
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 July 2018
Sec. Cytokines and Soluble Mediators in Immunity
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01653
This article is part of the Research Topic Sex Hormones and Gender Differences in Immune Responses View all 16 articles
 Sapana Kadel1,2
Sapana Kadel1,2 Susan Kovats1,2*
Susan Kovats1,2*
Sex differences in the incidence and severity of respiratory virus infection are widely documented in humans and murine models and correlate with sex biases in numbers and/or functional responses of innate immune cells in homeostasis and lung infection. Similarly, changes in sex hormone levels upon puberty, pregnancy, and menopause/aging are associated with qualitative and quantitative differences in innate immunity. Immune cells express receptors for estrogens (ERα and ERβ), androgens (AR), and progesterone (PR), and experimental manipulation of sex hormone levels or receptors has revealed that sex hormone receptor activity often underlies sex differences in immune cell numbers and/or functional responses in the respiratory tract. While elegant studies have defined mechanistic roles for sex hormones and receptors in innate immune cells, much remains to be learned about the cellular and molecular mechanisms of action of ER, PR, and AR in myeloid cells and innate lymphocytes to promote the initiation and resolution of antiviral immunity in the lung. Here, we review the literature on sex differences and sex hormone regulation in innate immune cells in the lung in homeostasis and upon respiratory virus infection.
Respiratory virus infections lead to significant health problems worldwide (1). Humans show marked sex differences in the severity, prevalence, and outcome of inflammatory lung diseases including viral infection (2, 3). Innate immune responses have crucial roles in early defense against viruses but also shape antigen-specific adaptive immune responses and promote tissue repair. A number of recent reviews highlight sex differences in innate immune pathways during infectious disease (4–6). Here, we review literature reports on the sex differences in numbers and functional responses of innate immune cells in the lung and their regulation by sex hormones in homeostasis and during viral lung infection. Specifically, we highlight ways in which sex differences in innate cells may influence both the proinflammatory/effector phase and the resolution/tissue repair phase important in the host response to respiratory virus infection (Figure 1).
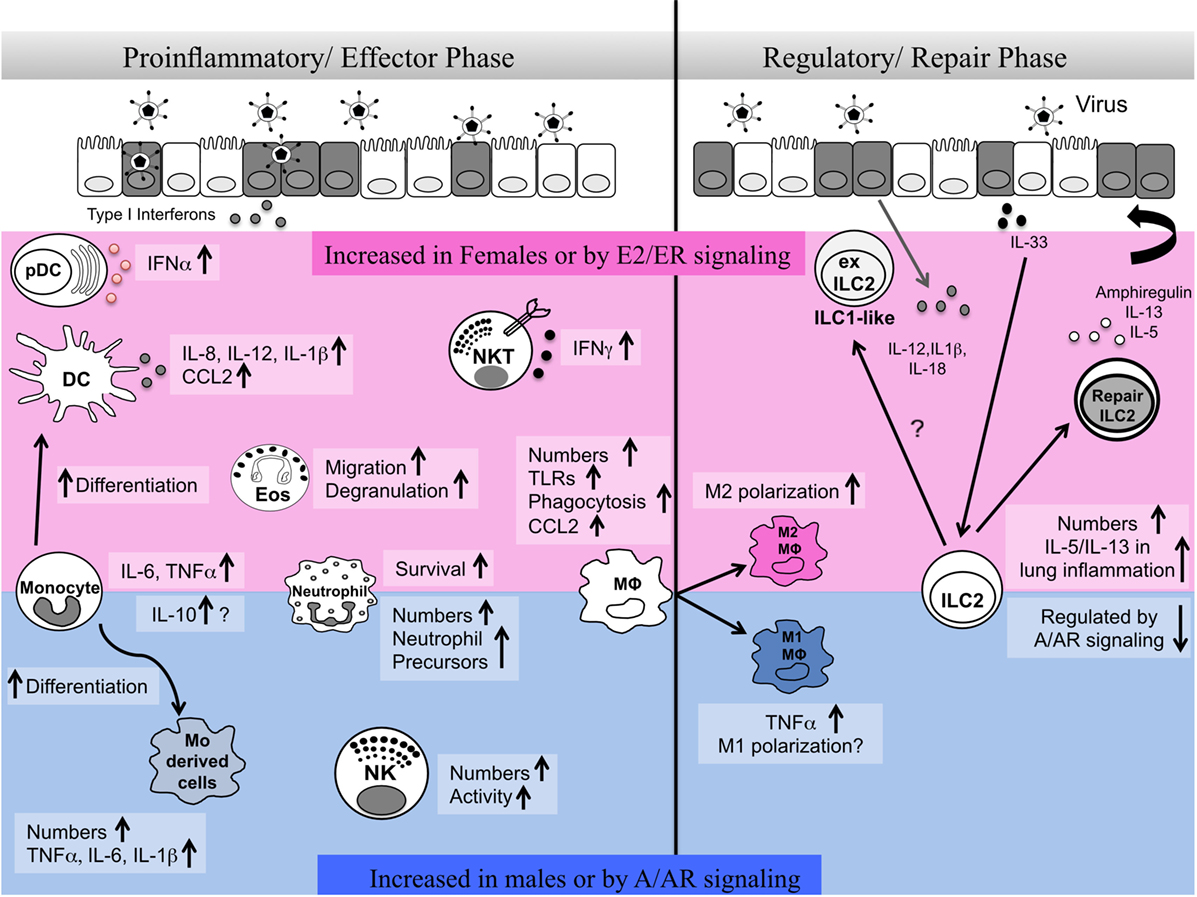
Figure 1. Sex differences in innate immune responses during the effector and repair phases of respiratory virus infection. Here, we summarize reports of sex differences or sex hormone receptor regulation of innate immune cells. The pink shaded area indicates cells and pathways reported to be elevated in females and/or upon estrogen/ER activity. The blue shaded area indicates cells and pathways reported to be increased in males and/or upon androgen/AR activity. A balanced type 1 immune response involving different innate immune cells is required early post-infection in the lung for viral clearance. At later stages of infection, regulatory immune responses mediated by alveolar macrophages and innate lymphoid cells are important for the repair of damaged tissues and renewal of barrier integrity. Sex differences in numbers, functional responses, plasticity, and survival of innate immune cells regulate the proinflammatory/effector and regulatory/repair phases of infection.
Endogenous estrogens include estrone (E1), 17-β-estradiol (E2), and estriol (E3), with E2 being the major form present in adult females and males while E3 is present at high levels in pregnancy (7). Testosterone is synthesized in both females and males and is converted by 5α-reductase to the physiologically active metabolite dihydrotestosterone (DHT) or by aromatase to estradiol (8). Progesterone (P4) is present in both sexes and, in females, varies during the menstrual cycle and is produced by the placenta at high levels during pregnancy (9). The levels of circulating sex hormones vary in both sexes throughout the lifespan, with the highest levels of estrogens and progesterone in females and testosterone in males during the reproductive years (10, 11). In pregnancy, estrogens and progesterone reach significantly higher levels (8, 12). Sex hormone levels in humans in phases of the menstrual cycle and pregnancy are nicely summarized in a recent review (13). The dramatic changes in sex hormone levels at puberty are correlated with changes in immune function and susceptibility to immune-mediated disease. Sex hormones also are present in utero and immediately post-birth, and this may influence immune cell differentiation and neonatal immunity. The developing testes in male fetuses produce testosterone, and both sexes are exposed to high levels of maternal estrogens in utero (14, 15). In the first weeks after birth, both human and rodent males have a “mini-puberty,” in which testosterone levels approach those of adults (15–17).
Sex steroids are synthesized in the gonads and adrenal cortex, and in peripheral tissues such as liver, fat, and kidney (8, 18). Little information is available about local synthesis in the lung (8). Activated macrophages may increase local estrogen levels since cytokine receptor signaling induces their synthesis of aromatase, the enzyme that converts testosterone to estradiol (19). Few studies of immune cells in tissues have correlated tissue levels of sex hormones with immune function.
Sex hormones mediate their effects through estrogen receptors (ERα and ERβ), androgen receptor (AR), and progesterone receptors (PR-A and PR-B) (20–22). Splice variants of ER leading to truncated but functional proteins such as ERα46 have been identified in myeloid cells (23). Sex steroid receptors are ligand-dependent transcription factors that recruit transcriptional coregulators such as SRC1 and histone-modifying enzymes such as p300/CBP into multi-protein complexes that bind DNA [reviewed in Ref. (20, 24)]. ERs, PRs, and AR bind to their respective response elements at specific DNA sites leading to epigenetic modifications of chromatin and changes in transcription of target genes. Nuclear sex hormone receptors also may be tethered indirectly to DNA via their ability to bind transcription factors such as SP1. Ligand-free receptors also can recruit corepressors such as NCOR and histone deacetylases to repress transcription. Rapid “nongenomic” sex steroid signaling occurs via inner plasma membrane-localized ER or AR, and possibly via the G protein-coupled receptor GPR30 (also termed GPER) (20, 25).
Innate immune cells express ERs (Esr1, Esr2), AR (Ar), and PRs (Pgr) to varying degrees. Esr1 and Ar RNAs also are expressed at high levels in hematopoietic progenitors in bone marrow (BM), consistent with documented effects of sex hormones on immune cell differentiation and numbers in homeostasis (26). Based on our literature review and data from the Immunological Genome Project (www.immgen.org), Table 1 summarizes the relative expression of sex steroid receptor RNA or protein in hematopoietic progenitors and innate cells of the lymphoid and myeloid lineages. Since limited information is available about sex steroid receptor expression in lung-resident immune cells, Table 1 includes information for the cell type regardless of tissue location or activation state. Patterns of receptor expression may underlie the effects of the sex hormones on numbers and functional responses of innate immune cells. Some mature innate cells do not apparently express significant levels of the sex hormone receptors, but they may still function differently in the sexes due to epigenetic imprinting of developmental precursors or because their responses are influenced indirectly via other cell types responding to sex hormones.
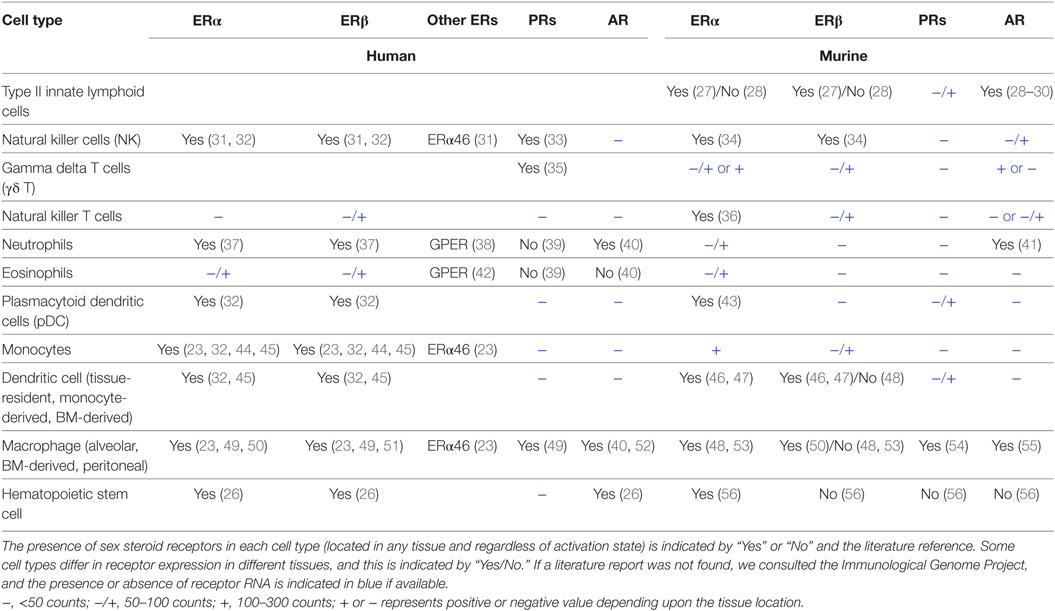
Table 1. Expression of sex steroid receptors in human and murine innate immune cells.
