
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 11 July 2018
Sec. Inflammation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01555
This article is part of the Research Topic Oxidants and Redox Signaling in Inflammation View all 12 articles
 Julien Cachat1
Julien Cachat1 Christine Deffert2
Christine Deffert2 Marco Alessandrini1
Marco Alessandrini1 Pascale Roux-Lombard2
Pascale Roux-Lombard2 Audrey Le Gouellec3,4
Audrey Le Gouellec3,4 Marie-José Stasia3,4
Marie-José Stasia3,4 Stéphanie Hugues1Karl-Heinz Krause1*
Stéphanie Hugues1Karl-Heinz Krause1*
Chronic granulomatous disease (CGD) is a primary immunodeficiency resulting from loss of function mutations in the reactive oxygen species generating phagocyte NADPH oxidase (NOX2). CGD patients are prone to infection, but also have an increased susceptibility to autoimmune diseases. The aim of this study was to investigate the role of NOX2 in the regulation of specific immunity. In both CGD patients and NOX2-deficient mice, we observed an alteration in the basal proportions of IgG subtypes. Upon immunization with curdlan—a dectin 1 agonist—NOX2-deficient mice showed increased production of IgG2c compared to controls, and restimulation of lymph node-derived cells led to increased production of IFNγ, but not IL-5, indicative hallmark of an enhanced Th1 response. T cell activation was increased in NOX2-deficient mice and a similar trend was observed in vitro when T cells were co-cultured with NOX2-deficient bone marrow-derived cells. In contrast, no difference in T cell activation was observed when NOX2-deficient T cells were co-cultured with wild-type BMDC. Following stimulation of NOX2-deficient dendritic cells (DCs), no difference in costimulatory molecules was observed, while there was an increase in the release of Th1-driving cytokines. In summary, both CGD patients and CGD mice have an altered IgG subtype distribution, which is associated with an increased IFNγ production. Thus, NOX2 within DCs appears to be an important regulator at the interface of innate and specific immunity, especially after activation of the dectin 1 pathway, limiting immune activation and the development of autoimmunity.
Chronic granulomatous disease (CGD) is a primary immunodeficiency disorder leading to life-threatening bacterial and fungal infections. The genetic cause of CGD is the loss of function mutations in genes coding for the phagocyte NADPH oxidase NOX2. NOX2 is a multi-subunit enzyme generating reactive oxygen species (ROS) and is an essential component of the neutrophil oxidative burst leading to microbial killing (1). While the role of NOX2-derived ROS in innate neutrophil activity is well established, NOX2 has other important functions in immune system regulation. Early reports of CGD had described an increase in immunoglobulin levels (2), but this feat has since received little attention. B cells express a functional NOX2 (3). NOX2 may have a role in antibody production, but only a few studies reported an impact of NOX2 deficiency on B cell biology and immunoglobulin production. CGD mice show increased immunoglobulin responses to injection of collagen (4) and to UV-inactivated bacteria (5). One study in NOX2-deficient mice observed an enhanced antibody production in response to T cell-independent antigens (6). To date, no study has carefully analyzed the effect of NOX2 on IgG subclass production.
In addition, recent observations argue in favor of an excessive inflammatory response and autoimmunity in CGD (7). CGD patient show increased prevalence of autoimmune diseases, including systemic lupus erythematosus, juvenile rheumatoid arthritis, and idiopathic thrombocytopenic purpura (8–11). Autoantibodies play a role in the pathogenesis of these immune diseases (12). The sera of patients with CGD also show high levels of anti-Saccharomyces cerevisiae, anti-OmpC, or anti-CBir1 antibodies, which are associated with Crohn disease (13). In rodents, a seminal paper by the team of Rikard Holmdahl demonstrated that a loss of function polymorphism in the Ncf1 gene—which codes for the p47phox subunit of NOX2—is a main driver of experimental rheumatoid arthritis (14, 15). Since then, observation converges toward a role of NOX2-derived ROS in T cell activation. Indeed adoptive T cell transfer from arthritic NOX2-deficient mice is sufficient to induce the disease in healthy wild-type (WT) mice (14). Thus, NOX2-derived ROS limit T cell activation, although the underlying mechanisms are still incompletely understood. NOX2-derived ROS, generated either by T cells themselves or antigen-presenting cells (APCs), might directly inhibit T cells, possibly through surface oxidation (16), ROS inhibition of lymphocyte ion channels (17), or other redox-sensitive signaling elements (18). Alternatively, NOX2-derived ROS might play a role in APCs and indirectly affect T cell function. For example, a recent study reported altered antigen processing, resulting in a different epitope repertoire in NOX2-deficient dendritic cells (DCs) (19), while another study has shown that oxidative modification of presented autoantigens enhances T cell response (20). NOX2-derived ROS appear to fundamentally control specific immune responses as mice deficient in Ncf1 also exhibit an increased sensitivity to autoimmune encephalitis (EAE) (21) and NOX2-deficient mice an increased sensitivity to lupus erythematous (22). Interestingly, a recent human genetic study also found that a missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases (23). Altogether, these studies suggest that the link between NOX2 and autoimmune disease is not limited to CGD patients, but also exists for less severe polymorphisms of the NOX2 system. Nevertheless, although patients with NOX2 deficiency present with increased risk to infection due to the impaired neutrophil oxidative burst, autoimmune features are not always visible and probably require specific additional stimuli. We have previously shown that dectin-1 activation strongly induces a CGD-associated hyperinflammation. Injection of curdlan, a β-glucan, which is a potent activator of dectin-1, results in a massive subcutaneous swelling and high levels of IL-6 and IFNγ in NOX2-deficient mice, while lipopolysaccharide was inactive (24). Altogether, the current literature indicates clinical and experimental links between NOX2-dependent ROS generation, production of immunoglobulins, specific hyperinflammatory states, and the development of autoimmune diseases.
In the present study, we measured IgG subclasses in the sera of NOX2-deficient mice and in CGD patients and detected altered IgG subtype production in NOX2 deficiency. We also addressed experimentally the activation of T cells following immunization with an ovalbumin-derived peptide (OVA323–339) and the impact of specific adjuvants in vivo and in BMDC and T cell co-culture experiments. Our results point toward a key role of dectin-1-dependent NOX2 in DCs in limiting T cell activation, IFNγ release, and the production of Th1-driving cytokines. This suggests that NOX2-deficient DCs release increased amount of Th1-driving cytokines, leading to the release of an increased amount of IFNγ, which in turn may drive a higher IgG2c generation by B cells.
C57Bl/6j (WT), B6.129S-Cybbtm1Din/J (NOX2KO), and B6.Cg-Tg(TcraTcrb)425Cbn/J (OTII) were purchased from The Jackson Laboratory and bred at the Animal Production facilities of the University of Geneva. Double OTII/NOX2KO-mutant mice were obtained by breeding B6.129S-Cybbtm1Din/J mice with B6.Cg-Tg(TcraTcrb)425Cbn/J mice. For the experiments, mice of age 8–12 months were used. The protocol was approved by the office cantonal vétérinaire du Canton de Genève, Switzerland (authorization no. 23624).
Patients were diagnosed as having CGD on the basis of their clinical symptoms and the inability of their phagocytes to generate ROS detectable by the dihydrorhodamine (DHR) flow cytometric test and the nitroblue tetrazolium dye reduction slide test. Blood samples were obtained from the CGD patients with appropriate institutional informed consent. Peripheral blood samples taken from healthy donors were obtained from the "Etablissement Français du sang" at the Grenoble University Hospital, France after their informed consent.
Cells were suspended at 106/ml in FACS buffer (PBS with 0.5% bovine serum albumin (BSA) and 5 mM ethylenediaminetetraacetate (EDTA)). Fc receptors were blocked by a 10 min incubation at 4°C with the mouse BD Fc block (BD Biosciences, USA) at a dilution of 1:100. The cells were then washed with FACS buffer and centrifuged at 5,000 rpm for 5 min. Cells were then resuspended in FACS buffer with the antibody of interest and incubated for 15 min at 4°C. After incubation, the cells were washed with FACS buffer, centrifuged at 5,000 rpm for 5 min and resuspended in FACS buffer for flow cytometry analysis.
Wild-type and NOX2KO mice were immunized by subcutaneous injection into the outer ear, using 50 µl of 1 µg ovalbumin protein and curdlan (Sigma-Aldrich, USA) (100 µg/ml) or Alum (ThermoFisher Scientific, USA) (50%) as adjuvant, which was diluted in PBS. After 10 and 14 days, blood was collected from the caudal vein. Serum was obtained by coagulation and centrifugation for 2 min at 2,500 rpm, and stocked at −20°C until ELISA was performed. Serum from non-immunized mice was also collected to determine basal levels of the IgG subgroup.
Ear thickness was measured using a caliper before immunization (basal ear thickness), at day 10 and 14 after immunization. The change in ear thickness (Δ ear thickness) was obtained by subtracting the basal ear thickness to the values obtained on days 10 and 14.
