 Lavida R. K. Brooks
Lavida R. K. Brooks George I. Mias
George I. Mias- 1Microbiology and Molecular Genetics, Michigan State University, East Lansing, MI, United States
- 2Institute for Quantitative Health Science & Engineering, Michigan State University, East Lansing, MI, United States
- 3Biochemistry and Molecular Biology, Michigan State University, East Lansing, MI, United States
Streptococcus pneumoniae is an infectious pathogen responsible for millions of deaths worldwide. Diseases caused by this bacterium are classified as pneumococcal diseases. This pathogen colonizes the nasopharynx of its host asymptomatically, but overtime can migrate to sterile tissues and organs and cause infections. Pneumonia is currently the most common pneumococcal disease. Pneumococcal pneumonia is a global health concern and vastly affects children under the age of five as well as the elderly and individuals with pre-existing health conditions. S. pneumoniae has a large selection of virulence factors that promote adherence, invasion of host tissues, and allows it to escape host immune defenses. A clear understanding of S. pneumoniae’s virulence factors, host immune responses, and examining the current techniques available for diagnosis, treatment, and disease prevention will allow for better regulation of the pathogen and its diseases. In terms of disease prevention, other considerations must include the effects of age on responses to vaccines and vaccine efficacy. Ongoing work aims to improve on current vaccination paradigms by including the use of serotype-independent vaccines, such as protein and whole cell vaccines. Extending our knowledge of the biology of, and associated host immune response to S. pneumoniae is paramount for our improvement of pneumococcal disease diagnosis, treatment, and improvement of patient outlook.
Introduction
Infectious diseases present a significant global burden affecting society (1, 2). Most of these diseases are due to exposure to or the invasion of host cells and organs by microorganisms (1–3). These pathogens disrupt the normal function of the human body by hindering immune responses and producing harmful toxins. Infectious diseases can easily spread from person-to-person via contact with body fluids, indirect contact or through animal vectors such as mosquitoes and ticks (4). Common widespread diseases of the respiratory system occur when microorganisms invade the respiratory tract. Infectious respiratory diseases are globally seen as a major health concern because they can rapidly become severe and lead to death. Respiratory diseases are categorized into upper and lower respiratory tract infections (LRIs). LRIs are more severe because pathogens infect sterile parts of the respiratory system such as the lungs, trachea, and bronchi (5). In 2013, an estimated 2.6 million deaths worldwide were attributed to LRIs, while by 2015, this increased to 2.74 million (6). Higher burden of LRIs is associated with low sociodemographic status, poor access to healthcare and nutrition (Figure 1) (6, 7).
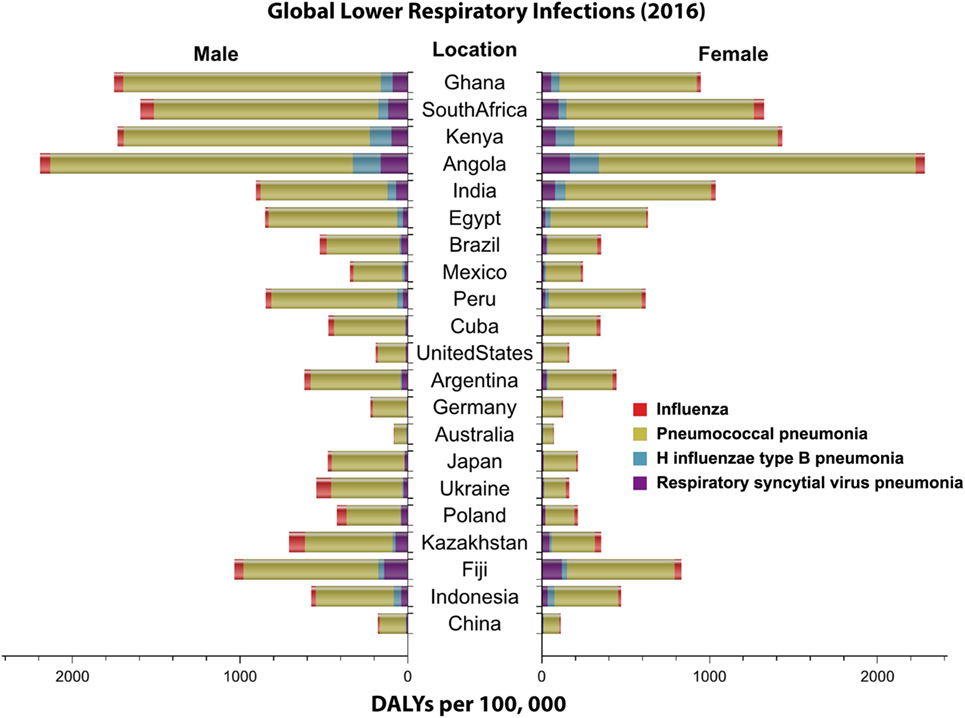
Figure 1. Global distribution of lower respiratory infections by sex. Highlighted in this figure is the distribution of the disability adjusted life year (DALY) per 100,000 (2016) for four major lower respiratory infections worldwide by sex. Data obtained from Institute for Health Metrics and Evaluation (7).
Immune system function is important in a host’s defense to pathogens. A host with a healthy and well-developed immune system is able to clear pathogens before they can become infectious and cause diseases (8–11). The ability to clear pathogens before they can become infectious depends on the quality of the immune system and its effectiveness, which is linked strongly to age (8, 12). The immune system continues to develop from infancy to adulthood, while later in life a fully developed immune system begins to deteriorate with aging. Infants and the elderly are at higher risks for contracting infectious diseases due to their weakened immune system and the inability to clear the pathogens before they become pathogenic (8–11, 13–17).
Streptococcus pneumoniae is a bacterium that has been widely linked to causing respiratory infections in individuals with a weakened immune system (9, 12, 16). S. pneumoniae is spread through airborne droplets, and it is estimated to cause about four million illnesses within the United States (US) and about 450,000 hospitalizations per year (18, 19). Studies indicate that 10% of patients with invasive pneumococcal diseases die of their illnesses (20, 21). S. pneumoniae invades its host by colonizing the nasopharynx asymptomatically as it has been found to be part of the commensal microbiota of the upper respiratory tract (9, 22). After colonization, if the bacterium is not cleared by the immune system, the bacterium is spread via horizontal dissemination into the lower airways and other organs and tissues, and becomes pathogenic (22). A strong immune system and the balance between resident flora and invaders can help to clear S. pneumoniae before it becomes pathogenic. With poor defense mechanisms, the host becomes subject to frequent and long-lasting colonization of S. pneumoniae, which can later lead to diseases (23, 24). The bacterium has several properties which allow it to go unnoticed by the host immune system, and defend against the resident flora within the nasopharynx that would try to clear it (17, 25, 26). Thus, decreasing the burden of this bacterium and preventing further infections is very important to the healthcare field (26, 27). Furthermore, S. pneumoniae is an opportunistic pathogen that takes advantage of hosts with underdeveloped, weakened, and or deteriorating immune systems. Because of this, S. pneumoniae has greater incidence rates in children under the age of two, the immunocompromised, and the elderly (28). Figure 2 depicts that disease burden for major LRIs are highest in young children and the elderly (7, 20, 29–31). Understanding how the immune system changes with age is important in providing appropriate treatments to hinder colonization of weaker hosts.
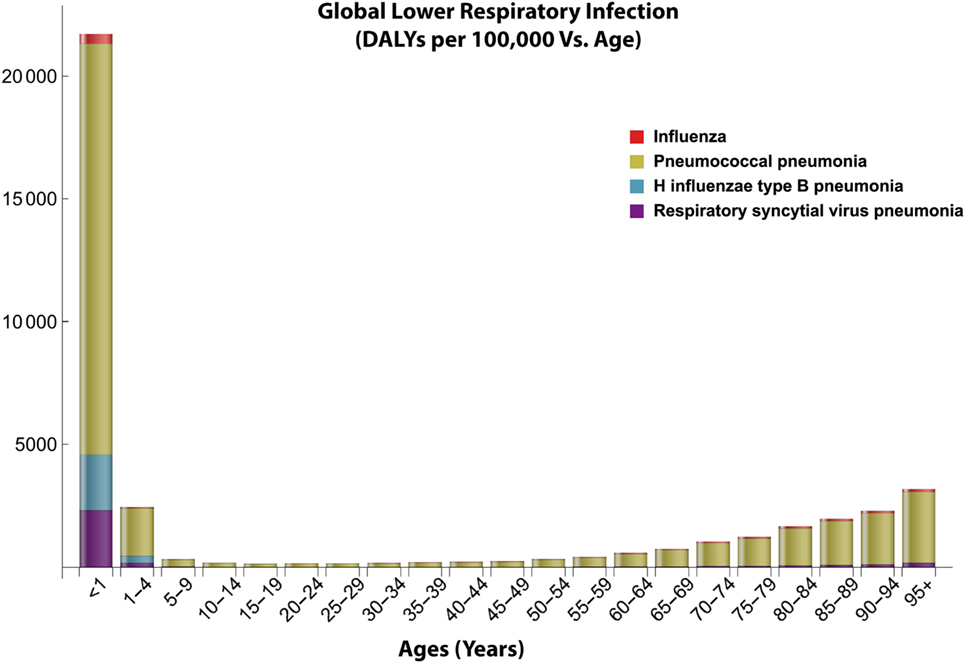
Figure 2. Global distribution of lower respiratory infections with age. This figure shows the age-dependent disease burden to lower respiratory infections especially pneumococcal pneumonia based on the disability adjusted life year (DALY) data from 2016. Data obtained from Institute for Health Metrics and Evaluation (7).
In this review, we provide a concise introduction to the expanding literature on S. pneumoniae, and focus on exploring the characteristics of S. pneumoniae, its pathogenesis, its virulence factors, and pathology. We will also delve into the general host immune response to S. pneumoniae, with a focus on pneumonia, and connect the severity of this disease to varying host immune responses with age. In addition, we will explore the medications available to prevent or treat pneumococcal diseases such as pneumonia, disease prognosis, and finally discuss what the future holds for pneumococcal diseases.
Pneumococcal Disease, Epidemiology, and Transmission
Streptococcus pneumoniae, a Gram-positive bacterium (Figure 3), also known as pneumococcus, can survive in both aerobic and anaerobic conditions (32). It is a facultative anaerobe that is often found as diplococci (32). Pasteur and Sternberg first isolated S. pneumoniae from saliva in 1881 (33–35). Currently, there are varying reports on the number of identified serotypes of S. pneumoniae (24, 34, 36, 37). However, there are at least 97 serotypes of S. pneumoniae that have been identified and characterized to date (34, 38). All of these serotypes are independently recognized by the host (9, 24, 39–41).
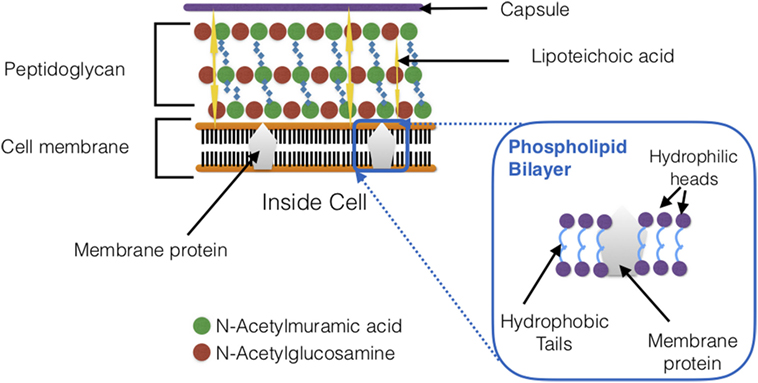
Figure 3. Schematic cross section of Streptococcus pneumoniae cell wall. The bacterial cell wall composes of teichoic acids, a thick peptidoglycan layer, and a phospholipid bilayer.
Pneumococcal diseases occur worldwide (24, 26, 42) and are more prevalent in young children, the elderly, and immunocompromised individuals (Table 1) (9, 22, 32, 41, 43, 44). S. pneumoniae causes many pneumococcal diseases such as meningitis, bacteremia, pneumonia, acute otitis media, and sinusitis (24). S. pneumoniae causes about 40,000 fatal pneumococcal infections per year within the US (23, 32, 45, 46). S. pneumoniae colonizes the upper respiratory tract—specifically the nasopharynx (9, 47), and is able to asymptomatically reside in the upper respiratory tract—this is known as carriage (9). Carriage is more prevalent in children (20–50%) compared to adults (5–20%) (47–49). Carriage can lead to further transmission of S. pneumoniae within the community or can advance to pneumococcal diseases (9). Biofilms form in the nasopharynx during colonization (50). S. pneumoniae has many virulence factors (Table 2; Figure 4) that allow for adherence to host cells, reduce the host’s immune system’s ability to clear the bacterium, and promote invasion of epithelial cells (17). If the host is unable to clear S. pneumoniae immediately after colonization of the upper respiratory tract, the bacterium multiplies, disrupts the regular non-pathogenic flora of the respiratory system (22, 51), and is able to migrate to the tissues and organs and cause infections. The migration of S. pneumoniae to sterile tissues and organs is the main cause of all pneumococcal diseases. For example, when meninges, the protective membranes surrounding the spinal cord and brain, become inflamed due to S. pneumoniae infection, this is known as bacterial meningitis (24, 52). Bacterial meningitis is predominantly seen in young children and is mostly caused by S. pneumoniae (53). S. pneumoniae causes more than 50% of bacterial meningitis within the US (24, 53). Bacteremia refers to infection of the blood by pneumococcus (24) which causes about 12,000 cases per year and usually accompanies other pneumococcal infections (24). S. pneumoniae can also colonize the middle ear of infants and young children causing acute otitis media (24). The Centers for Disease Control (CDC) estimates that approximately 60% of young children would have at least one ear infection (24). Sinusitis occurs when S. pneumoniae infects fluid trapped in the sinuses (24).
