 Pilar Sánchez-Corral
Pilar Sánchez-Corral Richard B. Pouw
Richard B. Pouw Margarita López-Trascasa
Margarita López-Trascasa Mihály Józsi
Mihály Józsi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 July 2018
Sec. Molecular Innate Immunity
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01607
This article is part of the Research Topic The Role of Complement in Health and Disease View all 50 articles
The alternative pathway is a continuously active surveillance arm of the complement system, and it can also enhance complement activation initiated by the classical and the lectin pathways. Various membrane-bound and plasma regulatory proteins control the activation of the potentially deleterious complement system. Among the regulators, the plasma glycoprotein factor H (FH) is the main inhibitor of the alternative pathway and its powerful amplification loop. FH belongs to a protein family that also includes FH-like protein 1 and five factor H-related (FHR-1 to FHR-5) proteins. Genetic variants and abnormal rearrangements involving the FH protein family have been linked to numerous systemic and organ-specific diseases, including age-related macular degeneration, and the renal pathologies atypical hemolytic uremic syndrome, C3 glomerulopathies, and IgA nephropathy. This review covers the known and recently emerged ligands and interactions of the human FH family proteins associated with disease and discuss the very recent experimental data that suggest FH-antagonistic and complement-activating functions for the FHR proteins.
While initially only regarded as a supporting factor for the effectivity of immunoglobulins, the complement system is nowadays widely recognized as a crucial part of the innate immune system involved in many different processes (1). In addition to acting as a first line of defense by directly targeting and killing invading pathogens, with or without the help of immunoglobulins, its role in inflammation, immune cell recruitment, and clearance of immune complexes, apoptotic cells, and necrotic cells places complement at the center of the human immune system. The relevant role of complement is corroborated by the variety of pathological situations associated with complement deficiency or dysfunction.
Three complement activation pathways have been defined, each comprised of various proteins forming an intricate cascade of activation events (Figure 1). Both the classical and the lectin pathways are initiated when pattern recognition molecules (PRMs) that are complexed with zymogens of serine proteases, bind to their ligand. The classical pathway is activated by the binding of the C1 complex to immunoglobulins and pentraxins, while the lectin pathway uses various PRMs, including mannose-binding lectin and ficolins, which bind to specific carbohydrate moieties. These ligands are normally not present on healthy human cells. In contrast, the alternative activation pathway is initiated through the constitutive low rate hydrolysis of the internal thioester bond of C3, allowing binding of various activating complement proteins. All three pathways lead to the cleavage of C3 into C3a and C3b. C3b contains a highly reactive thioester group that is exposed upon C3 cleavage, resulting in the deposition of C3b onto virtually any molecule or cell surface in close proximity. When left unchecked, C3b on its own will again initiate the alternative pathway. As both the classical and the lectin pathway will also activate the alternative pathway once C3b is formed, thus enhancing complement activation, the alternative pathway has a pivotal role as an amplification loop within the complement system. Up to 80% of total complement activation has been ascribed to this amplification loop (2). Due to the spontaneous nature of the alternative pathway, it must be tightly controlled to prevent unwarranted and dangerous complement activation.
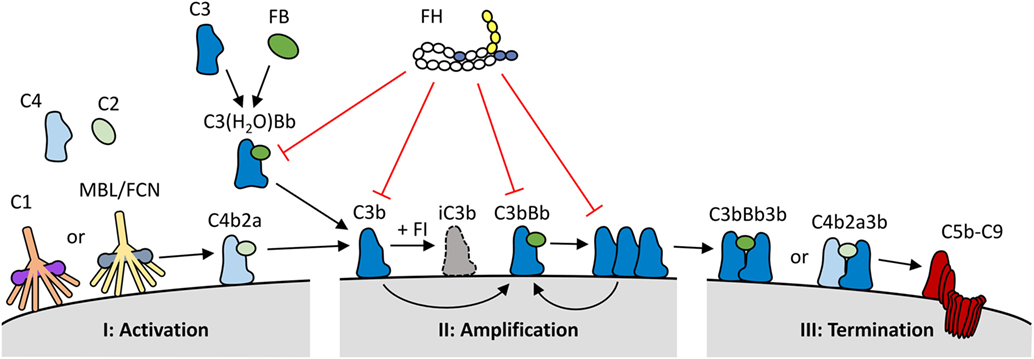
Figure 1. Overview of the role of factor H (FH) within the complement system. (I) The complement system is activated via binding of C1q (classical pathway), or mannan binding lectin/ficolins (MBL/FCN) (lectin pathway) in complex with serine proteases to specific molecules, or through the spontaneous activation of C3 into C3(H2O) (alternative pathway). Upon activation, the three pathways form C3 convertases (C4b2a or C3(H2O)Bb) resulting in the generation and deposition of C3b on the activating surface. (II) C3b forms new C3 convertase molecules (C3bBb) that enhance C3b deposition and amplify complement activation. (III) C3b can also bind to C3 convertases to generate C5 convertases (C4b2a3b or C3bBb3b); this process initiates the terminal pathway of complement activation, and the formation of the lytic C5b–C9 complex. FH keeps the spontaneous activation of C3 under control, and it also inhibits the complement system at both the activation and amplification stages. FH binds to deposited C3b and C3bBb complexes on human cell surfaces and inhibits further activation by three mechanisms: it competes with factor B (FB) for C3b binding and C3bBb generation; it increases the decay of C3bBb complexes, and it acts as a cofactor for factor I (FI), which in turn cleaves C3b into inactive C3b (iC3b).
Complement regulation takes places both on the human cell surface and in the fluid phase. Several regulators, like most complement components, are found in the circulation. In addition, human cells express a wide array of membrane-bound complement regulators that control the system at various steps. Especially due to the activating proteins of the alternative pathway, regulation in the fluid phase is crucial, as unchecked, spontaneous C3 activation would lead to complete consumption of C3 and loss of complement activity. The 155-kDa glycoprotein complement factor H (FH) is the major regulator of the alternative pathway, inhibiting C3 activation both in the fluid phase as well as on human cell surfaces (Figure 1) (3). Similar to other complement regulators encoded in the regulators of complement activation (RCA) gene cluster, FH is composed of complement control protein (CCP) domains, often also referred to as short consensus repeat domains. FH is composed of 20 CCPs (Figure 2) (4). The first four N-terminal domains contain the complement inhibiting activity, such as decay accelerating activity and co-factor activity (5). The two most C-terminal CCP domains (19 and 20), together with a region located in CCPs 6–8, are crucial for binding of FH to surfaces, such as human cell membranes, as well as for mediating binding to several host and non-host ligands (discussed below) (6, 7). FH is highly abundant in plasma, with circulating levels of 233–400 µg/mL on average, although it has to be noted that some of the assays used might detect other FH family members as well (8–11).
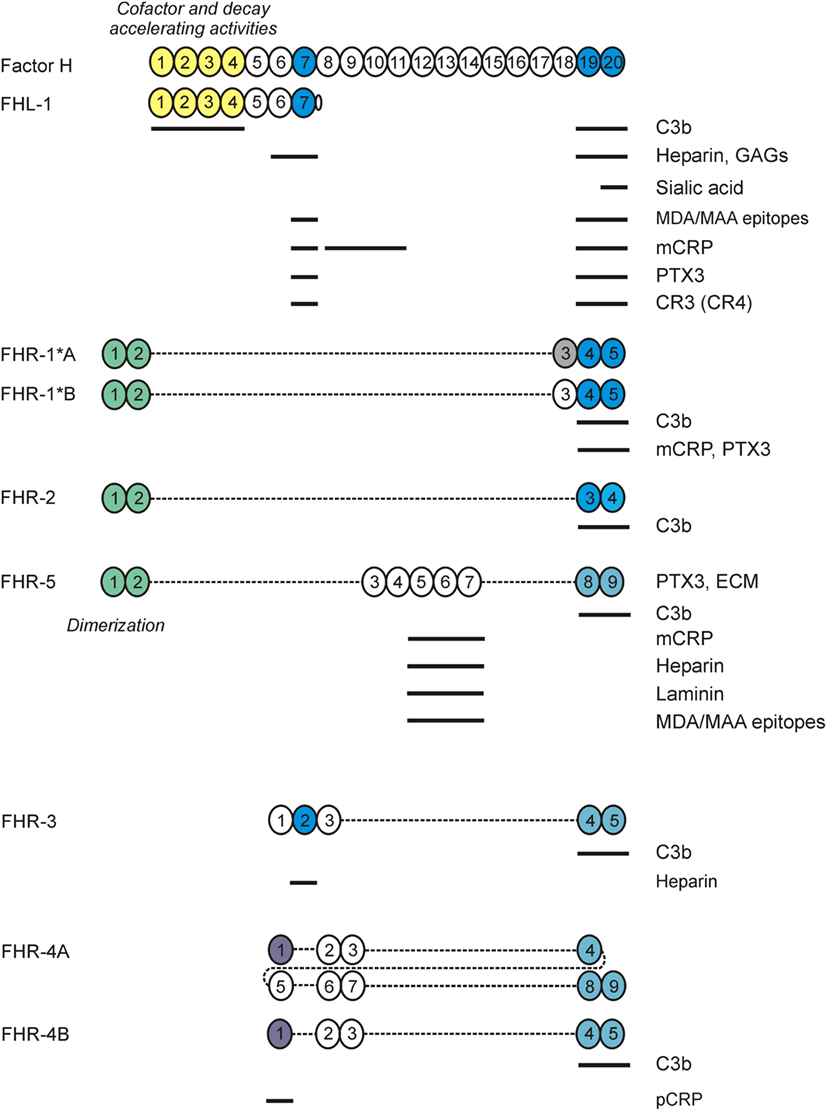
Figure 2. Factor H (FH) family proteins and their ligands. The schematic complement control protein (CCP) domain structure of FH, FHL-1, and the FHR proteins is shown, with CCPs aligned vertically to the homologous domains in FH. The N-terminal CCPs 1–4 of FH and FHL-1 mediate the complement regulatory functions of these proteins (shown in yellow). CCPs 7 and 19–20 (shown in blue) harbor the main ligand- and host surface-recognition sites; selected ligand binding sites are indicated by horizontal lines. The CCPs 1–2 of factor H related protein 1 (FHR)-1, FHR-2, and FHR-5 are closely related to each other and mediate dimerization of these FHRs. The CCPs in FHRs with high sequence identity to the homologous FH domains are indicated by identical/similar colors.
Factor H-like protein 1 (FHL-1) is a splice variant derived from the CFH gene. Serum levels of FHL-1 are estimated to be 10–50 µg/mL (12, 13). FHL-1 is identical to the first seven CCP domains of FH, with an unique, four amino acid long C-terminus (14, 15). Thus, FHL-1 shares the C3b binding and regulatory domains CCPs 1–4 with FH and, like FH, it has complement inhibiting activity (16). Likewise, due to the shared CCPs 6–7 domains, FHL-1 and FH bind some common ligands, such as heparin, the pentraxins C-reactive protein (CRP) and pentraxin 3 (PTX3), and malondialdehyde epitopes (Figure 2). However, there are also differences in ligand interactions between FHL-1 and FH, not only because of the extra domains in FH but also due to the difference in their conformation and the unique SFTL tail at the C-terminus of FHL-1. For example, it was recently reported that the SFTL tail increases the interaction of FHL-1 with CRP and PTX3 (17).
Next to FH and FHL-1, humans (and several non-human species; not discussed here) possess FH-related (FHR) proteins, homologous to FH. They are encoded separately, with their genes (CFHR1 to CFHR5) lying in tandem next to CFH at 1q31.3. The CFHR genes originate from CFH through gene duplication events (18). The CFH–CFHRs loci contain several segmental duplications, making them prone to genetic structural rearrangements due to nonallelic homologous recombination (NAHR) events. This has led to copy number polymorphisms (CNPs), with the very common 86.3-kb deletion (CNP147) that results in loss of CFHR3–CFHR1 (ΔCFHR3–CFHR1), and the very rare 122-kb deletion (CNP148) resulting in loss of CFHR1–CFHR4 (ΔCFHR1–CFHR4) (19). Like FH, the FHRs are entirely composed of CCP domains (Figure 2), which display high sequence similarity with CCP domains of FH known to be involved in ligand and surface binding. Remarkably, none of the human FHR proteins possess CCP domains homologous to FH CCPs 1–4. Thus, based on their primary structure, FHR proteins are not expected to have any direct complement inhibiting activity similar to FH. Nonetheless, several reports have observed direct complement inhibitory activity for some of the FHRs, albeit often weak compared to FH (20–24). However, other studies have not found such activity for FHRs, questioning whether this is truly the physiological role of the FHR proteins (25–30). Instead, the FHRs are currently hypothesized to have an antagonistic function over FH, competing with binding to FH ligands and cell surfaces. By lacking direct complement inhibiting activity, binding of FHRs instead of FH would allow complement activation to proceed (31). This process has also been termed complement de-regulation. Indeed, binding to various (FH) ligands has been reported for all FHRs, which will be discussed below. In addition, some FHRs were reported to promote alternative pathway activation by binding C3b and serving as a platform for the assembly of the C3 convertase (27, 32, 33). Recent characterization of some of the mouse FHRs supports a role of these proteins as positive regulators in the modulation of complement activation (34, 35).
In this review, we outline and provide an update on the recent developments regarding the FH protein family. New insights regarding circulating levels of FHRs, ligand binding, and disease associations allow re-assessing the role of FHRs in the complement system. Together, these results shed light on the balance of the FH–FHRs axis, and the role of FHRs in non-pathological and pathological conditions.
Factor H, FHL-1, and the FHR proteins are mainly synthesized by hepatocytes, but synthesis by other cells and tissues has also been reported, particularly for FH and FHL-1 (36–38). FH production has been detected in endothelial cells, platelets, mesangial cells, keratinocytes, fibroblasts, retinal pigment epithelial cells, monocytes, and dendritic cells, among others (39–46). On the other hand, little information on the extrahepatic expression of the FHR proteins is available. Both CFHR3 mRNA and FHR-3 protein have been identified in retinal macrophages, while no FHR-3 expression was found in other retinal cell types (47). Extrahepatic synthesis of FH/FHRs most likely contributes to an efficient control of complement activation locally, but a relevant contribution to the plasma levels of these proteins is unlikely, considering the relative low expression compared to the hepatic source.
Accurate quantification of the FHR proteins has been a great challenge since their discovery. Due to the high sequence similarity among FH and FHR proteins, it has proven to be very difficult to obtain specific reagents for each of the FHR proteins. For some time, only concentration estimates were available for most of the FHR proteins (21, 48). However, with recently renewed and successful efforts in generating highly specific antibodies, specific immunoassays for each of the FHR proteins are now becoming available, although some discrepancy about their actual physiological levels still remains (Table 1).
