 S. Harsha Krovi
S. Harsha Krovi Laurent Gapin
Laurent Gapin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 June 2018
Sec. T Cell Biology
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01393
This article is part of the Research Topic Role of CD1- and MR1-restricted T cells in Immunity and Disease View all 41 articles
Invariant natural killer T (iNKT) cells are a CD1d-restricted T cell population that can respond to lipid antigenic stimulation within minutes by secreting a wide variety of cytokines. This broad functional scope has placed iNKT cells at the frontlines of many kinds of immune responses. Although the diverse functional capacities of iNKT cells have long been acknowledged, only recently have distinct iNKT cell subsets, each with a marked functional predisposition, been appreciated. Furthermore, the subsets can frequently occupy distinct niches in different tissues and sometimes establish long-term tissue residency where they can impact homeostasis and respond quickly when they sense perturbations. In this review, we discuss the developmental origins of the iNKT cell subsets, their localization patterns, and detail what is known about how different subsets specifically influence their surroundings in conditions of steady and diseased states.
Adaptive immunity has long been appreciated as a chief means through which various jawed vertebrates stave off infectious pathogens. One of the main features differentiating the adaptive immune system from its innate counterpart is the generation and expression of diverse antigen receptors. Antigen sensors of the innate immune system are proteins with fixed sequences and pattern-recognition motifs encoded within the germline (1). By contrast, antigen receptor generation by adaptive immune cells involves complex somatic DNA rearrangements that juxtapose genes otherwise separated by thousands to millions of base pairs (2). Each cell randomly rearranges its antigen receptor locus to ensure that few cells express identical receptors. In so doing, the cells express clonal receptors and develop an antigen receptor repertoire diverse enough and with the potential to recognize the plethora of existing antigens that the host is likely to encounter. Because the antigen receptors in the adaptive immune system are heterodimers, with both subunits undergoing independent rearrangements, the combinatorial diversity has been estimated to exceed 1015 unique sequences (3).
The two types of cells belonging to the adaptive immune system, B and T lymphocytes, have each evolved in different ways to efficiently respond to infections. In particular, B cells produce and secrete antibodies that target and bind to different conformational epitopes on pathogens (4). T cells, on the other hand, express cell membrane-tethered antigen receptors (TCRs, composed of α-chains paired to β-chains), thereby necessitating their proximity to their targets in order to initiate an immune response. These TCRs primarily recognize their target ligands by interacting with major histocompatibility complex (MHC) proteins, which present linearized peptides, expressed on adjacent cells (5). The presented peptides are processed fragments from full length proteins and can be presented by MHC-I molecules interacting with TCRs expressed on CD8+ T cells or MHC-II proteins interacting with TCRs expressed on CD4+ T cells (6). Correspondingly, these T cells not only interact with different MHC molecules but also produce distinct responses.
To ensure that T cells are capable of mounting immune responses against all kinds of invading pathogens, T cells have further evolved to differentiate into functionally distinct subsets. Indeed, CD4+ T cells can differentiate into TH1, TH2, TH17, Treg, among others, upon exposure to their cognate antigens (7). Each of the subsets produces a distinct set of cytokines with the capacity to skew the immune response in a specific direction. For example, TH1 cells produce IFNγ, a pro-inflammatory cytokine that promotes increased antigen presentation by MHC molecules and increased phagocytosis by macrophages, to name a few of its effects. CD4+ T cell differentiation into TH1 cells is thought to be primarily due to intracellular pathogens (8). TH2 cells, though, produce a different set of cytokines, including IL-4, IL-5, and IL-13, and assist in combating extracellular pathogens such as parasites and helminths (9). Through this division of labor, functionally different T cell subsets can resolve infections by producing responses catered to the pathogen.
The substantial TCR repertoire diversity in the adaptive immune system serves as a double-edged sword. Many different TCRs are expressed by the T cell population en masse, but only a few T cells expressing TCRs specific for a given antigen exist within the population (10). Thus, T cells undergo extensive proliferation when they first encounter their antigen to generate enough cells with the proper antigen-specific TCRs. The activated cells can then travel to the site of infection and execute their appropriate functions upon antigen re-challenge. As this process usually takes several days, the adaptive immune system is considered a slow and deliberate yet specific form of response. After the immune response has been resolved, some of the cells differentiate into memory cells, which exhibit faster response times in the event that the pathogen reinfects the host (11).
Due to the delayed kinetics of this “conventional” arm of the adaptive immune system, other “innate-like” adaptive lymphocytes play a crucial role early during an infection (12). These cells are unique because they express markers associated with memory cells despite not having encountered their antigens previously. Additionally, they are functionally poised and capable of responding within hours as opposed to days, in line with their innate-like capabilities. Though they make up only a fraction of the overall T cell population, they still exert crucial and sometimes non-redundant functions. One such lymphocyte population, which will be the focus of this review, is the invariant natural killer T (iNKT) cell lineage. These cells straddle the innate-adaptive boundary because they respond quickly upon stimulation (within hours), yet, express a TCR that underwent somatic rearrangement (13). Indeed, the vast majority of iNKT cells express an identical TCRα chain (TRAV11-TRAJ18 in mice, TRAV10-TRAJ18 in humans) paired to a restricted set of TCRβ chains (TRBV1, TRBV13, and TRBV29 in mice, TRBV25 in humans), with some notable exceptions (14–16). Furthermore, instead of interacting with peptides presented by MHC molecules, the TCRs expressed by iNKT cells recognize (glyco)lipids presented by CD1d, a non-polymorphic MHC-I-like molecule (17).
Analogous to the aforementioned functionally different conventional T cell subsets, iNKT cells also come in different flavors, each of which exhibits a different functional profile (18–20). Such division of labor between functionally different iNKT cell subsets perhaps could explain why iNKT cells have been implicated in ameliorating or exacerbating a variety of diseases and illnesses ranging from autoimmunity to cancer. Historically, iNKT cells have been lumped into one category despite their varied roles in responses. However, with the recent identification of functionally distinct iNKT cell subsets, how and which iNKT cell subsets might affect the development of the immune system and its response need to be updated. In this review, we will focus on how the different iNKT cell subsets develop and consequently, to what extent each of these subsets actively participates in immune responses.
Initially, an intriguing population of mature T cells was identified in the thymus by their lack of expression of CD4 or CD8 coreceptors (double negative, DN) but with surface expression of a TCR, thereby distinguishing them from other immature thymocytes (21). These DN cells were functionally competent since they could produce IL-4 and IFNγ readily after stimulation and also expressed the natural killer (NK) cell marker NK1.1 (22–24). This was particularly novel because T cells were not traditionally considered to be cytokine secretion-competent in the thymus and suggested that functional competence by these cells might be acquired during their development. Sequencing the TCRs from these cells repeatedly provided investigators with the same TCRα chain sequence (25, 26), and it was eventually determined that the cells required CD1d expression for their development, suggesting that they recognized lipids instead of peptides (27). Due to the expression of a TCR as well as NK markers by these cells, the name iNKT took preferential hold as a label for these cells.
With the discovery of the marine sponge-derived lipid α-galactosylceramide (αGC) that when bound to CD1d strongly stimulated these cells and the advent of MHC-loaded tetramer technology, iNKT cells could now be tracked with profound resolution (28–30). Interestingly, it became readily apparent that not all the cells that were identified by αGC-loaded CD1d tetramers were NK1.1+, suggesting phenotypic heterogeneity within the iNKT compartment. Because the NK1.1+ cells composed the overwhelming majority of the total tetramer+ population in the thymus in C57BL/6 (B6) mice, the NK1.1− cells were thought to perhaps represent developmental intermediates. Indeed, support for this idea came from experiments in which intrathymic transfers of NK1.1− cells could generate NK1.1+ cells (31, 32). Interestingly, stimulating the NK1.1− cells led to the production of larger amounts of IL-4 compared to IFNγ, in stark contrast to what the NK1.1+ cells produced, which was primarily IFNγ and little IL-4 (31–33). Additionally, the iNKT cells that were primarily exported from the thymus were NK1.1− cells while the NK1.1+ cells were retained in the thymus (34, 35). Thus, it was unclear why the intermediates had a different cytokine secretion profile compared to the terminally matured population and furthermore, why/how the immature cells emigrated from the thymus if they were truly meant to give rise to the mature iNKT cells (36).
Only recently has this conundrum been resolved due in large part to the work by the Hogquist group. Instead of identifying iNKT cells simply by the tetramer and NK1.1, they also stained the cells with transcription factors known to endow specific fates (19). Because of years of work in understanding conventional CD4+ T cell differentiation, it was known that the master transcription factors engendering the TH1, TH2, and TH17 fates were T-bet, GATA-3, and RORγt, respectively (8). In addition, large scale screens by two groups had recently identified that all iNKT cells require the expression of the zinc finger transcription factor promyelotic leukemia zinc finger (PLZF) (37, 38). In fact, the few remaining iNKT cells (as identified by the tetramer) found in PLZF-deficient mice resembled naïve T cells that secreted IL-2 upon stimulation but not significant levels of IL-4 or IFNγ, highlighting the transcription factor’s importance (37, 38). By using antibodies targeting T-bet, GATA-3, RORγt, and PLZF, Hogquist and colleagues had the surprising finding that not all thymic iNKT cells expressed each of the transcription factors. Instead, three distinct thymic subpopulations were identified based on their staining: PLZFhi GATA-3hi (iNKT2), PLZFint RORγt+ (iNKT17), and PLZFlo, T-bet+ (iNKT1) (19). Moreover, NK1.1 primarily stained the iNKT1 cells. Thus, although a few iNKT1 precursors were present in this pool, many of the NK1.1− cells were terminally differentiated cells themselves. This was further confirmed by stimulating the different subsets in vitro, with iNKT1 cells producing large amounts of IFNγ and a little IL-4, iNKT2 cells producing large amounts of IL-4, and iNKT17 cells secreting IL-17, putting to rest the functional discrepancy between NK1.1+ and NK1.1− cells (19).
Interestingly, not all thymic iNKT subsets are equally represented in different strains of mice. Certain strains, such as the BALB/c, have large proportions of iNKT2 and iNKT17 cells with a correspondingly reduced proportion of iNKT1 cells. On the other extreme, B6 mice instead possess largely iNKT1 cells and few iNKT2/iNKT17 cells (19). This is particularly important because previous work in these mice had led to the prevalent idea that, generally speaking, B6 mice tended to be predisposed to a “TH1 phenotype” while BALB/c mice displayed a “TH2 phenotype” (39, 40). Whether the iNKT cell compositions in either of these strains are a cause or a consequence of these phenotypes is unknown. Another point of note is that the reason for the overrepresentation of NK1.1+ iNKT cells in previous experiments was the predominant use of the B6 mouse for the study of iNKT cell development. In fact, the antibody targeting NK1.1 in B6 mice does not recognize the epitope present in BALB/c mice due to an allelic variance (41). The use of this antibody necessitated experiments in B6 mice since the cells were initially characterized by their expression of NK1.1 (22). Now, however, iNKT cells in all strains are identified by their ability to interact with the αGC (or its analog PBS57) loaded CD1d tetramer and their transcription factor profile (or surface proteins known to be specifically upregulated by these transcription factors) serves as a readout of the subset proportions.
The terminal differentiation status of these subsets has also been challenged due to the discovery of new iNKT cell subsets in the periphery. Although only the three aforementioned subsets are largely represented in the thymus, analysis of other tissues in both steady-state and immunization conditions has revealed the presence of novel iNKT subsets. In the adipose tissue, a special iNKT cell population, named iNKT10, has been identified that depends on expression of the transcription factor E4BP4 for its role in maintaining adipose tissue homeostasis (42). Similarly, an iNKTFH population expressing the transcription factor Bcl6 has been observed in the peripheral lymphoid organs of immunized mice (43). This population secreted IL-21 and provided cognate help for B cells undergoing affinity maturation, much like conventional TFH cells in germinal centers (43–45). Ongoing work should help determine whether these additional subsets are indeed generated at low frequencies in the thymus or if they differentiate into their observed subsets within other tissues.
CD4+ CD8+ [double positive (DP)] thymocytes serve as the progenitors for all cells belonging to the αβ T cell lineage (46, 47). iNKT cells are no different as they also principally originate from DP precursors (48, 49), with a minor proportion utilizing an alternative pathway (Figure 1) (50). DP cells randomly rearrange their TCRα loci to generate the invariant TCRα chains that pair with suitable TCRβ chains (49). While DP precursors of conventional T cells are selected by MHC I/II on thymic epithelial cells (TECs), iNKT cell DP precursors are instead positively selected by self-lipids presented by CD1d expressed on fellow DP thymocytes (51, 52). The DP–DP interaction provides the iNKT precursor with the obligate lipid/CD1d ligand along with a distinctive homotypic co-stimulation through members of the signaling lymphocytic activated molecules (SLAM) family of receptors. Signals derived from SLAM family receptor interactions are required to produce mature iNKT cells because iNKT cells are notably absent in mice in which an adapter downstream of SLAM receptors (SAP) has been deleted (38, 53). Interestingly, different mouse strains also express different alleles of the SLAM receptors. For example, BALB/c mice possess an allele of SLAMF6 (Ly108) that is hyperphosphorylated upon engagement compared to the B6 Ly108 allele (54, 55). This hyperphosphorylation has a functional effect because a stronger signal is consequently transduced in BALB/c DP thymocytes (56), suggesting that signals received by iNKT cell precursors during development in the thymus might not be equivalent across mouse strains.