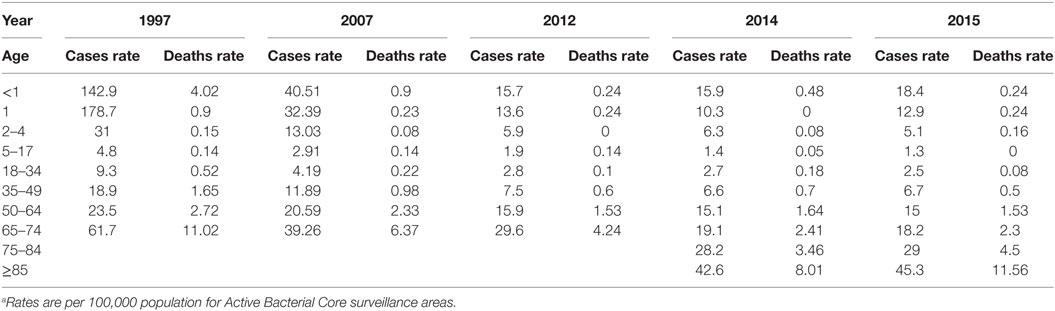
Table 1. Occurrence of pneumococcal diseases from 1995 to 2015 as reporteda by the Centers for Disease Control.
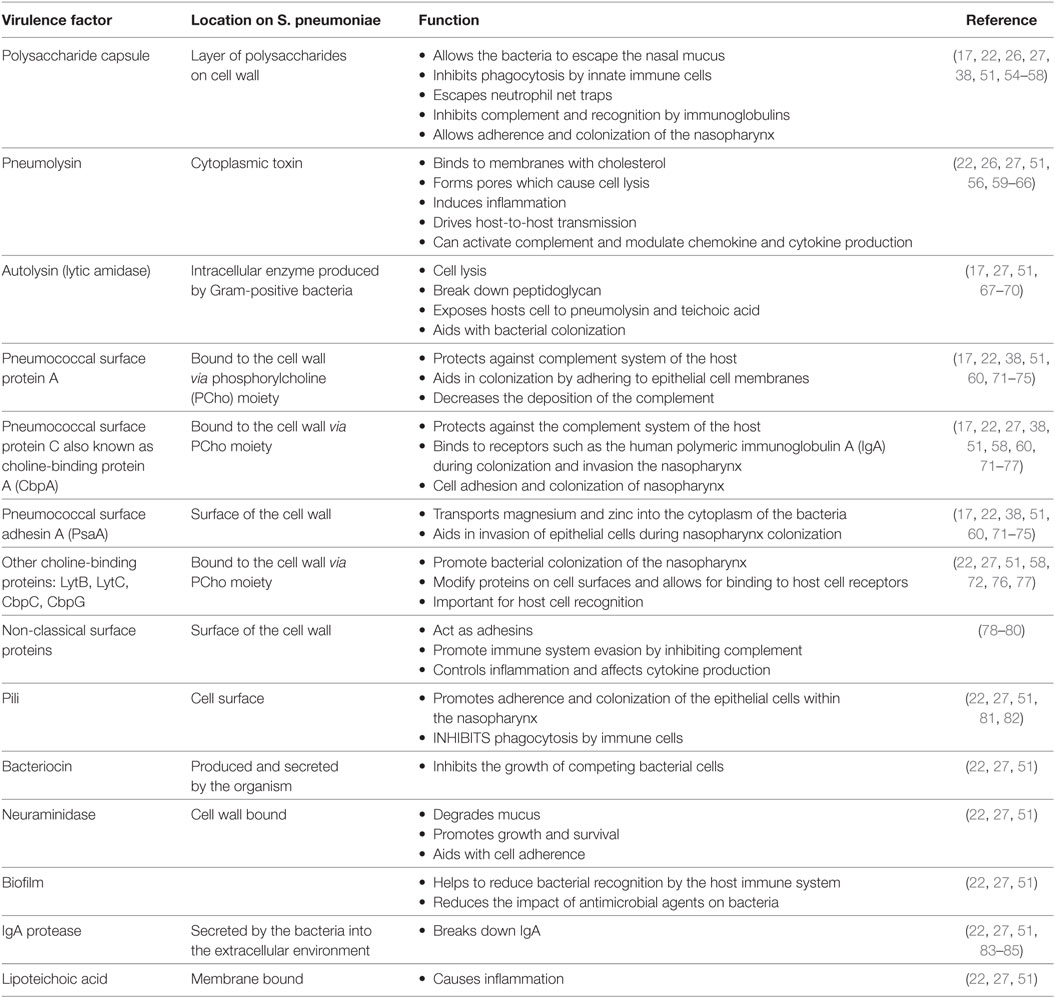
Table 2. Selected virulence factors of S. pneumoniae, their location, and function.
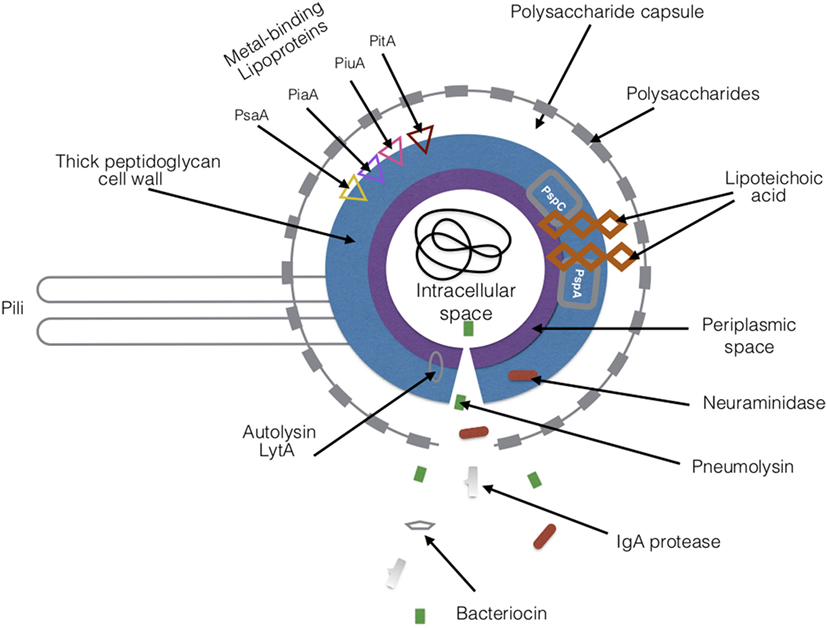
Figure 4. Virulence factors of Streptococcus pneumoniae. There are a variety of proteins and toxins that are expressed by S. pneumoniae that drive its pathogenesis. The major virulence factors are highlighted in the figure. Abbreviations: PsaA, pneumococcal surface adhesin A; PspA, pneumococcal surface protein A; PspC, pneumococcal surface protein C; PiaA, pneumococcal iron acquisition A; PiuA, pneumococcal iron uptake A; PitA, pneumococcal iron transporter.
Streptococcus pneumoniae, which initially inhabits the mucosal surfaces of the nasopharynx in its hosts (17), can migrate to the lungs, where it causes pneumococcal pneumonia (17). This is an infection of the lungs that leads to inflammation of the air sacs causing them to fill with fluid, and making it difficult to breathe. Individuals who have pneumonia usually suffer with high heart rates, shortness of breath, frequent coughing, and high fevers (86). Thus, despite S. pneumoniae’s asymptomatic colonization of the nasopharynx, having a poor immune response and lack of clearance, may develop into pneumococcal pneumonia, which can be a serious health risk for those with reduced host defenses.
Pneumococcal pneumonia dominates as the main type of pneumococcal disease within the US and worldwide (24) (Figure 5). Overall, pneumonia is the eighth leading cause of death in the US (87), and is mainly caused by bacteria, but can also be caused by other pathogens such as viruses and fungi (22). For example, Haemophilus influenzae type b, respiratory syncytial virus (RSV), and influenza can also cause pneumonia, but pneumococcal pneumonia is the most prevalent (Figures 1, 2 and 6) (7). Over time the global disease burden of LRIs such as pneumonia has decreased, but they remain a healthcare concern for specific high-risk populations (Figures 2 and 6) (7). Worldwide pneumonia is the leading cause of death in children under the age of five (31, 88). The World Health Organization reported that a child dies from pneumonia every 20 s (89). There are approximately 900,000 cases of pneumococcal pneumonia (33, 90) that occur annually within the US (32, 90). In addition, United Nations Children Fund stated that in 2016, pneumonia accounted for 16% of the fatalities observed among young children under the age of five worldwide (92). Pneumococcal pneumonia leads to about 300,000–600,000 elderly hospitalizations annually in the US, and the elderly have reduced survival rates (93, 94). There are different types of pneumonia: community-acquired pneumonia (CAP), atypical pneumonia, hospital acquired pneumonia, and aspiration pneumonia (24). These differ based on where someone contracts the infection and what bacteria cause the disease. Currently, the most common form of pneumonia is CAP (which is mostly pneumococcal). This type of pneumonia spreads via person-to-person contact in the community, but outside of healthcare facilities, by breathing in aerosol droplets from a carrier or infected person (51, 95). Worldwide, CAP is currently the leading cause of death for young children who are under the age of five (29, 96). In 2015, 920,136 children died from CAP (97). Infants, young children, the elderly, smokers, and immunocompromised individuals are all at a higher risk of developing pneumonia due to a weakened immune system (22). CAP has a higher occurrence rate in the elderly compared to younger populations, and is also the fifth leading cause of death in the elderly population (93, 94).
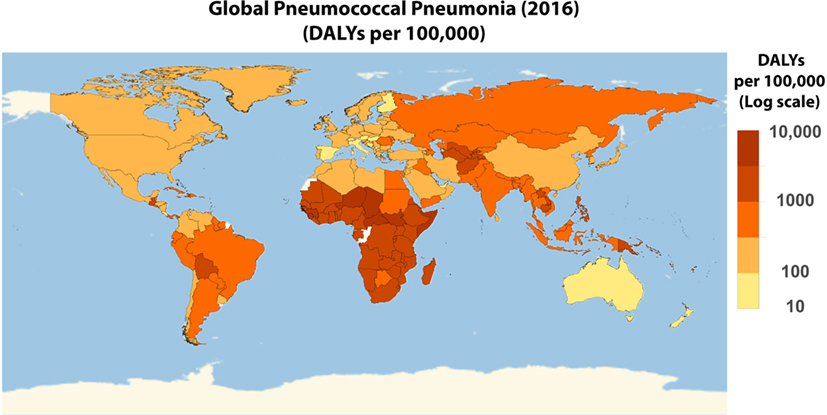
Figure 5. Worldwide disability adjusted life year (DALY) of pneumococcal pneumonia. Global distribution of pneumococcal pneumonia on a log10 scale of the 2016 DALY per 100,000 pneumococcal pneumonia data obtained from Institute for Health Metrics and Evaluation (7).
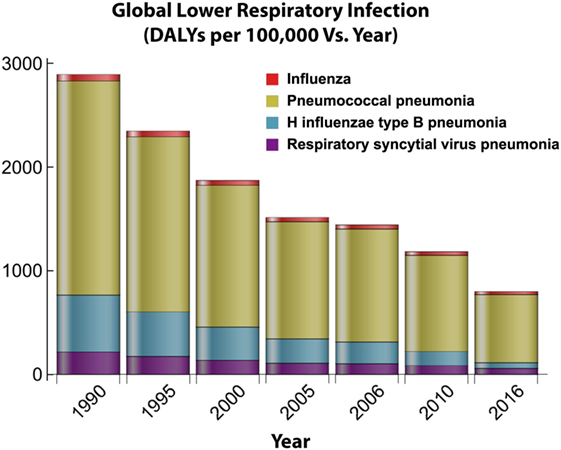
Figure 6. Global distribution of lower respiratory infections over time. This figure depicts how the burden for four major lower respiratory infections changes over time in response to the introduction of antibiotic treatments and vaccine implementation. Disability adjusted life year (DALY) data obtained from Institute for Health Metrics and Evaluation (7).
Transmission
The severity of pneumococcal diseases has led to multiple studies investigating how S. pneumoniae is transmitted. The nasopharynx has been classed as the main reservoir of S. pneumoniae. This is due to the nasopharynx of hosts being colonized without any symptoms (50). Following colonization, the spreading of the disease depends on carriers coming into close contact with healthy individuals within the community. The CDC has declared that the main source of S. pneumoniae transmission is direct contact with secretions of the respiratory system of a carrier (24). Le Polain de Waroux et al. (98) investigated transmission in 566 Ugandan subjects by studying nasopharyngeal samples, and determined that close interpersonal contact was necessary for the dissemination of S. pneumoniae.