High binding ELISA 96-well microplate (Corning®, Sigma-Aldrich, USA) were coated overnight at 4°C with either 10 µg/ml of ovalbumin (Sigma-Aldrich, USA) diluted in PBS to measure ovalbumin-specific antibody, or with purified goat anti-mouse total IgG (Biolegend, USA, dilution 1:100,000) to measure basal level of IgG subgroup. Plates were washed three times with PBS containing 0.05% tween-20 to remove unbound ovalbumin. A volume of 100 µl of PBS-BSA 1% was used for blocking for 1 h at room temperature. Fifty microliters per well of diluted serum sample were added to corresponding wells in duplicate. A pool of serum was used to obtain a standard curve by serial dilution. Sample dilutions were chosen to obtain a signal within the linear phase of the standard curve. The plates were incubated at room temperature for 1 h and washed three times. To measure ovalbumin-specific antibody, anti-mouse IgG2c, anti-mouse IgG2b, and anti-mouse IgG3 (Biolegend, USA) or anti-mouse IgG1 (ThermoFisher Scientific, USA) coupled to peroxidase was added to wells at the dilution of 1:1,000 for anti-mouse IgG2c and IgG1 and 1:500 for anti-mouse IgG2b and IgG3. Those secondary antibodies were incubated for 1 h at room temperature. For the measure of basal level of IgG subgroups, anti-mouse IgG2c (Biolegend, USA) or anti-mouse IgG1 (ThermoFisher Scientific, USA) or anti-mouse IgG3 (ThermoFisher Scientific, USA) or anti-mouse IgG2b (ThermoFisher Scientific, USA) coupled to peroxidase was added to wells at a dilution of 1:1,000 and incubated for 1 h at room temperature. Plates were washed three times and 50 µl of streptavidin coupled with horse-peroxidase was added to the plates, and incubated at room temperature for 20 min. After three washes, the signal was revealed by adding 50 µl of tetramethylbenzidine (TMB) and the plates were incubated at room temperature for 15 min in the dark. The reaction was stopped by adding 25 µl of H2SO4 2N. Optical density at 492 nm was measured using the fluoSTAR OPTIMA (BMG Labtech, Germany) plate reader.
Sera from normal (n = 6) and CGD patients (n = 16) were collected and stored at −80°C before testing. Levels of serum immunoglobulin G and A were measured on a Dimension VISTA® system (Siemens, Germany) with IGG Flex® and IGA Flex® reagent cartridges according to the dimension VISTA® Operator’s Guide. Serum IgG subclasses were measured by nephelometric assay with human IgG subclass liquid reagent kits (Siemens, Germany, including N Latex IgG1, N Latex IgG2, N Latex IgG3, and N Latex IgG4) on the automatic protein analyzer BN Prospect-II (Siemens, Germany) according to the manufacturer’s instructions.
Anti-nuclear antibody was determined by indirect immunofluorescence (IFI) on Hep2 slides and ANCA by IFI on ethanol- and formalin-fixed neutrophils (Inova Diagnostics, USA). Briefly, for both types of autoantibodies, the corresponding slides were incubated with serial dilutions of serum in phosphate buffered saline with 10% Tween for 30 min, washed with PBS-Tween, and incubated with fluorescein-isothiocyanate (FITC)-conjugated goat polyclonal anti-human IgG containing Evans blue (Inova Diagnostics, USA). After washing steps, slides were mounted with glycerine mounting medium and cover slips and read using a fluorescence microscope Axioscope 2plus (Carl Zeiss Microscopy GmBh, Germany). Sera were considered positive for autoantibodies when a staining was observed for a dilution equal to or greater than 1/80 for ANA and 1/40 for ANCA.
Immunized mice were sacrificed 14 days after immunization and draining lymph nodes (cervical lymph node) and non-draining lymph nodes (inguinal lymph node) were dissected. Lymph nodes were digested with 1 mg/ml collagenase D (ThermoFisher Scientific, USA) and 10 µg/ml DNAse I (ThermoFisher Scientific, USA) in 2 ml HBSS at 37°C for 40 min. Digestion was stopped by adding 15 ml of a PBS-BSA 1% plus 5 mM EDTA solution and digested lymph nodes were passed through a 70 µm cell strainer. The total number of cells obtained after lymph node digestion and cell subgroup were analyzed by flow cytometry with the following antibodies: anti-CD4 Percp (1:200), anti-CD8 PE (1:100), and anti-IgD fitc (1:100) (ThermoFisher Scientific, USA).
Lymph node cells were plated in a 96-well plate at a concentration of 2 × 106 cells per well and restimulated with 100 µg/ml of ovalbumin. 3 days after restimulation, supernatants were analyzed for the presence of IFNγ and IL5 using commercially available ELISA kits (ThermoFisher Scientific, USA), according to the manufacturer’s instructions.
Hind leg bones of WT and NOX2KO mice were dissected and the bone marrow flushed with RPMI medium, complemented with 1% streptomycin–penicillin and 10% fetal bovine serum, and plated in a petri dish. Bone marrow cells were centrifuged at 5,000 rpm for 5 min and the pellet resuspended in a solution of 155 mM NH4Cl and 12 mM NaHCO3 for 1 min to lyse red blood cells. 20 ng/ml of GM-CSF (Peprotech) was added to the medium to drive differentiation to DCs. Medium and GM-CSF were renewed at day 3 and 6. At day 10, purity of BMDCs was verified by flow cytometry using anti-CD11c APC (1:100) antibody.
Lymph node of OTII or OTII/NOX2 mice were dissected and passed through a 15 µm cell strainer to obtain a single cell suspension of lymph node cells. T cells were isolated using a microbead-based T cell isolation kit (Miltenyi Biotec, Germany), according to the manufacturer’s instruction. Purity of the isolated T cells was verified by flow cytometry using anti-CD4 PE antibody (1:200).
Wild-type and NOX2KO BMDCs were plated in RPMI (completed with 10% fetal bovine serum, 1% streptomycin–penicillin, 50 µM β-mercaptoethanol, and 1 mM sodium pyruvate) in a 96-well plate at a concentration of 104 cells per well. Curdlan at 5 µg/ml was added to the wells for BMDC activation. Finally, increasing concentrations (5–500 nM) of OVA323–339 peptide were added to the culture. After 1 day, BMDCs were harvested and washed twice with the prepared RPMI medium to remove the curdlan and peptide. Then, freshly isolated OTII or OTII/NOX2 T cells were added to the wells with the same medium preparation at a concentration of 105 T cells per well. For the experiment on T cell proliferation, freshly isolated T cells were first labeled with 5 µM carboxyfluorescein succinimidyl ester (CFSE) for 10 min at 37°C and washed three times. T cell activation was assessed by flow cytometry after 16 h of co-culture and after staining with anti-CD4 PercP (1:200) and anti-CD69 PE (1:200) antibody. T cell proliferation was assessed by flow cytometry after 4 days of co-culture and after staining with anti-CD4 PercP. The proliferation index and percent of dividing cells were calculated with FlowJo Software.
Wild-type and NOX2KO mice were immunized with 0.5 µg of OVA(323–339) and 100 µg/ml of curdlan in PBS subcutaneously in the outer ear (50 µl of the solution was injected by ear). 1 day after immunization, freshly isolated OTII or OTII/NOX2KO T cells were labeled with 5 µM CFSE and injected intravenously in immunized mice. For T cell activation, draining lymph nodes were dissected 16 h after T cell adoptive transfer. Lymph nodes were then scratched and lymph node cells were stained with anti-CD4 PercP (1:200) and anti-CD69 PE antibody (1:200) for flow cytometry analysis. For T cell proliferation, lymph node dissection was performed 3 days after immunization and was stained with anti-CD4 PercP antibody. For comparison of T cell activation and proliferation between OTII and OTII/NOX2KO T cells, the experiment were performed similarly except that OTII T cells were labeled with CFSE as described above and OTII/NOX2KO T cells were labeled with 1 µM CellTracker™ Orange Cmtmr ((5-(and-6)-(((4-chloromethyl)benzoyl)amino)tetramethyl-rhodamine)) (ThermoFisher scientific, USA) at 37°C for 15 min, and then washed three times.
Freshly isolated T cells were plated in a 96-well plate at a concentration of 105 T cell per well in the same culture medium as for co-culture experiments. Anti-CD3 (0.5 µg/ml) and anti-CD28 (2 µg/ml) antibody were then added to the culture to activate T cells. At the same time, increasing concentrations of H2O2 from 16 to 150 µM were added. After 16 h of treatment, T cells were stained with anti-CD4 PercP (1:200) antibody and anti-CD69 (1:200) PE antibody, and analyzed via flow cytometry. For T cell survival, activated T cell was stained with propidium iodide.
Bone marrow-derived dendritic cells were plated in a 96-well plate in same medium than for co-culture experiments at a concentration of 105 cells per well. Curdlan at concentration of 50, 25, and 5 µg/ml was added to the culture for BMDCs activation. After 24 h, BMDCs were harvested and supernatants were kept for further analysis at −20°C. BMDCs were stained with anti-CD11c Percp (1:200) or anti-CD11c APC (1:100), anti-DC-Sign APC (1:100), anti-ICOSL PE (1:100), anti-PD-L1 PE (1:100), anti-CD80 PE (1:400), anti-CD86 fitc (1:200), and anti-MHCII PE (1:1000). IL1β, IL10, and IL6 were measured by ELISA kit (eBioscience) in the supernatant, according to the manufacturer’s instructions.
All data were analyzed using GraphPad Prism software. For sample sizes of n ≥ 7, data were first tested for normality using the Shapiro–Wilk test. If normally distributed, the Student’s t-test was used for statistical comparison. For datasets not normally distributed and where sample sizes n ≤ 7, the Mann–Whitney U test was used.
We first analyzed immunoglobulin levels in serum samples from healthy donor and CGD patients. As expected, higher levels of immunoglobulin where found in patients with CGD (Table S1 in Supplementary Material). Six CGD patients had increased IgA serum levels of which four were adults. In contrast, of the six CGD patients that had higher levels of serum IgM, four were below the age of two. For serum IgG, five CGD patients had levels above the normal range, of which three were adults and two children. When investigating the presence of autoantibodies, 6 out of 16 CGD samples (37%) were positive for ANCA, while 3 out of 16 samples (19%) were positive for ANA (Table S1 in Supplementary Material). Thus, we confirmed previous observations that CGD patients are prone to hypergammaglobulinemia and a higher frequency of autoantibodies.
