 Edoardo Troncone
Edoardo Troncone Irene Marafini
Irene Marafini Carmine Stolfi
Carmine Stolfi Giovanni Monteleone
Giovanni Monteleone
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 June 2018
Sec. Mucosal Immunity
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01407
This article is part of the Research Topic Cytokines and Intestinal Mucosal Immunity View all 12 articles
In physiological conditions, the activity of the intestinal immune system is tightly regulated to prevent tissue-damaging reactions directed against components of the luminal flora. Various factors contribute to maintain immune homeostasis and diminished production and/or function of such molecules trigger and/or propagate detrimental signals, which can eventually lead to chronic colitis and colon cancer. One such a molecule is transforming growth factor-β1 (TGF-β1), a cytokine produced by many inflammatory and non-inflammatory cells and targeting virtually all the intestinal mucosal cell types, with the down-stream effect of activating intracellular Smad2/3 proteins and suppressing immune reactions. In patients with inflammatory bowel diseases (IBD), there is defective TGF-β1/Smad signaling due to high Smad7, an inhibitor of TGF-β1 activity. Indeed, knockdown of Smad7 with a specific antisense oligonucleotide restores endogenous TGF-β1 activity, thereby inhibiting inflammatory pathways in patients with IBD and colitic mice. Consistently, mice over-expressing Smad7 in T cells develop severe intestinal inflammation in various experimental models. Smad7 expression is also upregulated in colon cancer cells, in which such a protein controls positively intracellular pathways that sustain neoplastic cell growth and survival. We here review the role of TGF-β1 and Smad7 in intestinal immunity, inflammation, and cancer.
The gastrointestinal tract harbors a large number of commensal bacterial, viral, and fungal species, which trigger maturation of the mucosal immune system. There are over 300 Peyer’s patches and more than 30,000 isolated lymphoid follicles in the small and large intestines, and the lamina propria compartment is infiltrated with many CD4+ and CD8+ T cells, B cells, plasma cells, dendritic cells (DCs), and macrophages; T cells and various subsets of innate lymphoid cells (ILCs) are also present in the gut epithelium (1, 2). This state of “physiological inflammation” contributes to provide resistance to invading pathogens, while preserving barrier integrity and allowing normal absorptive and digestive functions. The maintenance of gut integrity and intestinal homeostasis depends also on the barrier effect of the epithelium, which limits translocation of luminal antigens and promotes immune regulation (3). Elegant studies in animal models of inflammation and observations in patients with chronic colitis strongly support this notion. Indeed, defects in epithelial barrier and/or lack of expression/function of counter-regulatory molecules can break the immune tolerance toward the luminal flora thus resulting in colitis and colorectal cancer (CRC) (2, 4–9). Data emerging from such studies indicate clearly that transforming growth factor (TGF)-β1 is one of the key molecules involved in the regulation of the epithelial cell biology and immunity in the gut (10, 11). TGF-β1 is a member of the TGF-β superfamily, which includes also TGF-β2, TGF-β3, bone morphogenetic proteins, and several growth and differentiation factors (12). In the gut, many immune and non-immune cells produce TGF-β1 and almost all the mucosal cells are targeted by this cytokine. TGF-β1 is secreted as part of a latent complex, which comprises latency-associated peptide (LAP) and latent TGF-β binding protein. In this form, TGF-β1 cannot bind to its receptor (13). TGF-β1 can be activated upon being released from the complex due to the proteolytic action of a number of proteinases or upon the interaction between the tripeptide integrin-binding motif on LAP and the correspondent binding sequence on αvβ3, αvβ5, αvβ6, or αvβ8 integrins expressed on the surface of epithelial cells, myofibroblasts, and DCs (14, 15). TGF-β1 signals through two transmembrane receptors with serine/threonine kinase activity, named TGF-β1 type 1 receptor (TβR1) and TGF-β1 type 2 receptor (TβR2) (13). Binding of TGF-β1 to TβR2 promotes auto-phosphorylation of the receptor and subsequent recruitment of TβR1 to form a transmembrane heterodimer. Then, the kinase activity of TβR2 determines phosphorylation of the regulatory glycine/serine-rich domain of TβR1 and, hence, the activated TβR1–TβR2 complex promotes phosphorylation of Smad2 and Smad3. Phosphorylated Smad2/3 proteins heterodimerize with Smad4 to generate a complex that moves to the nucleus, where it regulates the expression of target genes (13, 16) (Figure 1). TGF-β1-associated Smad signaling is a tightly controlled phenomenon, and another intracellular Smad protein, termed Smad7, acts as a negative regulator of such a pathway through various mechanisms. Smad7 can bind to TβR1 and compete with Smad2/3 for the catalytic site of phosphorylation, thus preventing the phosphorylation of Smad2/3 (Figure 1) (17, 18). Smad7 can recruit phosphatases to TβR1 thereby promoting de-phosphorylation and inactivation of the site (19) and can promote ubiquitination-driven proteasome-mediated degradation of TβR1 in association with E3 ubiquitin ligases SMURF1/2 (20, 21). Moreover, Smad7 can localize into the nucleus and inhibit the association of Smad2-3/Smad4 complex with target genes (22). Besides its inhibitory effect on TGFβ1 signaling, Smad7 regulates the expression and function of several molecules involved in the control of inflammation and carcinogenesis in a TGF-β1-independent manner (Figure 1).
