+CD62+ central memory phenotype compared with other vaccination groups (Figure 8). This was paralleled by decrease in CD44+CD62− (effector memory) among those responding CD4+ T cells (Figure 8). Thus, our data strongly suggest that the enhanced immune protection induced by SeV85AB boosting was associated with establishment of an increased capacity to recall central memory CD4+ T cell responses.
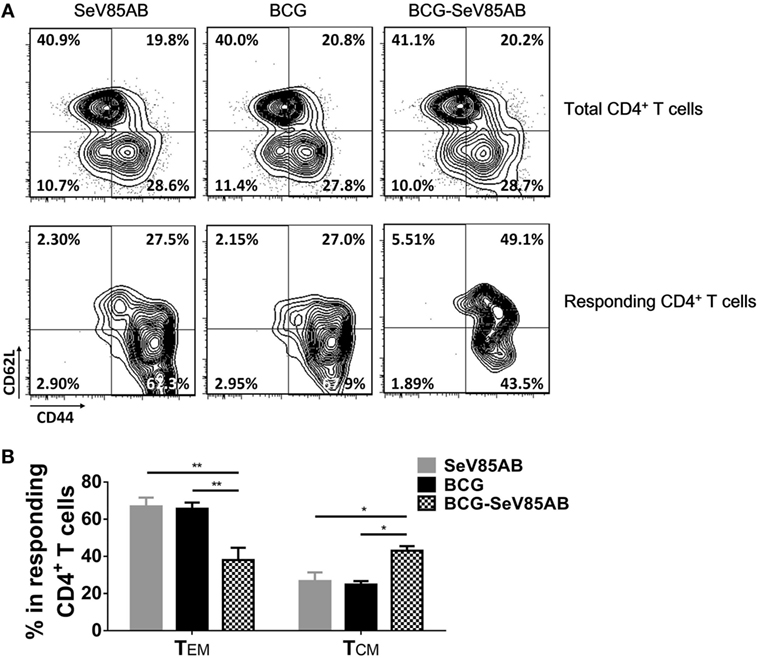
Figure 8. Memory phenotypes of recall T cell responses against Mycobacterium tuberculosis challenge. The memory phenotypes in total CD4+ T cells and Ag85AB-responding CD4+ T cells were assessed. The responding cells were defined as the cells that secreted at least one of the cytokines IFN-γ, IL-2, and TNF-α after Ag85AB peptide stimulation. Central memory T cells (TCM) and effector memory T cells (TEM) were defined as CD44+CD62L+ and CD44+CD62L−, respectively. Representative flow cytometric plots (A) and summarized data (B) are shown. Data are representative of two independent experiments with three mice per group. *P < 0.05 and **P < 0.01.
Up to now, most successful vaccines against microbial pathogens have depended on humoral immunity to achieve protection or even sterile eradication. However, as is the case for many intracellular bacteria, Mtb is able to resist most antibody-mediated antibacterial effects by growing inside macrophages (23). Thus, T cell-mediated immune responses are crucial for the development of anti-TB vaccines.
The only available anti-TB vaccine remains the almost 100-year-old BCG. The routine administration of BCG to infants provides significant protection against miliary and meningeal TB. However, the protective efficacy is inconsistent in adults. One explanation of BCG’s inadequate protection is a lack of an effective stimulation of an optimal blend of T cell populations (24). Considering that both CD4+ and CD8+ T cells play important roles in protective immunity against Mtb (3, 25, 26), the development of efficient memory CD4+ and CD8+ T cell responses is one of the main goals of the novel vaccine strategies against Mtb.