We further characterized different IgG subtypes and compared the relative amounts of IgG1 and IgG2. Since no reference values for the ratios of IgG1/IgG and IgG2/IgG exist, we compared these values for CGD patients relative to a control group of comparable ages. In the control group, IgG1 represented ~70% of total IgG. In CGD patients, these levels were statistically significantly decreased (p = 0.026; Figure 1A). In contrast, IgG2 represented ~30% of total IgG in controls, and were significantly increased to ~40% in CGD patients (p = 0.01; Figure 1B).
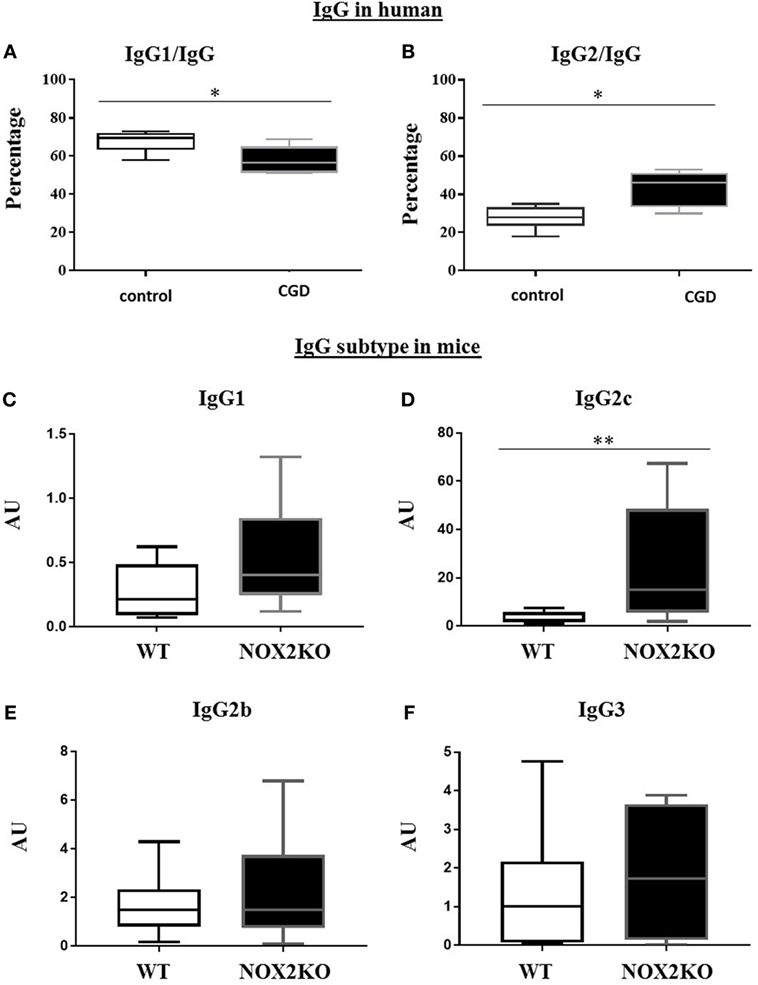
Figure 1. Relative levels of IgG1 and IgG2 in CGD patients and healthy donors and basal levels of immunoglobulin subtypes in wild-type (WT) and NOX2-deficient mice. (A,B) Proportion of serum IgG1 and IgG2 subtypes relative to total IgG. Basal level of IgG1 (C), IgG2c (D), IgG2b (E) and IgG3 (F) in WT and NOX2 deficient mice (NOX2KO). *p < 0.05 and **p < 0.01.
To determine whether the differences in IgG subtypes were also present in NOX2-deficient mice, we measured the basal levels of each IgG subtype (IgG1, IgG2b, IgG2c, and IgG3) in NOX2-deficient and WT mice (Figures 1C–F). Similar to our human data, levels of some, but not all, IgG subtypes were impacted by the presence of NOX2. We did not find significant differences in IgG1, IgG2b, and IgG3 levels between WT and NOX2-deficient mice (Figures 1C,E,F). However, levels of IgG2c were significantly increased in NOX2-deficient mice (p < 0.008; Figure 1D). Thus, an alteration of IgG subtypes was observed in human CGD samples and in NOX2-deficient mice.
In order to examine how NOX2 deficiency affects the production of antibodies, we compared the levels of antigen-specific IgG in response to immunization of control and NOX2-deficient mice. As a model antigen, we used ovalbumin, and either alum or curdlan as adjuvants. The two different adjuvants were chosen for the following reasons. Alum is a standard adjuvant used in most routine experiments, and it mostly induces IgG1 response. In contrast, curdlan is an adjuvant that privileges IgG2c responses. Immunization was performed by injection in the ear skin. Different investigations were performed 10 and/or 14 days after immunization, including ear thickness, serum IgG levels, and lymph node analysis (cellularity and cytokines). Anti-ovalbumin IgG subtypes were examined. Prior to immunization, no anti-ovalbumin IgG was detected (data not shown). After immunization, the level of anti-ovalbumin IgG3 was still below detection level (data not shown). In control mice, immunization with either alum or curdlan as adjuvant led to the generation of modest, but clearly detectable, amounts of anti-ovalbumin IgG1. In immunized NOX2-deficient mice, the IgG1 levels were broadly increased in comparison to control mice (Figures 2A,D). The situation of anti-ovalbumin IgG2b was slightly more complex. With alum as adjuvant, the level of anti-ovalbumin IgG2b remained low for all samples (Figure 2B). With curdlan as adjuvant, even though most of the samples had a low level of IgG2b, one sample in WT mice and two samples in NOX2KO mice exerted levels at least 10 times higher (Figure 2E). In contrast, there was a notable impact of NOX2 deficiency on the pattern of IgG2c production, which was primarily influenced by the choice of adjuvant. With alum as adjuvant, IgG2c responses were completely absent in both WT and NOX2-deficient mice (Figure 2C). However, with curdlan as adjuvant, while only minor elevated IgG2c levels were observed in WT mice, considerable and statistically significant IgG2c elevations were detected in the sera of NOX2-deficient mice (Figure 2F).
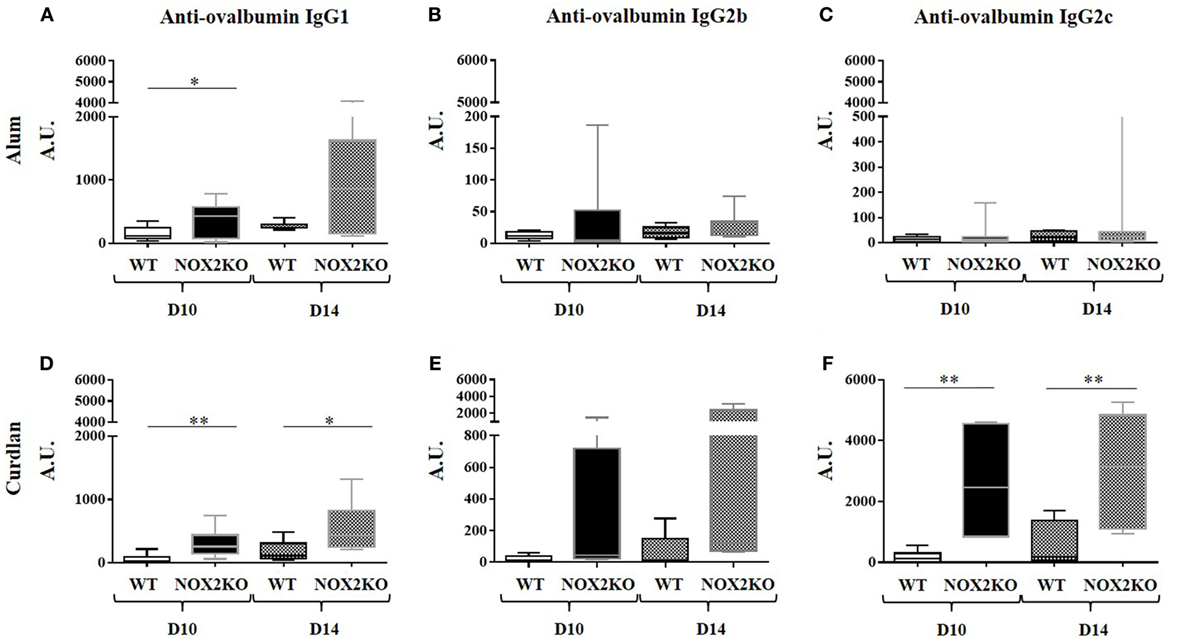
Figure 2. Enhanced immunoglobulin generation in CGD mice: IgG subtype and adjuvant dependence. Mice were immunized by subcutaneous injection into the outer ear of ovalbumin with either alum (A–C) or curdlan (D–F) as adjuvant. Levels of Anti-ovalbumin IgG1 (A,D), IgG2b (B,E) and IgG2c (C,F) were measured 10 and 14 days following immunization (n = 12). *p < 0.05; **p < 0.01; and ***p < 0.001.
The mechanisms leading to antibody production are complex and involve different cell types to reach a full humoral response. In order to address the mechanisms behind the marked increased IgG2c production in NOX2-deficient mice, we investigated inflammation resulting from the immunization, both locally and in draining lymph nodes. Measurement of ear thickness was performed as a read-out for the local inflammatory reaction. Ear thickness, prior to immunization, did not differ between WT and NOX2-deficient mice (data not shown). After intra-auricular immunization, a moderate increase in ear thickness was measured in WT mice (0.1–0.2 mm), which was similar when both alum and curdlan adjuvants were used. In NOX2-deficient mice, the ear thickness was strongly increased with curdlan as adjuvant, but not with alum (Figure 3A), indicating that curdlan induces an increased inflammatory reaction upon immunization.