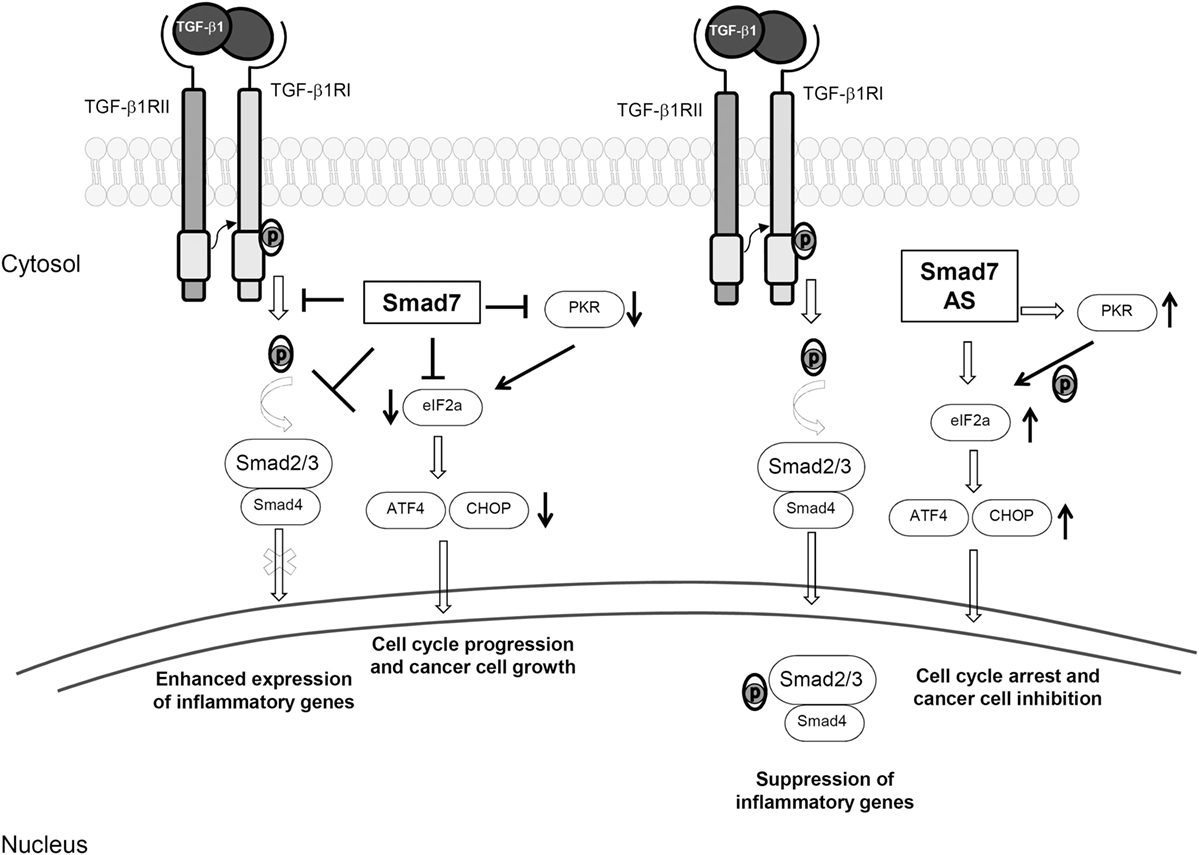
Figure 1. Smad7-induced biological effects. The left side of the figure shows the inhibitory effect of Smad7 on transforming growth factor (TGF)-β1 intracellular signaling. Smad7 binds to TGF-β receptor type I and prevents TGF-β1-driven Smad2/3 phosphorylation (p), thus sustaining inflammatory gene expression. High Smad7 prevents also eukaryotic translation initiation factor-2α (eIF2α) phosphorylation, either directly or through the inhibition of protein kinase RNA (PKR), thereby leading to downregulation of transcription factor 4 (ATF4) and CCAAT/enhancer binding protein homology protein (CHOP) with the down-stream effect of stimulating cell cycle progression and cancer cell growth. The right side of the figure shows the effect of Smad7 inhibition with a specific antisense oligonucleotide (AS). Smad7 inhibition restores TGF-β1-induced Smad2/3 phosphorylation, subsequent interaction of these two proteins with Smad4 and translocation of Smad2/3/4 complex to the nucleus, thereby suppressing inflammatory gene expression. Moreover, Smad7 knockdown causes TGF-β1-independent eIF2α phosphorylation, upregulation of ATF4 and CHOP with the down-stream effect of cell cycle arrest.
We here review the role of TGF-β1 and Smad7 in intestinal immunity, inflammation, and cancer.
TGF-β1 regulates the function of many mucosal cell types in an autocrine and paracrine manner (Figure 2). For instance, TGF-β1 suppresses proliferation and activation of CD4+ T helper (Th) lymphocytes (23). Mice with T-cell targeted deletion of TβR2 or transgenic mice expressing a dominant-negative of TβR2 are unable to respond to TGF-β1 and show a phenotype characterized by systemic autoimmunity and severe colitis (11). In both strains, activated T cells accumulate in multiple organs, including the gut, highlighting the role of TGF-β1 in blocking T cell activation, and maintaining intestinal immune tolerance. CD4+ T lymphocytes have a high grade of plasticity and can differentiate in various subsets depending on the specific cytokine milieu at the induction and effector sites (24). TGF-β1 strongly inhibits Th1 and Th2 differentiation. TGF-β1-induced inhibition of Th1 cell differentiation is mediated by a direct downregulation of the transcription factor Tbet, a master regulator of Th1 cell polarization (25). Moreover, TGF-β1 downregulates the expression of IL-12Rβ2 and prevents the expansion of Th1 responses driven by IL-12, a key cytokine in the induction of Th1-type immunity in humans (26). TGF-β1 can also directly downregulate GATA3, a transcription factor involved in Th2 cell differentiation (27, 28). In contrast, TGF-β1 promotes directly and indirectly T cells polarization toward a regulatory (T regulatory cells, Tregs) phenotype (29). Tregs express the transcription factor Foxp3 and exert regulatory functions by acting mainly on effector T cells (30). Mice with loss of TGF-β1 signaling have reduced numbers of circulating CD4+Foxp3+ Tregs, raising the possibility that TGF-β1 contributes to intestinal immune homeostasis in part by inducing Tregs differentiation (31, 32). This hypothesis is supported by studies demonstrating that TGF-β1 promotes generation of naturally occurring Tregs, a subset of Tregs, which are generated in the thymus early after birth, as well as differentiation of peripherally induced Tregs from naïve T cells (33–35). Interestingly, the number of CD4+Foxp3+ Tregs remains unaltered in mice lacking TGF-β1 in CD4+ T-cells, while TGF-β1-null mice have reduced numbers of Tregs. These findings suggest that peripheral differentiation of Tregs depends on TGF-β1 produced by other cell types rather than T cells. In the gut, CD103-expressing DCs are a major source of TGF-β1. These cells produce also elevated levels of retinoic acid (RA), which potentiates TGF-β1-induced expansion of Tregs because of a direct effect on Foxp3 promoter (36, 37). TGF-β1 and RA promote the in vitro differentiation of naïve T cells in another group of Foxp3-expressing Tregs, termed induced Tregs (38). TGF-β1 plays also a role in the interaction between intestinal immune system and gut microbiota. Clostridium butyricum promotes Tregs generation through the induction of TGF-β1 by colonic lamina propria DCs (39).