Who exactly the main carriers/reservoirs of S. pneumoniae are, is still heavily debated. There have been a variety of studies trying to pinpoint which age group acts mainly as carriers/reservoirs for S. pneumoniae (99–101). Some researchers have suggested infants (9, 101), while others suggest that older children actually transmit the pathogen to infants (99, 100). Lipsitch et al.’s longitudinal study suggests that infants are reservoirs due to the duration of carriage and colonization (101). In this study, they also observed that the carriage time of S. pneumoniae decreases with age (99, 101). On the other hand, a longitudinal study investigating transmission and colonization in a daycare setting showed that toddlers act as a reservoir for S. pneumoniae and spread to family members (99, 100). Another contradicting study that used pre-existing data and mathematical modeling suggests that older children introduce the pathogen to their homes and transmit S. pneumoniae to younger children, siblings, and adults (99). Althouse et al. did confirm that there is higher colonization in infants, however, their results show that S. pneumoniae’s direction of transmission is instead from older siblings to infants as opposed to transmission from infants or parents to others in the household (99, 102). The duration of carriage seems to affect how well S. pneumoniae is transmitted as well as close contact between carriers and healthy individuals (98, 99). Althouse et al. concluded that despite the larger percentage of carriage being in infants, their role in transmission is minimal compared to that of toddlers and older children (99). The differences between these findings suggest that the direction of transmission is still not yet fully understood and further research is required. Another possibility would be that multiple age groups are acting as reservoirs rather than one specific group under different conditions.
In addition to close contact with an S. pneumoniae carrier, the bacterium may also be transferred to healthy individuals via fomites (103). Chronic carriers of S. pneumoniae can contaminate inanimate objects with biofilms (103). S. pneumoniae biofilms are able to survive being in the environment because the biofilm’s structure provides protection from drying out (47, 104). S. pneumoniae was found in high concentration on items within a daycare center following bacterial cultures (50, 103). Pneumococcus can survive being in the environment for long periods of time (for example, up to 4 weeks) (103, 105). Because of this, fomites can serve as a reservoir. These findings indicate why it is important to improve hygiene and cleanliness in everyday-life, and at community-based facilities and daycare centers.
Streptococcus pneumoniae also makes a toxin, pneumolysin, that promotes shedding and in turn enhances bacterial transmission (59). Pneumolysin induces inflammation in hosts during colonization and this promotes bacterial shedding (59). Zafar et al. conducted a shedding assay which suggests that S. pneumoniae may be using the host’s inflammatory response as a signal for initiating its exit from the inhospitable host (59).
Transmission via Coinfections
Coinfection with S. pneumoniae is often seen during viral infections such as influenza, also the eighth cause of death within the US (87), and RSV. Coinfections by pathogenic bacteria such as S. pneumoniae increase the severity and mortality rates of viral infections (106, 107). For example, during the influenza pandemic of 1918, the analysis of lung samples from those infected indicated that a majority of the deaths were due to bacterial infections and not the influenza virus (107–109). Coinfection is possible due to the pre-existing damage on the epithelia of the respiratory tract which promotes bacterial colonization (110–113). More specifically, S. pneumoniae’s bacterial load increases during viral coinfections due to the bacteria’s attachment to cells that are already infected by the virus (114). Studies have also shown that colonization of S. pneumoniae is affected by flu vaccines, which also indicates that S. pneumoniae benefits from colonizing hosts that are already compromised (107, 115). Increased host colonization and bacterial cell density of S. pneumoniae during viral infections promote transmission (115). Khan et al. determined that there are higher risks of bacteremia, mortality, and spread to other tissues during coinfections (110, 115). Co-detection with S. pneumoniae has also been observed in RSV infections (116).
Virulence Factors
Streptococcus pneumoniae, like many other bacterial species, produces toxins that are harmful to its host, has several surface proteins and physical structures, which play a vital role in its pathogenesis (27). These virulence factors (Figure 4; Table 2) work by hindering the host’s immune system response, avoiding defense mechanisms, or by direct contact with host tissues and surface receptors, which in turn interferes with the host’s immune system activation and bacterial clearance (27). As discussed above, S. pneumoniae exploits hosts with weakened or compromised immune systems (13–15). S. pneumoniae’s effectiveness in causing infections is directly related to the host immune system’s developmental stage and possible deterioration with aging (see also Section “Host Immune System Responses to S. pneumoniae”).
Streptococcus pneumoniae’s virulence thrives because of the bacteria’s ability to acquire new genetic material via transformation and recombination (117). Investigating the level of genetic variation within S. pneumoniae is important for not only thoroughly understanding its virulence but also for developing effective treatments and vaccines. About 4,000 S. pneumoniae genomes have already been sequenced (117), with lengths ~2–2.2 million bp (54). More than 2,000 genes have been annotated, but novel genes are still regularly discovered as more sequences become available (117). Variation in gene content and single genes plays a role in defining the virulence profile of some of S. pneumoniae strains (117). Donati et al. describe genome diversification as S. pneumoniae’s ability to evolve in diverse host environments (117, 118). Genetic variation has been observed within identical S. pneumoniae clones, due to changes in gene content of their dispensable genes (117, 119). Dispensable genes are not needed for bacterial growth (117), but provide selective advantages to S. pneumoniae such as antibiotic resistance (120). Additional variants are introduced to the core genome of S. pneumoniae via allele replacement. This is because the bacteria lacks SOS genes and does not repair damaged DNA (121). Carriage can also influence genetic variation. In 2017, Lees et al. developed a model to assess carriage duration and assembled those findings with data from whole genome sequencing. The results indicated that pneumococcal genetic variation accounts for the phenotypic variation compared to host’s age and previous carriage (5%) (122).
The major virulence factors of S. pneumoniae that have been thoroughly characterized are summarized in Table 2. Below we further discuss virulence factors of particular interest:
1. Polysaccharide capsule: S. pneumoniae’s extracellular polysaccharide capsule, the most important virulence factor (55), helps to initiate infection by allowing the bacterium to adhere to host cells and cause inflammation, while also providing protection from the host’s immune system (54, 55). The capsule inhibits phagocytosis by innate immune cells, prevents the recognition of the bacterium by host receptors and complement factors, and also avoids neutrophil traps (17, 27, 55, 56, 71, 123). Many serotypes of S. pneumoniae are characterized by the polysaccharides that are on the outer coat of the capsule, and they are all pathogenic in their own unique manner—some more harmful than others (20, 56). For example, serotype 1 has been found in invasive infections which have lower fatalities, whereas serotype 3 is associated with colonization of the nasopharynx and serious infections which can lead to fatalities (39, 42, 56, 124, 125). The capsule manipulates how immunoglobulins recognize the bacteria (126) and inhibits the host’s defenses such as mucus layers and cilia from removing the bacterium, and is vital for pneumococcal bacterial cells’ colonization (57). The roles of the capsule in pathogenesis have been described to be due to its charge (57, 127). The capsule has a negative net charge which is in part due to the acidic polysaccharides and phosphates that make up this layer (57, 127). The charge is important because it defines how interactions with other cells take place, specifically host cells (57, 127). One explanation for S. pneumoniae’s ability to avoid being trapped by mucus layers and phagocytic cells is due to electrostatic repulsion (57, 127). Negatively charged mucus and phagocytic cells, such as macrophages, have led to a reduction in the clearance of S. pneumoniae because of this electrostatic interaction (57, 127).
Streptococcus pneumoniae’s virulence via its polysaccharide capsule is enhanced by its ability to undergo capsule switching (117, 128, 129). Mutations in the capsule polysaccharide synthesis genes (cps) promote serotype switching (117, 128, 129). Serotype switching in strains is increasingly being observed and it is often via recombination or polymorphisms based on antibiotic and vaccine selective pressures (further discussed in Section “Prevention, Antibiotic Response, and Age-Dependent Immune Responses”) (117, 128, 129). Currently, serotype switching is a healthcare concern as non-vaccine serotypes are being detected at higher rates compared to before vaccines were implemented (128). Moreover, mutations in novel genes or a disruption of the cps loci can lead to S. pneumoniae strains without capsules (130). Non-typeable S. pneumoniae cannot effectively colonize hosts, but novel genes such as pneumococcal surface protein K in the cps loci assist with adhesion (130). Serotype switching and capsule-free strains of S. pneumoniae together will add to the burden on the high-risk age groups (infants and the elderly) (130), and because of this vaccines and treatments should be improved.
2. S. pneumoniae’s cell wall components: S. pneumoniae is a Gram-positive bacterium with a thick cell wall. The cell wall is important because it provides protection and shapes the cell (131). Peptidoglycan, wall teichoic (WTA), and lipoteichoic acids (LTAs) are the main components of S. pneumoniae’s cell wall (131). WTAs are covalently attached to peptidoglycan whereas LTAs are non-covalently connected to the cytoplasmic membrane with a lipid anchor (131). The capsular and cell-surface proteins are all linked to the peptidoglycan (131). Alternating glycan chains of N-acetylglucosamine (GlcNac) and N-acetylmuramic (MurNac) acids crosslinked by peptides make up peptidoglycan (131, 132). These glycan chains can undergo secondary modifications such as deacetylation of GlcNac and O-acetylation of MurNac (131, 132). These modifications aid in S. pneumoniae’s virulence by making the cell resistant to lysozyme (132). Cell wall components, WTA, and LTA have phosphorylcholine (PCho) residues which serve as anchors for choline-binding proteins (CBPs). CBPs are important for host–pathogen interactions such as evasion of host immune responses (discussed later in this section) (131, 132). PCho in bacterial cells is unusual and S. pneumoniae are currently the only bacteria known to require it for growth (132). WTA, LTA, and peptidoglycan are pathogen-associated molecular patterns (PAMPs) that can cause an inflammatory response in hosts. Peptide synthesis, peptidoglycan structure, WTA, and LTA synthesis and modifications have been further discussed by Gisch et al. (131).
3. Pneumolysin: this toxin that is capable of forming pores in cell membranes (133) can be found in the cytoplasm of S. pneumoniae and other Gram-positive bacteria (27, 60, 133). Pneumolysin is released as a result of cell lysis and is toxic to host cells (27, 61, 134). Pneumolysin binds to membranes containing cholesterol (135), and forms pores which later lead to host cell lysis (27, 56, 62). In addition to causing cell lysis, pneumolysin plays a role in promoting the formation of biofilms (63), it reduces mucus clearance of the bacterium, and it can interfere with the host’s immune system (27, 60, 61, 134, 136). Pneumolysin regulates the complement system (54) and reduces phagocytosis by innate immune cells. It is also a pro-inflammatory toxin which causes damage to host cells. It can regulate cytokine and chemokine production (22, 51). This pro-inflammation has also been shown to assist with host-to-host transmission (59). By increasing cell inflammation, there is an increase in shedding and thus a higher rate of transmission of the bacteria (59).
Studies have also shown that pneumolysin can cause DNA damage by inducing double-stranded DNA breaks. One mechanism of DNA damage by pneumolysin was described by Rai et al. in 2016 (64). They showed that the toxin can dysregulate the production of reactive oxygen species (ROS) intracellularly (64). This is possible because of pneumolysin’s pore-forming properties—it creates ion channels that disrupt cell calcium levels, which leads to overproduction of ROS, that then causes DNA damage (64). Host DNA damage may lead to increased pneumolysin virulence in the elderly, who are already experiencing a compilation of DNA damage and telomere shortening due to aging (137).
Pneumolysin has different allelic forms that and can also affect the toxin’s hemolytic activity (54, 138). For example, genetic variation in allele 5 produces a non-hemolytic form of pneumolysin (138–141). Previously, a cysteine residue at amino acid position 428 was identified in the conserved sequence described to be important for the hemolytic activity of pneumolysin (142). However, cysteine was later substituted by alanine without affecting the toxin’s hemolytic activity (138, 143).
4. Autolysin: this enzyme is involved in autolysis of bacteria which results in the release of pneumolysin, teichoic acid, and other components from within the cell (22, 51). An example of this is lytic amidase (LytA) (67), a choline-binding amidase (17) (see below) that degrades peptidoglycan and causes cell lysis (27, 68, 69, 144). Autolysins promote colonization of nasopharyngeal cells due to the release of toxins such as pneumolysin during cell wall degradation (27).