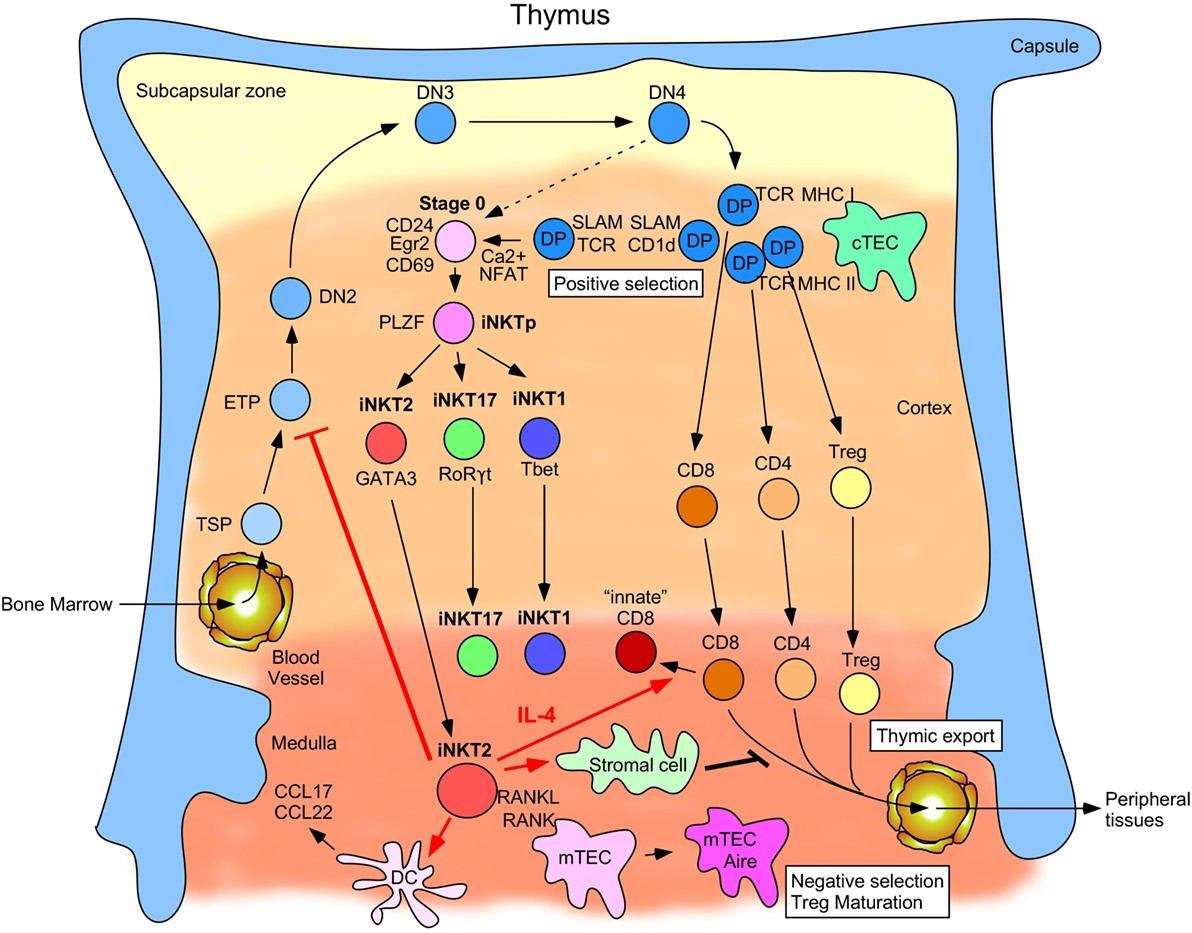
Figure 1. Schematic describing invariant natural killer T (iNKT) cell development and function in the thymus. Thymus-settling progenitors emigrate from the bone marrow and then mature into early thymic progenitors and progressively commit to the T cell lineage by maturing from the double negative 2 cell stage to DN3, DN4, and eventually to the double-positive (DP) cell stage. Here, they begin to rearrange their TCRα loci and can then be selected by MHC-I- and MHC-II-expressing cortical thymic epithelial cells (cTECs). Cells that successfully undergo positive selection by cTECs can become immature CD8+, CD4+, and Treg cells. By contrast, a small proportion of DP cells rearranges its TCRα locus to generate the iNKTα chain and is selected by other DP cells expressing CD1d and signaling lymphocytic activated molecules family receptors. Intracellular calcium levels are high in these post-selected stage 0 cells, which leads to NFAT translocation into the nucleus and upregulation of Egr2 and CD69. An alternative pathway (depicted by a dashed arrow) wherein a small number of DN4 thymocytes rearrange their TCRα loci to generate the iNKTα chain that can then give rise to stage 0 iNKT cells upon positive selection has also been described. These cells then transition through an uncommitted PLZFhi stage before diverging into the functionally distinct iNKT2, iNKT17, and iNKT1 subsets defined by the transcription factors GATA-3, RORγt, and T-bet, respectively. iNKT2 cells migrate from the cortex to the medulla where they begin to produce IL-4 at steady-state. This IL-4 production (as designated by red arrows) has been linked to conditioning surrounding CD8+ T cells to become innate-memory CD8+ T cells, promoting certain dendritic cell populations to secrete the chemokines CCL17 and CCL22, preventing ETP commitment to the T cell lineage and inhibiting thymic export of single positive (SP) thymocytes into peripheral tissues. RANKL expressed by medullary iNKT2 (and iNKT17) cells also promotes maturation of medullary thymic epithelial cells (mTECs) into Aire+ MHC-IIhi mTECs, which mediate negative selection of medullary SP thymocytes and Treg maturation.
Co-stimulations of DP cells via the TCR and the SLAM receptors elicit a strong signal in the iNKT precursors leading to a high expression of the transcription factor Egr2 (56). Without Egr2, thymocytes are arrested early during iNKT cell development (57–59). High expression of Egr2 is dispensable for conventional T cell development (57), suggesting that iNKT cells are unique in their requirement for stronger-than-normal agonistic signals to properly mature. Indeed, post-positive selection iNKT cells, commonly referred to as stage 0 iNKT cells, expressed the highest levels of Nur77 (encoded by Nr4a1), an immediate early protein induced upon TCR signaling, compared to all other thymocytes (60). Furthermore, Egr2 has been demonstrated to bind directly to the promoter and positively regulate the transcription of Zbtb16, the gene encoding PLZF. In the absence of Egr2 and its related protein Egr1, PLZF levels in the few remaining iNKT cells are significantly lower, further corroborating the link between strong TCR/SLAM signaling and proper iNKT development (59). iNKT cells are imbued with an activated/memory phenotype relatively early during their ontogeny, primarily due to their expression of PLZF (37, 38). In fact, ectopic expression of PLZF is sufficient to promote a memory phenotype even in conventional CD4+ T cells at steady state (38, 61).
One of the outstanding questions in iNKT cell biology currently is how iNKT cell subset differentiation occurs in the thymus. Conventional T cells in the periphery require antigen-induced priming as well as the appropriate cytokine milieu to be properly polarized into different functional subsets. For example, generating TH1 cells from naïve CD4+ T cells requires TCR-mediated signals in addition to the cytokine IL-12 that promotes commitment to the TH1 lineage through the actions of the transcription factor signal transducer and activator of transcription 4 (7). TH2 cells, though, require TCR-mediated signals in conjunction with IL-4 to be polarized into their lineage (9). Polarization into different lineages in this manner has been demonstrated to be dependent on specific factors both in vitro and in vivo. However, since iNKT cells differentiate in the thymus unlike conventional T cells that differentiate in the periphery, it is unclear if they follow a similar differentiation course.
Several factors, besides the canonical transcription factors, have been revealed to be relevant for specific iNKT cell subset development and maintenance, primarily through the use of knockout mice. The transcription factor lymphoid enhancer factor 1 (Lef-1) was shown to influence differentiation into the iNKT2 lineage (62). Lef-1 expression strongly correlates with PLZF expression and Lef-1−/− mice have significantly fewer iNKT2 cells capable of producing IL-4 (62). Mice lacking the serine protease Serpinb1 generate more iNKT17 cells in the thymus and more iNKT17 cells are found in peripheral tissues in KO mice as well (63). When the transcription factor Bcl11b was specifically knocked out of PLZF+ cells using a PLZF-Cre mouse, iNKT17 cells were preferentially maintained at the expense of iNKT1 and iNKT2 cells, suggesting that Bcl11b is required for iNKT1 and iNKT2 cell survival but is dispensable for iNKT17 cell survival (64). The transcription factor Th-POK appears to restrict the generation of iNKT17 cells because in its absence, iNKT17 cell numbers are increased and more iNKT cells are capable of producing IL-17 (65). Th-POK itself is epigenetically regulated by the micro-RNA miR133b and more iNKT17 cells are observed in mouse strains where iNKT cells express higher levels of this miRNA (66). In this manner, many other factors have similarly been described to be preferentially important for the development and/or maintenance of one subset over others. Despite the abundance of studies reporting proteins required for different subsets, what is unknown is the stage of development during which these proteins are first expressed. In other words, it is unclear if these factors themselves are the force behind the development of specific subsets or are instead a consequence of commitment to a given subset. If the former, then, different precursors should exist, each with a specific phenotype, which predispose cells into a given subset. Support for this hypothesis is currently lacking. Instead, it has been shown that DP precursors exist in a poised state on a population level. Approximately 1,000 genes in bulk DP cells are transcriptionally silent yet possess both permissive (H3K4me3) and repressive (H3K27me3) histone modifications at their transcriptional start sites (67, 68). About 14% of these genes code for transcription factors, some of which are implicated in influencing lineage diversification. These results suggest that a given DP precursor is capable of adopting various fates but only commits to one upon receiving specific cues (68). Thus, differential signals received by DP thymocytes consequently could drive the commitment of individual precursors into distinct lineages that are then preferentially dependent on specific proteins.
Several hypotheses, not all of which are mutually exclusive, can be formulated to explain how iNKT cell subsets might arise from differential signaling during development. First, the cells might enter distinct pathways, as observed in the periphery, due to stimulation by different cytokines that impose commitment to a specific lineage. In support of this idea, each of the thymic subsets expresses a unique composition of cytokine receptors (19, 69). Indeed, iNKT1 cells display a dependence on the cytokine IL-15 for survival while iNKT17 cells instead require IL-7 to maintain their numbers (70, 71). In addition, iNKT2 and iNKT17 cells also seem to require IL-25 for homeostasis and function (72). However, this idea pre-supposes that commitment only occurs after upregulation of cytokine receptors that can enforce lineage specification. Instead, cytokine receptor expression frequently occurs as a consequence of expression of particular transcription factors themselves (73–75). Because expression of the transcription factors would already signify commitment, this implies that an even earlier event drove the cells to differentially upregulate these proteins. Furthermore, stage 0 iNKT cells do not express any appreciable levels of transcripts coding for lineage-determining transcription factors, even at the single cell level (69). Instead, their transcriptomes are reminiscent of uncommitted DP cells having recently undergone positive selection, suggesting that although commitment to a subset can be reinforced by cytokine receptor signaling, it is unlikely to be the original signal driving diversification.
Another hypothesis is that recognition of specific self-ligands during selection has the potential to shape the subset ratio. Accordingly, in a recent study, antigen-specific iNKT cells were readily identified in the thymus when using CD1d tetramers loaded with a variety of lipids (76). Even though all iNKT cells could be identified using tetramers loaded with the potent antigen αGC, different subpopulations reacted exclusively to specific other lipids. Although the authors did not further categorize the responding cells based on their functional subsets, it remains an appealing idea that differential recognition of CD1d-presented lipids might have dramatic consequences for iNKT precursors. In this scenario, lineage commitment would be expected to occur early after positive selection for each cell. Different microenvironments within the thymus could present high levels of specific lipids, thereby specifically promoting selection of certain iNKT cell subsets. This seems an unlikely proposition because selection itself has been demonstrated to occur on cortical DP thymocytes while the majority of the mature subsets (approximately 70%) take up residence in the medulla of the thymus, implying that migration to different thymic niches occurs well after positive selection (77). It is instead plausible that the precursors encounter different antigens merely by chance in a homogenous cortical environment, although this possibility remains to be formally demonstrated.
Different ligands, though, are only distinguished by iNKT DP precursors through the use of a diverse TCR repertoire (19, 78, 79), raising a third non-mutually exclusive possibility that iNKT cell subsets might arise due to differential signaling transduced by their TCR during positive selection. As the cells undergo selection, the strength of the signal perceived by each cell due to the nature of the TCR as well as the specific ligand being recognized could instruct each cell to adopt and commit to a specific lineage. This is an intriguing idea since TCR signal strength influencing fate decisions has been demonstrated in a variety of contexts (80–82). In addition, at the population level, it is reasonable to postulate that the precursor cells express an even distribution for a variety of markers, precluding the predisposition of any one cell to enter a given pathway. However, the nature of the αβ TCR (the TCRβ chain in particular in the case of iNKT cells) does vary from cell to cell, making it likely that the signals transduced by the TCRs could similarly vary. The signals thus generated might have a small range of strengths but through co-stimulation by SLAM receptors (and perhaps other coreceptors), this range could be amplified and engender distinct fates to cells that land on either end of the spectrum. In agreement with this, mature iNKT subsets express different levels of Nur77, with iNKT2 cells expressing the highest, followed by iNKT17 cells, and finally with iNKT1 cells expressing the lowest levels (19). Using Egr2 as a marker for strength of TCR-mediated signals during positive selection, data generated in our lab also confirm this hierarchy (83). Thus, the cells within the subsets seem to retain a memory of the signals they received as precursors, with iNKT2 cells having received the strongest signals followed by iNKT17 and iNKT1 cells.
One way that cells might retain the signaling information could be through the transcription factor GATA-3. TCR signaling has been previously demonstrated to upregulate GATA-3 protein levels (84). Many of the genes coding for the components of the TCR complex, namely the Tcra, Cd3d, and Cd3g loci, are direct targets of GATA-3 (75, 85–87). In mice lacking GATA-3, expression of these different genes is significantly reduced. Furthermore, GATA-3 has also been previously shown to autoregulate its own expression in a positive feedback loop (88). Therefore, stronger signaling during positive selection could potentially lead to higher and sustained GATA-3 levels and consequently, higher TCR levels. In support of this, the TCR levels (and GATA-3 levels to some extent) on the different subsets follow the same pattern as Nur77 and Egr2 do, perhaps suggesting that signals received during selection could be maintained in this manner (19, 63).
