 Heng Yin1†
Heng Yin1† Qianjin Lu
Qianjin Lu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 July 2018
Sec. Immunological Tolerance and Regulation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01512
This article is part of the Research Topic Autophagy in Autoimmunity View all 13 articles
Autophagy is a complicated cellular mechanism that maintains cellular and tissue homeostasis and integrity via degradation of senescent, defective subcellular organelles, infectious agents, and misfolded proteins. Accumulating evidence has shown that autophagy is involved in numerous immune processes, such as removal of intracellular bacteria, cytokine production, autoantigen presentation, and survival of lymphocytes, indicating an apparent and important role in innate and adaptive immune responses. Indeed, in genome-wide association studies, autophagy-related gene polymorphisms have been suggested to be associated with the pathogenesis of several autoimmune and inflammatory disorders, such as systemic lupus erythematosus, psoriasis, rheumatoid arthritis, inflammatory bowel disease, and multiple sclerosis. In addition, conditional knockdown of autophagy-related genes in mice displayed therapeutic effects on several autoimmune disease models by reducing levels of inflammatory cytokines and autoreactive immune cells. However, the inhibition of autophagy accelerates the progress of some inflammatory and autoimmune diseases via promotion of inflammatory cytokine production. Therefore, this review will summarize the current knowledge of autophagy in immune regulation and discuss the therapeutic and pathogenic role of autophagy in autoimmune diseases to broaden our understanding of the etiopathogenesis of autoimmune diseases and shed light on autophagy-mediated therapies.
Autophagy was discovered more than 40 years ago and has recently become a topic of increasing interest since the Japanese biologist Yoshinori Ohsumi won the Nobel Prize in 2016 for the discovery of this “self-eating” mechanism. Autophagy refers to a survival mechanism that cells use to degrade unwanted and useless organelles, proteins, and infectious agents to maintain homeostasis (1). There are three types of autophagy: macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy. Among them, macroautophagy is the most intensively studied and is referred to as autophagy in general, and is the focus of this review. Macroautophagy occurs in all eukaryotic cells and initiates the recruitment of protein aggregates and misfolded proteins by phagophores. Then, vesicles undergo elongation and form double-membraned vesicles, called autophagosomes, and then the cytoplasmic components are enclosed via cargo sequestration and fuse with lysosomes for degradation and recycling (2) (Figure 1). Macroautophagy is capable of constitutively delivering cytosolic proteins for MHC-II presentation. Autophagosomes are capable of fusing with multi-vesicular bodies or enodosomes and MHC-II loading compartments (3). CMA is another type of autophagy, which is involved with the direct recognition, targeting, and degradation of substrates by lysosomes, rather than by autophagosome formation (4). As a substrate of CMA, a protein should contain the amino acid sequence of the polypeptide motif KRERQ (5). Target proteins are selectively recognized by cytosolic heat shock cognate 70/co-chaperones and then delivered to the lysosomal membrane. Proteins then bind to the integral lysosomal membrane protein (LAMP-2A) and unfold and reach the lumen via a LAMP-2A-enriched translocation complex. Then proteins undergo degradation in the autolysosome (6). In addition, microautophagy is the process of lysosomal engulfment of cytoplasmic cargo with the formation of autophagic tubes and vesicles (7). Cytosolic and soluble proteins and particulate cellular constituents are directly internalized in single-membrane vesicles into lysosome by invaginating, protrusion, and or septation of the lysosomal limiting membrane (8). Microautophagy is a process of detect invagination and fusion of the vacuolar/lysosomal membrane under the limited nutrient status. Therefore, microautophagy is critical for cell survival, particularly for cells that are under stress such as nutrient starvation (9).
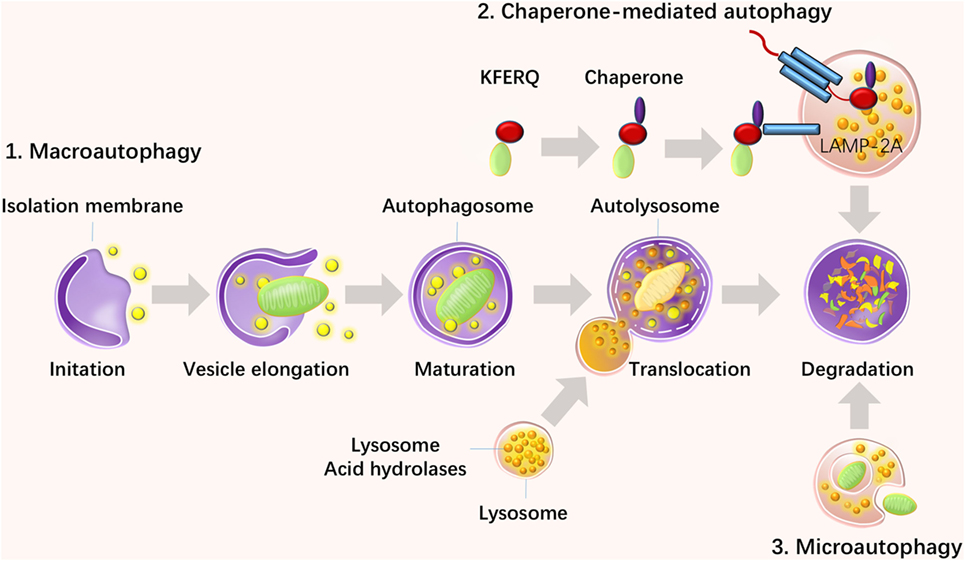
Figure 1. Three types of autophagy and their steps. There are three types of autophagy: macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy. Macroautophagy initiates with the recruitment of protein aggregates and misfolded proteins by phagophores. Then, vesicles undergo elongation and form double-membraned vesicles, called autophagosomes, and the cytoplasmic components are enclosed via cargo sequestration and fused with lysosomes for degradation and recycling. CMA is another type of autophagy, which is involved in the direct recognition, targeting, and degradation of substrates by lysosomes rather than autophagosome formation. Microautophagy is a process of lysosomal engulfment of cytoplasmic cargo with the formation of autophagic tubes and vesicles.
It is now widely accepted that macroautophagy (autophagy for short) is involved in several pathophysiological processes and complex diseases, such as autoimmune disorders, cancer, and metabolic disorders. Indeed, autophagy has been found to play four principal roles in immune responses: intracellular pathogen removal, lymphocyte development, pro-inflammatory signaling, and the secretory pathway (10–12). The combined data from genome-wide association studies (GWAS) and inhibition assays in mouse models have implicated autophagy in autoimmune diseases, especially in systemic lupus erythematosus (SLE), psoriasis, rheumatoid arthritis (RA), inflammatory bowel disease (IBD), and multiple sclerosis (MS). Therefore, this review will summarize the current understanding of the molecular regulation of autophagy and its roles in immunity and discuss the therapeutic and pathogenic role of autophagy in these autoimmune disorders, providing potential diagnostic targets and therapeutic strategies for autoimmune diseases. Although CMA and microautophagy has been found to be also related with immune responses (13), the regulations from these two pathways will not be discussed in this review.
It has been well established that autophagy is predominately regulated by the autophagy-related gene (Atg) family, which initiates the formation of autophagosomes (14). Energy depletion can activate AMP-activated protein kinase and further activate the mammalian target of rapamycin (mTOR) substrate complex, which consists of phosphorylated UNC-51-like kinase 1 (ULK1), ATG13, ATG101, and FIP200. In addition, this pathway positively regulates the formation of autophagosomes (15). Environmental signals, such as starvation, repress autophagy via inhibition of mTOR, which is located in the mTOR signaling complex 1, formed by the regulatory-associated protein of mTOR (Raptor), G protein beta subunit-like protein, and proline-rich Akt/PKB substrate 40 kDa (16). In addition, insulin and other growth factor signaling activates Class I PI3K–Akt, which inhibits autophagy via activation of the mTOR signaling complex 1 and inhibition of the Beclin 1 class III PI3K complex, which contains Beclin 1, class III phosphatidylinositol-3-kinase (PIK3C3), and ATG14L (17). It has been documented that autophagosomal elongation requires two ubiquitin-like conjugation systems: the ATG5–ATG12 conjugation system and light-chain 3 (LC3)–ATG8 conjugation system (16). When cells take up phagocytosed dead cells, LC3-associated phagocytosis (LAP) will take place to promote digestion (18). The molecular regulation of autophagy has been summarized in Figure 2.