5. Pneumococcal surface proteins: S. pneumoniae has a large variety of surface-exposed proteins (17, 72) that aid in its pathogenesis by acting as adhesins to host cells and hindering the host’s immune system, specifically the complement system (22, 27, 51, 145, 146). Pneumococcal surface proteins are categorized into four groups: CBPs, lipoproteins, non-classical proteins, and proteins that have an LPXTG motif (X represents any amino acid) and can be covalently bound through sortase cleavage of the motif (17, 72).
a. CBPs: many of S. pneumoniae’s surface proteins are classed as CBPs. For example, pneumococcal surface proteins (discussed below) are also classified as CBPs (27, 72, 76, 77). These proteins are known for binding to PCho on S. pneumoniae’s cell wall (27, 72, 76, 147) and are necessary for adhesion to host cells (27, 76, 147). CBPs affect the host’s complement system by blocking its activation and reducing the ability of immunoglobulins to eliminate the pathogen (27, 72). Some of these CBPs can also modify host cell surfaces to allow for binding interactions between to host cell receptors and S. pneumoniae (76). S. pneumoniae has approximately 10–16 identified CBPs (17, 148–150) including pneumococcal surface protein A (PspA), pneumococcal surface protein C (PspC), and LytA which are discussed below:
i. PspA is very electronegative, and this characteristic can block complement binding, which prevents opsonization of S. pneumoniae (17, 73). PspA can also bind to host lactoferrin (22, 27, 51), specifically apolactoferrin (iron-free), which in turn provides protection to S. pneumoniae against the bactericidal killing of apolactoferrin (151, 152).
ii. PspC, also known as CbpA (highly polymorphic), promotes adherence by binding to the polymeric immunoglobulin receptor (72, 153). It facilitates the colonization of S. pneumoniae into the nasopharynx and can prevent the formation of C3b (part of the complement system) by binding to factor H. This in turn interferes with opsonization of S. pneumoniae (17, 72, 154). PspC exists in multiple allelic forms with most alleles containing a C-terminal cell wall choline-binding motif. However, there are also 17 allelic variants that have the LPTXG motif (see LPXTG cell wall bound proteins) (54, 155, 156). In addition, allelic variant PspC 4.4 was characterized as a ligand for complement inhibitor C4b-binding protein (54, 157), which leads to an allele-dependent form of protection from the complement (157).
iii. LytA, an autolysin, was the first of three major lytic enzymes found in S. pneumoniae (76, 158). LytA degrades peptidoglycan by cleaving the N-acetyl-muramoyl-l-alanine bond (72, 159). This causes cell lysis and the release of pneumococcal antigens such as pneumolysin, peptidoglycan, and teichoic acids which are all harmful to host cells (72, 159, 160). The release of these harmful particles from S. pneumoniae cells is also capable of inhibiting cytokine [such as interleukin (IL)-12] production, which in turn blocks the activation of phagocytes (158, 161). This is thought to be due to the fact that cells are already decomposed so phagocytic activity is no longer necessary (158), and acts as a form of immune system evasion by S. pneumoniae (161–163). By blocking cell signaling via cytokine production, LytA has also been shown to hinder complement activation (76). How exactly this blockade might be happening, still needs to be further researched.
iv. LytB and LytC are two other lytic enzymes found in S. pneumoniae. Their roles in S. pneumoniae’s virulence are not as thoroughly understood as LytA. Studies have shown that LytB is necessary for separating daughter cells (164, 165). LytC on the other hand, is described as a lysozyme. Ramos-Sevillano et al. have indicated that LytB and LytC interact and are both involved in adhesion of S. pneumoniae to epithelial cells within the nasopharynx of hosts. Their results also suggest that LytC helps with evasion of the complement system via experiments with mutants. LytC mutants had larger amounts of C3b deposition and LytB and LytC double mutants all had a reduction in their ability to adhere to host cells (166). These findings shed light on the roles of LytB and LytC. They aid in virulence by playing a role in colonization and evasion of host immune responses (166, 167). In addition, LytC has also been described to play a role in cellular fratricide with LytA. These enzymes are released to lyse non-competent pneumococci in close proximity of competent cells (168). This is important for transformation of S. pneumoniae. Competent cells are able to uptake and incorporate free DNA from the lysed cells (168, 169). This promotes genetic exchange which in turn can improve bacterial survival. For example, the bacterium can take up genes for antibiotic resistance (168, 169). LytC’s activity is most active at 30°C which indicates it is probably most active in the upper respiratory tract (168, 169).
v. CbpF, the most abundant protein on S. pneumoniae’s cell wall is capable of regulating LytC (77, 170). CbpF regulates LytC’s activity by blocking LytC’s access to its substrate (77, 150, 170).
vi. Other CBP: there are about eight other CBPs: CbpD, CbpG, CbpI, CbpJ, CbpK, CbpL, CbpM, and CbpN. These have not been studied as extensively as the main CBPs previously discussed. There is not much known about their structure or function. CbpD has been shown to be involved in fratricide by working with LytA and LytC (150, 169, 171). The CbpD is able to provide a substrate for LytC that is more accessible by binding to target cells and breaking down the cross-links of the peptidoglycan (150). CbpG is necessary for adhesion and all others been reported to work as adhesins (77, 150).
b. Lipoproteins: these proteins are necessary for substrate transport. There are approximately 50 lipoproteins that have been characterized (72, 148, 150). The four main lipoproteins are the pneumococcal surface adhesin A (PsaA), pneumococcal iron acquisition A (PiaA), pneumococcal iron uptake A (PiuA), and pneumococcal iron transporter (PitA) (17, 72, 172, 173). They are all metal-binding proteins that combine with ATP-binding cassette (ABC) transporter complexes. ABC transporters transport substrates across membranes by utilizing energy generated from ATP binding and hydrolysis.
i. PsaA is involved in transporting magnesium and zinc into the cell (27, 74, 174, 175). Investigations have previously reported PsaA’s role in cell adhesion and promoting cell invasion of S. pneumoniae (74, 176). However, other studies on PsaA mutants have found that PsaA has no clear role in adhesion, but rather magnesium transport (175). This particular characteristic of adhesion needs to be further investigated (26). Also, genetic mutations can alter PsaA’s function which may lead to impaired ability to acquire manganese which results in decreased resistance to oxidative stress (54).
ii. PiaA, PiuA, and PitA are involved in regulating iron-uptake (177, 178). In addition to this, PiaA and PiuA have been described to be needed for full virulence of S. pneumoniae in mice (177, 179). Mutations in PiaA and PiuA affect growth and virulence of S. pneumoniae (180, 181). This indicates the importance of iron in the environment for growth. Furthermore, Cheng et al. in 2013 crystalized PiaA and discovered that PiaA is capable of binding to ferrichrome (182–184) despite previous findings suggesting pneumococci do not produce siderophores (185). Cheng et al. concluded that S. pneumoniae is probably able to acquire holo-siderophores from other bacteria within the host (181, 182). On the other hand, PiuA binds to both hemin and hemoglobin but has greater affinity for hemin (183, 186). PitA was later discovered and characterized to bind to ferric irons (172, 173, 183). A novel iron transport was discovered in 2016 by Yang et al. (187) via proteomics. In this study, they constructed a triple mutant by deleting PiaA, PiuA, and PitA (187). Using this mutant, they were able to identify potential iron transporters, such as putative protein SPD-1609, which functions similarly to PitA via translatomics and proteomics (187). These findings suggest that there are potentially more iron-binding proteins in S. pneumoniae to be discovered and that the bacteria have developed transport systems to ensure they have access to as much iron as possible for their survival.
c. LPXTG cell wall bound proteins are recognized by the sortase of the cell wall (54, 149, 188). Sortase recognizes the LPXTG sequence, cleaves at this site, and anchors the proteins to the cell wall (54, 188, 189). Mutating the sortase gene srtA caused a decrease in S. pneumoniae’s adhesion to host nasopharyngeal cells in vitro, and caused neuraminidase to be released from the cell well into the media (190). Neuraminidase is an example of an LPXTG cell wall bound protein and is known for cleaving sialic acid from glycoproteins. In the case of pathogenesis of S. pneumoniae, this activity can lead to the removal of sialic acid from lactoferrin which hinders lactoferrin’s bactericidal effect. Neuraminidase is secreted from S. pneumoniae cells and targets host cells (54, 188). It is also involved in colonization of the host and has been suggested to be involved with adhesion (150, 190).
d. Non-classical surface proteins (NCSPs) are found on S. pneumoniae’s surface, but do not have a membrane-anchoring motif nor a leader peptide (72). They are also known as moonlighting proteins for having multiple functions (72, 78, 148). NCSPs function as adhesins that are able to bind to host molecules which promotes pneumococcal host cell invasion (148). There are two main NCSPs: pneumococcal adherence and virulence factor A (PavA) and glycolytic enzymes [enolase and glyceraldehyde 3-phosphate dehydrogenase (GAPDH)] (150).
i. PavA attaches to fibronectin and assists with adherence to host cells (149). PavA also provides protection to pneumococci by controlling inflammation and inhibiting recognition by dendritic cells (191). PavA mutants were more susceptible to recognition and phagocytosis by dendritic cells compared to wild type (191). In addition to this, when the dendritic cells encountered PavA mutants there was a reduction in cytokine production, which affected the adaptive immune response. These findings characterize PavA’s potential function in immune system evasion by S. pneumoniae and cytokine production by dendritic cells (191).
ii. Enolase and GAPDH are both plasminogen-binding proteins. Enolase is an anchorless protein found at the surface of S. pneumoniae (192). It is important for proteolytic activity on the cell surface (193), which is necessary for the pathogenesis of S. pneumoniae (192). Enolase also promotes complement system evasion by binding to the complement inhibitor C4b-binding protein (194). In addition, studies suggest that enolase may cause host tissue damage by inducing the production of neutrophil extracellular traps by binding to neutrophils (195). GAPDH can be found on the surface and in the cytoplasm of S. pneumoniae (196). Although GAPDH binds to plasminogen, it has a higher affinity for plasmin (149, 196). GADPH is suggested to also play a role in iron acquisition due to its ability to bind to hemoglobin and heme (149). Like enolase, GADPH may also play a role in host cell invasion and evasion of the immune system. LytA has recently been identified to be involved in the delivery of GADPH to S. pneumoniae’s cell surface (79).
6. Pili: these hair-like structures are located on the cell surface of S. pneumoniae and many other bacteria (27, 51, 81). They assist with S. pneumoniae’s attachment and colonization of epithelial cells within the nasopharynx and lungs of hosts (51, 81, 82). These pili also help the bacteria avoid phagocytosis by host immune cells (22). There are two main types of pili found on S. pneumoniae: pilus-1 and pilus-2. Pilus-1 is found in 30% of clinical isolates (197) whereas pilus-2 is only in about 16% (198). Studies have shown that piliated S. pneumoniae induce higher tumor necrosis factor (TNF) responses than the non-piliated during pneumococcal infection (81). This suggests that pili are able to stimulate inflammatory responses of the host (81). Pancotto et al.’s findings indicated that pilus-1’s expression is regulated in vivo (82). High expression of pilus-1 is observed at early stages of colonization and reduced expression during later stages of infection. This downregulation may be necessary for avoiding host immune response but this needs to be further investigated as it is not clear why this might be happening (82). S. pneumoniae, like many other pathogenic bacteria have a type IV pilus that is necessary for transformation (199, 200). This pilus is formed on the surface of the bacterial cell and contains the major pilin ComGC. The operon that codes for ComGC also encodes for an ATPase which is needed for powering the pilus assembly. The structure of ComGC was recently discovered by Muschiol et al. in 2017 (199).
7. Immunoglobulin A1 (IgA1) protease: this enzyme is produced by S. pneumoniae and it works by cleaving the human IgA1 into fragments (83, 84). The IgA1 represents an isotype of immunoglobulin A (IgA) which has two isotypes: IgA1 and IgA2 (201). These two isotypes differ in hinge regions—IgA1 has an extended hinge region because of an insertion into this region of a set of duplicated amino acids (201). IgA1 proteases reduce the binding IgA’s effector region of the heavy chain and hinder killing of the bacterium by these antibodies (83, 85).
8. Hydrogen peroxide: S. pneumoniae secretes hydrogen peroxide (H2O2) which causes damage to host DNA (202). However, this is only observed in strains with pyruvate oxidase activity (SpxB gene) (202, 203). H2O2 production also has bactericidal effects. S. pneumoniae uses this to reduce the growth of bacteria it may be competing with (203). In addition, pneumococcal H2O2 induces an innate immune response by enhancing the release of pro-inflammatory cytokines, and targets cellular stress responses (204). As S. pneumoniae produces H2O2 via pyruvate oxidase, hydroxyl radicals form via the Fenton reaction (205). These radicals are often harmful to bacteria but do not affect S. pneumoniae. This is because of S. pneumoniae’s ability to reduce reactive OH before it comes into contact with DNA (206), by sequestering Fe2+ away from DNA (206). In addition to producing H2O2, the SpxB gene has also been found to increase resistance to H2O2 (207). SpxB mutants produced no H2O2 and were less resistant (207). In addition, S. pneumoniae has a variety of defense proteins involved in detoxification, repair, regulation, and cation homeostasis that provide protection against oxidative stress (206).
9. Pathogenicity islands (PAIs): these are parts of pathogenic bacterial genomes that were acquired via horizontal gene transfer (208). The genes on PAIs aid in the virulence of the pathogen (209). PAIs can code for iron-uptake systems and proteins involved in cell attachment (209). For example, the first PAI discovered in S. pneumoniae, pneumococcal PAI 1 codes for the PiaA iron transporter complex (180). In addition, the pilus-1 is encoded by another PAI, known as the rlrA islet (81). However, this PAI is not found in all of the S. pneumoniae clinical isolates (81). Pilus-2 is also encoded by a PAI, pilus islet 2 (198). Another important adhesin, pneumococcal serine-rich repeat protein (PsrP), is also coded for by a PAI. PsrP is important for S. pneumoniae’s attachment to cells within the lungs (210). High PsrP production is also linked to biofilm growth (211). PAIs promote genetic variation in species, and this may affect current treatment and vaccine targets.
10. Biofilms: these are structured communities that consist of aggregated microbial cells surrounded by an extracellular matrix of polysaccharides that attach to surfaces (47, 212). The extracellular matrix provides protection and enhances S. pneumoniae’s virulence (47, 212). Biofilms are formed in response to stress and harsh conditions to promote bacterial survival (47, 212, 213). To promote biofilm formation and competence, S. pneumoniae downregulates expression of capsular proteins (214). Within biofilms, horizontal gene transfer rates increase due to close cell proximity (47, 212, 215, 216). Studies indicate that S. pneumoniae biofilms are not effectively cleared during antimicrobial treatments due to increased antimicrobial resistance (217). In addition, S. pneumoniae biofilms are able to escape host immune responses such as mucociliary clearance (218).
Host Immune System Responses to S. pneumoniae
We have discussed above the virulence factors that aid in ensuring S. pneumoniae can evade the host’s immune system. On the other hand, there are several host defenses that recognize S. pneumoniae, act rapidly, and clear the pathogen before it can cause pneumococcal diseases. Protection from S. pneumoniae is dependent on the state of the host’s immune system. Age plays a role in how successful the immune system will be at clearing the infection by S. pneumoniae. Children under the age of five and the elderly are at higher risk for contracting pneumococcal diseases (Figure 2). This is due to infants having a naïve immune system, whereas the elderly are experiencing immunosenescence (28). A variety of immune cells are involved in the innate (first-line of defense) and adaptive immune responses. The most important immune cellular and humoral components for defending against pneumococcal infections (Figure 7) are summarized in the following sections including how aging may affect their ability to defend the host.
