 Li Yan
Li Yan Dejin Zheng
Dejin Zheng Ren-He Xu
Ren-He Xu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 20 July 2018
Sec. Immunological Tolerance and Regulation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01658
This article is part of the Research TopicThe Role of TNF-TNFR2 Signal in Immunosuppressive Cells and its Therapeutic ImplicationsView all 20 articles
Mesenchymal stem cells (MSCs) have been broadly used as a therapy for autoimmune disease in both animal models and clinical trials. MSCs inhibit T effector cells and many other immune cells, while activating regulatory T cells, thus reducing the production of pro-inflammatory cytokines, including tumor necrosis factor (TNF), and repressing inflammation. TNF can modify the MSC effects via two TNF receptors, i.e., TNFR1 in general mediates pro-inflammatory effects and TNFR2 mediates anti-inflammatory effects. In the central nervous system, TNF signaling plays a dual role, which enhances inflammation via TNFR1 on immune cells while providing cytoprotection via TNFR2 on neural cells. In addition, the soluble form of TNFR1 and membrane-bound TNF also participate in the regulation to fine-tune the functions of target cells. Other factors that impact TNF signaling and MSC functions include the gender of the host, disease course, cytokine concentrations, and the length of treatment time. This review will introduce the fascinating progress in this aspect of research and discuss remaining questions and future perspectives.
Among many multipotent stem cell types, mesenchymal stem cells (MSCs) are a unique cell type that possesses not only stem cell properties but also immunomodulatory capabilities. MSCs refer to multipotent cells derived from the mesenchyme—the embryonic connective tissue that originates from the mesoderm. MSCs can differentiate into a wide variety of cells from the mesoderm, including osteocytes, chondrocytes, adipocytes, and smooth muscle cells (1, 2), and some cell types from the other germ layers, such as neurons from the ectoderm (3, 4) and hepatocytes from the endoderm (5, 6). Recently, neural crest cells were identified as another source giving rise to mesenchymal progenitors, which, similar to MSCs, have a high potential to differentiate into osteocytes and chondrocytes (7, 8). MSCs can be isolated from many fetal and adult tissues or differentiated from human pluripotent stem cells (hPSCs). In vitro and in vivo studies have demonstrated that MSCs modulate immune responses and inflammation and execute cytoprotective and reparative effects mainly through cell–cell contact and paracrine mechanisms. Thus, MSCs have been used as a cell therapy for an increasing number of autoimmune, inflammatory, and degenerative diseases (1, 2).
Autoimmunity and chronic inflammation are known to share numerous factors, and thus, frequently coexist in the same patients. Autoimmune disease occurs when the immune system abnormally attacks a part of a normal body. Approximately 80 types of autoimmune diseases have been identified, and these diseases can involve almost any part of the body. The abnormal immune response is often associated with complicated genetic factors and the environment. Autoimmune disease is a common and often serious clinical problem due to the chronic nature, high incidence in human populations, especially in women, and rising cost of healthcare. Among the list of common autoimmune diseases, rheumatoid arthritis (RA) (9), inflammatory bowel disease (IBD) (10), and type-1 diabetes (T1D) (11) are on the top. Approximately 7% of people in the United States are affected by autoimmune disease. Tumor necrosis factor (TNF or TNFα), which is involved in a wide range of biological functions, is considered the master mediator of the pathogenesis of chronic inflammation and autoimmune diseases. Therefore, anti-TNF therapies have become mainstay treatments for autoimmune and inflammatory diseases.
Mesenchymal stem cells are susceptible to environmental changes, and their immunosuppressive functions can be modulated when exposed to an inflammatory milieu (12). TNF and other pro-inflammatory cytokines, such as interferon γ (IFNγ) and interleukin 1 (IL-1), determine the disease onset, severity, and relapse of autoimmune diseases and affect the efficacy of treatment, including MSC-based therapy. IFNγ, TNF, and IL-1 present in inflammatory tissues can augment the immunosuppressive functions of MSCs (13–15). Priming of MSCs with IFNγ can yield an augmented immunosuppressive population with a higher efficacy for anti-inflammatory treatment than non-primed MSCs (16). Primed MSCs have been broadly applied in both basic and clinical research (17). However, no focused review has discussed the role of TNF signaling in MSC-based therapy of autoimmune and inflammatory diseases, given the great progress in this area of research. TNF exerts its functions by binding to two receptors (TNFR1 and TNFR2) to regulate the survival, proliferation, migration, and differentiation of target cells, especially immune cells. This molecule also interacts with MSCs to modify or mediate their therapeutic effects. This review, aimed to introduce the progress in this area, will specifically discuss how TNF/TNFR and MSCs converge on the immune system to prevent autoimmune and inflammatory diseases.
Mesenchymal stem cells have tremendous potential as a cellular therapy for autoimmune and inflammatory diseases because of their strong immunomodulatory effects and tissue regenerative capability. A growing number of translational studies have been carried out on MSCs for the treatment of many autoimmune and inflammatory diseases, including T1D (18), RA (19), IBD (20), ulcerative colitis (21), systemic lupus erythematosus (SLE) (22), autoimmune uveitis (23, 24), and Sjogren’s syndrome (25). So far, over 5,000 MSC-related clinical trials have been registered at ClinicalTrials of the National Institutes of Health in the U.S. (https://clinicaltrials.gov/), of which over 1,900 trials have been completed. Both autologous and allogenic MSCs were used in these trials, in which bone marrow (BM), adipose tissue, umbilical cord, placenta, and dental pulp were the most common sources for MSCs. In addition, MSCs differentiated from hPSCs, including embryonic stem cells and induced pluripotent stem cells (iPSCs), have also been examined and demonstrated efficacy on a variety of animal disease models and may become new options for future clinical applications (21, 26–28).
Mesenchymal stem cells regulate the adaptive immune system by promoting the generation of regulatory T cells (Tregs) and repressing the functions of T effector (Teff) and B effector cells (29–31). These effects are mainly triggered by exposure to pro-inflammatory cytokines, such as TNF, IFNγ, and IL-1β, which are widely present in tissues affected by inflammatory and autoimmune diseases. For instance, TNF deregulates the balance between Tregs and pathogenic Th17 and Th1 cells in the synovium of RA patients and impairs Treg functions in RA and MS patients (32, 33). Systemically transplanting MSCs into patients leads to a decrease in the number of Teff cells and restoration of Treg functions (22, 34). Moreover, IFNγ-primed MSCs inhibit B cell differentiation by arresting the cell cycle and inducing apoptosis (35).
As for innate immunity, MSCs can inhibit natural killer (NK) cell cytotoxicity and block the differentiation and/or maturation of macrophages and dendritic cells (DCs). MSCs skew the polarization of macrophages from M1 to M2 in wound healing (36) and inhibit DC generation and migration to lymph nodes in vivo (37). Studies of the molecular mechanisms for the therapeutic effects of MSCs have revealed that MSCs modulate immune responses and promote tissue repair via secretion of soluble factors and direct cell–cell contact (29). MSCs exert immunosuppressive effect by secreting soluble factors, such as indoleamine 2,3-dioxygenase (IDO), prostaglandin E2 (PGE2), hepatocyte growth factor (HGF), transforming growth factor-β1, insulin-like growth factor-1 (IGF-1), nitric oxide, and human leukocyte antigen-G5 (38, 39). Inhibition of IDO or PGE2 synthesis results in reduction of MSC-mediated immunosuppression, and priming MSCs with pro-inflammatory cytokines, such as IFNγ and TNF, enhances the immunosuppressive effects by elevating the secretion of IDO, CXCR4, and PGE2 (29, 39–42). MSCs mixed with activated T cells have the strongest inhibition on the T cells via direct cell contact (43), and upregulated expression of intercellular adhesion molecule-1 and vascular adhesion molecule-1 in MSCs strengthens their interaction with T cells (44).
Although promising results have been obtained from MSC-based therapy, the outcomes are not always consistent and sometimes even contradictory, depending on the delivery strategies, MSC sources, and disease course (45–49). A phase I study reported that 7/10 patients with Crohn’s disease did not respond to autologous BM-MSC infusion, and three of them even manifested worsened symptoms (50). Site-specific administration of MSCs to patients with Crohn’s disease and mice with collagen-induced arthritis (CIA) appeared to be more effective than systemic injection (51, 52). It has been well documented that the functions of MSCs depend on the microenvironment. MSCs often manifest immunosuppressive effects in a strong inflammatory milieu, and this ability is reduced or lost and the immunogenicity of the cells increased in a weak inflammatory environment (2). Long-term exposure to IFNγ or TNF even converts MSCs from an immunosuppressive to pro-inflammatory status (53–55). Moreover, MSCs are effective at disease onset or when the symptoms reach peaks but fail to alleviate the symptoms after the disease stabilizes or during disease progression (46, 56).
In addition, the origin of MSCs also influences their immunomodulatory effects. For example, autologous BM-MSCs from patients with SLE or synovial-derived MSCs from patients with RA failed to improve the symptoms of the same donor patients (47, 57). Adipose-derived MSCs from mice with experimental autoimmune encephalomyelitis (EAE) had no therapeutic effect on the donor animals (58). MSCs isolated from obese mice or non-obese diabetic mice failed to alleviate the symptoms in EAE and T1D mice (18, 59). Thus, choosing MSCs from the right source and determining the immunomodulatory effects of MSCs are necessary before therapeutic applications.
Currently, 19 members have been identified in the TNF superfamily (TNFSF), including TNF, TNFβ, CD40L, FasL, and TRAIL, which participate in diverse cellular activities, including inflammation, cell proliferation, apoptosis, and morphogenesis (60). In particular, TNF is abundant in the serum and many other body fluids in patients with autoimmune disease. TNF is a trimeric type-II transmembrane protein that shares a TNF homology domain with the other TNFSF members and is produced mainly by activated macrophages, T, B, and NK cells. TNF is present in two different forms, the membrane-bound TNF (mTNF) and soluble TNF (sTNF or TNF), and TNF is cleaved from mTNF via metalloproteinases, such as TNF-converting enzyme (TACE) (61–63).
Tumor necrosis factor and sTNF bind to two structurally distinct transmembrane receptors, TNFR1 and TNFR2, both belonging to the TNFR superfamily, which comprises trimeric type-I transmembrane proteins with repeated extracellular cysteine-rich domains for ligand binding; the two receptors regulate gene expression via different signaling pathways (61). TNFR1 can be activated by both mTNF and TNF, whereas TNFR2 preferentially binds to mTNF to initiate the activation of the receptor (64). Moreover, TNFR1 is expressed on almost all cells of the body, whereas TNFR2 is expressed only on limited cells, e.g., immune cells, endothelial cells, nerve cells, and MSCs. TNFR also includes membrane-bound (mTNFR or TNFR) and soluble (sTNFR) forms, and sTNFR is cleaved from TNFR by TACE (63).