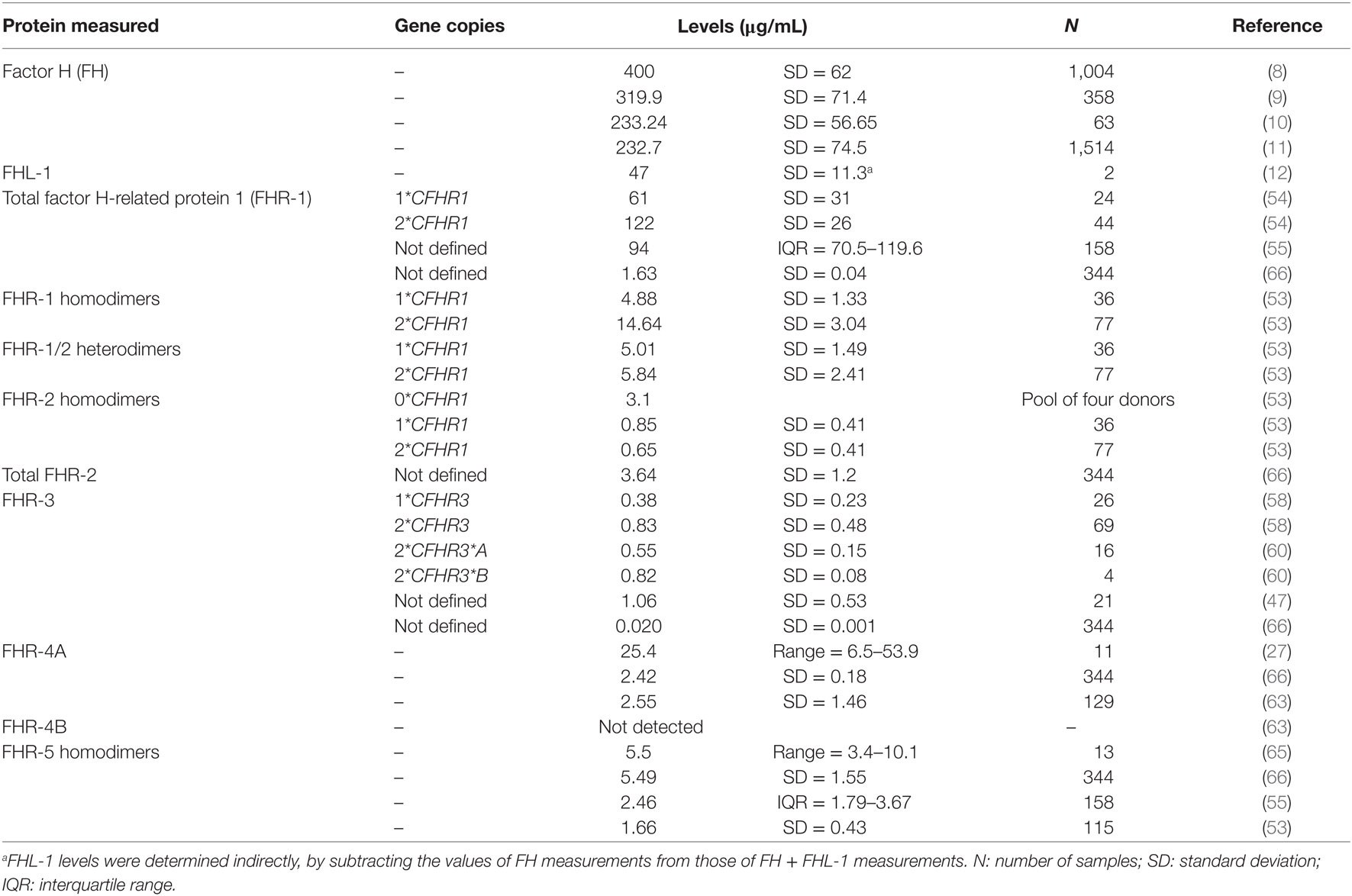
Table 1. Reported serum levels of the factor H-related (FHR) proteins.
Factor H-related protein 1 is composed of five CCP domains, and circulates in two forms (37 and 42 kDa), with either one or two N-linked carbohydrate moieties (30, 49, 50). Two genetic variants of FHR-1 have been described, FHR-1*A and FHR-1*B, the difference being three amino acids in CCP3 (51). FHR-1*B CCP3 is identical to FH CCP18, whereas FHR-1*A CCP3 shares 95% sequence identity with FH CCP18. FHR-1 CCPs 4 and 5 share high sequence identity (100 and 97%) with FH CCPs 19 and 20, respectively. FHR-1 has a dimerization motif located in CCPs 1–2 that are highly similar (>85% sequence identity) to CCPs 1–2 of FHR-2 and FHR-5, and allow the formation of FHR-1 homodimers and heterodimers with FHR-2 (26, 52, 53). While identified in vitro, the existence of FHR-1/FHR-5 heterodimers in vivo is still controversial (26, 52, 53). Similarly, FHR-1 quantification also remains controversial. In 2017, several groups determined FHR-1 levels. Tortajada et al. reported an average of 122 µg/mL in 44 healthy controls with two copies of CFHR1, and an overall average of 90.4 µg/mL in 76 controls (including eight homozygous ΔCFHR3–CFHR1 carriers and 24 heterozygous ΔCFHR3–CFHR1 carriers) (54). Using the same immunoassay, Medjeral-Thomas et al. reported 94.4 µg/mL FHR-1 in 158 controls (of whom 133 were genotyped: 3 ΔCFHR3–CFHR1 homozygous, 45 ΔCFHR3–CFHR1 heterozygous, and 85 without ΔCFHR3–CFHR1) (55). Of note, the immunoassay described by Tortajada et al. does not distinguish between FHR-1 homodimers or heterodimers. In contrast, using immunoassays specific for FHR-1 homodimers and FHR-1/-2 heterodimers, van Beek et al. reported ~10-fold lower levels (averages of 11.33 and 5.48 µg/mL, respectively), in 115 healthy donors (2 homozygous ΔCFHR3–CFHR1, 36 heterozygous ΔCFHR3–CFHR1 carriers, and 77 without ΔCFHR3–CFHR1) (53).
Factor H-related protein 2 is the smallest FHR protein, composed of four CCP domains (56). FHR-2 circulates either non-glycosylated (24 kDa) or with one N-linked carbohydrate moiety in CCP2 (29 kDa). FHR-2 CCP1 and CCP2 are nearly identical to FHR-1 CCP1 and CCP2 (100 and 98%), respectively, including all residues comprising the dimerization motif (26). Similar to the proposed FHR-1/FHR-5 dimers, FHR-2/FHR-5 dimers remain to be identified in vivo, while FHR-2 homodimers and FHR-1/FHR-2 heterodimers have been confirmed (52, 53). FHR-2 homodimer levels have been shown to be around 3 µg/mL; with these relatively low levels, FHR-2 seems to be the limiting factor in the formation of FHR-1/FHR-2 heterodimers and, indeed, most FHR-2 is found dimerized with FHR-1 (53).
Factor H-related protein 3 is composed of five CCP domains, of which CCP1 and CCP2 have high sequence similarity with FH CCP6 and CCP7 (94 and 86%), respectively (57). The C-terminal CCPs 3–5 are virtually identical to the C-terminal domains of FHR-4A and FHR-4B (93–100%). FHR-3 contains four N-linked glycosylation sites, and it circulates in plasma as multiple glycosylation variants ranging from 37 to 50 kDa. A quantitative FHR-3-specific immunoassay was first described by Pouw et al., reporting levels of 0.38 and 0.83 µg/mL for healthy individuals carrying either one or two CFHR3 copies, respectively (58). These results were later confirmed in a similar assay, reporting mean levels of 1.06 µg/mL (47). Two major genetic variants of CFHR3 (CFHR3*A and CFHR3*B) have been described (59); interestingly, these are quantitative variants, with CFHR3*B determining higher FHR-3 levels than CFHR3*A (60). The FHR-3*A and FHR-3*B allotypes differ at aminoacid 241 in CCP3 (Pro/Ser), but its functional relevance has not been determined.
CFHR4 is the only known CFHR gene that expresses two splice variants, FHR-4A and FHR-4B (61, 62). FHR-4A is composed of nine CCP domains (86 kDa), while FHR-4B has five CCP domains (43 kDa). All FHR-4B domains are also present in FHR-4A, with FHR-4B CCP1 being identical to FHR-4A CCP1, and FHR-4B CCPs 2–5 being identical to FHR-4A CCPs 6–9. FHR-4A CCPs 2–4 seems to have arisen from internal gene duplication, and have high sequence similarity (85–93% amino acid identity) with the other CCPs in FHR-4A/B (61). Thus, obtaining specific reagents to distinguish FHR-4A from FHR-4B is challenging on first sight. Quantification by using an immunoassay that in principle measures both FHR-4A and FHR-4B resulted in average levels of 25.4 µg/mL (27). However, FHR-4A-specific antibodies have been described recently and used in an FHR-4A-specific ELISA which shows 10-fold lower levels for FHR-4A (2.55 ± 1.46 µg/mL) (63). In line with the complete sequence identity of FHR-4B with several FHR-4A domains, no specific antibodies for FHR-4B could be obtained. Strikingly, FHR-4B was not detected in plasma using various antibodies that did react with recombinant FHR-4B (63). This indicates that free FHR-4B must be in an extremely low concentration or even absent from plasma.
Factor H-related protein 5 is composed of nine CCPs and is the only FHR with domains (CCPs 3–7) homologous to FH CCPs 10–14 (64). FHR-5 CCPs 1–2 are highly similar (85–93% amino acid identity) to CCPs 1–2 of FHR-1 and FHR-2, although not all residues identified in the FHR-1/2 dimerization motif are present in FHR-5 (26). This could explain why the presence of FHR-5 heterodimers in vivo is still controversial (26, 52, 53). FHR-5 seems to circulate predominantly as homodimer in vivo (53), making quantification a bit more straightforward. FHR-5 serum levels were reported to be 3–6 µg/mL (24), which was later confirmed in 13 healthy individuals, with median levels of 5.5 µg/mL (65). Similar FHR-5 levels (median 2.46 µg/mL) were found in a larger group of 158 healthy controls using the same immunoassay (55). More recently, an average concentration of 1.66 µg/mL was shown in 115 controls by using a newly developed FHR-5 ELISA (53).
In addition to the specific immunoassays described above, mass spectrometry has also been used to quantify the FHR proteins (66). While this approach allows specific measurement of FHRs based on unique peptide sequences, quantification of FHR dimers is not possible. Results similar to the immunoassays were obtained for FHR-2 (3.64 ± 1.2 µg/mL), FHR-4A (2.42 ± 0.18 µg/mL), and FHR-5 (5.49 ± 1.55 µg/mL). However, much lower levels were found for FHR-1 (1.63 ± 0.04 µg/mL) and FHR-3 (0.020 ± 0.001 µg/mL). It is unclear why such lower concentrations were found for FHR-1 and FHR-3, although the frequency of ΔCFHR3–CFHR1 in the studied population (n = 344, Icelandic origin) was not determined. Of note, the peptide used for FHR-4 quantification is only present in FHR-4A, thus providing no extra information whether FHR-4B exists in vivo.
Kopczynska et al. measured FHR-1, FHR-2, and FHR-5 altogether in one immunoassay, finding a total FHR-1/2/5 concentration of 10.67 µg/mL (±5.42) in 42 healthy individuals (67). This result is in great contrast to previously reported levels of approximately 100 µg/mL for FHR-1 (54, 55), but is comparable to a combined mean FHR-1/2/5 concentration of 19.27 (53) and 10.76 µg/mL (66).
The reasons for the huge differences in FHR levels outlined above are unclear. Moreover, the existence of homo- and heterodimers, and the fact that the frequency of the ΔCFHR3–CFHR1 polymorphism is highly population-dependent (19, 68, 69), further complicate the accuracy and assessment of measurements. To exclude any possible cross-reactivity that interferes with FH or FHR quantifications, it is crucial to extensively characterize antibodies generated against FH or any of the FHRs. FH immunoassays should ideally use at least one antibody targeting an epitope located in domains absent from the FHRs, such as CCPs 15–17. Furthermore, when quantifying FHR proteins, it is highly recommended to stratify protein levels based on CFHR CNPs, as well as distinguishing between hetero- and homodimers. This would aid in comparison of control and patient groups, as CNP frequencies and dimer formation might be altered in patients. CNPs should be determined at the genetic level, as stratification based only on protein levels seems not to be possible due to the wide range in protein concentration within each CNP group (53, 58). CNPs are most commonly determined using multiplex ligation-dependent probe amplification (MLPA), although there is currently no commercial kit available that also covers CFHR4. In addition, while normal levels of FHR proteins are now being reported, further data are necessary to reach consensus on their actual concentrations in circulation.
As outlined above, FH is a major inhibitor of the alternative pathway in plasma and when bound to cells and surfaces like the glomerular basement membrane. This complement regulatory activity is due to the interaction of FH with C3b (70). In addition, FH binds to several other ligands (Figure 2) and, when ligand-bound, in many cases maintains its complement inhibitory activity. These FH interactions ensure proper regulation of complement activation, as well as the resulting opsonization and inflammation.
Complement activation can be initiated on modified, dangerous self surfaces, which are recognized by PRMs within (C1q, ficolins, MBL, and properdin) and outside the complement system (e.g., pentraxins). FH along with other regulators may ensure targeted but restricted complement activation and an optimal degree of opsonization, while preventing overt inflammation and damage resulting from cascade over-activation (71, 72). The FHR proteins appear to counter-balance this activity of FH and enhance complement activation by binding to the same or similar ligands and outcompeting FH (Figure 3), and in some instances also by interacting with C3b and other ligands independent of FH (31). This section briefly summarizes the main ligand interactions of FH and the FHR proteins (Figure 2), and indicates their relevance in the regulation and modulation of complement activation.