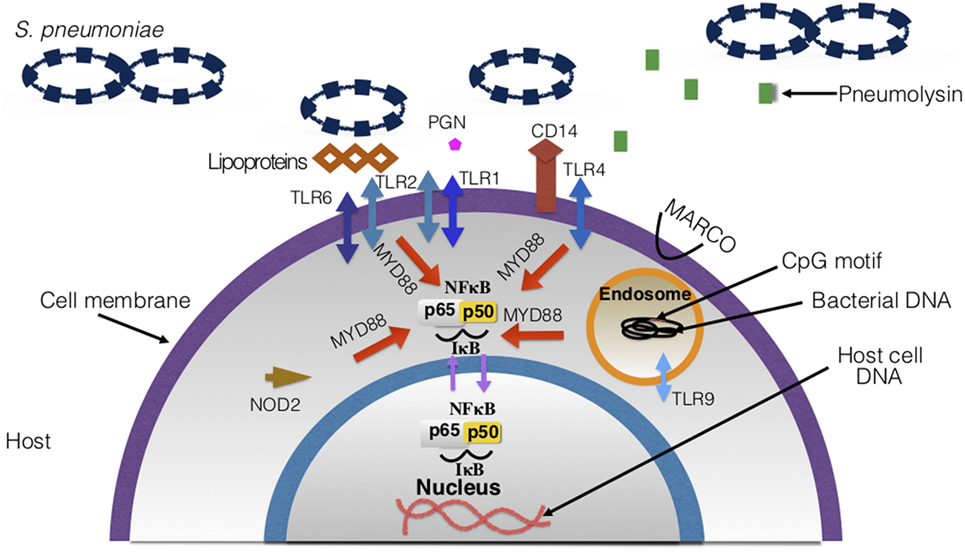
Figure 7. Host surface and intracellular receptors necessary for immune response to Streptococcus pneumoniae. Highlighted in this figure are the major pathogen recognition receptors necessary for binding to pneumococcal ligands and eliciting an immune response. Upon binding to the ligands, receptors and signaling pathways are activated, which leads to the overall production of inflammatory cytokines and recruitment of immune cells. There are 10 toll-like receptors (TLRs) that have been discovered in humans—TLRs involved in pneumococcal disease are depicted in the figure.
Innate Immune Responses
Innate immunity involves non-specific immune responses—cells and receptors recognize foreign particles and elicit immune responses to eliminate the invaders that can be harmful to the host (16, 159, 219). Cell-related innate immune responses against pneumococcal infection include:
i. Mucosa and respiratory epithelial cells: epithelial cells provide a protective barrier for tissues and organs (219). In this case, they line the respiratory tract and protect against pneumococcus (219). There are epithelial cells known as goblet cells, which secrete mucus (220). The negatively charged mucus is necessary for maintaining moisture and trapping foreign particles and pathogens. In addition, ciliated epithelial cells function simultaneously with the mucus to clear pathogens. This process is known as mucociliary clearance (57). Once the pathogen is trapped in the mucus, the cilia (hair-like structures) move together to direct the trapped pathogen and the mucus to the mouth, for expelling the pathogen via coughing or swallowing (221). The respiratory epithelial cells can recruit other cells by producing and releasing cytokines and chemokines (22, 220). They also can directly kill pneumococcus by secreting antimicrobial peptides such as defensins, human apolactoferrin, and lysozyme (Figure 8) (22, 220, 222). Human apolactoferrin sequesters iron and lyses cells. Lysozyme also lyses cells and acts as a bactericidal (222). d-alanine in teichoic acids of S. pneumoniae’s cell wall help to evade killing by antimicrobial proteins (positively charged) by reducing the negative charge (223, 224). The negatively charged capsule also promotes evasion of mucus via electrostatic repulsion (225). Mouse model experiments showed that encapsulated S. pneumoniae were easily trapped in mucus and unable to migrate to the epithelial cells when compared to capsulated S. pneumoniae (57). This again was due to the negatively charged capsule. In addition to this, S. pneumoniae’s neuraminidase degrades mucin and reduces the negative charge by removing sialic acids (225–227). As previously mentioned in Section “Virulence Factors,” the structure of peptidoglycan can be modified. This modification promotes resistance of S. pneumoniae to lysis via the lysozyme (228). Another impressive method of evasion by S. pneumoniae is its ability to undergo phase variation (229, 230). S. pneumoniae is able to express a thick and a thin capsule under certain conditions (229, 230). The thick capsule is necessary to avoid entrapment in mucus, and the thin capsule is necessary for binding directly to epithelial cells (229, 230). Once the thin capsule is expressed, adhesins are exposed for binding to the glycoconjugates on epithelial cells (229, 230).
Infants and the elderly both are challenged with mucociliary clearance due to different reasons: in infants, immature submucosal glands, surface epithelial secretory cells, and low numbers of ciliated epithelial cells can result in poor mucociliary clearance (231). In the elderly, as the host ages there is a deterioration of mucociliary clearance, with reduced mucin and slower cilia beat frequencies (28, 232), which promotes dissemination of the bacteria (28). As S. pneumoniae virulence factors can also degrade mucus and slow down cilia (28), immaturity and deterioration of mucociliary clearance could cause disease exacerbation through increased colonization and recurrent infections.
ii. Phagocytes:
a. Neutrophils: these are found in larger concentrations compared to any other white blood cells (WBC), and they are generally the first to travel to the infection (233, 234). Neutrophils are phagocytic cells (234) that also produce granules, which break down the cell walls of pathogens ultimately killing them (234). There are two main types of granules produced by neutrophils: primary and secondary, which differ based on age/maturity of the neutrophil (219, 235). Primary granules include defensins whereas secondary granules include enzymes necessary for digestion, such as lysosomes. Neutrophils can also trap S. pneumoniae extracellularly, by using extracellular fibers made up of DNA (236).
Neutrophil response changes with age: infants experience minimal protection by neutrophils in their early days of life due to poor bactericidal function, impaired phagocytotic activity, low response to inflammatory signals, and reduced chemotaxis (237–239). With age, neutrophil activity improves and strengthens in young adults but later starts to deteriorate. Elderly populations experience impaired chemotaxis, which may lead to the overproduction of proteases by neutrophils. This causes an increase in inflammation levels in older subjects (15, 240). Neutrophil extracellular traps generation, phagocytosis, and killing diminishes with age (15, 241).
b. Macrophages: macrophages are derived from monocytes (219) and function as phagocytic cells that engulf and directly kill S. pneumoniae (16, 219). These cells can recruit other immune cells, such as neutrophils via cytokine signaling (242), and remove dead neutrophils (159, 243) and other cells via phagocytosis and apoptosis. Macrophages attack cells that have been opsonized by the complement system and Fcγ receptors (225). The macrophage receptor with collagenous structure (MARCO) (244), found on the surface of macrophages, aids with the phagocytosis of non-opsonized antigens (244). Macrophage activation due to S. pneumoniae’s presence is dependent on pattern recognition receptors (PRRs) (225). For example, toll-like receptors (TLRs) 2 and 4 work together to activate macrophages in the presence of pneumococci (225).
At birth, macrophage levels are low with impaired phagocytosis, cell signaling, and TLR4 (discussed in Section “Innate Immune Responses” iii) expression (237, 245). Within days post birth, macrophage levels and function improve to reach adult levels (15). By contrast, with old age alveolar macrophage concentrations are depleted, cytokine production and phagocytotic activity are reduced, and lowered expression of MARCO contributes to poor killing of S. pneumoniae (15, 28, 237).
iii. PRRs: these receptors can be found on host cell surfaces that recognize PAMPs (16, 219), PRRs can also be located intracellularly or be secreted (16). PAMPs are structures found in bacteria and viruses. Many of these are necessary for virulence in pathogens. There are two main types of receptors that participate in the host’s immune response to pneumococcus: TLRs and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) as described below.
a. TLRs: TLRs are mostly found on cell surfaces as membrane-bound molecules that recognize PAMPs (246). Recognition of PAMPs activates TLR signaling pathways that cause the recruitment of immune cells and cytokines production (247). There are currently 10 identified TLRs in humans (248). The main TLRs involved in pneumococcal infections are TLR2, TLR4, and TLR9. TLR2 is necessary in pneumococcal infection because it recognizes bacterial cell wall constituents. Former findings suggested that TLR2 recognized LTAs (16, 249, 250). However, TLR2 is now found to be binding to pneumococcal lipoproteins and peptidoglycan (225, 251, 252). TLR2 also has a role in transmission of pneumococci. Mouse models with deficient TLR2 had increased inflammation and shedding (253). TLR2 forms dimers with TLR1 and TLR6 which assist in the recognition of microbial antigens (246). TLR4 was the first TLR to be characterized and is needed for recognition of pneumococcal pneumolysin (51, 249, 254). On the other hand, TLR9 is intracellular and senses bacterial DNA within endosomes. TLR9 binds to CpG motifs (246) on the DNA, and when activated it also has signaling pathways which result in the release of cytokines (255, 256). TLR1, 2, 4, 6, and 9 work in a myeloid differentiation primary response 88 (MYD88)-dependent manner. MYD88 is an intracellular protein necessary for signal transduction and activation of TLR signaling pathways (246, 257). In addition to cytokine production, the activation of these TLRs facilitates the secretion of co-stimulatory molecules (246) which are necessary for activating T cells (258). Thus, the functions of these TLRs also play a role in adaptive immunity (Figure 9) (246, 255, 256).
Aging greatly affects TLR function. Expression of TLR1 is reduced with age (28). TLR4 expression appears to remain normal but experiences a reduction of function (15, 28). This association has been made in mice, due to macrophages having a lowered production of pro-interleukin-1B (15, 28, 259, 260). This also indicates TLR4’s inability to respond to pneumoccocal cell wall components (261, 262). Overall TLR cell signaling impairment causes a reduction in cytokines produced, leading to poor defense against S. pneumoniae (28).
b. NLRs: NLRs are intracellular proteins that can stimulate nuclear factor-kappa B (NF-κB) (263), control inflammation, and activate inflammasome formation (56, 264). NOD2’s role in pneumococcal infections has been thoroughly investigated (263–265). This NLR recognizes muramyl-dipeptide which is a fragment of bacterial peptidoglycan in the cytosol (16, 22). It promotes the production of cytokines and activation of nucleotide-binding domain and leucine-rich-repeat-containing protein 3 (NLRP3) genes (56). For example, when NOD2 senses peptidoglycan, CCL2 is made and that recruits macrophages and monocytes to the infection (266). This is dependent on the lysozyme producing these peptidoglycan fragments (266). NLRs expression decreases with age and responses to S. pneumoniae’s PAMPs are weakened (261). Lack of NLR expression may contribute to the chronic low pro-inflammatory state observed in the elderly (discussed in Section “Additional Immune Response Considerations”).
c. CD14: this has been characterized as a PRR as it recognizes LTA and other cell wall components (247, 267). CD14 works by interacting with other PRRs such as TLR4 for signal transduction (267). It has been reported that, in the case of pneumococcal infections, CD14 promotes growth and dissemination of the bacteria (22, 267). Previous studies have found CD14 to be beneficial and protective to hosts against Gram-negative infections, but as for Gram-positive pathogens such as pneumococci, it instead enhances the pathogenesis of the bacteria and facilitates infection (267).
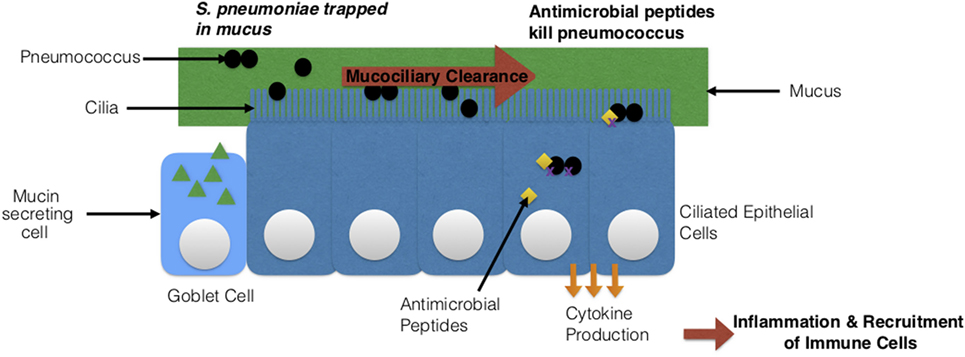
Figure 8. Streptococcus pneumoniae’s interaction with host epithelial cells. Two types of epithelial cells are depicted: goblet cells and ciliated epithelial cells. The cilia on the epithelial cells together with the mucus produced by goblet cells clear the pathogen via mucociliary clearance. Epithelial cells can also secrete antimicrobial peptides that directly kill S. pneumoniae or produce cytokines, which leads to a state of inflammation and the recruitment of immune cells.