Although BCG is a strong inducer of Th1 CD4+ T cell immune responses, the incidence of active TB disease increases with time after BCG immunization (27), suggesting that a decline of immunological memory after BCG vaccination is one of the causes of the vaccine’s low protective efficacy. However, the waning of BCG protection was not prevented by a BCG revaccination strategy (28). Since the majority of human beings are BCG inoculated, and this seems unlikely to change, a prime-boost regimen is an attractive strategy to counter the waning immune memory post BCG. Heterologous prime-boost vaccination strategy is known to be highly effective for enhancing anti-TB T cell-mediated immunity (17, 29). For example, a vaccinia virus-vectored vaccine (MVA85A) and two adenovirus-based vaccines (AdAg85A and Crucell Ad35) are candidate booster vaccines that are under clinical evaluation. Although MVA85A was shown to be effective in boosting BCG-primed immune protection in a variety of Mtb animal infection models, a phase IIb clinical trial indicated that MVA85A may not be effective in humans (30). This failure suggested that novel anti-TB vaccine platforms or optimized delivery systems are urgently needed (31). Improvement would benefit from knowing which cellular responses constitute optimum protective immunity. Our analysis of the cells engaged in the enhanced protective response in the lungs of Mtb-challenged mice after BCG/SeV85AB prime/boost is illuminating.
In Mtb natural infection, T cells are initially primed in the draining lymph nodes of the infected lung, phagocytosis leads to the presentation and cross-presentation of Mtb peptide-loaded MHC-I and -II complexes, which provides the priming “first signal” at the DC surface (32). The “second signal,” a costimulatory signal, tunes the T cell responses by decreasing the activation thresholds of T cells, and pathogen-specific inflammation provides the “third signal” that shapes the maturation of T cells (33–36). A deficiency in any of these signals might lead to the inadequate activation of anti-TB T cell immune memory (37). Sendai virus was chosen here to serve as vector of a booster anti-TB vaccine primarily because it was known that Sendai virus treatment matured dendritic cells and led to complete elimination of tumors cells in vivo (38). In addition, Sendai virus tests had demonstrated a potent induction of type I interferon (39) and a Sendai virus-derived RNA agonist of RIG-I had been used as an adjuvant to enhance vaccine-induced immune responses by providing an inflammatory microenvironment (40), thereby optimizing antigen-specific CD4+ and CD8+ T cell responses (41). Based on this, we initiated this program of investigation and found that the novel Sendai virus vectored vaccine, SeV85AB, did indeed induce robust T cell responses and substantial protection against Mtb challenge (15).
Bacille Calmette–Guérin is a strong inducer of systematic CD4+ T cell immunity but fails to induce efficient CD8+ T cells. In contrast, a single immunization of SeV85AB was able to establish antigen-specific T cells responses and CD8+ T cells mediated immune protection (15). Here, we found that the strong antigen-specific CD4+ and modest CD8+ T cells primed by BCG vaccination were both enhanced by SeV85AB in the spleen. Most notably, the weak antigen-specific CD8+ T cell responses that were primed by BCG in the lung were significantly enhanced by the SeV85AB boosting. Heterologous prime-boost and recombinant protein anti-TB vaccine models have shown that the vaccine-induced IL-2-secreting CD4+ T cell subsets could maintain the IL-2-producing ability for at least 26 weeks post challenge infection and were associated with enhanced control of bacterial growth in mouse models (42, 43). Increased TNF-α/IFN-γ/IL-2 and decreased TNF-α/IFN-γ responses induced by BCG have been associated with protection against bovine TB (44). In this study, although the IL-2 response was not dominated in the vaccine-induced primary responses in the spleen, IL-2-producing poly-functional T cells were significantly boosted by SeV85AB, supporting the protective role of IL-2 in anti-TB vaccine-induced protection.