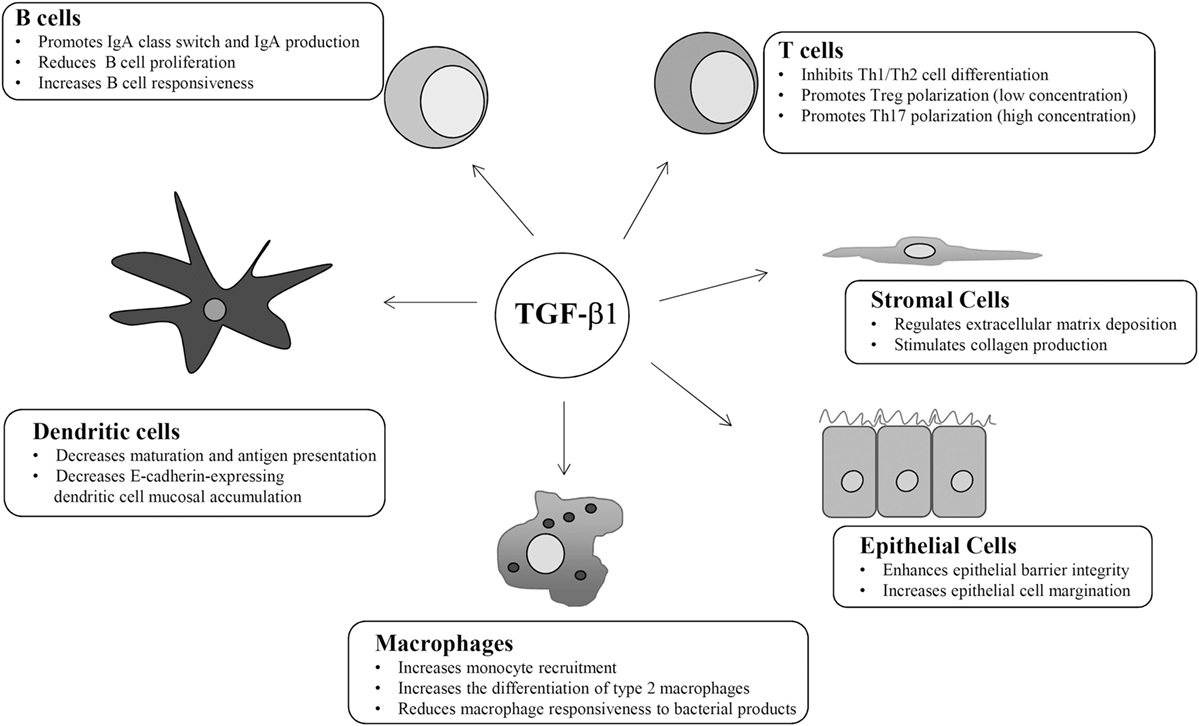
Figure 2. Schematic view of the main cell targets and biological function of transforming growth factor (TGF)-β1 in the gut.
TGF-β1, along with IL-6, IL-21, and IL-1β, contributes also to the differentiation of Th17 cells, a CD4+ T cell subset characterized by the expression and activity of the master regulator retinoid acid-related orphan receptor-γt and producing various cytokines, including IL-17A (40). It has also been demonstrated that commitment of naïve CD4+ T cells along the Th17 or Tregs phenotype depends on TGF-β1 concentration, given that low concentration of the cytokine promotes Tregs differentiation through downregulation of IL-23 receptor, while high concentration of TGF-β1, in conjunction with IL-6 and IL-21, upregulates IL-23 receptor and promotes Th17 polarization (41).
TGF-β1 controls memory CD8+ T cells. Mice with deletion of TβR2 show a reduced number of antigen-specific memory CD8+ T cells in the gut, and this phenomenon seems to be, at least in part, secondary to a reduced expression of integrins (42).
TGF-β1 is also a relevant regulator of B cell and plasm-cell biology. TGF-β1 in B cells mediates IgA class-switch and promotes IgA production (43, 44). Mice lacking TGF-β signaling in B cells do not develop intestinal inflammation, but deletion of TβR2 in CD19-expressing B cells associates with B cell hyperplasia in Peyer’s patches, modifications in B cell responsiveness and serum IgA deficiency (44, 45). Secretory IgAs control the bacterial composition, as they neutralize luminal bacteria by enhancing phagocytosis and improving the ability of DCs to present antigens. Moreover, IgAs block surface epitopes of luminal bacteria, thus inhibiting bacterial adhesion to the intestinal epithelium (46). In Peyer’s patches, B cells and DCs interact through TGF-β1-activated integrin αvβ8, and this interaction promotes IgA production (47). TGF-β1 regulates IgA production through the canonical Smad-mediated pathway as the absence of Smad2 results in IgA deficiency, while overexpression of Smad3 and 4 determines an increased IgA production (48, 49).
Innate lymphoid cells are a family of hematopoietic cells involved in host defense, immune homeostasis, and tissue remodeling. These cells belong to the innate immune system and are abundantly present at mucosal sites where they act as a first line of defense against pathogens (50). Although there is no clear evidence that TGF-β1 regulates the function of ILCs in the gut, it is known that TGF-β1 guides the differentiation of type 1 ILCs in salivary glands through a Smad4-independent pathway (51).
TGF-β1 is involved in the control of intestinal DC function. Mice with deletion of TGF-β signaling in DCs due to a selective lack of TβR2 develop systemic autoimmunity and colitis, the later being characterized by loss of goblet cells and marked mucosal lymphocytic infiltration with altered Tregs differentiation, T cells and B cells with an activated phenotype, and increased expression of pro-inflammatory cytokines (52, 53). As mentioned above, DCs produce TGF-β1 and at the same time contribute to TGF-β1 activation. Mice lacking integrin β8 in DCs fail to activate TGF-β1 and spontaneously develop colitis (54). TGF-β1 negatively regulates the mucosal accumulation of other DC subtypes, such as those expressing the adhesion molecule, E-cadherin. Interestingly, adoptive transfer of E-cadherin-expressing bone marrow DCs into T-cell-restored Rag1-deficient mice enhances Th17 cell responses and exacerbates colitis (55).
TGF-β1 is an important regulator of macrophage function. TGF-β1 produced by intestinal epithelial cells can act as a chemokine and stimulate recruitment of blood monocytes to the intestinal mucosa (56). Moreover, TGF-β1 promotes differentiation of type 2 macrophages, a subset of anti-inflammatory cells, and reduces the macrophage responsiveness to bacterial products, thus promoting an anergic state that is crucial to maintain intestinal homeostasis (57). Mice with selective knockout of TβR2 in macrophages do not show spontaneous inflammation in the gut, but develop a more severe colitis after DSS administration, with reduced levels of IL-10, further confirming the ability of the cytokine to trigger anti-inflammatory signals in macrophages (58).
TGF-β1 targets also non-immune cells, including epithelial cells and stromal cells. Both these cell types produce elevated amounts of the cytokine. TGF-β1 promotes the expression of tight junction protein (i.e., Claudin-1) and adhesion molecules, thus enhancing epithelial barrier integrity, and is a powerful inducer of intestinal epithelial cell margination, a phenomenon that facilitates wound healing (Figure 2) (59). Mice with selective deletion of TGFβ1 signaling in the intestinal epithelium do not develop inflammation but are more susceptible to DSS-colitis (60). TGF-β1 stimulates stromal cells (i.e., myofibroblasts) to produce collagen and is a crucial regulator of extra-cellular matrix deposition (Figure 2), a phenomenon that is relevant for wound healing processes (61). A poorly controlled TGF-β1-induced extra-cellular matrix deposition has been involved in the development of intestinal strictures, such as those complicating Crohn’s disease (CD) natural history (62, 63). In the gut, fibrogenesis is a complex and multifactorial process that involves several mediators, and TGF-β1 is supposed to be the most relevant pro-fibrogenic cytokine (64). TGF-β1 promotes fibronectin, type I collagen, and connective tissue growth factor production in fibroblasts isolated from CD strictures and enhances fibroblasts contractile activity (65).