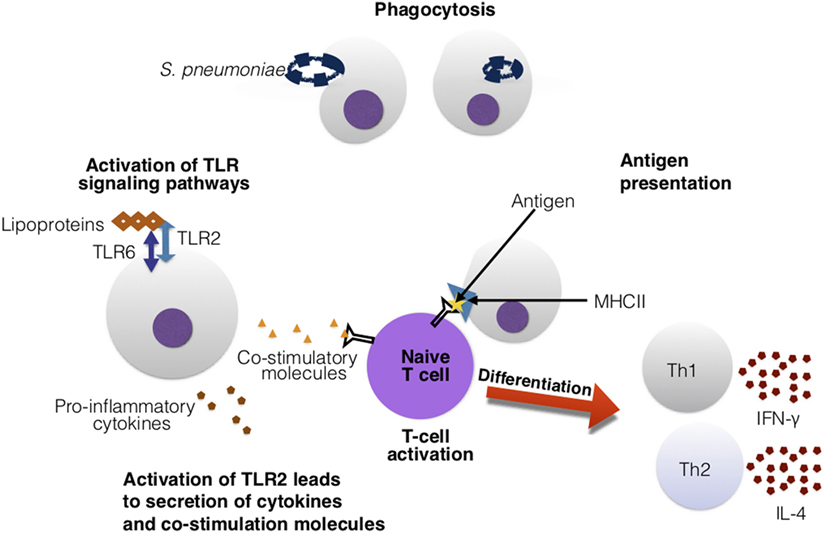
Figure 9. Toll-like receptors (TLRs) assist in the activation of adaptive immune cells. In this figure, TLR2 recognizes the Streptococcus pneumoniae’s lipoproteins. Upon activation, TLR2 secretes cytokines and co-stimulatory molecules. These co-stimulatory molecules are essential for co-stimulation and activation of T cells. The T cell is presented an antigen with major histocompatibility complex (MHC)II and antigen-presenting cell. The recognition of the antigen–MHCII complex and the co-stimulatory molecules activates the T cell and leads downstream to differentiation into Th1 and Th2 cells, that can release various cytokines such as interferon-gamma (IFN)-γ and interleukin (IL)-4.
Adaptive Immune Responses (B and T Cells)
Adaptive immune responses transpire a few to several days post-infection. The cells involved in adaptive immune responses respond to specific antigens from pathogens. Adaptive immunity can also be broken down into two types of responses: humoral and cell-mediated (268). Humoral immunity involves B cells that are activated by antigens, and production of antibodies that are specific to antigens. Cell-mediated immunity also involves T cells, including T cell activation and T cell-mediated recruitment, which involves the activation of other immune cells that can directly kill pathogenic cells (268).
These immune cells are formed in the bone marrow—B cells mature in the bone marrow into plasma cells that make antigen-specific antibodies (268). Infections at mucosal sites are controlled by the pneumococcal specific IgA antibody. IgA is observed in mucosal areas of the nose and saliva following S. pneumoniae colonization (269). Secretory IgA is important for opsonizing S. pneumoniae and promoting phagocytosis (230). S. pneumoniae on the other hand, possess an IgA1 protease the cleaves the IgA (discussed in Section “Virulence Factors”). This blocks opsonization. Following cleaving, the remaining Fab fragment binds to the cell wall (230, 270). This exposes CBPs, decreases negative charge of the capsule and increases cell adhesion (230, 270). Studies suggest that the Fab neutralizes the negative charge of the capsule and instead promotes cell adhesion (230, 270). Furthermore, the complement (C3) activates B cells. Following antigen stimulation, the naïve B cells differentiate into IgM+ memory B cells. Class switching produces other immunoglobulins needed for clearing the infection (269).
T cells instead migrate to the thymus for maturity into mature helper (CD4+) and cytotoxic (CD8+) T cells (268). Antigen-presenting cells (APCs) paired with the major histocompatibility complex (MHC) proteins present antigens (specifically, peptides) to T cells to stimulate an immune response (268). In pneumococcal infection, CD4+ T cells are stimulated via co-stimulatory molecules and APCs. Upon activation, helper T cells differentiate into Th1 and Th2 cells (Figure 9). Th1 helper cells stimulate a cellular-mediated immune response by producing cytokines such as interferon-gamma (IFN-γ), that activate and recruit other immune cells such as macrophages (271). Th2 helper cells release IL-4 cytokines, and are geared toward facilitating a humoral immune response (271) by interacting with B cells and aiding in antibody production (268). Cytotoxic T cells directly kill infected cells (268). Furthermore, upon activation of T and B cells, they can differentiate into memory B and T cells that can provide quicker clearance in reoccurring infection (268). Similarly, natural killer T-cells are also important for clearance of pneumococci (16, 22). More specifically, CD4+ T cells have been found to provide protection to S. pneumoniae in an antibody-dependent manner (272). T-helper 17 (Th17) and regulatory T cells (Tregs) are also very important for pneumococcal infections. Th17 cells release the cytokine IL-17 which is pro-inflammatory. IL-17 is needed for recruiting and activating macrophages, monocytes, and neutrophils to sites of infection and promotes clearance of S. pneumoniae (273). Increased production of IL-17 has been connected to reduced S. pneumoniae density in the nasopharynx of mice and children (273). Tregs are necessary for regulating Th17’s production of IL-17. Imbalance between Tregs and Th17 cells can lead to autoimmune disease due to over inflammation (273).
Infants experience poor T cell responses to foreign antigens because their exposure to non-maternal antigens was restricted prior to birth (237). Infants also display a skewed Th2 response to foreign antigens. To compensate for this, infants have a population of γδ T cells that generate IFN-γ, to provide a Th1 type response (237). As for B cells, in infants there is a limited response to antigens due to low expression of co-receptors (237). Infants also experience incomplete class switching for immunoglobulins and lower somatic hypermutations compared to adults (237). Immunoglobulin protection against S. pneumoniae’s capsular polysaccharides is developmentally regulated. At birth, maternal IgG antibodies protect infants until 27 days of age (based on the half-life of IgG) (28, 274). Once maternal antibodies have been depleted, the infant’s ability to protect itself via steady antibody generation experiences a delay until age two (28). By contrast, IgM has been detected in infants following S. pneumoniae infection and carriage (28, 275, 276). Encountering the pathogen again, also promotes antibody production similar to booster effects in vaccines (discussed below) (277). With development, the adaptive immune cells mature, develop memory, and the incidence of S. pneumoniae infections decrease.
In elderly populations, the efficacy of the adaptive immune cells diminishes. Aging leads to reduced production of antibodies, immunoglobulin class switching, and cell maturation, which promotes S. pneumoniae’s colonization (275). Antibodies specific for capsular polysaccharides decrease with age (275). In addition, there is an overall reduction in naïve T cells with age due to thymus involution (278). Previously Th17 levels were described to increase in elderly populations whereas most recently, in 2014, van der Geest et al. showed lower Th17 concentrations and increased concentrations of memory Tregs (15, 279). The ratio between CD4+ and Treg cell populations was also reported to increase toward more Tregs (15, 279). Diminished responses from the adaptive immune cells explain the higher incidence rates of pneumococcal diseases in these high-risk age groups.
Additional Immune Response Considerations
Chemokines and Cytokines
These are signaling molecules released by innate and adaptive immune cells and receptors to direct other immune cells to the infected tissues (242, 268). Chemokines are examples of cytokines that attract cells to the infected site. In addition to recruiting cells, they promote inflammation (242, 268). TNF-α, a well-studied pro-inflammatory cytokine, inhibits growth and dissemination of pneumococci (22). Together TNF-α and IFN-γ can enhance clearance of pathogens by activating phagocytes. T cells, monocytes, and macrophages produce TNF-α (158). The phagocyte-activating cytokines are suggested to be inhibited by autolysin activity in pneumococci (158).
The elderly experience chronic low-grade age-associated inflammation (inflammaging) (28). This involves constant low levels of pro-inflammatory cytokines such as TNF-α and IL-6. The inflammatory state of the elderly is worsened due to increased NF-κB activation and the secretion of pro-inflammatory cytokines such as TNF-α from senescent cells (28). High concentrations of TNF-α have been correlated with higher disease incidences (28). Inflammaging induces the expression of host proteins which enhances S. pneumoniae adhesion, and is often accompanied by other morbidities that increase risk of S. pneumoniae infections (28).
Inflammasome
This is a protein complex that consists of a sensor protein, caspase 1, and an apoptosis-associated Speck-like protein with a caspase recruitment domain (ASC) (280). The inflammasome is used by the host for indirectly recognizing bacterial or pathogenic molecules and DNA (56). Upon recognition, the inflammasome regulates cytokine production (56). NLRP3 plays a role in identifying pneumococcal infection, activating macrophages and has been shown to directly interact with pneumolysin during pneumococcal infection (225). Pneumolysin can directly activate NLRP3 (281), and when activated, inflammasomes secrete IL-1β and IL-18 (282). Although inflammasomes may aid in the recognition of pathogens, the activation of inflammasomes promote inflammation and this can be harmful to the host (56).
Cho et al. studied the effects of aging on NLRP3’s activation in mice (261), and reported that enhancement in ER stress with age leads to decreased NLRP3 inflammasome activation in S. pneumoniae infection. Ensuring that NLRP3 is activated appropriately in the elderly population will promote stronger immune defenses against S. pneumoniae.
Complement System
This is comprised of a set of small proteins that enhance the ability of antibodies and phagocytic cells to clear microbes and damaged cells (219). These proteins can mark antigens and cells by coating them with opsonins (16, 268). Complement activation involves three cascade pathways: classical, mannose-lectin, and alternative pathways. In the classical pathway, the complement proteins bind to an antibody-antigen complex (159), whereas the alternate and mannose-lectin pathways, bind directly to PAMPs and cell surface components. Which pathway plays the main role in response to pneumococcus has been debated. Brown et al. in 2002 stated that the classical pathway is most important in response to pneumococcal infection following investigations of complement pathways deficient mice (283). On the other hand, in 2012 Ali et al. stated that the mannose-lectin pathway is more important, after following the use of mannose-lectin pathway in deficient mice that could still use the classical and alternate pathways showed susceptibility to pneumococcal infection (284). The importance of the different complement pathways’ role in pneumococcal infections needs to be further investigated. However, irrespective of the specific pathway, the complement proteins also help to fight infections by pathogens such as pneumococcus by promoting inflammation, attacking pathogens, and rupturing their cell walls (16, 22, 219). For example, mice deficient in complement C3 infected with S. pneumoniae were unable to clear the infection and had short survival times in comparison to mice with complement C3 (285). S. pneumoniae can evade the host complement system in many ways—most of which were previously discussed in the Section “Virulence Factors.” Pneumolysin is able to divert the complement system away from S. pneumoniae by directly activating the classical complement pathway (285). PspA inhibits C1q binding and polyhistidine triad proteins are suggested to degrade C3. S. pneumoniae’s complement evasion has been detailed by Dockrell and Brown (225).
The effects of aging on the complement system are complex. Previous studies suggest that complement levels are low in infants (286, 287). In 2014, Grumach et al. also showed that in newborns complement activity is low with C1, factor H, and C3a levels being lower than adult levels (287). Studies have also indicated that complement activity is greater in the elderly compared to young adults (15, 241, 288).
Acute Phase Serum Proteins
These proteins increase in concentration within the blood during an acute inflammatory infection (289). The three main proteins that have been investigated and associated with pneumococcal infection include C-reactive protein (CRP), serum amyloid P (SAP), and mannose-binding lectin (MBL) (289). These proteins work to alleviate infections and can recognize and bind to bacterial surfaces (289). Acute phase proteins are made as a result of cytokine production from innate cells such as macrophages (289). For example, CRP production by the liver is increased in response to IL-6 (289). CRP and SAP bind to PCho which is part of the S. pneumoniae’s cell wall. Once bound to the PCho, CRP and SAP activate the complement deposition on the bacteria via the classical pathway (290). As for MBL, there are conflicting reports about its role in pneumococcus infection as discussed above in the description of the complement system. It has been shown to recognize and attach to sugars on the cell surface of S. pneumoniae (291), but more verification is needed on MBL’s role in pneumococcal infection.
Diagnosis, Age-Dependent Response, Prevention, and Disease Prognosis
Diagnosis
Currently, there are several methods utilized in pneumococcal disease diagnostics. Traditionally, diagnosis begins with physicians performing a physical exam. For example, in the case of an ear infection, an otoscope (24) is used to confirm infection, whereas for pneumonia physicians monitor breathing for cracking sounds and wheezing (1, 292, 293). More specifically, for pneumonia, based on the results of the physical exam, physicians can conduct a chest X-ray to examine the lungs and monitor inflammation to confirm the presence of infection (292, 293). This X-ray is also performed following signs of respiratory distress (293, 294). Blood oxygen levels are also measured via pulse oximetry in both children and adults to assess the severity of the infection (1, 292, 293). Pulse oximetry at the primary care level should be the future, and future technological developments might add respiratory rate and work of breathing to the parameters measured by oximetry (295). Body fluids are also processed to assess whether or not pneumococcus is present, and to confirm its identity (1, 292–294, 296). These fluids include blood, urine, cerebrospinal fluid, and saliva (1, 292–294, 296). The blood test allows physicians to examine complete blood cell count. This test confirms whether or not an infection is present by giving an estimate of the percentage of WBC that are circulating (1, 292–294, 296). A large concentration of WBC is indicative of an infection (1, 292–294, 296). This is expected in infection response. However, Gardner et al. in 2017 indicated that upon the time of admission, about 25% of subjects with pneumococcal pneumonia and roughly 38% with CAP actually have normal WBC counts (297). Studies have also shown that poor prognosis has been associated with low WBC (297). New findings associate low WBC rather than high WBC with poor prognosis (297). These conflicting results indicate that WBC count alone should not be used to diagnose pneumonia and should be better investigated as key indicator of pneumonia.