In general, TNF induces cell apoptosis or survival through at least five different signals, including caspase, NFκB, ERK, JNK, and P38 MAPK pathways, via TNFR1 and -R2 (60). TNFR1 contains 434 amino acids, and its intracellular region contains a death domain (DD), which recruits the TNF-associated death domain (TRADD), and the latter then recruits Fas-associated death domain to trigger the caspase cascades and apoptosis. In addition, TNFR1 also induces reactive oxygen species release from mitochondria to activate apoptotic events. Paradoxically, TRADD can also recruit the TNFR-associated factor (TRAF2) to initiate the NFκB, ERK, JNK, and p38 MAPK signaling pathways to regulate the cell survival and proliferation. By contrast, TNFR2 consists of 439 amino acids and does not include a cytoplasmic DD, which binds to TRAF2 directly and activates pro-survival genes through the NFκB, ERK, JNK, and p38 MAPK pathways (60). There is some degree of cross talk between the TNFR1 and -R2 signaling pathways.
Another key feature of TNF signaling is the phenomenon called “reverse signaling,” in which the signal transmits from the TNFRs (including their membrane-bound and soluble forms) to mTNF-bearing cells (outside to inside). Reverse signaling of TNF has been shown to be functional in macrophages and T, B, and NK cells in humans. For example, activation of mTNF reverse signaling enhances the cytotoxicity of CD8 T cells and NK cells and the survival of B cells (65–67). In addition, soluble TNFR1 (sTNFR1)-stimulated monocytes manifest pro-inflammatory effects without TNF treatment and anti-inflammatory effects after TNF treatment, as reflected by regulation of the pro-inflammatory cytokines IL1β and IL8 (68). Moreover, the mTNF reverse signaling renders macrophage resistant to LPS-induced effects by inducing TGFβ expression (69, 70). It has been shown that the cytoplasmic domain of mTNF contains a casein consensus sequence, which is dephosphorylated during activation of the mTNF reverse signal. mTNF then triggers the p38 MAPK and JNK pathways via interaction with protein kinases. Alternatively, a 10-kDa cytoplasmic domain of mTNF can be cleaved and translocated into the nucleus to regulate the expression of various cytokines, such as IL1β and IL12 (71). However, how the mTNF reverse signal works has not yet been fully understood.
The important role of TNF in autoimmune and inflammatory disease has been supported by large amounts of evidence from clinical studies. TNF and sTNFR1 are recognized as useful indicators for assessing disease activity. For example, they are often at high levels in patients with RA and ankylosing spondylitis (72). In SLE patients, TNF is also elevated, and circulating sTNFR is significantly higher than in patients with RA and spondyloarthropathies (73). Chronic progressive MS patients manifest elevated TNF in CSF and active lesions compared with serum (74). The TNF level correlates with the manifestation and degree of disability in patients.
A vast number of animal studies have uncovered much more knowledge than clinical trials about the pathogenesis mediated by TNF. Transgenic mice overproducing TNF develop severe inflammatory arthritis, and the disease onset depends on IL1 production (75). IL17 promotes osteoclastogenesis by stimulating TNF production (76). In IBD patients, TNF disrupts the intestinal epithelial barrier, which makes the intestines vulnerable to infections, thus promoting inflammation (77). Mice overexpressing TNF develop chronic inflammation resembling IBD (77). However, TNF/lymphotoxin knockout or ectopic expression of mTNF delays the disease onset of EAE in mice (78, 79).
Activation of TNF/TNFR1 signaling predominantly promotes inflammation and tissue degeneration. Interaction of TNF with TNFR1 activates Teff cells and guides the migration of Teff cells to inflammatory sites (80); for example, CD4+ Teff cells are preferably accumulated in synovial joints in RA patients (81). Meanwhile, TNFR1 knockout prevents the development of arthritis and IBD in mice (82) and shortens the disease course of EAE and T1D in mice (78, 83), indicating a pro-inflammatory role of TNFR1 signaling. Furthermore, TNFR1 signaling likely impairs Treg functions via induction of the dephosphorylation of FoxP3 by protein phosphatase 1 in the inflamed synovium of RA, accompanied by increased numbers of Th17 and IFN+ CD4 T cells (84). Thus, TNF and TNFR1 have been used as therapeutic targets for the treatment of autoimmune and inflammatory diseases. An anti-TNFR1 nanobody protects against EAE development in mice (85), and sTNFR1 has been used as a natural inhibitor of TNFR1 signaling by binding and saturating TNF to repress its signaling (64).
In contrast to the pro-inflammatory effects of TNFR1, the TNF/TNFR2 interaction preferentially mediates immunosuppressive effects (86–89). In mice with dextran sulfate sodium-induced colitis, TNFR1 ablation exacerbated the severity of the disease, while TNFR2 deficiency led to the opposite results (90). TNFR2 knockout in EAE mice accelerated the disease progression accompanied by severe demyelination (78), suggesting a repressive role of TNFR2 in the disease development. Similarly, polymorphisms in TNFR2 have been found in various autoimmune diseases, which might lead to deregulation of TNF signaling via upregulation or shedding of TNFR2 (91).
TNFR2 has been identified as a marker for activated Tregs. TNFR2 and its ligands can activate and stabilize Tregs in an inflammatory environment (92–94). A subset of Tregs with high TNFR2 expression exhibits maximally suppressive activities in both mouse and human, which makes them the most desirable cells for the treatment of autoimmune and inflammatory diseases (95, 96). Furthermore, TNFR2 agonists have proved effective for the treatment of autoimmune disease (91, 97). Upon stimulation, TNFR2 is rapidly upregulated in Tregs, which are empowered to exert stronger immunosuppressive effects on Teff cells than non-stimulated Tregs (93).
However, stimulation of TNFR2 on Teff (e.g., Th1, Th17, and CD8+) cells promotes the cells to proliferate, secrete cytokines, and develop resistance to Treg-mediated suppression (95, 98–100). For example, the CD25hi/TNFR2+ Treg subset induced upon TCR stimulation allows the identification of maximal cytokine-producing effectors (101). These lines of evidence indicate the complex effects of TNFR2 on T cells, which help balance between Treg and Teff cells and partially explain the reasons for the controversial responses of some patients to TNFR2 agonists.
Although beneficial effects of TNF therapies have been observed in patients with RA, Crohn’s disease, SLE, and psoriasis, clinical trials on MS patients showed the opposite effects, with worsening of their symptoms (102). Adverse effects have also been found in trials on patients with optic neuritis, MS, and other demyelinating diseases following anti-TNF medications (103, 104). The adverse effects occurred in 0.05–0.2% of patients treated with three licensed anti-TNF agents. The opposing outcomes of TNF therapies may result from the dual effects of TNF on inflammation in the CNS.
Circulating TNF in the periphery can cross the blood–brain barrier (BBB) and enter the CNS. Infiltrating immune cells such as macrophages as well as activated microglia in the CNS can produce TNF (105). Generally, binding of TNF to TNFR1 predominantly mediates pro-inflammatory effects of TNF accompanied by activation of the target cells. In murine models of ischemia and EAE, TNFR1-ablation reduced neuronal loss and demyelination (105, 106). In addition, TNFR1 signaling activates microglia to promote neural inflammation due to increased production of pro-inflammatory factors including TNF, IL-1β, and IL-6 (107). TNF also induces apoptosis of human adult oligodendrocytes by causing mitochondrial dysfunction via TNFR1/JNK-3 signaling pathway and inhibits differentiation of oligodendrocyte progenitor cells (OPC) via AMPK activation and mitochondrial impairment (108, 109). These results indicate the adverse effects of TNFR1 signaling on multiple cell types in the CNS during the disease progression.
By contrast, upregulation of TNFR2 in OPC, microglia, and astrocytes promotes neuroprotection and remyelination, as observed in TNFR1-ablated mice with cerebral ischemia and EAE (105). TNFR2 ablation impairs OPC differentiation and causes dysfunction of oligodendrocytes (110). TNFR2 signaling promotes OPC differentiation and remyelination by inducing secretion of CXCL12 and leukemia inhibitory factor from astrocytes (111) and protects oligodendrocytes from oxidative stress-induced damage (112).
In addition, TNFR2 ablation in microglia in the CNS accelerates the onset of EAE, whereas disruption of TNFR2 in monocytes/macrophages suppresses the disease progression accompanied by reduction of T cell activation and infiltration, and attenuated demyelination (113), indicating that TNFR2 plays opposite roles even in microglia and macrophages during development of EAE. Activated microglia enhance the myelin debris clearance and remyelination, which is likely mediated by TNFR2 signaling (113, 114). These findings are instrumental for developing tissue- and receptor-specific medications to target TNF signaling in the treatment of different autoimmune and inflammatory diseases.
Interferon γ affects MSC efficacy in a dose-dependent manner. At low concentrations, it completely abolishes the therapeutic effect of MSCs on EAE, accompanied by increased secretion of the pro-inflammatory chemokine CCL2 and elevated expression of major histocompatibility complex molecules (115). At higher concentrations, IFNγ strengthens the MSC efficacy to reduce the severity of induced colitis in mice (27, 41). Similarly, TNF also dose dependently alters MSC functions. For example, osteogenic differentiation from murine ST2 MSCs is promoted by TNF at lower concentrations as indicated by elevated expression of the osteogenic genes Runx2, Osx, OC, and ALP but inhibited by TNF at higher concentrations, which depends on NFκB signaling (116). Compared with non-primed controls, TNF-primed MSCs have stronger immunomodulatory and tissue-repair capacity, evidenced by increased secretion of immunosuppressive molecules, such as PGE2, sTNFR, and TSG-6 (42, 117–123); chemokines, such as IL-8, CXCL5, and CXCL6 (124, 125); growth factors, such as HGF, IGF1, and VEGF (126–128); and increased tunneling nanotube (TNT) formation (129) through the TNFR1 or TNFR2 signaling pathway. The important effects of MSC through TNF signaling are listed in Table 1.