Moreover, it was shown that the antigen-specific IL-2+CD4+ T cell subsets were negative for KLRG1, which is a surface marker of terminally differentiated T cells, during Mtb challenge infection (42). We showed previously that the inadequacy of T cell immunity during chronic human TB infection is associated with the less-protective terminally differentiated T cell state marked by KLRG1 expression. Furthermore, the blockade of KLRG1 signaling increased CD4+ T cell function through enhancement of Akt pathway in human TB infection (45, 46), supporting the protective role of IL-2 in TB infection. In this study, IL-2-producing T cells were significantly enhanced post Mtb infection in the BCG prime-SeV85AB boost regimen group, again showing the recall of IL-2 responses was associated with protective immunity. IL-2 enhances competitiveness for survival in CD4+ T cells, thereby facilitating the development of a memory population (47). The CD4+ T cells that produce IL-2 could be sustained over a prolonged period and developed into effector cells following antigen stimulation as a result of quick recall response (48). In human TB infection, active TB patients had decreased IL-2-producing CD4+ T cells compared with latent TB infection, and 6 months of anti-TB treatment increased specific IL-2-producing T cells (49). In contrast, although also essential for host resistance against Mtb, IFN-γ responses do not always correlate with immune protection (26). Recently, Barber et al. also found that IFN-γ accounts for only ~30% of the cumulative CD4+ T cell-mediated reduction in lung bacterial loads, but increasing the per capita production of IFN-γ led to the early death of the host in murine TB infection (20). Thus, although it can be regarded as a positive marker of vaccine-induced primary T cell responses, the degree of infection-induced IFN-γ production might not always be the factor limiting immune protection against TB. Instead, our data support a key positive role of IL-2 in anti-TB immune protection. Thus, perhaps recall IL-2-mediated, instead of IFN-γ-mediated T cell responses is the critical factor normally limiting protection against TB infection.
Cumulatively, our data suggest that boosting BCG with SeV85AB might compensate for the weak induction by BCG of IL-2-dependent recall T cell immune responses in the lung. Since we showed here that an SeV85AB boost significantly enhanced the T cell immune memory induced by BCG vaccination in mice, further studies of SeV85AB in other animal models are warranted before a clinical trial of safety in humans.
This study was approved by the Institutional Animal Care and Use Committee and was performed according to the guidelines of the Laboratory Animal Ethical Board of Shanghai Public Health Clinical Center.
X-YF and TS conceive the project. X-YF and ZH designed the study, analyzed the data, and wrote the manuscript. ZH, LG, and C-LL performed the experiments. DL provided editorial input and improved the writing.
X-YF, TS, ZH, and DL are co-inventors of a patent application on the novel SeV85AB vaccine.
This work was supported by Grants from Chinese National Mega Science and Technology Program on Infectious Diseases (2018ZX10731301, 2018ZX10302301), National Science Foundation of China (31771004, 81501365), and Shanghai Science and Technology Commission (17ZR1423900), and Shanghai Municipal Commission of Health and Family Planning (20144Y0072).
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01796/full#supplementary-material.
1. Rodrigues LC, Diwan VK, Wheeler JG. Protective effect of BCG against tuberculous meningitis and miliary tuberculosis: a meta-analysis. Int J Epidemiol (1993) 22:1154–8. doi:10.1093/ije/22.6.1154
2. Bloom BR, Fine PEM. The BCG experience: implications for future vaccines against tuberculosis. In: Bloom BR, editor. Tuberculosis: Pathogenesis, Protection, and Control. Washington, DC: ASM Press (1994). p. 531–57.