Sex differences in lung development, structure, and function have been identified (57). The lungs of human females are smaller than males of the same height; however, airway size and capacity do not always correlate with lung size, and the extent and type of sex differences in lung function vary throughout the lifespan (58). Smooth muscle, fibroblasts, and epithelial cells express sex steroid receptors (8), and their functional responses in different sex hormone environments may alter the immune response or modulate infection severity.
Investigators have taken multiple approaches to understand the impact of sex differences and sex hormone receptor signaling on immunity. Diverse approaches in different experimental models have often led to conflicting results. Age should be carefully considered when studying sex differences in immune cell numbers or functional responses in humans and rodents since sex hormone levels vary over the lifespan (2). Littermate or colony matching will reduce variables such as diet and microbiome and help to identify sex differences. Studies of sex differences in human immunity would be improved by direct measurements of hormone levels in each individual (particularly in women) because age alone does not predict hormone levels modulated by oral contraceptives or hormone replacement therapy. A common approach is to gonadectomize young mice and replace estrogens or androgens by implantation of time release pellets. While this strategy has led to many answers, the absence of sex hormones in young gonadectomized mice may alter immune cell development and numbers prior to infection. In addition, replacement of hormones to a constant level does not mimic the cyclic variation that occurs particularly in females. Similarly, it is difficult to recapitulate accurate in vivo exposures of sex hormones in cell culture models. Another approach is to impose male levels of DHT in a female mouse (or female levels of estradiol in a male mouse) to help elucidate sex hormone interactions and their effects independent of chromosomal sex and developmental programming.
Mice lacking sex hormone receptors also have informed our understanding of sex differences in immunity. However, global deletion of sex hormone receptors can lead to abnormal levels of estrogens and androgens; for example, global Esr1 deficiency leads to high levels of circulating testosterone (59–61). Furthermore, global loss of receptor signaling may alter the function of non-immune cell types in ways that impact immune responses. To circumvent this issue, investigators are beginning to study mice bearing conditional deletion of Esr1 or Ar along with lineage-restricted Cre drivers to understand the effects of sex hormone receptor deficiency on numbers and function of specific cell types. This approach will help to identify direct effects of sex hormone receptor signaling in immune cells. Use of emerging technologies such as single cell RNA-sequencing, assay of transposase-accessible chromatin-sequencing, multiplex mass cytometry, or chip cytometry (62–64) to monitor the transcriptome, epigenome, or proteome at the single cell level will help us to understand sex differences in immune function and how sex hormone receptors regulate immune cells in homeostasis and during viral infection. These approaches will be especially valuable to dissect the diversity of responses of rare immune cell types in peripheral tissues such as the lung. More precise methods and attention to age and hormonal cycles and levels will help to clarify the roles of sex hormones and receptors in immune responses.
Despite the lung’s structural and chemical barriers to pathogen entry, many viruses subvert these barriers and efficiently infect and replicate within lung epithelial cells [reviewed in Ref. (65)]. Damage to host lungs may be directly induced by the virus or be secondary to a strong immune response. Upon respiratory virus infection, immune cells typically participate in three phases: (i) innate immune cells sense presence of the virus and initiate early antiviral responses and prime the adaptive response; (ii) effector or adaptive immune cells clear virus by killing infected cells and producing antiviral antibodies, followed by conversion of a subset to memory lymphocytes; and (iii) innate immune cells act in concert with epithelial regeneration pathways to repair injured tissue and produce mediators that return the immune system to homeostasis (65). Herein, we focus on responses of innate immune cells in the initiation and repair phases of respiratory virus infection.
Epidemiological studies of humans and experimental models with rodents show that it is difficult to arrive at a universal paradigm regarding effects of sex or sex hormones on immune responses to respiratory viruses. vom Steeg and Klein hypothesize that sex differences in infection outcome are a function of the strength of the immune response and resulting host tissue damage (66). In this model, a male bias in risk occurs when weak immune responses underlie significant host damage, while a female bias in risk occurs when strong immune responses promote host damage. Experimental manipulation of sex hormones and their receptors in rodents has shown that sex differences in systemic estrogen and androgen levels often underlie differential immune function and infection outcome. Depending on the role of the sex hormone to promote or inhibit inflammation or immunity, sex differences may arise due to either the predominance of, or the absence of, estrogen or androgen in one sex.
In the initiation phase, lung-resident dendritic cells (DCs) and macrophages (alveolar and interstitial) respond to viral molecules (nucleic acids and glycoproteins) via cell surface or intracellular receptors that are linked to signaling pathways resulting in production of interferons (IFN), cytokines, and chemokines (65, 67). Coupled with these viral “pathogen-associated molecular patterns,” damage to host cells results in the release of host molecules such as ATP, heat shock proteins, or HMGB1, termed “danger-associated molecular patterns,” which also trigger innate immune receptors and inflammasomes. Innate lymphocytes respond to cytokines produced by activated myeloid cells or alarmins released by damaged tissue and in turn produce type 1 (IFNs, IL-12, IL-1β, TNFα) or type 2 or regulatory (IL-5, IL-10) cytokines that direct subsequent innate or adaptive responses. Type I and III IFNs elicit expression of molecules that are directly antiviral. DCs acquire and present viral antigens, migrate to draining lymph nodes and prime adaptive responses through interactions with naïve T. Activated T cells then return to the lung where they interact again with resident or recruited myeloid cells, produce pro- or anti-inflammatory cytokines, and lyse infected cells.
Respiratory viruses typically elicit strong type 1 immune responses involving myeloid cell production of type I and III IFN and proinflammatory mediators such as IL-12, TNFα, and CCL2 and lymphocyte production of IFNγ (67). As described in detail in later sections, there is some evidence for sex differences in (or sex hormone regulation of) the function of myeloid cells and innate lymphocytes during respiratory virus infection. However, more often, reports of sex differences or sex hormone regulation involve studies of immune cells at other tissue sites, in autoimmune or other pathogen models or performed in vitro. In brief, type I IFN synthesis is promoted by estrogen and ERα signaling, and multiple reports show that female plasmacytoid DCs (pDCs) produce more type I IFN than male pDCs (32, 43, 68, 69). Sex hormone regulation of proinflammatory cytokines (IL-12, IL-6, IL-1β) seems more complex, but a number of studies show that lower physiological levels of estrogens enhance their production while higher physiological levels dampen their production and instead promote regulatory cytokines such as IL-10 [reviewed in Ref. (24)]. In contrast, reports show that testosterone decreases cytokines such as IFNγ and TNFα and increases IL-10 (21).
Regulatory and type 2 immune responses are important in later stages of respiratory viral infection. It is now recognized that while type 1 responses are important for viral clearance, type 2 responses also are elicited and promote repair of injured tissue and resolution of the immune response upon influenza virus and respiratory syncytial virus (RSV) infection (70–72). In murine models of allergic asthma, estrogen and ERα signaling promote type 2 responses of alveolar macrophages (AM) (73), while androgens and AR signaling attenuate type 2 responses promoted by innate lymphoid cells (ILC2s) and myeloid cells (74). These ER and AR regulated pathways also may be important in respiratory virus infection. Indeed, the chronic elevation of type 2 responses in asthmatic individuals can lead to a milder course of influenza virus infection and reduced lung injury (75), while ILC2 activity in influenza virus infection can exacerbate asthma (76).
Sex differences in the incidence and severity of IAV infection in the human population have been well documented (13, 77). However, given the strong impact of age on morbidity and mortality, it is often difficult to separate effects of sex and age since sex hormone levels change dramatically with age (78). Furthermore, social and cultural differences in gender norms also may influence ascertainment or self-reporting of infection symptoms or access to medical care. While the incidence of infection is often higher in males, females often show greater morbidity. Increased infection severity in females may result from stronger innate and adaptive responses in females that lead to more extensive immunopathology. Epidemiological studies from the 1957 H2N2, 2005 H1N1, and 2009 H1N1 pandemic IAV infections showed that the mortality and hospitalization of patients following viral infection was higher for females than males during their reproductive years (77, 79–81). This suggests that adult levels of sex hormones modulate immunity to IAV infection; however, these studies did not measure immune responses at the molecular or cellular level. Females in their reproductive years also have increased asthma incidence (58), which may alter immune responses and increase IAV-induced pathology. In contrast, infection of young males (<age 20) and elderly adults (>age 80) led to greater hospitalization or mortality (80). While this might suggest that lower levels of androgens in young boys and elderly men correlate with increased infection severity, information about comorbidities and measurement of androgen levels coupled with more precise information regarding susceptibility of males pre- and post-puberty would be needed to make this correlation.
We also lack information regarding differential susceptibility to IAV infection in distinct phases of the menstrual cycle and in women taking oral contraceptives. These hormonal variables may modulate susceptibility or severity of IAV infection, as epidemiological data from asthmatic women and girls show premenstrual aggravation of asthma symptoms and alleviation of this cyclical effect while taking the oral combined contraceptive pill (58). Pregnancy was highly associated with increased mortality and morbidity following IAV infection, and one factor may be immune suppression by elevated estrogens and progesterone (82–84).
Studies of mice infected with mouse adapted and pandemic H1N1, and avian H3N1 and H7N9, viruses have provided valuable insights into sex differences in susceptibility and immunity to IAV. Morbidity, mortality, and the associated inflammatory response is greater in female than male mice at moderate IAV loads, but mortality of both sexes is similar at higher loads (85–87). At sublethal doses, females showed higher levels of TNFα, IFNγ, and CCL2 (85, 88) and neutralizing anti-influenza antibodies, which correlated with greater protection upon heterosubtypic virus challenge (86). At viral doses lethal in females, but not males, estrogen protected from mortality, as shown by comparing ovariectomized mice supplemented with estradiol or placebo (85). Estrogen replacement correlated with reduced TNFα and CCL2 production, yet increased numbers of neutrophils and CD8+ viral antigen-specific T cells producing IFNγ (89). Overall, gonadally intact and gonadectomized females produced greater inflammatory responses and showed increased morbidity following infection, suggesting that low levels of estrogens promote excessive inflammatory responses. In contrast, replacement of higher levels of estradiol to gonadectomized mice ameliorated inflammation and promoted adaptive immunity. This is consistent with anti-inflammatory effects of replaced estrogen in autoimmune disease models (90) and the ability of ERα to negatively regulate NF-κB signaling [reviewed in Ref. (20)].
Ovariectomy of females followed by progesterone replacement to luteal phase levels also reduced morbidity upon IAV infection (91). Progesterone led to increased tissue repair due to upregulation of the epidermal growth factor amphiregulin (Areg) in the lung (91). These studies suggest that progesterone-based contraceptives may promote recovery from respiratory virus infection (9).
Gonadectomy of young males increased morbidity and pathology upon IAV infection, and replacement of testosterone or DHT, which cannot be metabolized to estradiol, reduced morbidity, mortality, and inflammation (85, 92). In contrast, testosterone treatment of old male mice, which have decreased testosterone levels as in humans, increased survival but did not alter pathology (92). These data are consistent with the ability of testosterone to suppress inflammation (21, 93).
In murine models of sublethal IAV infection, morbidity typically is most related to immune-mediated pathology rather than failure to clear virus (67). Thus, data from the above studies suggest that the increased morbidity and mortality of females is secondary to a strong proinflammatory response that leads to extensive tissue damage, while the lesser morbidity in males is the result of a more balanced immune response that clears virus with less tissue damage. Sex hormones that suppress inflammation (testosterone, progesterone, or high levels of estrogens) may attenuate antiviral immune responses to an optimal level, while lower levels of estrogens and androgens may permit excessive inflammation in some cases. The evolutionary benefit of this disparate effect of female and male sex hormones on immunity remains unclear.