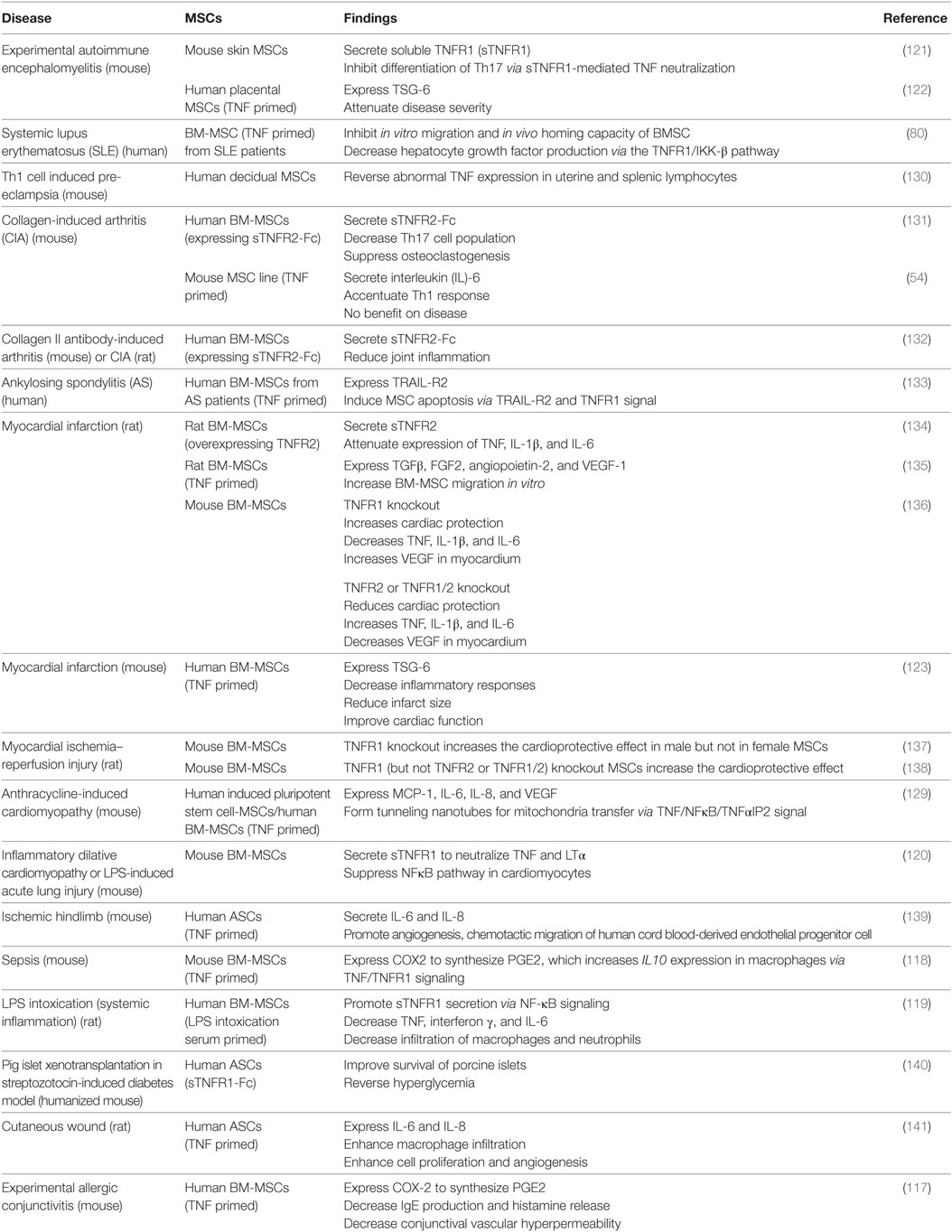
Table 1. Tumor necrosis factor (TNF) regulation of mesenchymal stem cell (MSC) efficacy on autoimmune and inflammatory diseases.
Generally, TNFR1-mediated signaling reduces the MSC efficacy. For example, BM-MSCs derived from mice with TNFR1 knockout caused greater recovery of myocardial functions in a rat model of acute ischemia than wild-type MSCs, which was associated with increased production of VEGF and decreased production of the pro-inflammatory factors TNF, IL-1β, IL-6, etc., in the myocardium (136, 138). Interestingly, another study found that TNFR1 knockout only increased the cardioprotective effect of male, but not female, MSCs in a murine ischemic injury model (137), indicating that the effect of TNFR1 signaling is gender dependent.
TNFR1 signaling reduces MSC efficacy by inhibiting the production of immunosuppressive molecules and growth factors. For example, TNF-priming reversed the immunosuppressive effect of mouse MSCs on T cell proliferation, accompanied by increased secretion of the pro-inflammatory cytokine IL-6 and failure of the MSCs in the treatment of murine CIA (54). In addition, ablation of TNFR1 remarkably increased TNF-stimulated HGF production from human BM-MSCs (142), indicating the inhibitory effect of TNFR1 signaling in HGF production. Similar effects have been observed on MSCs derived from patients with autoimmune diseases. For instance, it has been shown that TNF treatment decreased the HGF production by BM-MSCs derived from SLE patients via the TNFR1/IKK-β pathway (80) and induced apoptosis in BM-MSCs from ankylosing spondylitis patients via TNFR1-mediated upregulation of TRAIL-R2 (133).
Interestingly, in some scenarios, TNFR1 signaling can enhance MSC efficacy by inducing production of immunomodulatory molecules. For example, TNFR1 knockdown in mouse skin-derived MSCs abrogated their therapeutic effects on EAE accompanied by reduced inhibition on the polarization of Th17 cells (121), which might be partially explained by the loss of beneficial effects of sTNFR1 produced by MSC under the inflammatory situation. In addition, in dilative cardiomyopathy, acute lung injury, and LPS-induced intoxication, both murine and human BM-MSCs primed by TNF or inflammatory serum secreted more sTNFR1 than the non-primed controls, which promotes disease recovery (119, 120). In addition, human adipose-derived MSCs engineered to express sTNFR1-Fc improved the survival of porcine islets and reversed the hyperglycemia in a mouse model of streptozotocin-induced diabetes (140). sTNFR1 may act by neutralizing circulating TNF and activating mTNF-mediated reverse signaling in immune cells during diseases progression.
TNFR1 signaling can also increase PGE2 secretion by inducing COX2 expression in mouse or human BM-MSCs, which in turn reprograms host macrophages to increase IL-10 production thus inhibiting inflammation in a mouse sepsis model and experimental allergic conjunctivitis (117, 118). In addition, it has been shown that other immunosuppressive molecules, growth factors, and chemokines such as TSG-6, TGFβ, and IL-8 were produced by TNF-primed MSCs to attenuate the symptoms in diseases including EAE, myocardial infarction, ischemic hind limb, and cutaneous wound probably via TNFR1 signaling pathway (122, 135, 139, 141). TNF can also induce TNT formation between iPSC-derived MSC and cardiomyocytes for mitochondria transfer to attenuate the damage in mouse anthracycline-induced cardiomyopathy, which is regulated by TNF/NFκB/TNF-IP2 signaling pathway (129). Thus, TNFR1 signaling can exert dual effects on MSC-based therapy in autoimmune and inflammatory diseases, depending on the type and stage of the diseases.
In contrast to the dual effects of TNFR1, TNFR2-mediated signaling enhances MSC efficacy in general. For example, compared with wild-type controls, both male and female murine BM-MSCs with TNFR2 knockout showed less or no myocardial functional recovery in a rat model of acute ischemia accompanied by increased production of pro-inflammatory factors and a reduced level of VEGF in the myocardium (136, 138). These results are consistent with the in vitro observations that production of VEGF, IGF-1, and HGF by TNF-primed human BM-MSCs is mediated through the TNFR2 signaling (126–128). Consistently, TNFR2 knockout reduced the secretion of VEGF and IGF-1 by TNF-primed BM-MSCs, but this only happened on BM-MSC from female mice. By contrast, secretion of these growth factors increased in TNF-primed TNFR2−/− BM-MSCs from male mice (143, 144), and TNFR2−/− BM-MSCs from male mice failed to promote myocardial functional recovery (136, 138). The opposite outcomes implicate that the effects of TNFR2 signaling, like TNFR1 signaling, on MSC functions are also gender dependent. In support of this, the male sex hormone testosterone has been reported to exert deleterious effect on myocardial recovery in a rat model (145, 146).
Furthermore, overexpression of sTNFR2 or TNFR2 in human or rat BM-MSCs enhanced their therapeutic effects in mice and rats with RA (131, 132) and rats with cardiac ischemia (134, 147), which was associated with reduced TNF level and attenuated expression of IL1β and IL6. Macrophages are a major cell type that secretes TNF. Treating activated macrophages with culture supernatant of human sTNFR2-expressing MSCs reduced osteoclast formation in vitro (131). Similar to sTNFR1, sTNFR2 may also execute cytoprotective effect via neutralization of circulating TNF or induction of mTNF-mediated reverse signaling in immune cells.
The expression of TNFR2 is highly upregulated in oligodendrocytes, microglia, astrocytes, and several subsets of neurons in neurological diseases (105, 148). TNFR2 on astrocytes mediates beneficial activities to protect oligodendrocytes in co-culture (111). Upregulated TNFR2 on activated microglia promotes the clearance of myelin debris and remyelination (149). In addition, MSCs that infiltrate into the CNS can exert immunomodulatory effects by regulating the local microglia and astrocytes as well as infiltrating immune cells, e.g., suppressing the functions of Teff cells and macrophages and promoting the proliferation of Tregs (150). Moreover, TNF in inflamed CNS induces MSC to secrete immunomodulatory factors and neural tropic factors such as BDNF and HGF (151), which exert pleiotropic effects to attenuate the brain inflammation, reduce brain damage, and promote neural regeneration.
Regulatory T cells play a central role in the maintenance of the immune balance to tolerate self-antigens and prevent autoimmunity (152). In general, they refer to CD4+/FOXP3+ T cells, including two major subtypes: natural Treg (nTreg) cells and induced adaptive Treg (iTreg) cells. nTreg cells are generated and selected in the thymus and then migrate to peripheral tissues (153), while iTreg cells acquire CD25 (IL-2Rα) expression outside of the thymus and are typically induced by inflammation and during disease processes, such as autoimmunity and cancer (152). T cell receptor stimulation and the cytokines TGFβ and IL-2 are required for iTreg cell generation in vitro and in vivo (95, 154, 155). In contrast to the pro-inflammatory effects of TNF/TNFR1 signaling (156), TNF/TNFR2 signaling preferentially activates, stabilizes, and expands Tregs to mediate their immunosuppressive effects and contribute to the treatment of autoimmune disease (86–89). TNFR2 is an expression marker relevant to Treg functions. TNFR2 agonists have been shown to be effective for the treatment of autoimmune and inflammatory diseases (91, 97).