CD and ulcerative colitis (UC), are chronic, relapsing inflammatory disorders of the gastrointestinal tract and represent the two most relevant forms of IBD in humans (66). The etiology of IBD is unknown but accumulating evidence suggests that IBD are multifactorial diseases in which environmental and genetic factors trigger an abnormal immune response against component of intestinal microflora (67). In both disorders, defects of counter-regulatory factors/mechanisms contribute to amplify mucosal inflammatory signals. One such defect involves the TGF-β1/Smad pathway. In the intestine of healthy individuals, TGF-β1 functions are properly suggested by the constitutive elevated levels of phosphorylated Smad3. Moreover, in vitro treatment of normal intestinal mucosal samples with an anti-TGF-β1 antibody increases expression of pro-inflammatory molecules such as T-bet and IFN-γ and stimulation of normal lamina propria mononuclear cells (LPMC) with exogenous TGF-β1 inhibits NF-kB activity and reduces IL-8 production (68–70). In contrast, stimulation of IBD LPMC with TGF-β1 neither inhibits NF-kB activation nor reduces the production of pro-inflammatory mediators, highlighting the possibility that IBD cells are resistant to TGF-β1-mediated immune suppression (70). This is consistent with the demonstration that in inflamed intestine of IBD patients, there is reduced Smad2/3 phosphorylation (69). This finding associates with enhanced expression of Smad7 (69). Interestingly, analysis of Smad7 content in mucosal samples of IBD patients revealed that Smad7 is upregulated at protein but not RNA level, suggesting a post-transcriptional regulation of Smad7 (71). Indeed, we have previously shown that, in both CD and UC mucosa, Smad7 protein stability is sustained by post-translational mechanisms, which enhance Smad7 acetylation thereby inhibiting ubiquitination-driven proteasomal-mediated degradation. Such modifications are partly due to p300, as silencing of this transcription coactivator, which is over-produced in CD mucosa, reduces Smad7 acetylation thereby stimulating ubiquitination-driven proteasomal-mediated degradation (71). Additional factors are supposed to stabilize Smad7 expression in IBD. In this context, our data indicate that, in IBD tissue, cells over-expressing Smad7 have reduced levels of SIRT1, a component of the mammalian Sirtuin family proteins that deacetylates the lysine residues of Smad7 with the down-stream effect of reducing Smad7 expression (72).
Various approaches have been used to assess the role of Smad7 in IBD. Initially, IBD LPMC and mucosal explants were treated with a specific Smad7 antisense oligonucleotide (AS). Inhibition of Smad7 associated with enhanced Smad3 phosphorylation and reduced production of inflammatory cytokines (Figure 1) (69). Pre-incubation of IBD LPMC with a blocking TGF-β1 antibody abrogated the Smad7 AS-mediated effects, indicating that the anti-inflammatory function of Smad7 AS is mediated by TGF-β1. As pointed out above, TGF-β1 promotes differentiation of Foxp3-expressing Tregs and the activity of the cytokine is required for the function of such cells (73, 74). Mucosal IBD CD4+ T cells show resistance to Foxp3-expressing Tregs-mediated suppression and this has been associated with Smad7, as knockdown of Smad7 restores the responsiveness of effector CD4+ T cells to Foxp3-expressing Tregs (75). Like IBD patients, mice with trinitrobenzene sulfonic acid (TNBS-) and oxazolone-mediated colitis, two mouse models of colitis, which resemble CD and UC, respectively, express elevated levels of TGF-β1 in the inflamed colons (76, 77). In both these models, Smad7 expression is upregulated and p-Smad3 is reduced. Colitic mice given oral Smad7 AS exhibit enhanced p-Smad3 expression, reduced expression of inflammatory cytokines and a less severe colitis (78). We generated a T cell-specific Smad7 transgenic (Tg) mouse on C57B6 genetic background, which does not develop spontaneously colitis (75). However, using the T cell-transfer model of colitis, we showed that adoptive transfer of Smad7 Tg naïve CD4+ T cells into immunodeficient mice produced a more severe intestinal inflammation than that documented in mice reconstituted with wild-type cells, and colitis induced by Smad7 Tg cells was only partly inhibited by co-transfer of Tregs (75). Finally, we showed that Smad7 Tg mice develop a more severe colitis in comparison to wild-type mice after DSS administration (79).
T cells of Smad7 transgenic mice have reduced levels of aryl hydrocarbon receptor (AhR), a transcription factor that stimulates IL-22 production and promotes regulatory mechanisms in the gut (80). In the T-cell transfer colitis model, AhR activation significantly ameliorates the course of colitis driven by wild-type T cells, but does not influence colitis induced by Smad7 transgenic T cells. Consistently, in normal but not in IBD LPMC, TGF-β1 enhances AhR expression (81). Altogether, these data show an inverse correlation between AhR and Smad7 expression in the gut.
The demonstration that Smad7 is upregulated in IBD mucosa and inhibition of this protein allows endogenous TGF-β1 to dampen the ongoing mucosal inflammation paved the way for the development of an oral Smad7 AS-containing pharmaceutical compound. This drug, initially named GED0301 and later on mongersen, was formulated in order to facilitate the deliver of the active molecule in the terminal ileum and right colon, which are the primarily affected sites in CD (82). A phase 1, open label, dose-escalating clinical trial conducted in 15 patients with active, steroid-dependent/resistant CD showed that 7-day treatment was safe and well-tolerated by the patients and associated with clinical benefit (82). Patients enrolled in the trial were monitored for the development of intestinal strictures. Six months after the end of the treatment, no patient developed strictures (83). To further assess the impact of Smad7 inhibition on the development of intestinal fibrosis, we used a mouse model of TNBS-mediated colitis-driven intestinal fibrosis. Interestingly, treatment of colitic mice with mongersen reduced the degree of intestinal inflammation and limited the development of intestinal fibrosis (84).
A subsequent phase 2, multicenter double-blind, placebo-controlled clinical trial was conducted in steroid-resistant/dependent CD patients with inflammatory lesions confined to the terminal ileum and/or right colon. The study confirmed the safety profile of mongersen and showed that 55 and 65% of the patients treated with the highest doses of the drug (i.e., 40 or 160 mg/day for 2 weeks) achieved clinical remission (primary end-point) as compared to 10% in the placebo group (85). Responders to mongersen exhibited reduction in the serum levels of CCL20, a chemokine that contributes to recruit immune cells to the intestine ad is over-produced in the epithelium of CD patients (86). Next, an exploratory, phase 2, multicenter study confirmed the clinical efficacy of the drug and documented an endoscopic improvement in nearly one-third of the patients treated with mongersen for 4–12 weeks (87). Next, a phase III clinical trial was conducted in steroid-resistant/dependent CD patients with inflammatory lesions of the terminal ileum and/or colon and endoscopic evidence of active inflammation. The trial was discontinued in October 2017 as an interim analysis documented a lack of efficacy of the drug.