3. Prezzemolo T, Guggino G, La Manna MP, Di Liberto D, Dieli F, Caccamo N. Functional signatures of human CD4 and CD8 T cell responses to Mycobacterium tuberculosis. Front Immunol (2014) 5:180. doi:10.3389/fimmu.2014.00180
4. Jasenosky LD, Scriba TJ, Hanekom WA, Goldfeld AE. T cells and adaptive immunity to Mycobacterium tuberculosis in humans. Immunol Rev (2015) 264:74–87. doi:10.1111/imr.12274
5. Kaufmann SH. Tuberculosis vaccines: time to think about the next generation. Semin Immunol (2013) 25:172–81. doi:10.1016/j.smim.2013.04.006
6. Urdahl KB. Understanding and overcoming the barriers to T cell-mediated immunity against tuberculosis. Semin Immunol (2014) 26:578–87. doi:10.1016/j.smim.2014.10.003
7. van Pinxteren LA, Cassidy JP, Smedegaard BH, Agger EM, Andersen P. Control of latent Mycobacterium tuberculosis infection is dependent on CD8 T cells. Eur J Immunol (2000) 30:3689–98. doi:10.1002/1521-4141(200012)30:12<3689::AID-IMMU3689>3.0.CO;2-4
8. Sousa AO, Mazzaccaro RJ, Russell RG, Lee FK, Turner OC, Hong S, et al. Relative contributions of distinct MHC class I-dependent cell populations in protection to tuberculosis infection in mice. Proc Natl Acad Sci U S A (2000) 97:4204–8. doi:10.1073/pnas.97.8.4204
9. Villarreal-Ramos B, McAulay M, Chance V, Martin M, Morgan J, Howard CJ. Investigation of the role of CD8+ T cells in bovine tuberculosis in vivo. Infect Immun (2003) 71:4297–303. doi:10.1128/IAI.71.8.4297-4303.2003
10. Chen CY, Huang D, Wang RC, Shen L, Zeng G, Yao S, et al. A critical role for CD8 T cells in a nonhuman primate model of tuberculosis. PLoS Pathog (2009) 5:e1000392. doi:10.1371/journal.ppat.1000392
11. Wu Y, Woodworth JS, Shin DS, Morris S, Behar SM. Vaccine-elicited 10-kilodalton culture filtrate protein-specific CD8+ T cells are sufficient to mediate protection against Mycobacterium tuberculosis infection. Infect Immun (2008) 76:2249–55. doi:10.1128/IAI.00024-08
12. Grode L, Seiler P, Baumann S, Hess J, Brinkmann V, Nasser EA, et al. Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis bacille Calmette-Guerin mutants that secrete listeriolysin. J Clin Invest (2005) 115:2472–9. doi:10.1172/JCI24617
13. Wang J, Santosuosso M, Ngai P, Zganiacz A, Xing Z. Activation of CD8 T cells by mycobacterial vaccination protects against pulmonary tuberculosis in the absence of CD4 T cells. J Immunol (2004) 173:4590–7. doi:10.4049/jimmunol.173.7.4590
14. Moliva JI, Hossfeld AP, Canan CH, Dwivedi V, Wewers MD, Beamer G, et al. Exposure to human alveolar lining fluid enhances Mycobacterium bovis BCG vaccine efficacy against Mycobacterium tuberculosis infection in a CD8(+) T-cell-dependent manner. Mucosal Immunol (2017) 11:968–78. doi:10.1038/mi.2017.80
15. Hu Z, Wong KW, Zhao HM, Wen HL, Ji P, Ma H, et al. Sendai virus mucosal vaccination establishes lung-resident memory CD8 T cell immunity and boosts BCG-primed protection against TB in mice. Mol Ther (2017) 25:1222–33. doi:10.1016/j.ymthe.2017.02.018
16. Nieuwenhuizen NE, Kaufmann S. Next-generation vaccines based on bacille Calmette-Guerin. Front Immunol (2018) 9:121. doi:10.3389/fimmu.2018.00121
17. Dalmia N, Ramsay AJ. Prime-boost approaches to tuberculosis vaccine development. Expert Rev Vaccines (2012) 11:1221–33. doi:10.1586/erv.12.94
18. Lewinsohn DA, Lewinsohn DM, Scriba TJ. Polyfunctional CD4(+) T cells as targets for tuberculosis vaccination. Front Immunol (2017) 8:1262. doi:10.3389/fimmu.2017.01262