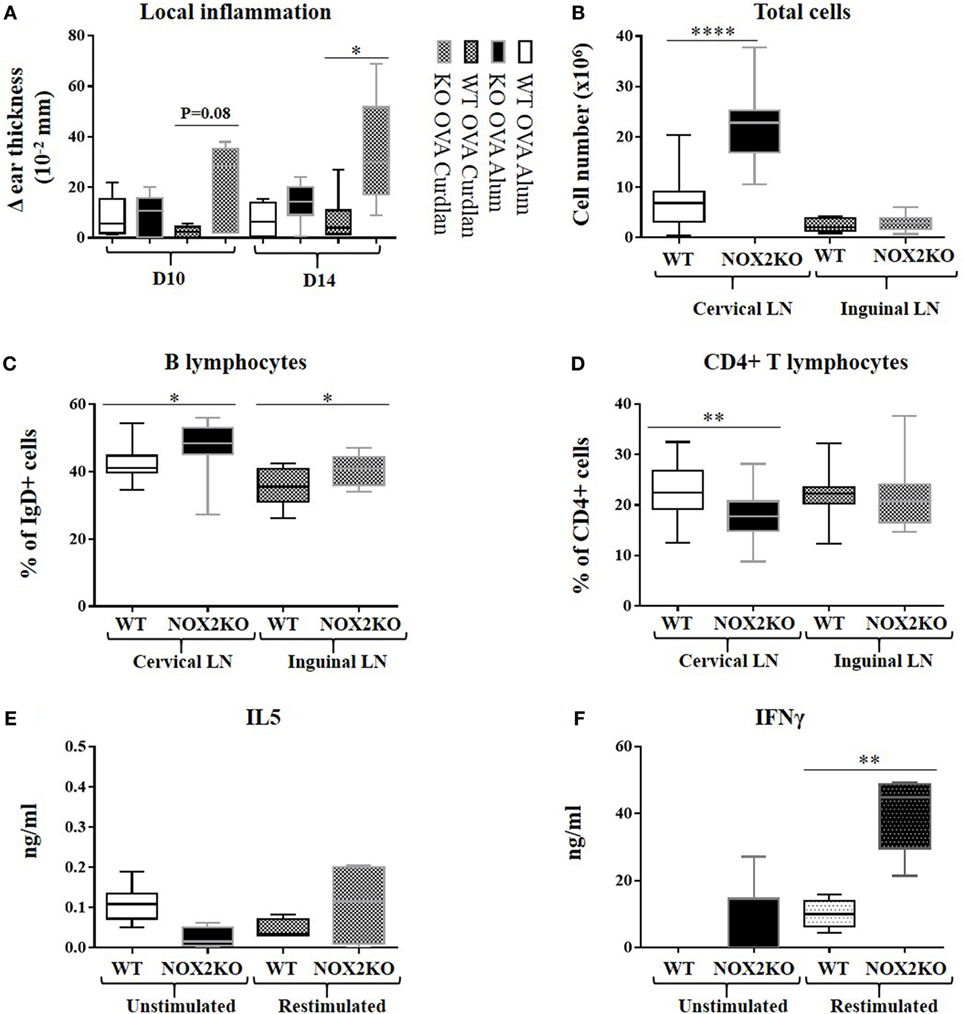
Figure 3. Enhanced inflammation and release of inflammatory mediators upon immunization of CGD mice. Mice were immunized by subcutaneous injection into the outer ear of ovalbumin with either alum or curdlan as adjuvant. (A) Ear thickness, reflecting the inflammatory response, was measured 10 and 14 days following immunization. (B–D) Draining cervical and non-draining inguinal lymph nodes were removed 14 days post immunization, dissociated, and analyzed by flow cytometry: (B) total cell number; (C) percentage of IgD-positive B cells; (D) percentage of CD4-positive T cells (n = 10). (E,F) Draining lymph nodes were isolated 10 days post immunization with ovalbumin and curdlan, and release of IL5 (E) and IFNγ (F) was measured 24 h after restimulation of lymph node homogenates with ovalbumin (n = 10). *p < 0.05; **p < 0.01; and ****p < 0.0001. The legend next to panel (A) refers only to this panel.
To characterize the inflammatory response, we investigated the total cell numbers of draining (cervical) and non-draining (inguinal) lymph nodes after immunization with ovalbumin and curdlan. In draining lymph nodes, there was a threefold median increase in cellularity (p < 0.0001) in NOX2-deficient mice when compared to WT mice (Figure 3B). In non-draining lymph nodes, the total number of cells was relatively low and there was no difference between WT and NOX2-deficient mice. We next analyzed the different subpopulations of lymph node cells. The proportion of CD4 positive T cells in draining lymph nodes of NOX2-deficient mice was significantly lower (p = 0.001) when compared to control WT mice (Figure 3D), whereas the proportion of IgD-positive B cell population was significantly higher (Figure 3C). Eventhough the proportion of CD4-positive T cells is decreased in NOX2-deficient immunized mice, it is important to notice that the absolute numbers of both CD4-positive T cells and IgD-positive B cells are increased in NOX2-deficient mice compared to WT (Figure S1 in Supplementary Material). Taken together, our data demonstrate that immunization of NOX2-deficient mice with curdlan as adjuvant leads to an increase of the total cell number in the draining lymph nodes, which is predominantly accounted for by an increase in IgD-positive B cells.
We next investigated the levels of cytokine release by cells isolated from draining lymph nodes, as an indirect measure of T cell differentiation and/or proliferation in response to immunization. For these experiments, mice were immunized with ovalbumin and curdlan, as described above. 10 days post immunization, draining lymph nodes were mechanically dissociated and cultured for 72 h in the presence or absence of ovalbumin. Low-level IL-5 production persisted for all conditions (Figure 3E), suggesting limited Th2 response. However, significant differences were observed with respect to the IFNγ levels (Th1): restimulation with ovalbumin led to a moderate increase in IFNγ levels from WT lymph node cells, while the production of IFNγ was strongly increased in NOX2-deficient lymph node cells (Figure 3F). These results suggest that immunization of NOX2-deficient mice with curdlan as adjuvant preferentially leads to an antigen-specific Th1 response.
To perform a more in-depth analysis of T cell responses, we used an in vitro co-culture system with OVA(323–339)-specific T cells (OTII T cells) and WT or NOX2-deficient (NOX2KO) BMDCs. T cell activation was assessed by the upregulation of CD69 at the surface of CD4 T cells after 16 h of co-culture. CD69 is an early T cell activation marker. In absence of OVA(323–339) peptide, barely no CD69hi CD4+ T cells were detectable (Figure S2 in Supplementary Material). Addition of different OVA(323–339) concentrations led to low to moderate T cell activation (identified as CD69hi CD4+ T cells) in a dose-dependent manner (Figure S2 in Supplementary Material). For each OVA(323–339) peptide concentration tested, there was a trend toward a higher percentage of CD69hi T cells when T cells were co-cultured with NOX2KO BMDCs compared to WT BMDCs. This difference was highest at 500 nM of OVA(323–339) peptide, where a 2.5-fold higher T cell activation was observed for NOX2KO BMDCs compared to WT DCs (Figure 4A).