Pairing the invariant TCRα chain with different TCRβ chains can also affect the affinity with which the TCR heterodimer interacts with antigen/CD1d and consequently, how efficiently the TCR can initiate and propagate a signal intracellularly (89). Interestingly, in retrogenic mice generated with distinct TCRβ chains, the proportions of each of the subsets could be linked to the avidity of the TCR for its ligand (90). Similarly, when clonal mice were generated using nuclei from iNKT cells expressing different TCRs, the proportion of PLZFhi iNKT cells in the thymus directly correlated with the avidity of the TCR for lipid/CD1d (91). Finally, different studies have revealed that TCR signaling regulates the expression levels of several proteins involved in chromatin remodeling and in whose absence, the subset ratios are vastly altered (68, 92, 93). With the advent of myriad technologies allowing immunologists to assess transcriptomic and epigenomic signatures at the resolution of a single cell, it will become paramount in the future to pursue single cell analyses on the stage 0 iNKT cells immediately following positive selection and determine if TCR signaling-mediated differences can already be identified within these cells. Although a recent study did conduct single-cell RNA-sequencing analysis on stage 0 iNKT cells, the study concluded that these cells were similar to other positively selected conventional cells (69). As this study only analyzed 45 stage 0 iNKT cells, obtaining greater depth by sequencing more stage 0 iNKT cells could potentially provide more information on otherwise non-sampled low-abundance transcripts and/or accessible loci in different cells. With this information, perhaps an early signature can be identified that correlates with eventual iNKT cell subset.
After developing in the thymus, iNKT cells have been observed in various tissues throughout the body (13). Unfortunately, due to an incomplete understanding of iNKT cell subsets, only their presence or absence in various tissues could be ascertained until recently. Some studies had identified iNKT cells in different tissues by αGC-CD1d tetramer staining, which remains the gold standard (30, 94, 95). This staining, however, was rarely done in conjunction with staining for the master transcription factors associated with the subsets, precluding their identification. In other studies, cells were frequently identified by their co-expression of NK1.1 and TCRβ (78, 96, 97). This strategy is perhaps problematic for multiple reasons. First, since staining for NK1.1 is not successful in all strains (41), it is entirely possible that observations made using the B6 mouse model are not generalizable to all mouse strains, as demonstrated in BALB/c and non-obese diabetic (NOD) NK1.1-congenic mice (98). Second, NK1.1 does not exclusively mark iNKT cells as conventional CD8+ T cells can also co-express NK1.1, potentially obfuscating the real iNKT population (99, 100). Indeed, cytokine stimulation can lead to upregulation of NK1.1 and other NK cell-related markers in CD8+ T cells, perhaps suggesting that iNKT1 cells acquire NK1.1 expression in a similar manner (101). And finally, since iNKT1 cells are primarily the only cells expressing NK1.1, learning about iNKT cell tissue localization through the use of this marker is by necessity restricted to this subset. Despite these drawbacks, some aspects of the tissue distribution patterns of iNKT cell subsets could be gleaned from early studies.
Of the subsets, iNKT1 cells have been indirectly demonstrated to remain long-term thymic residents and accumulate over time. When congenically marked thymic lobes were transplanted in recipient mice, while different kinds of iNKT cells were observed early after transplantation, only NK1.1+ iNKT cells persisted in the thymus as time progressed (34). In fact, over 50% of the mature αβ TCR+ cells remaining of donor origin were these likely iNKT1 cells that were maintained for a long period of time. This finding is in stark contrast to the conventional T cell population that is rapidly turned over in the thymus (102, 103). One possible explanation for thymic retention of iNKT1 cells is that T-bet drives expression of the chemokine receptor CXCR3, allowing them to be maintained in the thymus due to high levels of the cognate CXCR3 ligand CXCL10 (35). Another explanation for this phenomenon could be that T-bet in iNKT1 cells induces the expression of the gene Il2rb coding for the protein CD122 (73), thereby supporting the response to the trans-presented IL-15 cytokine (104). This cytokine is produced by cells in the thymic medulla and not only serves as a survival cytokine for iNKT1 cells but also help stabilize T-bet itself in those cells (70, 105). In addition, IL-15 has been previously shown to upregulate CD69 in cells sensitive to this cytokine and indeed, iNKT1 cells do express high levels of CD69 (106, 107). Because ectopic overexpression of CD69 prevents thymic egress of conventional T cells (108), this IL-15-induced CD69 could potentially also play a role in iNKT1 cell thymic retention.
Despite the thymic retention, NK1.1+ iNKT cells are also found in the periphery. Interestingly, large numbers of NK1.1+ iNKT cells are found in the liver (109). This could be linked back to iNKT1 cells expressing T-bet and their sensitivity to IL-15. As previously mentioned, T-bet expressing cells also concomitantly express CXCR3 while IL-15 has been shown to condition cells to express CXCR6 in humans (110). The ligands for both these chemokine receptors (CXCL9/CXCL10 for CXCR3 and CXCL16 for CXCR6) are present in abundant quantities in the liver (111–113). Thus, by following their chemotactic gradients, it is not surprising that iNKT cells compose 20–30% of T-lymphocytes in the liver (30, 94). Moreover, liver iNKT cells also establish strong residency upon arrival, as evidenced by their reduced circulation in parabiotic mice (94). Long-term residency by these lymphocytes has been proposed to be due to the high expression of the transcription factor Hobit (114). Induced by both T-bet and IL-15, Hobit has been shown to be preferentially expressed in liver iNKT cells, preventing their egress from the liver (114), although a recent study disputes this finding (115). Nevertheless, thymic iNKT1 cells also express high levels of Hobit perhaps suggesting they might maintain their residency in a similar manner (116).
With B6 mice remaining a popular mouse model to study iNKT cells, iNKT2 and iNKT17 cell localization has been largely understudied. While in some cases, there has been some direct evidence of a specific subset, iNKT2/iNKT17 presence in peripheral tissues has instead been frequently inferred, either by chemokine/cytokine receptor expression or by their cytokine secretion profile. iNKT17 cells, in particular, were initially identified as IL-17 producing iNKT cells within the NK1.1− population by several groups (117, 118). Thereafter, using RORγt-GFP reporter mice, a unique population of iNKT cells was identified in the thymus that was dependent on RORγt for secretion of IL-17 (119). Since RORγt expression is strongly correlated with expression of the chemokine receptor CCR6, iNKT17 cells are specifically directed to the skin (120, 121). Additionally, expression of this chemokine receptor also endows some iNKT17 cells to enter lymph nodes as they are enriched in peripheral lymph nodes compared to the other sub-lineages. Similar to CCR6, expression of CD103 is also high on iNKT17 cells, leading to preferential retention of these cells in the skin, where epithelial cells express the CD103 ligand, E-cadherin (121, 122). iNKT17 cells also uniformly express high levels of the protein Syndecan-1 (CD138), although the reason for why they express this is unknown (123).
Insight into iNKT2 cell localization, however, has been further hindered by the lack of unique markers defining this subset. Unlike iNKT1 and iNKT17 cells, cytokine secretion is insufficient to specifically identify iNKT2 cells since IL-4 is also secreted by iNKT1 cells. Additionally, while iNKT2 cells express high levels of GATA-3, iNKT1, and iNKT17 cells also express this transcription factor, albeit at slightly lower levels (19). And finally, although expression of the cytokine receptor IL-17RB (specific for the cytokine IL-25) on iNKT cells has been demonstrated to enrich for IL-4/IL-13-secreting cells, iNKT17 cells also express this receptor, thereby preventing the use of this marker to specifically distinguish iNKT2 cells in tissues (72).
Through the use of reporter mice and transcription factor staining, a recent study has resolved these ambiguities by shedding substantial light on iNKT cell tissue distribution as well as location within tissues (77). In this seminal study, iNKT cell subsets were identified by their transcription factor expression and analyzed in many different tissues. Additionally, by developing a technique called histocytometry, the authors were able to identify the intra-tissue localization of the iNKT cell subsets. For example, it can now be appreciated that approximately 70% of the thymic iNKT cells, irrespective of subset, reside in the medullary space. This could be due to greater accessibility to homeostatic/survival cytokines (IL-15 for iNKT1 and IL-25 for iNKT2/iNKT17) in the medulla as well as chemokine-mediated trafficking. Remarkably, the relative iNKT subset distribution within tissues is not equivalent across different strains of mice as evidenced by strain-specific iNKT cell subset distribution patterns (77). For example, skin-draining lymph nodes were largely enriched for iNKT17 cells in the NOD background and to a lesser extent in B6 mice. However, iNKT2 cells were the principal subset present in these lymph nodes in BALB/c mice. Other tissues also similarly contained different ratios of the subsets across the strains. It is currently unclear if this corresponds merely to the proportion of each subset generated in the thymus in different strains, since BALB/c mice generate significantly more iNKT2 cells, or if strain-specific tissue-homing biases also exist.
New subsets of iNKT cells besides the three described here have also been identified. iNKT cells producing IL-10 are abundant in adipose tissues, where they make up approximately 30% of all T cells (124). Acquiring the moniker iNKT10 due to their ability to produce IL-10, these cells express low levels of PLZF and are dependent on the transcription factor E4BP4 for their functional competence (42). Although these cells are thymically derived as they are absent in adipose tissues of athymic nude mice, they could not be identified in detectable numbers in the thymus of a WT mouse (42). However, in mice expressing a transgene with a modified TCRβ chain that results in fewer iNKT cells due to improper signaling, more iNKT10-like cells were observed in the thymus that preferentially homed to adipose tissue (125). Currently, though, how they arise in a WT mouse is unknown. Therefore, it is possible that one of the three thymic subsets gives rise to this new subset that differentiates in the periphery. What and how specific cells home and differentiate within adipose tissue is uncertain. It is conceivable that due to their expression of T-bet and ability to produce IFNγ after stimulation with PMA/ionomycin, they are cells that deviate from the iNKT1 lineage due to the adipose tissue microenvironment (42).
Another subset that has also received attention of late is the iNKTFH subset, which, analogous to the conventional TFH population, expresses Bcl6 and helps in antibody class-switching and somatic hypermutation (43, 45). This population was initially described in secondary lymphoid organs upon immunization with antigen in conjunction with αGC, prompting these cells to form stable contacts with B cells and induce germinal centers through the secretion of IL-21. Since this subset has been found in the spleen and iNKT1 cells are also found in higher numbers in the B cell zone (77), perhaps iNKTFH cells represent another branch-off subset from the iNKT1 lineage. This would suggest that iNKT1 cells are somewhat plastic in the periphery and can adopt other fates based on the inflammatory cues they receive.
Although T cells commonly circulate throughout the host body, so, they can properly survey all sites for any perturbations, many cells also establish long-term residency in various tissues (126). After a primary immune response has been cleared, a proportion of the antigen-specific cells are retained in the tissue to guarantee a faster response in the future. In the absence of any immune response, however, these cells are not quiescent and sessile but rather dynamically interact with other cells in the tissues to shape their microenvironment in crucial ways. Perhaps owing in part to their memory phenotype, iNKT cells similarly establish long-term residency in several different tissues (42, 94, 127). Beyond that, even without establishing residency, they play important roles in maintaining homeostasis even in steady-state conditions. For instance, their effector status enables them to readily secrete cytokines upon stimulation, which can have dramatic consequences for their surroundings (19, 77). Thus, they serve as a rheostat for how nearby cells acquire phenotypes that correspondingly influence tissue equilibrium.
Ample evidence exists that iNKT cells in the thymus skew the thymic microenvironment in substantial ways (Figure 1). Importantly, the ratio of the subsets affects the phenotypes of other conventional cells. For example, iNKT2 cells in the thymus affect the phenotype and functionality of CD8+ T cells. Usually, thymic CD8+ T cells exhibit naïve characteristics and display antigen-response kinetics that are delayed compared to memory CD8+ T cells. Through the use of IL-4 reporter mice, it was discovered that thymic iNKT2 cells constitutively produce IL-4 (19). This IL-4 conditions the surrounding CD8+ T cells to upregulate CXCR3 and Eomes and exhibit memory traits (128, 129). These “innate” memory CD8+ T cells display antigen-response kinetics reminiscent of memory cells despite never having encountered antigen previously (130). In so doing, they can play a major role in combating chronic viral infections by mounting rapid and robust responses (131, 132). Mutant mice with larger numbers of iNKT2 cells compared to wild-type mice or different strains of mice that endogenously produce large numbers of iNKT2 cells consequently have larger numbers of innate memory CD8+ T cells. For example, only ~15% of the total iNKT cells in 8-week-old B6 mice thymi are iNKT2 cells while similarly aged CBA and BALB/c mice have ~40 and ~50%, respectively (19). The IL-4 produced by these iNKT2 cells has been directly demonstrated to affect the numbers of innate memory CD8+ T cells, with B6 mice thymi possessing <4% while CBA and BALB/c mice thymi contain ~30 and ~60% innate memory CD8+ T cells, respectively.
In addition to affecting the CD8+ T cells, the IL-4 produced at steady state by iNKT2 cells also conditions SIRPα+ thymic dendritic cells (DCs) to upregulate and produce the chemokines CCL17 and CCL22 (19). These chemokines interact with CCR4, also expressed by iNKT2 cells, perhaps implying a positive feedback loop whereby iNKT2 cells are drawn to the medulla by these chemokines where they enact their effects and further ensure their continued presence due to their sustained production of IL-4. Regulatory T cells (Tregs) appear to also be increased in number and proportion by the iNKT2-produced IL-4 (133). These Tregs exhibit more of an activated phenotype and, in fact, have a greater suppressive capacity in an immune response. Recent data have also identified IL-4 as an inhibitory cytokine for early thymic progenitors (ETPs) to commit to the T cell lineage (134). ETPs stimulated through the IL-4 receptor upregulated the myeloid-specific transcription factor C/EBPα, presumably halting their development into T cells. It would be curious to see if mice with a higher frequency of iNKT2 cells had correspondingly fewer ETPs seeding the T cell pool in the thymus. Finally, IL-4 promotes thymic egress of SP4 thymocytes in a S1P/S1PR1-independent manner (135). Although how IL-4 leads to an accumulation of SP thymocytes is currently unknown, it is clear that the pleiotropic effects of IL-4 by iNKT2 cells markedly change the thymic landscape, reinforcing their importance in tissue maintenance.