Infection by two other respiratory viruses leads to increased morbidity in males. RSV is a common respiratory tract infection that most often progresses to the lower respiratory tract with severe consequences in infants and the elderly. The overall incidence of RSV is higher in young boys than girls (94, 95); however, the possible immunological basis of this sex difference and the role of sex hormones remains unknown. In outbreaks of pathogenic coronaviruses including the severe acute respiratory syndrome (SARS-CoV) and the Middle East respiratory syndrome (MERS-CoV), males showed increased infection incidence and severity (96, 97). Male mice showed enhanced susceptibility to SARS-CoV including elevated viral titers and increased accumulation of inflammatory monocytes and neutrophils in the lungs (98). ER signaling in females may be protective in this infection since ovariectomy or treatment with an ER antagonist increased mortality, while male gonadectomy did not alter disease outcome.
During respiratory viral infection, responses of lymphoid and myeloid innate cells play a crucial role in early antiviral protection and promote the generation of adaptive immune responses including effector and memory T and B cells. Here, we highlight studies demonstrating sex differences and effects of sex hormones in the number, function, and development of innate cells in the respiratory tract (Figure 1). We also review reports of sex differences and sex hormone regulation in innate cells in other tissues, which may inform our understanding of sex-dependent regulatory mechanisms in the respiratory tract. This topic is the subject of excellent recent reviews (74, 93, 99, 100).
Innate lymphoid cells are tissue-resident cells that develop from lymphoid progenitors but lack antigen specific receptors. Like T cells, ILCs are divided into the ILC1, ILC2, ILC3, and natural killer cell (NK) subsets based on expression of fate-determining transcription factors and cytokine production (101). In homeostasis, ILC2s are the predominant ILC subset in the murine lung (102), and both ILC2s and ILC3s are predominant in human lung (103). NKs (see below), ILC1s, and ILC2s generate innate responses during IAV infection while the role of ILC3s has not been investigated. Notably, ILC2s in murine lung, BM, and small intestine express high levels of Ar but little Esr1 or Esr2 (27–30).
Respiratory viral infections cause death of lung epithelium mediated by viral toxicity and immune cell activation, and appropriate remodeling of lung tissue to maintain barrier integrity is crucial (70). ILC2s are important for tissue repair following IAV infection as they expand and secrete Areg, IL-13, and IL-5 (76, 104, 105). Areg promotes regeneration of the bronchial epithelium, and IL-13 promotes barrier integrity by inducing epithelial cell proliferation and survival (91, 106). IL-5 recruits eosinophils that promote antiviral immunity and lung tissue regeneration in the resolution phase (105, 107, 108). Via these pathways, ILC2s facilitate tissue repair in IAV and RSV infection (104, 109).
We and others reported sex differences in murine lung ILC2 numbers, with female mice harboring more lung ILC2s compared to males in homeostasis (28, 30, 110). A functional subset of lung ILC2s that lack the inhibitory E-cadherin-binding receptor KLRG1 is uniquely present in females (28, 110). Experiments involving hormone replacement in gonadectomized mice and mice bearing global or conditional deficiency in Esr1 or Ar showed that the sex difference in ILC2s is regulated by androgens and AR but not estrogens or progesterone (28, 30, 110). Males have increased numbers of ILC precursors in BM, suggesting that androgens attenuate the progression from ILC precursor to mature ILC2 (110). In humans, sex differences in lung ILC2s have not yet been investigated; however, increased numbers of ILC2s are present in the blood of asthmatic females compared to males (30). Interestingly, sex hormones may regulate ILC2s differently in each tissue. Estrogen and ERα signaling sustain uterine ILC2s, which express high levels of Esr1 compared to lung ILC2s (27). Fewer ILC2s accumulate in the central nervous system of female mice in the EAE model of multiple sclerosis (111). A lower proportion of ILC2s are present in cord blood of human female neonates compared to males (112).
Innate lymphoid cells in gonadectomized males produce more IL-5 and IL-13 after stimulation (28). Similarly, DHT treatment in vivo decreases IL-5 and IL-13 production by ILC2s (30), although a direct role for AR was not tested in these studies. Together with the finding that progesterone increases Areg expression (91), these data suggest that IAV-infected females may show superior tissue repair due to increased numbers of ILC2s capable of producing IL-13, IL-5, and Areg.
Alternately, the higher number of ILC2s in females may induce more pathology due to their functional plasticity. ILC2s convert to ILC1-like cells producing IFNγ in response to IL-12 and IL-18 produced during IAV infection and lung inflammation triggered by smoking or chronic obstructive pulmonary disease (113–115). Although sex differences in ILC2 plasticity during IAV infection have not been reported, higher numbers of ILC2s that are capable of converting to ILC1s at the peak of infection may contribute to more severe immunopathology in females.
Natural killers are cytotoxic innate lymphocytes that control viral burden via their early production of IFNγ (116). NKs enhance DC migration and T cell recruitment upon infection with a sublethal IAV dose, but depletion of NKs was protective after infection with a lethal dose (116). Human and murine NKs express ERs and PR but not AR (Table 1). Human studies revealed higher numbers and cytotoxic activity of blood NK cells in males compared to females (117–119). This sex difference was reversed in old age (120). Studies show that NK numbers in blood correlate with stages of the menstrual cycle, suggesting regulation by sex hormones (121–123). In pregnancy, the recruitment of NKs from the blood to the uterine mucosa coincides with the dramatic rise in estriol and progesterone (124, 125). However, the effect of estrogen or progesterone on NK cell activity is unclear since some studies showed that in vitro (human) or in vivo exposure to estrogen or progesterone decreases NK cell activity while others found no effect (33, 126–129). Sex differences in NK numbers or function during IAV infection have not been reported.
Innate γδ T cells bear TCRs with limited junctional diversity that recognize intact protein antigens and small phosphate or amine containing molecules (130). γδ T cells are divided into different tissue-specific subsets based on predominant pairings of particular Vγ or Vδ genes (131). γδ T cells show important functional responses during infection by RSV and IAV. In murine RSV infection, γδ T cells are recruited to the lungs and produce IFNγ, IL-17A, IL-10, and IL-4 resulting in the activation of other innate cells (132). Vγ4+ T cells also secrete IL-17A during IAV H1N1 infection to aggravate acute lung immunopathology (133). Levels of circulating Vγ9/Vδ2 T cells in adult women were significantly higher than in men (134); however, another study showed the opposite trend (135). Sex differences in γδ T cells in mice have not been reported.
Natural killer T cells facilitate cross-talk between the innate and adaptive immune system during viral infection. NKT cells are a subset of T lymphocytes expressing a restricted αβ TCR that recognizes CD1d-bound lipids. NKT cells play a protective role in IAV infection through their secretion of IL-22 and IFNγ to activate NK cells and CD8+ T cells (67). The absence of NKTs in a murine model of RSV infection led to a delay in viral clearance, suggesting a protective role in infection (136). In mice, estrogen acting via ERα regulates a sexual dimorphism in NKT function. Administration of estradiol to ovariectomized mice increased NKT IFNγ production upon in vivo stimulation by IL-12 + IL-18 and a CD1d ligand, and NKTs in ERα−/− mice produced less IFNγ (36). Reports of sex differences in human NKTs are inconsistent, and data from the Immunological Genome Project show only low levels of sex hormone receptor RNA in human NKTs (Table 1). Increased blood NKT cell numbers in women relative to men was reported in some studies (137–139). Sex differences in NKT cells in respiratory virus infection have not been reported, but in view of the above studies, analyses of possible sex disparate responses in NKT function in murine models of IAV or RSV may yield important insights.
Innate lymphocytes express sex hormone receptor RNAs at varying levels depending on the subset and tissue location (Table 1). While NK and NKT cells primarily express ERs, lung ILC2s predominantly express AR, suggesting regulation of the classes of innate lymphocytes by distinct sex hormone-mediated mechanisms. However, we lack information about sex differences in numbers and function of these diverse subsets in the murine lung during respiratory virus infection. Recent studies have shown a profound sex difference in numbers and functional responses of murine lung-resident ILC2s, and AR signaling decreases numbers of ILC2s in males. Future work will determine if this numerical disparity in ILC2s leads to sex differences in the resolution of respiratory virus infection. Reports of sex differences in numbers or function of innate lymphocytes in human blood are often conflicting, and more studies that carefully correlate gender, age, and sex hormone status with lymphocyte numbers and function in blood or tissues are needed to clarify the field.
Neutrophils are the predominant infiltrating innate cell type during respiratory viral infection in both humans and mice. Neutrophils mediate antiviral defense via their production of proinflammatory cytokines and reactive oxygen species (140). Their role in respiratory viral infection remains unclear, as they cause pathology and susceptibility to secondary infections in mice. Neutrophil numbers and neutrophil extracellular trap (NET) formation directly correlate with the severity of RSV infection (141, 142).
Neutrophils express ER and AR (Table 1), and sex differences in the number and function of neutrophils in humans have been reported. Neutrophil numbers in blood are increased during pregnancy and the luteal phase of the menstrual cycle, suggesting that higher levels of progesterone or estrogens promote neutrophil numbers (143–145). Neutrophils from healthy women of reproductive age show improved survival in vitro compared to those of healthy men. Estradiol and progesterone contribute to the delay in neutrophil apoptosis by decreasing expression of the pro-apoptotic protein caspase 3 (146). Other studies showed that sex hormones modulate neutrophil function in vitro, including chemotaxis and nitric oxide and superoxide production (147–149).
Sex hormones also regulate neutrophil numbers in homeostasis and infection in murine models. AR-deficient mice show reduced numbers of neutrophils and neutrophil precursors in BM (41). Consistent with regulation of neutrophil numbers by AR signaling, the enhanced susceptibility of male mice to SARS-CoV infection was associated with increased accumulation of neutrophils in the lung (98). In contrast, estradiol treatment of ovariectomized females elevated neutrophil chemoattractants and recruitment of neutrophils into the lungs, thereby increasing protection in IAV infection (89).
Eosinophils enhance antiviral immunity during RSV infection by sensing viral RNA via TLR7 and producing nitric oxide (150). In IAV infection, eosinophil degranulation and activation of viral antigen specific CD8+ T cells increases protection against infection (151). Estrogen increases eosinophil migration adhesion, survival, and degranulation both in vitro (42, 152) and in vivo (153). Furthermore, the number of eosinophils in female rats peaks with higher levels of estrogen during estrus, and ovariectomy significantly reduces uterine eosinophils (154, 155). These studies suggest that female sex hormones regulate eosinophil numbers, but sex differences in the numbers or function of eosinophil during respiratory viral infection have not been reported. Since eosinophils were reported to express very little Esr1 and no Ar or Pgr RNA (Table 1), sex differences in eosinophil numbers may be secondary to the sex differences in numbers of IL-5-producing ILC2s.
Alveolar macrophages are lung-resident phagocytic cells that induce protective antiviral immune responses via production of soluble mediators (156). In viral infection, AMs produce high levels of type I IFN important for viral clearance and chemokines that recruit inflammatory monocyte into the lung (156). Sex differences or the effect of sex hormones in AM function during respiratory virus infection have not been described, although murine AMs express both ERα and AR (73). Studies of peritoneal macrophages, which also express ERβ, offer some insight into how AMs may be regulated by sex hormones during virus-induced inflammation. Increased numbers of macrophages were present in the pleural and peritoneal cavities of female mice, and they showed higher levels of TLRs and phagocytic capacity, which was associated with stronger acute inflammatory responses (157). Consistent with this, inflammatory TLR-mediated responses of human monocyte-derived macrophages and murine peritoneal macrophages were enhanced by estrogen and reduced by testosterone exposure (158–161).