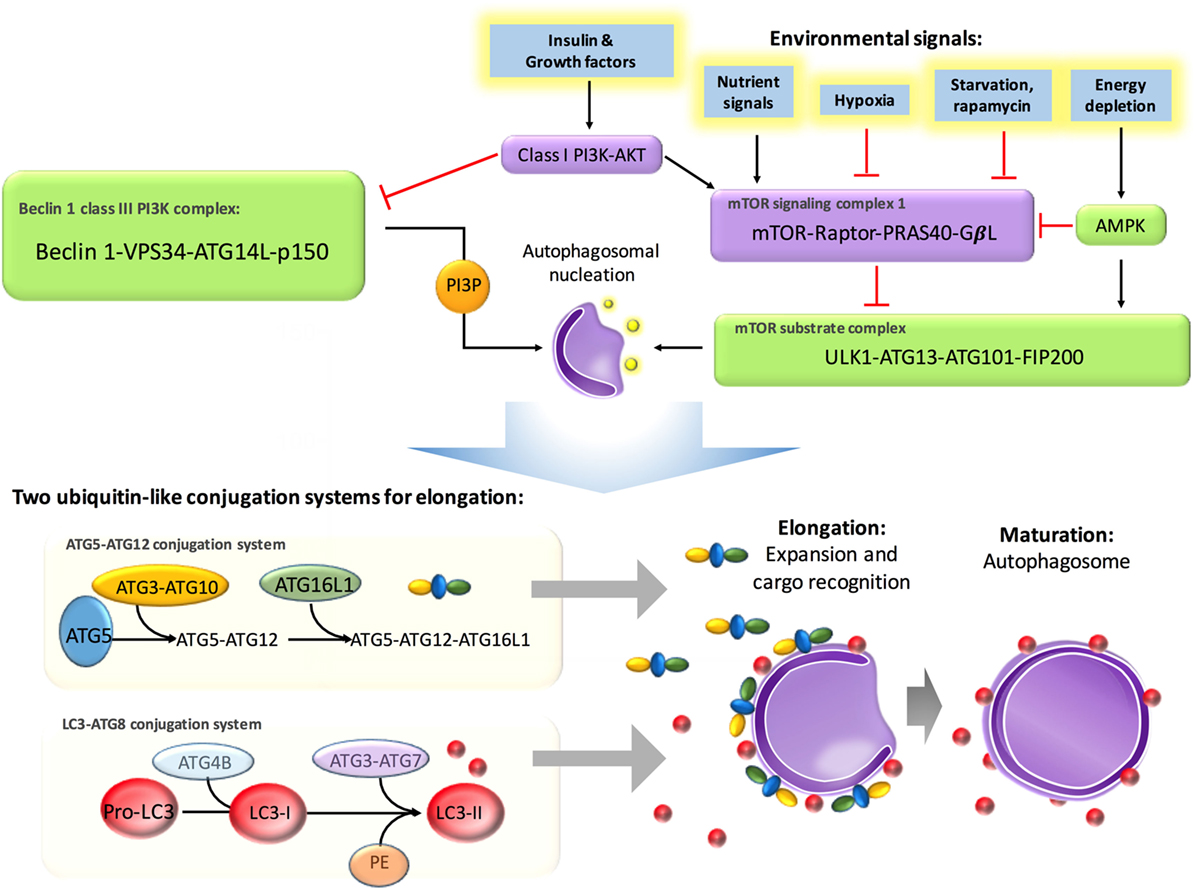
Figure 2. Molecular regulation in autophagy. Autophagy is suppressed by insulin and other growth factor signaling, starvation, and nutrient signals but activated by energy depletion and AMBRAs, BIF1, and UVRAG. Energy depletion can activate AMP-activated protein kinase (AMPK) and further activate the mammalian target of rapamycin (mTOR) substrate complex, which consists of phosphorylated UNC-51-like kinase 1 (ULK1), ATG13, ATG101, and FIP200. In addition, this pathway positively regulates the formation of autophagosomes. Environmental signals, such as starvation, repress autophagy via inhibition of mTOR, which is located in the mTOR signaling complex 1, formed by the regulatory-associated protein of mTOR (Raptor), G protein beta subunit-like protein (GβL), and proline-rich Akt/PKB substrate 40 kDa (PRAS40). Insulin and other growth factor signaling activates class I PI3K–Akt, which inhibits autophagy via activation of mTOR signaling complex 1 and inhibition of the Beclin 1 class III PI3K complex, which contains Beclin 1, class III phosphatidylinositol-3-kinase (PIK3C3), and ATG14L. Autophagosomal elongation requires two ubiquitin-like conjugation systems: the ATG5–ATG12 conjugation system and light-chain 3–ATG8 conjugation system.
It has been reported that autophagy is initiated by different families of pathogen-recognition receptors [such as toll-like receptors (TLRs)], damage-associated molecular patterns (such as HMGB1 and misfolded proteins), pathogen receptors, IFN-gamma, DAP kinase, JNK, CD40, TNF-alpha, and NF-κB (19). And it is repressed by Th2 cytokines, Bcl-2, and canonical nutrient sensing insulin–AKT–TOR pathway. Therefore, it is no surprise that autophagy involves in innate and adaptive immune response.
Autophagy participates in innate immunity via removing intracellular microbial pathogens and protecting the cytosol. There are two major ways to remove intracellular pathogens by autophagy in dendritic cells (DCs), macrophages, and other phagocytes. One is called xenophagy, which eliminates pathogens by engulfing them in double-membrane autophagosomes. This process is activated during infection by TLRs (20). Many bacteria and parasites can be removed by xenophagy. The other way is mediated by microtubule-associated protein LC3 and named LAP, which encloses pathogens in single-membrane phagosomes and involves LC3 (18). LAP has been found to be activated by TLR agonists and immune complexes (21), which are abundant in SLE. LAP is triggered by bacteria via surface markers (TLRs, FcγR, NOD, and SLAM) expressed by host cells, or by cytosolic pathogen sensing signals via direct induction or binding to autophagy component proteins. LAP shows capacity in dead cell clearance, which requires PtdSer receptor TIM4 to induce the recruitment of LC3 to the phagosome (22).
However, several pathogens have been found to have some strategies to avoid autophagy process. Listeria monocytogenes and Shigella flexneri have been found to express several proteins which are modified to inhibit recognition by the autophagic machinery. Human immunodeficiency virus 1 (HIV-1) can express Nef protein to interact with Beclin-1, thereby blocking the fusion of autophagosomes with lysosomes (23). This is the reason why we can be infected with bacteria and viruses even though we have normal innate immune responses.
In physiological conditions, apoptotic cells, with ATG and Bclin-1expression, expresses phosphatidylserine on cell surfaces, which is referred to as “eat-me” signals. Then they release lysophosphatidylcoline, which is considered as “come-get-me” signals. Then, these apoptotic cells can be efficiently cleared by phagocytes. However, in ATG5 or Beclin-1-deficient cells, apoptotic cells cannot express “eat-me” and “come-get-me” signals, which will lead to impaired clearance of apoptotic bodies, thereby contributing to autoimmune disorders (24).