Significantly fewer MHC-IIhi Aire+ medullary thymic epithelial cells (mTECs) exist within the thymus of CD1d−/− mice compared to the B6 control mice (105). Aire is a transcription factor exclusively expressed in mTECs that promotes the expression of peripheral tissue antigens and tolerance of developing SP thymocytes. Both central tolerance of SP4 thymocytes and generation of Tregs depends on MHC-II and Aire expression by mTECs. Reduction of the number of cells capable of carrying out these tasks compromises both of these functions (136). mTECs in a CD1d−/− mouse are enriched for an immature phenotype (MHC-IIlo Aire−). Interestingly, this mTEC developmental arrest is critically dependent on RANKL expression by NK1.1− cells iNKT cells, suggesting a potential other role for iNKT2 (and possibly iNKT17) cells in the thymus beyond their production of IL-4. It would be of further relevance to identify if BALB/c mice, which have much higher numbers of iNKT2 and iNKT17 cells in the thymus, have an even more profound defect in mTEC maturation in the absence of CD1d than was described in B6 mice.
It remains unclear why iNKT2 cells play such key roles in influencing different thymic compartments when iNKT1 cells have been identified as long-term thymic residents. What role(s), if any, iNKT1 and iNKT17 might have in maintaining thymic homeostasis is currently unknown.
Substantially less evidence exists for iNKT subsets impacting steady-state functions of other tissues. Production of IL-4 by iNKT2 cells continues to condition the peripheral tissues by contributing to the high IgE levels found in the sera of BALB/c mice as well as promoting a proportion of CD4+ T cells in the mesenteric lymph nodes (mLNs) to constitutively express the activated form of the transcription factor STAT6 (phospho-STAT6) (19, 77). Activated STAT6 translocates to the nucleus from the cytosol and promotes expression of GATA-3, implicating iNKT2 cells in potentially influencing the “TH2-bias” observed in BALB/c mice (137). Beyond this IL-4-mediated role of iNKT2 cells, our understanding of iNKT subset functions at steady-state in peripheral tissues is limited. Parabiosis experiments have determined that iNKT cells establish long-term residency in hosts in the liver and the lung (94, 127). In the liver, based on their expression of NK1.1, the resident cells are largely iNKT1 while expression of IL-17RB suggests an enrichment of iNKT2/iNKT17 cells in the lung. Although it is possible that the reason for their tissue residency is simply to act as sentinels that kickstart the overall immune response during an infection, tissue-resident lymphocytes quite frequently have roles beyond that. Thus, future experiments where iNKT cells are prevented from accumulating in those tissues, perhaps by conditional deletion of chemokine ligands in those tissues, should help illuminate how iNKT subsets are affecting tissues in non-infectious settings.
Recently, though, the increased attention paid to iNKT10 cells has uncovered some interesting functions of these cells in maintaining adipose tissue homeostasis. Experiments conducted using parabiotic mice demonstrated that iNKT10 cells establish long-term residency in adipose tissue where they support an immunosuppressive environment (42). Upon stimulation, over half of them secrete IL-10, which helps induce an anti-inflammatory “M2” macrophage phenotype. Moreover, in contrast to other peripheral iNKT cell subsets, these cells produce high amounts of IL-2 upon stimulation. This, in conjunction with the IL-10, also promotes Treg expansion with a highly suppressive phenotype. This supports the idea that iNKT cells in the adipose tissue might also be producing these two cytokines at steady state, but this remains to be formally demonstrated. Although many iNKT10 functional features have primarily been uncovered by stimulating these cells with αGC, the cells also express high levels of PD-1 and Nr4a1 even at steady state. This could indicate that the iNKT cells perhaps receive continuous TCR-mediated signals in the adipose tissue (42). Indeed, adipocytes themselves display high levels of CD1d molecules. Yet, the nature of the lipids that might be presented to iNKT10 cells by adipocytes remains to be discovered.
Because of their varied responses, iNKT cells have been demonstrated to be involved in myriad immune responses in which they can be either protective or pathogenic (138, 139). In mice infected with Streptococcus pneumoniae, iNKT cells produce IFNγ within hours of infection (140, 141). Preventing iNKT cells from getting activated by using an antibody that blocks CD1d recognition by iNKT TCRs significantly increased bacterial loads, suggesting that iNKT cell activation contributed to bacterial clearance. Similar findings have been observed in other models in which mice have been infected with Pseudomonas aeruginosa or Mycobacterium tuberculosis, where iNKT cell deficiency also correlated strongly with increased bacterial burdens, hinting that iNKT cells are perhaps involved in helping clear different kinds of pathogenic bacteria (142–144). When mice deficient in iNKT cells were injected with fibrosarcoma cells, tumor progression was inhibited significantly only upon transfer of iNKT cells (145). Yet again, this protective effect was evident only when the recipient mice expressed CD1d, perhaps implying that the fibrosarcoma cells expressed lipids capable of activating iNKT cells when presented by CD1d. On the other hand, in a model of implanted osteosarcoma, 88% of CD1d−/− mice rejected the tumors compared to only 24% of WT mice (146). The reasons for the contradicting roles of iNKT cells in tumor models remain unclear. Finally, transferring iNKT cells into the diabetes-prone NOD mouse conferred resistance to diabetes and, in one study, reduced the incidence of diabetes from 90 to 10% (147). In contrast, anti-CD1d treatment of (NZBxNZW)F1 mice led to increased protection from lupus (148, 149). Indeed, transferring iNKT cells from (NZBxNZW)F1 mice into healthy recipients was sufficient to transfer disease (150). Thus, iNKT cells can modulate the course of the immune response in a variety of manners, depending on the models being studied.
Although iNKT cell responses have been characterized in different diseased-state conditions, the specific iNKT cell subsets contributing to the response are largely unknown. Usually, the contribution of iNKT cells to an immune response is determined through the use of a CD1d−/− mouse model and/or a TRAJ18−/− mouse model, both of which lack iNKT cells. However, both of these mouse models have drawbacks. The original TRAJ18−/− mouse was generated by introducing a neomycin resistance gene into the Traj18 locus (151). Interestingly, these mice lack approximately 60% of their overall TCR repertoire due to an inability to express TCR rearrangements involving TRAJ genes upstream of Traj18 (152), potentially due to the presence of the neomycin resistance gene (153). Thus, these animals not only lack iNKT cells but also a substantial proportion of their conventional TCR repertoire, potentially obfuscating some of the findings discovered in studies using these mice. Repeating these experiments in mice where Traj18 was deleted without the presence of a neomycin resistance gene should help clarify the original results (154–156). In the case of the CD1d−/− mouse model, new data have revealed that one of the four widely distributed knockout strains (157) continues to possess a small number of iNKT cells (158). Therefore, this specific knockout strain cannot be considered iNKT cell-deficient mice and conclusions obtained using these mice should be reassessed and instead be reevaluated using mice in which iNKT cells are completely absent (159, 160). Besides the use of these mouse models, the iNKT cell contribution to an immune response is further characterized only by the cytokines that affect the progression of the disease, frequently IFNγ and IL-4, but not the phenotype of the iNKT cells secreting those factors. Although this cytokine-secretion profile is more indicative of the specific subsets involved, it is often insufficient since iNKT1 cells are also capable of producing IL-4. Thus, a greater effort needs to be put forth to identify the subsets involved in any disease based on not just their cytokines produced but also by the transcription factors expressed.
A few studies have shed some light on the roles of specific iNKT cell subsets in diseases, albeit indirectly. One study used a transplantable tumor model to determine that CD4+ T cells negatively regulated tumor rejection. Upon further examination, it was discovered that CD4+ iNKT cells were the primary source of IL-13, creating an immunosuppressive environment that prevented tumor rejection (161). When these iNKT cells were depleted, either through the use of depleting antibodies targeting CD4 or CD1d−/− mice, tumors were rejected at a significantly higher frequency. It is possible that the responding iNKT cells were iNKT2 cells due to their production of IL-13 and the fact that iNKT2 cells would have been abundant in the BALB/c mice in which these experiments were conducted (19, 72, 162). In addition, iNKT2 cells are enriched in the CD4+ population (19, 63), further lending credence to this idea, although it is perhaps worth revisiting these experiments using currently available tools.
Interestingly, there appears to be a tissue-specific bias associated with iNKT cells capable of mediating tumor rejection. When bulk iNKT cells from the liver were transferred into TRAJ18−/− mice harboring a sarcoma, they were capable of halting tumor progression (163). However, bulk splenic and thymic iNKT cells were not similarly capable of rejecting the tumor growth. It was further determined that the DN hepatic iNKT cells were significantly better at tumor rejection compared to the CD4+ hepatic iNKT cells. This latter finding could perhaps be because upon stimulation, CD4+ iNKT cells tend to produce more TH2 cytokines that would be immunosuppressive compared to the TH1 cytokines that DN iNKT cells are more biased to produce (79). Indeed, when IL-4−/− iNKT cells, irrespective of their tissue origin, were transferred into mice with tumors, they more potently rejected tumors compared to WT iNKT cells (163). Curiously, however, CD4+ and DN iNKT cells from different tissues all produced similar levels of IFNγ and IL-4, suggesting that although IL-4 does have an impact on tumor rejection, other differences between the subsets and their tissue-origin also likely affect the functions of the iNKT cells in vivo. The specific functional subsets associated with these findings, though, remain unknown because iNKT1 cells, for example, can be found in both the CD4+ and DN compartments (63). Thus, in light of what is known today, these experiments bear repeating with the use of transcription factor staining to identify any differences between the various sources of iNKT cells. It would be especially interesting to identify gene signature differences between the same iNKT subsets but from different tissues to understand if certain tissues impose functional variations.
In other studies, iNKT cells were also discovered to be relevant in airway hyperreactivity (AHR). One of the hallmarks of asthma, AHR features eosinophilia in the airways, enhanced mast cell growth, and increased levels of serum IgE (164). Conventional TH2 cells play a major role in exacerbating antigen-induced AHR. However, it appears that iNKT cells can prime the immune system initially to bias the response toward a TH2 phenotype. In fact, in several models of AHR, iNKT-deficient mice do not develop AHR (165, 166). Further, intratracheal administration of a lipid agonist and a protein antigen strongly activated the pulmonary iNKT cells to prime CD4+ cells specific for the protein antigen to polarize into TH2 cells (127). Indeed, IL-4/IL-13 produced by iNKT cells was discovered to be fundamental for mice to succumb to AHR in these studies, although this remains controversial since a later study found that iNKT cells were dispensable for airway inflammation (167). Despite this, the iNKT cells secreting the type 2 cytokines were subsequently identified as expressing IL-17RB, providing some evidence to suggest that the cells potentially promoting AHR were possibly iNKT2 cells (168, 169). Only IL-17RB+ cells, usually expressed by iNKT2 and iNKT17 cells (72), were capable of recapitulating the symptoms upon transfer into a iNKT cell-deficient mouse by secreting IL-4 and IL-13 (168, 169). Interestingly, in a different model of AHR in which mice were exposed to ozone instead of an allergen/antigen, IL-17 production by iNKT cells was also required in addition to IL-4/IL-13, perhaps implicating that iNKT17 cells also can contribute to AHR in certain contexts (170). The IL-17 produced by iNKT cells led to increased neutrophilia instead of eosinophilia in the airways and has been demonstrated in a separate study to be dependent on c-Maf, a transcription factor also involved in promoting the proper function of TH17 cells (171–173).
Administration of αGC intravenously in mice can activate the vascular-localized iNKT cells. In this fashion, the hepatic and the red-pulp splenic iNKT cells, which are primarily iNKT1 cells, respond within minutes by producing IFNγ and IL-4. Serum increases of both these cytokines can easily be detected in these conditions (77) and the IL-4 secreted under these conditions appears to have long-range effects as demonstrated by the increased phosphorylation of STAT6 in CD4+ T cells in other tissues, such as LNs, despite the iNKT cells in those tissues remaining unstimulated. Thus, blood-borne pathogens that are capable of activating iNKT cells could possibly activate iNKT1 cells due to their localization that could then condition T cells in distal tissues. Analogously, since iNKT2 cells are present in high numbers in the mLNs of certain mouse strains, oral administration of αGC largely activated these cells and caused them to secrete IL-4 in large quantities (77). However, perhaps due to a lack of proximity to the circulation, the IL-4 produced in this setting had primarily local effects, with only the CD4+ T cells in mLNs increasing their phospho-STAT6 levels while T cells in other tissues were unaffected.
Bacteria express their own lipids, some of which might serve as stimulatory antigens to iNKT cells. Viruses, however, hijack host machinery for their own purposes and thus, are devoid of any lipids themselves and thought to not activate iNKT cells directly. However, viral infections can lead to upregulation of CD1d by triggering toll-like receptors (TLRs) (174). Additionally, infection can also lead to activation of hypoxia-inducible factor, which in turn could alter the lipid metabolism and allow antigenic self-lipids to be presented to iNKT cells (175). Thus, viral infections could lead to activation of iNKT cells in a CD1d-dependent manner (176). Alternatively, activation of innate cells such as DCs through TLRs could prompt them to secrete pro-inflammatory cytokines such as IL-12 and IL-18 that consequently activate iNKT cells (177, 178). These activated iNKT cells can secrete IFNγ that promotes an antiviral response (179, 180). Thus, iNKT cells can participate in viral infections, potentially in a protective role. However, the specific subsets involved in viral clearance are unknown. Although the production of IFNγ by iNKT cells strongly suggests that the subset involved is the iNKT1 subset, this remains to be formally demonstrated.
Interestingly, a new study has highlighted that iNKT cells influence humoral immunity during Influenza A virus infection (181). A previous study had identified iNKT cells as important in curbing myeloid-derived suppressor cell (MDSCs) function in influenza infection (182). The MDSCs in influenza-infected mice suppressed influenza-specific immune responses, leading to high titers of the virus. In a CD1d-dependent manner, iNKT cells were able to restrict the activity of MDSCs and consequently boost the immune responses directed against influenza. Thus, a role for iNKT cells in combating influenza virus infection had already been established. In the recent study, the authors focused on how iNKT cells affect B cell responses in influenza infection. These cells influence B cell germinal center formation and antibody class switching despite not being iNKTFH cells. The iNKT cells are the primary secretors of IL-4 early during the infection and express CXCR3, suggesting that they are possibly iNKT1 cells. CD1d-mediated interactions with CD169+ macrophages were critical for the production of IL-4 by the iNKT cells. This response underscores a novel role that iNKT1 cells potentially play in mounting an immune response against viral pathogens. Although why iNKT1 (if the cells are indeed iNKT1) cells are producing IL-4 and the iNKT2 cells are not is unknown. It could be due to a possible abundance of iNKT1 cells in the mediastinal LNs or it could be that the macrophages present lipids on CD1d only capable of activating iNKT1 cells. Confirming the specific subset involved in this immune response and why these cells are preferentially activated is paramount.