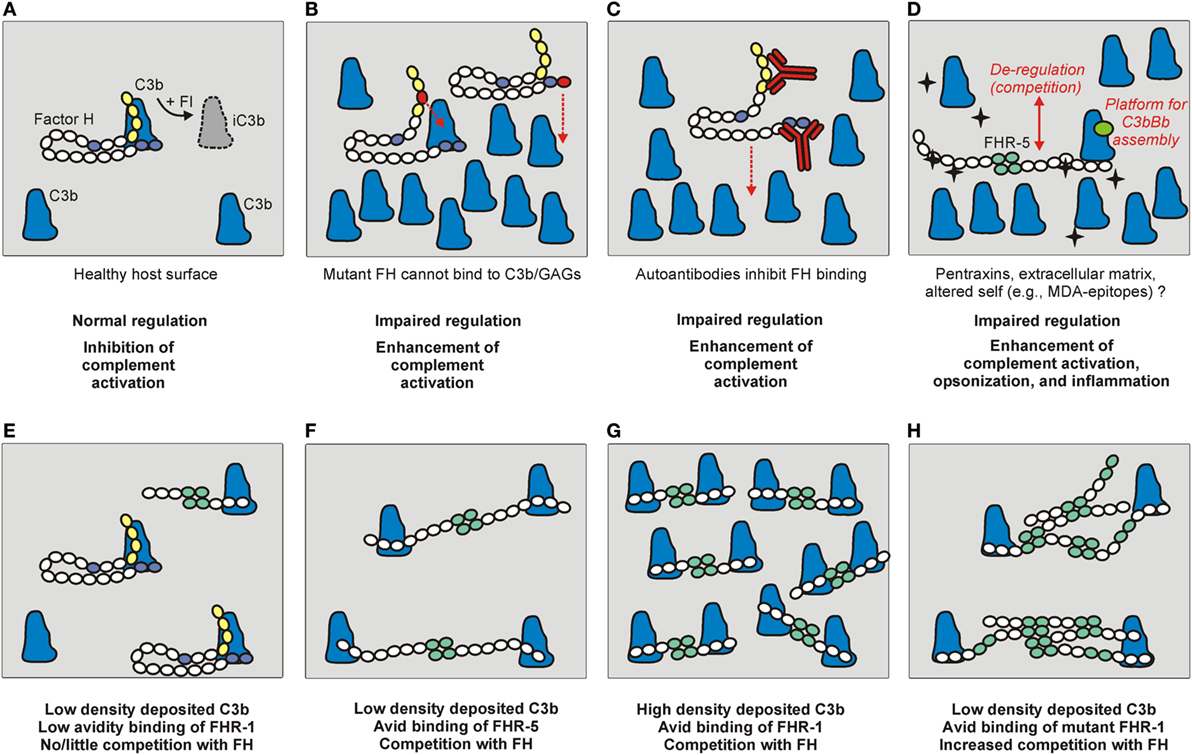
Figure 3. The role of factor H (FH) and the FHRs under physiological and pathological conditions. (A) Normal complement control on a healthy host surface by FH bound to surface-deposited C3b. FH recognizes self surfaces under complement attack by binding to a complex of GAGs/sialic acid and C3b. This recognition is mediated by complement control proteins (CCPs) domain 19–20 indicated in blue. Through the activity of the N-terminal regulatory domains (CCPs 1–4, in yellow), FH assists factor I (FI) in the proteolytic cleavage of C3b into inactive C3b (iC3b). (B) Mutations in the regulatory or recognition domains, and (C) autoantibodies bound to these domains can cause functional FH defect and result in impaired surface complement control. (D) FHRs can also interact with similar surfaces and ligands as FH, and compete with FH for binding (de-regulation), and/or they can directly activate the alternative pathway by binding C3b and serving as a platform for the assembly of an active C3bBb convertase. The black star-shape indicates newly exposed ligand/altered self. (E–H) The relative FH/FHR concentrations, the ligand density (avidity), and the dimeric/oligomeric states of the FHRs influence surface complement regulation. (E) At low ligand (e.g., deposited C3b) density and relative FH surplus, FH can potently regulate complement activation on the surface. (F) Increased FHR levels and/or (G) ligand densities, and (H) formation of higher order oligomers (e.g., due to duplicated dimerization domains) can cause enhanced competition with FH and tip the balance to increased complement activation.
We would like to briefly note that tumor cells and microbes can bind FH in an attempt to avoid their destruction by host complement. In addition, the main microbial ligand binding sites of FH are in CCPs 6–7 and 19–20, and homologous domains are conserved in the FHRs, thus these proteins may modulate opsonization/killing of microbes. These aspects have been reviewed in detail (6, 31, 73–75).
The main ligand of FH is the active C3 fragment C3b, which can be generated by fluid phase and surface-bound C3 convertases. Since C3b is the central component that promotes complement amplification via the alternative pathway, and is also required for the assembly of C5 convertases and the initiation of the terminal pathway, its regulation is key to maintain the proper balance of complement activation and inhibition. FH interacts with C3b at two main sites, harbored by CCPs 1–4 and 19–20 (76). The N-terminal C3b binding site is active when FH is in the fluid phase (e.g., in blood plasma) and also when FH is bound to cells or other surfaces [via glycosaminoglycans (GAGs), sialic acid, or a specific receptor—see below] (Figure 3A). FH may also bind C3b by CCPs 1–4 when already bound to other ligands, such as pentraxins, because these interactions typically involve CCPs 6–7 and 19–20 (74, 77–79). Thus, FH maintains its complement regulatory activity when bound to cells or other ligands.
Structural studies revealed that FH engages surface-deposited C3b in the context of host GAGs/sialic acid, i.e., CCPs 19–20 bind to these ligands at the same time, which allows avid interaction of FH with a host surface under complement attack. The FH C-terminal site also binds C3d, the final C3b degradation product that remains covalently attached to the surface (80, 81).
The FHR proteins also bind to C3b, but the nature of these interactions is inherently different from that of FH because FHRs lack domains homologous to FH CCPs 1–4. Thus, FHRs lack FH-like cofactor activity and decay accelerating activity, although some residual activity may be present due to the interaction of the C-terminal domains of these proteins with C3b. This should be investigated in detail in the future to clarify the currently contradicting reports in this regard (20, 23, 24, 26, 27, 32, 33).
In contrast to possible inhibitory activities, FHR-1, FHR-4, and FHR-5 were reported to activate the alternative pathway, by binding C3b through their C-terminal domains and forming a platform for the assembly of an active C3bBb convertase (27, 32, 33). This activity could take place on surfaces where these FHRs are bound directly, or via another ligand, such as pentraxins (33). FHR-1 and FHR-5 were shown to enhance complement activation on the extracellular matrix (ECM) and on the surface of apoptotic or necrotic cells (32, 33, 82).
Additionally, FHRs may compete with FH for binding to C3b deposited on surfaces, a process termed complement de-regulation, because FHRs can enhance complement activation by inhibiting FH binding (Figure 3D). This activity of the FHRs may only be significant—considering their relative serum concentrations and avidity for C3b—if increased amounts of FHRs or altered FHR forms (such as higher order oligomers) are present (Figures 3E–H) (25, 26, 52, 83, 84). For FHR-2, it was described that, despite binding to C3b, it cannot effectively compete with FH for binding to surface-bound C3b (20).
Altogether, based on these data the FHRs can be regarded as positive complement regulators.
While interacting sites for other C3 fragments were described, current evidence strongly supports the physiologically relevant binding of FH to C3b via CCPs 1–4 and 19–20, as well as to C3d via CCPs 19–20 (76). Interaction of FHR-1 and FHR-2 with C3d was also shown, but without functional analyses (20, 84). Binding of FHR-3, but not of FHR-1, to C3d was shown to prevent the binding of C3d to its receptor on B cells, thus modulating B cell activation (85). FHR-5 was reported to bind to iC3b and C3d with affinities similar to C3b; in contrast, FH bound very weakly to iC3b and C3d compared with FHR-5, indicating that despite its lower serum concentration FHR-5 can be an efficient competitor of FH for binding to deposited C3 fragments (26).
Distinction between self, non-self, or altered self surfaces relies in part on the recognition of host-specific GAGs and sialic acid by FH (and FHL-1). This allows complement activation to proceed unhindered on microbial (“activator”) surfaces, but prevents activation on host (“non-activator”) surfaces (86). This has been a subject of intensive research, often using heparin as a model for polyanionic molecules. The main heparin-binding sites were identified in FH (and FHL-1) CCP7 and FH CCP20 (87, 88). This allows recognition of, and attachment to, host glycomatrix and cells, such as platelets and endothelial cells (89, 90). Recent studies revealed some functional differences indicating that while some GAGs are recognized by FH and FHL-1 via CCP7, the sialic acid binding site is in CCP20 (91), also targeting these host regulators to different surfaces and explaining the different consequences of mutations affecting these domains (89, 92, 93).
Factor H-related protein 1 can also bind to host surfaces via its FH-homologous C-terminus (22, 29), and FHR-3 binds heparin through CCP2, which is homologous to CCP7 of FH (23, 87). In addition, FHR-5 has a heparin-binding site in CCPs 5–7 (24, 94). The functional relevance of these interactions needs to be investigated further, but they could anchor these proteins on certain cells and surfaces.
Extracellular matrices occur in many tissues and can have different functions, the most important ones being the physical support of cells and acting as barriers and filters. The composition of ECMs differs at distinct anatomic sites and is dynamically regulated. Under certain conditions, e.g., endothelial cell activation or injury, ECMs can be exposed to body fluids and plasma proteins; in addition, the Bruch’s membrane in the eye and the kidney glomerular basement membrane are also exposed because the lining cell layer is fenestrated. To prevent overt complement activation, such ECMs rely largely on soluble complement regulators, such as FH and FHL-1, which can bind via their GAG binding sites and locally regulate complement (95). As noted above, differences in ECMs and in domain composition of the FH family proteins may target FH and FHRs toward distinct sites, such as FH to the glomerular basement membrane (via CCPs 19–20) and FHL-1 to the Bruch’s membrane (via CCP7) (95). FHR-5 was shown to bind to MaxGel, an ECM extract, and de-regulate complement on this surface (32); a recent study identified laminin as an ECM ligand of FHR-5 (94).
Complement is largely involved in the immunologically safe and silent disposal of apoptotic and necrotic cells via opsonophagocytosis (96). The soluble regulators FH and C4b-binding protein bind to dead cells and prevent excessive complement activation and potential deleterious effects when membrane-anchored regulators are down-regulated on the cells (97). FH can bind to Annexin-II, DNA, and histones (98), as well as malondialdehyde epitopes on apoptotic cells (94, 99). In addition, the pentraxins CRP and pentraxin 3 (PTX3) also bind to dead cells and recruit FH (77, 100). For FHR-1 and FHR-5, binding to necrotic cells and enhancement of complement activation have been shown (33, 82), suggesting that these FHRs modulate opsonization of dead cells.
The pentraxins are soluble PRMs of the innate immune system and, based on their structure, categorized as short and long pentraxins. Pentraxins have numerous ligands and functions, reviewed in detail elsewhere (101); of note, they participate in the opsonization of microbes and dead cells, and they also bind to components of the ECM. For the prototypic short pentraxin CRP and the long pentraxin PTX3, interactions with both complement activators (C1q, MBL) and inhibitors (FH, C4b-binding protein) were described (74, 77, 79, 101–107).
C-reactive protein circulates in its native, pentameric form (pCRP) in body fluids, but it can adopt an altered conformation exposing neoepitopes upon pH change or binding to membranes, and it can even decay to its monomeric form (mCRP) in vitro by chelation of the Ca2+ ions or adsorption on plastic. FH was described to bind primarily to mCRP via CCPs 7, 8–11, and 19–20 (79, 108, 109), but interaction with pCRP via CCPs 7 and 19–20 at acute phase concentrations was also reported (110). The binding to mCRP allows targeting of the complement inhibitor FH to certain surfaces, including apoptotic cells (71, 100, 109). Among the FHRs, FHR-1 binds to mCRP via CCPs 4–5 (33) and FHR-5 via CCPs 5–7 (24, 32). The FHR-1/mCRP interaction enhanced classical and alternative pathway activation, and FHR-5 efficiently competed with FH for mCRP binding, resulting in enhanced complement activation on mCRP (32, 33). In contrast, FHR-4 binds to pCRP via CCP1, and this interaction results in enhanced classical pathway activation (111, 112).
PTX3 forms a complex, octameric structure stabilized in part by covalent bonds (113). PTX3 binds to FH via CCPs 7 and 19–20, and recruits it to apoptotic cells to downregulate complement activation (77). PTX3 also binds to FHR-1 (weaker than FH) and FHR-5 (stronger than FH); FHR-5 competes with FH and enhance complement activation on PTX3 (32, 33, 74).
Malondialdehyde (MDA) and malondialdehyde-acetaldehyde (MAA) adducts of proteins and lipids may be generated upon oxidation as oxidation-related neoepitopes, and induce inflammatory responses. FH was shown to bind to MDA/MAA epitopes and inhibit complement activation and the proinflammatory effects of such MDA/MAA epitopes (99). Two binding sites, within CCP7 and CCPs 19–20 of FH, were identified to bind to MDA/MAA epitopes (99, 114). Recently, FHR-5 was also shown to bind to MAA epitopes (MAA-BSA) via CCPs 5–7 and to compete with FH for MAA-BSA binding, thus increasing complement activation. In addition, binding of FHR-5 to necrotic cells was mediated by the same domains, possibly in part via the MDA/MAA epitopes that appear on dead cells (94).
Factor H binds to other ligands that are implicated in certain diseases, particularly in the thrombotic microangiopathy atypical hemolytic uremic syndrome (aHUS). One of these ligands is thrombomodulin, a transmembrane glycoprotein present in endothelial cells, which is involved in the regulation of coagulation and inflammation; thrombomodulin soluble fragments can also be released upon endothelial cell activation or injury. Thrombomodulin was shown to bind to FH and the FH–C3b complex with nanomolar affinity and to enhance FH cofactor activity, which would be reduced in the case of thrombomodulin mutations in aHUS (115–117). These data suggest a role for thrombomodulin in inhibiting alternative pathway activation locally via its interaction with FH, but thrombomodulin was also found to inhibit complement hemolytic activity in a FH-independent mechanism (116). An additional, complement-activating function of thrombomodulin by enhancing C3 cleavage into C3b has also been described (117).
Similarly, binding of von Willebrand factor (vWF) to FH enhances FH cofactor activity and also modulates the vWF prothrombotic status (118–120). FH was found co-localized with vWF in the Weibel–Palade bodies in human umbilical vein endothelial cells, and the complex was also detected in human plasma. Purified FH and vWF were shown to interact with nanomolar affinity, and to influence their respective functions; vWF enhanced the cofactor activity of FH, whereas FH inhibited ADAMTS13-mediated cleavage of vWF and facilitated platelet aggregation (120). However, another investigation found that FH binds via its C-terminus to the vWF A2 domain, and enhances its cleavage by ADAMTS13 (118). FH was also reported to reduce large soluble vWF multimers (119). Thus, further studies are needed to clarify the functional relevance of the complex interaction between FH and vWF, and its potential role in disease.
Recently, hemolysis-derived heme was shown to activate the alternative pathway in serum and on endothelial cells, and to bind both C3 and FH. Heme-exposed C3 and endothelial cells displayed increased FH binding, and FH was shown to be a major serum factor that regulates C3 deposition on heme-treated endothelial cells (121).
Factor H was also reported to bind to apolipoprotein E via domains CCPs 5–7, and to regulate alternative pathway activation on high density lipoprotein particles (122). Complement regulation by FH on such lipoprotein particles could be potentially impaired in diseases characterized by immune deposits containing also apolipoprotein E, such as age-related macular degeneration (AMD) and dense deposit disease (DDD) (122).
In addition, FH binds to myeloperoxidase (MPO) released from activated neutrophil granulocytes, and FH and MPO co-localize in neutrophil extracellular traps. Interestingly, the binding site for MPO in FH was determined to be CCPs 1–4 and, thus, MPO inhibited FH binding to C3b, as well as FH decay accelerating activity and cofactor activity (123).
Factor H and some of the FHRs can also bind to cells via specific receptors, and may modulate the cell activation and response, as well as inflammatory processes. These aspects are reviewed in detail elsewhere (124); here, we summarize only some major FH/FHR-receptor interactions and their role, particularly those described very recently.
Complement receptor type 3 (CR3; CD11b/CD18; or integrin αMβ2) was identified as a main FH receptor on neutrophils and macrophages (125, 126). FH maintains its cofactor activity when receptor bound, but it also directly affects cellular functions, such as adhesion, cell spreading, migration, and cytokine production (125–127). Interestingly, FH was able to inhibit the release of extracellular traps by human neutrophils activated with immobilized fibronectin plus fungal β-glucan, or with phorbol 12-myristate 13-acetate (127). FH can also enhance the interaction of certain pathogens with human macrophages and neutrophils, and modulate the response of the phagocytes (128, 129). This was also shown for FHR-1 which, by binding to CR3, could enhance neutrophil responses to Candida albicans (129). In addition, FHL-1 was shown to mediate cell adhesion and spreading (129, 130).