Mesenchymal stem cells regulate both innate and adaptive immune systems partially by promoting the generation of Tregs (29–31). In the presence of high levels of inflammatory cytokines, e.g., TNF and IFNγ, MSCs produce various soluble factors, such as IDO, TGFβ, PGE2, and IGF, to inhibit Teff cells and increase the expression of FOXP3, CTLA4, and GITR in Tregs to enhance their immunosuppressive effects (53). Cell-to-cell contact also mediates the induction of Tregs by cytokine-primed MSCs (53). Overexpression of inducible co-stimulator ligands in MSCs promotes the induction of functional Tregs (157).
In addition, MSCs also modulate antigen-presenting cells, such as DCs and macrophages, by converting them to anti-inflammatory phenotypes (M2), which then promote Treg expansion and suppress Teff cell functions (30). Recently, Miyagawa et al. reported that MSCs control Treg proliferation by releasing IGFBP4, an inhibitor of IGF (53). Moreover, some studies have shown that low levels of IFNγ and TNF or long-term exposure to these cytokines converts MSC from an immunosuppressive to pro-inflammatory status (53–55). Thus, these pro-inflammatory cytokines can modify MSC effects on Tregs, altering their efficacy on autoimmune and inflammatory diseases.
Mesenchymal stem cells have demonstrated immunosuppressive effects against various autoimmune and inflammatory diseases. However, the efficacy of MSC on many of the diseases remains controversial, which can be attributed to many reasons. The first is the challenge MSCs encounter when adapting to a new microenvironment following delivery into the body. They have to first survive in the new and often harsh conditions, during which the MSC effects can be reduced or even lost. Thus, improvement of the MSC efficacy should focus on achieving high delivery efficiency, long-term retention, and specific modification to target different inflammatory diseases.
Genetically modified MSCs can gain remarkably enhanced therapeutic capability, in which MSCs serve as a carrier to deliver cytokines or verified biological drugs for target-oriented therapies. For example, compared with unmodified MSCs, MSCs transduced with TGFβ suppressed CIA in a mouse model (158). MSCs expressing IL-12p40 alleviate murine colitis more effectively than a wild-type control (159). Overexpressing IL-10 in MSCs suppressed the development of graft-versus-host disease (160), and MSCs overexpressing TNFR2 treat CIA in mouse more effectively than controls (131). MSCs can also be engineered to release abundant amounts of sTNFR1 to neutralize TNF in the circulation (121, 140). In addition, since MSCs promote activation and proliferation of Tregs, combined therapy of MSCs and Tregs further enhances the number and functions of Tregs and achieves much stronger efficacy than each alone, which has been observed in GVHD (161, 162) and ischemic myocardium (163).
In this review, we describe the progress in research on how TNF signaling interacts with MSCs in the treatment of autoimmune and inflammatory diseases (Figure 1). At appropriate concentrations and timing, TNF promotes secretion of immunosuppressive molecules from MSCs, which inhibit Teff cells and activate Tregs. In the periphery, TNFR2 signaling also stimulates Tregs; thus, it may synergize with MSCs to repress inflammation. In the CNS, TNFR2 signaling protects the survival of astrocytes, OPC, microglia, and neurons. Activated MSCs secrete immunosuppressive molecules to inhibit inflammation and neurotropic molecules to protect neural cells and promote remyelination. Some of the TNF functions mediated by either TNFR1 or -R2 in MSCs can vary in different genders. Together, these findings suggest that TNF signaling plays a pivotal role in MSC-based therapy of autoimmune disease, which is highly dependent on the context, timing, concentration, gender, etc.
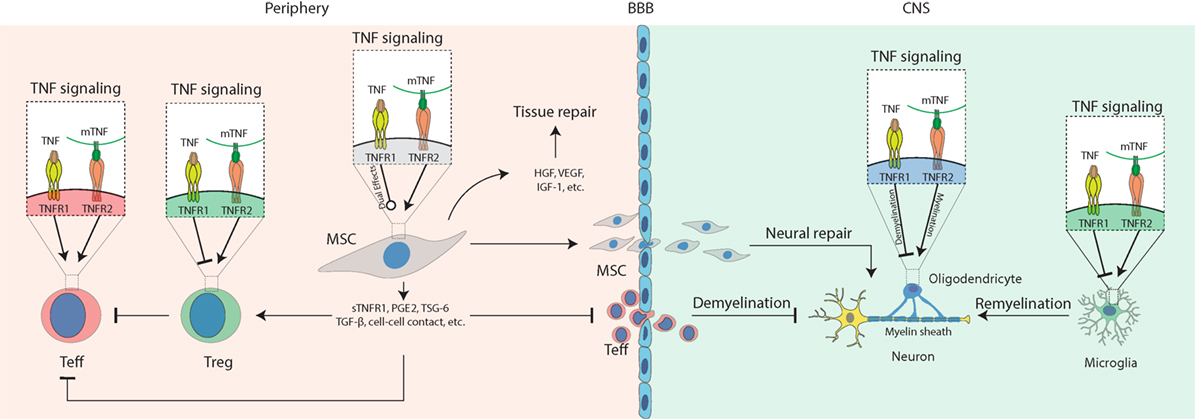
Figure 1. Schematic diagram for the role of tumor necrosis factor (TNF) signaling in mesenchymal stem cell (MSC)-based therapy on autoimmune and inflammatory diseases. Under inflammatory conditions, TNF binds TNFR1 to activate T effector (Teff) cells while impairing regulatory T cells (Tregs); mTNF mostly binds TNFR2 to activate Teff cells and also activate Tregs to mediate their immunosuppressive effects in the periphery. In the central nervous system (CNS), TNFR1 signaling induces cytotoxic effects on oligodendrocytes resulting in neural demyelination and activates microglia to produce pro-inflammatory molecules such as TNF. TNFR2 signaling protects the survival of oligodendrocyte and microglia and promotes myelin clearance and remyelination mediated by microglia. In addition, TNFR1 or -R2 signaling can enhance the immunosuppressive effects of MSCs to alleviate autoimmune and inflammatory diseases. Compared to non-primed controls, TNF-primed MSC produce more soluble TNFR1 (sTNFR1), PGE2, TSG-6, and TGF-β, enhance Treg functions, neutralize TNF via sTNFR, prevent Teff cell infiltration into the CNS, release growth factors such as hepatocyte growth factor (HGF), insulin-like growth factor-1 (IGF-1), and VEGF to promote tissue or neural repair, infiltrate the CNS to mediate neural protection by regulating oligodendrocytes and microglia, suppressing Teff cells that have infiltrated the CNS.
Despite these interesting findings, many more questions remain to be addressed than have been solved. For example, how do transplanted MSCs respond to TNF, function in the periphery and infiltrate the inflamed CNS in patients. Why does gender affect TNF functions? Would genetic variations among different individuals affect TNF functions? Can inflammatory factors also epigenetically modify and alter the expression of the genes involved in TNF signaling? Future studies are needed to address these and many new challenging questions. Continuous progress in this field will most likely lead to the identification of new targets for more precise and effective therapies of autoimmune and inflammatory diseases.
LY, DZ, and R-HX conceived, designed, and wrote the manuscript. R-HX gave the final approval of the manuscript.
RH-X is a founder of ImStem Biotechnology, Inc., a stem cell company. He declares competing financial interests. No financial conflicts of interest exist for any of the authors.
This work was supported by University of Macau Research Committee funds MYRG #2015-00169-FHS, #2016-00070-FHS, and #2017-00124-FHS, and Macau Science and Technology Development Fund (FDCT) #028/2015/A1 and #095/2017/A1 to R-HX.
1. Ankrum JA, Ong JF, Karp JM. Mesenchymal stem cells: immune evasive, not immune privileged. Nat Biotechnol (2014) 32:252–60. doi:10.1038/nbt.2816
2. Wang Y, Chen X, Cao W, Shi Y. Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat Immunol (2014) 15:1009–16. doi:10.1038/ni.3002
3. Tropel P, Platet N, Platel JC, Noel D, Albrieux M, Benabid AL, et al. Functional neuronal differentiation of bone marrow-derived mesenchymal stem cells. Stem Cells (2006) 24:2868–76. doi:10.1634/stemcells.2005-0636
4. Jang S, Cho HH, Cho YB, Park JS, Jeong HS. Functional neural differentiation of human adipose tissue-derived stem cells using bFGF and forskolin. BMC Cell Biol (2010) 11:25. doi:10.1186/1471-2121-11-25
5. Aurich H, Sgodda M, Kaltwasser P, Vetter M, Weise A, Liehr T, et al. Hepatocyte differentiation of mesenchymal stem cells from human adipose tissue in vitro promotes hepatic integration in vivo. Gut (2009) 58:570–81. doi:10.1136/gut.2008.154880
6. Stock P, Bruckner S, Ebensing S, Hempel M, Dollinger MM, Christ B. The generation of hepatocytes from mesenchymal stem cells and engraftment into murine liver. Nat Protoc (2010) 5:617–27. doi:10.1038/nprot.2010.7
7. Fukuta M, Nakai Y, Kirino K, Nakagawa M, Sekiguchi K, Nagata S, et al. Derivation of mesenchymal stromal cells from pluripotent stem cells through a neural crest lineage using small molecule compounds with defined media. PLoS One (2014) 9:e112291. doi:10.1371/journal.pone.0112291
8. Menendez L, Kulik MJ, Page AT, Park SS, Lauderdale JD, Cunningham ML, et al. Directed differentiation of human pluripotent cells to neural crest stem cells. Nat Protoc (2013) 8:203–12. doi:10.1038/nprot.2012.156
9. Manicourt DH, Triki R, Fukuda K, Devogelaer JP, Nagant de Deuxchaisnes C, Thonar EJ. Levels of circulating tumor necrosis factor alpha and interleukin-6 in patients with rheumatoid arthritis. Relationship to serum levels of hyaluronan and antigenic keratan sulfate. Arthritis Rheum (1993) 36:490–9. doi:10.1002/art.1780360409
10. Hagel AF, de Rossi T, Konturek PC, Albrecht H, Walker S, Hahn EG, et al. Plasma histamine and tumour necrosis factor-alpha levels in Crohn’s disease and ulcerative colitis at various stages of disease. J Physiol Pharmacol (2015) 66:549–56.