More recently, with iNKT10 cells entering the fold, a new role has been added to the growing list of iNKT cell roles (183). In a model of diet-induced obesity in which mice were fed with high-fat diets (HFD), iNKT cells were depleted from adipose tissues, although this was reversible once their diets were switched to standard fat diets (124). Mice lacking iNKT cells fed HFD weighed more, had large adipocytes, elevated fasting blood glucose levels, and increased insulin resistance. Furthermore, there was an increased infiltration of proinflammatory macrophages into the adipose tissues, an important intermediary step in the inflammation and pathogenesis associated with obesity. As mentioned previously, it is unknown what cytokines the adipose tissue-resident iNKT cells secrete at steady state to maintain healthy adipocytes. However, these iNKT10 cells are different from other tissue localized iNKT cells because of their expression of E4BP4 (42). When iNKT cells transfected with E4BP4 were stimulated, they secreted more IL-10 than E4BP4− stimulated iNKT cells, directly correlating E4BP4 to IL-10 production in iNKT cells. In addition, upon stimulation, the adipose-resident iNKT cells were capable of expanding Tregs in the adipose tissue in an IL-2-dependent manner and the adipose Treg population is substantially reduced in iNKT cell-deficient mice (42). Thus, the immunoregulatory role that the iNKT10 subset plays in adipose tissue to prevent obesity-related illnesses could be due to direct secretion of IL-10 (and possibly IL-2) at steady state.
In humans, iNKT cell numbers are substantially more variable compared to the inbred mouse strains routinely used. Usually, their frequencies are lower in human blood compared to mouse blood (around 0.01–0.1% compared to 0.2–0.5% in mice), and their frequencies are more variable in other tissues when compared to the analogous mouse tissues (184, 185). Humans instead have larger proportions of other innate-like T lymphocyte populations, such as mucosal-associated invariant T cells and the group I CD1-restricted T cells (12). Despite the reduced frequencies, human iNKT cells can be isolated from healthy individuals and patients and analyzed for function and phenotype. Unfortunately, a rigorous manner of identifying human iNKT cells from clinical samples has not always been consistently employed. Many times, the cells were identified by staining for human NK-cell markers such as CD56 and CD161 (the human counterpart of NK1.1) but as in mice, these markers are also expressed on other T cell populations (186, 187). In other studies, iNKT cells were identified by using antibodies targeting the TCRβ chain used by these cells (TRBV25), but this is also problematic since other T cell populations also express this TCRβ chain (188). More recently, iNKT cells have been identified either through the use of αGC-loaded human CD1d tetramers or by using an antibody targeting the invariant TCRα rearrangement unique to iNKT cells (189, 190). Both tools have provided greater resolution into understanding iNKT cell function in humans.
Invariant natural killer T cells in humans can be broadly categorized as DN, CD4+, and a small percentage of CD8+ cells (185). There appears to be some functional conservation of these subpopulations between species since the DN cells tend to have a TH1 bias while the CD4+ cells have a TH2 bias, although the CD4+ cells are also capable of secreting TH1 cytokines (184, 191). This suggests that perhaps human iNKT1 cells are present in both the CD4+ and DN fractions while iNKT2 cells are primarily present within the CD4+ fraction. Further evidence to support this hypothesis stems from the fact that the DN cells express higher levels of several NK receptors compared to the CD4+ cells, similar to how murine iNKT1 cells primarily express the NK receptors (185, 191).
Interestingly, it has been shown that human iNKT cells also express high levels of PLZF compared to other T cell populations (37, 38). Additionally, the CD4+ iNKT cell population appears to express higher levels of PLZF as identified by mRNA levels, perhaps because more iNKT2 cells are present within this population. Indeed, iNKT cell numbers and phenotype appeared to be significantly altered when a patient with biallelic PLZF deficiency was analyzed (192). Human iNKT cells also require SLAM receptor-mediated signals for proper development because humans lacking the adapter SAP lack any observable iNKT cells (193). Despite these studies, whether or not functional iNKT cell subsets follow a similar developmental path in humans and mice has not been formally addressed.
The identification of functionally distinct human iNKT cell subsets with differential expression of master transcription factors, similar to what is observed in mice, is currently limited. Instead, the cells are usually sub-divided based on their cytokine-secretion profile and/or their expression of the CD4 coreceptor. For example, iNKT cells found in the cord blood of humans appear to have an intrinsic bias to secrete IL-17 and cannot produce IFNγ (194). Furthermore, a RORγt inhibitor selectively impaired IL-17 production by iNKT cells in different tissues, suggesting that some iNKT cells could indeed constitutively express RORγt (although this was not formally tested) that endows them with the ability to secrete IL-17 upon stimulation (195). Different iNKT subsets identified by their differential expression of CD4 could induce secretion of different isotypes of antibodies by B cells. In particular, CD4+ iNKT cells were unique in their ability to induce expansion of a CD1dhi CD5hi Breg population (196). These cells, however, did not express CXCR5 or PD-1 at high levels when placed in co-culture with the B cells, suggesting that they are not likely to be iNKTFH cells. As in mice, immunosuppressive functions were associated with CD4+ iNKT cells in various tumor settings, further implicating a functional distinction between CD4+ and CD4− iNKT populations (197). In other models of autoimmunity, iNKT cell numbers were found to be reduced and functionally impaired in their ability to secrete IL-4, although whether this reflects the loss of a specific subset is unknown (138). Reduction in iNKT cell numbers was also observed in obese patients in the peripheral blood, and these numbers appeared to increase once the patients underwent bariatric surgery (124). Whether these cells are the human equivalents of the murine iNKT10 population remains to be explored. Thus, overall, there are primarily tidbits of information regarding functional diversity in human iNKT cells without a cohesive paradigm comparable to the one established in mice. Future work should focus on understanding iNKT cell function in diseased states with increased granularity, with special attention paid to linking function to transcription factors expressed by different cells.
Subset differentiation of iNKT cells is a complex and multifaceted process. Despite this complexity, the final subsets are surprisingly similar phenotypically to other cell types belonging to their corresponding functional group. For example, there is a striking similarity between iNKT1 cells, NK cells, TH1 cells, and ILC1 cells (198). Similarly, iNKT2 and iNKT17 cells share similarities to their γδ and ILC counterparts. What then makes iNKT cell subsets special when other cells occupy similar niches and respond similarly? Two different aspects provide iNKT cells with a unique ability to influence the immune response. First, their ability to recognize lipids in an antigen-specific manner allows these cells to sample an antigen space that would otherwise be unmonitored by conventional T cells. Second, the kinetics of their responses to antigenic stimulation allow the iNKT subsets to rapidly skew the course of the immune response in directed ways. By establishing an initial path for the immune response, iNKT cells have the potential to dictate how downstream adaptive cells are polarized and, consequently, how they respond. Thus, understanding the functional diversity within iNKT cells is essential to be able to manipulate the immune system. By gaining a greater understanding about iNKT cell subsets and their functions, one can hope to target specific subsets in an effort to influence various immune responses in the future.
Both authors wrote and edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SJ and handling Editor declared their shared affiliation.
This work was supported by National Institutes of Health Grants AI121761 and AI124076 (to LG).
1. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol (2015) 33:257–90. doi:10.1146/annurev-immunol-032414-112240
2. Schatz DG, Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol (2011) 11(4):251–63. doi:10.1038/nri2941
3. Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature (1988) 334(6181):395–402. doi:10.1038/334395a0
4. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol (2015) 15(3):160–71. doi:10.1038/nri3795
5. Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol (2006) 24:419–66. doi:10.1146/annurev.immunol.23.021704.115658
6. Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol (2011) 11(12):823–36. doi:10.1038/nri3084
7. Caza T, Landas S. Functional and phenotypic plasticity of CD4(+) T cell subsets. Biomed Res Int (2015) 2015:521957. doi:10.1155/2015/521957
8. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine (2015) 74(1):5–17. doi:10.1016/j.cyto.2014.09.011
9. Walker JA, McKenzie ANJ. TH2 cell development and function. Nat Rev Immunol (2018) 18(2):121–33. doi:10.1038/nri.2017.118
10. Jenkins MK, Moon JJ. The role of naive T cell precursor frequency and recruitment in dictating immune response magnitude. J Immunol (2012) 188(9):4135–40. doi:10.4049/jimmunol.1102661
11. Jameson SC, Masopust D. Diversity in T cell memory: an embarrassment of riches. Immunity (2009) 31(6):859–71. doi:10.1016/j.immuni.2009.11.007
12. Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB. The burgeoning family of unconventional T cells. Nat Immunol (2015) 16(11):1114–23. doi:10.1038/ni.3298
13. Salio M, Silk JD, Jones EY, Cerundolo V. Biology of CD1- and MR1-restricted T cells. Annu Rev Immunol (2014) 32:323–66. doi:10.1146/annurev-immunol-032713-120243
14. Gadola SD, Dulphy N, Salio M, Cerundolo V. Valpha24-JalphaQ-independent, CD1d-restricted recognition of alpha-galactosylceramide by human CD4(+) and CD8alphabeta(+) T lymphocytes. J Immunol (2002) 168(11):5514–20. doi:10.4049/jimmunol.168.11.5514
15. Brigl M, van den Elzen P, Chen X, Meyers JH, Wu D, Wong CH, et al. Conserved and heterogeneous lipid antigen specificities of CD1d-restricted NKT cell receptors. J Immunol (2006) 176(6):3625–34. doi:10.4049/jimmunol.176.6.3625
16. Uldrich AP, Patel O, Cameron G, Pellicci DG, Day EB, Sullivan LC, et al. A semi-invariant Valpha10+ T cell antigen receptor defines a population of natural killer T cells with distinct glycolipid antigen-recognition properties. Nat Immunol (2011) 12(7):616–23. doi:10.1038/ni.2051
17. Beckman EM, Porcelli SA, Morita CT, Behar SM, Furlong ST, Brenner MB. Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature (1994) 372(6507):691–4. doi:10.1038/372691a0
18. Constantinides MG, Bendelac A. Transcriptional regulation of the NKT cell lineage. Curr Opin Immunol (2013) 25(2):161–7. doi:10.1016/j.coi.2013.01.003
19. Lee YJ, Holzapfel KL, Zhu J, Jameson SC, Hogquist KA. Steady-state production of IL-4 modulates immunity in mouse strains and is determined by lineage diversity of iNKT cells. Nat Immunol (2013) 14(11):1146–54. doi:10.1038/ni.2731
20. Gapin L. Development of invariant natural killer T cells. Curr Opin Immunol (2016) 39:68–74. doi:10.1016/j.coi.2016.01.001
21. Fowlkes BJ, Kruisbeek AM, Ton-That H, Weston MA, Coligan JE, Schwartz RH, et al. A novel population of T-cell receptor alpha beta-bearing thymocytes which predominantly expresses a single V beta gene family. Nature (1987) 329(6136):251–4. doi:10.1038/329251a0
22. Sykes M. Unusual T cell populations in adult murine bone marrow. Prevalence of CD3+CD4-CD8- and alpha beta TCR+NK1.1+ cells. J Immunol (1990) 145(10):3209–15.
23. Levitsky HI, Golumbek PT, Pardoll DM. The fate of CD4-8- T cell receptor-alpha beta+ thymocytes. J Immunol (1991) 146(4):1113–7.
24. Zlotnik A, Godfrey DI, Fischer M, Suda T. Cytokine production by mature and immature CD4-CD8- T cells. Alpha beta-T cell receptor+ CD4-CD8- T cells produce IL-4. J Immunol (1992) 149(4):1211–5.