The roles of sex hormones in AM function during allergic asthma may provide insight into sex differences in AM functional responses in viral infection. AMs are polarized to an M1 phenotype in a type 1 environment involving IFN or to an M2 phenotype in a type 2 environment involving IL-4/IL-13. In allergy models, female mice show an increased type 2 polarized AM response, and estrogen signaling via ERα in AMs was an important driver of the allergic response in vivo (73, 99, 162). This is consistent with other reports that female sex and/or ERα signaling promotes M2 macrophage function in cutaneous wound healing (163), Coxsackievirus-induced myocarditis (164), and atherosclerosis (53). In contrast, AR activity in macrophages suppresses wound healing by enhancing local TNFα expression (165). These data suggest that estrogens and ERα may promote, while AR may attenuate, the type 2 responses that promote tissue repair in the resolution phase of a viral infection.
Monocytes respond to viral infection by secreting cytokines and chemokines. They also are precursors to “inflammatory” macrophages or DCs in tissues. Following virus infection, CCR2+ monocytes are recruited via the chemokine CCL2 from blood to the lung, where they differentiate into DC- or macrophage-like cells often producing proinflammatory cytokines such as TNFα and IL-12 (166). Physiological levels of estradiol decrease expression of CCR2 and CXCR3 on murine monocytes in vivo, suggesting that ER signaling might reduce monocyte recruitment to tissues (167). Indeed, systemic estradiol treatment of ovariectomized mice reduced CCL2 induction and numbers of infiltrating monocytes during IAV infection, although no differences in numbers of inflammatory monocyte-derived DCs (Mo-DCs) were noted (89). Consistent with this, SARS-CoV infection of more susceptible male mice led to increased accumulation of monocyte-derived cells (Ly6C+ CD11b+) producing proinflammatory cytokines relative to female mice, and depletion of the monocyte-derived cells partially protected mice from a lethal infection (98). In this model, ovarian hormones and ER signaling in female mice were protective while orchidectomy of male mice had no effect, suggesting estrogens rather than androgens regulate pathogenic monocyte responses.
Reports of sex differences in human monocyte numbers and cytokine production are inconsistent and may reflect the diversity of the human population. Postmenopausal women showed increased numbers of monocytes compared to premenopausal women (168). Other work showed that monocyte counts were higher in the luteal phase associated with higher progesterone levels than in the follicular phase (143). Pregnancy also was associated with higher monocyte numbers, yet reduced capacity for IL-12 and TNFα production (169). Peripheral monocytes from healthy females produced more IL-6 upon LPS stimulation as compared to males (170). However, studies to determine if estrogens regulate pro-inflammatory cytokine production by female monocytes and monocyte-derived macrophages showed either negative (167, 171) or positive regulation (172). Macrophages and monocytes exposed to testosterone decreased their production of proinflammatory cytokines and increased synthesis of IL-10 (173–175).
Dendritic cells are professional antigen-presenting cells classified by phenotype and functional capacity into distinct subsets including (pDCs), conventional DCs (cDCs), and Mo-DCs. While the lung harbors at least three subsets of tissue-resident cDCs (176), pDCs and Mo-DCs enter the lung in significant numbers upon infection. Murine lung-resident DCs express Esr1 but little Ar (Table 1). The direct effect of sex hormone receptor signaling in these DC subsets in the lung during respiratory virus infection has not been reported. However, studies of sex differences and sex hormone effects on DCs in other tissues may provide some clues about lung DC subsets (100, 177).
Upon infection, lung-resident cDCs migrate to the draining mediastinal lymph nodes and prime naïve T cells. While sex differences in the numbers or function of these DCs during virus infection have not been reported, no differences in lung cDC numbers were found in ovariectomized mice treated with placebo or estradiol and infected with IAV (89). Functional studies with murine BM-derived DCs showed that estradiol and ERα signaling promote the TLR dependent production of proinflammatory cytokines of cDCs in the Flt3L-driven model and inflammatory DCs in the GM-CSF model (178–181). Estradiol also increased the production of IL-8 and CCL2 from human Mo-DCs (182). Other studies have shown that estradiol promotes GM-CSF-driven DC differentiation in vitro [reviewed in Ref. (177)]. Estradiol acts via ERα in murine myeloid progenitors to promote DC differentiation by upregulating the transcription factor IRF4 (183). In contrast, progesterone decreased TNFα and IL-1β but not IL-10 production by rat BM-derived DCs (184) and reversed estradiol-mediated changes in differentiation and function of BM-derived murine DCs (185). Progesterone modulated TLR-induced activation and cytokine production by murine BM-derived DCs (186).
Plasmacytoid DCs rapidly respond to viral particles via endosomal and cytosolic sensors of viral nucleic acids and produce type I IFN and IFN-induced proteins that are directly antiviral. Female pDCs produce significantly more IFNα in response to viral nucleic acids or synthetic TLR7 ligands than male pDCs (68, 69), and this correlates with higher levels of ERα-regulated IRF5 in female cells (187). Estrogen signaling and XX chromosome dosage promoted sex differences in TLR7-mediated IFNα production by human pDC (32), and estradiol treatment of postmenopausal women enhanced their production of IFNα (43). Models of conditional Esr1 deficiency in DCs showed that ERα signaling drives sex differences in pDC functions (43, 188). Consistent with greater production of type I IFN by pDCs or other innate cells, female rats infected with respiratory Hantavirus showed greater expression of genes encoding viral nucleic acid sensors and type I IFN compared to males (189).
Testosterone and progesterone may suppress pDC responses, although pDCs do not apparently express significant levels of Ar or Pgr RNA in homeostasis (Table 1). Progesterone inhibits IFNα production by pDCs (190). Upon stimulation with a TLR7/8 agonist, human infant male infant pDC responses were significantly lower than those of females (191), which may be due to increased testosterone (or lower estrogen) levels in infants post-birth. Male PBMCs produced similar amounts of IFNα, yet greater amounts of IL-10 than female PBMCs upon IAV stimulation, and the IL-10 may dampen type 1 inflammation in males (192, 193). Taken together, these studies show that female pDCs produce higher levels of type I IFNs, consistent with stronger antiviral immune responses, yet more immunopathology in females.
Sex differences in the numbers or functional responses of myeloid cells in murine models of IAV and coronavirus infection have been reported. Manipulation of sex hormone signaling through gonadectomy −/+ sex hormone replacement, or ER or AR deficiency, has provided evidence for sex hormone-mediated regulation of neutrophils, pDCs, monocytes, and monocyte-derived cells in the lung during infection. Sex differences in lung-resident cDCs during infection have not been reported, but these DCs do express Esr1 suggesting estrogens may regulate their important role in initiation of innate and adaptive responses to viruses. In asthma models in which females exhibit more disease, sex hormones regulate AM type 2 responses, suggesting that sex differences in AM function during the resolution phase of respiratory virus infection also may occur. Overall, more research is needed to fully understand mechanisms of sex hormone regulation of myeloid cells during respiratory virus infection and how these may contribute to sex differences in antiviral defense.
Sex differences in immunity to respiratory viruses are evident in humans and experimental rodent models. Sex hormones may act directly in innate immune cells or their precursors to promote or attenuate their function, but it is also probable that innate cells are indirectly modulated by actions of other immune or non-immune cells responding to sex hormones. Differential regulation of innate cells by sex hormones during the proinflammatory/effector phase and resolution/repair phase is likely to shape the mechanisms of viral clearance and the host capacity to resolve inflammation and repair damaged tissue. For example, estrogens and ER signaling may promote IFN production by pDCs and NKT cells early post-infection, but also type 2 or regulatory responses of AMs important for optimal resolution of the infection. Sex or sex hormones may not have universal effects during respiratory virus infection. Indeed, although endogenous estrogens in gonad-intact murine females promoted inflammation during IAV, they were protective in coronavirus infection.
While elegant studies of sex differences and the role of sex hormones have informed the field of innate antiviral immunity, we still lack information on how sex hormone receptors act in individual cell types to regulate functional responses. Many reports of sex differences or sex hormone effects in immunity are conflicting, most likely because of experimental approaches that do not fully take into account sex hormone levels varying due to age or cycle, difficulty in reproducing natural sex hormone levels via manipulation in vitro or in vivo, or hormone imbalances in globally Ar or Esr1 deficient mice. Our understanding of sex biases in the antiviral responses of innate lymphoid and myeloid cells of the respiratory tract will be greatly facilitated by more precise approaches and measurements enabled by emerging technologies. When possible, careful studies of innate immune cells in the respiratory tract of infected humans would also contribute greatly to our understanding of sex-specific molecular and cellular pathways that underlie population data on incidence and severity of viral infections.
Whether sex differences in immunity confer an advantage at the population level remains unclear. Ideally, the capacity for strong immune responses to infection or tumors would be balanced by a lesser propensity for autoimmunity. Studies suggest this continuum differs between the sexes, with females often capable of superior immunity to pathogens but more susceptible to autoimmunity (2), although not all reported data fit into this simple model. Sex differences in immune function may arise as a byproduct of the distinct levels of androgens and estrogens that specify biological sex and gonad development. Consistent with their ability to bind DNA and regulate chromatin conformation, sex hormone receptors may act early in the pre- or postnatal period or during puberty to imprint sex-specific epigenetic patterns in the genome (5, 20). Epigenetically imprinted regions of open or closed chromatin in hematopoietic progenitors may differ between the sexes, and a sex divergent epigenome may be reinforced in mature immune cells in response to the sex hormone environment. The challenge of the field is to understand how sex hormones and their receptors regulate the epigenome and transcriptome in innate immune cells to mediate sex-divergent pathways that govern antiviral immune responses.
S Kadel and S Kovats reviewed the literature and wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by NIH HL119501 and the Presbyterian Health Foundation (to SKovats) and by an OMRF Patricia and Don Capra Predoctoral Fellowship (to SKadel).
1. Mizgerd JP. Lung infection – a public health priority. PLoS Med (2006) 3(2):e76. doi:10.1371/journal.pmed.0030076
2. Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol (2016) 16(10):626–38. doi:10.1038/nri.2016.90
3. Chamekh M, Deny M, Romano M, Lefevre N, Corazza F, Duchateau J, et al. Differential susceptibility to infectious respiratory diseases between males and females linked to sex-specific innate immune inflammatory response. Front Immunol (2017) 8:1806. doi:10.3389/fimmu.2017.01806
4. Fischer J, Jung N, Robinson N, Lehmann C. Sex differences in immune responses to infectious diseases. Infection (2015) 43(4):399–403. doi:10.1007/s15010-015-0791-9
5. Ghosh S, Klein RS. Sex drives dimorphic immune responses to viral infections. J Immunol (2017) 198(5):1782–90. doi:10.4049/jimmunol.1601166
6. Jaillon S, Berthenet K, Garlanda C. Sexual dimorphism in innate immunity. Clin Rev Allergy Immunol (2017). doi:10.1007/s12016-017-8648-x
7. Frump AL, Lahm T. Sex hormone signaling in the lung in health and disease: airways, parenchyma, and pulmonary vasculature. In: Hemnes AR, editor. Gender, Sex Hormones and Respiratory Disease: A Comprehensive Guide. Cham: Springer International Publishing (2016). p. 27–62.