11. Qiao YC, Chen YL, Pan YH, Tian F, Xu Y, Zhang XX, et al. The change of serum tumor necrosis factor alpha in patients with type 1 diabetes mellitus: a systematic review and meta-analysis. PLoS One (2017) 12:e0176157. doi:10.1371/journal.pone.0176157
12. Papadopoulou A, Yiangou M, Athanasiou E, Zogas N, Kaloyannidis P, Batsis I, et al. Mesenchymal stem cells are conditionally therapeutic in preclinical models of rheumatoid arthritis. Ann Rheum Dis (2012) 71:1733–40. doi:10.1136/annrheumdis-2011-200985
13. Sheng H, Wang Y, Jin Y, Zhang Q, Zhang Y, Wang L, et al. A critical role of IFNgamma in priming MSC-mediated suppression of T cell proliferation through up-regulation of B7-H1. Cell Res (2008) 18:846–57. doi:10.1038/cr.2008.80
14. Polchert D, Sobinsky J, Douglas G, Kidd M, Moadsiri A, Reina E, et al. IFN-gamma activation of mesenchymal stem cells for treatment and prevention of graft versus host disease. Eur J Immunol (2008) 38:1745–55. doi:10.1002/eji.200738129
15. Krampera M, Cosmi L, Angeli R, Pasini A, Liotta F, Andreini A, et al. Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells (2006) 24:386–98. doi:10.1634/stemcells.2005-0008
16. Shi Y, Su J, Roberts AI, Shou P, Rabson AB, Ren G. How mesenchymal stem cells interact with tissue immune responses. Trends Immunol (2012) 33:136–43. doi:10.1016/j.it.2011.11.004
17. Sivanathan KN, Gronthos S, Rojas-Canales D, Thierry B, Coates PT. Interferon-gamma modification of mesenchymal stem cells: implications of autologous and allogeneic mesenchymal stem cell therapy in allotransplantation. Stem Cell Rev (2014) 10:351–75. doi:10.1007/s12015-014-9495-2
18. Fiorina P, Jurewicz M, Augello A, Vergani A, Dada S, La Rosa S, et al. Immunomodulatory function of bone marrow-derived mesenchymal stem cells in experimental autoimmune type 1 diabetes. J Immunol (2009) 183:993–1004. doi:10.4049/jimmunol.0900803
19. Yan X, Cen Y, Wang Q. Mesenchymal stem cells alleviate experimental rheumatoid arthritis through microRNA-regulated IkappaB expression. Sci Rep (2016) 6:28915. doi:10.1038/srep28915
20. Molendijk I, Bonsing BA, Roelofs H, Peeters KC, Wasser MN, Dijkstra G, et al. Allogeneic bone marrow-derived mesenchymal stromal cells promote healing of refractory perianal fistulas in patients with Crohn’s disease. Gastroenterology (2015) 149:918–27.e6. doi:10.1053/j.gastro.2015.06.014
21. Wang X, Lazorchak AS, Song L, Li E, Zhang Z, Jiang B, et al. Immune modulatory mesenchymal stem cells derived from human embryonic stem cells through a trophoblast-like stage. Stem Cells (2016) 34:380–91. doi:10.1002/stem.2242
22. Sun L, Akiyama K, Zhang H, Yamaza T, Hou Y, Zhao S, et al. Mesenchymal stem cell transplantation reverses multiorgan dysfunction in systemic lupus erythematosus mice and humans. Stem Cells (2009) 27:1421–32. doi:10.1002/stem.68
23. Zhang L, Zheng H, Shao H, Nian H, Zhang Y, Bai L, et al. Long-term therapeutic effects of mesenchymal stem cells compared to dexamethasone on recurrent experimental autoimmune uveitis of rats. Invest Ophthalmol Vis Sci (2014) 55:5561–71. doi:10.1167/iovs.14-14788
24. Kimbrel EA, Kouris NA, Yavanian GJ, Chu J, Qin Y, Chan A, et al. Mesenchymal stem cell population derived from human pluripotent stem cells displays potent immunomodulatory and therapeutic properties. Stem Cells Dev (2014) 23:1611–24. doi:10.1089/scd.2013.0554
25. Xu J, Wang D, Liu D, Fan Z, Zhang H, Liu O, et al. Allogeneic mesenchymal stem cell treatment alleviates experimental and clinical Sjogren syndrome. Blood (2012) 120:3142–51. doi:10.1182/blood-2011-11-391144
26. Wang X, Kimbrel EA, Ijichi K, Paul D, Lazorchak AS, Chu J, et al. Human ESC-derived MSCs outperform bone marrow MSCs in the treatment of an EAE model of multiple sclerosis. Stem Cell Reports (2014) 3:115–30. doi:10.1016/j.stemcr.2014.04.020
27. Jiang B, Yan L, Miao Z, Li E, Wong KH, Xu RH. Spheroidal formation preserves human stem cells for prolonged time under ambient conditions for facile storage and transportation. Biomaterials (2017) 133:275–86. doi:10.1016/j.biomaterials.2017.03.050
28. Yan L, Jiang B, Li E, Wang X, Ling Q, Zheng D, et al. Scalable generation of mesenchymal stem cells from human embryonic stem cells in 3D. Int J Biol Sci (2018) 14(10):1196–210. doi:10.7150/ijbs.25023
29. Gebler A, Zabel O, Seliger B. The immunomodulatory capacity of mesenchymal stem cells. Trends Mol Med (2012) 18:128–34. doi:10.1016/j.molmed.2011.10.004
30. Le Blanc K, Mougiakakos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol (2012) 12:383–96. doi:10.1038/nri3209
31. Rosado MM, Bernardo ME, Scarsella M, Conforti A, Giorda E, Biagini S, et al. Inhibition of B-cell proliferation and antibody production by mesenchymal stromal cells is mediated by T cells. Stem Cells Dev (2015) 24:93–103. doi:10.1089/scd.2014.0155
32. Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood (2006) 108:253–61. doi:10.1182/blood-2005-11-4567
33. Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med (2007) 13:423–31. doi:10.1038/nm1564
34. Sun L, Wang D, Liang J, Zhang H, Feng X, Wang H, et al. Umbilical cord mesenchymal stem cell transplantation in severe and refractory systemic lupus erythematosus. Arthritis Rheum (2010) 62:2467–75. doi:10.1002/art.27548
35. Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, et al. Human mesenchymal stem cells modulate B-cell functions. Blood (2006) 107:367–72. doi:10.1182/blood-2005-07-2657
36. Zhang QZ, Su WR, Shi SH, Wilder-Smith P, Xiang AP, Wong A, et al. Human gingiva-derived mesenchymal stem cells elicit polarization of m2 macrophages and enhance cutaneous wound healing. Stem Cells (2010) 28:1856–68. doi:10.1002/stem.503
37. Ramasamy R, Fazekasova H, Lam EW, Soeiro I, Lombardi G, Dazzi F. Mesenchymal stem cells inhibit dendritic cell differentiation and function by preventing entry into the cell cycle. Transplantation (2007) 83:71–6. doi:10.1097/01.tp.0000244572.24780.54
38. Selmani Z, Naji A, Zidi I, Favier B, Gaiffe E, Obert L, et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4+CD25highFOXP3+ regulatory T cells. Stem Cells (2008) 26:212–22. doi:10.1634/stemcells.2007-0554
39. Chinnadurai R, Copland IB, Patel SR, Galipeau J. IDO-independent suppression of T cell effector function by IFN-gamma-licensed human mesenchymal stromal cells. J Immunol (2014) 192:1491–501. doi:10.4049/jimmunol.1301828
40. Fan H, Zhao G, Liu L, Liu F, Gong W, Liu X, et al. Pre-treatment with IL-1beta enhances the efficacy of MSC transplantation in DSS-induced colitis. Cell Mol Immunol (2012) 9:473–81. doi:10.1038/cmi.2012.40
41. Duijvestein M, Wildenberg ME, Welling MM, Hennink S, Molendijk I, van Zuylen VL, et al. Pretreatment with interferon-gamma enhances the therapeutic activity of mesenchymal stromal cells in animal models of colitis. Stem Cells (2011) 29:1549–58. doi:10.1002/stem.698
42. English K, Barry FP, Field-Corbett CP, Mahon BP. IFN-gamma and TNF-alpha differentially regulate immunomodulation by murine mesenchymal stem cells. Immunol Lett (2007) 110:91–100. doi:10.1016/j.imlet.2007.04.001
43. Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood (2002) 99:3838–43. doi:10.1182/blood.V99.10.3838
44. Ren G, Zhao X, Zhang L, Zhang J, L’Huillier A, Ling W, et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J Immunol (2010) 184:2321–8. doi:10.4049/jimmunol.0902023
45. Schurgers E, Kelchtermans H, Mitera T, Geboes L, Matthys P. Discrepancy between the in vitro and in vivo effects of murine mesenchymal stem cells on T-cell proliferation and collagen-induced arthritis. Arthritis Res Ther (2010) 12:R31. doi:10.1186/ar2939
46. Zappia E, Casazza S, Pedemonte E, Benvenuto F, Bonanni I, Gerdoni E, et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood (2005) 106:1755–61. doi:10.1182/blood-2005-04-1496
47. Carrion F, Nova E, Ruiz C, Diaz F, Inostroza C, Rojo D, et al. Autologous mesenchymal stem cell treatment increased T regulatory cells with no effect on disease activity in two systemic lupus erythematosus patients. Lupus (2010) 19:317–22. doi:10.1177/0961203309348983
48. Youd M, Blickarz C, Woodworth L, Touzjian T, Edling A, Tedstone J, et al. Allogeneic mesenchymal stem cells do not protect NZBxNZW F1 mice from developing lupus disease. Clin Exp Immunol (2010) 161:176–86. doi:10.1111/j.1365-2249.2010.04158.x
49. Taddio A, Tommasini A, Valencic E, Biagi E, Decorti G, De Iudicibus S, et al. Failure of interferon-gamma pre-treated mesenchymal stem cell treatment in a patient with Crohn’s disease. World J Gastroenterol (2015) 21:4379–84. doi:10.3748/wjg.v21.i14.4379
50. Duijvestein M, Vos AC, Roelofs H, Wildenberg ME, Wendrich BB, Verspaget HW, et al. Autologous bone marrow-derived mesenchymal stromal cell treatment for refractory luminal Crohn’s disease: results of a phase I study. Gut (2010) 59:1662–9. doi:10.1136/gut.2010.215152