Described functional effects of FH on monocytes include enhancement of IL-1β secretion, respiratory burst, and chemotactic effect (131–134). FH was shown to induce an anti-inflammatory and tolerogenic phenotype in monocyte-derived dendritic cells in vitro (135). Very recently, in the context of inflammation in AMD, FH, and its two variants Y402 and H402 were investigated in a mouse model. FH was shown to inhibit the resolution of inflammation by binding to CR3 and thus blocking thrombospondin-1–CD47 signaling that would normally promote the elimination of macrophages. The AMD-associated H402 FH variant displayed a stronger inhibitory effect compared to FH Y402, causing increased accumulation of macrophages in the inflamed tissue (136).
Factor H was shown to bind to B cells and may modulate some B cell functions, such as proliferation and immunoglobulin secretion, but no specific receptor has been identified to date (137–140). A recent report described an indirect modulation of B cell activation by FHR-3, which was shown to bind to C3d and inhibit its binding to complement receptor type 2, a co-receptor of the B cell receptor complex; FH and FHR-1 had no such effect (85).
These non-canonical functions of the FH family proteins deserve further investigation, because they may play roles in inflammation and anti-microbial defense that are currently underappreciated. Clarification of their cell-mediated effects may provide additional insights into disease mechanisms.
Studies in patients and controls have shown a variety of common CFH/CFHRs genetic variants that predispose to autologous damage, which is predominantly organ-specific. Prevalent kidney damage occurs in the rare diseases aHUS and C3 glomerulopathies (C3G), and in the more frequent IgA nephropathy (IgAN), while destruction of the retinal pigment epithelium by autologous complement contributes to AMD. A defective regulation of complement activation on the renal microvasculature endothelium occurs in aHUS, while in C3G uncontrolled complement activation in plasma gives rise to massive deposition of C3b breakdown products (iC3b, C3dg, and C3c) in the glomeruli (141–143). IgAN is characterized by mesangial cell proliferation and hypoglycosylated IgA1 deposits in the glomeruli, and it is likely that complement defects contribute, at least in part, to its clinical heterogeneity (144, 145). A defective control of complement activation in the retina is most relevant in AMD pathogenesis, and enhances the inflammatory response (146).
Extremely rare and pathogenic CFH/CFHRs variants have been mainly found in aHUS and C3G patients. Some of these variants result from gene conversion events between CFH and CFHR1, and they give rise to mutated FH or FHR-1. Other variants are intragenic duplications or hybrid genes resulting from gene rearrangements, and generate abnormal proteins; some of these proteins have distinct molecular weights and can be detected by Western blot analysis. It is interesting that abnormal rearrangements involving FH/FHRs associate with aHUS, while in C3G patients only FHR proteins are affected.
Common SNPs in CFH give rise to different haplotypes that can be disease neutral, predisposing, or protecting. Thus, haplotype CFH(H1) predisposes to membranoproliferative glomerulonephritis (MPGN) and AMD, haplotype CFH(H3) predisposes to aHUS, and haplotype CFH(H2) is protective against these three diseases (147–149). Haplotype CFH(H2) generates the FH62Ile variant, which shows increased binding to C3b and cofactor activity in the fluid phase and on cellular surfaces (150), thus favoring protection against autologous complement damage.
The common variant FH402His, which is present in FH and its shorter isoform FHL-1, is a major predisposing factor in AMD (151). The functional relevance of FH402His in C3b, CRP, or heparin binding has been analyzed in several studies. Reduced binding of FH402His to polyanionic surfaces has been found (152), but the pathogenic mechanism may also depend on FHL-1, which can regulate complement activation similarly to FH. It has been shown that FHL-1, but not FH, is present in the retinal Bruch’s membrane, a major target in AMD pathogenesis, and that binding of the AMD-FHL-1402His variant was lower than binding of the FHL-1402Tyr variant (93). Nonetheless, CFH intronic variants show stronger association with AMD than FH402His (153). In an analysis of seven common CFH haplotypes, haplotypes H1, H6, and H7 were found to confer increased risk to AMD; these haplotypes share a 32-kb region downstream of rs1061170 (FH Tyr402His) that must be critical for AMD development (19), and that includes a 12-kb block 89% similar to a noncoding region in CNP148 (see below).
Other disease-predisposing FH variants are very rare. One of the most relevant is Arg1210Cys (FH1210C), which was initially identified in aHUS patients (154), and shown to be covalently bound to albumin in plasma (155); the presence of albumin most likely prevents the interaction of FH1210C with its physiological ligands, generating a partial, pathogenic FH deficiency. FH1210C has been also associated with C3G (156), and it highly increases AMD-risk and predisposes to early disease onset (157, 158). It has been suggested that in individuals with the FH1210C variant it is the concurrence of other genetic predisposing factors what ultimately determines the clinical phenotype (159).
As happens with the common CFH haplotypes, the two main CFHR1 alleles show differential disease associations. CFHR1*B, displaying increased similarity with CFH, increases aHUS risk (51), and CFHR1*A predisposes to AMD (160). The molecular bases for these associations have not been determined, but they will most likely depend on subtle functional differences among the FHR-1*A and FHR-1*B allotypes. CFHR1*A is in strong linkage disequilibrium with the AMD-risk CFH402His allele, and CFHR1 genotyping has similar predictive value of developing AMD as CFH402His;ΔCFHR3–CFHR1 genotyping (160); these findings are suggestive of a direct role of FHR-1 in AMD pathogenesis, most likely by interfering with the interaction of FH with specific ligands and promoting complement activation (33).
The CFHR3 gene also has two major variants, CFHR3*A, more frequent in healthy controls, and CFHR3*B, which predisposes to aHUS but not to C3G (59). Because the aHUS risk CFHR3*B allele generates higher FHR-3 levels than the non-risk CFHR3*A allele (60), it seems that increased competition of FHR-3 and FH for certain ligands could favor aHUS development. FHR-3 is also produced in the retina, and its contribution to retinal degeneration by inhibiting FH binding to C3b and modified surfaces has been suggested (47); nonetheless, the relevance of the CFHR3*A and CFHR3*B variants in AMD has not been addressed.
The two CNPs in the CFHR genes have been shown to be disease-relevant (19). The common variant ΔCFHR3–CFHR1 is protective against AMD (161), and IgAN (162), but it predisposes to aHUS (163) and to systemic lupus erythematosus (SLE) (69) because it is associated with generation of anti-FH autoantibodies (discussed on page 13). The rare variant ΔCFHR1–CFHR4 was initially identified in a few aHUS patients with anti-FH autoantibodies (51), and is present in 1.4% of aHUS patients and 0.9% of controls (164).
The protective effect of the ΔCFHR3–CFHR1 haplotype against AMD was first described in 2006 (161), and it is the more common copy number variation in the CFH/CFHRs region (165). ΔCFHR3–CFHR1 is tagged by CFH rs6677604A with 99% accuracy (166), and strongly correlates with the 86.4-kb deletion CNP147 and high FH levels (8). Because protection conferred by ΔCFHR3–CFHR1 was independent of the FH Tyr402His polymorphism, a direct effect of FHR-1 and FHR-3 in AMD pathogenesis was suggested (21). Nonetheless, the strong association of ΔCFHR3–CFHR1 with high FH levels, together with the finding that FHR-1 levels were lower in AMD patients than in control individuals, suggests that ΔCFHR3–CFHR1 is actually tagging an allele expressing high FH levels, but it is not causal in protection against AMD (8). The much rarer ΔCFHR1–CFHR4 deletion (also referred to as CNP148) also confers protection against AMD independent of SNPs in CFH (19); because ΔCFHR1–CFHR4 also removes non-coding flanking regions, its protective effect against AMD could either be due to the reduction of FHR-1 and/or FHR-4A levels, or to the absence of regulatory regions relevant for disease pathogenesis.
The first evidence for a direct complement role in IgAN pathogenesis was the finding that the common variant ΔCFHR3–CFHR1 protects against IgAN when it is in homozygosis (162), pointing out to a possible role of FHR-3 and/or FHR-1 levels in the pathogenic mechanism. However, because the ΔCFHR3–CFHR1 allele generates high FH levels which associate with lower mesangial C3 deposition, the actual contribution of FHR-1 and/or FHR-3 levels to IgAN is unclear (167). Two studies in different IgAN cohorts have recently shown that FHR-1 levels and FHR-1/FH ratios are increased in patients with disease progression, thus providing evidence for a direct role of FHR-1 in the disease mechanism. One of these studies reported that high FHR-5 levels were also slightly elevated in the IgAN patients, but without any correlation with progressive disease (55). The other study also reported low FH levels associated with CFH or CFI mutations in a few IgAN patients (54).
CFH exons 18–20 and CFHR1 exons 4–6 have a high degree of sequence similarity, that result in only five amino acid difference between CCPs 18–20 of FH (Y1040-V1042-Q1058-S1191-V1197) and CCPs 3–5 of FHR-1 (H157-L159-E175-L290-A296). Studies in aHUS patients have revealed that these differences determine higher binding of FH than FHR-1 to cell surfaces. Amino acids S1191 and V1197 in FH seem to be particularly important, and single mutations involving these amino acids (FHS1191L and FHV1197A) have been found in a number of aHUS patients from different geographical origins. The double mutant (FHS1191L-V1197A) was observed in two unrelated aHUS patients with early disease onset, showed a defective capacity to control complement activation on cellular surfaces, and had been generated by gene conversion (168).
FHS1191L-V1197A can also be generated by NAHR events that give rise to CFH::CFHR1 hybrid genes. A CFH(Ex1–21)::CFHR1(Ex5–6) hybrid gene was first described in a family with many cases of aHUS along several generations, and a clinical history of disease recurrence in affected individuals (169), demonstrating that FHS1191L-V1197A is highly pathogenic. This hybrid gene has also been found in other non-related aHUS patients. A slightly different CFH(Ex1–22)::CFHR1(Ex6) gene which also generates FHS1191L-V1197A has been found in another patient with a prompt aHUS onset (170). The reverse situation (i.e., the existence of CFHR1::CFH hybrid genes) has also been reported. A CFHR1(Ex1–3)::CFH(Ex19–20) hybrid gene generated by “de novo” NAHR was identified in one sporadic case of aHUS (171), and a CFHR1(Ex1–4)::CFH(Ex20) hybrid gene was found in a family with two members affected with aHUS (172). These two CFHR1::CFH hybrid genes generated a double-mutated FHR-1 protein that carries the homologous amino acids in FH CCP20 domain (FHR-1L290S-A296V); these amino acids most likely confer the mutated FHR-1 increased competition with FH for endothelial cell binding, and result in reduced protection against complement damage (173). Screening of CFH::CFHR1 and CFHR1::CFH hybrid genes is normally done by MLPA analysis of copy number variations. The CFH::CFHR1 alleles lack a normal copy of CFHR3 and CFHR1, while the CFHR1::CFH allele contains two copies of CFHR3; it cannot be ruled out that these additional factors also contribute to the pathogenic mechanism.
A FH::FHR-3 hybrid protein containing CCPs 1–19 of FH and the five CCPs of FHR-3 was identified in a large family with aHUS (174). This protein resulted from an abnormal rearrangement that deleted the last exon of CFH, which was then fused to the adjacent CFHR3 gene by the genetic mechanism microhomology mediated end joining (MMEJ). The absence of FH CCP20 domain in the hybrid protein and/or the presence of the FHR-3 CCPs does not affect complement regulation in the fluid phase, but cellular surface regulation seemed to be highly reduced. Estimation of aHUS penetrance in carriers of the hybrid gene is 33%. Another FH::FHR-3 hybrid protein containing CCPs 1–17 of FH and the five CCPs of FHR-3 was found in an aHUS patient with a very early disease onset (175). The hybrid protein resulted from a “de novo” 6.3 kb-deletion of exons 21–23 of the CFH gene through a MMEJ mechanism, and it showed impaired cell surface complement regulation.
The abnormal rearrangements that predispose to C3G thus far described involve exclusively the CFHR genes, but not the CFH gene. This is a distinctive feature from aHUS that suggests a more important contribution of FHRs in the protection of the glomerular basement membrane and mesangium than in protection of endothelial cells. Abnormal rearrangements include intragenic duplications in CFHR1 or CFHR5, and CFHR2::CFHR5 and CFHR3::CFHR1 hybrid genes.
Larger forms of FHR-1 and FHR-5 with duplicated dimerization domains have been observed in a few C3G patients. These proteins circulate in plasma together with the normal FHR-1 and FHR-5 proteins, but disease penetrance in mutation carriers is very high, strongly suggesting a dominant negative effect of the larger, abnormal protein. This is particularly evident for a partially duplicated FHR-5 protein initially observed in two families of Cypriot ancestry in which renal disease was consistent with autosomal dominant transmission (176). All affected individuals were heterozygous for a CFHR5 gene in which exon 2 (coding for CCP1) and exon 3 (coding for CCP2) were duplicated, giving rise to an abnormal FHR-5 protein containing two extra dimerization domains (FHR-512123-9). In vitro studies with patient’s sera showed reduced binding of the FHR-512123-9 to the cell surface, and increased FI cofactor activity, but the relevance of these findings for the pathogenic mechanism is unknown. Patients carrying FHR-512123-9 had a high risk of progressive renal disease, particularly males. This renal phenotype, which histologically corresponds to a C3 glomerulonephritis, is clinically characterized by continuous macroscopic hematuria, and was denominated as “CFHR5 nephropathy.” These seminal observations were further extended to 16 pedigrees of Cypriot origin in a study that also provided a thorough description of histological, molecular, and clinical findings (177). Recurrence of CFHR5 nephropathy in a kidney allograft has been reported in one patient, although it did not occur in two other cases (178). The same duplicated FHR-5 protein observed in patients of Cypriot ancestry was found in a familial case of C3 glomerulonephritis with a different ethnic origin (179). Of note, this protein was generated from a different genomic rearrangement, reinforcing the relevance of the duplicated FHR-5 protein for the pathogenic mechanism, and the authors proposed that all patients with clinical suspicion of CFHR5 nephropathy should be screened for the abnormal protein by Western blot.
Another FHR-5 protein with two additional dimerization domains was found in a familial case of C3G with DDD (C3G-DDD) (83). In this family, a genomic 24.8 kb-deletion from intron III of the CFHR2 gene to CFHR5 gives rise to a hybrid CFHR2::CFHR5 gene which generates a so-called FHR-21,2-FHR-5-hybrid protein very similar to the FHR-512123-9 protein previously described. This hybrid protein shows increased binding to C3b and stabilization of the AP C3 convertase, which would explain the low C3 and increased Ba levels detected in the patients’ sera; in addition, reduced regulation of the AP C3 convertase by FH will result in increased generation of iC3b molecules which will deposit on the glomerular basement membrane and favor the pathogenic mechanism.