8. Sathish V, Martin YN, Prakash YS. Sex steroid signaling: implications for lung diseases. Pharmacol Ther (2015) 150:94–108. doi:10.1016/j.pharmthera.2015.01.007
9. Hall OJ, Klein SL. Progesterone-based compounds affect immune responses and susceptibility to infections at diverse mucosal sites. Mucosal Immunol (2017) 10(5):1097–107. doi:10.1038/mi.2017.35
10. Decaroli MC, Rochira V. Aging and sex hormones in males. Virulence (2017) 8(5):545–70. doi:10.1080/21505594.2016.1259053
11. Bouman A, Heineman MJ, Faas MM. Sex hormones and the immune response in humans. Hum Reprod Update (2005) 11(4):411–23. doi:10.1093/humupd/dmi008
12. Tulchinsky D, Hobel CJ, Yeager E, Marshall JR. Plasma estrone, estradiol, estriol, progesterone, and 17-hydroxyprogesterone in human pregnancy. I. Normal pregnancy. Am J Obstet Gynecol (1972) 112(8):1095–100. doi:10.1016/0002-9378(72)90185-8
13. Gabriel G, Arck PC. Sex, immunity and influenza. J Infect Dis (2014) 209(Suppl_3):S93–9. doi:10.1093/infdis/jiu020
14. vom Saal FS. Sexual differentiation in litter-bearing mammals: influence of sex of adjacent fetuses in utero. J Anim Sci (1989) 67(7):1824–40. doi:10.2527/jas1989.6771824x
15. Pang SF, Tang F. Sex differences in the serum concentrations of testosterone in mice and hamsters during their critical periods of neural sexual differentiation. J Endocrinol (1984) 100(1):7–11. doi:10.1677/joe.0.1000007
16. Quigley CA. Editorial: the postnatal gonadotropin and sex steroid surge-insights from the androgen insensitivity syndrome. J Clin Endocrinol Metab (2002) 87(1):24–8. doi:10.1210/jc.87.1.24
17. Motelica-Heino I, Castanier M, Corbier P, Edwards DA, Roffi J. Testosterone levels in plasma and testes of neonatal mice. J Steroid Biochem (1988) 31(3):283–6. doi:10.1016/0022-4731(88)90351-2
18. Labrie F, Luu-The V, Belanger A, Lin SX, Simard J, Pelletier G, et al. Is dehydroepiandrosterone a hormone? J Endocrinol (2005) 187(2):169–96. doi:10.1677/joe.1.06264
19. Capellino S, Straub RH, Cutolo M. Aromatase and regulation of the estrogen-to-androgen ratio in synovial tissue inflammation: common pathway in both sexes. Ann N Y Acad Sci (2014) 1317:24–31. doi:10.1111/nyas.12398
20. Kovats S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol (2015) 294(2):63–9. doi:10.1016/j.cellimm.2015.01.018
21. Trigunaite A, Dimo J, Jorgensen TN. Suppressive effects of androgens on the immune system. Cell Immunol (2015) 294(2):87–94. doi:10.1016/j.cellimm.2015.02.004
22. Buskiewicz IA, Huber SA, Fairweather D. Chapter 4 – sex hormone receptor expression in the immune system A2. In: Neigh GN, Mitzelfelt MM, editors. Sex Differences in Physiology. Boston: Academic Press (2016). p. 45–60.
23. Murphy AJ, Guyre PM, Wira CR, Pioli PA. Estradiol regulates expression of estrogen receptor ERalpha46 in human macrophages. PLoS One (2009) 4(5):e5539. doi:10.1371/journal.pone.0005539
24. Kovats S, Carreras E, Agrawal H. Sex steroid receptors in immune cells. In: Klein SL, Roberts C, editors. Sex Hormones and Immunity to Infection. Berlin, Heidelberg: Springer Berlin Heidelberg (2010). p. 53–91.
25. Langer G, Bader B, Meoli L, Isensee J, Delbeck M, Noppinger PR, et al. A critical review of fundamental controversies in the field of GPR30 research. Steroids (2010) 75(8–9):603–10. doi:10.1016/j.steroids.2009.12.006
26. Igarashi H, Kouro T, Yokota T, Comp PC, Kincade PW. Age and stage dependency of estrogen receptor expression by lymphocyte precursors. Proc Natl Acad Sci U S A (2001) 98(26):15131–6. doi:10.1073/pnas.011513098
27. Bartemes K, Chen CC, Iijima K, Drake L, Kita H. IL-33-responsive group 2 innate lymphoid cells are regulated by female sex hormones in the uterus. J Immunol (2018) 200(1):229–36. doi:10.4049/jimmunol.1602085
28. Laffont S, Blanquart E, Savignac M, Cenac C, Laverny G, Metzger D, et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J Exp Med (2017) 214(6):1581–92. doi:10.1084/jem.20161807
29. Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol (2015) 16(3):306–17. doi:10.1038/ni.3094
30. Cephus JY, Stier MT, Fuseini H, Yung JA, Toki S, Bloodworth MH, et al. Testosterone attenuates group 2 innate lymphoid cell-mediated airway inflammation. Cell Rep (2017) 21(9):2487–99. doi:10.1016/j.celrep.2017.10.110
31. Pierdominici M, Maselli A, Colasanti T, Giammarioli AM, Delunardo F, Vacirca D, et al. Estrogen receptor profiles in human peripheral blood lymphocytes. Immunol Lett (2010) 132(1):79–85. doi:10.1016/j.imlet.2010.06.003
32. Laffont S, Rouquie N, Azar P, Seillet C, Plumas J, Aspord C, et al. X-Chromosome complement and estrogen receptor signaling independently contribute to the enhanced TLR7-mediated IFN-alpha production of plasmacytoid dendritic cells from women. J Immunol (2014) 193(11):5444–52. doi:10.4049/jimmunol.1303400
33. Arruvito L, Giulianelli S, Flores AC, Paladino N, Barboza M, Lanari C, et al. NK cells expressing a progesterone receptor are susceptible to progesterone-induced apoptosis. J Immunol (2008) 180(8):5746–53. doi:10.4049/jimmunol.180.8.5746
34. Curran EM, Berghaus LJ, Vernetti NJ, Saporita AJ, Lubahn DB, Estes DM. Natural killer cells express estrogen receptor-α and estrogen receptor-β and can respond to estrogen via a non-estrogen receptor-α-mediated pathway. Cell Immunol (2001) 214(1):12–20. doi:10.1006/cimm.2002.1886
35. Polgar B, Barakonyi A, Xynos I, Szekeres-Bartho J. The role of gamma/delta T cell receptor positive cells in pregnancy. Am J Reprod Immunol (1999) 41(4):239–44. doi:10.1111/j.1600-0897.1999.tb00433.x
36. Gourdy P, Araujo LM, Zhu R, Garmy-Susini B, Diem S, Laurell H, et al. Relevance of sexual dimorphism to regulatory T cells: estradiol promotes IFN-gamma production by invariant natural killer T cells. Blood (2005) 105(6):2415–20. doi:10.1182/blood-2004-07-2819
37. Molero L, Garcia-Duran M, Diaz-Recasens J, Rico L, Casado S, Lopez-Farre A. Expression of estrogen receptor subtypes and neuronal nitric oxide synthase in neutrophils from women and men: regulation by estrogen. Cardiovasc Res (2002) 56(1):43–51. doi:10.1016/S0008-6363(02)00505-9
38. Rodenas MC, Tamassia N, Cabas I, Calzetti F, Meseguer J, Cassatella MA, et al. G protein-coupled estrogen receptor 1 regulates human neutrophil functions. Biomed Hub (2017) 2(1):2. doi:10.1159/000454981
39. Aerts JL, Christiaens MR, Vandekerckhove P. Evaluation of progesterone receptor expression in eosinophils using real-time quantitative PCR. Biochim Biophys Acta (2002) 1571(3):167–72. doi:10.1016/S0304-4165(02)00192-7
40. Mantalaris A, Panoskaltsis N, Sakai Y, Bourne P, Chang C, Messing EM, et al. Localization of androgen receptor expression in human bone marrow. J Pathol (2001) 193(3):361–6. doi:10.1002/1096-9896(0000)9999:9999<::AID-PATH803>3.0.CO;2-W
41. Chuang K-H, Altuwaijri S, Li G, Lai J-J, Chu C-Y, Lai K-P, et al. Neutropenia with impaired host defense against microbial infection in mice lacking androgen receptor. J Exp Med (2009) 206(5):1181–99. doi:10.1084/jem.20082521
42. Tamaki M, Konno Y, Kobayashi Y, Takeda M, Itoga M, Moritoki Y, et al. Expression and functional roles of G-protein-coupled estrogen receptor (GPER) in human eosinophils. Immunol Lett (2014) 160(1):72–8. doi:10.1016/j.imlet.2014.03.012
43. Seillet C, Laffont S, Tremollieres F, Rouquie N, Ribot C, Arnal JF, et al. The TLR-mediated response of plasmacytoid dendritic cells is positively regulated by estradiol in vivo through cell-intrinsic estrogen receptor alpha signaling. Blood (2012) 119(2):454–64. doi:10.1182/blood-2011-08-371831
44. Pioli PA, Jensen AL, Weaver LK, Amiel E, Shen Z, Shen L, et al. Estradiol attenuates lipopolysaccharide-induced CXC chemokine ligand 8 production by human peripheral blood monocytes. J Immunol (2007) 179(9):6284–90. doi:10.4049/jimmunol.179.9.6284
45. Komi J, Lassila O. Nonsteroidal anti-estrogens inhibit the functional differentiation of human monocyte-derived dendritic cells. Blood (2000) 95(9):2875–82.