Bacterial cultures and Gram-staining tests using body fluids are important for determining the strain of bacteria and confirming its identity (1, 292–294, 296). Currently, physicians are investigating other means of diagnosing pneumococcal infections due to the poor yield and quality of sample when conducting cultures. This process is also dependent on bacterial growth which can be time consuming. One useful tool that is being developed is the urinary antigen detection test (298), which is only currently used in adults. This test monitors the levels of the C-polysaccharide antigen of pneumococcus in the urine. It appears to be quicker, can allow for targeted treatment with better results than culture-based methods of diagnosis (1, 292, 293, 298). In addition to testing for pneumococcus, physicians also test for other bacteria which may be causing the infection, and other viruses such as influenza which can coinfect patients (293). Once all the tests confirm the presence of an infection, the cause of the infection and the severity of the disease patients are treated accordingly.
Currently, thoracic ultrasounds are being investigated as a method for diagnosing CAP (299). When compared to chest X-rays, thoracic ultrasounds identified 73.5% of the lung consolidations confirmed by chest X-rays, with about 27% false negative results. D’Amato et al. suggest using ultrasound as a monitoring tool. Lung ultrasound has been tested for its diagnostic potential and it was found to be a sensitive tool for confirming CAP in children (300). 96% children with pneumonia were detected, however, given the small sample size, further investigation is necessary. Chest computed tomography is not used for children due to radiation (300). Recently, a computer-aided differential diagnosis system was tested for distinguishing types of pneumonia, using high-resolution computed tomography. This method was compared to radiologists’ classification of interstitial and non-specific interstitial pneumonia, and was concluded to be a robust method for diagnostics (301). In addition, researchers have proposed combining clinical signs and laboratory markers to assess an individual’s risk of contracting pneumonia. For example, high levels of CRP and procalcitonin accompanied by unilateral hyperventilation and grunting were associated with pneumonia (302). On the other hand, children with no clinical signs of pneumonia and low CRP results were at a lower risk for pneumonia. The use of PCR for diagnosis is also being developed. A positive blood pneumococcal PCR can more accurately confirm the diagnosis of pneumonia (302). PCR has been used to detect pneumolysin in whole blood samples (303). The sensitivity of PCR tests varied from 68 to 100% and had poor specificity (303). By contrast, assessment of quantitative real-time PCR indicated that it is more successful in achieving greater speed, specificity, and sensitivity compared to multiplex PCR (304).
Prevention, Antibiotic Response, and Age-Dependent Immune Responses
The two main modes of preventing pneumococcal infections are using antibiotics and vaccinations against pneumococcus (24). Antibiotics are essential in reducing bacterial load (305). Such treatment can work by killing the bacteria or hindering their growth (305). The first antibiotic to be created was penicillin which was discovered in 1928 by Alexander Fleming (306), and antibiotics have been used widely since. However, misuse of antibiotics can cause bacteria to become resistant (39, 305, 307). Resistant bacteria are then able to survive post antibiotic treatment and they can grow, multiply, and share antibiotic-resistant genes with each other. Pneumococcal strains that were penicillin-resistant were first recorded in the 1970s (22). Currently, penicillin-resistant strains have spread worldwide with pneumococcus also being resistant to other types of antibiotics: erythromycin, tetracycline, and chloramphenicol (305). S. pneumoniae acquires multiple antibiotic resistance genes via transformation and evolution with the increase in antibiotic use (308). Mutations in penicillin-binding proteins (pbp) affect binding of penicillin which acts by blocking cell wall synthesis (309). Erythromycin resistance gene erm(B) blocks the binding of macrolides (antibiotics targeting protein synthesis) and mefA and mefE genes produce an efflux pump which regulates entry of the antibiotics (308–312). Resistant S. pneumoniae strains have rapidly spread, and infections are harder to treat. In 2013, the CDC estimated that about 30% of pneumococcal cases were due to antibiotic resistance to one or more antibiotics (307). This resistance increases the number of doctor visits and hospitalizations (307). For example, the CDC reports that the resistance can lead to 1,200,000 more illness and 7,000 deaths annually (307). This reduction in ability to treat and clear the pathogen led to the development of vaccines that would provide protection prior to infection and thus reduce the need for antibiotics (305).
Currently, there are two types of inactivated vaccinations that protect against S. pneumoniae (313–315). The pneumococcal polysaccharide vaccine 23 (PPSV23) (316) uses purified capsular polysaccharides and is routinely given to adults who are 65 and older (313–315). It protects against 23 serotypes of S. pneumoniae and is effective in 50–70% of cases in adults (126). This vaccine works in a T-cell independent manner. The polysaccharide antigens are recognized by B cells which differentiate into plasma cells that produce antibodies specific for the polysaccharide antigens (317). PPSV23 provides T-cell-independent immunity and requires revaccination 5 years after the first vaccination because the immunity is transient (316, 318). The pneumococcal conjugate vaccine (PCV) (319) was developed after noticing the low efficacy and poor immunogenicity of PPSV23 in infants and young children (91, 320). In the conjugate vaccine, the purified polysaccharides covalently conjugated to a carrier protein, specifically CRM197 (318, 319). The current FDA approved conjugate vaccine is PCV13 which protects against 13 serotypes of the S. pneumoniae (319). PCV13 replaced PCV7 in 2010 and protects against six additional serotypes (321). This elicits a T-cell-dependent response which provides mucosal immunity and immunologic memory in children (126). PCV13 provides long-lasting immunity by causing B and T cells to interact (317). B cells recognize and process the carrier protein (317). The MHCII needed for antigen-presentation to T cells, binds to the peptide produced following B cell breakdown of the carrier protein (317). The peptide is presented to the T cells by MHCII at the surface of the APC, providing co-stimulation necessary for producing plasma cells and memory B cells (317). The use of this vaccine has led to a decrease in pneumonia cases in young children by more than 90%, and is most effective in children younger than five (126).
When it comes to high-risk individuals, the CDC recommends the prime-boost method of vaccination. This involves priming the immune system to a specific antigen, and enhancing this antigen-specific immune response by re-administering the antigen (322). The prime-boost strategy increases immunity to antigens and is recommended for high-risk individuals (323, 324). There are two ways to prime and boost the immune system: homologous, in which the same vaccine is received twice, and heterologous, which utilizes different types of vaccines (322). The heterologous method has been shown to be more immunogenic (325). Currently, children and adults who are at high risk for pneumococcal disease and have pre-existing conditions undergo the prime-boost strategy prevention by receiving the PCV13 followed by the PPSV23 (326, 327). This is also due to the poor immunological response seen in HIV patients who receive the PPSV23. Prime-boost vaccinated HIV-infected groups have been shown to be more likely to display a twofold increase in IgG geometric mean concentrations (328). PCV13 provides a longer and stronger level of protection against S. pneumoniae (323). Within 4–8 weeks, PCV13’s IgG levels can equal or exceed PPSV23 in high-risk individuals (323, 324).
Despite the availability of pneumococcal vaccines, it is important to note that these vaccinations are both serotype and age dependent (23, 91, 313–315, 320). Understanding the role that age plays in host immune system activation is essential for better prognosis and treatment of diseases. As stated previously, young children and the elderly are at higher risk for contracting pneumococcal diseases (237). This is due to immunosenescence within the elderly population, whereas for infants, it is due to their underdeveloped immune systems (237). In addition to age recommendations, the CDC also recommends the use of either vaccine in high-risk individuals with pre-existing health conditions. For example, both vaccines are recommended in young children and adults ages 19–64 with pre-existing health conditions (23, 24, 91). PPSV23 is also recommended by the CDC for use in adults that smoke or have asthma (24). These vaccine recommendations are reevaluated regularly based on vaccine efficacy and changes within the bacteria serotypes (326, 329).
Vaccines have drastically reduced invasive pneumococcal diseases, especially CAP in young children and adults (Table 2) (18). However, these vaccines have pitfalls. First, there have been at least 97 serotypes identified but these vaccines protect against 14–25% of these. The current vaccines only protect against S. pneumoniae serotypes that are mainly associated with causing the disease. Some studies suggest that there is little evidence that PPSV23 protects against non-invasive pneumococcal diseases, which are more prevalent in adults (330). The CDC also confirms that PPSV23’s efficacy in non-bacteremic pneumonia has led to contradicting findings, but nevertheless, it has shown sufficient efficacy in invasive pneumococcal diseases (331). Weinberger et al. discuss the challenges of vaccinating the elderly with PCV13 and PPSV23. These researchers argue that PPSV23 does not show a real benefit to the elderly (330). As for, PCV13 they argue that it is already used in children and thus adults should be partially protected from serotypes in PCV13 due to herd immunity (330). They also state that herd immunity should provide partial protection and thus will lead to reduction of efficacy of PCV13 (330). Other studies also discuss herd immunity from PCV13 due to infants and toddlers being vaccinated (332, 333). Due to PCV13, disease serotypes rates within this vaccine will decrease by 50% (332, 333). This becomes a problem because of serotype replacement. The serotypes that are not in the vaccine can colonize young children and spread to adults (334, 335). In addition, with serotype vaccines, the serotypes that are popular and commonly cause CAP and other diseases may not necessarily do so in the future and so these vaccines would need to be reevaluated. PCV13’s replacement of PCV7 was a prime example of changes to serotypes that cause pneumococcal diseases. Recently researchers at Merck Sharp and Dohme Corp. completed a phase 1 clinical trial (NCT01215175) investigating a new conjugate vaccine, PCV15, immunogenic, and safety properties compared to PCV13 (336). This contains two extra serotypes (22F and 33F), which were previously identified for the cause of approximately 10% of invasive pneumococcal diseases in adults in 2007 (337). Another concern for current vaccines is that 3–19% of pneumococcal diseases are due to non-encapsulated S. pneumoniae (38). Current vaccines are ineffective against non-encapsulated S. pneumoniae due to serotype specificity (38). Further developments of vaccinations are vital for eliminating the burden of S. pneumoniae and reduce the number of infections.
Post-Infection Prognosis
Following pneumococcal diseases such as pneumonia, high-risk individuals may experience longer recovery times and complications due to the disease (1, 292, 293). About 1.6 million deaths from pneumococcal diseases occur worldwide (36). According to the CDC, there were over 50,000 deaths within the US during 2014 (29) and the majority of these deaths were seen in the elderly (29). Older adults have lower survival rates than other age groups (94, 338). The elderly may recover from pneumococcal diseases such as CAP, but they face higher death rates due to the high possibility of developing other health problems and the reoccurrence of the disease (1, 94, 338). Infants and young children who recover from CAP have an increased risk for developing respiratory problems (339). For example, research indicates that young children face a greater risk for reduced lung function and developing chronic obstructive pulmonary disease (44, 339). In some cases, increased death rates and complications are due to delays in diagnosis. Such delays in turn hamper timely treatment, which also increases the severity of the disease. For example, meningitis can progress quickly and cause permanent disabilities such as brain damage, hearing loss, and seizures (24, 52, 53). Timely treatment can reduce the risk of neurological damage and death due to this infection (53). In addition, ear and sinus infections can lead to hearing loss and respiratory problems respectively (24). The environment also plays a role in affecting recovery rates and reoccurrence of the disease especially for smokers and those residing in nursing homes and crowded areas (1). Furthermore, Tiewsoh et al. study investigating the outcome of children with severe pneumonia showed that children who were not breastfed had a low birth weight and were within crowded homes had longer hospital stays and the initial antibiotics were not helpful and required new antibiotics (340). Nutrition also plays a vital role in how well someone will recover from these diseases (341).
Some complications due to pneumococcal pneumonia include respiratory failure, lowered oxygen levels, and collapsed lungs (4). It is also possible for the lungs to fill with fluid and this fluid can become infected. S. pneumoniae may also migrate to the blood (4, 24). This is called bacteremia which is the most common complication for pneumonia (4, 24). Pneumonia and other pneumococcal diseases are classified as invasive if the bacteria migrate to the blood. In addition, individuals with this disease can develop pericarditis which is inflammation of the sac around the heart, lung abscess, empyema, and blockage of airways (4, 24). It is also highly probable for coinfections to occur when suffering with pneumonia. An example of this is influenza—66% of CAP cases also present coinfection with influenza (113). Most of these health complications are seen in elderly subjects, and this also points to the increasing importance of improved diagnostics, treatments, and vaccinations for this age group.
Discussion
Pneumococcal diseases cause millions of deaths worldwide (36, 97). In this review, we have characterized S. pneumoniae, explored its virulence factors, and how hosts respond to its presence. We have discussed the host defenses against S. pneumoniae, and how individuals with weakened immune systems may experience a harder time clearing the pathogen. We have also indicated that young children, elders, and individuals who are immunocompromised all have an increased risk for contracting pneumococcal diseases. The majority of previous efforts have provided an extensive characterization of S. pneumoniae features and began probing the interactions of the bacteria with the host in the context of pneumococcal disorders. However, in terms of treatment and prevention there remain substantial open questions that need to be addressed as discussed below.
There are a variety of methods available for pneumococcal disease diagnostics. Many of the current tests needed to confirm S. pneumoniae’s identity are culture-dependent (1, 292, 293). Culture-independent methods that take advantage of the latest technologies are being developed, such as the use of a lung ultrasound to assess pneumonia (342). Chavez et al. and Long et al. discuss the possibility of lung ultrasound use in pneumonia diagnosis (343, 344) indicating high diagnostic accuracy, while at the same time providing a radiation free method of examining the lungs (343, 344). Similarly, the use of mass spectrometry to examine metabolites from the saliva (345), breath (346), and urine (298, 347) of patients being tested for pneumococcal diseases is under development. The urine antigen test discussed above also provides rapid results that will allow for quicker diagnosis and treatment once S. pneumoniae antigens are detected in the urine (298, 347).