Colorectal cancer represents a leading cause of cancer-related morbidity and mortality, with 1.65 million new cases and almost 835,000 deaths estimated worldwide in 2015 (88). In 70% of cases, CRC arises as sporadic disease, with several environmental and genetic factors involved in the pathogenesis, most of which are still unknown (89). Instead, in 2% of cases, CRC arises in patients with long-standing UC or extensive CD (colitis-associated cancer, CAC), with a cumulative risk that has been related with disease duration, extension, and severity of inflammation (90, 91). TGF-β1 seems to play both pro-tumorigenic and anti-tumorigenic roles in CRC depending on the tumor stage and probably reflecting the complexity of TGF-β1 function and the large number of biological processes in which the cytokine is involved. While at early stages of tumorigenesis, TGF-β1 contributes to maintain cell differentiation and restricts epithelial cell growth, thus acting as a tumor suppressor, at later stages, it promotes epithelial–mesenchymal transition, neo-angiogenesis, cancer progression, and metastasis (12, 92). Similarly, a dual role of Smad7 has been described in various types of cancer with pro-tumorigenic or anti-tumorigenic effects according to the cancer site and biology (93). Single nucleotide polymorphisms of Smad7 gene associate with CRC (i.e., rs4939827, rs12953717) (93, 94). Moreover, in a study from Boulay and colleagues, Smad7 mutations were analyzed in 264 CRC biopsies, and amplification of Smad7 gene was associated with a poor prognosis, with a possible dose effect, while deletion of Smad7 gene associated with a better outcome (95). Our studies have recently evidenced a link between Smad7 expression in immune cells and CAC. In particular, a reduced number of Smad7-expressing CD4+ T lymphocytes were documented in the colonic mucosa of IBD patients who developed CAC compared to IBD patients with uncomplicated disease (79). In line with this observation, in an experimental model of CAC, Tg mice that overexpress Smad7 in T cells developed a more severe intestinal inflammation, characterized by an abundant infiltrate of cytotoxic CD8+ T cells and NKT cells, compared to control mice, and were largely protected from tumors. The anti-tumorigenic effect of Smad7 over-expression in T cells appeared related to the action of IFN-γ, as genetic deletion of such a cytokine abolished the protective effect of Smad7 on colon carcinogenesis (79). Consistent with these observations is the demonstration that Smad7 Tg mice develop less tumors than wild-type littermates following the subcutaneous injection of syngenic MC38 colon carcinoma cells (96). Altogether, these data suggest that high Smad7 in immune cells promotes the amplification of Th1-cytokine responses, which protect against colon carcinogenesis. However, in line with the above-referenced genetic studies, a different scenario emerges when the role of Smad7 is analyzed in sporadic CRC. Indeed, CRC cells produce huge amounts of Smad7 and knockdown of Smad7 with a specific AS inhibits the in vitro and in vivo growth of CRC cells (97). This effect of Smad7 AS relies on the modulation of cell cycle-related proteins and ultimately results in S phase arrest and cell death. Since these findings are seen in CRC cells unresponsive to TGFβ1 and the anti-proliferative effect of Smad7 AS is not affected by stimulation of CRC cells with TGF-β1 or anti-TGF-β1, it is highly likely that Smad7 exerts pro-tumorigenic effects in a TGFβ1-independent manner (Figure 1) (97). Analysis of the basic mechanisms underlying the mitogenic effect of Smad7 in CRC cells showed that Smad7 knockdown causes phosphorylation of eukaryotic translation initiation factor 2α (eIF2α), a transcription factor that regulates cell cycle arrest, and consequent upregulation of activating transcription factor 4 (ATF4) and CCAAT/enhancer binding protein homology protein (CHOP) (98). Silencing of the serine–threonine protein kinase RNA abrogates Smad7 AS induced eIF2α phosphorylation and ATF4/CHOP induction, thus preventing cell death (Figure 1) (98). In contrast, an anti-tumorigenic role for Smad7 has been recently reported by Wang and colleagues, who showed that nuclear reporter subfamily 2, group F, and member 2 (NR2F2), a molecule involved in many cancers, induces a TGFβ1-dependent epithelial–mesenchymal transition by inhibiting Smad7, thus promoting CRC metastasizing process (99).
Altogether, these data highlight the complex role of Smad7/TGFβ1 signaling in colon carcinogenesis.
The findings discussed in this review underline the crucial role of TGF-β1 in the maintenance of intestinal homeostasis and suggest that defective function of this cytokine can contribute to trigger and/or amplify detrimental signals in the gut. There is also discussion on the expression of Smad7 in patients with IBD and the crucial role played by this protein in inhibiting TGF-β1 function and sustaining IBD-related inflammation. Knockdown of Smad7 allows endogenous TGF-β to suppress effector responses. In line with this, CD patients treated with oral compound containing Smad7 AS showed clinical and endoscopic improvement during phase 1 and phase 2 clinical trials. However, a recent phase 3 clinical trial has been discontinued apparently due to the lack of efficacy of the drug, but the reasons for the discrepancy between phase 2 and phase 3 studies remain unknown. Similarly, some issues regarding the expression/function of Smad7 in IBD and CRC remain to be addressed. For instance, we still do not known whether, in IBD, Smad7 is regulated in a cell-specific manner and which factors/mechanisms contribute to maintain the elevated levels of Smad7. It would also be relevant to know whether molecular profiling of IBD patients can help to identify better candidates to treatment with Smad7 inhibitors. Further experimentation is also needed to clarify whether and which regulatory effects of Smad7 on the ongoing mucosal inflammation and colon carcinogenesis are in part independent on TGF-β1, as it is known that Smad7 controls some biological functions in a TGF-β1-independent manner.
ET and IM wrote the manuscript. CS supervised parts of the project. GM designed the paper, supervised the project, and wrote the manuscript.
GM has filed a patent related to the treatment of inflammatory bowel diseases with Smad7 antisense oligonucleotides, while the remaining authors have no conflict of interest.
1. Macdonald TT, Monteleone G. Immunity, inflammation, and allergy in the gut. Science (2005) 307(5717):1920–5. doi:10.1126/science.1106442
2. Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature (2011) 474(7351):298–306. doi:10.1038/nature10208
3. Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol (2008) 8(6):411–20. doi:10.1038/nri2316
4. Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology (2012) 143(2):347–55. doi:10.1053/j.gastro.2012.04.045
5. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med (2009) 361(21):2033–45. doi:10.1056/NEJMoa0907206
6. Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med (1999) 190(7):995–1004. doi:10.1084/jem.190.7.995
7. Kullberg MC, Ward JM, Gorelick PL, Caspar P, Hieny S, Cheever A, et al. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect Immun (1998) 66(11):5157–66.
8. Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell (1993) 75(2):263–74. doi:10.1016/0092-8674(93)80068-P
9. Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature (2007) 446(7135):557–61. doi:10.1038/nature05698
10. Kulkarni AB, Karlsson S. Transforming growth factor-beta 1 knockout mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am J Pathol (1993) 143(1):3–9.
11. Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity (2000) 12(2):171–81. doi:10.1016/S1074-7613(00)80170-3
12. Skeen VR, Paterson I, Paraskeva C, Williams AC.TGF-beta1 signalling, connecting aberrant inflammation and colorectal tumorigenesis. Curr Pharm Des (2012) 18(26):3874–88. doi:10.2174/138161212802083734
13. Piek E, Heldin CH, Ten Dijke P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J (1999) 13(15):2105–24. doi:10.1096/fasebj.13.15.2105
14. Munger JS, Harpel JG, Gleizes PE, Mazzieri R, Nunes I, Rifkin DB. Latent transforming growth factor-beta: structural features and mechanisms of activation. Kidney Int (1997) 51(5):1376–82. doi:10.1038/ki.1997.188
15. Munger JS, Harpel JG, Giancotti FG, Rifkin DB. Interactions between growth factors and integrins: latent forms of transforming growth factor-beta are ligands for the integrin alphavbeta1. Mol Biol Cell (1998) 9(9):2627–38. doi:10.1091/mbc.9.9.2627
16. Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev (2005) 19(23):2783–810. doi:10.1101/gad.1350705
17. Nakao A, Afrakhte M, Morén A, Nakayama T, Christian JL, Heuchel R, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature (1997) 389(6651):631–5. doi:10.1038/39369
18. Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell (1997) 89(7):1165–73. doi:10.1016/S0092-8674(00)80303-7
19. Shi W, Sun C, He B, Xiong W, Shi X, Yao D, et al. GADD34-PP1c recruited by Smad7 dephosphorylates TGFbeta type I receptor. J Cell Biol (2004) 164(2):291–300. doi:10.1083/jcb.200307151
20. Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, et al. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J Biol Chem (2001) 276(16):12477–80. doi:10.1074/jbc.C100008200
21. Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell (2000) 6(6):1365–75. doi:10.1016/S1097-2765(00)00134-9
22. Zhang S, Fei T, Zhang L, Zhang R, Chen F, Ning Y, et al. Smad7 antagonizes transforming growth factor beta signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol Cell Biol (2007) 27(12):4488–99. doi:10.1128/MCB.01636-06
23. Travis MA, Sheppard D. TGF-beta activation and function in immunity. Annu Rev Immunol (2014) 32:51–82. doi:10.1146/annurev-immunol-032713-120257
24. Fang D, Zhu J. Dynamic balance between master transcription factors determines the fates and functions of CD4 T cell and innate lymphoid cell subsets. J Exp Med (2017) 214(7):1861–76. doi:10.1084/jem.20170494
25. Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J Exp Med (2002) 195(11):1499–505. doi:10.1084/jem.20012076
26. Gorham JD, Güler ML, Fenoglio D, Gubler U, Murphy KM. Low dose TGF-beta attenuates IL-12 responsiveness in murine Th cells. J Immunol (1998) 161(4):1664–70.
27. Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol (2000) 165(9):4773–7. doi:10.4049/jimmunol.165.9.4773
28. Heath VL, Murphy EE, Crain C, Tomlinson MG, O’Garra A. TGF-beta1 down-regulates Th2 development and results in decreased IL-4-induced STAT6 activation and GATA-3 expression. Eur J Immunol (2000) 30(9):2639–49. doi:10.1002/1521-4141(200009)30:9<2639::AID-IMMU2639>3.0.CO;2-7
29. Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol (2004) 172(9):5149–53. doi:10.4049/jimmunol.172.9.5149
30. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi:10.1146/annurev.immunol.25.022106.141623
31. Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med (2005) 201(7):1061–7. doi:10.1084/jem.20042276
32. Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity (2006) 25(3):455–71. doi:10.1016/j.immuni.2006.07.011
33. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol (2008) 9(6):632–40. doi:10.1038/ni.1607
34. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi:10.1084/jem.20030152
35. Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J Immunol (2007) 178(7):4022–6. doi:10.4049/jimmunol.178.7.4022
36. Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med (2007) 204(8):1765–74. doi:10.1084/jem.20070719
37. Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med (2007) 204(8):1775–85. doi:10.1084/jem.20070602
38. Coombes JL, Siddiqui KR, Arancibia-Cárcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med (2007) 204(8):1757–64. doi:10.1084/jem.20070590
39. Kashiwagi I, Morita R, Schichita T, Komai K, Saeki K, Matsumoto M, et al. Smad2 and Smad3 inversely regulate TGF-beta autoinduction in Clostridium butyricum-activated dendritic cells. Immunity (2015) 43(1):65–79. doi:10.1016/j.immuni.2015.06.010
40. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol (2009) 27:485–517. doi:10.1146/annurev.immunol.021908.132710
41. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature (2008) 453(7192):236–40. doi:10.1038/nature06878
42. Zhang N, Bevan MJ. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity (2013) 39(4):687–96. doi:10.1016/j.immuni.2013.08.019
43. Ruane D, Chorny A, Lee H, Faith J, Pandey G, Shan M, et al. Microbiota regulate the ability of lung dendritic cells to induce IgA class-switch recombination and generate protective gastrointestinal immune responses. J Exp Med (2016) 213(1):53–73. doi:10.1084/jem.20150567
44. Cazac BB, Roes J. TGF-beta receptor controls B cell responsiveness and induction of IgA in vivo. Immunity (2000) 13(4):443–51. doi:10.1016/S1074-7613(00)00044-3
45. Roes J, Choi BK, Cazac BB. Redirection of B cell responsiveness by transforming growth factor beta receptor. Proc Natl Acad Sci U S A (2003) 100(12):7241–6. doi:10.1073/pnas.0731875100
46. Kubinak JL, Round JL. Do antibodies select a healthy microbiota? Nat Rev Immunol (2016) 16(12):767–74. doi:10.1038/nri.2016.114
47. Reboldi A, Arnon TI, Rodda LB, Atakilit A, Sheppard D, Cyster JG. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science (2016) 352(6287):aaf4822. doi:10.1126/science.aaf4822