51. de la Portilla F, Alba F, Garcia-Olmo D, Herrerias JM, Gonzalez FX, Galindo A. Expanded allogeneic adipose-derived stem cells (eASCs) for the treatment of complex perianal fistula in Crohn’s disease: results from a multicenter phase I/IIa clinical trial. Int J Colorectal Dis (2013) 28:313–23. doi:10.1007/s00384-012-1581-9
52. Augello A, Tasso R, Negrini SM, Cancedda R, Pennesi G. Cell therapy using allogeneic bone marrow mesenchymal stem cells prevents tissue damage in collagen-induced arthritis. Arthritis Rheum (2007) 56:1175–86. doi:10.1002/art.22511
53. Miyagawa I, Nakayamada S, Nakano K, Yamagata K, Sakata K, Yamaoka K, et al. Induction of regulatory T cells and its regulation with insulin-like growth factor/insulin-like growth factor binding protein-4 by human mesenchymal stem cells. J Immunol (2017) 199:1616–25. doi:10.4049/jimmunol.1600230
54. Djouad F, Fritz V, Apparailly F, Louis-Plence P, Bony C, Sany J, et al. Reversal of the immunosuppressive properties of mesenchymal stem cells by tumor necrosis factor alpha in collagen-induced arthritis. Arthritis Rheum (2005) 52:1595–603. doi:10.1002/art.21012
55. Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell (2013) 13:392–402. doi:10.1016/j.stem.2013.09.006
56. Gonzalez-Rey E, Anderson P, Gonzalez MA, Rico L, Buscher D, Delgado M. Human adult stem cells derived from adipose tissue protect against experimental colitis and sepsis. Gut (2009) 58:929–39. doi:10.1136/gut.2008.168534
57. El-Jawhari JJ, El-Sherbiny YM, Jones EA, McGonagle D. Mesenchymal stem cells, autoimmunity and rheumatoid arthritis. QJM (2014) 107:505–14. doi:10.1093/qjmed/hcu033
58. Zhang X, Bowles AC, Semon JA, Scruggs BA, Zhang S, Strong AL, et al. Transplantation of autologous adipose stem cells lacks therapeutic efficacy in the experimental autoimmune encephalomyelitis model. PLoS One (2014) 9:e85007. doi:10.1371/journal.pone.0085007
59. Strong AL, Bowles AC, Wise RM, Morand JP, Dutreil MF, Gimble JM, et al. Human adipose stromal/stem cells from obese donors show reduced efficacy in halting disease progression in the experimental autoimmune encephalomyelitis model of multiple sclerosis. Stem Cells (2016) 34:614–26. doi:10.1002/stem.2272
60. Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood (2012) 119:651–65. doi:10.1182/blood-2011-04-325225
61. Croft M, Siegel RM. Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat Rev Rheumatol (2017) 13:217–33. doi:10.1038/nrrheum.2017.22
62. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature (1997) 385:729–33. doi:10.1038/385729a0
63. Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, et al. Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J Biol Chem (2000) 275:14608–14. doi:10.1074/jbc.275.19.14608
64. Fischer R, Kontermann RE, Maier O. Targeting sTNF/TNFR1 signaling as a new therapeutic strategy. Antibodies (2015) 4:48–70. doi:10.3390/antib4010048
65. Vudattu NK, Holler E, Ewing P, Schulz U, Haffner S, Burger V, et al. Reverse signalling of membrane-integrated tumour necrosis factor differentially regulates alloresponses of CD4(+) and CD8(+) T cells against human microvascular endothelial cells. Immunology (2005) 115:536–43. doi:10.1111/j.1365-2567.2005.02190.x
66. Zhang H, Yan D, Shi X, Liang HF, Pang Y, Qin NL, et al. Transmembrane TNF-alpha mediates “forward” and “reverse” signaling, inducing cell death or survival via the NF-kappa B pathway in Raji Burkitt lymphoma cells. J Leukoc Biol (2008) 84:789–97. doi:10.1189/jlb.0208078
67. Yu MX, Shi WF, Zhang J, Niu L, Chen QZ, Yan D, et al. Influence of reverse signaling via membrane TNF-alpha on cytotoxicity of NK92 cells. Eur J Cell Biol (2009) 88:181–91. doi:10.1016/j.ejcb.2008.09.001
68. Xin LJ, Wang J, Zhang HL, Shi WF, Yu MX, Li QF, et al. Dual regulation of soluble tumor necrosis factor-alpha induced activation of human monocytic cells via modulating transmembrane TNF-alpha-mediated ‘reverse signaling’. Int J Mol Med (2006) 18:885–92. doi:10.3892/ijmm.18.5.885
69. Eissner G, Kirchner S, Lindner H, Kolch W, Janosch P, Grell M, et al. Reverse signaling through transmembrane TNF confers resistance to lipopolysaccharide in human monocytes and macrophages. J Immunol (2000) 164:6193–8. doi:10.4049/jimmunol.164.12.6193
70. Pallai A, Kiss B, Vereb G, Armaka M, Kollias G, Szekanecz Z, et al. Transmembrane TNF-alpha reverse signaling inhibits lipopolysaccharide-induced proinflammatory cytokine formation in macrophages by inducing TGF-beta: therapeutic implications. J Immunol (2016) 196:1146–57. doi:10.4049/jimmunol.1501573
71. Szondy Z, Pallai A. Transmembrane TNF-alpha reverse signaling leading to TGF-beta production is selectively activated by TNF targeting molecules: therapeutic implications. Pharmacol Res (2017) 115:124–32. doi:10.1016/j.phrs.2016.11.025
72. Schulz M, Dotzlaw H, Neeck G. Ankylosing spondylitis and rheumatoid arthritis: serum levels of TNF-alpha and its soluble receptors during the course of therapy with etanercept and infliximab. Biomed Res Int (2014) 2014:675108. doi:10.1155/2014/675108
73. Gabay C, Cakir N, Moral F, Roux-Lombard P, Meyer O, Dayer JM, et al. Circulating levels of tumor necrosis factor soluble receptors in systemic lupus erythematosus are significantly higher than in other rheumatic diseases and correlate with disease activity. J Rheumatol (1997) 24:303–8.
74. Sharief MK, Hentges R. Association between tumor-necrosis-factor-alpha and disease progression in patients with multiple-sclerosis. N Engl J Med (1991) 325:467–72. doi:10.1056/NEJM199108153250704
75. Probert L, Plows D, Kontogeorgos G, Kollias G. The type I interleukin-1 receptor acts in series with tumor necrosis factor (TNF) to induce arthritis in TNF-transgenic mice. Eur J Immunol (1995) 25:1794–7. doi:10.1002/eji.1830250647
76. Kotake S, Yago T, Kobashigawa T, Nanke Y. The plasticity of Th17 cells in the pathogenesis of rheumatoid arthritis. J Clin Med (2017) 6:E67. doi:10.3390/jcm6070067
77. Slebioda TJ, Kmiec Z. Tumour necrosis factor superfamily members in the pathogenesis of inflammatory bowel disease. Mediators Inflamm (2014) 2014:325129. doi:10.1155/2014/325129
78. Eugster HP, Frei K, Bachmann R, Bluethmann H, Lassmann H, Fontana A. Severity of symptoms and demyelination in MOG-induced EAE depends on TNFR1. Eur J Immunol (1999) 29:626–32. doi:10.1002/(SICI)1521-4141(199902)29:02<626::AID-IMMU626>3.0.CO;2-A
79. Alexopoulou L, Kranidioti K, Xanthoulea S, Denis M, Kotanidou A, Douni E, et al. Transmembrane TNF protects mutant mice against intracellular bacterial infections, chronic inflammation and autoimmunity. Eur J Immunol (2006) 36:2768–80. doi:10.1002/eji.200635921
80. Geng L, Li X, Feng X, Zhang J, Wang D, Chen J, et al. Association of TNF-alpha with impaired migration capacity of mesenchymal stem cells in patients with systemic lupus erythematosus. J Immunol Res (2014) 2014:169082. doi:10.1155/2014/169082
81. Rossol M, Schubert K, Meusch U, Schulz A, Biedermann B, Grosche J, et al. Tumor necrosis factor receptor type I expression of CD4+ T cells in rheumatoid arthritis enables them to follow tumor necrosis factor gradients into the rheumatoid synovium. Arthritis Rheum (2013) 65:1468–76. doi:10.1002/art.37927
82. Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity (1999) 10:387–98. doi:10.1016/S1074-7613(00)80038-2
83. Chee J, Angstetra E, Mariana L, Graham KL, Carrington EM, Bluethmann H, et al. TNF receptor 1 deficiency increases regulatory T cell function in nonobese diabetic mice. J Immunol (2011) 187:1702–12. doi:10.4049/jimmunol.1100511
84. Nie H, Zheng YX, Li RS, Guo TB, He DY, Fang L, et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med (2013) 19:322–8. doi:10.1038/nm.3085
85. Steeland S, Van Ryckeghem S, Van Imschoot G, De Rycke R, Toussaint W, Vanhoutte L, et al. TNFR1 inhibition with a nanobody protects against EAE development in mice. Sci Rep (2017) 7:13646. doi:10.1038/s41598-017-13984-y
86. Vasanthakumar A, Liao Y, Teh P, Pascutti MF, Oja AE, Garnham AL, et al. The TNF receptor superfamily-NF-kappaB axis is critical to maintain effector regulatory T cells in lymphoid and non-lymphoid tissues. Cell Rep (2017) 20:2906–20. doi:10.1016/j.celrep.2017.08.068
87. Okubo Y, Torrey H, Butterworth J, Zheng H, Faustman DL. Treg activation defect in type 1 diabetes: correction with TNFR2 agonism. Clin Transl Immunology (2016) 5:e56. doi:10.1038/cti.2015.43
88. Chen X, Nie Y, Xiao H, Bian Z, Scarzello AJ, Song NY, et al. TNFR2 expression by CD4 effector T cells is required to induce full-fledged experimental colitis. Sci Rep (2016) 6:32834. doi:10.1038/srep32834