46. Mao A, Paharkova-Vatchkova V, Hardy J, Miller MM, Kovats S. Estrogen selectively promotes the differentiation of dendritic cells with characteristics of Langerhans cells. J Immunol (2005) 175(8):5146–51. doi:10.4049/jimmunol.175.8.5146
47. Paharkova-Vatchkova V, Maldonado R, Kovats S. Estrogen preferentially promotes the differentiation of CD11c+ CD11b(intermediate) dendritic cells from bone marrow precursors. J Immunol (2004) 172(3):1426–36. doi:10.4049/jimmunol.172.3.1426
48. Lambert KC, Curran EM, Judy BM, Lubahn DB, Estes DM. Estrogen receptor-alpha deficiency promotes increased TNF-alpha secretion and bacterial killing by murine macrophages in response to microbial stimuli in vitro. J Leukoc Biol (2004) 75(6):1166–72. doi:10.1189/jlb.1103589
49. Khan KN, Masuzaki H, Fujishita A, Kitajima M, Sekine I, Matsuyama T, et al. Estrogen and progesterone receptor expression in macrophages and regulation of hepatocyte growth factor by ovarian steroids in women with endometriosis. Hum Reprod (2005) 20(7):2004–13. doi:10.1093/humrep/deh897
50. Vegeto E, Bonincontro C, Pollio G, Sala A, Viappiani S, Nardi F, et al. Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J Neurosci (2001) 21(6):1809–18. doi:10.1523/JNEUROSCI.21-06-01809.2001
51. Kramer PR, Wray S. 17-Beta-estradiol regulates expression of genes that function in macrophage activation and cholesterol homeostasis. J Steroid Biochem Mol Biol (2002) 81(3):203–16. doi:10.1016/S0960-0760(02)00065-1
52. McCrohon JA, Death AK, Nakhla S, Jessup W, Handelsman DJ, Stanley KK, et al. Androgen receptor expression is greater in macrophages from male than from female donors. A sex difference with implications for atherogenesis. Circulation (2000) 101(3):224–6. doi:10.1161/01.CIR.101.3.224
53. Ribas V, Drew BG, Le JA, Soleymani T, Daraei P, Sitz D, et al. Myeloid-specific estrogen receptor α deficiency impairs metabolic homeostasis and accelerates atherosclerotic lesion development. Proc Natl Acad Sci U S A (2011) 108(39):16457–62. doi:10.1073/pnas.1104533108
54. Lu J, Reese J, Zhou Y, Hirsch E. Progesterone-induced activation of membrane-bound progesterone receptors in murine macrophage cells. J Endocrinol (2015) 224(2):183–94. doi:10.1530/JOE-14-0470
55. Ashcroft GS, Mills SJ. Androgen receptor-mediated inhibition of cutaneous wound healing. J Clin Invest (2002) 110(5):615–24. doi:10.1172/JCI0215704
56. Nakada D, Oguro H, Levi BP, Ryan N, Kitano A, Saitoh Y, et al. Oestrogen increases haematopoietic stem-cell self-renewal in females and during pregnancy. Nature (2014) 505(7484):555–8. doi:10.1038/nature12932
58. Becklake MR, Kauffmann F. Gender differences in airway behaviour over the human life span. Thorax (1999) 54(12):1119–38. doi:10.1136/thx.54.12.1119
59. Elliot SJ, Berho M, Korach K, Doublier S, Lupia E, Striker GE, et al. Gender-specific effects of endogenous testosterone: female alpha-estrogen receptor-deficient C57Bl/6J mice develop glomerulosclerosis. Kidney Int (2007) 72(4):464–72. doi:10.1038/sj.ki.5002328
60. Akingbemi BT, Ge R, Rosenfeld CS, Newton LG, Hardy DO, Catterall JF, et al. Estrogen receptor-alpha gene deficiency enhances androgen biosynthesis in the mouse Leydig cell. Endocrinology (2003) 144(1):84–93. doi:10.1210/en.2002-220292
61. Sims NA, Dupont S, Krust A, Clement-Lacroix P, Minet D, Resche-Rigon M, et al. Deletion of estrogen receptors reveals a regulatory role for estrogen receptors-beta in bone remodeling in females but not in males. Bone (2002) 30(1):18–25. doi:10.1016/S8756-3282(01)00643-3
62. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods (2013) 10(12):1213–8. doi:10.1038/nmeth.2688
63. Cheung P, Vallania F, Warsinske HC, Donato M, Schaffert S, Chang SE, et al. Single-cell chromatin modification profiling reveals increased epigenetic variations with aging. Cell (2018) 173(6):1385–97.e14. doi:10.1016/j.cell.2018.03.079
64. Happle C, Lachmann N, Skuljec J, Wetzke M, Ackermann M, Brennig S, et al. Pulmonary transplantation of macrophage progenitors as effective and long-lasting therapy for hereditary pulmonary alveolar proteinosis. Sci Transl Med (2014) 6(250):250ra113. doi:10.1126/scitranslmed.3009750
65. Yoo JK, Kim TS, Hufford MM, Braciale TJ. Viral infection of the lung: host response and sequelae. J Allergy Clin Immunol (2013) 132(6):1263–76; quiz 77. doi:10.1016/j.jaci.2013.06.006
66. vom Steeg LG, Klein SL. SeXX matters in infectious disease pathogenesis. PLoS Pathog (2016) 12(2):e1005374. doi:10.1371/journal.ppat.1005374
67. Newton AH, Cardani A, Braciale TJ. The host immune response in respiratory virus infection: balancing virus clearance and immunopathology. Semin Immunopathol (2016) 38(4):471–82. doi:10.1007/s00281-016-0558-0
68. Berghofer B, Frommer T, Haley G, Fink L, Bein G, Hackstein H. TLR7 ligands induce higher IFN-alpha production in females. J Immunol (2006) 177(4):2088–96. doi:10.4049/jimmunol.177.4.2088
69. Meier A, Chang JJ, Chan ES, Pollard RB, Sidhu HK, Kulkarni S, et al. Sex differences in the toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med (2009) 15(8):955–9. doi:10.1038/nm.2004
70. Gorski SA, Hufford MM, Braciale TJ. Recent insights into pulmonary repair following virus-induced inflammation of the respiratory tract. Curr Opin Virol (2012) 2(3):233–41. doi:10.1016/j.coviro.2012.04.006
71. Guo XJ, Thomas PG. New fronts emerge in the influenza cytokine storm. Semin Immunopathol (2017) 39(5):541–50. doi:10.1007/s00281-017-0636-y
72. Sang Y, Miller LC, Blecha F. Macrophage polarization in virus-host interactions. J Clin Cell Immunol (2015) 6(2):311–21. doi:10.4172/2155-9899.1000311
73. Keselman A, Fang X, White PB, Heller NM. Estrogen signaling contributes to sex differences in macrophage polarization during asthma. J Immunol (2017) 199(5):1573–83. doi:10.4049/jimmunol.1601975
74. Laffont S, Blanquart E, Guery JC. Sex differences in asthma: a key role of androgen-signaling in group 2 innate lymphoid cells. Front Immunol (2017) 8:1069. doi:10.3389/fimmu.2017.01069
75. Samarasinghe AE, Woolard SN, Boyd KL, Hoselton SA, Schuh JM, McCullers JA. The immune profile associated with acute allergic asthma accelerates clearance of influenza virus. Immunol Cell Biol (2014) 92(5):449–59. doi:10.1038/icb.2013.113
76. Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol (2011) 12(7):631–8. doi:10.1038/ni.2045
77. Klein SL, Hodgson A, Robinson DP. Mechanisms of sex disparities in influenza pathogenesis. J Leukoc Biol (2012) 92(1):67–73. doi:10.1189/jlb.0811427
78. Gubbels Bupp MR, Potluri T, Fink AL, Klein SL. The confluence of sex hormones and aging on immunity. Front Immunol (2018) 9:1269. doi:10.3389/fimmu.2018.01269
79. Serfung RE, Sherman IL, Houseworth WJ. Excess pneumonia-influenza mortality by age and sex in three major influenza A2 epidemics, United States, 1957–58, 1960 and 1963. Am J Epidemiol (1967) 86(2):433–41. doi:10.1093/oxfordjournals.aje.a120753
80. Eshima N, Tokumaru O, Hara S, Bacal K, Korematsu S, Tabata M, et al. Sex- and age-related differences in morbidity rates of 2009 pandemic influenza A H1N1 virus of swine origin in Japan. PLoS One (2011) 6(4):e19409. doi:10.1371/journal.pone.0019409
81. Kumar A, Zarychanski R, Pinto R, Cook DJ, Marshall J, Lacroix J, et al. Critically ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA (2009) 302(17):1872–9. doi:10.1001/jama.2009.1496
82. Satpathy HK, Lindsay M, Kawwass JF. Novel H1N1 virus infection and pregnancy. Postgrad Med (2009) 121(6):106–12. doi:10.3810/pgm.2009.11.2080
83. Louie JK, Acosta M, Jamieson DJ, Honein MA. Severe 2009 H1N1 influenza in pregnant and postpartum women in California. N Engl J Med (2010) 362(1):27–35. doi:10.1056/NEJMoa0910444
84. Jamieson DJ, Honein MA, Rasmussen SA, Williams JL, Swerdlow DL, Biggerstaff MS, et al. H1N1 2009 influenza virus infection during pregnancy in the USA. Lancet (2009) 374(9688):451–8. doi:10.1016/S0140-6736(09)61304-0
85. Robinson DP, Lorenzo ME, Jian W, Klein SL. Elevated 17beta-estradiol protects females from influenza A virus pathogenesis by suppressing inflammatory responses. PLoS Pathog (2011) 7(7):e1002149. doi:10.1371/journal.ppat.1002149
86. Lorenzo ME, Hodgson A, Robinson DP, Kaplan JB, Pekosz A, Klein SL. Antibody responses and cross protection against lethal influenza A viruses differ between the sexes in C57BL/6 mice. Vaccine (2011) 29(49):9246–55. doi:10.1016/j.vaccine.2011.09.110
87. Hoffmann J, Otte A, Thiele S, Lotter H, Shu Y, Gabriel G. Sex differences in H7N9 influenza A virus pathogenesis. Vaccine (2015) 33(49):6949–54. doi:10.1016/j.vaccine.2015.08.044
88. Larcombe AN, Foong RE, Bozanich EM, Berry LJ, Garratt LW, Gualano RC, et al. Sexual dimorphism in lung function responses to acute influenza A infection. Influenza Other Respi Viruses (2011) 5(5):334–42. doi:10.1111/j.1750-2659.2011.00236.x
89. Robinson DP, Hall OJ, Nilles TL, Bream JH, Klein SL. 17beta-estradiol protects females against influenza by recruiting neutrophils and increasing virus-specific CD8 T cell responses in the lungs. J Virol (2014) 88(9):4711–20. doi:10.1128/JVI.02081-13
90. Offner H, Polanczyk M. A potential role for estrogen in experimental autoimmune encephalomyelitis and multiple sclerosis. Ann N Y Acad Sci (2006) 1089:343–72. doi:10.1196/annals.1386.021
91. Hall OJ, Limjunyawong N, Vermillion MS, Robinson DP, Wohlgemuth N, Pekosz A, et al. Progesterone-based therapy protects against influenza by promoting lung repair and recovery in females. PLoS Pathog (2016) 12(9):e1005840. doi:10.1371/journal.ppat.1005840
92. vom Steeg LG, Vermillion MS, Hall OJ, Alam O, McFarland R, Chen H, et al. Age and testosterone mediate influenza pathogenesis in male mice. Am J Physiol Lung Cell Mol Physiol (2016) 311(6):L1234–44. doi:10.1152/ajplung.00352.2016
93. Gubbels Bupp MR, Jorgensen TN. Androgen-induced immunosuppression. Front Immunol (2018) 9:794. doi:10.3389/fimmu.2018.00794
94. Glezen WP, Loda FA, Clyde WA Jr, Senior RJ, Sheaffer CI, Conley WG, et al. Epidemiologic patterns of acute lower respiratory disease of children in a pediatric group practice. J Pediatr (1971) 78(3):397–406. doi:10.1016/S0022-3476(71)80218-4