To understand how the presence of two extra dimerization domains in FHR-512123-9 has pathogenic consequences it is necessary to recapitulate that the dimerization domains in FHR-1, FHR-2, and FHR-5 confer these proteins the ability to generate homo- and hetero-dimers physiologically (26). Although a recent study has not found evidence of the presence of FHR-5 heterodimers with FHR-1 or FHR-2 (53), the additional dimerization domains in FHR-512123-9 and FHR-21,2-FHR-5 will most likely give rise to higher order oligomeric forms with increased avidity for surface-bound C3b, and these multimeric proteins will compete more efficiently with FH and favor autologous tissue damage, as illustrated in Figure 3. In this context, it is intriguing that two very similar FHR-5 proteins result in different clinical entities (CFHR5 nephropathy or DDD). FHR-512123-9 and FHR-21,2-FHR-5 contain the nine CCPs of FHR-5 preceded by the two dimerization domains of FHR-5 or FHR-2, respectively, that present 85% aminoacid identity. Functional studies with the recombinant forms of these two proteins (referred to as FHR-5Dup and FHR-2-FHR-5Hyb) revealed that they exacerbate local complement activation by recruiting the complement-activating protein properdin, and that properdin binding is mediated by the FHR-5 dimerization domains, and not by the FHR-2 dimerization domains (82). Therefore, local complement activation would be higher in patients with FHR-5Dup than in patients with FHR-2-FHR-5Hyb, and this could explain the different clinical phenotype. Another, non-exclusive, explanation is that the pathogenic mechanism is much dependent on the plasma levels of these FHR-5 proteins. In line with this hypothesis, Western blot analyses of patient serum samples showed that the FHR-5 band has similar intensity to a normal serum, while the intensity of the FHR-512123-9 (32) or the FHR-21,2-FHR-5 band is much higher, suggesting highly increased levels (82). The latter study also showed that FHR-5 binds to necrotic human endothelial cells, but not to normal endothelial cells, strongly suggesting a role for FHR-5 in complement-mediated elimination of damaged cells.
An abnormal, large FHR-1 protein was identified in a Spanish family with C3G by Western blot analysis (52). This protein was generated by an internal duplication of the CFHR1 gene, and contains two copies of domains CCPs 1–4. Purification of the normal and the duplicated FHR-1 proteins allowed biochemical, functional, and structural studies that illustrated that normal FHR-1 circulates in plasma as homo- and hetero-oligomers (with FHR-2 and FHR-5), and that the duplicated FHR-1 (containing nine CCP domains) organized into much larger oligomers with increased binding to C3b, iC3b, and C3dg. These findings provided the first evidence for the existence of oligomeric forms of FHR-1, FHR-2, and FHR-5 in normal plasma, and confirmed that duplication of their homologous CCPs 1–2 is pathogenic and associates with C3G. The authors proposed that multimerization of FHR-1 strongly inhibits FH binding to certain cell surfaces, but not to endothelial cells, the target surface in aHUS. A different FHR-1 protein containing two copies of domains CCPs 1–2 has been described in another Spanish patient with a C3G clinical phenotype, but further characterization of this duplicated FHR-1 (containing seven domains) has not been provided (180).
A hybrid CFHR3::CFHR1 gene associated with C3G-MPGN III has been described in an Irish family (181). This hybrid gene contains exons 1–3 of CFHR3 and exons 2–6 of CFHR1, and generates a protein containing CCPs 1–2 of FHR-3 followed by the five CCPs of FHR-1. The protein was detected in the patients’ plasma by Western blot, and it was apparently at a much lower concentration than normal FHR-1. Because patients with the CFHR3::CFHR1 gene also has two copies of CFHR3 and CFHR1, the authors propose a dominant effect of the hybrid FHR-3::FHR-1 protein in the pathogenic mechanism. It is of note that plasma C3 levels in all affected individuals were normal, as opposed to the reduced levels observed in the C3G-DDD patient with FHR-21,2-FHR-5-hybrid protein (83); this fact suggests that the potential pathogenic effect of FHR-3::FHR-1 on complement activation or regulation is surface-restricted. The clinical data and outcome of the five patients from this family who received renal transplantation has been reported (182); disease recurrence in the kidney allograft was high, but the overall graft survival was good.
Disorders related with these autoantibodies are mainly present in aHUS and C3 glomerulonephritis patients and secondary in other autoimmune diseases. The anti-FH autoantibodies cause a functional FH defect, resulting in impaired complement regulation by FH (Figure 3C).
The existence of anti-factor H autoantibodies in aHUS and the resulting functional deficiency of FH were first described in 2005 (183). The frequency of the anti-FH antibodies associated with aHUS is approximately 10% of the pediatric patients in the European series and occasionally in patients with adult onset (184). These autoantibodies form complexes with FH and induce a functional FH deficiency. Characterization of these autoantibodies showed that they recognized the C-terminal region of FH, involved in the binding to cell surfaces (185, 186). Moreover, it has been shown that, especially in the acute phase, these antibodies are also capable of blocking the activity of FH as cofactor of FI and the acceleration of the dissociation of the convertases of the alternative pathway (187).
The presence of anti-FH autoantibodies is associated with homozygous ΔCFHR3–CFHR1 in several aHUS cohorts (51, 164, 188, 189). The ΔCFHR1–CFHR4 has also been found in a few patients (51, 164), suggesting a relevant role for the absence of FHR-1 in autoantibody generation. In this context, it has been found that most anti-FH autoantibodies also bind to FHR-1, which presents high similarity with FH CCPs 19–20 (29, 164).
The anti-FH autoantibodies in aHUS patients are able of forming immune complexes that can be detected in serum. The amount of these complexes correlates better with the clinical evolution than the total autoantibody titer (187), because FH bound to the complexes cannot regulate the AP on cell surfaces. The use of two monoclonal antibodies binding to different parts of FH allowed the quantitation of total and free FH, which depends on the concentration of circulating anti-FH immune complexes (29, 186, 190). In some cases, the concentration of total FH was within the normal range, but the amount of free FH was practically undetectable, indicating that the anti-FH autoantibodies almost completely blocked the ability of FH to protect cell surfaces from complement activation, although its regulatory activity in the fluid phase was conserved (190).
The epitope recognized by anti-FH autoantibodies has been defined more precisely using recombinant fragments of CCPs 19–20 containing point mutations (191). In this work, it was found that in patients with FHR-1 deficiency, anti-FH antibodies recognize a region that acquires a different conformation in FH and FHR-1 after binding to certain ligands, including various bacterial proteins. This suggests a model in which the absence of FHR-1 plays a role in the loss of tolerance to FH and in the generation of anti-FH autoantibodies, thus explaining the frequent association between the presence of anti-FH antibodies and homozygous ΔCFHR3–CFHR1 in aHUS. By using the same mutated FH recombinant fragments in our series of patients with anti-FH autoantibodies, we have obtained concordant results, at least in the patients with FHR-1 deficiency, which supports the proposed model for the generation of autoantibodies in these patients (192). However, the mechanism of anti-FH autoantibody generation in aHUS patients without FHR-1 deficiency remains to be determined.
Anti-FH autoantibodies have also been described in patients with C3G (193–197). This association is much less frequent than in the case of aHUS despite having been described for the first time (196). In cases in which the effect of these anti-FH autoantibodies has been studied, it has been shown that they inhibit the regulatory activity of FH by recognizing and blocking its N-terminal region (193, 194, 197), which is a difference with the anti-FH autoantibodies from aHUS patients.
In patients with SLE and other autoimmune diseases, a greater frequency of anti-FH autoantibodies has been described with respect to healthy controls (198). Unlike the anti-FH autoantibodies present in aHUS, the epitopes that are recognized by the autoantibodies seem to be distributed throughout the entire protein, and they are not associated with FHR-1 deficiency.
The FH protein family remains an intriguing group of proteins. FH is well-known for its protecting role against self-damage from complement, and the FHRs are emerging as FH antagonists that act as an additional regulatory mechanism to control where and when FH protects human cells and/or surfaces. With the recent development of FHR-specific assays, quantification of the whole protein family has now become possible. This has elucidated the intricate balance between FH and the FHR proteins, showing that overall the balance is in favor of FH. However, this balance can shift on altered self, and also genetic variations have a major impact on FH and FHRs. This includes decreased FH function due to mutations, altered expression levels, as well as hybrid FH::FHR and FHR::FHR proteins and unusual FHR multimers with abnormal function that disturb complement regulation. Associations of increased FHR levels, as a result of genetic variations, with diseases like aHUS and IgAN are highly suggestive of a pathological role for the FHRs. It remains to be seen whether the FHRs are indeed causative in these diseases, but it is likely that they at least contribute to altered complement regulation on host surfaces.
PS-C, RBP, ML-T, and MJ prepared the text and the figures. All authors have revised and approved the manuscript.
RBP is co-inventor of a patent describing potentiating anti-FH antibodies and uses thereof. The other authors declare no conflict of interest.
PS-C and ML-T are funded by grants PI16/00723 and PI15/00255 (Spanish Ministerio de Economía y Competitividad/ISCIII, and European Program FEDER) and B2017/BMD3673 (Complement II-CM network from the Comunidad de Madrid). MJ is supported by the National Research, Development and Innovation Fund of Hungary (NKFIA grants K 109055 and K 125219), the Kidneeds Foundation, Iowa, US, and by the Institutional Excellence Program of the Ministry of Human Capacities of Hungary.
1. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol (2017) 14(1):26–47. doi:10.1038/nrneph.2017.156
2. Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol (2004) 138(3):439–46. doi:10.1111/j.1365-2249.2004.02627.x
3. Rodriguez de Cordoba S, Esparza-Gordillo J, Goicoechea de Jorge E, Lopez-Trascasa M, Sanchez-Corral P. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol (2004) 41(4):355–67. doi:10.1016/j.molimm.2004.02.005
4. Ripoche J, Day AJ, Harris TJ, Sim RB. The complete amino acid sequence of human complement factor H. Biochem J (1988) 249(2):593–602. doi:10.1042/bj2490593
5. Gordon DL, Kaufman RM, Blackmore TK, Kwong J, Lublin DM. Identification of complement regulatory domains in human factor H. J Immunol (1995) 155(1):348–56.
6. Kopp A, Hebecker M, Svobodova E, Jozsi M. Factor h: a complement regulator in health and disease, and a mediator of cellular interactions. Biomolecules (2012) 2(1):46–75. doi:10.3390/biom2010046
7. Schmidt CQ, Herbert AP, Hocking HG, Uhrin D, Barlow PN. Translational mini-review series on complement factor H: structural and functional correlations for factor H. Clin Exp Immunol (2008) 151(1):14–24. doi:10.1111/j.1365-2249.2007.03553.x
8. Ansari M, McKeigue PM, Skerka C, Hayward C, Rudan I, Vitart V, et al. Genetic influences on plasma CFH and CFHR1 concentrations and their role in susceptibility to age-related macular degeneration. Hum Mol Genet (2013) 22(23):4857–69. doi:10.1093/hmg/ddt336
9. Esparza-Gordillo J, Soria JM, Buil A, Almasy L, Blangero J, Fontcuberta J, et al. Genetic and environmental factors influencing the human factor H plasma levels. Immunogenetics (2004) 56(2):77–82. doi:10.1007/s00251-004-0660-7
10. Hakobyan S, Harris CL, Tortajada A, Goicochea de Jorge E, Garcia-Layana A, Fernandez-Robredo P, et al. Measurement of factor H variants in plasma using variant-specific monoclonal antibodies: application to assessing risk of age-related macular degeneration. Invest Ophthalmol Vis Sci (2008) 49(5):1983–90. doi:10.1167/iovs.07-1523
11. Sofat R, Mangione PP, Gallimore JR, Hakobyan S, Hughes TR, Shah T, et al. Distribution and determinants of circulating complement factor H concentration determined by a high-throughput immunonephelometric assay. J Immunol Methods (2013) 390(1–2):63–73. doi:10.1016/j.jim.2013.01.009
12. Friese MA, Hellwage J, Jokiranta TS, Meri S, Muller-Quernheim HJ, Peter HH, et al. Different regulation of factor H and FHL-1/reconectin by inflammatory mediators and expression of the two proteins in rheumatoid arthritis (RA). Clin Exp Immunol (2000) 121(2):406–15. doi:10.1046/j.1365-2249.2000.01285.x
13. Zipfel PF, Skerka C. FHL-1/reconectin: a human complement and immune regulator with cell-adhesive function. Immunol Today (1999) 20(3):135–40. doi:10.1016/S0167-5699(98)01432-7
14. Misasi R, Huemer HP, Schwaeble W, Solder E, Larcher C, Dierich MP. Human complement factor H: an additional gene product of 43 kDa isolated from human plasma shows cofactor activity for the cleavage of the third component of complement. Eur J Immunol (1989) 19(9):1765–8. doi:10.1002/eji.1830190936
15. Schwaeble W, Zwirner J, Schulz TF, Linke RP, Dierich MP, Weiss EH. Human complement factor H: expression of an additional truncated gene product of 43 kDa in human liver. Eur J Immunol (1987) 17(10):1485–9. doi:10.1002/eji.1830171015
16. Kuhn S, Skerka C, Zipfel PF. Mapping of the complement regulatory domains in the human factor H-like protein 1 and in factor H1. J Immunol (1995) 155(12):5663–70.
17. Swinkels M, Zhang JH, Tilakaratna V, Black G, Perveen R, McHarg S, et al. C-reactive protein and pentraxin-3 binding of factor H-like protein 1 differs from complement factor H: implications for retinal inflammation. Sci Rep (2018) 8(1):1643. doi:10.1038/s41598-017-18395-7
18. Cantsilieris S, Nelson BJ, Huddleston J, Baker C, Harshman L, Penewit K, et al. Recurrent structural variation, clustered sites of selection, and disease risk for the complement factor H (CFH) gene family. Proc Natl Acad Sci U S A (2018) 115(19):E4433–42. doi:10.1073/pnas.1717600115
19. Sivakumaran TA, Igo RP Jr, Kidd JM, Itsara A, Kopplin LJ, Chen W, et al. A 32 kb critical region excluding Y402H in CFH mediates risk for age-related macular degeneration. PLoS One (2011) 6(10):e25598. doi:10.1371/journal.pone.0025598
20. Eberhardt HU, Buhlmann D, Hortschansky P, Chen Q, Bohm S, Kemper MJ, et al. Human factor H-related protein 2 (CFHR2) regulates complement activation. PLoS One (2013) 8(11):e78617. doi:10.1371/journal.pone.0078617
21. Fritsche LG, Lauer N, Hartmann A, Stippa S, Keilhauer CN, Oppermann M, et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Hum Mol Genet (2010) 19(23):4694–704. doi:10.1093/hmg/ddq399
22. Heinen S, Hartmann A, Lauer N, Wiehl U, Dahse HM, Schirmer S, et al. Factor H-related protein 1 (CFHR-1) inhibits complement C5 convertase activity and terminal complex formation. Blood (2009) 114(12):2439–47. doi:10.1182/blood-2009-02-205641
23. Hellwage J, Jokiranta TS, Koistinen V, Vaarala O, Meri S, Zipfel PF. Functional properties of complement factor H-related proteins FHR-3 and FHR-4: binding to the C3d region of C3b and differential regulation by heparin. FEBS Lett (1999) 462(3):345–52. doi:10.1016/S0014-5793(99)01554-9
24. McRae JL, Duthy TG, Griggs KM, Ormsby RJ, Cowan PJ, Cromer BA, et al. Human factor H-related protein 5 has cofactor activity, inhibits C3 convertase activity, binds heparin and C-reactive protein, and associates with lipoprotein. J Immunol (2005) 174(10):6250–6. doi:10.4049/jimmunol.174.10.6250
25. Caesar JJ, Lavender H, Ward PN, Exley RM, Eaton J, Chittock E, et al. Competition between antagonistic complement factors for a single protein on N. meningitidis rules disease susceptibility. Elife (2014) 3:e04008. doi:10.7554/eLife.04008
26. Goicoechea de Jorge E, Caesar JJ, Malik TH, Patel M, Colledge M, Johnson S, et al. Dimerization of complement factor H-related proteins modulates complement activation in vivo. Proc Natl Acad Sci U S A (2013) 110(12):4685–90. doi:10.1073/pnas.1219260110
27. Hebecker M, Jozsi M. Factor H-related protein 4 activates complement by serving as a platform for the assembly of alternative pathway C3 convertase via its interaction with C3b protein. J Biol Chem (2012) 287(23):19528–36. doi:10.1074/jbc.M112.364471
28. Meszaros T, Csincsi AI, Uzonyi B, Hebecker M, Fulop TG, Erdei A, et al. Factor H inhibits complement activation induced by liposomal and micellar drugs and the therapeutic antibody rituximab in vitro. Nanomedicine (2016) 12(4):1023–31. doi:10.1016/j.nano.2015.11.019
29. Strobel S, Abarrategui-Garrido C, Fariza-Requejo E, Seeberger H, Sanchez-Corral P, Jozsi M. Factor H-related protein 1 neutralizes anti-factor H autoantibodies in autoimmune hemolytic uremic syndrome. Kidney Int (2011) 80(4):397–404. doi:10.1038/ki.2011.152
30. Timmann C, Leippe M, Horstmann RD. Two major serum components antigenically related to complement factor H are different glycosylation forms of a single protein with no factor H-like complement regulatory functions. J Immunol (1991) 146(4):1265–70.