89. Punit S, Dube PE, Liu CY, Girish N, Washington MK, Polk DB. Tumor necrosis factor receptor 2 restricts the pathogenicity of CD8(+) T cells in mice with colitis. Gastroenterology (2015) 149:993–1005.e2. doi:10.1053/j.gastro.2015.06.004
90. Wang K, Han G, Dou Y, Wang Y, Liu G, Wang R, et al. Opposite role of tumor necrosis factor receptors in dextran sulfate sodium-induced colitis in mice. PLoS One (2012) 7:e52924. doi:10.1371/journal.pone.0052924
91. Faustman D, Davis M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov (2010) 9:482–93. doi:10.1038/nrd3030
92. Schmid T, Falter L, Weber S, Muller N, Molitor K, Zeller D, et al. Chronic inflammation increases the sensitivity of mouse Treg for TNFR2 costimulation. Front Immunol (2017) 8:1471. doi:10.3389/fimmu.2017.01471
93. Chen X, Wu X, Zhou Q, Howard OM, Netea MG, Oppenheim JJ. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. J Immunol (2013) 190:1076–84. doi:10.4049/jimmunol.1202659
94. Faustman DL, Davis M. TNF receptor 2 and disease: autoimmunity and regenerative medicine. Front Immunol (2013) 4:478. doi:10.3389/fimmu.2013.00478
95. Zaragoza B, Chen X, Oppenheim JJ, Baeyens A, Gregoire S, Chader D, et al. Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat Med (2016) 22:16–7. doi:10.1038/nm.4019
96. Chen X, Subleski JJ, Kopf H, Howard OM, Mannel DN, Oppenheim JJ. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol (2008) 180:6467–71. doi:10.4049/jimmunol.180.10.6467
97. Nguyen DX, Ehrenstein MR. Anti-TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF-TNF-RII binding in rheumatoid arthritis. J Exp Med (2016) 213:1241–53. doi:10.1084/jem.20151255
98. Chen X, Oppenheim JJ. Contrasting effects of TNF and anti-TNF on the activation of effector T cells and regulatory T cells in autoimmunity. FEBS Lett (2011) 585:3611–8. doi:10.1016/j.febslet.2011.04.025
99. Chen X, Hamano R, Subleski JJ, Hurwitz AA, Howard OM, Oppenheim JJ. Expression of costimulatory TNFR2 induces resistance of CD4+FoxP3- conventional T cells to suppression by CD4+FoxP3+ regulatory T cells. J Immunol (2010) 185:174–82. doi:10.4049/jimmunol.0903548
100. Ye LL, Wei XS, Zhang M, Niu YR, Zhou Q. The significance of tumor necrosis factor receptor type II in CD8(+) regulatory T cells and CD8(+) effector T cells. Front Immunol (2018) 9:583. doi:10.3389/fimmu.2018.00583
101. Govindaraj C, Scalzo-Inguanti K, Scholzen A, Li S, Plebanski M. TNFR2 expression on CD25(hi)FOXP3(+) T cells induced upon TCR stimulation of CD4 T cells identifies maximal cytokine-producing effectors. Front Immunol (2013) 4:233. doi:10.3389/fimmu.2013.00233
102. van Oosten BW, Barkhof F, Truyen L, Boringa JB, Bertelsmann FW, von Blomberg BM, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology (1996) 47:1531–4. doi:10.1212/WNL.47.6.1531
103. Kaltsonoudis E, Zikou AK, Voulgari PV, Konitsiotis S, Argyropoulou MI, Drosos AA. Neurological adverse events in patients receiving anti-TNF therapy: a prospective imaging and electrophysiological study. Arthritis Res Ther (2014) 16:R125. doi:10.1186/ar4582
104. Kaltsonoudis E, Voulgari PV, Konitsiotis S, Drosos AA. Demyelination and other neurological adverse events after anti-TNF therapy. Autoimmun Rev (2014) 13:54–8. doi:10.1016/j.autrev.2013.09.002
105. Probert L. TNF and its receptors in the CNS: the essential, the desirable and the deleterious effects. Neuroscience (2015) 302:2–22. doi:10.1016/j.neuroscience.2015.06.038
106. McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation (2008) 5:45. doi:10.1186/1742-2094-5-45
107. Kuno R, Wang JY, Kawanokuchi J, Takeuchi H, Mizuno T, Suzumura A. Autocrine activation of microglia by tumor necrosis factor-alpha. J Neuroimmunol (2005) 162:89–96. doi:10.1016/j.jneuroim.2005.01.015
108. Jurewicz A, Matysiak M, Tybor K, Selmaj K. TNF-induced death of adult human oligodendrocytes is mediated by c-Jun NH2-terminal kinase-3. Brain (2003) 126:1358–70. doi:10.1093/brain/awg146
109. Bonora M, De Marchi E, Patergnani S, Suski JM, Celsi F, Bononi A, et al. Tumor necrosis factor-alpha impairs oligodendroglial differentiation through a mitochondria-dependent process. Cell Death Differ (2014) 21:1198–208. doi:10.1038/cdd.2014.35
110. Madsen PM, Motti D, Karmally S, Szymkowski DE, Lambertsen KL, Bethea JR, et al. Oligodendroglial TNFR2 mediates membrane TNF-dependent repair in experimental autoimmune encephalomyelitis by promoting oligodendrocyte differentiation and remyelination. J Neurosci (2016) 36:5128–43. doi:10.1523/JNEUROSCI.0211-16.2016
111. Fischer R, Wajant H, Kontermann R, Pfizenmaier K, Maier O. Astrocyte-specific activation of TNFR2 promotes oligodendrocyte maturation by secretion of leukemia inhibitory factor. Glia (2014) 62:272–83. doi:10.1002/glia.22605
112. Maier O, Fischer R, Agresti C, Pfizenmaier K. TNF receptor 2 protects oligodendrocyte progenitor cells against oxidative stress. Biochem Biophys Res Commun (2013) 440:336–41. doi:10.1016/j.bbrc.2013.09.083
113. Gao H, Danzi MC, Choi CS, Taherian M, Dalby-Hansen C, Ellman DG, et al. Opposing functions of microglial and macrophagic TNFR2 in the pathogenesis of experimental autoimmune encephalomyelitis. Cell Rep (2017) 18:198–212. doi:10.1016/j.celrep.2016.11.083
114. Yamasaki R, Lu HY, Butovsky O, Ohno N, Rietsch AM, Cialic R, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med (2014) 211:1533–49. doi:10.1084/jem.20132477
115. Rafei M, Birman E, Forner K, Galipeau J. Allogeneic mesenchymal stem cells for treatment of experimental autoimmune encephalomyelitis. Mol Ther (2009) 17:1799–803. doi:10.1038/mt.2009.157
116. Huang H, Zhao N, Xu X, Xu Y, Li S, Zhang J, et al. Dose-specific effects of tumor necrosis factor alpha on osteogenic differentiation of mesenchymal stem cells. Cell Prolif (2011) 44:420–7. doi:10.1111/j.1365-2184.2011.00769.x
117. Su W, Wan Q, Huang J, Han L, Chen X, Chen G, et al. Culture medium from TNF-alpha-stimulated mesenchymal stem cells attenuates allergic conjunctivitis through multiple antiallergic mechanisms. J Allergy Clin Immunol (2015) 136:423–32.e8. doi:10.1016/j.jaci.2014.12.1926
118. Nemeth K, Leelahavanichkul A, Yuen PST, Mayer B, Parmelee A, Doi K, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E-2-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med (2009) 15:42–9. doi:10.1038/nm0409-462b
119. Yagi H, Soto-Gutierrez A, Navarro-Alvarez N, Nahmias Y, Goldwasser Y, Kitagawa Y, et al. Reactive bone marrow stromal cells attenuate systemic inflammation via sTNFR1. Mol Ther (2010) 18:1857–64. doi:10.1038/mt.2010.155
120. Martire A, Bedada FB, Uchida S, Poling J, Kruger M, Warnecke H, et al. Mesenchymal stem cells attenuate inflammatory processes in the heart and lung via inhibition of TNF signaling. Basic Res Cardiol (2016) 111:54. doi:10.1007/s00395-016-0573-2
121. Ke F, Zhang L, Liu Z, Yan S, Xu Z, Bai J, et al. Soluble tumor necrosis factor receptor 1 released by skin-derived mesenchymal stem cells is critical for inhibiting Th17 cell differentiation. Stem Cells Transl Med (2016) 5:301–13. doi:10.5966/sctm.2015-0179
122. Fisher-Shoval Y, Barhum Y, Sadan O, Yust-Katz S, Ben-Zur T, Lev N, et al. Transplantation of placenta-derived mesenchymal stem cells in the EAE mouse model of MS. J Mol Neurosci (2012) 48:176–84. doi:10.1007/s12031-012-9805-6
123. Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL, et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell (2009) 5:54–63. doi:10.1016/j.stem.2009.05.003
124. Lee MJ, Kim J, Kim MY, Bae YS, Ryu SH, Lee TG, et al. Proteomic analysis of tumor necrosis factor-alpha-induced secretome of human adipose tissue-derived mesenchymal stem cells. J Proteome Res (2010) 9:1754–62. doi:10.1021/pr900898n
125. Francois M, Romieu-Mourez R, Li M, Galipeau J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol Ther (2012) 20:187–95. doi:10.1038/mt.2011.189
126. Zhang A, Wang Y, Ye Z, Xie H, Zhou L, Zheng S. Mechanism of TNF-alpha-induced migration and hepatocyte growth factor production in human mesenchymal stem cells. J Cell Biochem (2010) 111:469–75. doi:10.1002/jcb.22729
127. Wang MJ, Crisostomo PR, Herring C, Meldrum KK, Meldrum DR. Human progenitor cells from bone marrow or adipose tissue produce VEGF, HGF, and IGF-I in response to TNF by a p38 MAPK-dependent mechanism. Am J Physiol Regul Integr Comp Physiol (2006) 291:R880–4. doi:10.1152/ajpregu.00280.2006
128. Crisostomo PR, Wang Y, Markel TA, Wang M, Lahm T, Meldrum DR. Human mesenchymal stem cells stimulated by TNF-alpha, LPS, or hypoxia produce growth factors by an NF kappa B- but not JNK-dependent mechanism. Am J Physiol Cell Physiol (2008) 294:C675–82. doi:10.1152/ajpcell.00437.2007
129. Zhang YL, Yu ZD, Jiang D, Liang XT, Liao SY, Zhang Z, et al. iPSC-MSCs with high intrinsic MIRO1 and sensitivity to TNF-alpha yield efficacious mitochondrial transfer to rescue anthracycline-induced cardiomyopathy. Stem Cell Reports (2016) 7:749–63. doi:10.1016/j.stemcr.2016.08.009