Antigen-presenting cells (APCs), including DCs, macrophages, and B cells, are the bridges between innate and adaptive immunity. MHC-I molecules present endogenous antigens to CD8+ T cells via processing them by proteasome and translocating them into ER. Whereas MHC-II presents extracellular antigens on lysosome-derived organelles to CD4+ T cells. Before presentation with MHC-II by APCs, extracellular pathogens need to be properly digested and processed by APCs via degradation pathways, which include the ubiquitin–proteasome system and autophagy. MHC-II molecules can also present intracellular antigens, such as cytosolic or nuclear antigens, via the fusion of autophagosomes with MHC-II rather than the lysosome (8). In addition, the process of MHC-I presentation can be enhanced by autophagy (25). This urgent MHC-I presentation which is promoted by autophagy seems to need more time and more antigens, which means that this alternative process in the “cellular emergency” situation occurs when the classical pathway is imparied.
It has been reported that autophagy plays a critical role for the thymic selection, T cell development, survival, and proliferation. Atg5−/− mice show reduced thymocyts and peripheral lymphocytes and increased cell death in CD8+ T cells. And these Atg5−/− T cells fail to proliferate under TCR stimulation (26). Atg7−/− T cells show deficiency in cell survival, with the expanded ER content and mitochondria (27). The similar results have been observed in the Atg3−/− mice, which display impaired autophagy (28). On the other hand, some studies have claimed that autophagy promote the cell death during virus infection. For example, HIV Env-mediated autophagy induces apoptosis of CD4+ T cells via CXCR4 (29), and autophagy is involved in RIPK1-dependent necroptotic cell death when Fas-associated death domain activity and caspase-8 is insufficient (30). In addition, Beclin-1−/− CD4+ T cells, which are deficiency in autophagy, show preference in apoptosis under the TCR stimulation. Accumulation of pro-caspase-3, pro-caspase-3, and Bim might be one of the mechanisms (31). Therefore, the role of autophagy in T cell survival remains unclear. Future studies are needed to address this issue.
There are two subsets of conventional DCs (cDCs): CD8α (CD103+) cDCs and CD4+ (CD11b+) cDCs. CD8α cDCs efficiently cross-present exogenous antigens on MHC-I to CD8+ T cells, whereas CD4+ cDCs more efficiently polarize CD4+ T cells into Th1, Th2, Th17, or regulatory T cells (Tregs) by MHC-II-restricted presentation (32). Autophagy has been reported to be essential for the CD4+ T-cell response by DCs (33, 34). Indeed, a deficiency of autophagy in DCs results in a mild EAE phenotype in mice (35), which is a Th17-mediated mouse model. Furthermore, autophagy has also been linked with DC-derived cytokine production, such as IL-6 and IL-12p40 (36), which is critical for T-cell activation and polarization. This evidence indicates autophagy might involve in the Th cell differentiation. Besides, Tregs have been reported to be regulated by autophagy. For example, during chronic hepatitis B virus infection, HMGB-1-induced autophagy maintains Treg cell functions (37). And Agt16l1gene has been found to differentially regulate Treg and Th2 cell and further control intestinal inflammation (38). However, autophagy can be also regulated by Treg cells. For example, Foxp3+ Treg cells have been found to suppress immune response via inhibiting autophagic machinery in DCs depending on CTLA4. The binding of CTLA-4 promotes activation of PI3K/Akt/mTOR axis and FoxO1 nuclear exclusion in DCs, resulting in reduced expression of autophagy component microtubule-associated protein 1 light chan 3 beta (39).
In addition, autophagy also regulates B cell survival and development. ATG5 has been identified to be critical for B-cell survival and subset maintenance, such as pre-B and mature B1a B cells (40). Autophagy has been found to be required for immunoglobulin production by plasma cells (41). Indeed, the degradation of misfolded protein is particularly important for antibody-secreting cells, in which protein synthesis and degradation must be balanced. It has been observed that increased autophagosome formation and degradation occur in activated mouse B cells during plasma cell differentiation (41), as well as in human B cells activated by CpG (42). Genetic studies have identified several autophagy-related genes essential for antibody responses and plasma cell homeostasis (43), and dysregulated autophagy contributes to the plasma cell pathology in antibody-mediated autoimmune diseases such as SLE (44, 45). In addition, autophagy has been found to contribute to IL-17-dependent plasma differentiation via regulating Blimp-1 expression and Beclin-1/p62-associated B cell apoptosis (46, 47). These findings show that autophagy in B cells might play a pathogenic role in antibody-mediated autoimmune disease, such as lupus.
Increasing evidence has demonstrated the interplay between autophagy and the NF-κB signaling pathway (48). The NF-κB family of transcription factors regulates transcription of a broad range of genes, which are engaged in cell proliferation, survival, differentiation, and development. These transcription factors are also essential in inflammation and immune responses (49). The mammalian NF-κB family contains five members: RelA, c-Rel, Rel-B, p50, and p52 (50), while inhibitors of the NF-κB protein family consist of IκBα, IκBβ, IκBε, and the IκB-like inhibitors p100 and p105 (51). It has been well documented that activation of the inhibitor of NF-κB (IκBα) kinase complex is required for the induction of autophagy. Conversely, in an Atg5- and Atg7-deficient system, autophagy has been proven to be critical for the activation of NF-κB (52). In addition, the cross talk between NF-κB and autophagy has been observed in immune cells. The regulation of T-cell receptor-mediated NF-κB activation by B-cell lymphoma/leukemia 10 is associated with autophagy adaptor p62/SQSTM1 (53), which is also found to be a modulator of NLRP3-inflammasome activation and IL-1 beta production in macrophages (54). However, in tissue-specific macrophages, autophagy has been revealed to promote NF-κB activation to boost the antifungal immune response (55). This evidence indicates a pathogenic or therapeutic role of autophagy depending on different microenvironments and signals.
It is no surprise that autophagy-regulated cytokine secretion by the secretory pathway shares some common functions with phagocytosis, such as vesicle trafficking and membrane fusion, which facilitates the important role of autophagy in immune regulation. ATG5 deficiency, for example, results in elevated IL-1 alpha secretion by macrophages (56), while inhibition of autophagy leads to promotion of IL-1 beta via reducing degradation (57) by APCs and increases IL-23 secretion as a consequence (58), which can further promote Th17-mediated inflammatory responses. On the other hand, cytokines can also regulate autophagy. IL-10, which is an anti-inflammatory cytokine, has been found to inhibit autophagy in murine macrophages via activation of mTOR complex 1 (59). Another example is IL-6, which is a universal inflammatory cytokine involved in many autoimmune and inflammatory diseases. IL-6 has been illustrated to inhibit starvation-induced (60) and IFN-gamma-induced autophagy (61) by regulating Bcl-2 and Beclin1. However, IL-6 has also been found to be required for autophagy by promoting autophagosomal maturation (62, 63). The functions of autophagy have been summarized in Figure 3. Further studies are necessary to validate the interplay between autophagy and cytokines.