31. Jozsi M, Tortajada A, Uzonyi B, Goicoechea de Jorge E, Rodriguez de Cordoba S. Factor H-related proteins determine complement-activating surfaces. Trends Immunol (2015) 36(6):374–84. doi:10.1016/j.it.2015.04.008
32. Csincsi AI, Kopp A, Zoldi M, Banlaki Z, Uzonyi B, Hebecker M, et al. Factor H-related protein 5 interacts with pentraxin 3 and the extracellular matrix and modulates complement activation. J Immunol (2015) 194(10):4963–73. doi:10.4049/jimmunol.1403121
33. Csincsi AI, Szabo Z, Banlaki Z, Uzonyi B, Cserhalmi M, Karpati E, et al. FHR-1 binds to C-reactive protein and enhances rather than inhibits complement activation. J Immunol (2017) 199(1):292–303. doi:10.4049/jimmunol.1600483
34. Antonioli AH, White J, Crawford F, Renner B, Marchbank KJ, Hannan JP, et al. Modulation of the alternative pathway of complement by murine factor H-related proteins. J Immunol (2018) 200(1):316–26. doi:10.4049/jimmunol.1602017
35. Cserhalmi M, Csincsi AI, Mezei Z, Kopp A, Hebecker M, Uzonyi B, et al. The murine factor H-related protein FHR-B promotes complement activation. Front Immunol (2017) 8:1145. doi:10.3389/fimmu.2017.01145
36. de Cordoba SR, de Jorge EG. Translational mini-review series on complement factor H: genetics and disease associations of human complement factor H. Clin Exp Immunol (2008) 151(1):1–13. doi:10.1111/j.1365-2249.2007.03552.x
37. Friese MA, Hellwage J, Jokiranta TS, Meri S, Peter HH, Eibel H, et al. FHL-1/reconectin and factor H: two human complement regulators which are encoded by the same gene are differently expressed and regulated. Mol Immunol (1999) 36(13–14):809–18. doi:10.1016/S0161-5890(99)00101-7
38. Roumenina LT, Rayes J, Frimat M, Fremeaux-Bacchi V. Endothelial cells: source, barrier, and target of defensive mediators. Immunol Rev (2016) 274(1):307–29. doi:10.1111/imr.12479
39. Brooimans RA, van der Ark AA, Buurman WA, van Es LA, Daha MR. Differential regulation of complement factor H and C3 production in human umbilical vein endothelial cells by IFN-gamma and IL-1. J Immunol (1990) 144(10):3835–40.
40. Chen M, Forrester JV, Xu H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp Eye Res (2007) 84(4):635–45. doi:10.1016/j.exer.2006.11.015
41. Devine DV, Rosse WF. Regulation of the activity of platelet-bound C3 convertase of the alternative pathway of complement by platelet factor H. Proc Natl Acad Sci U S A (1987) 84(16):5873–7. doi:10.1073/pnas.84.16.5873
42. Dixon KO, O’Flynn J, Klar-Mohamad N, Daha MR, van Kooten C. Properdin and factor H production by human dendritic cells modulates their T-cell stimulatory capacity and is regulated by IFN-gamma. Eur J Immunol (2017) 47(3):470–80. doi:10.1002/eji.201646703
43. Katz Y, Strunk RC. Synthesis and regulation of complement protein factor H in human skin fibroblasts. J Immunol (1988) 141(2):559–63.
44. Timar KK, Pasch MC, van den Bosch NH, Jarva H, Junnikkala S, Meri S, et al. Human keratinocytes produce the complement inhibitor factor H: synthesis is regulated by interferon-gamma. Mol Immunol (2006) 43(4):317–25. doi:10.1016/j.molimm.2005.02.009
45. van den Dobbelsteen ME, Verhasselt V, Kaashoek JG, Timmerman JJ, Schroeijers WE, Verweij CL, et al. Regulation of C3 and factor H synthesis of human glomerular mesangial cells by IL-1 and interferon-gamma. Clin Exp Immunol (1994) 95(1):173–80. doi:10.1111/j.1365-2249.1994.tb06033.x
46. Whaley K. Biosynthesis of the complement components and the regulatory proteins of the alternative complement pathway by human peripheral blood monocytes. J Exp Med (1980) 151(3):501–16. doi:10.1084/jem.151.3.501
47. Schafer N, Grosche A, Reinders J, Hauck SM, Pouw RB, Kuijpers TW, et al. Complement regulator FHR-3 is elevated either locally or systemically in a selection of autoimmune diseases. Front Immunol (2016) 7:542. doi:10.3389/fimmu.2016.00542
48. Skerka C, Chen Q, Fremeaux-Bacchi V, Roumenina LT. Complement factor H related proteins (CFHRs). Mol Immunol (2013) 56(3):170–80. doi:10.1016/j.molimm.2013.06.001
49. Estaller C, Koistinen V, Schwaeble W, Dierich MP, Weiss EH. Cloning of the 1.4-kb mRNA species of human complement factor H reveals a novel member of the short consensus repeat family related to the carboxy terminal of the classical 150-kDa molecule. J Immunol (1991) 146(9):3190–6.
50. Skerka C, Horstmann RD, Zipfel PF. Molecular cloning of a human serum protein structurally related to complement factor H. J Biol Chem (1991) 266(18):12015–20.
51. Abarrategui-Garrido C, Martinez-Barricarte R, Lopez-Trascasa M, de Cordoba SR, Sanchez-Corral P. Characterization of complement factor H-related (CFHR) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. Blood (2009) 114(19):4261–71. doi:10.1182/blood-2009-05-223834
52. Tortajada A, Yebenes H, Abarrategui-Garrido C, Anter J, Garcia-Fernandez JM, Martinez-Barricarte R, et al. C3 glomerulopathy-associated CFHR1 mutation alters FHR oligomerization and complement regulation. J Clin Invest (2013) 123(6):2434–46. doi:10.1172/JCI68280
53. van Beek AE, Pouw RB, Brouwer MC, van Mierlo G, Geissler J, Ooijevaar-de Heer P, et al. Factor H-related (FHR)-1 and FHR-2 Form homo- and heterodimers, while FHR-5 circulates only as homodimer in human plasma. Front Immunol (2017) 8:1328. doi:10.3389/fimmu.2017.01328
54. Tortajada A, Gutierrez E, Goicoechea de Jorge E, Anter J, Segarra A, Espinosa M, et al. Elevated factor H-related protein 1 and factor H pathogenic variants decrease complement regulation in IgA nephropathy. Kidney Int (2017) 92(4):953–63. doi:10.1016/j.kint.2017.03.041
55. Medjeral-Thomas NR, Lomax-Browne HJ, Beckwith H, Willicombe M, McLean AG, Brookes P, et al. Circulating complement factor H-related proteins 1 and 5 correlate with disease activity in IgA nephropathy. Kidney Int (2017) 92(4):942–52. doi:10.1016/j.kint.2017.03.043
56. Skerka C, Timmann C, Horstmann RD, Zipfel PF. Two additional human serum proteins structurally related to complement factor H. Evidence for a family of factor H-related genes. J Immunol (1992) 148(10):3313–8.
57. Skerka C, Kuhn S, Gunther K, Lingelbach K, Zipfel PF. A novel short consensus repeat-containing molecule is related to human complement factor H. J Biol Chem (1993) 268(4):2904–8.
58. Pouw RB, Brouwer MC, Geissler J, van Herpen LV, Zeerleder SS, Wuillemin WA, et al. Complement factor H-related protein 3 serum levels are low compared to factor H and mainly determined by gene copy number variation in CFHR3. PLoS One (2016) 11(3):e0152164. doi:10.1371/journal.pone.0152164
59. Bernabeu-Herrero ME, Jimenez-Alcazar M, Anter J, Pinto S, Sanchez Chinchilla D, Garrido S, et al. Complement factor H, FHR-3 and FHR-1 variants associate in an extended haplotype conferring increased risk of atypical hemolytic uremic syndrome. Mol Immunol (2015) 67(2 Pt B):276–86. doi:10.1016/j.molimm.2015.06.021
60. Pouw RB, Gomez Delgado I, Lopez Lera A, Rodriguez de Cordoba S, Wouters D, Kuijpers TW, et al. High complement factor H-related (FHR)-3 levels are associated with the atypical hemolytic-uremic syndrome-risk allele CFHR3*B. Front Immunol (2018) 9:848. doi:10.3389/fimmu.2018.00848
61. Jozsi M, Richter H, Loschmann I, Skerka C, Buck F, Beisiegel U, et al. FHR-4A: a new factor H-related protein is encoded by the human FHR-4 gene. Eur J Hum Genet (2005) 13(3):321–9. doi:10.1038/sj.ejhg.5201324
62. Skerka C, Hellwage J, Weber W, Tilkorn A, Buck F, Marti T, et al. The human factor H-related protein 4 (FHR-4). A novel short consensus repeat-containing protein is associated with human triglyceride-rich lipoproteins. J Biol Chem (1997) 272(9):5627–34. doi:10.1074/jbc.272.9.5627
63. Pouw RB, Brouwer MC, van Beek AE, Jozsi M, Wouters D, Kuijpers TW. Complement factor H-related protein 4A is the dominant circulating splice variant of CFHR4. Front Immunol (2018) 9:729. doi:10.3389/fimmu.2018.00729
64. McRae JL, Cowan PJ, Power DA, Mitchelhill KI, Kemp BE, Morgan BP, et al. Human factor H-related protein 5 (FHR-5). A new complement-associated protein. J Biol Chem (2001) 276(9):6747–54. doi:10.1074/jbc.M007495200
65. Vernon KA, Goicoechea de Jorge E, Hall AE, Fremeaux-Bacchi V, Aitman TJ, Cook HT, et al. Acute presentation and persistent glomerulonephritis following streptococcal infection in a patient with heterozygous complement factor H-related protein 5 deficiency. Am J Kidney Dis (2012) 60(1):121–5. doi:10.1053/j.ajkd.2012.02.329
66. Zhang P, Zhu M, Geng-Spyropoulos M, Shardell M, Gonzalez-Freire M, Gudnason V, et al. A novel, multiplexed targeted mass spectrometry assay for quantification of complement factor H (CFH) variants and CFH-related proteins 1-5 in human plasma. Proteomics (2017) 17(6):1600237. doi:10.1002/pmic.201600237
67. Kopczynska M, Zelek W, Touchard S, Gaughran F, Di Forti M, Mondelli V, et al. Complement system biomarkers in first episode psychosis. Schizophr Res (2017). doi:10.1016/j.schres.2017.12.012
68. Holmes LV, Strain L, Staniforth SJ, Moore I, Marchbank K, Kavanagh D, et al. Determining the population frequency of the CFHR3/CFHR1 deletion at 1q32. PLoS One (2013) 8(4):e60352. doi:10.1371/journal.pone.0060352
69. Zhao J, Wu H, Khosravi M, Cui H, Qian X, Kelly JA, et al. Association of genetic variants in complement factor H and factor H-related genes with systemic lupus erythematosus susceptibility. PLoS Genet (2011) 7(5):e1002079. doi:10.1371/journal.pgen.1002079
70. Xue X, Wu J, Ricklin D, Forneris F, Di Crescenzio P, Schmidt CQ, et al. Regulator-dependent mechanisms of C3b processing by factor I allow differentiation of immune responses. Nat Struct Mol Biol (2017) 24(8):643–51. doi:10.1038/nsmb.3427
71. Riley-Vargas RC, Lanzendorf S, Atkinson JP. Targeted and restricted complement activation on acrosome-reacted spermatozoa. J Clin Invest (2005) 115(5):1241–9. doi:10.1172/JCI23213
72. Sjoberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol (2009) 30(2):83–90. doi:10.1016/j.it.2008.11.003
73. Jozsi M. Factor H family proteins in complement evasion of microorganisms. Front Immunol (2017) 8:571. doi:10.3389/fimmu.2017.00571
74. Kopp A, Strobel S, Tortajada A, Rodriguez de Cordoba S, Sanchez-Corral P, Prohaszka Z, et al. Atypical hemolytic uremic syndrome-associated variants and autoantibodies impair binding of factor h and factor h-related protein 1 to pentraxin 3. J Immunol (2012) 189(4):1858–67. doi:10.4049/jimmunol.1200357
75. Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol (2008) 6(2):132–42. doi:10.1038/nrmicro1824
76. Schmidt CQ, Herbert AP, Kavanagh D, Gandy C, Fenton CJ, Blaum BS, et al. A new map of glycosaminoglycan and C3b binding sites on factor H. J Immunol (2008) 181(4):2610–9. doi:10.4049/jimmunol.181.4.2610
77. Deban L, Jarva H, Lehtinen MJ, Bottazzi B, Bastone A, Doni A, et al. Binding of the long pentraxin PTX3 to factor H: interacting domains and function in the regulation of complement activation. J Immunol (2008) 181(12):8433–40. doi:10.4049/jimmunol.181.12.8433
78. Hebecker M, Alba-Dominguez M, Roumenina LT, Reuter S, Hyvarinen S, Dragon-Durey MA, et al. An engineered construct combining complement regulatory and surface-recognition domains represents a minimal-size functional factor H. J Immunol (2013) 191(2):912–21. doi:10.4049/jimmunol.1300269
79. Jarva H, Jokiranta TS, Hellwage J, Zipfel PF, Meri S. Regulation of complement activation by C-reactive protein: targeting the complement inhibitory activity of factor H by an interaction with short consensus repeat domains 7 and 8–11. J Immunol (1999) 163(7):3957–62.