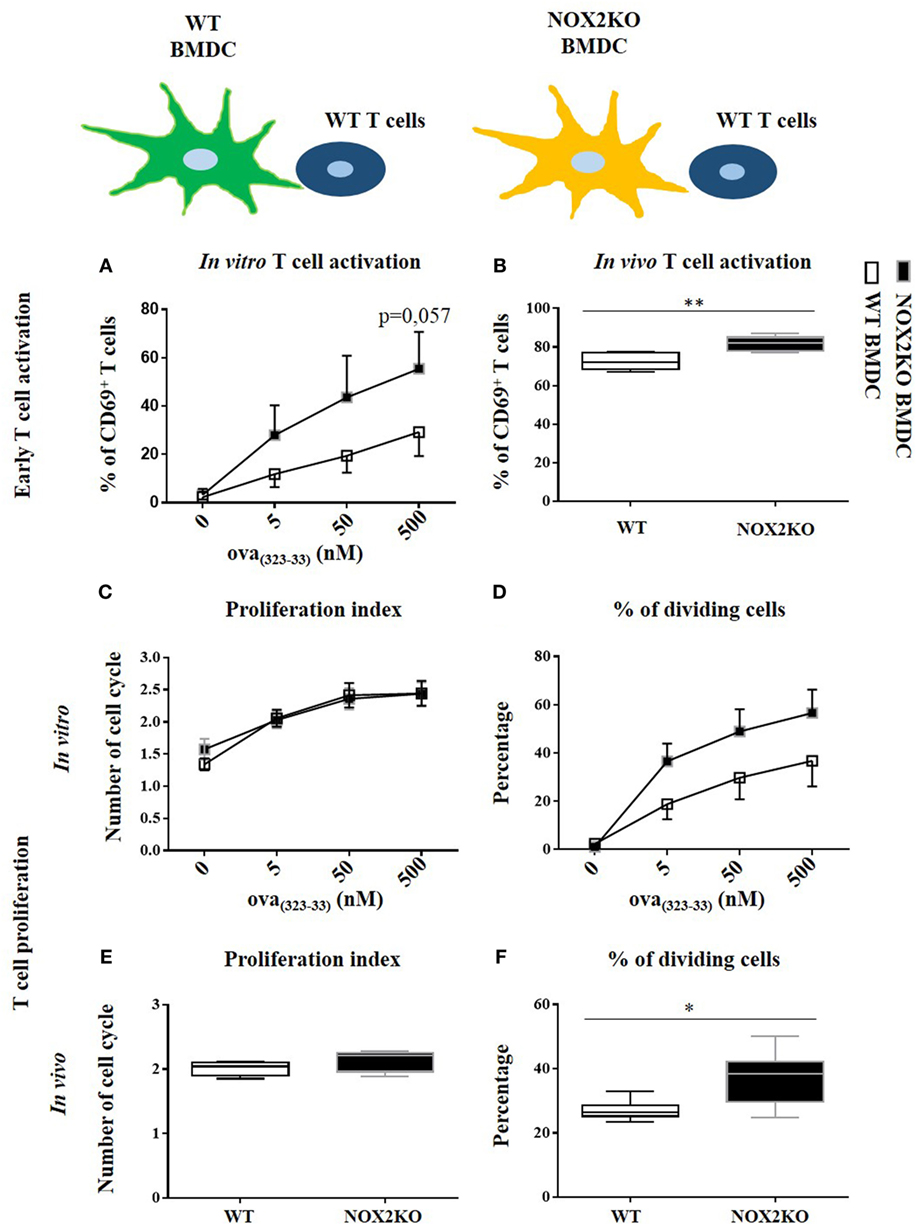
Figure 4. Effect of NOX2 in dendritic cells on T cell activation and proliferation in vitro and in vivo. OTII T cells were co-cultured with wild-type (WT) or NOX2KO bone marrow-derived dendritic cells (BMDCs) in the presence of different concentrations of OVA(323–339) peptide (5, 50, and 500 nM) for in vitro experiments (A,C,D), or were labeled with carboxyfluorescein succinimidyl ester (CFSE) and injected in OVA(323–339) pre-immunized WT or NOX2KO mice for in vivo experiments (B,E,F). T cell activation was determined 16 h after co-culture or 16 h after T cell injection by measuring the upregulation of CD69 by flow cytometry. (A) Percentage of CD69hiCD4hi T cells in vitro. (B) Percentage of CD69hiCD4hiCFSEhi T cells in vivo. For T cell proliferation, T cells were labeled with CFSE for the in vivo and in vitro experiments. The proliferation index and the percent of dividing cells were determined by the dilution of the CFSE signal in the OTII T cell population: (C,E) proliferation index and (D,F) percent of dividing cells in vitro (C,D) (n = 7) and in vivo (E,F). *p < 0.05 and **p < 0.01.
To further characterize the effect of NOX2 in BMDCs on T cell responses, we measured T cell proliferation. For that purpose, we used the property of the CFSE vital dye, for which fluorescence is equally divided by two in daughter cells after each cell division, allowing to study cell division. In the absence of antigen, T cells were not activated and did not enter into cell division. There was, therefore, only one peak of CFSE fluorescence (Figure S3 in Supplementary Material). With the addition of OVA(323–339) peptide to the co-culture, T cells started to divide and different peaks of CFSE fluorescence were detected. The percentage of cells that entered into division increased as a function of OVA(323–339) peptide concentration (Figure S3 in Supplementary Material). Co-culture of T cells with either WT or NOX2KO BMDCs did not change their proliferation index (Figure 4C). In contrast, there was also a trend toward a higher percentage of T cells that entered into cell cycle when T cells were incubated with NOX2KO BMDCs compared to WT BMDCs (Figure 4D). Therefore, the absence of NOX2 in BMDCs might facilitate the entry of T cells into cell division but once T cells went into cell division, they behaved similarly when incubated with either WT or NOX2KO DCs. These results confirm the above observations, and suggest an increase in early T cell activation with NOX2KO DCs compared to WT DCs.
Given the observed trends in our in vitro data, we wanted to independently confirm our results and further address the question in vivo. WT or NOX2KO mice were immunized subcutaneously in the outer ear with curdlan and OVA(323–339) peptide. 1 day later, CFSE-labeled OTII T cells were injected intravenously. The T cell response was analyzed in the draining and the non-draining lymph nodes at day 1 for early T cell activation (CD69 expression) and at day 3 for T cell proliferation (CFSE dilution). The upregulation of CD69 on CFSEhi CD4+ T cells in the draining lymph nodes demonstrated an activation of T cells 1 day after immunization, which was not the case in non-draining lymph nodes (data not shown). Recapitulating our in vitro co-culture assays, the percent of CD69hi CD4+ CFSEhi T cell was higher 1 day after immunization of NOX2KO mice compared to WT controls (Figure 4B). At day 3, the percentage of CFSE-labeled OTII T cells that had initiated proliferation was markedly higher in CGD mice, as compared to WT (Figure 4F), whereas there was no difference in the proliferation index (Figure 4E). Thus, both in vitro and in vivo, the number of CD4+ T cells upregulating the early activation marker and entering into cell division is increased when the APCs do not express NOX2.
To determine whether NOX2 expression in T cells contribute to the above observations, we assessed whether NOX2-deficient and WT T cells behave differently in vitro and in vivo. For that purpose, we crossed OTII mice with NOX2KO mice, as a source of T cells that were OVA-specific (OTII) and NOX2-deficient. For in vitro experiments, OTII WT and OTII NOX2KO T cells were co-cultured with curdlan-activated OVA(323–339) peptide loaded WT BMDCs. For in vivo experiments, OTII WT and OTII NOX2KO T cells were labeled with two different fluorescent vital dyes: a mixture of OTII WT and OTII NOX2KO T cells (ratio 1:1) was injected intravenously in WT mice that had been previously immunized with the OVA(323–339) peptide and curldan. Using this system, we were able to analyze and compare the activation of both WT and NOX2KO T cells in the same recipient mice. Both in vitro and in vivo experiments showed neither difference in T cell activation nor T cell proliferation between WT and NOX2KO CD4+ T cells. Percentages of CD69hi CD4+ T cells were identical when OTII WT and OTII NOX2KO where used, in vivo and in vitro (Figures 5A,B), and both the proliferation index and the percent of dividing cells were identical in vitro (Figures 5C,D). Thus, T cells from control and NOX2-deficient mice responded similarly to activation by DCs, refuting claims of a cell autonomous effect of NOX2 in T cells.
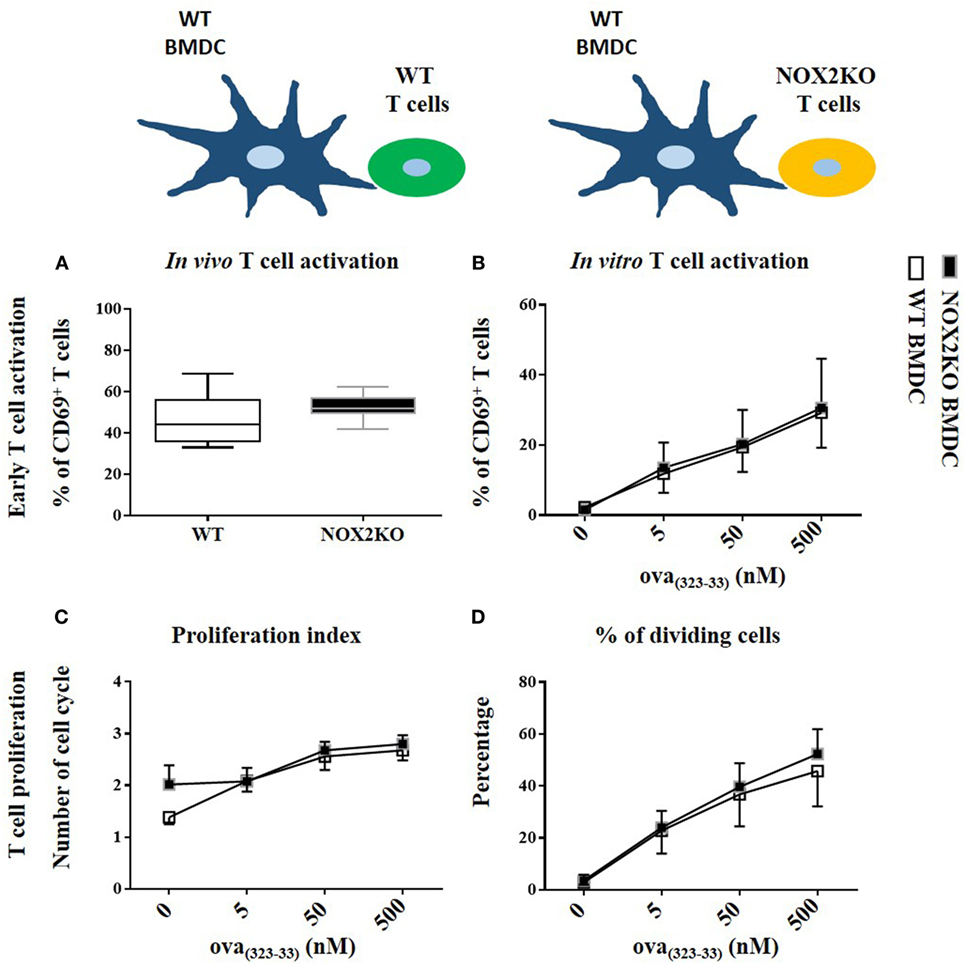
Figure 5. NOX2 in T cells does not impact T cell activation and proliferation in vitro and in vivo. Percent of CD69hi in NOX2 and wild-type CD4+ T cells in vitro (A) and in vivo (B). Proliferation index (C) and percent of dividing cells (D) at the different concentration of OVA323–339 peptide in vitro (n = 7).