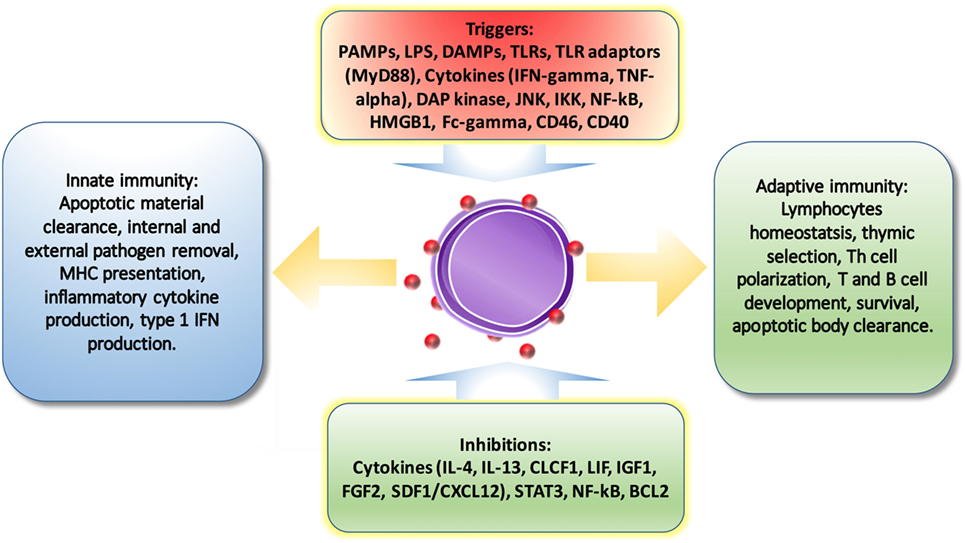
Figure 3. The regulations of autophagy on immune system. Autophagy is triggered and inhibited by cytokines and molecules from immune system. And also, autophagy is involved in pathogen removal, cytokine secretion, lymphocyte survival and differentiation, MHC presentation, apoptotic cell clearance, and pro-inflammatory signaling.
Based on its functions in the immune system, autophagy might display a pathogenic and/or therapeutic role in autoimmune diseases, depending on the pathogenesis of the disease and the key players in disease development. The detailed findings for each autoimmune disease are elaborated upon in the following paragraphs.
Systemic lupus erythematosus is a typical autoimmune disease, which is characterized by abnormal APCs and T and B cells, with abundant autoantibodies (64–66). Dysregulated production of IL-17 (67) and abnormally differentiated follicular helper T (Tfh) cells (68, 69), aberrant DCs, and plasma B cells (70) have been identified to play an essential role in the pathogenesis of SLE. Although autophagy negatively regulates IL-17 production, the inhibition of autophagy in a lupus mouse model reduced the disease phenotype by partially suppressing plasma cell differentiation and antibody production (44). In addition, GWAS in lupus cohorts have identified that several SNPs in the Atg5 gene confer genetic susceptibility to lupus (71, 72). In addition, in a follow-up study, a SNP in the Atg5 gene, rs573775, was identified to be related to IL-10 production and higher risk of lupus (73). In APCs, LAP has been found to be required for the trafficking of immune complexes and TLR9 into the interferon signaling pathway and to promote type 1 interferon production (21, 74), which is a key player in lupus pathogenesis. In addition, autophagy has been reported to deliver viral ligands to TLR7 in plasmacytoid DCs during vesicular stomatitis virus and Sendai virus infection and contribute to type 1 interferon production (75), which might be associated with lupus. However, defects in LAP, rather than canonical autophagy, can cause SLE-like phenotypes (76) with IL-10 production (77). Increased autophagy has been observed in T and B cells from a lupus mouse model, as well as in PBMCs from patients with SLE (44). In addition, autophagy-related genes, including mTOR, Beclin-1, LC3, and p62, have been found to be expressed differentially by lupus PBMCs (45). Blockade of macrophage autophagy ameliorates activated lymphocyte-derived DNA-induced murine lupus possibly via inhibition of proinflammatory cytokine production, such as IL-6 and TNF-alpha (78). Recently, IL-21, which is a key cytokine produced by Tfh cells, has been found to induce mTOR activation and further eliminate autophagy and differentiation of Treg cells (79). To summarize, ATG5 deficiency and mTOR elevation in innate immunity leads to insufficient autophagy, which results in reduced dead cells clearance, enhanced levels of cytoplasmic nucleic acid and autoantigens. As a consequence, increased type 1 IFN by DCs can induce B cell hyper-differentiation and antibody production. In adaptive immunity, high LC3 and accumulation of autophagic vacuoles can increase autophagy and promote the survival of T and B cells (8).
In addition, environmental factors, such as UV light and Epstein–Barr virus (EBV) infection, which have been shown to contribute to the initiation of lupus, have been linked to autophagy. For example, UV-induced DNA damage has been found to result in decreased expression of AMBRA1 and ULK1, which are important mechanisms in autophagy (80). In addition, autophagy has been reported to be involved in MHC-II presentation of EB nuclear antigen 1 to T cells (81) and to participate in EBV infections (82). Furthermore, severe vitamin D deficiency affected the expression of ATGs in PBMCs and T-cell subsets in active SLE patients, indicating that vitamin D may affect T-cell subsets via regulating autophagy (83).
In SLE treatment, autophagy has been reported to be a therapeutic target. Rapamycin is a FDA approved immunosuppressive agent for organ transplantation. It excuses its inhibitory effects on T cells via blocking mTORC1 (84), which is also the key player of autophagy. Rapamycin has been shown to be efficient in treating lupus mice and patients, with the decreased levels of autoantibodies, proteinuria, and prolonged survival in mice and patients (85, 86). In an off-label clinical study, refractory SLE patients were treated with rapamycin. Compared with standard treatment, the rapamycin-treated group showed decreased disease activity and prednisone requirement (87). And this suppressive effect might be conducted through inhibition on HRES 1/Rab4 and Rab5A and limiting the production of type I IFN by DCs (88). Besides, other treatments also have effects on autophagy. In a clinical observation, hydroxychloroquine, which is the most common treatment for SLE, has been found to inhibit autophagy, particularly LAP-mediated autophagy (89). Glucocorticoids can induce autophagy via inhibiting IP3-mediated calcium signaling and mTOR (90). Anti-CD20 mAbs can trigger autophagy by caspase-independent cell death induction (91). And anti-TNF alpha mAbs can inhibit autophagy by limiting proautophagic cytokine production (92). In a recent study, inhibition of Treg cell differentiation and IL-21 has been found to repress rapamycin axis via suppression of autophagy in lupus patients (79). Although different drugs have various effects on autophagy in lupus, and the role of autophagy in lupus can be friend or foe, the balance between innate immunity and adaptive immune response should be considered when consider autophagy as therapeutic target.
Psoriasis is an inflammatory autoimmune skin disease, which can also affect other organs. Approximately 6–42% of psoriasis patients will develop psoriatic arthritis (93). The pathogenesis of psoriasis is unclear. However, increased epithelial keratinocyte proliferation is an essential characteristic of psoriasis (94), and IL-17 and other inflammatory cytokines have been revealed to play an important role in the development of psoriasis. There is no direct evidence to show whether ATG16L1 contributes to psoriasis. However, defects in autophagy have been found to result in proinflammatory cytokine production and keratinocyte proliferation via increased p62 expression (95). Enhanced expression of the autophagy-related gene SQSTM1 has been observed in psoriatic skin lesions (95). In addition, mutation of the psoriasis risk gene AP1S3 has been found to result in impaired autophagy and increased skin inflammation (96), and increased expression of ATG16L1 has been observed in DCs from psoriatic arthritic patients (97). The inhibition of autophagy via activation of PI3K/AKT/mTOR has been suggested as a therapeutic method for the treatment of IL-17a-mediated psoriasis (98). In addition, other studies have shown the therapeutic role of autophagy in psoriasis via inhibition of IL-17a production (58). In addition, the inhibition of autophagy by chloroquine may accelerate psoriasis via promoting IL-23 production (99). In addition, vitamin D, sirolimus, retinoids, and UVB therapy, which can promote psoriasis, can induce the activation of autophagy (100–103). Taken together, these findings indicate that autophagy shows a therapeutic role in this disease.
Multiple sclerosis is an inflammatory disorder that is characterized by immune system reactivity against myelin in the central nervous system, resulting in varying degrees of either relapsing or progressive neurological degeneration. ATG5 (104) and immune-related GTPase M (IRGM) 1 are increased, while ATG16L2 is decreased, in autoreactive T cells in EAE and actively relapsing-remitting MS brains (105). Inhibition of autophagy by conditional knockout of Beclin-1 in CD4+ T cells has shown a protective role in the EAE model (31). Similar effects have been observed in an ATG7 conditional knockout system (35). Administration of rapamycin reduces relapsing–remitting EAE via inhibition of autophagy (106), indicating a pathological role of autophagy in MS.