130. Liu L, Zhao G, Fan H, Zhao X, Li P, Wang Z, et al. Mesenchymal stem cells ameliorate Th1-induced pre-eclampsia-like symptoms in mice via the suppression of TNF-alpha expression. PLoS One (2014) 9:e88036. doi:10.1371/journal.pone.0088036
131. Park N, Rim YA, Jung H, Kim J, Yi H, Kim Y, et al. Etanercept-synthesising mesenchymal stem cells efficiently ameliorate collagen-induced arthritis. Sci Rep (2017) 7:39593. doi:10.1038/srep39593
132. Liu LN, Wang G, Hendricks K, Lee K, Bohnlein E, Junker U, et al. Comparison of drug and cell-based delivery: engineered adult mesenchymal stem cells expressing soluble tumor necrosis factor receptor II prevent arthritis in mouse and rat animal models. Stem Cells Transl Med (2013) 2:362–75. doi:10.5966/sctm.2012-0135
133. Liu Z, Gao L, Wang P, Xie Z, Cen S, Li Y, et al. TNF-alpha induced the enhanced apoptosis of mesenchymal stem cells in ankylosing spondylitis by overexpressing TRAIL-R2. Stem Cells Int (2017) 2017:4521324. doi:10.1155/2017/4521324
134. Bao CY, Guo J, Lin GS, Hu MY, Hu ZM. TNFR gene-modified mesenchymal stem cells attenuate inflammation and cardiac dysfunction following MI. Scand Cardiovasc J (2008) 42:56–62. doi:10.1080/14017430701543556
135. Selvasandran K, Makhoul G, Jaiswal PK, Jurakhan R, Li L, Ridwan K, et al. A tumor necrosis factor-alpha and therapy for myocardial hypoxia-induced secretome repair. Ann Thorac Surg (2018) 105:715–23. doi:10.1016/j.athoracsur.2017.09.005
136. Tan J, Weil BR, Abarbanell AM, Wang Y, Herrmann JL, Dake ML, et al. Ablation of TNF-alpha receptors influences mesenchymal stem cell-mediated cardiac protection against ischemia. Shock (2010) 34:236–42. doi:10.1097/SHK.0b013e3181d75ae3
137. Zeller CN, Wang Y, Markel TA, Weil B, Abarbanell A, Herrmann JL, et al. Role of tumor necrosis factor receptor 1 in sex differences of stem cell mediated cardioprotection. Ann Thorac Surg (2009) 87:812–9. doi:10.1016/j.athoracsur.2008.12.033
138. Kelly ML, Wang M, Crisostomo PR, Abarbanell AM, Herrmann JL, Weil BR, et al. TNF receptor 2, not TNF receptor 1, enhances mesenchymal stem cell-mediated cardiac protection following acute ischemia. Shock (2010) 33:602–7. doi:10.1097/SHK.0b013e3181cc0913
139. Kwon YW, Heo SC, Jeong GO, Yoon JW, Mo WM, Lee MJ, et al. Tumor necrosis factor-alpha-activated mesenchymal stem cells promote endothelial progenitor cell homing and angiogenesis. Biochim Biophys Acta (2013) 1832:2136–44. doi:10.1016/j.bbadis.2013.08.002
140. Lee HS, Song S, Shin DY, Kim GS, Lee JH, Cho CW, et al. Enhanced effect of human mesenchymal stem cells expressing human TNF-R-Fc and HO-1 gene on porcine islet xenotransplantation in humanized mice. Xenotransplantation (2018) 25:e12342. doi:10.1111/xen.12342
141. Heo SC, Jeon ES, Lee IH, Kim HS, Kim MB, Kim JH. Tumor necrosis factor-alpha-activated human adipose tissue-derived mesenchymal stem cells accelerate cutaneous wound healing through paracrine mechanisms. J Invest Dermatol (2011) 131:1559–67. doi:10.1038/jid.2011.64
142. Wang Y, Weil BR, Herrmann JL, Abarbanell AM, Tan JN, Markel TA, et al. MEK, p38, and PI-3K mediate cross talk between EGFR and TNFR in enhancing hepatocyte growth factor production from human mesenchymal stem cells. Am J Physiol Cell Physiol (2009) 297:C1284–93. doi:10.1152/ajpcell.00183.2009
143. Markel TA, Crisostomo PR, Wang M, Wang Y, Lahm T, Novotny NM, et al. TNFR1 signaling resistance associated with female stem cell cytokine production is independent of TNFR2-mediated pathways. Am J Physiol Regul Integr Comp Physiol (2008) 295:R1124–30. doi:10.1152/ajpregu.90508.2008
144. Markel TA, Crisostomo PR, Wang M, Herring CM, Meldrum DR. Activation of individual tumor necrosis factor receptors differentially affects stem cell growth factor and cytokine production. Am J Physiol Gastrointest Liver Physiol (2007) 293:G657–62. doi:10.1152/ajpgi.00230.2007
145. Ray R, Herring CM, Markel TA, Crisostomo PR, Wang M, Weil B, et al. Deleterious effects of endogenous and exogenous testosterone on mesenchymal stem cell VEGF production. Am J Physiol Regul Integr Comp Physiol (2008) 294:R1498–503. doi:10.1152/ajpregu.00897.2007
146. Wang M, Tsai BM, Kher A, Baker LB, Wairiuko GM, Meldrum DR. Role of endogenous testosterone in myocardial proinflammatory and proapoptotic signaling after acute ischemia-reperfusion. Am J Physiol Heart Circ Physiol (2005) 288:H221–6. doi:10.1152/ajpheart.00784.2004
147. Bao C, Guo J, Zheng M, Chen Y, Lin G, Hu M. Enhancement of the survival of engrafted mesenchymal stem cells in the ischemic heart by TNFR gene transfection. Biochem Cell Biol (2010) 88:629–34. doi:10.1139/O10-018
148. Brambilla R, Ashbaugh JJ, Magliozzi R, Dellarole A, Karmally S, Szymkowski DE, et al. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain (2011) 134:2736–54. doi:10.1093/brain/awr199
149. Shemer A, Jung S. Differential roles of resident microglia and infiltrating monocytes in murine CNS autoimmunity. Semin Immunopathol (2015) 37:613–23. doi:10.1007/s00281-015-0519-z
150. Drago D, Cossetti C, Iraci N, Gaude E, Musco G, Bachi A, et al. The stem cell secretome and its role in brain repair. Biochimie (2013) 95:2271–85. doi:10.1016/j.biochi.2013.06.020
151. Azari MF, Mathias L, Ozturk E, Cram DS, Boyd RL, Petratos S. Mesenchymal stem cells for treatment of CNS injury. Curr Neuropharmacol (2010) 8:316–23. doi:10.2174/157015910793358204
152. Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu Rev Immunol (2012) 30:733–58. doi:10.1146/annurev-immunol-020711-075043
153. Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends Mol Med (2007) 13:108–16. doi:10.1016/j.molmed.2007.01.003
154. Miller PG, Bonn MB, McKarns SC. Transmembrane TNF-TNFR2 impairs Th17 differentiation by promoting Il2 expression. J Immunol (2015) 195:2633–47. doi:10.4049/jimmunol.1500286
155. Baeyens A, Saadoun D, Billiard F, Rouers A, Gregoire S, Zaragoza B, et al. Effector T cells boost regulatory T cell expansion by IL-2, TNF, OX40, and plasmacytoid dendritic cells depending on the immune context. J Immunol (2015) 194:999–1010. doi:10.4049/jimmunol.1400504
156. Nagashima H, Okuyama Y, Hayashi T, Ishii N, So T. TNFR-associated factors 2 and 5 differentially regulate the instructive IL-6 receptor signaling required for Th17 development. J Immunol (2016) 196:4082–9. doi:10.4049/jimmunol.1501610
157. Lee HJ, Kim SN, Jeon MS, Yi T, Song SU. ICOSL expression in human bone marrow-derived mesenchymal stem cells promotes induction of regulatory T cells. Sci Rep (2017) 7:44486. doi:10.1038/srep44486
158. Park MJ, Park HS, Cho ML, Oh HJ, Cho YG, Min SY, et al. Transforming growth factor beta-transduced mesenchymal stem cells ameliorate experimental autoimmune arthritis through reciprocal regulation of Treg/Th17 cells and osteoclastogenesis. Arthritis Rheum (2011) 63:1668–80. doi:10.1002/art.30326
159. Kim DJ, Kim KS, Song MY, Seo SH, Kim SJ, Yang BG, et al. Delivery of IL-12p40 ameliorates DSS-induced colitis by suppressing IL-17A expression and inflammation in the intestinal mucosa. Clin Immunol (2012) 144:190–9. doi:10.1016/j.clim.2012.06.009
160. Min CK, Kim BG, Park G, Cho B, Oh IH. IL-10-transduced bone marrow mesenchymal stem cells can attenuate the severity of acute graft-versus-host disease after experimental allogeneic stem cell transplantation. Bone Marrow Transplant (2007) 39:637–45. doi:10.1038/sj.bmt.1705644
161. Lim JY, Park MJ, Im KI, Kim N, Jeon EJ, Kim EJ, et al. Combination cell therapy using mesenchymal stem cells and regulatory T-cells provides a synergistic immunomodulatory effect associated with reciprocal regulation of Th1/Th2 and Th17/Treg cells in a murine acute graft-versus-host disease model. Cell Transplant (2014) 23:703–14. doi:10.3727/096368913X664577
162. Lee ES, Lim JY, Im KI, Kim N, Nam YS, Jeon YW, et al. Adoptive transfer of Treg cells combined with mesenchymal stem cells facilitates repopulation of endogenous Treg cells in a murine acute GVHD model. PLoS One (2015) 10:e0138846. doi:10.1371/journal.pone.0138846
Keywords: mesenchymal stem cells, tumor necrosis factor, TNFR, regulatory T, autoimmune and inflammatory diseases
Citation: Yan L, Zheng D and Xu R-H (2018) Critical Role of Tumor Necrosis Factor Signaling in Mesenchymal Stem Cell-Based Therapy for Autoimmune and Inflammatory Diseases. Front. Immunol. 9:1658. doi: 10.3389/fimmu.2018.01658
Received: 03 February 2018; Accepted: 04 July 2018;
Published: 20 July 2018
Edited by:
Magdalena Plebanski, RMIT University, AustraliaReviewed by:
Masahide Tone, Cedars-Sinai Medical Center, United StatesCopyright: © 2018 Yan, Zheng and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ren-He Xu, cmVuaGV4dUB1bWFjLm1v
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.