Finally, several NOX2-dependent mechanisms limiting T cell activation by BMDCs were tested: (i) a paracrine effect of BMDC-derived H2O2 on T cells, (ii) a NOX2-dependent modulation of costimulatory molecules in BMDCs, which are known to influence the strength of T cell activation, and (iii) an impact of NOX2 on the release of T cell modulatory cytokines by BMDCs. For the first mechanism, the possible effect of BMDC-derived H2O2 on T cells was assessed by activating T cells from WT mice with anti-CD3/anti-CD28 antibodies and exposing them to increasing concentrations of exogenous H2O2. The percentage of CD69hi CD4+ T cells was not affected by H2O2, even at cytotoxic concentrations of H2O2 (Figures 6A,B). This excludes a direct role of BMDC-derived H2O2 on T cells. To test the second possibility, co-stimulatory molecule expression at the BMDC surface was analyzed by measuring the level of expression of MHCII, CD80, CD86, ICOSL, PDL1, and DC-SIGN after exposure of BMDCs to different concentrations of curdlan. All these molecules were similarly expressed by NOX2KO and WT BMDCs (Figures 6C,E,G; Figures S4A–C in Supplementary Material), ruling out an effect of NOX2 on the phenotype of BMDCs. To test the third mechanism, the production of T cell modulatory cytokines by NOX2 and WT BMDCs was measured after stimulation with curdlan. Whereas no effect of NOX2 deficiency was observed for the production of Th1 unrelated cytokines (IL-6 and IL-10) (Figure 6H; Figure S4D in Supplementary Material), NOX2-deficient BMDCs produced increased amounts of the pro-Th1 cytokines IL-12 and IL-1β compared to WT BMDCs (Figures 6D,F). In conclusion, our data neither favor a direct impact of NOX2-derived H2O2 on lymphocytes, nor a signaling through costimulatory receptors. In contrast, our data indicate an important role of NOX2 in BMDCs for limiting the release of pro-Th1 cytokines.
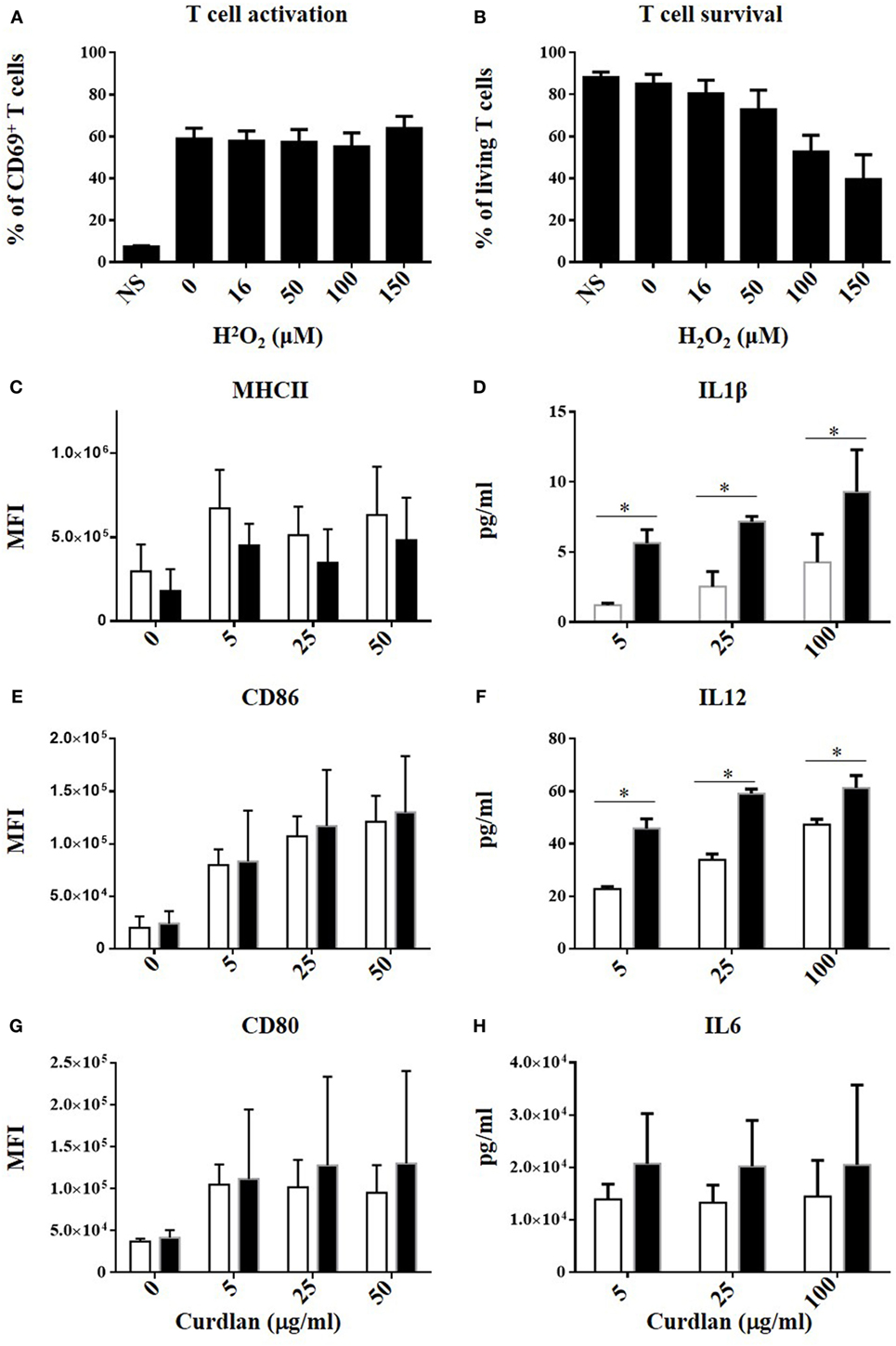
Figure 6. No direct effect of H2O2 on T cell activation but increased IL12 and IL1β production by NOX2KO bone marrow-derived dendritic cells (BMDCs) after activation by curdlan. Effect of increasing concentration of H2O2 on CD69 upregulation (A) and survival (B) in anti-CD3/anti-CD28 activated CD4+ T cell (n = 4). Expression level of MHCII (C), CD86 (E), and CD80 (G) in curdlan-activated wild-type (WT) and NOX2 BMDC was analyzed by flow cytometry (n = 4; MFI, mean fluorescent intensity). Level of IL-1β (D), IL-12 (F), and IL-6 (H) present in the supernatant of curdlan-activated WT and NOX2KO BMDC (n = 3).
Our study represents, to the best of our knowledge, the first analysis of the impact of the phagocyte NADPH oxidase (NOX2) on the pattern of IgG subtype repartition. The results show that NOX2 not only limits the amount of circulating antibodies, but also contributes to the determination of the composition of IgG subtypes. Indeed, upon immunization, NOX2-deficient mice show increased IgG2c levels. As underlying mechanisms, we propose that the increased release of Th1-driving cytokines from NOX2-deficient APCs leads to enhanced IFNγ production by activated T cells, which may in turn promote IgG2c production by B cells.
First reports describing CGD in the late 1950s had already described increased immunoglobulin levels (2), even before the lack of microbicidal activity of phagocytes in CGD was discovered (25). Subsequently, however, the increased immunoglobulin levels in CGD patients received little attention and were mostly attributed to repeated infections. Our study confirms that even in the absence of an experimental immunization, CGD mice have altered immunoglobulins levels. We found that serum levels of IgG2c were increased in a statistically significant manner. Note that these mice never developed a clinical apparent infection. Thus, we assume that the daily host–pathogen interaction, even in the absence of infection or controlled immunization is sufficient to reveal the tendency of CGD mice to produce a different subset of IgG. However, the controlled immunization performed in this study allowed us to further characterize the immune phenotype. Interestingly, the impact of NOX2 deficiency on IgG subtype production depends on the nature of the adjuvant. Alum (known to drive a Th2 response) promotes a modest IgG1 production in both WT and NOX2-deficient mice. Neither WT nor NOX2-deficient mice produced IgG2c with alum as adjuvant. Curdlan has been described to induce both Th1 and Th17 responses, with the Th1 response being predominant (26). However, in a previous study, our laboratory showed that IFNγ but not IL-17 is increased in NOX2-deficient mice after injection of curdlan in the outer ear of mice (27). Accordingly, we found a low level of the Th2-driven anti-ovalbumin IgG1 subtype in both WT and NOX2-deficient mice. In contrast, curdlan induces a marked increase in the production of IgG2c in WT mice, which was strikingly enhanced in NOX2-deficient mice. Thus, NOX2 deficiency by itself does not alter the type of the immune response elicited by a given adjuvant, but rather amplifies its response. Our results suggest the following scenario: in WT mice, curdlan activation of the dectin-1 pathway in DCs leads to production of Th1-driving cytokines, such as IL-12 and IL-1β, the magnitude of these cytokines being limited by NOX2-dependent ROS generation. In the absence of NOX2, the Th1 response is not kept under control, resulting in an enhancement of this pathway, and a subsequent deviation toward IgG2c responses. It will be of major interest to identify the ROS target in DCs. Typically, ROS signaling may occur through oxidation of redox-sensitive cysteines, for example, within the catalytic site of protein phosphatases (28). The identification of the ROS target would potentially pave the way toward innovative immunomodulatory treatments. Interestingly, a study showed an enhanced IL12 production after stimulation of NOX2-deficient BMDCs with IFNγ/LPS of NOX2 and this effect was correlated with a decreased p38-MAPK (29). Therefore, it would be interesting to investigate this pathway in order to elucidate the mechanism by which NOX2 controls IL12 production.