95. Simoes EA. Environmental and demographic risk factors for respiratory syncytial virus lower respiratory tract disease. J Pediatr (2003) 143(5 Suppl):S118–26. doi:10.1067/S0022-3476(03)00511-0
96. Karlberg J, Chong DS, Lai WY. Do men have a higher case fatality rate of severe acute respiratory syndrome than women do? Am J Epidemiol (2004) 159(3):229–31. doi:10.1093/aje/kwh056
97. Alghamdi IG, Hussain II, Almalki SS, Alghamdi MS, Alghamdi MM, El-Sheemy MA. The pattern of Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive epidemiological analysis of data from the Saudi Ministry of Health. Int J Gen Med (2014) 7:417–23. doi:10.2147/IJGM.S67061
98. Channappanavar R, Fett C, Mack M, Ten Eyck PP, Meyerholz DK, Perlman S. Sex-based differences in susceptibility to severe acute respiratory syndrome coronavirus infection. J Immunol (2017) 198(10):4046–53. doi:10.4049/jimmunol.1601896
99. Keselman A, Heller N. Estrogen signaling modulates allergic inflammation and contributes to sex differences in asthma. Front Immunol (2015) 6:568. doi:10.3389/fimmu.2015.00568
100. Laffont S, Seillet C, Guéry J-C. Estrogen receptor-dependent regulation of dendritic cell development and function. Front Immunol (2017) 8:108. doi:10.3389/fimmu.2017.00108
101. Artis D, Spits H. The biology of innate lymphoid cells. Nature (2015) 517(7534):293–301. doi:10.1038/nature14189
102. Lai D-M, Shu Q, Fan J. The origin and role of innate lymphoid cells in the lung. Military Med Res (2016) 3:25. doi:10.1186/s40779-016-0093-2
103. De Grove KC, Provoost S, Verhamme FM, Bracke KR, Joos GF, Maes T, et al. Characterization and quantification of innate lymphoid cell subsets in human lung. PLoS One (2016) 11(1):e0145961. doi:10.1371/journal.pone.0145961
104. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol (2011) 12(11):1045–54. doi:10.1031/ni.2131
105. Califano D, Furuya Y, Roberts S, Avram D, McKenzie ANJ, Metzger DW. IFN-gamma increases susceptibility to influenza A infection through suppression of group II innate lymphoid cells. Mucosal Immunol (2018) 11(1):209–19. doi:10.1038/mi.2017.41
106. Zaiss DMW, Gause WC, Osborne LC, Artis D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity (2015) 42(2):216–26. doi:10.1016/j.immuni.2015.01.020
107. Gorski SA, Hahn YS, Braciale TJ. Group 2 innate lymphoid cell production of IL-5 is regulated by NKT cells during influenza virus infection. PLoS Pathog (2013) 9(9):e1003615. doi:10.1371/journal.ppat.1003615
108. Stevens RL. Viral infections: beneficial role of eosinophils. Blood (2007) 110(5):1406. doi:10.1182/blood-2007-05-091389
109. Liu J, Wu J, Qi F, Zeng S, Xu L, Hu H, et al. Natural helper cells contribute to pulmonary eosinophilia by producing IL-13 via IL-33/ST2 pathway in a murine model of respiratory syncytial virus infection. Int Immunopharmacol (2015) 28(1):337–43. doi:10.1016/j.intimp.2015.05.035
110. Kadel S, Ainsua-Enrich E, Hatipoglu I, Turner S, Singh S, Khan S, et al. A major population of functional KLRG1(-) ILC2s in female lungs contributes to a sex bias in ILC2 numbers. Immunohorizons (2018) 2(2):74–86. doi:10.4049/immunohorizons.1800008
111. Russi AE, Walker-Caulfield ME, Ebel ME, Brown MA. Cutting edge: c-Kit signaling differentially regulates type 2 innate lymphoid cell accumulation and susceptibility to central nervous system demyelination in male and female SJL mice. J Immunol (2015) 194(12):5609–13. doi:10.4049/jimmunol.1500068
112. Forsberg A, Bengtsson M, Eringfalt A, Ernerudh J, Mjosberg J, Jenmalm MC. GATA binding protein 3(+) group 2 innate lymphoid cells are present in cord blood and in higher proportions in male than in female neonates. J Allergy Clin Immunol (2014) 134(1):228–30. doi:10.1016/j.jaci.2014.01.027
113. Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol (2016) 17(6):626–35. doi:10.1038/ni.3443
114. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1beta, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol (2016) 17(6):636–45. doi:10.1038/ni.3444
115. Ohne Y, Silver JS, Thompson-Snipes L, Collet MA, Blanck JP, Cantarel BL, et al. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat Immunol (2016) 17(6):646–55. doi:10.1038/ni.3447
116. Lam VC, Lanier LL. NK cells in host responses to viral infections. Curr Opin Immunol (2017) 44:43–51. doi:10.1016/j.coi.2016.11.003
117. Abdullah M, Chai PS, Chong MY, Tohit ER, Ramasamy R, Pei CP, et al. Gender effect on in vitro lymphocyte subset levels of healthy individuals. Cell Immunol (2012) 272(2):214–9. doi:10.1016/j.cellimm.2011.10.009
118. Lee BW, Yap HK, Chew FT, Quah TC, Prabhakaran K, Chan GS, et al. Age- and sex-related changes in lymphocyte subpopulations of healthy Asian subjects: from birth to adulthood. Cytometry (1996) 26(1):8–15. doi:10.1002/(SICI)1097-0320(19960315)26:1<8::AID-CYTO2>3.0.CO;2-E
119. Chng WJ, Tan GB, Kuperan P. Establishment of adult peripheral blood lymphocyte subset reference range for an Asian population by single-platform flow cytometry: influence of age, sex, and race and comparison with other published studies. Clin Diagn Lab Immunol (2004) 11(1):168–73. doi:10.1128/CDLI.11.1.168-173.2004
120. Al-Attar A, Presnell S, Peterson CA, Travis Thomas D, Lutz CT. The effect of sex on immune cells in healthy aging: elderly women have more robust natural killer lymphocytes than do elderly men. Mech Ageing Dev (2016) 156:25–33. doi:10.1016/j.mad.2016.04.001
121. McDonald FG, Ferguson MM. Variation in natural killer activity in peripheral blood during the menstrual cycle. Br Med J (Clin Res Ed) (1985) 290(6480):1514–5. doi:10.1136/bmj.290.6480.1514-b
122. Souza SS, Castro FA, Mendonca HC, Palma PV, Morais FR, Ferriani RA, et al. Influence of menstrual cycle on NK activity. J Reprod Immunol (2001) 50(2):151–9. doi:10.1016/S0165-0378(00)00091-7
123. Yovel G, Shakhar K, Ben-Eliyahu S. The effects of sex, menstrual cycle, and oral contraceptives on the number and activity of natural killer cells. Gynecol Oncol (2001) 81(2):254–62. doi:10.1006/gyno.2001.6153
124. King A, Jokhi PP, Burrows TD, Gardner L, Sharkey AM, Loke YW. Functions of human decidual NK cells. Am J Reprod Immunol (1996) 35(3):258–60. doi:10.1111/j.1600-0897.1996.tb00041.x
125. Carlino C, Stabile H, Morrone S, Bulla R, Soriani A, Agostinis C, et al. Recruitment of circulating NK cells through decidual tissues: a possible mechanism controlling NK cell accumulation in the uterus during early pregnancy. Blood (2008) 111(6):3108–15. doi:10.1182/blood-2007-08-105965
126. Sulke AN, Jones DB, Wood PJ. Hormonal modulation of human natural killer cell activity in vitro. J Reprod Immunol (1985) 7(2):105–10. doi:10.1016/0165-0378(85)90064-6
127. Hou J, Zheng WF. Effect of sex hormones on NK and ADCC activity of mice. Int J Immunopharmacol (1988) 10(1):15–22. doi:10.1016/0192-0561(88)90145-2
128. Ferguson MM, McDonald FG. Oestrogen as an inhibitor of human NK cell cytolysis. FEBS Lett (1985) 191(1):145–8. doi:10.1016/0014-5793(85)81011-5
129. Hao S, Zhao J, Zhou J, Zhao S, Hu Y, Hou Y. Modulation of 17beta-estradiol on the number and cytotoxicity of NK cells in vivo related to MCM and activating receptors. Int Immunopharmacol (2007) 7(13):1765–75. doi:10.1016/j.intimp.2007.09.017
130. Allison TJ, Garboczi DN. Structure of γδ T cell receptors and their recognition of non-peptide antigens. Mol Immunol (2002) 38(14):1051–61. doi:10.1016/S0161-5890(02)00034-2
131. Hayday AC. [gamma][delta] cells: a right time and a right place for a conserved third way of protection. Annu Rev Immunol (2000) 18:975–1026. doi:10.1146/annurev.immunol.18.1.975
132. Dodd J, Riffault S, Kodituwakku JS, Hayday AC, Openshaw PJ. Pulmonary V gamma 4+ gamma delta T cells have proinflammatory and antiviral effects in viral lung disease. J Immunol (2009) 182(2):1174–81. doi:10.4049/jimmunol.182.2.1174
133. Xue C, Wen M, Bao L, Li H, Li F, Liu M, et al. Vgamma4(+)gammadelta T cells aggravate severe H1N1 influenza virus infection-induced acute pulmonary immunopathological injury via secreting interleukin-17A. Front Immunol (2017) 8:1054. doi:10.3389/fimmu.2017.01054
134. Caccamo N, Dieli F, Wesch D, Jomaa H, Eberl M. Sex-specific phenotypical and functional differences in peripheral human Vgamma9/Vdelta2 T cells. J Leukoc Biol (2006) 79(4):663–6. doi:10.1189/jlb.1105640
135. Michishita Y, Hirokawa M, Guo YM, Abe Y, Liu J, Ubukawa K, et al. Age-associated alteration of gammadelta T-cell repertoire and different profiles of activation-induced death of Vdelta1 and Vdelta2 T cells. Int J Hematol (2011) 94(3):230–40. doi:10.1007/s12185-011-0907-7
136. Johnson TR, Hong S, Van Kaer L, Koezuka Y, Graham BS. NK T cells contribute to expansion of CD8(+) T cells and amplification of antiviral immune responses to respiratory syncytial virus. J Virol (2002) 76(9):4294–303. doi:10.1128/JVI.76.9.4294-4303.2002
137. Bernin H, Fehling H, Marggraff C, Tannich E, Lotter H. The cytokine profile of human NKT cells and PBMCs is dependent on donor sex and stimulus. Med Microbiol Immunol (2016) 205(4):321–32. doi:10.1007/s00430-016-0449-y
138. Kee SJ, Park YW, Cho YN, Jin HM, Kim MJ, Lee SJ, et al. Age- and gender-related differences in circulating natural killer T cells and their subset levels in healthy Korean adults. Hum Immunol (2012) 73(10):1011–6. doi:10.1016/j.humimm.2012.07.335
139. Sandberg JK, Bhardwaj N, Nixon DF. Dominant effector memory characteristics, capacity for dynamic adaptive expansion, and sex bias in the innate Valpha24 NKT cell compartment. Eur J Immunol (2003) 33(3):588–96. doi:10.1002/eji.200323707
140. Camp JV, Jonsson CB. A role for neutrophils in viral respiratory disease. Front Immunol (2017) 8:550. doi:10.3389/fimmu.2017.00550
141. Funchal GA, Jaeger N, Czepielewski RS, Machado MS, Muraro SP, Stein RT, et al. Respiratory syncytial virus fusion protein promotes TLR-4-dependent neutrophil extracellular trap formation by human neutrophils. PLoS One (2015) 10(4):e0124082. doi:10.1371/journal.pone.0124082
142. Emboriadou M, Hatzistilianou M, Magnisali C, Sakelaropoulou A, Exintari M, Conti P, et al. Human neutrophil elastase in RSV bronchiolitis. Ann Clin Lab Sci (2007) 37(1):79–84.