48. Park SR, Lee JH, Kim PH. Smad3 and Smad4 mediate transforming growth factor-beta1-induced IgA expression in murine B lymphocytes. Eur J Immunol (2001) 31(6):1706–15. doi:10.1002/1521-4141(200106)31:6<1706::AID-IMMU1706>3.0.CO;2-Z
49. Klein J, Ju W, Heyer J, Wittek B, Haneke T, Knaus P, et al. B cell-specific deficiency for Smad2 in vivo leads to defects in TGF-beta-directed IgA switching and changes in B cell fate. J Immunol (2006) 176(4):2389–96. doi:10.4049/jimmunol.176.4.2389
50. Sedda S, Marafini I, Figliuzzi MM, Pallone F, Monteleone G. An overview of the role of innate lymphoid cells in gut infections and inflammation. Mediators Inflamm (2014) 2014:235460. doi:10.1155/2014/235460
51. Cortez VS, Cervantes-Barragan L, Robinette ML, Bando JK, Wang Y, Geiger TL, et al. Transforming growth factor-beta signaling guides the differentiation of innate lymphoid cells in salivary glands. Immunity (2016) 44(5):1127–39. doi:10.1016/j.immuni.2016.03.007
52. Ramalingam R, Larmonier CB, Thurston RD, Midura-Kiela MT, Zheng SG, Ghishan FK, et al. Dendritic cell-specific disruption of TGF-beta receptor II leads to altered regulatory T cell phenotype and spontaneous multiorgan autoimmunity. J Immunol (2012) 189(8):3878–93. doi:10.4049/jimmunol.1201029
53. Ihara S, Hirata Y, Serizawa T, Suzuki N, Sakitani K, Kinoshita H, et al. TGF-beta signaling in dendritic cells governs colonic homeostasis by controlling epithelial differentiation and the luminal microbiota. J Immunol (2016) 196(11):4603–13. doi:10.4049/jimmunol.1502548
54. Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature (2007) 449(7160):361–5. doi:10.1038/nature06110
55. Siddiqui KR, Laffont S, Powrie F. E-cadherin marks a subset of inflammatory dendritic cells that promote T cell-mediated colitis. Immunity (2010) 32(4):557–67. doi:10.1016/j.immuni.2010.03.017
56. Smythies LE, Maheshwari A, Clements R, Eckhoff D, Novak L, Vu HL, et al. Mucosal IL-8 and TGF-beta recruit blood monocytes: evidence for cross-talk between the lamina propria stroma and myeloid cells. J Leukoc Biol (2006) 80(3):492–9. doi:10.1189/jlb.1005566
57. Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol (2015) 33:643–75. doi:10.1146/annurev-immunol-032414-112220
58. Rani R, Smulian AG, Greaves DR, Hogan SP, Herbert DR. TGF-beta limits IL-33 production and promotes the resolution of colitis through regulation of macrophage function. Eur J Immunol (2011) 41(7):2000–9. doi:10.1002/eji.201041135
59. Howe KL, Reardon C, Wang A, Nazli A, McKay DM. Transforming growth factor-beta regulation of epithelial tight junction proteins enhances barrier function and blocks enterohemorrhagic Escherichia coli O157:H7-induced increased permeability. Am J Pathol (2005) 167(6):1587–97. doi:10.1016/S0002-9440(10)61243-6
60. Oshima H, Nakayama M, Han TS, Naoi K, Ju X, Maeda Y, et al. Suppressing TGFbeta signaling in regenerating epithelia in an inflammatory microenvironment is sufficient to cause invasive intestinal cancer. Cancer Res (2015) 75(4):766–76. doi:10.1158/0008-5472.CAN-14-2036
61. Biancheri P, Giuffrida P, Docena GH, MacDonald TT, Corazza GR, Di Sabatino A. The role of transforming growth factor (TGF)-beta in modulating the immune response and fibrogenesis in the gut. Cytokine Growth Factor Rev (2014) 25(1):45–55. doi:10.1016/j.cytogfr.2013.11.001
62. Stallmach A, Schuppan D, Riese HH, Matthes H, Riecken EO. Increased collagen type III synthesis by fibroblasts isolated from strictures of patients with Crohn’s disease. Gastroenterology (1992) 102(6):1920–9. doi:10.1016/0016-5085(92)90314-O
63. Di Sabatino A, Jackson CL, Pickard KM, Buckley M, Rovedatti L, Leakey NA, et al. Transforming growth factor beta signalling and matrix metalloproteinases in the mucosa overlying Crohn’s disease strictures. Gut (2009) 58(6):777–89. doi:10.1136/gut.2008.149096
64. Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J (2004) 18(7):816–27. doi:10.1096/fj.03-1273rev
65. Mulsow JJ, Watson RW, Fitzpatrick JM, O’Connell PR. Transforming growth factor-beta promotes pro-fibrotic behavior by serosal fibroblasts via PKC and ERK1/2 mitogen activated protein kinase cell signaling. Ann Surg (2005) 242(6):880–7, discussion 887–9. doi:10.1097/01.sla.0000189606.58343.cd
66. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature (2007) 448(7152):427–34. doi:10.1038/nature06005
67. Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol (2003) 3(7):521–33. doi:10.1038/nri1132
68. Di Sabatino A, Pickard KM, Rampton D, Kruidenier L, Rovedatti L, Leakey NA, et al. Blockade of transforming growth factor beta upregulates T-box transcription factor T-bet, and increases T helper cell type 1 cytokine and matrix metalloproteinase-3 production in the human gut mucosa. Gut (2008) 57(5):605–12. doi:10.1136/gut.2007.130922
69. Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J Clin Invest (2001) 108(4):601–9. doi:10.1172/JCI12821
70. Monteleone G, Mann J, Monteleone I, Vavassori P, Bremner R, Fantini M, et al. A failure of transforming growth factor-beta1 negative regulation maintains sustained NF-kappaB activation in gut inflammation. J Biol Chem (2004) 279(6):3925–32. doi:10.1074/jbc.M303654200
71. Monteleone G, Del Vecchio Blanco G, Monteleone I, Fina D, Caruso R, Gioia V, et al. Post-transcriptional regulation of Smad7 in the gut of patients with inflammatory bowel disease. Gastroenterology (2005) 129(5):1420–9. doi:10.1053/j.gastro.2005.09.005
72. Caruso R, Marafini I, Franzè E, Stolfi C, Zorzi F, Monteleone I, et al. Defective expression of SIRT1 contributes to sustain inflammatory pathways in the gut. Mucosal Immunol (2014) 7(6):1467–79. doi:10.1038/mi.2014.35
73. Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H, et al. TGF-beta 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J Immunol (2004) 172(2):834–42. doi:10.4049/jimmunol.172.2.834
74. Maloy KJ, Salaun L, Cahill R, Dougan G, Saunders NJ, Powrie F. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med (2003) 197(1):111–9. doi:10.1084/jem.20021345