25. Koseki H, Imai K, Nakayama F, Sado T, Moriwaki K, Taniguchi M. Homogenous junctional sequence of the V14+ T-cell antigen receptor alpha chain expanded in unprimed mice. Proc Natl Acad Sci U S A (1990) 87(14):5248–52. doi:10.1073/pnas.87.14.5248
26. Lantz O, Bendelac A. An invariant T cell receptor alpha chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4-8- T cells in mice and humans. J Exp Med (1994) 180(3):1097–106. doi:10.1084/jem.180.3.1097
27. Bendelac A, Lantz O, Quimby ME, Yewdell JW, Bennink JR, Brutkiewicz RR. CD1 recognition by mouse NK1+ T lymphocytes. Science (1995) 268(5212):863–5. doi:10.1126/science.7538697
28. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science (1997) 278(5343):1626–9. doi:10.1126/science.278.5343.1626
29. Benlagha K, Weiss A, Beavis A, Teyton L, Bendelac A. In vivo identification of glycolipid antigen-specific T cells using fluorescent CD1d tetramers. J Exp Med (2000) 191(11):1895–903. doi:10.1084/jem.191.11.1895
30. Matsuda JL, Naidenko OV, Gapin L, Nakayama T, Taniguchi M, Wang CR, et al. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J Exp Med (2000) 192(5):741–54. doi:10.1084/jem.192.5.741
31. Benlagha K, Kyin T, Beavis A, Teyton L, Bendelac A. A thymic precursor to the NK T cell lineage. Science (2002) 296(5567):553–5. doi:10.1126/science.1069017
32. Pellicci DG, Hammond KJ, Uldrich AP, Baxter AG, Smyth MJ, Godfrey DI. A natural killer T (NKT) cell developmental pathway iInvolving a thymus-dependent NK1.1(-)CD4(+) CD1d-dependent precursor stage. J Exp Med (2002) 195(7):835–44. doi:10.1084/jem.20011544
33. Gadue P, Stein PL. NK T cell precursors exhibit differential cytokine regulation and require Itk for efficient maturation. J Immunol (2002) 169(5):2397–406. doi:10.4049/jimmunol.169.5.2397
34. Berzins SP, McNab FW, Jones CM, Smyth MJ, Godfrey DI. Long-term retention of mature NK1.1+ NKT cells in the thymus. J Immunol (2006) 176(7):4059–65. doi:10.4049/jimmunol.176.7.4059
35. Drennan MB, Franki AS, Dewint P, Van Beneden K, Seeuws S, van de Pavert SA, et al. Cutting edge: the chemokine receptor CXCR3 retains invariant NK T cells in the thymus. J Immunol (2009) 183(4):2213–6. doi:10.4049/jimmunol.0901213
36. Matsuda JL, Gapin L. Does the developmental status of Valpha14i NKT cells play a role in disease? Int Rev Immunol (2007) 26(1–2):5–29. doi:10.1080/08830180601070211
37. Kovalovsky D, Uche OU, Eladad S, Hobbs RM, Yi W, Alonzo E, et al. The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nat Immunol (2008) 9(9):1055–64. doi:10.1038/ni.1641
38. Savage AK, Constantinides MG, Han J, Picard D, Martin E, Li B, et al. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity (2008) 29(3):391–403. doi:10.1016/j.immuni.2008.07.011
39. Hsieh CS, Macatonia SE, O’Garra A, Murphy KM. T cell genetic background determines default T helper phenotype development in vitro. J Exp Med (1995) 181(2):713–21. doi:10.1084/jem.181.2.713
40. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol (2000) 164(12):6166–73. doi:10.4049/jimmunol.164.12.6166
41. Carlyle JR, Mesci A, Ljutic B, Belanger S, Tai LH, Rousselle E, et al. Molecular and genetic basis for strain-dependent NK1.1 alloreactivity of mouse NK cells. J Immunol (2006) 176(12):7511–24. doi:10.4049/jimmunol.176.12.7511
42. Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat Immunol (2015) 16(1):85–95. doi:10.1038/ni.3047
43. Chang PP, Barral P, Fitch J, Pratama A, Ma CS, Kallies A, et al. Identification of Bcl-6-dependent follicular helper NKT cells that provide cognate help for B cell responses. Nat Immunol (2011) 13(1):35–43. doi:10.1038/ni.2166
44. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi:10.1146/annurev-immunol-031210-101400
45. King IL, Fortier A, Tighe M, Dibble J, Watts GF, Veerapen N, et al. Invariant natural killer T cells direct B cell responses to cognate lipid antigen in an IL-21-dependent manner. Nat Immunol (2011) 13(1):44–50. doi:10.1038/ni.2172
46. Singer A, Adoro S, Park JH. Lineage fate and intense debate: myths, models and mechanisms of CD4- versus CD8-lineage choice. Nat Rev Immunol (2008) 8(10):788–801. doi:10.1038/nri2416
47. Stritesky GL, Jameson SC, Hogquist KA. Selection of self-reactive T cells in the thymus. Annu Rev Immunol (2012) 30:95–114. doi:10.1146/annurev-immunol-020711-075035
48. Gapin L, Matsuda JL, Surh CD, Kronenberg M. NKT cells derive from double-positive thymocytes that are positively selected by CD1d. Nat Immunol (2001) 2(10):971–8. doi:10.1038/ni710
49. Egawa T, Eberl G, Taniuchi I, Benlagha K, Geissmann F, Hennighausen L, et al. Genetic evidence supporting selection of the Valpha14i NKT cell lineage from double-positive thymocyte precursors. Immunity (2005) 22(6):705–16. doi:10.1016/j.immuni.2005.03.011
50. Dashtsoodol N, Shigeura T, Aihara M, Ozawa R, Kojo S, Harada M, et al. Alternative pathway for the development of Valpha14(+) NKT cells directly from CD4(-)CD8(-) thymocytes that bypasses the CD4(+)CD8(+) stage. Nat Immunol (2017) 18(3):274–82. doi:10.1038/ni.3668
51. Bendelac A. Positive selection of mouse NK1+ T cells by CD1-expressing cortical thymocytes. J Exp Med (1995) 182(6):2091–6. doi:10.1084/jem.182.6.2091
52. Coles MC, Raulet DH. NK1.1+ T cells in the liver arise in the thymus and are selected by interactions with class I molecules on CD4+CD8+ cells. J Immunol (2000) 164(5):2412–8. doi:10.4049/jimmunol.164.5.2412
53. Griewank K, Borowski C, Rietdijk S, Wang N, Julien A, Wei DG, et al. Homotypic interactions mediated by Slamf1 and Slamf6 receptors control NKT cell lineage development. Immunity (2007) 27(5):751–62. doi:10.1016/j.immuni.2007.08.020
54. Peck SR, Ruley HE. Ly108: a new member of the mouse CD2 family of cell surface proteins. Immunogenetics (2000) 52(1–2):63–72. doi:10.1007/s002510000252
55. Zhong MC, Veillette A. Control of T lymphocyte signaling by Ly108, a signaling lymphocytic activation molecule family receptor implicated in autoimmunity. J Biol Chem (2008) 283(28):19255–64. doi:10.1074/jbc.M800209200
56. Dutta M, Kraus ZJ, Gomez-Rodriguez J, Hwang SH, Cannons JL, Cheng J, et al. A role for Ly108 in the induction of promyelocytic zinc finger transcription factor in developing thymocytes. J Immunol (2013) 190(5):2121–8. doi:10.4049/jimmunol.1202145
57. Lazarevic V, Zullo AJ, Schweitzer MN, Staton TL, Gallo EM, Crabtree GR, et al. The gene encoding early growth response 2, a target of the transcription factor NFAT, is required for the development and maturation of natural killer T cells. Nat Immunol (2009) 10(3):306–13. doi:10.1038/ni.1696
58. Hu T, Gimferrer I, Simmons A, Wiest D, Alberola-Ila J. The Ras/MAPK pathway is required for generation of iNKT cells. PLoS One (2011) 6(5):e19890. doi:10.1371/journal.pone.0019890
59. Seiler MP, Mathew R, Liszewski MK, Spooner CJ, Barr K, Meng F, et al. Elevated and sustained expression of the transcription factors Egr1 and Egr2 controls NKT lineage differentiation in response to TCR signaling. Nat Immunol (2012) 13(3):264–71. doi:10.1038/ni.2230
60. Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med (2011) 208(6):1279–89. doi:10.1084/jem.20110308
61. Li W, Sofi MH, Yeh N, Sehra S, McCarthy BP, Patel DR, et al. Thymic selection pathway regulates the effector function of CD4 T cells. J Exp Med (2007) 204(9):2145–57. doi:10.1084/jem.20070321
62. Carr T, Krishnamoorthy V, Yu S, Xue HH, Kee BL, Verykokakis M. The transcription factor lymphoid enhancer factor 1 controls invariant natural killer T cell expansion and Th2-type effector differentiation. J Exp Med (2015) 212(5):793–807. doi:10.1084/jem.20141849
63. Georgiev H, Ravens I, Benarafa C, Forster R, Bernhardt G. Distinct gene expression patterns correlate with developmental and functional traits of iNKT subsets. Nat Commun (2016) 7:13116. doi:10.1038/ncomms13116
64. Uddin MN, Sultana DA, Lorentsen KJ, Cho JJ, Kirst ME, Brantly ML, et al. Transcription factor Bcl11b sustains iNKT1 and iNKT2 cell programs, restricts iNKT17 cell program, and governs iNKT cell survival. Proc Natl Acad Sci U S A (2016) 113(27):7608–13. doi:10.1073/pnas.1521846113
65. Engel I, Zhao M, Kappes D, Taniuchi I, Kronenberg M. The transcription factor Th-POK negatively regulates Th17 differentiation in Valpha14i NKT cells. Blood (2012) 120(23):4524–32. doi:10.1182/blood-2012-01-406280
66. Di Pietro C, De Giorgi L, Cosorich I, Sorini C, Fedeli M, Falcone M. MicroRNA-133b regulation of Th-POK expression and dendritic cell signals affect NKT17 cell differentiation in the thymus. J Immunol (2016) 197(8):3271–80. doi:10.4049/jimmunol.1502238
67. Zhang JA, Mortazavi A, Williams BA, Wold BJ, Rothenberg EV. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell (2012) 149(2):467–82. doi:10.1016/j.cell.2012.01.056
68. Dobenecker MW, Kim JK, Marcello J, Fang TC, Prinjha R, Bosselut R, et al. Coupling of T cell receptor specificity to natural killer T cell development by bivalent histone H3 methylation. J Exp Med (2015) 212(3):297–306. doi:10.1084/jem.20141499
69. Engel I, Seumois G, Chavez L, Samaniego-Castruita D, White B, Chawla A, et al. Innate-like functions of natural killer T cell subsets result from highly divergent gene programs. Nat Immunol (2016) 17(6):728–39. doi:10.1038/ni.3437
70. Gordy LE, Bezbradica JS, Flyak AI, Spencer CT, Dunkle A, Sun J, et al. IL-15 regulates homeostasis and terminal maturation of NKT cells. J Immunol (2011) 187(12):6335–45. doi:10.4049/jimmunol.1003965
71. Webster KE, Kim HO, Kyparissoudis K, Corpuz TM, Pinget GV, Uldrich AP, et al. IL-17-producing NKT cells depend exclusively on IL-7 for homeostasis and survival. Mucosal Immunol (2014) 7(5):1058–67. doi:10.1038/mi.2013.122
72. Watarai H, Sekine-Kondo E, Shigeura T, Motomura Y, Yasuda T, Satoh R, et al. Development and function of invariant natural killer T cells producing T(h)2- and T(h)17-cytokines. PLoS Biol (2012) 10(2):e1001255. doi:10.1371/journal.pbio.1001255
73. Matsuda JL, George TC, Hagman J, Gapin L. Temporal dissection of T-bet functions. J Immunol (2007) 178(6):3457–65. doi:10.4049/jimmunol.178.6.3457
74. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol (2008) 9(6):641–9. doi:10.1038/ni.1610
75. Wei G, Abraham BJ, Yagi R, Jothi R, Cui K, Sharma S, et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity (2011) 35(2):299–311. doi:10.1016/j.immuni.2011.08.007
76. Cameron G, Pellicci DG, Uldrich AP, Besra GS, Illarionov P, Williams SJ, et al. Antigen Specificity of type I NKT cells is governed by TCR beta-chain diversity. J Immunol (2015) 195(10):4604–14. doi:10.4049/jimmunol.1501222
77. Lee YJ, Wang H, Starrett GJ, Phuong V, Jameson SC, Hogquist KA. Tissue-specific distribution of iNKT cells impacts their cytokine response. Immunity (2015) 43(3):566–78. doi:10.1016/j.immuni.2015.06.025
78. Eberl G, Lees R, Smiley ST, Taniguchi M, Grusby MJ, MacDonald HR. Tissue-specific segregation of CD1d-dependent and CD1d-independent NK T cells. J Immunol (1999) 162(11):6410–9.
79. Hammond KJ, Pelikan SB, Crowe NY, Randle-Barrett E, Nakayama T, Taniguchi M, et al. NKT cells are phenotypically and functionally diverse. Eur J Immunol (1999) 29(11):3768–81. doi:10.1002/(SICI)1521-4141(199911)29:11<3768::AID-IMMU3768>3.0.CO;2-G
80. Constant S, Pfeiffer C, Woodard A, Pasqualini T, Bottomly K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. J Exp Med (1995) 182(5):1591–6. doi:10.1084/jem.182.5.1591
81. Hayes SM, Laird RM, Love PE. Beyond alphabeta/gammadelta lineage commitment: TCR signal strength regulates gammadelta T cell maturation and effector fate. Semin Immunol (2010) 22(4):247–51. doi:10.1016/j.smim.2010.04.006
82. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi:10.1146/annurev.immunol.25.022106.141623
83. Tuttle KD, Krovi SH, Zhang J, Bedel R, Harmacek L, Peterson LK, et al. TCR signal strength controls thymic differentiation of iNKT cell subsets. Nat Commun (2018) (in press).
84. Hernandez-Hoyos G, Anderson MK, Wang C, Rothenberg EV, Alberola-Ila J. GATA-3 expression is controlled by TCR signals and regulates CD4/CD8 differentiation. Immunity (2003) 19(1):83–94. doi:10.1016/S1074-7613(03)00176-6
85. Ho IC, Vorhees P, Marin N, Oakley BK, Tsai SF, Orkin SH, et al. Human GATA-3: a lineage-restricted transcription factor that regulates the expression of the T cell receptor alpha gene. EMBO J (1991) 10(5):1187–92.