Rheumatoid arthritis is a chronic and systemic inflammatory autoimmune condition that primarily affects the joints and is characterized by progressive destruction of the joints. The pathogenesis of RA remains unclear. However, dysregulated immune cells, such as Th17 cells, Tfh cells, macrophages, B cells, and fibroblast-like synoviocytes have been identified to contribute to this disorder (107). Fibroblasts are a key player, and their survival has been found to be regulated by autophagy induction and CHOP underexpression under endoplasmic reticulum stress (108). Increased levels of autophagy have been observed in the synovial tissues from patients with active RA and are correlated with disease activity (109). However, the effect of autophagy on the survival of RA synovial fibroblasts is controversial (110). In another study, reduced expression of ALFY and the formation of p62-positive polyubiquitinated protein aggregates promote cell death in RA synovial fibroblasts under severe ER stress (111). Moreover, IL-17-mediated mitochondrial dysfunction has been found to impair apoptosis in RA synovial fibroblasts through activation of autophagy (112).
In immune cells, increased autophagy has been observed in RA CD4+ T cells, resulting in T-cell hyperactivation and resistance to apoptosis (113). The inhibition of autophagy via an ATG7 knockdown system showed impaired bone destruction in TNF-mediated arthritis (114), partially resulting from the reduced production of IL-6 and IL-1 by inhibition of autophagy. The autophagy-related protein optineurin has also been found to negatively regulate osteoclastogenesis by modulating NF-κB and IFN-β signaling (115). Taken together, these findings indicate that autophagy shows a pathological role in RA by regulating inflammatory cytokines and bone destruction.
Inflammatory bowel disease refers to two different chronic conditions or diseases that may be related, Crohn’s disease and ulcerative colitis, which consist of inflammation of the wall of the bowel or intestines. GWAS have identified ATG16L1 and immunity-related IRGM in Crohn’s disease (116), indicating a role of autophagy in the pathogenesis of IBD. Altered expression of IRGM leads to Crohn’s disease with defective autophagy (117). In addition, IBD is an IL-17a- and IL-1 beta-mediated disease. The deletion of ATG16L1 also leads to increased IL-1 beta production in macrophages (118), which might contribute to IBD. Moreover, ATG16L1 and nucleotide-binding oligomerization domain-containing protein 2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis (119).
The interplay between autophagy and the immune system emphasizes an important role of autophagy in the pathogenesis of autoimmune diseases. Autophagy is involved in pathogen removal, cytokine secretion, lymphocyte survival and differentiation, MHC presentation, apoptotic cell clearance, and pro-inflammatory signaling. In both in vivo and in vitro systems, inhibition of autophagy ameliorates diseases including SLE, MS, and RA. However, in other cases, it seems to exacerbate diseases such as psoriasis, psoriatic arthritis, and IBD. Even in RA, autophagy shows a therapeutic and pathogenesis role in the survival of RAFLS (Table 1). In addition, the regulation of autophagy varies in different tissues and cells. This evidence means that extreme care should be exercised if autophagy is to be utilized as a therapeutic target. Individual differences, even in the same types of diseases, should be considered. For example, SLE is a heterogeneous disease, and lupus patients might be either predominated by IL-17a and/or IL-21 expression. The outcome might be totally different if autophagy inhibition is applied in these two different types of patients. Therefore, further investigation is needed to clarify the regulation of autophagy in each autoimmune disease, and personalized therapy is strongly recommended in the future.
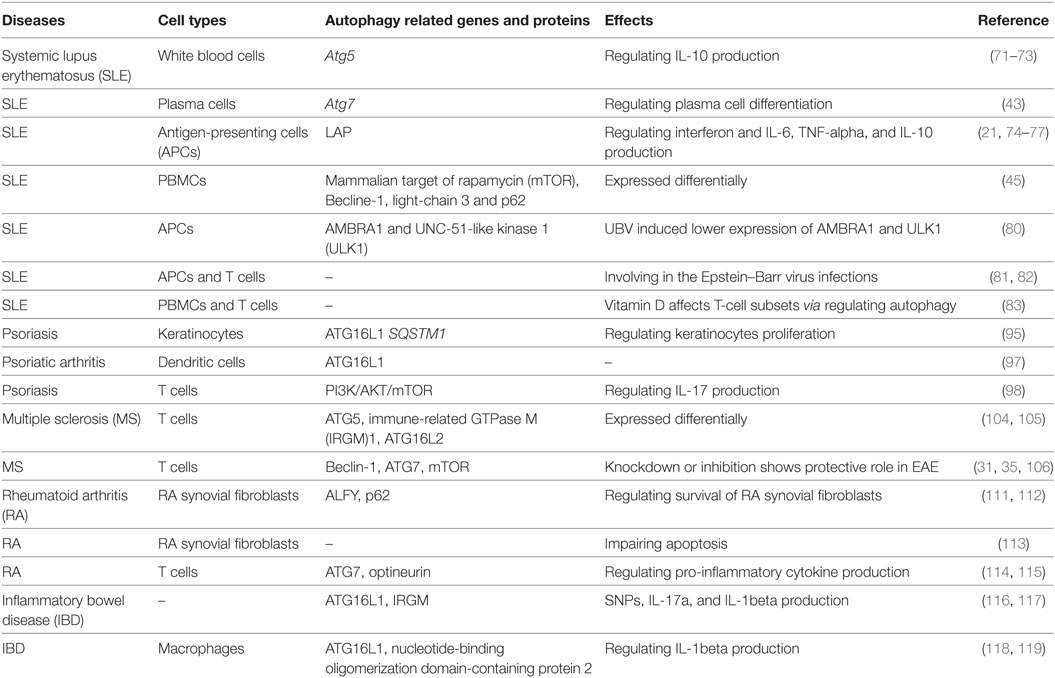
Table 1. The regulation of autophagy in autoimmune diseases.
HY and HW wrote the manuscript. YC, JZ, MZ, LL, and GC edited the manuscript. QL revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer PS and the handling Editor declared their shared affiliation.
This work was supported by the National Natural Science Foundation of China (No. 81773332, No. 81602767, No. 81430074, No. 91442116, No. 81373195, and No. 81771761), National Basic Research Program of China (No. 2014CB541904), the Programs of Science-Technology Commission of Hunan Province (2013F J4202), the Natural Science Foundation of Hunan Province (2017JJ3453, 2017SK2042), the National Key research and Development Program of China (2016YFC0903900), and the Natural Key Clinical Specialty Construction Project of National Health and Family Planning Commission of the People’s Republic of China.