Understanding which NOX2 expressing cells influence the antibody responses is essential, the more relevant candidates being APCs, T cells, and B cells. Our results argue against a role for NOX2 in T cells. This is supported by previous publications suggesting that NOX2 is virtually not expressed in T cells (30). Our results rather support a role for APCs: when studying in vitro and in vivo T cell priming by DCs, we observed a trend toward an enhanced T cell activation by NOX2-deficient DCs in vitro, which was confirmed by an enhanced T cell activation in vivo in NOX2-deficient mice. However, we cannot exclude that NOX2 in other cell types might contribute to increase the T cell activation and altered IgG production. Indeed, fully functional NOX2 is expressed in B cells.
There is now increasing consensus that specific immunity, including humoral immune responses are enhanced in CGD patients. For most parts, these findings have also been documented in CGD mice. However, there are certain differences between the results reported in different studies that should be noted. Indeed, studies using the TLR4 ligand LPS as adjuvant, did not find increased T cell activation by phagocyte NADPH oxidase-deficient DCs (4, 31). Note that in our study, we have also observed differences between adjuvants, namely the use of alum vs. the dectin-1 ligand curdlan. Thus, different ways of DC activation yields distinct T and B cell responses, adding to the complexity of the specific immunity in CGD patients. At least two observations argue in favor of the in vivo relevance of our observations: (i) dectin-1 is increasingly recognized as a relevant activator of DCs (32) and (ii) altered IgG subtypes in CGD patients were identified in our study.
The enhanced T cell activation by NOX2-deficient DCs might be explained through at least four different mechanisms:
(i) Direct impact of ROS on T cells. Our study did not find direct impact of hydrogen peroxide on T cell activation (Figure 6A). Interestingly, other studies using adaptive transfer of athritogenic T cells derived from Ncf1-deficient rodents report that hydrogen peroxide attenuates the athritogenic properties of such T cells (30, 33). Thus, depending on the experimental set-up there might be a direct effect of hydrogen peroxide. Also, the primary product of NOX2 activation is the superoxide radical anion and these experiments do not address the impact of NOX2-derived oxidants, other than H2O2.
(ii) Antigen processing in NOX2KO DCs. Some studies have described an altered antigen processing in NOX2KO DCs, resulting in an altered cross-presentation or in a different epitopic repertoire (19, 34–37). However, we have used the OVA(323–339) peptide for our experiment on T cell activation, which does not require further peptidic cleavage to be presented through MHC class II molecules. Although, we cannot exclude that mechanism such as peptide transport and post-proteolytic modifications might be impacted by NOX2 deficiency, our results cannot be explained by a difference in proteases activity.
(iii) Alteration of surface proteins (in particular costimulatory molecules). We have investigated expression of costimulatory molecules by WT and NOX2-deficient DCs and did not observe any differences.
(iv) Altered release of soluble mediators (in particular cytokines): this mechanism is strongly supported by our data, as we find important differences in the release of two important T cell modulatory cytokines, namely IL1-β and IL-12, which polarize T cell toward a Th1 effector phenotype. Thus, in our hands, the release of soluble mediators from DCs is important for the understanding of the enhanced immune response in the CGD situation.
Another novel and unexpected result of both our in vitro and in vivo studies is the observation that the enhanced immune activation in NOX2-deficient mice concurred with an increased entry of T cells into the cell cycle, rather than an increased T cell proliferation rate. This observation is in line with our results suggesting that there is no cell autonomous effect of NOX2 in T cells, but rather that the initiation of T cell activation is controlled by NOX2 in DCs.
The human situation is more complex, due to first, the limitation of our knowledge on the IgG isotype switch, and second, the difficulty to perform clinical studies in CGD patients investigating the response to a specific antigen after immunization. Although the nomenclature is close between human and mice IgG subclass, their function differ in many aspects and direct comparison between high mouse IgG2c in mice and altered IgG1/IgG and IgG2/IgG ratio in human should be interpreted with caution. Nevertheless, our study has identified a remarkable, hitherto not described, pattern of IgG subtype distribution. The proportion of IgG2 was increased in adult CGD patients, while there was a relative decrease in IgG1. The increased levels of IgG2 were not observed at birth, but rather developed over the first 12 years of life. This suggests that there might be similarities between our experimental results obtained in NOX2-deficient mice and the condition of CGD children exposed to antigens. The age-dependent increase in IgG2 in human CGD patients might possibly reflect exposure to encapsulated bacteria (38). The increased IgG, and in particular IgG2, levels might provide compensatory mechanisms for the host defense against infections in CGD patients. However, such an increased propensity toward antibody generation is likely to come at a price. Indeed, as discussed in the Section "Introduction," CGD patients are prone to autoimmune diseases. In our serum samples from CGD patients, we found an increased proportion of positivity for the ANCA autoantibody (31%). In adult controls, only 6% (58 out of 924) serum samples were found positive using a similar IFI method (39). However, the results are largely variable among laboratories and methods. In addition, a further confounding factor is that ANCA formation is triggered by infection, and since CGD patients often encounter repeated infection, it would not be possible to exclude this as a cause of increased ANCA prevalence in these patients (40). ANA antibodies were positive in 3 out of 16 CGD patients (19%) while the percentage is 13.3% in control adults (41). The percentage of ANCA and ANA positivity is unknown in children, but autoantibody frequency usually increases with age and positivity is, therefore, likely to be decreased in children compared to adults. Altogether, our results confirm the increased prevalence of autoantibodies in CGD patients.
In summary, our results provide novel insights into the mechanisms underlying increased specific immune responses in CGD patients. Augmented cytokine production by NOX2-deficient DCs appears to be a crucial mechanism implicated in enhanced T cell activation and autoantibody production. Thus, targeting the overshooting release of T cell modulatory cytokines might be a promising approach for the treatment of hyperimmune disorders in CGD patients. It would, however, be beneficial to better understand how ROS dampen the release of cytokines in DCs. The identification of NOX2-derived ROS targets would open the path to novel immunomodulatory strategies at the interface of innate and specific immunity.
Blood samples were obtained from the CGD patients with appropriate institutional informed consent. This study also includes peripheral blood samples taken from healthy donors obtained from the "Establishment Français du sang" at the Grenoble University Hospital, France after their informed consent. For animal subject, the protocol was approved by the office cantonal vétérinaire du Canton de Genève, Switzerland (authorization no. 23624).
JC conceived the study, designed and performed experiments, analyzed the data, and wrote the manuscript. CD helped in the design of the study and the analysis of data. MA edited the manuscript. PR-L performed the autoantibody titer in human. AG performed the measurement of serum immunoglobulin in human. MJS diagnosed and characterized the mutation of CGD patients. SH helped in the design of T cell experiment in vivo and in vitro. K-HK conceived the study, wrote and edited the manuscript. All authors read and approved the final manuscript.
K-HK holds shares of Genkyotex SA, a company aiming at developing NOX inhibitors. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by the Swiss National Science Foundation ProDoc Research Module, PDFMP3-127302. The authors would like to thank the collaborators at the animal core facility, Cécile Gameiro at the flow cytometry core facility, and Yves Alexandre Cambet at READS units at the CMU of the University of Geneva. The authors would also like to thank Lesly Guery for discussions and help for the co-culture experiments. We are grateful to Olivier Plastre, for technical help and Vincent Jaquet for extensive editing of the manuscript.
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01555/full#supplementary-material.
Table S1. Demographic and blood biochemistry values for CGD patients and healthy controls. CGD patient samples are listed from 1–16, while healthy controls are assigned A–F. Values exceeding laboratory reference values are indicated in red shading. Reference values: IgG = 7–10 g/l; IgA = 0.9–2.3 g/l; IgM = 0.4–0.9 g/l; anti-MPO < 6 U; anti-PR3 < 5 U; and ANA < 80. Abbreviations: F, female; M, male; NA, not available; ND, not detected (below detection level); CGD, chronic granulomatous disease; A470, p47phox-deficient autosomal recessive CGD; A670, p67phox-deficient autosomal recessive CGD; A220, p22phox-deficient autosomal recessive CGD; ANCA, anti-neutrophil cytoplasmic antibodies; ANA, anti-nuclear antibody.
Figure S1. Enhanced inflammation of draining lymph node: draining cervical and non-draining inguinal lymph nodes were removed 14 days post immunization, dissociated, and analyzed by flow cytometry: (A) absolute number of IgD+ B cells and (B) absolute number of CD4+ T cells.
Figure S2. Representative dot plot graphs showing CD69 expression in CD4-positive T cells after co-culture with different concentration of OVA(323–339) peptide.
Figure S3. Representative histogram of carboxyfluorescein succinimidyl ester dilution at different concentration of OVA323–339 peptide after 3 days of co-culture with wild-type or NOX2KO BMDC.
Figure S4. NOX2 deficiency has no impact on the expression level of ICOSL (A), PDL1 (B) and DC-SIGN (C) or the level of IL10 (D) present in the supernatant, after activation of BMDCs by increasing concentration of curdlan.
1. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev (2007) 87:245–313. doi:10.1152/physrev.00044.2005
2. Janeway CA, Craig J, Davidson M, Downey W, Gitlin D. Hypergammaglobulinemia associated with severe recurrent and chronic nonspecific infection. Am J Dis Child (1954) 88:388–92.
3. Maly FE, Nakamura M, Gauchat JF, Urwyler A, Walker C, Dahinden CA, et al. Superoxide-dependent nitroblue tetrazolium reduction and expression of cytochrome b-245 components by human tonsillar B lymphocytes and B cell lines. J Immunol (1989) 142:1260–7.