143. Mathur S, Mathur RS, Goust JM, Williamson HO, Fudenberg HH. Cyclic variations in white cell subpopulations in the human menstrual cycle: correlations with progesterone and estradiol. Clin Immunol Immunopathol (1979) 13(3):246–53. doi:10.1016/0090-1229(79)90069-2
144. Bain BJ, England JM. Variations in leucocyte count during menstrual cycle. Br Med J (1975) 2(5969):473–5. doi:10.1136/bmj.2.5969.473
145. Chandra S, Tripathi AK, Mishra S, Amzarul M, Vaish AK. Physiological changes in hematological parameters during pregnancy. Indian J Hematol Blood Transfus (2012) 28(3):144–6. doi:10.1007/s12288-012-0175-6
146. Molloy EJ, O’Neill AJ, Grantham JJ, Sheridan-Pereira M, Fitzpatrick JM, Webb DW, et al. Sex-specific alterations in neutrophil apoptosis: the role of estradiol and progesterone. Blood (2003) 102(7):2653–9. doi:10.1182/blood-2003-02-0649
147. Bekesi G, Kakucs R, Varbiro S, Racz K, Sprintz D, Feher J, et al. In vitro effects of different steroid hormones on superoxide anion production of human neutrophil granulocytes. Steroids (2000) 65(12):889–94. doi:10.1016/S0039-128X(00)00183-5
148. Marczell I, Hrabak A, Nyiro G, Patocs A, Stark J, Dinya E, et al. 17-beta-estradiol decreases neutrophil superoxide production through Rac1. Exp Clin Endocrinol Diabetes (2016) 124(10):588–92. doi:10.1055/s-0042-105556
149. Garcia-Duran M, de Frutos T, Diaz-Recasens J, Garcia-Galvez G, Jimenez A, Monton M, et al. Estrogen stimulates neuronal nitric oxide synthase protein expression in human neutrophils. Circ Res (1999) 85(11):1020–6. doi:10.1161/01.RES.85.11.1020
150. Phipps S, Lam CE, Mahalingam S, Newhouse M, Ramirez R, Rosenberg HF, et al. Eosinophils contribute to innate antiviral immunity and promote clearance of respiratory syncytial virus. Blood (2007) 110(5):1578–86. doi:10.1182/blood-2007-01-071340
151. Samarasinghe AE, Melo RC, Duan S, LeMessurier KS, Liedmann S, Surman SL, et al. Eosinophils promote antiviral immunity in mice infected with influenza A virus. J Immunol (2017) 198(8):3214–26. doi:10.4049/jimmunol.1600787
152. Hamano N, Terada N, Maesako K, Numata T, Konno A. Effect of sex hormones on eosinophilic inflammation in nasal mucosa. Allergy Asthma Proc (1998) 19(5):263–9. doi:10.2500/108854198778557773
153. Katayama ML, Federico MH, Brentani RR, Brentani MM. Eosinophil accumulation in rat uterus following estradiol administration is modulated by laminin and its integrin receptors. Cell Adhes Commun (1998) 5(5):409–24. doi:10.3109/15419069809010785
154. Perez MC, Furth EE, Matzumura PD, Lyttle CR. Role of eosinophils in uterine responses to estrogen. Biol Reprod (1996) 54(1):249–54. doi:10.1095/biolreprod54.1.249
155. Luque EH, Munoz de Toro MM, Ramos JG, Rodriguez HA, Sherwood OD. Role of relaxin and estrogen in the control of eosinophilic invasion and collagen remodeling in rat cervical tissue at term. Biol Reprod (1998) 59(4):795–800. doi:10.1095/biolreprod59.4.795
156. Goritzka M, Makris S, Kausar F, Durant LR, Pereira C, Kumagai Y, et al. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J Exp Med (2015) 212(5):699–714. doi:10.1084/jem.20140825
157. Scotland RS, Stables MJ, Madalli S, Watson P, Gilroy DW. Sex differences in resident immune cell phenotype underlie more efficient acute inflammatory responses in female mice. Blood (2011) 118(22):5918–27. doi:10.1182/blood-2011-03-340281
158. Rettew JA, Huet-Hudson YM, Marriott I. Testosterone reduces macrophage expression in the mouse of toll-like receptor 4, a trigger for inflammation and innate immunity. Biol Reprod (2008) 78(3):432–7. doi:10.1095/biolreprod.107.063545
159. Chao TC, Chao HH, Chen MF, Greager JA, Walter RJ. Female sex hormones modulate the function of LPS-treated macrophages. Am J Reprod Immunol (2000) 44(5):310–8. doi:10.1111/j.8755-8920.2000.440511.x
160. Calippe B, Douin-Echinard V, Laffargue M, Laurell H, Rana-Poussine V, Pipy B, et al. Chronic estradiol administration in vivo promotes the proinflammatory response of macrophages to TLR4 activation: involvement of the phosphatidylinositol 3-kinase pathway. J Immunol (2008) 180(12):7980–8. doi:10.4049/jimmunol.180.12.7980
161. Corcoran MP, Meydani M, Lichtenstein AH, Schaefer EJ, Dillard A, Lamon-Fava S. Sex hormone modulation of proinflammatory cytokine and C-reactive protein expression in macrophages from older men and postmenopausal women. J Endocrinol (2010) 206(2):217–24. doi:10.1677/JOE-10-0057
162. Melgert BN, Oriss TB, Qi Z, Dixon-McCarthy B, Geerlings M, Hylkema MN, et al. Macrophages: regulators of sex differences in asthma? Am J Respir Cell Mol Biol (2010) 42(5):595–603. doi:10.1165/rcmb.2009-0016OC
163. Campbell L, Emmerson E, Williams H, Saville CR, Krust A, Chambon P, et al. Estrogen receptor-alpha promotes alternative macrophage activation during cutaneous repair. J Invest Dermatol (2014) 134(9):2447–57. doi:10.1038/jid.2014.175
164. Li K, Xu W, Guo Q, Jiang Z, Wang P, Yue Y, et al. Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis. Circ Res (2009) 105(4):353–64. doi:10.1161/CIRCRESAHA.109.195230
165. Lai JJ, Lai KP, Chuang KH, Chang P, Yu IC, Lin WJ, et al. Monocyte/macrophage androgen receptor suppresses cutaneous wound healing in mice by enhancing local TNF-alpha expression. J Clin Invest (2009) 119(12):3739–51. doi:10.1172/JCI39335
166. Lauvau G, Chorro L, Spaulding E, Soudja SMH. Inflammatory monocyte effector mechanisms. Cell Immunol (2014) 291(1-2):32–40. doi:10.1016/j.cellimm.2014.07.007
167. Janis K, Hoeltke J, Nazareth M, Fanti P, Poppenberg K, Aronica SM. Estrogen decreases expression of chemokine receptors, and suppresses chemokine bioactivity in murine monocytes. Am J Reprod Immunol (2004) 51(1):22–31. doi:10.1046/j.8755-8920.2003.00117.x
168. Ben-Hur H, Mor G, Insler V, Blickstein I, Amir-Zaltsman Y, Sharp A, et al. Menopause is associated with a significant increase in blood monocyte number and a relative decrease in the expression of estrogen receptors in human peripheral monocytes. Am J Reprod Immunol (1995) 34(6):363–9. doi:10.1111/j.1600-0897.1995.tb00965.x
169. Elenkov IJ, Wilder RL, Bakalov VK, Link AA, Dimitrov MA, Fisher S, et al. IL-12, TNF-α, and hormonal changes during late pregnancy and early postpartum: implications for autoimmune disease activity during these times. J Clin Endocrinol Metab (2001) 86(10):4933–8. doi:10.1210/jc.86.10.4933
170. O’Connor MF, Motivala SJ, Valladares EM, Olmstead R, Irwin MR. Sex differences in monocyte expression of IL-6: role of autonomic mechanisms. Am J Physiol Regul Integr Comp Physiol (2007) 293(1):R145–51. doi:10.1152/ajpregu.00752.2006
171. Kramer PR, Kramer SF, Guan G. 17 beta-estradiol regulates cytokine release through modulation of CD16 expression in monocytes and monocyte-derived macrophages. Arthritis Rheum (2004) 50(6):1967–75. doi:10.1002/art.20309
172. Miyagi M, Aoyama H, Morishita M, Iwamoto Y. Effects of sex hormones on chemotaxis of human peripheral polymorphonuclear leukocytes and monocytes. J Periodontol (1992) 63(1):28–32. doi:10.1902/jop.1992.63.1.28
173. Angele MK, Knoferl MW, Schwacha MG, Ayala A, Cioffi WG, Bland KI, et al. Sex steroids regulate pro- and anti-inflammatory cytokine release by macrophages after trauma-hemorrhage. Am J Physiol (1999) 277(1 Pt 1):C35–42. doi:10.1152/ajpcell.1999.277.1.C35
174. D’Agostino P, Milano S, Barbera C, Di Bella G, La Rosa M, Ferlazzo V, et al. Sex hormones modulate inflammatory mediators produced by macrophages. Ann N Y Acad Sci (1999) 876:426–9. doi:10.1111/j.1749-6632.1999.tb07667.x
175. Li ZG, Danis VA, Brooks PM. Effect of gonadal steroids on the production of IL-1 and IL-6 by blood mononuclear cells in vitro. Clin Exp Rheumatol (1993) 11(2):157–62.
176. Bajana S, Turner S, Paul J, Ainsua-Enrich E, Kovats S. IRF4 and IRF8 act in CD11c+ cells to regulate terminal differentiation of lung tissue dendritic cells. J Immunol (2016) 196(4):1666–77. doi:10.4049/jimmunol.1501870
177. Kovats S. Estrogen receptors regulate an inflammatory pathway of dendritic cell differentiation: mechanisms and implications for immunity. Horm Behav (2012) 62(3):254–62. doi:10.1016/j.yhbeh.2012.04.011
178. Seillet C, Rouquie N, Foulon E, Douin-Echinard V, Krust A, Chambon P, et al. Estradiol promotes functional responses in inflammatory and steady-state dendritic cells through differential requirement for activation function-1 of estrogen receptor alpha. J Immunol (2013) 190(11):5459–70. doi:10.4049/jimmunol.1203312
179. Siracusa MC, Overstreet MG, Housseau F, Scott AL, Klein SL. 17beta-estradiol alters the activity of conventional and IFN-producing killer dendritic cells. J Immunol (2008) 180(3):1423–31. doi:10.4049/jimmunol.180.3.1423
180. Carreras E, Turner S, Paharkova-Vatchkova V, Mao A, Dascher C, Kovats S. Estradiol acts directly on bone marrow myeloid progenitors to differentially regulate GM-CSF or Flt3 ligand-mediated dendritic cell differentiation. J Immunol (2008) 180(2):727–38. doi:10.4049/jimmunol.180.2.727
181. Cunningham MA, Naga OS, Eudaly JG, Scott JL, Gilkeson GS. Estrogen receptor alpha modulates toll-like receptor signaling in murine lupus. Clin Immunol (2012) 144(1):1–12. doi:10.1016/j.clim.2012.04.001
182. Bengtsson AK, Ryan EJ, Giordano D, Magaletti DM, Clark EA. 17beta-estradiol (E2) modulates cytokine and chemokine expression in human monocyte-derived dendritic cells. Blood (2004) 104(5):1404–10. doi:10.1182/blood-2003-10-3380
183. Carreras E, Turner S, Frank MB, Knowlton N, Osban J, Centola M, et al. Estrogen receptor signaling promotes dendritic cell differentiation by increasing expression of the transcription factor IRF4. Blood (2010) 115(2):238–46. doi:10.1182/blood-2009-08-236935
184. Butts CL, Shukair SA, Duncan KM, Bowers E, Horn C, Belyavskaya E, et al. Progesterone inhibits mature rat dendritic cells in a receptor-mediated fashion. Int Immunol (2007) 19(3):287–96. doi:10.1093/intimm/dxl145
185. Xiu F, Anipindi VC, Nguyen PV, Boudreau J, Liang H, Wan Y, et al. High physiological concentrations of progesterone reverse estradiol-mediated changes in differentiation and functions of bone marrow derived dendritic cells. PLoS One (2016) 11(4):e0153304. doi:10.1371/journal.pone.0153304
186. Jones LA, Kreem S, Shweash M, Paul A, Alexander J, Roberts CW. Differential modulation of TLR3- and TLR4-mediated dendritic cell maturation and function by progesterone. J Immunol (2010) 185(8):4525–34. doi:10.4049/jimmunol.0901155
187. Griesbeck M, Ziegler S, Laffont S, Smith N, Chauveau L, Tomezsko P, et al. Sex differences in plasmacytoid dendritic cell levels of IRF5 drive higher IFN-alpha production in women. J Immunol (2015) 195(11):5327–36. doi:10.4049/jimmunol.1501684
188. Scott JL, Cunningham MA, Naga OS, Wirth JR, Eudaly JG, Gilkeson GS. Estrogen receptor alpha deficiency modulates TLR ligand-mediated PDC-TREM expression in plasmacytoid dendritic cells in lupus-prone mice. J Immunol (2015) 195(12):5561–71. doi:10.4049/jimmunol.1500315
189. Hannah MF, Bajic VB, Klein SL. Sex differences in the recognition of and innate antiviral responses to Seoul virus in Norway rats. Brain Behav Immun (2008) 22(4):503–16. doi:10.1016/j.bbi.2007.10.005
190. Hughes GC, Thomas S, Li C, Kaja MK, Clark EA. Cutting edge: progesterone regulates IFN-alpha production by plasmacytoid dendritic cells. J Immunol (2008) 180(4):2029–33. doi:10.4049/jimmunol.180.4.2029
191. Wang JP, Zhang L, Madera RF, Woda M, Libraty DH. Plasmacytoid dendritic cell interferon-α production to R-848 stimulation is decreased in male infants. BMC Immunol (2012) 13:35. doi:10.1186/1471-2172-13-35
192. Torcia MG, Nencioni L, Clemente AM, Civitelli L, Celestino I, Limongi D, et al. Sex differences in the response to viral infections: TLR8 and TLR9 ligand stimulation induce higher IL10 production in males. PLoS One (2012) 7(6):e39853. doi:10.1371/journal.pone.0039853
Keywords: sex hormones, respiratory virus, lung, estrogen, androgen, innate immunity
Citation: Kadel S and Kovats S (2018) Sex Hormones Regulate Innate Immune Cells and Promote Sex Differences in Respiratory Virus Infection. Front. Immunol. 9:1653. doi: 10.3389/fimmu.2018.01653
Received: 16 April 2018; Accepted: 04 July 2018;
Published: 20 July 2018
Edited by:
Virginia Rider, Pittsburg State University, United StatesReviewed by:
Jonatan Leffler, Telethon Kids Institute, AustraliaCopyright: © 2018 Kadel and Kovats. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan Kovats, c3VzYW4ta292YXRzQG9tcmYub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.