1. Doria A, Gatto M, Punzi L. Autophagy in human health and disease. N Engl J Med (2013) 368(19):1845. doi:10.1056/NEJMc1303158
2. Lamark T, Johansen T. Autophagy: links with the proteasome. Curr Opin Cell Biol (2010) 22(2):192–8. doi:10.1016/j.ceb.2009.11.002
3. Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity (2007) 26(1):79–92. doi:10.1016/j.immuni.2006.10.018
4. Catarino S, Pereira P, Girao H. Molecular control of chaperone-mediated autophagy. Essays Biochem (2017) 61(6):663–74. doi:10.1042/EBC20170057
5. Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci (1990) 15(8):305–9. doi:10.1016/0968-0004(90)90019-8
6. Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol (2012) 22(8):407–17. doi:10.1016/j.tcb.2012.05.006
7. Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci (2012) 69(7):1125–36. doi:10.1007/s00018-011-0865-5
8. Yang Z, Goronzy JJ, Weyand CM. Autophagy in autoimmune disease. J Mol Med (Berl) (2015) 93(7):707–17. doi:10.1007/s00109-015-1297-8
9. Kalachev AV, Yurchenko OV. Microautophagy in nutritive phagocytes of sea urchins. Protoplasma (2017) 254(1):609–14. doi:10.1007/s00709-016-0963-1
10. Brest P, Corcelle EA, Cesaro A, Chargui A, Belaid A, Klionsky DJ, et al. Autophagy and Crohn’s disease: at the crossroads of infection, inflammation, immunity, and cancer. Curr Mol Med (2010) 10(5):486–502. doi:10.2174/156652410791608252
11. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol (2013) 13(10):722–37. doi:10.1038/nri3532
12. Law AH, Lee DC, Yuen KY, Peiris M, Lau AS. Cellular response to influenza virus infection: a potential role for autophagy in CXCL10 and interferon-alpha induction. Cell Mol Immunol (2010) 7(4):263–70. doi:10.1038/cmi.2010.25
13. Li W, Yang Q, Mao Z. Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mol Life Sci (2011) 68(5):749–63. doi:10.1007/s00018-010-0565-6
14. Noda NN, Inagaki F. Mechanisms of autophagy. Annu Rev Biophys (2015) 44:101–22. doi:10.1146/annurev-biophys-060414-034248
15. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011) 13(2):132–41. doi:10.1038/ncb2152
16. Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev (2010) 90(4):1383–435. doi:10.1152/physrev.00030.2009
17. Salminen A, Kaarniranta K, Kauppinen A. Beclin 1 interactome controls the crosstalk between apoptosis, autophagy and inflammasome activation: impact on the aging process. Ageing Res Rev (2013) 12(2):520–34. doi:10.1016/j.arr.2012.11.004
18. Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A (2011) 108(42):17396–401. doi:10.1073/pnas.1113421108
19. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature (2011) 469(7330):323–35. doi:10.1038/nature09782
20. Delgado MA, Deretic V. Toll-like receptors in control of immunological autophagy. Cell Death Differ (2009) 16(7):976–83. doi:10.1038/cdd.2009.40
21. Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity (2012) 37(6):986–97. doi:10.1016/j.immuni.2012.09.014
22. Lai SC, Devenish RJ. LC3-associated phagocytosis (LAP): connections with host autophagy. Cells (2012) 1(3):396–408. doi:10.3390/cells1030396
23. Deretic V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol Rev (2011) 240(1):92–104. doi:10.1111/j.1600-065X.2010.00995.x
24. Pierdominici M, Vomero M, Barbati C, Colasanti T, Maselli A, Vacirca D, et al. Role of autophagy in immunity and autoimmunity, with a special focus on systemic lupus erythematosus. FASEB J (2012) 26(4):1400–12. doi:10.1096/fj.11-194175
25. Chemali M, Radtke K, Desjardins M, English L. Alternative pathways for MHC class I presentation: a new function for autophagy. Cell Mol Life Sci (2011) 68(9):1533–41. doi:10.1007/s00018-011-0660-3
26. Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med (2007) 204(1):25–31. doi:10.1084/jem.20061303
27. Jia W, Pua HH, Li QJ, He YW. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol (2011) 186(3):1564–74. doi:10.4049/jimmunol.1001822
28. Jia W, He YW. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J Immunol (2011) 186(9):5313–22. doi:10.4049/jimmunol.1002404
29. Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, et al. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest (2006) 116(8):2161–72. doi:10.1172/JCI26185
30. Bell BD, Leverrier S, Weist BM, Newton RH, Arechiga AF, Luhrs KA, et al. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci U S A (2008) 105(43):16677–82. doi:10.1073/pnas.0808597105
31. Kovacs JR, Li C, Yang Q, Li G, Garcia IG, Ju S, et al. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ (2012) 19(1):144–52. doi:10.1038/cdd.2011.78
32. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science (2007) 315(5808):107–11. doi:10.1126/science.1136080
33. Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity (2010) 32(2):227–39. doi:10.1016/j.immuni.2009.12.006
34. Ghislat G, Lawrence T. Autophagy in dendritic cells. Cell Mol Immunol (2018) 15:1–9. doi:10.1038/cmi.2018.2
35. Bhattacharya A, Parillon X, Zeng S, Han S, Eissa NT. Deficiency of autophagy in dendritic cells protects against experimental autoimmune encephalomyelitis. J Biol Chem (2014) 289(38):26525–32. doi:10.1074/jbc.M114.575860
36. Morris S, Swanson MS, Lieberman A, Reed M, Yue Z, Lindell DM, et al. Autophagy-mediated dendritic cell activation is essential for innate cytokine production and APC function with respiratory syncytial virus responses. J Immunol (2011) 187(8):3953–61. doi:10.4049/jimmunol.1100524
37. Cheng LS, Li J, Liu Y, Wang FP, Wang SQ, She WM, et al. HMGB1-induced autophagy: a new pathway to maintain Treg function during chronic hepatitis B virus infection. Clin Sci (Lond) (2017) 131(5):381–94. doi:10.1042/CS20160704
38. Kabat AM, Harrison OJ, Riffelmacher T, Moghaddam AE, Pearson CF, Laing A, et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. Elife (2016) 5:e12444. doi:10.7554/eLife.12444
39. Alissafi T, Banos A, Boon L, Sparwasser T, Ghigo A, Wing K, et al. Tregs restrain dendritic cell autophagy to ameliorate autoimmunity. J Clin Invest (2017) 127(7):2789–804. doi:10.1172/JCI92079
40. Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy (2008) 4(3):309–14. doi:10.4161/auto.5474
41. Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol (2013) 14(3):298–305. doi:10.1038/ni.2524
42. Eriksen AB, Torgersen ML, Holm KL, Abrahamsen G, Spurkland A, Moskaug JO, et al. Retinoic acid-induced IgG production in TLR-activated human primary B cells involves ULK1-mediated autophagy. Autophagy (2015) 11(3):460–71. doi:10.1080/15548627.2015.1009797
43. Milan E, Perini T, Resnati M, Orfanelli U, Oliva L, Raimondi A, et al. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy (2015) 11(7):1161–78. doi:10.1080/15548627.2015.1052928
44. Clarke AJ, Ellinghaus U, Cortini A, Stranks A, Simon AK, Botto M, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis (2015) 74(5):912–20. doi:10.1136/annrheumdis-2013-204343
45. Wu ZZ, Zhang JJ, Gao CC, Zhao M, Liu SY, Gao GM, et al. Expression of autophagy related genes mTOR, Becline-1, LC3 and p62 in the peripheral blood mononuclear cells of systemic lupus erythematosus. Am J Clin Exp Immunol (2017) 6(1):1–8.
46. Yuan J, Yu M, Li HH, Long Q, Liang W, Wen S, et al. Autophagy contributes to IL-17-induced plasma cell differentiation in experimental autoimmune myocarditis. Int Immunopharmacol (2014) 18(1):98–105. doi:10.1016/j.intimp.2013.11.008
47. McLeod IX, He Y. Roles of autophagy in lymphocytes: reflections and directions. Cell Mol Immunol (2010) 7(2):104–7. doi:10.1038/cmi.2009.115
48. Trocoli A, Djavaheri-Mergny M. The complex interplay between autophagy and NF-kappaB signaling pathways in cancer cells. Am J Cancer Res (2011) 1(5):629–49.