4. Gelderman KA, Hultqvist M, Pizzolla A, Zhao M, Nandakumar KS, Mattsson R, et al. Macrophages suppress T cell responses and arthritis development in mice by producing reactive oxygen species. J Clin Invest (2007) 117:3020–8. doi:10.1172/JCI31935
5. Vasilevsky S, Liu Q, Koontz SM, Kastenmayer R, Shea K, Jackson SH. Role of p47phox in antigen-presenting cell-mediated regulation of humoral immunity in mice. Am J Pathol (2011) 178:2774–82. doi:10.1016/j.ajpath.2011.02.038
6. Richards SM, Clark EA. BCR-induced superoxide negatively regulates B-cell proliferation and T-cell-independent type 2 Ab responses. Eur J Immunol (2009) 39:3395–403. doi:10.1002/eji.200939587
7. Cachat J, Deffert C, Hugues S, Krause KH. Phagocyte NADPH oxidase and specific immunity. Clin Sci (Lond) (2015) 128:635–48. doi:10.1042/CS20140635
8. Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease – report on a national registry of 368 patients. Medicine (2000) 79:155–69. doi:10.1097/00005792-200005000-00003
9. Foster CB, Lehrnbecher T, Mol F, Steinberg SM, Venzon DJ, Walsh TJ, et al. Host defense molecule polymorphisms influence the risk for immune-mediated complications in chronic granulomatous disease. J Clin Invest (1998) 102:2146–55. doi:10.1172/JCI5084
10. De Ravin SS, Naumann N, Cowen EW, Friend J, Hilligoss D, Marquesen M, et al. Chronic granulomatous disease as a risk factor for autoimmune disease. J Allergy Clin Immunol (2008) 122:1097–103. doi:10.1016/j.jaci.2008.07.050
11. Van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PLoS One (2009) 4(4):e5234. doi:10.1371/journal.pone.0005234
12. Jacob N, Stohl W. Autoantibody-dependent and autoantibody-independent roles for B cells in systemic lupus erythematosus: past, present, and future. Autoimmunity (2010) 43:84–97. doi:10.3109/08916930903374600
13. Yu JE, De Ravin SS, Uzel G, Landers C, Targan S, Malech HL, et al. High levels of Crohn’s disease-associated anti-microbial antibodies are present and independent of colitis in chronic granulomatous disease. Clin Immunol (2011) 138:14–22. doi:10.1016/j.clim.2010.08.003
14. Olofsson P, Holmberg J, Tordsson J, Lu S, Akerström B, Holmdahl R. Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat Genet (2003) 33:25–32. doi:10.1038/ng1058
15. Hultqvist M, Holmdahl R. Ncf1 (p47phox) polymorphism determines oxidative burst and the severity of arthritis in rats and mice. Cell Immunol (2005) 233:97–101. doi:10.1016/j.cellimm.2005.04.008
16. Gelderman KA, Hultqvist M, Holmberg J, Olofsson P, Holmdahl R. T cell surface redox levels determine T cell reactivity and arthritis susceptibility. Proc Natl Acad Sci U S A (2006) 103:12831–6. doi:10.1073/pnas.0604571103
17. Bogeski I, Kummerow C, Al-Ansary D, Schwarz EC, Koehler R, Kozai D, et al. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal (2010) 3. doi:10.1126/scisignal.2000672
18. Kwon J, Devadas S, Williams M. T cell receptor-stimulated generation of hydrogen peroxide inhibits MEK-ERK activation and lck serine phosphorylation. Free Radic Biol Med (2003) 35:406–17. doi:10.1016/S0891-5849(03)00318-6
19. Allan ER, Tailor P, Balce DR, Pirzadeh P, McKenna NT, Renaux B, et al. NADPH oxidase modifies patterns of MHC class II-restricted epitopic repertoires through redox control of antigen processing. J Immunol (2014) 192:4989–5001. doi:10.4049/jimmunol.1302896
20. Yang M, Haase C, Viljanen J, Xu B, Ge C, Kihlberg J, et al. Cutting edge: processing of oxidized peptides in macrophages regulates T cell activation and development of autoimmune arthritis. J Immunol (2017) 199:3937–42. doi:10.4049/jimmunol.1700774
21. Hultqvist M, Olofsson P, Holmberg J, Bäckström BT, Tordsson J, Holmdahl R. Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci U S A (2004) 101:12646–51. doi:10.1073/pnas.0403831101
22. Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med (2012) 4(157):157ra141. doi:10.1126/scitranslmed.3004801
23. Zhao J, Ma J, Deng Y, Kelly JA, Kim K, Bang SY, et al. A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat Genet (2017) 49:433–7. doi:10.1038/ng.3782
24. Schäppi M, Deffert C, Fiette L, Gavazzi G, Herrmann F, Belli D, et al. Branched fungal beta-glucan causes hyperinflammation and necrosis in phagocyte NADPH oxidase-deficient mice. J Pathol (2008) 214:434–44. doi:10.1002/path.2298
25. Baehner RL, Nathan DG. Leukocyte oxidase: defective activity in chronic granulomatous disease. Science (1967) 155:835–6. doi:10.1126/science.155.3764.835
26. LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol (2007) 8:630–8. doi:10.1038/ni1460
27. Deffert C, Carnesecchi S, Yuan H, Rougemont AL, Kelkka T, Holmdahl R, et al. Hyperinflammation of chronic granulomatous disease is abolished by NOX2 reconstitution in macrophages and dendritic cells. J Pathol (2012) 228:341–50. doi:10.1002/path.4061
28. Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell (2002) 9:387–99. doi:10.1016/S1097-2765(02)00445-8
29. Jendrysik MA, Vasilevsky S, Yi L, Wood A, Zhu N, Zhao Y, et al. NADPH oxidase-2 derived ROS dictates murine DC cytokine-mediated cell fate decisions during CD4 T helper-cell commitment. PLoS One (2011) 6:e28198. doi:10.1371/journal.pone.0028198
30. Hultqvist M, Olofsson P, Gelderman KA, Holmberg J, Holmdahl R. A new arthritis therapy with oxidative burst inducers. PLoS Med (2006) 3:e348. doi:10.1371/journal.pmed.0030348
31. Padgett LE, Tse HM. NADPH oxidase-derived superoxide provides a third signal for CD4 T cell effector responses. J Immunol (2016) 197:1733–42. doi:10.4049/jimmunol.1502581
32. Agrawal S, Gupta S, Agrawal A. Human dendritic cells activated via dectin-1 are efficient at priming Th17, cytotoxic CD8 T and B cell responses. PLoS One (2010) 5:e13418. doi:10.1371/journal.pone.0013418
33. Yang Z, Shen Y, Oishi H, Matteson EL, Tian L, Goronzy JJ, et al. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med (2016) 8:331ra338. doi:10.1126/scitranslmed.aad7151
34. Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell (2006) 126:205–18. doi:10.1016/j.cell.2006.05.035
35. Rybicka JM, Balce DR, Chaudhuri S, Allan ER, Yates RM. Phagosomal proteolysis in dendritic cells is modulated by NADPH oxidase in a pH-independent manner. EMBO J (2012) 31:932–44. doi:10.1038/emboj.2011.440
36. Crotzer VL, Matute JD, Arias AA, Zhao H, Quilliam LA, Dinauer MC, et al. Cutting edge: NADPH oxidase modulates MHC class II antigen presentation by B cells. J Immunol (2012) 189:3800–4. doi:10.4049/jimmunol.1103080
37. Balce DR, Li B, Allan ER, Rybicka JM, Krohn RM, Yates RM. Alternative activation of macrophages by IL-4 enhances the proteolytic capacity of their phagosomes through synergistic mechanisms. Blood (2011) 118:4199–208. doi:10.1182/blood-2011-01-328906
38. Kuijpers TW, Weening RS, Out TA. IgG subclass deficiencies and recurrent pyogenic infections, unresponsiveness against bacterial polysaccharide antigens. Allergol Immunopathol (Madr) (1992) 20:28–34.
39. Damoiseaux J, Csernok E, Rasmussen N, Moosig F, van Paassen P, Baslund B, et al. Detection of antineutrophil cytoplasmic antibodies (ANCAs): a multicentre European Vasculitis Study Group (EUVAS) evaluation of the value of indirect immunofluorescence (IIF) versus antigen-specific immunoassays. Ann Rheum Dis (2016) 76(4):647–53. doi:10.1136/annrheumdis-2016-209507
40. Konstantinov KN, Ulff-Moller CJ, Tzamaloukas AH. Infections and antineutrophil cytoplasmic antibodies: triggering mechanisms. Autoimmun Rev (2015) 14:201–3. doi:10.1016/j.autrev.2014.10.020
Keywords: NOX2, B cells, T cells, immunoglobulin, antigen-presenting cells, curdlan, IgG subtype
Citation: Cachat J, Deffert C, Alessandrini M, Roux-Lombard P, Le Gouellec A, Stasia M-J, Hugues S and Krause K-H (2018) Altered Humoral Immune Responses and IgG Subtypes in NOX2-Deficient Mice and Patients: A Key Role for NOX2 in Antigen-Presenting Cells. Front. Immunol. 9:1555. doi: 10.3389/fimmu.2018.01555
Received: 31 January 2018; Accepted: 25 June 2018;
Published: 11 July 2018
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Selinda Jane Orr, Cardiff University, United KingdomCopyright: © 2018 Cachat, Deffert, Alessandrini, Roux-Lombard, Le Gouellec, Stasia, Hugues and Krause. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karl-Heinz Krause, a2FybC1oZWluei5rcmF1c2VAdW5pZ2UuY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.