19. Zeng G, Zhang G, Chen X. Th1 cytokines, true functional signatures for protective immunity against TB? Cell Mol Immunol (2018) 15:206–15. doi:10.1038/cmi.2017.113
20. Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, et al. CD4 T cell-derived IFN-gamma plays a minimal role in control of pulmonary Mycobacterium tuberculosis infection and must be actively repressed by PD-1 to prevent lethal disease. PLoS Pathog (2016) 12:e1005667. doi:10.1371/journal.ppat.1005667
21. Kumar P. IFNgamma-producing CD4(+) T lymphocytes: the double-edged swords in tuberculosis. Clin Transl Med (2017) 6:21. doi:10.1186/s40169-017-0151-8
22. Mourik BC, Lubberts E, de Steenwinkel J, Ottenhoff T, Leenen P. Interactions between type 1 interferons and the Th17 response in tuberculosis: lessons learned from autoimmune diseases. Front Immunol (2017) 8:294. doi:10.3389/fimmu.2017.00294
23. Nunes-Alves C, Booty MG, Carpenter SM, Jayaraman P, Rothchild AC, Behar SM. In search of a new paradigm for protective immunity to TB. Nat Rev Microbiol (2014) 12(4):289–99. doi:10.1038/nrmicro3230
24. Agger EM, Andersen P. A novel TB vaccine; towards a strategy based on our understanding of BCG failure. Vaccine (2002) 21:7–14. doi:10.1016/S0264-410X(02)00447-4
25. Zuniga J, Torres-Garcia D, Santos-Mendoza T, Rodriguez-Reyna TS, Granados J, Yunis EJ. Cellular and humoral mechanisms involved in the control of tuberculosis. Clin Dev Immunol (2012) 2012:193923. doi:10.1155/2012/193923
26. Sakai S, Mayer-Barber KD, Barber DL. Defining features of protective CD4 T cell responses to Mycobacterium tuberculosis. Curr Opin Immunol (2014) 29:137–42. doi:10.1016/j.coi.2014.06.003
27. Sterne JA, Rodrigues LC, Guedes IN. Does the efficacy of BCG decline with time since vaccination? Int J Tuberc Lung Dis (1998) 2:200–7.
28. Karonga Prevention Trial Group. Randomised controlled trial of single BCG, repeated BCG, or combined BCG and killed Mycobacterium leprae vaccine for prevention of leprosy and tuberculosis in Malawi. Lancet (1996) 348:17–24. doi:10.1016/S0140-6736(96)02166-6
29. Brennan MJ, Clagett B, Fitzgerald H, Chen V, Williams A, Izzo AA, et al. Preclinical evidence for implementing a prime-boost vaccine strategy for tuberculosis. Vaccine (2012) 30:2811–23. doi:10.1016/j.vaccine.2012.02.036
30. Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet (2013) 381:1021–8. doi:10.1016/S0140-6736(13)60177-4
31. Meyer J, McShane H. The next 10 years for tuberculosis vaccines: do we have the right plans in place? Expert Rev Vaccines (2013) 12:443–51. doi:10.1586/erv.13.19
32. Ottenhoff TH, Kaufmann SH. Vaccines against tuberculosis: where are we and where do we need to go? PLoS Pathog (2012) 8:e1002607. doi:10.1371/journal.ppat.1002607
33. Hu Z, Wang J, Wan Y, Zhu L, Ren X, Qiu S, et al. Boosting functional avidity of CD8+ T cells by vaccinia virus vaccination depends on intrinsic T-cell MyD88 expression but not the inflammatory milieu. J Virol (2014) 88:5356–68. doi:10.1128/JVI.03664-13
34. Khan N, Gowthaman U, Pahari S, Agrewala JN. Manipulation of costimulatory molecules by intracellular pathogens: veni, vidi, vici!! PLoS Pathog (2012) 8:e1002676. doi:10.1371/journal.ppat.1002676
35. Etna MP, Giacomini E, Severa M, Coccia EM. Pro- and anti-inflammatory cytokines in tuberculosis: a two-edged sword in TB pathogenesis. Semin Immunol (2014) 26:543–51. doi:10.1016/j.smim.2014.09.011