49. Hayden MS, Ghosh S. NF-kappaB in immunobiology. Cell Res (2011) 21(2):223–44. doi:10.1038/cr.2011.13
50. Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol (2009) 27:693–733. doi:10.1146/annurev.immunol.021908.132641
51. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell (2008) 132(3):344–62. doi:10.1016/j.cell.2008.01.020
52. Criollo A, Chereau F, Malik SA, Niso-Santano M, Marino G, Galluzzi L, et al. Autophagy is required for the activation of NFkappaB. Cell Cycle (2012) 11(1):194–9. doi:10.4161/cc.11.1.18669
53. Paul S, Kashyap AK, Jia W, He YW, Schaefer BC. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-kappaB. Immunity (2012) 36(6):947–58. doi:10.1016/j.immuni.2012.04.008
54. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell (2016) 164(5):896–910. doi:10.1016/j.cell.2015.12.057
55. Kanayama M, Inoue M, Danzaki K, Hammer G, He YW, Shinohara ML. Autophagy enhances NFkappaB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat Commun (2015) 6:5779. doi:10.1038/ncomms6779
56. Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A (2012) 109(46):E3168–76. doi:10.1073/pnas.1210500109
57. Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem (2011) 286(11):9587–97. doi:10.1074/jbc.M110.202911
58. Peral de Castro C, Jones SA, Ni Cheallaigh C, Hearnden CA, Williams L, Winter J, et al. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J Immunol (2012) 189(8):4144–53. doi:10.4049/jimmunol.1201946
59. Park HJ, Lee SJ, Kim SH, Han J, Bae J, Kim SJ, et al. IL-10 inhibits the starvation induced autophagy in macrophages via class I phosphatidylinositol 3-kinase (PI3K) pathway. Mol Immunol (2011) 48(4):720–7. doi:10.1016/j.molimm.2010.10.020
60. Qin B, Zhou Z, He J, Yan C, Ding S. IL-6 inhibits starvation-induced autophagy via the STAT3/Bcl-2 signaling pathway. Sci Rep (2015) 5:15701. doi:10.1038/srep15701
61. Dutta RK, Kathania M, Raje M, Majumdar S. IL-6 inhibits IFN-gamma induced autophagy in Mycobacterium tuberculosis H37Rv infected macrophages. Int J Biochem Cell Biol (2012) 44(6):942–54. doi:10.1016/j.biocel.2012.02.021
62. Li XZ, Sui CY, Chen Q, Chen XP, Zhang H, Zhou XP. Promotion of autophagy at the maturation step by IL-6 is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Mol Cell Biochem (2013) 380(1–2):219–27. doi:10.1007/s11010-013-1676-9
63. Wu DJ, Adamopoulos IE. Autophagy and autoimmunity. Clin Immunol (2017) 176:55–62. doi:10.1016/j.clim.2017.01.007
64. Wu H, Zeng J, Yin J, Peng Q, Zhao M, Lu Q. Organ-specific biomarkers in lupus. Autoimmun Rev (2017) 16(4):391–7. doi:10.1016/j.autrev.2017.02.011
65. Wu H, Zhao M, Chang C, Lu Q. The real culprit in systemic lupus erythematosus: abnormal epigenetic regulation. Int J Mol Sci (2015) 16(5):11013–33. doi:10.3390/ijms160511013
66. Wu H, Zhao M, Tan L, Lu Q. The key culprit in the pathogenesis of systemic lupus erythematosus: aberrant DNA methylation. Autoimmun Rev (2016) 15(7):684–9. doi:10.1016/j.autrev.2016.03.002
67. Mok MY, Wu HJ, Lo Y, Lau CS. The relation of interleukin 17 (IL-17) and IL-23 to Th1/Th2 cytokines and disease activity in systemic lupus erythematosus. J Rheumatol (2010) 37(10):2046–52. doi:10.3899/jrheum.100293
68. Wu H, Huang X, Qiu H, Zhao M, Liao W, Yuan S, et al. High salt promotes autoimmunity by TET2-induced DNA demethylation and driving the differentiation of Tfh cells. Sci Rep (2016) 6:28065. doi:10.1038/srep28065
69. Huang X, Wu H, Qiu H, Yang H, Deng Y, Zhao M, et al. The expression of Bcl-6 in circulating follicular helper-like T cells positively correlates with the disease activity in systemic lupus erythematosus. Clin Immunol (2016) 173:161–70. doi:10.1016/j.clim.2016.10.017
70. Wu H, Deng Y, Feng Y, Long D, Ma K, Wang X, et al. Epigenetic regulation in B-cell maturation and its dysregulation in autoimmunity. Cell Mol Immunol (2018) 14:1–9. doi:10.1038/cmi.2017.133
71. Zhou XJ, Lu XL, Lv JC, Yang HZ, Qin LX, Zhao MH, et al. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis (2011) 70(7):1330–7. doi:10.1136/ard.2010.140111
72. International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet (2008) 40(2):204–10. doi:10.1038/ng.81
73. Lopez P, Alonso-Perez E, Rodriguez-Carrio J, Suarez A. Influence of Atg5 mutation in SLE depends on functional IL-10 genotype. PLoS One (2013) 8(10):e78756. doi:10.1371/journal.pone.0078756
74. Simon AK, Clarke AJ. Non-canonical autophagy LAPs lupus. Cell Death Differ (2016) 23(8):1267–8. doi:10.1038/cdd.2016.55
75. Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science (2007) 315(5817):1398–401. doi:10.1126/science.1136880
76. Allison SJ. Systemic lupus erythematosus: defective noncanonical autophagy in SLE-like disease. Nat Rev Rheumatol (2016) 12(6):311. doi:10.1038/nrrheum.2016.73
77. Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature (2016) 533(7601):115–9. doi:10.1038/nature17950
78. Li B, Yue Y, Dong C, Shi Y, Xiong S. Blockade of macrophage autophagy ameliorates activated lymphocytes-derived DNA induced murine lupus possibly via inhibition of proinflammatory cytokine production. Clin Exp Rheumatol (2014) 32(5):705–14.
79. Kato H, Perl A. Blockade of Treg cell differentiation and function by the Interleukin-21-mechanistic target of rapamycin axis via suppression of autophagy in patients with systemic lupus erythematosus. Arthritis Rheumatol (2018) 70(3):427–38. doi:10.1002/art.40380
80. Kemp MG, Lindsey-Boltz LA, Sancar A. UV light potentiates STING (stimulator of interferon genes)-dependent innate immune signaling through deregulation of ULK1 (Unc51-like Kinase 1). J Biol Chem (2015) 290(19):12184–94. doi:10.1074/jbc.M115.649301
81. Leung CS, Haigh TA, Mackay LK, Rickinson AB, Taylor GS. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc Natl Acad Sci U S A (2010) 107(5):2165–70. doi:10.1073/pnas.0909448107
82. Lee DY, Sugden B. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene (2008) 27(20):2833–42. doi:10.1038/sj.onc.1210946
83. Zhao M, Duan XH, Wu ZZ, Gao CC, Wang N, Zheng ZH. Severe vitamin D deficiency affects the expression of autophagy related genes in PBMCs and T-cell subsets in active systemic lupus erythematosus. Am J Clin Exp Immunol (2017) 6(4):43–51.