80. Kajander T, Lehtinen MJ, Hyvarinen S, Bhattacharjee A, Leung E, Isenman DE, et al. Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc Natl Acad Sci U S A (2011) 108(7):2897–902. doi:10.1073/pnas.1017087108
81. Morgan HP, Schmidt CQ, Guariento M, Blaum BS, Gillespie D, Herbert AP, et al. Structural basis for engagement by complement factor H of C3b on a self surface. Nat Struct Mol Biol (2011) 18(4):463–70. doi:10.1038/nsmb.2018
82. Chen Q, Manzke M, Hartmann A, Buttner M, Amann K, Pauly D, et al. Complement factor H-related 5-hybrid proteins anchor properdin and activate complement at self-surfaces. J Am Soc Nephrol (2016) 27(5):1413–25. doi:10.1681/ASN.2015020212
83. Chen Q, Wiesener M, Eberhardt HU, Hartmann A, Uzonyi B, Kirschfink M, et al. Complement factor H-related hybrid protein deregulates complement in dense deposit disease. J Clin Invest (2014) 124(1):145–55. doi:10.1172/JCI71866
84. Hannan JP, Laskowski J, Thurman JM, Hageman GS, Holers VM. Mapping the complement factor H-related protein 1 (CFHR1):C3b/C3d interactions. PLoS One (2016) 11(11):e0166200. doi:10.1371/journal.pone.0166200
85. Buhlmann D, Eberhardt HU, Medyukhina A, Prodinger WM, Figge MT, Zipfel PF, et al. FHR3 blocks C3d-mediated coactivation of human B cells. J Immunol (2016) 197(2):620–9. doi:10.4049/jimmunol.1600053
86. Meri S. Self-nonself discrimination by the complement system. FEBS Lett (2016) 590(15):2418–34. doi:10.1002/1873-3468.12284
87. Blackmore TK, Hellwage J, Sadlon TA, Higgs N, Zipfel PF, Ward HM, et al. Identification of the second heparin-binding domain in human complement factor H. J Immunol (1998) 160(7):3342–8.
88. Blackmore TK, Sadlon TA, Ward HM, Lublin DM, Gordon DL. Identification of a heparin binding domain in the seventh short consensus repeat of complement factor H. J Immunol (1996) 157(12):5422–7.
89. Hyvarinen S, Meri S, Jokiranta TS. Disturbed sialic acid recognition on endothelial cells and platelets in complement attack causes atypical hemolytic uremic syndrome. Blood (2016) 127(22):2701–10. doi:10.1182/blood-2015-11-680009
90. Jokiranta TS, Cheng ZZ, Seeberger H, Jozsi M, Heinen S, Noris M, et al. Binding of complement factor H to endothelial cells is mediated by the carboxy-terminal glycosaminoglycan binding site. Am J Pathol (2005) 167(4):1173–81. doi:10.1016/S0002-9440(10)61205-9
91. Blaum BS, Hannan JP, Herbert AP, Kavanagh D, Uhrin D, Stehle T. Structural basis for sialic acid-mediated self-recognition by complement factor H. Nat Chem Biol (2015) 11(1):77–82. doi:10.1038/nchembio.1696
92. Clark SJ, Ridge LA, Herbert AP, Hakobyan S, Mulloy B, Lennon R, et al. Tissue-specific host recognition by complement factor H is mediated by differential activities of its glycosaminoglycan-binding regions. J Immunol (2013) 190(5):2049–57. doi:10.4049/jimmunol.1201751
93. Clark SJ, Schmidt CQ, White AM, Hakobyan S, Morgan BP, Bishop PN. Identification of factor H-like protein 1 as the predominant complement regulator in Bruch’s membrane: implications for age-related macular degeneration. J Immunol (2014) 193(10):4962–70. doi:10.4049/jimmunol.1401613
94. Rudnick RB, Chen Q, Stea ED, Hartmann A, Papac-Milicevic N, Person F, et al. FHR5 binds to laminins, uses separate C3b and surface-binding sites, and activates complement on malondialdehyde-acetaldehyde surfaces. J Immunol (2018) 200(7):2280–90. doi:10.4049/jimmunol.1701641
95. Clark SJ, Bishop PN, Day AJ. The proteoglycan glycomatrix: a sugar microenvironment essential for complement regulation. Front Immunol (2013) 4:412. doi:10.3389/fimmu.2013.00412
96. Martin M, Blom AM. Complement in removal of the dead – balancing inflammation. Immunol Rev (2016) 274(1):218–32. doi:10.1111/imr.12462
97. Trouw LA, Bengtsson AA, Gelderman KA, Dahlback B, Sturfelt G, Blom AM. C4b-binding protein and factor H compensate for the loss of membrane-bound complement inhibitors to protect apoptotic cells against excessive complement attack. J Biol Chem (2007) 282(39):28540–8. doi:10.1074/jbc.M704354200
98. Leffler J, Herbert AP, Norstrom E, Schmidt CQ, Barlow PN, Blom AM, et al. Annexin-II, DNA, and histones serve as factor H ligands on the surface of apoptotic cells. J Biol Chem (2010) 285(6):3766–76. doi:10.1074/jbc.M109.045427
99. Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HP, Charbel Issa P, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature (2011) 478(7367):76–81. doi:10.1038/nature10449
100. Gershov D, Kim S, Brot N, Elkon KB. C-Reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. J Exp Med (2000) 192(9):1353–64. doi:10.1084/jem.192.9.1353
101. Bottazzi B, Doni A, Garlanda C, Mantovani A. An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu Rev Immunol (2010) 28:157–83. doi:10.1146/annurev-immunol-030409-101305
102. Biro A, Rovo Z, Papp D, Cervenak L, Varga L, Fust G, et al. Studies on the interactions between C-reactive protein and complement proteins. Immunology (2007) 121(1):40–50. doi:10.1111/j.1365-2567.2007.02535.x
103. Braunschweig A, Jozsi M. Human pentraxin 3 binds to the complement regulator c4b-binding protein. PLoS One (2011) 6(8):e23991. doi:10.1371/journal.pone.0023991
104. Deban L, Jaillon S, Garlanda C, Bottazzi B, Mantovani A. Pentraxins in innate immunity: lessons from PTX3. Cell Tissue Res (2011) 343(1):237–49. doi:10.1007/s00441-010-1018-0
105. Ma YJ, Doni A, Skjoedt MO, Honore C, Arendrup M, Mantovani A, et al. Heterocomplexes of mannose-binding lectin and the pentraxins PTX3 or serum amyloid P component trigger cross-activation of the complement system. J Biol Chem (2011) 286(5):3405–17. doi:10.1074/jbc.M110.190637
106. Nauta AJ, Bottazzi B, Mantovani A, Salvatori G, Kishore U, Schwaeble WJ, et al. Biochemical and functional characterization of the interaction between pentraxin 3 and C1q. Eur J Immunol (2003) 33(2):465–73. doi:10.1002/immu.200310022
107. Sjoberg AP, Trouw LA, McGrath FD, Hack CE, Blom AM. Regulation of complement activation by C-reactive protein: targeting of the inhibitory activity of C4b-binding protein. J Immunol (2006) 176(12):7612–20. doi:10.4049/jimmunol.176.12.7612
108. Hakobyan S, Harris CL, van den Berg CW, Fernandez-Alonso MC, de Jorge EG, de Cordoba SR, et al. Complement factor H binds to denatured rather than to native pentameric C-reactive protein. J Biol Chem (2008) 283(45):30451–60. doi:10.1074/jbc.M803648200
109. Mihlan M, Stippa S, Jozsi M, Zipfel PF. Monomeric CRP contributes to complement control in fluid phase and on cellular surfaces and increases phagocytosis by recruiting factor H. Cell Death Differ (2009) 16(12):1630–40. doi:10.1038/cdd.2009.103
110. Okemefuna AI, Nan R, Miller A, Gor J, Perkins SJ. Complement factor H binds at two independent sites to C-reactive protein in acute phase concentrations. J Biol Chem (2010) 285(2):1053–65. doi:10.1074/jbc.M109.044529
111. Hebecker M, Okemefuna AI, Perkins SJ, Mihlan M, Huber-Lang M, Jozsi M. Molecular basis of C-reactive protein binding and modulation of complement activation by factor H-related protein 4. Mol Immunol (2010) 47(6):1347–55. doi:10.1016/j.molimm.2009.12.005
112. Mihlan M, Hebecker M, Dahse HM, Halbich S, Huber-Lang M, Dahse R, et al. Human complement factor H-related protein 4 binds and recruits native pentameric C-reactive protein to necrotic cells. Mol Immunol (2009) 46(3):335–44. doi:10.1016/j.molimm.2008.10.029
113. Inforzato A, Baldock C, Jowitt TA, Holmes DF, Lindstedt R, Marcellini M, et al. The angiogenic inhibitor long pentraxin PTX3 forms an asymmetric octamer with two binding sites for FGF2. J Biol Chem (2010) 285(23):17681–92. doi:10.1074/jbc.M109.085639
114. Hyvarinen S, Uchida K, Varjosalo M, Jokela R, Jokiranta TS. Recognition of malondialdehyde-modified proteins by the C terminus of complement factor H is mediated via the polyanion binding site and impaired by mutations found in atypical hemolytic uremic syndrome. J Biol Chem (2014) 289(7):4295–306. doi:10.1074/jbc.M113.527416
115. Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med (2009) 361(4):345–57. doi:10.1056/NEJMoa0810739
116. Heurich M, Preston RJ, O’Donnell VB, Morgan BP, Collins PW. Thrombomodulin enhances complement regulation through strong affinity interactions with factor H and C3b-Factor H complex. Thromb Res (2016) 145:84–92. doi:10.1016/j.thromres.2016.07.017
117. Tateishi K, Imaoka M, Matsushita M. Dual modulating functions of thrombomodulin in the alternative complement pathway. Biosci Trends (2016) 10(3):231–4. doi:10.5582/bst.2016.01052
118. Feng S, Liang X, Cruz MA, Vu H, Zhou Z, Pemmaraju N, et al. The interaction between factor H and Von Willebrand factor. PLoS One (2013) 8(8):e73715. doi:10.1371/journal.pone.0073715
119. Nolasco L, Nolasco J, Feng S, Afshar-Kharghan V, Moake J. Human complement factor H is a reductase for large soluble von Willebrand factor multimers – brief report. Arterioscler Thromb Vasc Biol (2013) 33(11):2524–8. doi:10.1161ATVBAHA.113.302280
120. Rayes J, Roumenina LT, Dimitrov JD, Repesse Y, Ing M, Christophe O, et al. The interaction between factor H and VWF increases factor H cofactor activity and regulates VWF prothrombotic status. Blood (2014) 123(1):121–5. doi:10.1182/blood-2013-04-495853
121. Frimat M, Tabarin F, Dimitrov JD, Poitou C, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, et al. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood (2013) 122(2):282–92. doi:10.1182/blood-2013-03-489245
122. Haapasalo K, van Kessel K, Nissila E, Metso J, Johansson T, Miettinen S, et al. Complement factor H binds to human serum apolipoprotein E and mediates complement regulation on high density lipoprotein particles. J Biol Chem (2015) 290(48):28977–87. doi:10.1074/jbc.M115.669226
123. Chen SF, Wang FM, Li ZY, Yu F, Chen M, Zhao MH. Myeloperoxidase influences the complement regulatory activity of complement factor H. Rheumatology (Oxford) (2018). doi:10.1093/rheumatology/kex529
124. Jozsi M, Schneider AE, Karpati E, Sandor N. Complement factor H family proteins in their non-canonical role as modulators of cellular functions. Semin Cell Dev Biol (2018). doi:10.1016/j.semcdb.2017.12.018
125. DiScipio RG, Daffern PJ, Schraufstatter IU, Sriramarao P. Human polymorphonuclear leukocytes adhere to complement factor H through an interaction that involves alphaMbeta2 (CD11b/CD18). J Immunol (1998) 160(8):4057–66.
126. Svoboda E, Schneider AE, Sandor N, Lermann U, Staib P, Kremlitzka M, et al. Secreted aspartic protease 2 of Candida albicans inactivates factor H and the macrophage factor H-receptors CR3 (CD11b/CD18) and CR4 (CD11c/CD18). Immunol Lett (2015) 168(1):13–21. doi:10.1016/j.imlet.2015.08.009
127. Schneider AE, Sandor N, Karpati E, Jozsi M. Complement factor H modulates the activation of human neutrophil granulocytes and the generation of neutrophil extracellular traps. Mol Immunol (2016) 72:37–48. doi:10.1016/j.molimm.2016.02.011
128. Agarwal V, Asmat TM, Luo S, Jensch I, Zipfel PF, Hammerschmidt S. Complement regulator Factor H mediates a two-step uptake of Streptococcus pneumoniae by human cells. J Biol Chem (2010) 285(30):23486–95. doi:10.1074/jbc.M110.142703
129. Losse J, Zipfel PF, Jozsi M. Factor H and factor H-related protein 1 bind to human neutrophils via complement receptor 3, mediate attachment to Candida albicans, and enhance neutrophil antimicrobial activity. J Immunol (2010) 184(2):912–21. doi:10.4049/jimmunol.0901702
130. Hellwage J, Kuhn S, Zipfel PF. The human complement regulatory factor-H-like protein 1, which represents a truncated form of factor H, displays cell-attachment activity. Biochem J (1997) 326(Pt 2):321–7. doi:10.1042/bj3260321
131. Iferroudjene D, Schouft MT, Lemercier C, Gilbert D, Fontaine M. Evidence for an active hydrophobic form of factor H that is able to induce secretion of interleukin 1-beta or by human monocytes. Eur J Immunol (1991) 21(4):967–72. doi:10.1002/eji.1830210416
132. Nabil K, Rihn B, Jaurand MC, Vignaud JM, Ripoche J, Martinet Y, et al. Identification of human complement factor H as a chemotactic protein for monocytes. Biochem J (1997) 326(Pt 2):377–83. doi:10.1042/bj3260377
133. Ohtsuka H, Imamura T, Matsushita M, Tanase S, Okada H, Ogawa M, et al. Thrombin generates monocyte chemotactic activity from complement factor H. Immunology (1993) 80(1):140–5.
134. Schopf RE, Hammann KP, Scheiner O, Lemmel EM, Dierich MP. Activation of human monocytes by both human beta 1H and C3b. Immunology (1982) 46(2):307–12.
135. Olivar R, Luque A, Cardenas-Brito S, Naranjo-Gomez M, Blom AM, Borras FE, et al. The complement inhibitor factor H generates an anti-inflammatory and tolerogenic state in monocyte-derived dendritic cells. J Immunol (2016) 196(10):4274–90. doi:10.4049/jimmunol.1500455
136. Calippe B, Augustin S, Beguier F, Charles-Messance H, Poupel L, Conart JB, et al. Complement factor H inhibits CD47-mediated resolution of inflammation. Immunity (2017) 46(2):261–72. doi:10.1016/j.immuni.2017.01.006
137. Erdei A, Sim RB. Complement factor H-binding protein of Raji cells and tonsil B lymphocytes. Biochem J (1987) 246(1):149–56. doi:10.1042/bj2460149
138. Hammann KP, Raile A, Schmitt M, Mussel HH, Peters H, Scheiner O, et al. beta 1H stimulates mouse-spleen B lymphocytes as demonstrated by increased thymidine incorporation and formation of B cell blasts. Immunobiology (1981) 160(3–4):289–301. doi:10.1016/S0171-2985(81)80055-1
139. Lambris JD, Dobson NJ, Ross GD. Release of endogenous C3b inactivator from lymphocytes in response to triggering membrane receptors for beta 1H globulin. J Exp Med (1980) 152(6):1625–44. doi:10.1084/jem.152.6.1625
140. Tsokos GC, Inghirami G, Tsoukas CD, Balow JE, Lambris JD. Regulation of immunoglobulin secretion by factor H of human complement. Immunology (1985) 55(3):419–26.
141. Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Fremeaux-Bacchi V, Kavanagh D, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a "kidney disease: improving global outcomes" (KDIGO) Controversies Conference. Kidney Int (2017) 91(3):539–51. doi:10.1016/j.kint.2016.10.005
142. Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, et al. Atypical aHUS: state of the art. Mol Immunol (2015) 67(1):31–42. doi:10.1016/j.molimm.2015.03.246
143. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med (2009) 361(17):1676–87. doi:10.1056/NEJMra0902814
144. Maillard N, Wyatt RJ, Julian BA, Kiryluk K, Gharavi A, Fremeaux-Bacchi V, et al. Current understanding of the role of complement in IgA nephropathy. J Am Soc Nephrol (2015) 26(7):1503–12. doi:10.1681/ASN.2014101000
145. Suzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol (2011) 22(10):1795–803. doi:10.1681/ASN.2011050464
146. Geerlings MJ, de Jong EK, den Hollander AI. The complement system in age-related macular degeneration: a review of rare genetic variants and implications for personalized treatment. Mol Immunol (2017) 84:65–76. doi:10.1016/j.molimm.2016.11.016
147. Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A (2005) 102(20):7227–32. doi:10.1073/pnas.0501536102
148. Hageman GS, Hancox LS, Taiber AJ, Gehrs KM, Anderson DH, Johnson LV, et al. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications. Ann Med (2006) 38(8):592–604. doi:10.1080/07853890601097030
149. Pickering MC, de Jorge EG, Martinez-Barricarte R, Recalde S, Garcia-Layana A, Rose KL, et al. Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med (2007) 204(6):1249–56. doi:10.1084/jem.20070301
150. Tortajada A, Montes T, Martinez-Barricarte R, Morgan BP, Harris CL, de Cordoba SR. The disease-protective complement factor H allotypic variant Ile62 shows increased binding affinity for C3b and enhanced cofactor activity. Hum Mol Genet (2009) 18(18):3452–61. doi:10.1093/hmg/ddp289
151. Zareparsi S, Branham KE, Li M, Shah S, Klein RJ, Ott J, et al. Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age-related macular degeneration. Am J Hum Genet (2005) 77(1):149–53. doi:10.1086/431426
152. Clark SJ, Perveen R, Hakobyan S, Morgan BP, Sim RB, Bishop PN, et al. Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch’s membrane in human retina. J Biol Chem (2010) 285(39):30192–202. doi:10.1074/jbc.M110.103986
153. Li M, Atmaca-Sonmez P, Othman M, Branham KE, Khanna R, Wade MS, et al. CFH haplotypes without the Y402H coding variant show strong association with susceptibility to age-related macular degeneration. Nat Genet (2006) 38(9):1049–54. doi:10.1038/ng1871
154. Caprioli J, Bettinaglio P, Zipfel PF, Amadei B, Daina E, Gamba S, et al. The molecular basis of familial hemolytic uremic syndrome: mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J Am Soc Nephrol (2001) 12(2):297–307.
155. Sanchez-Corral P, Perez-Caballero D, Huarte O, Simckes AM, Goicoechea E, Lopez-Trascasa M, et al. Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am J Hum Genet (2002) 71(6):1285–95. doi:10.1086/344515
156. Servais A, Noel LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int (2012) 82(4):454–64. doi:10.1038/ki.2012.63
157. Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, Bragg-Gresham JL, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet (2016) 48(2):134–43. doi:10.1038/ng.3448
158. Raychaudhuri S, Iartchouk O, Chin K, Tan PL, Tai AK, Ripke S, et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat Genet (2011) 43(12):1232–6. doi:10.1038/ng.976
159. Recalde S, Tortajada A, Subias M, Anter J, Blasco M, Maranta R, et al. Molecular basis of factor H R1210C association with ocular and renal diseases. J Am Soc Nephrol (2016) 27(5):1305–11. doi:10.1681/ASN.2015050580
160. Martinez-Barricarte R, Recalde S, Fernandez-Robredo P, Millan I, Olavarrieta L, Vinuela A, et al. Relevance of complement factor H-related 1 (CFHR1) genotypes in age-related macular degeneration. Invest Ophthalmol Vis Sci (2012) 53(3):1087–94. doi:10.1167/iovs.11-8709
161. Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet (2006) 38(10):1173–7. doi:10.1038/ng1890
162. Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet (2011) 43(4):321–7. doi:10.1038/ng.787
163. Zipfel PF, Edey M, Heinen S, Jozsi M, Richter H, Misselwitz J, et al. Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet (2007) 3(3):e41. doi:10.1371/journal.pgen.0030041
164. Moore I, Strain L, Pappworth I, Kavanagh D, Barlow PN, Herbert AP, et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood (2010) 115(2):379–87. doi:10.1182/blood-2009-05-221549
165. Kubista KE, Tosakulwong N, Wu Y, Ryu E, Roeder JL, Hecker LA, et al. Copy number variation in the complement factor H-related genes and age-related macular degeneration. Mol Vis (2011) 17:2080–92.
166. Schmid-Kubista KE, Tosakulwong N, Wu Y, Ryu E, Hecker LA, Baratz KH, et al. Contribution of copy number variation in the regulation of complement activation locus to development of age-related macular degeneration. Invest Ophthalmol Vis Sci (2009) 50(11):5070–9. doi:10.1167/iovs.09-3975
167. Zhu L, Zhai YL, Wang FM, Hou P, Lv JC, Xu DM, et al. Variants in complement factor H and complement factor H-related protein genes, CFHR3 and CFHR1, affect complement activation in IgA nephropathy. J Am Soc Nephrol (2015) 26(5):1195–204. doi:10.1681/ASN.2014010096
168. Heinen S, Sanchez-Corral P, Jackson MS, Strain L, Goodship JA, Kemp EJ, et al. De novo gene conversion in the RCA gene cluster (1q32) causes mutations in complement factor H associated with atypical hemolytic uremic syndrome. Hum Mutat (2006) 27(3):292–3. doi:10.1002/humu.9408
169. Venables JP, Strain L, Routledge D, Bourn D, Powell HM, Warwicker P, et al. Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PLoS Med (2006) 3(10):e431. doi:10.1371/journal.pmed.0030431
170. Maga TK, Meyer NC, Belsha C, Nishimura CJ, Zhang Y, Smith RJ. A novel deletion in the RCA gene cluster causes atypical hemolytic uremic syndrome. Nephrol Dial Transplant (2011) 26(2):739–41. doi:10.1093/ndt/gfq658
171. Eyler SJ, Meyer NC, Zhang Y, Xiao X, Nester CM, Smith RJ. A novel hybrid CFHR1/CFH gene causes atypical hemolytic uremic syndrome. Pediatr Nephrol (2013) 28(11):2221–5. doi:10.1007/s00467-013-2560-2
172. Valoti E, Alberti M, Tortajada A, Garcia-Fernandez J, Gastoldi S, Besso L, et al. A novel atypical hemolytic uremic syndrome-associated hybrid CFHR1/CFH gene encoding a fusion protein that antagonizes factor H-dependent complement regulation. J Am Soc Nephrol (2015) 26(1):209–19. doi:10.1681/ASN.2013121339
173. Goicoechea de Jorge E, Tortajada A, Garcia SP, Gastoldi S, Merinero HM, Garcia-Fernandez J, et al. Factor H competitor generated by gene conversion events associates with atypical hemolytic uremic syndrome. J Am Soc Nephrol (2018) 29(1):240–9. doi:10.1681/ASN.2017050518
174. Francis NJ, McNicholas B, Awan A, Waldron M, Reddan D, Sadlier D, et al. A novel hybrid CFH/CFHR3 gene generated by a microhomology-mediated deletion in familial atypical hemolytic uremic syndrome. Blood (2012) 119(2):591–601. doi:10.1182/blood-2011-03-339903
175. Challis RC, Araujo GS, Wong EK, Anderson HE, Awan A, Dorman AM, et al. A De Novo deletion in the regulators of complement activation cluster producing a hybrid complement factor H/complement factor H-related 3 gene in atypical hemolytic uremic syndrome. J Am Soc Nephrol (2016) 27(6):1617–24. doi:10.1681/ASN.2015010100
176. Gale DP, de Jorge EG, Cook HT, Martinez-Barricarte R, Hadjisavvas A, McLean AG, et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet (2010) 376(9743):794–801. doi:10.1016/S0140-6736(10)60670-8
177. Athanasiou Y, Voskarides K, Gale DP, Damianou L, Patsias C, Zavros M, et al. Familial C3 glomerulopathy associated with CFHR5 mutations: clinical characteristics of 91 patients in 16 pedigrees. Clin J Am Soc Nephrol (2011) 6(6):1436–46. doi:10.2215/CJN.09541010
178. Vernon KA, Gale DP, de Jorge EG, McLean AG, Galliford J, Pierides A, et al. Recurrence of complement factor H-related protein 5 nephropathy in a renal transplant. Am J Transplant (2011) 11(1):152–5. doi:10.1111/j.1600-6143.2010.03333.x
179. Medjeral-Thomas N, Malik TH, Patel MP, Toth T, Cook HT, Tomson C, et al. A novel CFHR5 fusion protein causes C3 glomerulopathy in a family without Cypriot ancestry. Kidney Int (2014) 85(4):933–7. doi:10.1038/ki.2013.348
180. Tortajada A, Gutierrez-Tenorio J, Saiz Gonzalez A, Marcen Letosa R, Bouthelier A, Sanchez-Corral P, et al. Novel duplication of the FHRs dimerization domain associated with C3G. Mol Immunol (2017) 89:181. doi:10.1016/j.molimm.2017.06.169
181. Malik TH, Lavin PJ, Goicoechea de Jorge E, Vernon KA, Rose KL, Patel MP, et al. A hybrid CFHR3-1 gene causes familial C3 glomerulopathy. J Am Soc Nephrol (2012) 23(7):1155–60. doi:10.1681/ASN.2012020166
182. Wong L, Moran S, Lavin PJ, Dorman AM, Conlon PJ. Kidney transplant outcomes in familial C3 glomerulopathy. Clin Kidney J (2016) 9(3):403–7. doi:10.1093/ckj/sfw020
183. Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H, et al. Anti-factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol (2005) 16(2):555–63. doi:10.1681/ASN.2004050380
184. Dragon-Durey MA, Blanc C, Garnier A, Hofer J, Sethi SK, Zimmerhackl LB. Anti-factor H autoantibody-associated hemolytic uremic syndrome: review of literature of the autoimmune form of HUS. Semin Thromb Hemost (2010) 36(6):633–40. doi:10.1055/s-0030-1262885
185. Jozsi M, Oppermann M, Lambris JD, Zipfel PF. The C-terminus of complement factor H is essential for host cell protection. Mol Immunol (2007) 44(10):2697–706. doi:10.1016/j.molimm.2006.12.001
186. Strobel S, Hoyer PF, Mache CJ, Sulyok E, Liu WS, Richter H, et al. Functional analyses indicate a pathogenic role of factor H autoantibodies in atypical haemolytic uraemic syndrome. Nephrol Dial Transplant (2010) 25(1):136–44. doi:10.1093/ndt/gfp388
187. Blanc C, Roumenina LT, Ashraf Y, Hyvarinen S, Sethi SK, Ranchin B, et al. Overall neutralization of complement factor H by autoantibodies in the acute phase of the autoimmune form of atypical hemolytic uremic syndrome. J Immunol (2012) 189(7):3528–37. doi:10.4049/jimmunol.1200679
188. Dragon-Durey MA, Blanc C, Marliot F, Loirat C, Blouin J, Sautes-Fridman C, et al. The high frequency of complement factor H related CFHR1 gene deletion is restricted to specific subgroups of patients with atypical haemolytic uraemic syndrome. J Med Genet (2009) 46(7):447–50. doi:10.1136/jmg.2008.064766
189. Jozsi M, Licht C, Strobel S, Zipfel SL, Richter H, Heinen S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood (2008) 111(3):1512–4. doi:10.1182/blood-2007-09-109876
190. Nozal P, Garrido S, Alba-Dominguez M, Espinosa L, Pena A, Cordoba SR, et al. An ELISA assay with two monoclonal antibodies allows the estimation of free factor H and identifies patients with acquired deficiency of this complement regulator. Mol Immunol (2014) 58(2):194–200. doi:10.1016/j.molimm.2013.11.021
191. Bhattacharjee A, Reuter S, Trojnar E, Kolodziejczyk R, Seeberger H, Hyvarinen S, et al. The major autoantibody epitope on factor H in atypical hemolytic uremic syndrome is structurally different from its homologous site in factor H-related protein 1, supporting a novel model for induction of autoimmunity in this disease. J Biol Chem (2015) 290(15):9500–10. doi:10.1074/jbc.M114.630871
192. Nozal P, Bernabeu-Herrero ME, Uzonyi B, Szilagyi A, Hyvarinen S, Prohaszka Z, et al. Heterogeneity but individual constancy of epitopes, isotypes and avidity of factor H autoantibodies in atypical hemolytic uremic syndrome. Mol Immunol (2016) 70:47–55. doi:10.1016/j.molimm.2015.12.005
193. Blanc C, Togarsimalemath SK, Chauvet S, Le Quintrec M, Moulin B, Buchler M, et al. Anti-factor H autoantibodies in C3 glomerulopathies and in atypical hemolytic uremic syndrome: one target, two diseases. J Immunol (2015) 194(11):5129–38. doi:10.4049/jimmunol.1402770
194. Goodship TH, Pappworth IY, Toth T, Denton M, Houlberg K, McCormick F, et al. Factor H autoantibodies in membranoproliferative glomerulonephritis. Mol Immunol (2012) 52(3–4):200–6. doi:10.1016/j.molimm.2012.05.009
195. Jokiranta TS, Solomon A, Pangburn MK, Zipfel PF, Meri S. Nephritogenic lambda light chain dimer: a unique human miniautoantibody against complement factor H. J Immunol (1999) 163(8):4590–6.
196. Meri S, Koistinen V, Miettinen A, Tornroth T, Seppala IJ. Activation of the alternative pathway of complement by monoclonal lambda light chains in membranoproliferative glomerulonephritis. J Exp Med (1992) 175(4):939–50. doi:10.1084/jem.175.4.939 Epub 1992/04/01.,
197. Nozal P, Strobel S, Ibernon M, López D, Sánchez-Corral P, Rodriguez de Cordoba S, et al. Anti-factor H antibody affecting factor H cofactor activity in a patient with dense deposit disease. Clin Kidney J (2012) 5:133–6. doi:10.1093/ckj/sfs002
Keywords: age-related macular degeneration, atypical hemolytic uremic syndrome, C3 glomerulopathy, complement activation, complement de-regulation, factor H, factor H-related protein, opsonization
Citation: Sánchez-Corral P, Pouw RB, López-Trascasa M and Józsi M (2018) Self-Damage Caused by Dysregulation of the Complement Alternative Pathway: Relevance of the Factor H Protein Family. Front. Immunol. 9:1607. doi: 10.3389/fimmu.2018.01607
Received: 31 May 2018; Accepted: 28 June 2018;
Published: 12 July 2018
Edited by:
Maciej Cedzynski, Institute for Medical Biology (PAN), PolandReviewed by:
Cees Van Kooten, Leiden University, NetherlandsCopyright: © 2018 Sánchez-Corral, Pouw, López-Trascasa and Józsi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mihály Józsi, bWloYWx5LmpvenNpQHR0ay5lbHRlLmh1
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.