86. Ko LJ, Yamamoto M, Leonard MW, George KM, Ting P, Engel JD. Murine and human T-lymphocyte GATA-3 factors mediate transcription through a cis-regulatory element within the human T-cell receptor delta gene enhancer. Mol Cell Biol (1991) 11(5):2778–84. doi:10.1128/MCB.11.5.2778
87. Ling KW, van Hamburg JP, de Bruijn MJ, Kurek D, Dingjan GM, Hendriks RW. GATA3 controls the expression of CD5 and the T cell receptor during CD4 T cell lineage development. Eur J Immunol (2007) 37(4):1043–52. doi:10.1002/eji.200636485
88. Ouyang W, Lohning M, Gao Z, Assenmacher M, Ranganath S, Radbruch A, et al. Stat6-independent GATA-3 autoactivation directs IL-4-independent Th2 development and commitment. Immunity (2000) 12(1):27–37. doi:10.1016/S1074-7613(00)80156-9
89. Mallevaey T, Clarke AJ, Scott-Browne JP, Young MH, Roisman LC, Pellicci DG, et al. A molecular basis for NKT cell recognition of CD1d-self-antigen. Immunity (2011) 34(3):315–26. doi:10.1016/j.immuni.2011.01.013
90. Cruz Tleugabulova M, Escalante NK, Deng S, Fieve S, Ereno-Orbea J, Savage PB, et al. Discrete TCR binding kinetics control invariant NKT cell selection and central priming. J Immunol (2016) 197(10):3959–69. doi:10.4049/jimmunol.1601382
91. Clancy-Thompson E, Chen GZ, Tyler PM, Servos MM, Barisa M, Brennan PJ, et al. Monoclonal invariant NKT (iNKT) cell mice reveal a role for both tissue of origin and the TCR in development of iNKT functional subsets. J Immunol (2017) 199(1):159–71. doi:10.4049/jimmunol.1700214
92. Pereira RM, Martinez GJ, Engel I, Cruz-Guilloty F, Barboza BA, Tsagaratou A, et al. Jarid2 is induced by TCR signalling and controls iNKT cell maturation. Nat Commun (2014) 5:4540. doi:10.1038/ncomms5540
93. Beyaz S, Kim JH, Pinello L, Xifaras ME, Hu Y, Huang J, et al. The histone demethylase UTX regulates the lineage-specific epigenetic program of invariant natural killer T cells. Nat Immunol (2017) 18(2):184–95. doi:10.1038/ni.3644
94. Thomas SY, Scanlon ST, Griewank KG, Constantinides MG, Savage AK, Barr KA, et al. PLZF induces an intravascular surveillance program mediated by long-lived LFA-1-ICAM-1 interactions. J Exp Med (2011) 208(6):1179–88. doi:10.1084/jem.20102630
95. Wingender G, Stepniak D, Krebs P, Lin L, McBride S, Wei B, et al. Intestinal microbes affect phenotypes and functions of invariant natural killer T cells in mice. Gastroenterology (2012) 143(2):418–28. doi:10.1053/j.gastro.2012.04.017
96. Laloux V, Beaudoin L, Ronet C, Lehuen A. Phenotypic and functional differences between NKT cells colonizing splanchnic and peripheral lymph nodes. J Immunol (2002) 168(7):3251–8. doi:10.4049/jimmunol.168.7.3251
97. Barral P, Sanchez-Nino MD, van Rooijen N, Cerundolo V, Batista FD. The location of splenic NKT cells favours their rapid activation by blood-borne antigen. EMBO J (2012) 31(10):2378–90. doi:10.1038/emboj.2012.87
98. Hammond KJ, Pellicci DG, Poulton LD, Naidenko OV, Scalzo AA, Baxter AG, et al. CD1d-restricted NKT cells: an interstrain comparison. J Immunol (2001) 167(3):1164–73. doi:10.4049/jimmunol.167.3.1164
99. Kambayashi T, Assarsson E, Michaelsson J, Berglund P, Diehl AD, Chambers BJ, et al. Emergence of CD8+ T cells expressing NK cell receptors in influenza A virus-infected mice. J Immunol (2000) 165(9):4964–9. doi:10.4049/jimmunol.165.9.4964
100. Slifka MK, Pagarigan RR, Whitton JL. NK markers are expressed on a high percentage of virus-specific CD8+ and CD4+ T cells. J Immunol (2000) 164(4):2009–15. doi:10.4049/jimmunol.164.4.2009
101. Assarsson E, Kambayashi T, Sandberg JK, Hong S, Taniguchi M, Van Kaer L, et al. CD8+ T cells rapidly acquire NK1.1 and NK cell-associated molecules upon stimulation in vitro and in vivo. J Immunol (2000) 165(7):3673–9. doi:10.4049/jimmunol.165.7.3673
102. McCaughtry TM, Wilken MS, Hogquist KA. Thymic emigration revisited. J Exp Med (2007) 204(11):2513–20. doi:10.1084/jem.20070601
103. Stritesky GL, Xing Y, Erickson JR, Kalekar LA, Wang X, Mueller DL, et al. Murine thymic selection quantified using a unique method to capture deleted T cells. Proc Natl Acad Sci U S A (2013) 110(12):4679–84. doi:10.1073/pnas.1217532110
104. Matsuda JL, Gapin L, Sidobre S, Kieper WC, Tan JT, Ceredig R, et al. Homeostasis of V alpha 14i NKT cells. Nat Immunol (2002) 3(10):966–74. doi:10.1038/ni837
105. White AJ, Jenkinson WE, Cowan JE, Parnell SM, Bacon A, Jones ND, et al. An essential role for medullary thymic epithelial cells during the intrathymic development of invariant NKT cells. J Immunol (2014) 192(6):2659–66. doi:10.4049/jimmunol.1303057
106. Lin SJ, Chao HC, Kuo ML. The effect of interleukin-12 and interleukin-15 on CD69 expression of T-lymphocytes and natural killer cells from umbilical cord blood. Biol Neonate (2000) 78(3):181–5. doi:10.1159/000014268
107. Drennan MB, Govindarajan S, Verheugen E, Coquet JM, Staal J, McGuire C, et al. NKT sublineage specification and survival requires the ubiquitin-modifying enzyme TNFAIP3/A20. J Exp Med (2016) 213(10):1973–81. doi:10.1084/jem.20151065
108. Nakayama T, Kasprowicz DJ, Yamashita M, Schubert LA, Gillard G, Kimura M, et al. The generation of mature, single-positive thymocytes in vivo is dysregulated by CD69 blockade or overexpression. J Immunol (2002) 168(1):87–94. doi:10.4049/jimmunol.168.1.87
109. Slauenwhite D, Johnston B. Regulation of NKT cell localization in homeostasis and infection. Front Immunol (2015) 6:255. doi:10.3389/fimmu.2015.00255
110. Unutmaz D, Xiang W, Sunshine MJ, Campbell J, Butcher E, Littman DR. The primate lentiviral receptor Bonzo/STRL33 is coordinately regulated with CCR5 and its expression pattern is conserved between human and mouse. J Immunol (2000) 165(6):3284–92. doi:10.4049/jimmunol.165.6.3284
111. Geissmann F, Cameron TO, Sidobre S, Manlongat N, Kronenberg M, Briskin MJ, et al. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol (2005) 3(4):e113. doi:10.1371/journal.pbio.0030113
112. Saiman Y, Friedman SL. The role of chemokines in acute liver injury. Front Physiol (2012) 3:213. doi:10.3389/fphys.2012.00213
113. Heymann F, Tacke F. Immunology in the liver – from homeostasis to disease. Nat Rev Gastroenterol Hepatol (2016) 13(2):88–110. doi:10.1038/nrgastro.2015.200
114. Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science (2016) 352(6284):459–63. doi:10.1126/science.aad2035
115. Weizman OE, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, et al. ILC1 confer early host protection at initial sites of viral infection. Cell (2017) 171(4):795–808.e12. doi:10.1016/j.cell.2017.09.052
116. van Gisbergen KP, Kragten NA, Hertoghs KM, Wensveen FM, Jonjic S, Hamann J, et al. Mouse Hobit is a homolog of the transcriptional repressor Blimp-1 that regulates NKT cell effector differentiation. Nat Immunol (2012) 13(9):864–71. doi:10.1038/ni.2393
117. Michel ML, Keller AC, Paget C, Fujio M, Trottein F, Savage PB, et al. Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med (2007) 204(5):995–1001. doi:10.1084/jem.20061551
118. Rachitskaya AV, Hansen AM, Horai R, Li Z, Villasmil R, Luger D, et al. Cutting edge: NKT cells constitutively express IL-23 receptor and RORgammat and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J Immunol (2008) 180(8):5167–71. doi:10.4049/jimmunol.180.8.5167
119. Michel ML, Mendes-da-Cruz D, Keller AC, Lochner M, Schneider E, Dy M, et al. Critical role of ROR-gammat in a new thymic pathway leading to IL-17-producing invariant NKT cell differentiation. Proc Natl Acad Sci U S A (2008) 105(50):19845–50. doi:10.1073/pnas.0806472105
120. Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B, et al. CCR6 regulates the migration of inflammatory and regulatory T cells. J Immunol (2008) 181(12):8391–401. doi:10.4049/jimmunol.181.12.8391
121. Doisne JM, Becourt C, Amniai L, Duarte N, Le Luduec JB, Eberl G, et al. Skin and peripheral lymph node invariant NKT cells are mainly retinoic acid receptor-related orphan receptor (gamma)t+ and respond preferentially under inflammatory conditions. J Immunol (2009) 183(3):2142–9. doi:10.4049/jimmunol.0901059
122. Cepek KL, Shaw SK, Parker CM, Russell GJ, Morrow JS, Rimm DL, et al. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature (1994) 372(6502):190–3. doi:10.1038/372190a0
123. Dai H, Rahman A, Saxena A, Jaiswal AK, Mohamood A, Ramirez L, et al. Syndecan-1 identifies and controls the frequency of IL-17-producing naive natural killer T (NKT17) cells in mice. Eur J Immunol (2015) 45(11):3045–51. doi:10.1002/eji.201545532
124. Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V, et al. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity (2012) 37(3):574–87. doi:10.1016/j.immuni.2012.06.016
125. Vieth JA, Das J, Ranaivoson FM, Comoletti D, Denzin LK, Sant’Angelo DB. TCRalpha-TCRbeta pairing controls recognition of CD1d and directs the development of adipose NKT cells. Nat Immunol (2017) 18(1):36–44. doi:10.1038/ni.3622
126. Fan X, Rudensky AY. Hallmarks of tissue-resident lymphocytes. Cell (2016) 164(6):1198–211. doi:10.1016/j.cell.2016.02.048
127. Scanlon ST, Thomas SY, Ferreira CM, Bai L, Krausz T, Savage PB, et al. Airborne lipid antigens mobilize resident intravascular NKT cells to induce allergic airway inflammation. J Exp Med (2011) 208(10):2113–24. doi:10.1084/jem.20110522
128. Weinreich MA, Takada K, Skon C, Reiner SL, Jameson SC, Hogquist KA. KLF2 transcription-factor deficiency in T cells results in unrestrained cytokine production and upregulation of bystander chemokine receptors. Immunity (2009) 31(1):122–30. doi:10.1016/j.immuni.2009.05.011
129. Weinreich MA, Odumade OA, Jameson SC, Hogquist KA. T cells expressing the transcription factor PLZF regulate the development of memory-like CD8+ T cells. Nat Immunol (2010) 11(8):709–16. doi:10.1038/ni.1898
130. Berg LJ. Signalling through TEC kinases regulates conventional versus innate CD8(+) T-cell development. Nat Rev Immunol (2007) 7(6):479–85. doi:10.1038/nri2091
131. Lee A, Park SP, Park CH, Kang BH, Park SH, Ha SJ, et al. IL-4 induced innate CD8+ T cells control persistent viral infection. PLoS Pathog (2015) 11(10):e1005193. doi:10.1371/journal.ppat.1005193
132. Renkema KR, Lee JY, Lee YJ, Hamilton SE, Hogquist KA, Jameson SC. IL-4 sensitivity shapes the peripheral CD8+ T cell pool and response to infection. J Exp Med (2016) 213(7):1319–29. doi:10.1084/jem.20151359
133. Kang BH, Park HJ, Park HJ, Lee JI, Park SH, Jung KC. PLZF(+) innate T cells support the TGF-beta-dependent generation of activated/memory-like regulatory T cells. Mol Cells (2016) 39(6):468–76. doi:10.14348/molcells.2016.0004
134. Barik S, Miller MM, Cattin-Roy AN, Ukah TK, Chen W, Zaghouani H. IL-4/IL-13 Signaling inhibits the potential of early thymic progenitors to commit to the T cell lineage. J Immunol (2017) 199(8):2767–76. doi:10.4049/jimmunol.1700498
135. White AJ, Baik S, Parnell SM, Holland AM, Brombacher F, Jenkinson WE, et al. A type 2 cytokine axis for thymus emigration. J Exp Med (2017) 214(8):2205–16. doi:10.1084/jem.20170271
136. Anderson MS, Su MA. AIRE expands: new roles in immune tolerance and beyond. Nat Rev Immunol (2016) 16(4):247–58. doi:10.1038/nri.2016.9
137. Maier E, Duschl A, Horejs-Hoeck J. STAT6-dependent and -independent mechanisms in Th2 polarization. Eur J Immunol (2012) 42(11):2827–33. doi:10.1002/eji.201242433
138. Novak J, Lehuen A. Mechanism of regulation of autoimmunity by iNKT cells. Cytokine (2011) 53(3):263–70. doi:10.1016/j.cyto.2010.11.001
139. Crosby CM, Kronenberg M. Invariant natural killer T cells: front line fighters in the war against pathogenic microbes. Immunogenetics (2016) 68(8):639–48. doi:10.1007/s00251-016-0933-y
140. Kawakami K, Yamamoto N, Kinjo Y, Miyagi K, Nakasone C, Uezu K, et al. Critical role of Valpha14+ natural killer T cells in the innate phase of host protection against Streptococcus pneumoniae infection. Eur J Immunol (2003) 33(12):3322–30. doi:10.1002/eji.200324254
141. Kinjo Y, Illarionov P, Vela JL, Pei B, Girardi E, Li X, et al. Invariant natural killer T cells recognize glycolipids from pathogenic gram-positive bacteria. Nat Immunol (2011) 12(10):966–74. doi:10.1038/ni.2096
142. Nieuwenhuis EE, Matsumoto T, Exley M, Schleipman RA, Glickman J, Bailey DT, et al. CD1d-dependent macrophage-mediated clearance of Pseudomonas aeruginosa from lung. Nat Med (2002) 8(6):588–93. doi:10.1038/nm0602-588
143. Hazlett LD, Li Q, Liu J, McClellan S, Du W, Barrett RP. NKT cells are critical to initiate an inflammatory response after Pseudomonas aeruginosa ocular infection in susceptible mice. J Immunol (2007) 179(2):1138–46. doi:10.4049/jimmunol.179.2.1138
144. Sada-Ovalle I, Chiba A, Gonzales A, Brenner MB, Behar SM. Innate invariant NKT cells recognize Mycobacterium tuberculosis-infected macrophages, produce interferon-gamma, and kill intracellular bacteria. PLoS Pathog (2008) 4(12):e1000239. doi:10.1371/journal.ppat.1000239
145. Crowe NY, Smyth MJ, Godfrey DI. A critical role for natural killer T cells in immunosurveillance of methylcholanthrene-induced sarcomas. J Exp Med (2002) 196(1):119–27. doi:10.1084/jem.20020092
146. Terabe M, Khanna C, Bose S, Melchionda F, Mendoza A, Mackall CL, et al. CD1d-restricted natural killer T cells can down-regulate tumor immunosurveillance independent of interleukin-4 receptor-signal transducer and activator of transcription 6 or transforming growth factor-beta. Cancer Res (2006) 66(7):3869–75. doi:10.1158/0008-5472.CAN-05-3421
147. Beaudoin L, Laloux V, Novak J, Lucas B, Lehuen A. NKT cells inhibit the onset of diabetes by impairing the development of pathogenic T cells specific for pancreatic beta cells. Immunity (2002) 17(6):725–36. doi:10.1016/S1074-7613(02)00473-9
148. Zeng D, Lee MK, Tung J, Brendolan A, Strober S. Cutting edge: a role for CD1 in the pathogenesis of lupus in NZB/NZW mice. J Immunol (2000) 164(10):5000–4. doi:10.4049/jimmunol.164.10.5000
149. Zeng D, Liu Y, Sidobre S, Kronenberg M, Strober S. Activation of natural killer T cells in NZB/W mice induces Th1-type immune responses exacerbating lupus. J Clin Invest (2003) 112(8):1211–22. doi:10.1172/JCI200317165
150. Morshed SR, Mannoor K, Halder RC, Kawamura H, Bannai M, Sekikawa H, et al. Tissue-specific expansion of NKT and CD5+B cells at the onset of autoimmune disease in (NZBxNZW)F1 mice. Eur J Immunol (2002) 32(9):2551–61. doi:10.1002/1521-4141(200209)32:9<2551::AID-IMMU2551>3.0.CO;2-C
151. Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, et al. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science (1997) 278(5343):1623–6. doi:10.1126/science.278.5343.1623
152. Bedel R, Matsuda JL, Brigl M, White J, Kappler J, Marrack P, et al. Lower TCR repertoire diversity in Traj18-deficient mice. Nat Immunol (2012) 13(8):705–6. doi:10.1038/ni.2347
153. Riegert P, Gilfillan S. A conserved sequence block in the murine and human TCR J alpha region: assessment of regulatory function in vivo. J Immunol (1999) 162(6):3471–80.