84. Walsh CM, Edinger AL. The complex interplay between autophagy, apoptosis, and necrotic signals promotes T-cell homeostasis. Immunol Rev (2010) 236:95–109. doi:10.1111/j.1600-065X.2010.00919.x
85. Lai ZW, Kelly R, Winans T, Marchena I, Shadakshari A, Yu J, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet (2018) 391(10126):1186–96. doi:10.1016/S0140-6736(18)30485-9
86. Ramos-Barron A, Pinera-Haces C, Gomez-Alamillo C, Santiuste-Torcida I, Ruiz JC, Buelta-Carrillo L, et al. Prevention of murine lupus disease in (NZBxNZW)F1 mice by sirolimus treatment. Lupus (2007) 16(10):775–81. doi:10.1177/0961203307081401
87. Fernandez D, Bonilla E, Mirza N, Niland B, Perl A. Rapamycin reduces disease activity and normalizes T cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheum (2006) 54(9):2983–8. doi:10.1002/art.22085
88. Cao W, Manicassamy S, Tang H, Kasturi SP, Pirani A, Murthy N, et al. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat Immunol (2008) 9(10):1157–64. doi:10.1038/ni.1645
89. Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol (2005) 25(3):1025–40. doi:10.1128/MCB.25.3.1025-1040.2005
90. Harr MW, McColl KS, Zhong F, Molitoris JK, Distelhorst CW. Glucocorticoids downregulate Fyn and inhibit IP(3)-mediated calcium signaling to promote autophagy in T lymphocytes. Autophagy (2010) 6(7):912–21. doi:10.4161/auto.6.7.13290
91. Turzanski J, Daniels I, Haynes AP. Involvement of macroautophagy in the caspase-independent killing of Burkitt lymphoma cell lines by rituximab. Br J Haematol (2009) 145(1):137–40. doi:10.1111/j.1365-2141.2008.07555.x
92. Ni Cheallaigh C, Keane J, Lavelle EC, Hope JC, Harris J. Autophagy in the immune response to tuberculosis: clinical perspectives. Clin Exp Immunol (2011) 164(3):291–300. doi:10.1111/j.1365-2249.2011.04381.x
93. Gladman DD, Antoni C, Mease P, Clegg DO, Nash P. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis (2005) 64(Suppl 2):ii14–ii17. doi:10.1136/ard.2004.032482
95. Lee HM, Shin DM, Yuk JM, Shi G, Choi DK, Lee SH, et al. Autophagy negatively regulates keratinocyte inflammatory responses via scaffolding protein p62/SQSTM1. J Immunol (2011) 186(2):1248–58. doi:10.4049/jimmunol.1001954
96. Mahil SK, Twelves S, Farkas K, Setta-Kaffetzi N, Burden AD, Gach JE, et al. AP1S3 mutations cause skin autoinflammation by disrupting keratinocyte autophagy and up-regulating IL-36 production. J Invest Dermatol (2016) 136(11):2251–9. doi:10.1016/j.jid.2016.06.618
97. Wenink MH, Santegoets KC, Butcher J, van Bon L, Lamers-Karnebeek FG, van den Berg WB, et al. Impaired dendritic cell proinflammatory cytokine production in psoriatic arthritis. Arthritis Rheum (2011) 63(11):3313–22. doi:10.1002/art.30577
98. Varshney P, Saini N. PI3K/AKT/mTOR activation and autophagy inhibition plays a key role in increased cholesterol during IL-17A mediated inflammatory response in psoriasis. Biochim Biophys Acta (2018) 1864(5 Pt A):1795–803. doi:10.1016/j.bbadis.2018.02.003
99. Said A, Bock S, Lajqi T, Muller G, Weindl G. Chloroquine promotes IL-17 production by CD4+ T cells via p38-dependent IL-23 release by monocyte-derived Langerhans-like cells. J Immunol (2014) 193(12):6135–43. doi:10.4049/jimmunol.1303276
100. Wang RC, Levine B. Calcipotriol induces autophagy in HeLa cells and keratinocytes. J Invest Dermatol (2011) 131(4):990–3. doi:10.1038/jid.2010.423
101. Rajawat Y, Hilioti Z, Bossis I. Autophagy: a target for retinoic acids. Autophagy (2010) 6(8):1224–6. doi:10.4161/auto.6.8.13793
102. Anguiano J, Garner TP, Mahalingam M, Das BC, Gavathiotis E, Cuervo AM. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol (2013) 9(6):374–82. doi:10.1038/nchembio.1230
103. Yang Y, Wang H, Wang S, Xu M, Liu M, Liao M, et al. GSK3beta signaling is involved in ultraviolet B-induced activation of autophagy in epidermal cells. Int J Oncol (2012) 41(5):1782–8. doi:10.3892/ijo.2012.1620
104. Paunovic V, Petrovic IV, Milenkovic M, Janjetovic K, Pravica V, Dujmovic I, et al. Autophagy-independent increase of ATG5 expression in T cells of multiple sclerosis patients. J Neuroimmunol (2018) 319:100–5. doi:10.1016/j.jneuroim.2018.03.001
105. Igci M, Baysan M, Yigiter R, Ulasli M, Geyik S, Bayraktar R, et al. Gene expression profiles of autophagy-related genes in multiple sclerosis. Gene (2016) 588(1):38–46. doi:10.1016/j.gene.2016.04.042
106. Liang P, Le W. Role of autophagy in the pathogenesis of multiple sclerosis. Neurosci Bull (2015) 31(4):435–44. doi:10.1007/s12264-015-1545-5
107. Firestein GS. Evolving concepts of rheumatoid arthritis. Nature (2003) 423(6937):356–61. doi:10.1038/nature01661
108. Shin YJ, Han SH, Kim DS, Lee GH, Yoo WH, Kang YM, et al. Autophagy induction and CHOP under-expression promotes survival of fibroblasts from rheumatoid arthritis patients under endoplasmic reticulum stress. Arthritis Res Ther (2010) 12(1):R19. doi:10.1186/ar2921
109. Zhu L, Wang H, Wu Y, He Z, Qin Y, Shen Q. The autophagy level is increased in the synovial tissues of patients with active rheumatoid arthritis and is correlated with disease severity. Mediators Inflamm (2017) 2017:7623145. doi:10.1155/2017/7623145
110. Buckland J. Rheumatoid arthritis: autophagy: a dual role in the life and death of RASFs. Nat Rev Rheumatol (2013) 9(11):637. doi:10.1038/nrrheum.2013.148
111. Kato M, Ospelt C, Gay RE, Gay S, Klein K. Dual role of autophagy in stress-induced cell death in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol (2014) 66(1):40–8. doi:10.1002/art.38190
112. Kim EK, Kwon JE, Lee SY, Lee EJ, Kim DS, Moon SJ, et al. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis (2017) 8(1):e2565. doi:10.1038/cddis.2016.490
113. van Loosdregt J, Rossetti M, Spreafico R, Moshref M, Olmer M, Williams GW, et al. Increased autophagy in CD4(+) T cells of rheumatoid arthritis patients results in T-cell hyperactivation and apoptosis resistance. Eur J Immunol (2016) 46(12):2862–70. doi:10.1002/eji.201646375
114. Lin NY, Beyer C, Giessl A, Kireva T, Scholtysek C, Uderhardt S, et al. Autophagy regulates TNFalpha-mediated joint destruction in experimental arthritis. Ann Rheum Dis (2013) 72(5):761–8. doi:10.1136/annrheumdis-2012-201671
115. Obaid R, Wani SE, Azfer A, Hurd T, Jones R, Cohen P, et al. Optineurin negatively regulates osteoclast differentiation by modulating NF-kappaB and interferon signaling: implications for Paget’s disease. Cell Rep (2015) 13(6):1096–102. doi:10.1016/j.celrep.2015.09.071
116. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet (2007) 39(5):596–604. doi:10.1038/ng2032
117. McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet (2008) 40(9):1107–12. doi:10.1038/ng.215
118. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature (2008) 456(7219):264–8. doi:10.1038/nature07383
Keywords: autophagy, light-chain 3, Agt, autoimmunity, LAP
Citation: Yin H, Wu H, Chen Y, Zhang J, Zheng M, Chen G, Li L and Lu Q (2018) The Therapeutic and Pathogenic Role of Autophagy in Autoimmune Diseases. Front. Immunol. 9:1512. doi: 10.3389/fimmu.2018.01512
Received: 02 April 2018; Accepted: 18 June 2018;
Published: 31 July 2018
Edited by:
Jennifer Martinez, National Institute of Environmental Health Sciences (NIEHS), United StatesReviewed by:
Yisong Wan, University of North Carolina at Chapel Hill, United StatesCopyright: © 2018 Yin, Wu, Chen, Zhang, Zheng, Chen, Li and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qianjin Lu, cWlhbmx1NTg2MEBjc3UuZWR1, cWlhbmx1NTg2MEBnbWFpbC5jb20=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.