75. Fantini MC, Rizzo A, Fina D, Caruso R, Sarra M, Stolfi C, et al. Smad7 controls resistance of colitogenic T cells to regulatory T cell-mediated suppression. Gastroenterology (2009) 136(4):1308–16, e1–3. doi:10.1053/j.gastro.2008.12.053
76. Neurath MF, Fuss I, Kelsall BL, Stüber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med (1995) 182(5):1281–90. doi:10.1084/jem.182.5.1281
77. Boirivant M, Fuss IJ, Chu A, Strober W. Oxazolone colitis: a murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J Exp Med (1998) 188(10):1929–39. doi:10.1084/jem.188.10.1929
78. Boirivant M, Pallone F, Di Giacinto C, Fina D, Monteleone I, Marinaro M, et al. Inhibition of Smad7 with a specific antisense oligonucleotide facilitates TGF-beta1-mediated suppression of colitis. Gastroenterology (2006) 131(6):1786–98. doi:10.1053/j.gastro.2006.09.016
79. Rizzo A, Waldner MJ, Stolfi C, Sarra M, Fina D, Becker C, et al. Smad7 expression in T cells prevents colitis-associated cancer. Cancer Res (2011) 71(24):7423–32. doi:10.1158/0008-5472.CAN-11-1895
80. Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology (2011) 141(1):237–48, 248.e1. doi:10.1053/j.gastro.2011.04.007
81. Monteleone I, Marafini I, Zorzi F, Di Fusco D, Dinallo V, Rizzo A, et al. Smad7 knockdown restores aryl hydrocarbon receptor-mediated protective signals in the gut. J Crohns Colitis (2016) 10(6):670–7. doi:10.1093/ecco-jcc/jjw030
82. Monteleone G, Fantini MC, Onali S, Zorzi F, Sancesario G, Bernardini S, et al. Phase I clinical trial of Smad7 knockdown using antisense oligonucleotide in patients with active Crohn’s disease. Mol Ther (2012) 20(4):870–6. doi:10.1038/mt.2011.290
83. Zorzi F, Calabrese E, Monteleone I, Fantini M, Onali S, Biancone L, et al. A phase 1 open-label trial shows that smad7 antisense oligonucleotide (GED0301) does not increase the risk of small bowel strictures in Crohn’s disease. Aliment Pharmacol Ther (2012) 36(9):850–7. doi:10.1111/apt.12051
84. Izzo R, Bevivino G, De Simone V, Sedda S, Monteleone I, Marafini I, et al. Knockdown of Smad7 with a specific antisense oligonucleotide attenuates colitis and colitis-driven colonic fibrosis in mice. Inflamm Bowel Dis (2018) 24(6):1213–24. doi:10.1093/ibd/izy062
85. Monteleone G, Neurath MF, Ardizzone S, Di Sabatino A, Fantini MC, Castiglione F, et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N Engl J Med (2015) 372(12):1104–13. doi:10.1056/NEJMoa1407250
86. Marafini I, Monteleone I, Dinallo V, Di Fusco D, De Simone V, Laudisi F, et al. CCL20 is negatively regulated by TGF-beta1 in intestinal epithelial cells and reduced in Crohn’s disease patients with a successful response to mongersen, a Smad7 antisense oligonucleotide. J Crohns Colitis (2017) 11(5):603–9. doi:10.1093/ecco-jcc/jjw191
87. Feagan BG, Sands BE, Rossiter G, Li X, Usiskin K, Zhan X, et al. Effects of mongersen (GED-0301) on endoscopic and clinical outcomes in patients with active Crohn’s disease. Gastroenterology (2018) 154(1):61–64.e6. doi:10.1053/j.gastro.2017.08.035
88. Global Burden of Disease Cancer Collaboration, Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the Global Burden of Disease Study. JAMA Oncol (2017) 3(4):524–48. doi:10.1001/jamaoncol.2016.5688
89. Amersi F, Agustin M, Ko CY. Colorectal cancer: epidemiology, risk factors, and health services. Clin Colon Rectal Surg (2005) 18(3):133–40. doi:10.1055/s-2005-916274
90. Francescone R, Hou V, Grivennikov SI. Cytokines, IBD, and colitis-associated cancer. Inflamm Bowel Dis (2015) 21(2):409–18. doi:10.1097/MIB.0000000000000236
91. Rutter M, Saunders B, Wilkinson K, Rumbles S, Schofield G, Kamm M, et al. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology (2004) 126(2):451–9. doi:10.1053/j.gastro.2003.11.010
92. Katsuno Y, Lamouille S, Derynck R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol (2013) 25(1):76–84. doi:10.1097/CCO.0b013e32835b6371
93. Stolfi C, Marafini I, De Simone V, Pallone F, Monteleone G. The dual role of Smad7 in the control of cancer growth and metastasis. Int J Mol Sci (2013) 14(12):23774–90. doi:10.3390/ijms141223774
94. Huang Y, Wu W, Nie M, Li C, Wang L. SMAD7 polymorphisms and colorectal cancer risk: a meta-analysis of case-control studies. Oncotarget (2016) 7(46):75561–70. doi:10.18632/oncotarget.12285
95. Boulay JL, Mild G, Lowy A, Reuter J, Lagrange M, Terracciano L, et al. SMAD7 is a prognostic marker in patients with colorectal cancer. Int J Cancer (2003) 104(4):446–9. doi:10.1002/ijc.10908
96. Rizzo A, De Mare V, Rocchi C, Stolfi C, Colantoni A, Neurath MF, et al. Smad7 induces plasticity in tumor-infiltrating Th17 cells and enables TNF-alpha-mediated killing of colorectal cancer cells. Carcinogenesis (2014) 35(7):1536–46. doi:10.1093/carcin/bgu027
97. Stolfi C, De Simone V, Colantoni A, Franzè E, Ribichini E, Fantini MC, et al. A functional role for Smad7 in sustaining colon cancer cell growth and survival. Cell Death Dis (2014) 5:e1073. doi:10.1038/cddis.2014.49
98. De Simone V, Bevivino G, Sedda S, Izzo R, Laudisi F, Dinallo V, et al. Smad7 knockdown activates protein kinase RNA-associated eIF2alpha pathway leading to colon cancer cell death. Cell Death Dis (2017) 8(3):e2681. doi:10.1038/cddis.2017.103
Keywords: mucosal immunity, cytokines, inflammatory bowel diseases, colon cancer, antisense oligonucleotides
Citation: Troncone E, Marafini I, Stolfi C and Monteleone G (2018) Transforming Growth Factor-β1/Smad7 in Intestinal Immunity, Inflammation, and Cancer. Front. Immunol. 9:1407. doi: 10.3389/fimmu.2018.01407
Received: 26 February 2018; Accepted: 06 June 2018;
Published: 20 June 2018
Edited by:
Fabio Cominelli, Case Western Reserve University, United StatesReviewed by:
Luca Pastorelli, Università degli Studi di Milano, ItalyCopyright: © 2018 Troncone, Marafini, Stolfi and Monteleone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanni Monteleone, Z2kubW9udGVsZW9uZUBtZWQudW5pcm9tYTIuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.