36. Hu Z, Zhu L, Wang J, Wan Y, Yuan S, Chen J, et al. Immune signature of enhanced functional avidity CD8(+) T cells in vivo induced by vaccinia vectored vaccine. Sci Rep (2017) 7:41558. doi:10.1038/srep41558
37. Egen JG, Rothfuchs AG, Feng CG, Horwitz MA, Sher A, Germain RN. Intravital imaging reveals limited antigen presentation and T cell effector function in mycobacterial granulomas. Immunity (2011) 34:807–19. doi:10.1016/j.immuni.2011.03.022
38. Shibata S, Okano S, Yonemitsu Y, Onimaru M, Sata S, Nagata-Takeshita H, et al. Induction of efficient antitumor immunity using dendritic cells activated by recombinant Sendai virus and its modulation by exogenous IFN-beta gene. J Immunol (2006) 177:3564–76. doi:10.4049/jimmunol.177.6.3564
39. Buggele WA, Horvath CM. MicroRNA profiling of Sendai virus-infected A549 cells identifies miR-203 as an interferon-inducible regulator of IFIT1/ISG56. J Virol (2013) 87:9260–70. doi:10.1128/JVI.01064-13
40. Martinez-Gil L, Goff PH, Hai R, Garcia-Sastre A, Shaw ML, Palese P. A Sendai virus-derived RNA agonist of RIG-I as a virus vaccine adjuvant. J Virol (2013) 87:1290–300. doi:10.1128/JVI.02338-12
41. Seki S, Matano T. Development of a Sendai virus vector-based AIDS vaccine inducing T cell responses. Expert Rev Vaccines (2016) 15:119–27. doi:10.1586/14760584.2016.1105747
42. Lindenstrom T, Knudsen NP, Agger EM, Andersen P. Control of chronic Mycobacterium tuberculosis infection by CD4 KLRG1-IL-2-secreting central memory cells. J Immunol (2013) 190:6311–9. doi:10.4049/jimmunol.1300248
43. Woodworth JS, Aagaard CS, Hansen PR, Cassidy JP, Agger EM, Andersen P. Protective CD4 T cells targeting cryptic epitopes of Mycobacterium tuberculosis resist infection-driven terminal differentiation. J Immunol (2014) 192:3247–58. doi:10.4049/jimmunol.1300283
44. Maggioli MF, Palmer MV, Thacker TC, Vordermeier HM, McGill JL, Whelan AO, et al. Increased TNF-alpha/IFN-gamma/IL-2 and decreased TNF-alpha/IFN-gamma production by central memory T cells are associated with protective responses against bovine tuberculosis following BCG vaccination. Front Immunol (2016) 7:421. doi:10.3389/fimmu.2016.00421
45. Hu Z, Zhao HM, Li CL, Liu XH, Barkan D, Lowrie DB, et al. The role of KLRG1 in human CD4+ T-cell immunity against tuberculosis. J Infect Dis (2018) 217:1491–503. doi:10.1093/infdis/jiy046
47. Dooms H, Kahn E, Knoechel B, Abbas AK. IL-2 induces a competitive survival advantage in T lymphocytes. J Immunol (2004) 172:5973–9. doi:10.4049/jimmunol.172.10.5973
48. Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol (2008) 8:247–58. doi:10.1038/nri2355
Keywords: tuberculosis, Sendai virus, vaccine, T cell responses, prime-boost, IL-2
Citation: Hu Z, Gu L, Li C-L, Shu T, Lowrie DB and Fan X-Y (2018) The Profile of T Cell Responses in Bacille Calmette–Guérin-Primed Mice Boosted by a Novel Sendai Virus Vectored Anti-Tuberculosis Vaccine. Front. Immunol. 9:1796. doi: 10.3389/fimmu.2018.01796
Received: 28 April 2018; Accepted: 20 July 2018;
Published: 03 August 2018
Edited by:
Christophe Chevalier, Institut National de la Recherche Agronomique (INRA), FranceReviewed by:
Juraj Ivanyi, King’s College London, United KingdomCopyright: © 2018 Hu, Gu, Li, Shu, Lowrie and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Yong Fan, eHlmYW4wMDhAZnVkYW4uZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.