154. Chandra S, Zhao M, Budelsky A, de Mingo Pulido A, Day J, Fu Z, et al. A new mouse strain for the analysis of invariant NKT cell function. Nat Immunol (2015) 16(8):799–800. doi:10.1038/ni.3203
155. Dashtsoodol N, Shigeura T, Ozawa R, Harada M, Kojo S, Watanabe T, et al. Generation of novel Traj18-deficient mice lacking Valpha14 natural killer T cells with an undisturbed T cell receptor alpha-chain repertoire. PLoS One (2016) 11(4):e0153347. doi:10.1371/journal.pone.0153347
156. Zhang J, Bedel R, Krovi SH, Tuttle KD, Zhang B, Gross J, et al. Mutation of the Traj18 gene segment using TALENs to generate natural killer T cell deficient mice. Sci Rep (2016) 6:27375. doi:10.1038/srep27375
157. Mendiratta SK, Martin WD, Hong S, Boesteanu A, Joyce S, Van Kaer L. CD1d1 mutant mice are deficient in natural T cells that promptly produce IL-4. Immunity (1997) 6(4):469–77. doi:10.1016/S1074-7613(00)80290-3
158. Sundararaj S, Zhang J, Krovi SH, Bedel R, Tuttle KD, Veerapen N, et al. Differing roles of CD1d2 and CD1d1 proteins in type I natural killer T cell development and function. Proc Natl Acad Sci U S A (2018) 115(6):E1204–13. doi:10.1073/pnas.1716669115
159. Chen YH, Chiu NM, Mandal M, Wang N, Wang CR. Impaired NK1+ T cell development and early IL-4 production in CD1-deficient mice. Immunity (1997) 6(4):459–67. doi:10.1016/S1074-7613(00)80289-7
160. Smiley ST, Kaplan MH, Grusby MJ. Immunoglobulin E production in the absence of interleukin-4-secreting CD1-dependent cells. Science (1997) 275(5302):977–9. doi:10.1126/science.275.5302.977
161. Terabe M, Matsui S, Noben-Trauth N, Chen H, Watson C, Donaldson DD, et al. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat Immunol (2000) 1(6):515–20. doi:10.1038/82771
162. O’Brien TF, Bao K, Dell’Aringa M, Ang WX, Abraham S, Reinhardt RL. Cytokine expression by invariant natural killer T cells is tightly regulated throughout development and settings of type-2 inflammation. Mucosal Immunol (2016) 9(3):597–609. doi:10.1038/mi.2015.78
163. Crowe NY, Coquet JM, Berzins SP, Kyparissoudis K, Keating R, Pellicci DG, et al. Differential antitumor immunity mediated by NKT cell subsets in vivo. J Exp Med (2005) 202(9):1279–88. doi:10.1084/jem.20050953
164. Umetsu DT, DeKruyff RH. The regulation of allergy and asthma. Immunol Rev (2006) 212:238–55. doi:10.1111/j.0105-2896.2006.00413.x
165. Akbari O, Stock P, Meyer E, Kronenberg M, Sidobre S, Nakayama T, et al. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nat Med (2003) 9(5):582–8. doi:10.1038/nm851
166. Lisbonne M, Diem S, de Castro Keller A, Lefort J, Araujo LM, Hachem P, et al. Cutting edge: invariant V alpha 14 NKT cells are required for allergen-induced airway inflammation and hyperreactivity in an experimental asthma model. J Immunol (2003) 171(4):1637–41. doi:10.4049/jimmunol.171.4.1637
167. Das J, Eynott P, Jupp R, Bothwell A, Van Kaer L, Shi Y, et al. Natural killer T cells and CD8+ T cells are dispensable for T cell-dependent allergic airway inflammation. Nat Med (2006) 12(12):1345–6; author reply 1347.
168. Terashima A, Watarai H, Inoue S, Sekine E, Nakagawa R, Hase K, et al. A novel subset of mouse NKT cells bearing the IL-17 receptor B responds to IL-25 and contributes to airway hyperreactivity. J Exp Med (2008) 205(12):2727–33. doi:10.1084/jem.20080698
169. Stock P, Lombardi V, Kohlrautz V, Akbari O. Induction of airway hyperreactivity by IL-25 is dependent on a subset of invariant NKT cells expressing IL-17RB. J Immunol (2009) 182(8):5116–22. doi:10.4049/jimmunol.0804213
170. Pichavant M, Goya S, Meyer EH, Johnston RA, Kim HY, Matangkasombut P, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med (2008) 205(2):385–93. doi:10.1084/jem.20071507
171. Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, et al. A validated regulatory network for Th17 cell specification. Cell (2012) 151(2):289–303. doi:10.1016/j.cell.2012.09.016
172. Tanaka S, Suto A, Iwamoto T, Kashiwakuma D, Kagami S, Suzuki K, et al. Sox5 and c-Maf cooperatively induce Th17 cell differentiation via RORgammat induction as downstream targets of Stat3. J Exp Med (2014) 211(9):1857–74. doi:10.1084/jem.20130791
173. Yu JS, Hamada M, Ohtsuka S, Yoh K, Takahashi S, Miaw SC. Differentiation of IL-17-producing invariant natural killer T cells requires expression of the transcription factor c-Maf. Front Immunol (2017) 8:1399. doi:10.3389/fimmu.2017.01399
174. Raftery MJ, Winau F, Giese T, Kaufmann SH, Schaible UE, Schonrich G. Viral danger signals control CD1d de novo synthesis and NKT cell activation. Eur J Immunol (2008) 38(3):668–79. doi:10.1002/eji.200737233
175. Webb TJ, Carey GB, East JE, Sun W, Bollino DR, Kimball AS, et al. Alterations in cellular metabolism modulate CD1d-mediated NKT-cell responses. Pathog Dis (2016) 74(6):1–9. doi:10.1093/femspd/ftw055
176. Tessmer MS, Fatima A, Paget C, Trottein F, Brossay L. NKT cell immune responses to viral infection. Expert Opin Ther Targets (2009) 13(2):153–62. doi:10.1517/14712590802653601
177. Holzapfel KL, Tyznik AJ, Kronenberg M, Hogquist KA. Antigen-dependent versus -independent activation of invariant NKT cells during infection. J Immunol (2014) 192(12):5490–8. doi:10.4049/jimmunol.1400722
178. Tyznik AJ, Verma S, Wang Q, Kronenberg M, Benedict CA. Distinct requirements for activation of NKT and NK cells during viral infection. J Immunol (2014) 192(8):3676–85. doi:10.4049/jimmunol.1300837
179. Kakimi K, Guidotti LG, Koezuka Y, Chisari FV. Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J Exp Med (2000) 192(7):921–30. doi:10.1084/jem.192.7.921
180. Ishikawa H, Tanaka K, Kutsukake E, Fukui T, Sasaki H, Hata A, et al. IFN-gamma production downstream of NKT cell activation in mice infected with influenza virus enhances the cytolytic activities of both NK cells and viral antigen-specific CD8+ T cells. Virology (2010) 407(2):325–32. doi:10.1016/j.virol.2010.08.030
181. Gaya M, Barral P, Burbage M, Aggarwal S, Montaner B, Warren Navia A, et al. Initiation of antiviral B cell immunity relies on innate signals from spatially positioned NKT cells. Cell (2018) 172(3):517–533.e20. doi:10.1016/j.cell.2017.11.036
182. De Santo C, Salio M, Masri SH, Lee LY, Dong T, Speak AO, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest (2008) 118(12):4036–48. doi:10.1172/JCI36264
183. Lynch L. Adipose invariant natural killer T cells. Immunology (2014) 142(3):337–46. doi:10.1111/imm.12269
184. Lee PT, Benlagha K, Teyton L, Bendelac A. Distinct functional lineages of human V(alpha)24 natural killer T cells. J Exp Med (2002) 195(5):637–41. doi:10.1084/jem.20011908
185. Berzins SP, Smyth MJ, Baxter AG. Presumed guilty: natural killer T cell defects and human disease. Nat Rev Immunol (2011) 11(2):131–42. doi:10.1038/nri2904
186. Kim CH, Johnston B, Butcher EC. Trafficking machinery of NKT cells: shared and differential chemokine receptor expression among V alpha 24(+)V beta 11(+) NKT cell subsets with distinct cytokine-producing capacity. Blood (2002) 100(1):11–6. doi:10.1182/blood-2001-12-0196
187. Thomas SY, Hou R, Boyson JE, Means TK, Hess C, Olson DP, et al. CD1d-restricted NKT cells express a chemokine receptor profile indicative of Th1-type inflammatory homing cells. J Immunol (2003) 171(5):2571–80. doi:10.4049/jimmunol.171.5.2571
188. Wu C, Li Z, Fu X, Yu S, Lao S, Yang B. Antigen-specific human NKT cells from tuberculosis patients produce IL-21 to help B cells for the production of immunoglobulins. Oncotarget (2015) 6(30):28633–45. doi:10.18632/oncotarget.5764
189. Lee PT, Putnam A, Benlagha K, Teyton L, Gottlieb PA, Bendelac A. Testing the NKT cell hypothesis of human IDDM pathogenesis. J Clin Invest (2002) 110(6):793–800. doi:10.1172/JCI0215832
190. Montoya CJ, Pollard D, Martinson J, Kumari K, Wasserfall C, Mulder CB, et al. Characterization of human invariant natural killer T subsets in health and disease using a novel invariant natural killer T cell-clonotypic monoclonal antibody, 6B11. Immunology (2007) 122(1):1–14. doi:10.1111/j.1365-2567.2007.02647.x
191. Gumperz JE, Miyake S, Yamamura T, Brenner MB. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J Exp Med (2002) 195(5):625–36. doi:10.1084/jem.20011786
192. Eidson M, Wahlstrom J, Beaulieu AM, Zaidi B, Carsons SE, Crow PK, et al. Altered development of NKT cells, gammadelta T cells, CD8 T cells and NK cells in a PLZF deficient patient. PLoS One (2011) 6(9):e24441. doi:10.1371/journal.pone.0024441
193. Pasquier B, Yin L, Fondaneche MC, Relouzat F, Bloch-Queyrat C, Lambert N, et al. Defective NKT cell development in mice and humans lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product. J Exp Med (2005) 201(5):695–701. doi:10.1084/jem.20042432
194. Moreira-Teixeira L, Resende M, Coffre M, Devergne O, Herbeuval JP, Hermine O, et al. Proinflammatory environment dictates the IL-17-producing capacity of human invariant NKT cells. J Immunol (2011) 186(10):5758–65. doi:10.4049/jimmunol.1003043
195. Xue X, Soroosh P, De Leon-Tabaldo A, Luna-Roman R, Sablad M, Rozenkrants N, et al. Pharmacologic modulation of RORgammat translates to efficacy in preclinical and translational models of psoriasis and inflammatory arthritis. Sci Rep (2016) 6:37977. doi:10.1038/srep37977
196. Zeng SG, Ghnewa YG, O’Reilly VP, Lyons VG, Atzberger A, Hogan AE, et al. Human invariant NKT cell subsets differentially promote differentiation, antibody production, and T cell stimulation by B cells in vitro. J Immunol (2013) 191(4):1666–76. doi:10.4049/jimmunol.1202223
197. Berzofsky JA, Terabe M. The contrasting roles of NKT cells in tumor immunity. Curr Mol Med (2009) 9(6):667–72. doi:10.2174/156652409788970706
Keywords: invariant natural killer T cells, subsets, development, homeostasis, cytokine secretion
Citation: Krovi SH and Gapin L (2018) Invariant Natural Killer T Cell Subsets—More Than Just Developmental Intermediates. Front. Immunol. 9:1393. doi: 10.3389/fimmu.2018.01393
Received: 02 May 2018; Accepted: 05 June 2018;
Published: 20 June 2018
Edited by:
Luc Van Kaer, Vanderbilt University, United StatesReviewed by:
Sebastian Joyce, Vanderbilt University, United StatesCopyright: © 2018 Krovi and Gapin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurent Gapin, bGF1cmVudC5nYXBpbkB1Y2RlbnZlci5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.