With diagnostic methods improving, pneumococcal disease treatments are also being updated. Antibiotics are available to reduce the colonization of S. pneumoniae, however, the efficacy of antibiotics is being reduced due to the increase in antibiotic resistance (305, 307). Broad-spectrum antibiotics are no longer as effective (305, 307). Inhaled therapeutics are underdeveloped but can be beneficial for treating pneumonia and other pneumococcal diseases. This method can provide a mode of delivering antibiotics and antimicrobials (348) in a more targeted manner, improve mucociliary clearance via hypertonic saline solutions (348) and inhalation of cytokines to stimulate the immune system (348). On the other hand, to also reduce the effect of antibiotic resistance, S. pneumoniae strains may also be studied via RNA-sequencing and other high throughput technologies to detect antibiotic resistance genes and thoroughly characterize serotypes.
Treatment and prevention of pneumonia and other pneumococcal diseases are of major concern for the clinical field due to the high death rates and low efficacy of current vaccines due to aging differences and serotype replacement. Some alternative vaccination methods have been proposed and are also being developed. For instance, Weinberger et al. propose the use of a conjugate vaccine that is specific for elderly subjects, which targets the serotypes not in current vaccines but other serotypes that are mostly seen in elderly patients with pneumococcal diseases (330). Some researchers have proposed creating a conjugate vaccine that targets all or more of the identified serotypes of S. pneumoniae (330, 349). However, the impact on the immune system and immunogenicity of this vaccine would need to be thoroughly investigated (330). This vaccine would also need to demonstrate better efficacy than existing vaccines (330). In addition to this, conjugate vaccines are expensive [currently, the PCV13 costs about $160 per dose (350)], and a true benefit will need to be clearly identified. In addition, observing how pneumococcal disease incidence rates are changing as more and more people are getting vaccinated will lead to accurate assessment of pneumococcal disease burden and vaccine efficacy (341). Vaccination policies and cost-effect analyses can benefit from information on vaccine disease reduction (341).
Serotype-independent vaccines are also being investigated. These include protein, protein and polysaccharide combination, and whole cell vaccines (330, 349, 351, 352). Protein vaccines would contain surface proteins that are highly conserved in S. pneumoniae (353, 354). For example, PspA and inactivated pneumolysin have been tested in phase 1 clinical trials as protein antigens (354). They both demonstrated safety (354), but PspA antigen’s immunogenicity was low (355) whereas the inactivated pneumolysin was found to be immunogenic and effective in eliciting protective immune response (356). PspA is considered an ideal protein candidate because reports indicate that PspA family 2 is commonly found in S. pneumoniae strains (357). For example, in Pakistan most strains of pneumococci have PspA genes (357). These protein vaccines can provide an extra preventative method once developed and will require thorough analysis of regulation and what regulatory issues may be faced (358). In addition, as a form of combination therapy, a vaccine with protein antigens as well as conjugated polysaccharide antigens may also provide a broader range of protection against pneumococcal diseases (353, 354). On the other hand, whole cell vaccinations would introduce a dead S. pneumoniae cell to hosts with the potential to provide broader protection to S. pneumoniae (359, 360). HogenEsch et al. investigated the use of whole cell vaccines in mice by using a capsule deficient and autolysin mutant cell (359). This exposed the host to multiple parts of S. pneumoniae. They found that the vaccine led to the productions of antibodies and IL-17 which defend against S. pneumoniae colonization of the nasopharynx in mice (359). Researchers have also started developing live attenuated pneumococcal vaccines (361, 362). The SPY1 strain is a live attenuated strain of pneumococci that does not have a capsule (362). Xu et al. in 2015 experimented with delivering this vaccine intranasally in mice and observed that it elicited a humoral response (362). More recently, Zhang et al. added a mineralized shell to SPY1 to improve its stability and test if it can elicit a stronger immune response (361). The modified strain (SPY1ΔlytA) also did not have autolysin activity (361). This modified SPY1 vaccine led to higher stability, more production of IgG, and an overall increase in protection when compared to the SPY1 vaccine (361). Additional concerns of serotype-independent vaccines include determining if the vaccines will be immunogenic in all ages, whether or not the vaccines would elicit a strong immune response, and ensuring that they can induce a pro-inflammatory state while not leading to an over activation of the immune system. All of these novel methods show great promise, but they require further assessments.
Overall, there has been progress in our understanding of pneumococcal diseases over the last three decades, however, the diseases still constitute a big burden on health care. There has been a great decrease in pneumococcal diseases since the implementation of purified polysaccharide and polysaccharide conjugate vaccines, but over time due to serotype replacement, antibiotic resistance, and changes in immunity with age, the treatments, and vaccines in place may prove ineffective. Therefore, ongoing research to improve vaccinations and treatments must continue toward alleviating the ill effects of S. pneumoniae.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
LB is funded through a University Enrichment Fellowship at Michigan State University. GM is funded by Jean P. Schultz Endowed Biomedical Research Fund and previously the National Human Genome Research Institute of the National Institutes of Health under Award Number R00 HG007065. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
1. Mandell LA, Wunderink RG, Anzueto A, Bartlett JG, Campbell GD, Dean NC, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis (2007) 44(Suppl 2):S27–72. doi:10.1086/511159
2. Wolfe ND, Dunavan CP, Diamond J. Origins of major human infectious diseases. Nature (2007) 447(7142):279–83. doi:10.1038/nature05775
3. Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis (2009) 48(1):1–12. doi:10.1086/595011
4. Mayo Clinic Staff. Infectious Diseases. Mayo Foundation for Medical Education and Research (2016). Available from: https://www.mayoclinic.org/diseases-conditions/infectious-diseases/symptoms-causes/syc-20351173 (Accessed: March 26, 2017).
5. Dasaraju PV, Liu C. Chapter 93 – Infections of the respiratory system. In: Baron S, editor. Med Microbiol 4th ed. Galveston, TX: University of Texas Medical Branch (1996).
6. GBD 2015 LRI Collaborators. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory tract infections in 195 countries: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect Dis (2017) 17(11):1133–61. doi:10.1016/S1473-3099(17)30396-1
7. Institute for Health Metrics and Evaluation (IHME). GBD Compare Data Visualization. Seattle, WA: IHME, University of Washington (2017). Available from: http://vizhub.healthdata.org/gbd-compare (Accessed: May, 2018).
8. Bandaranayake T, Shaw AC. Host resistance and immune aging. Clin Geriatr Med (2016) 32(3):415–32. doi:10.1016/j.cger.2016.02.007
9. Bogaert D, de Groot R, Hermans PWM. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis (2004) 4(3):144–54. doi:10.1016/S1473-3099(04)00938-7
10. Burel JG, Apte SH, Doolan DL. Systems approaches towards molecular profiling of human immunity. Trends Immunol (2016) 37(1):53–67. doi:10.1016/j.it.2015.11.006
11. Castelo-Branco C, Soveral I. The immune system and aging: a review. Gynecol Endocrinol (2014) 30(1):16–22. doi:10.3109/09513590.2013.852531
12. Pinti M, Appay V, Campisi J, Frasca D, Fulop T, Sauce D, et al. Aging of the immune system – focus on inflammation and vaccination. Eur J Immunol (2016) 46(10):2286–301. doi:10.1002/eji.201546178
13. Aw D, Silva AB, Palmer DB. Immunosenescence: emerging challenges for an ageing population. Immunology (2007) 120(4):435–46. doi:10.1111/j.1365-2567.2007.02555.x
14. Caruso C, Buffa S, Candore G, Colonna-Romano G, Dunn-Walters D, Kipling D, et al. Mechanisms of immunosenescence. Immun Ageing (2009) 6:10. doi:10.1186/1742-4933-6-10
15. Goncalves MT, Mitchell TJ, Lord JM. Immune ageing and susceptibility to Streptococcus pneumoniae. Biogerontology (2016) 17(3):449–65. doi:10.1007/s10522-015-9614-8
16. Paterson GK, Mitchell TJ. Innate immunity and the pneumococcus. Microbiology (2006) 152(Pt 2):285–93. doi:10.1099/mic.0.28551-0
17. Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol (2008) 6(4):288–301. doi:10.1038/nrmicro1871
18. Centers for Disease Control and Prevention. Chapter 11 – Pneumococcal. In: Roush SW, Baldy LM, editors. Manual for the Surveillance of Vaccine-Preventable Diseases. Atlanta, GA: Centers for Disease Control and Prevention (2017). Available from: https://www.cdc.gov/vaccines/pubs/surv-manual/chpt11-pneumo.html (Accessed: March 19, 2018).
19. Centers for Disease Control Prevention. Active Bacterial Core Surveillance Report, Emerging Infections Program Network, Streptococcus pneumoniae. (2017). Available from: https://www.cdc.gov/abcs/reports-findings/survreports/spneu16.html (Accessed: March 24, 2017).
20. Centers for Disease Control and Prevention. Active Bacterial Core Surveillance Report, Emerging Infections Program Network, Streptococcus pneumoniae. Atlanta, GA: Centers for Disease Control and Prevention (2015). Available from: https://www.cdc.gov/abcs/reports-findings/survreports/spneu15.html (Accessed: May 30, 2018).
21. Centers for Disease Control Prevention. ABCs Report: Streptococcus pneumoniae. Centers for Disease Control and Prevention (2010). Available from: https://www.cdc.gov/abcs/reports-findings/survreports/spneu10.html (Accessed: February 18, 2017).
22. van der Poll T, Opal SM. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet (2009) 374:1543–56. doi:10.1016/S0140-6736(09)61114-4
23. Centers for Disease Control and Prevention. Prevention of pneumococcal disease: recommendations of the advisory committee on immunization practices (ACIP). MMWR Morb Mortal Wkly Rep (1997) 46(RR–8):1–24.
24. Centers for Disease Control Prevention. Pneumococcal Disease. (2015). Available from: https://www.cdc.gov/pneumococcal/index.html (Accessed: March 20, 2017).
25. Blumental S, Granger-Farbos A, Moisi JC, Soullie B, Leroy P, Njanpop-Lafourcade BM, et al. Virulence factors of Streptococcus pneumoniae. Comparison between African and French invasive isolates and implication for future vaccines. PLoS One (2015) 10(7):e0133885. doi:10.1371/journal.pone.0133885
26. Feldman C, Anderson R. Epidemiology, virulence factors and management of pneumococcus. F1000Research (2016) 5(F1000 Faculty Rev):2320. doi:10.12688/f1000research.9283.1
27. Jedrzejas MJ. Pneumococcal virulence factors: structure and function. Microbiol Mol Biol Rev (2001) 65(2):187–207. doi:10.1128/MMBR.65.2.187-207.2001
28. Infante AJ, McCullers JA, Orihuela CJ. Chapter 19 – mechanisms of predisposition to pneumonia: infants, the elderly, and viral infections. In: Brown J, Hammerschmidt S, Orihuela C, editors. Streptococcus pneumoniae. Amsterdam: Academic Press (2015). p. 363–82.
29. Centers for Disease Control and Prevention. Fast Stats: Leading Causes of Death. Atlanta, GA: Centers for Disease Control and Prevention (2013). Available from: https://www.cdc.gov/nchs/fastats/leading-causes-of-death.htm (Accessed: February 7, 2017).
30. Centers for Disease Control and Prevention. Influenza (Flu): Flu Vaccines & Preventing Flu Illness. Atlanta, GA: Centers for Disease Control and Prevention (2016). Available from: https://www.cdc.gov/flu/faq/preventing-flu-illness.htm (Accessed: February 7, 2017).
31. American Lung Association. Trends in Pneumonia and Influenza Morbidity and Mortality. American Lung Association Epidemiology and Statistics Unit Research and Health Education Division (2015). Available from: http://www.lung.org/assets/documents/research/pi-trend-report.pdf (Accessed: February 7, 2017).
32. Bridy-Pappas AE, Margolis MB, Center KJ, Isaacman DJ. Streptococcus pneumoniae: description of the pathogen, disease epidemiology, treatment, and prevention. J Hum Pharmacol Drug Ther (2005) 25(9):1193–212. doi:10.1592/phco.2005.25.9.1193
33. Centers for Disease Control and Prevention. Epidemiology and Prevention of Vaccine-Preventable Diseases. Washington, DC: Public Health Foundation (2011). 12 p.
34. Geno KA, Gilbert GL, Song JY, Skovsted IC, Klugman KP, Jones C, et al. Pneumococcal capsules and their types: past, present, and future. Clin Microbiol Rev (2015) 28(3):871–99. doi:10.1128/CMR.00024-15
35. White B, Robinson ES, Barnes LA. The Biology of Pneumococcus: The Bacteriological, Biochemical, and Immunological Characters and Activities of Diplococcus Pneumoniae. New York: Harvard University Press (1938).
36. World Health Organization. 23-valent pneumococcal polysaccharide vaccine - WHO position paper. Weekly Epidemiological Record. (2008). p. 373–84. Available from: http://www.who.int/wer/2008/wer8342.pdf; https://www.ncbi.nlm.nih.gov/pubmed/18927997 (Accessed: May 30, 2018).
37. Sugimoto N, Yamagishi Y, Hirai J, Sakanashi D, Suematsu H, Nishiyama N, et al. Invasive pneumococcal disease caused by mucoid serotype 3 Streptococcus pneumoniae: a case report and literature review. BMC Res Notes (2017) 10(1):21. doi:10.1186/s13104-016-2353-3
38. Keller LE, Robinson DA, McDaniel LS. Nonencapsulated Streptococcus pneumoniae: emergence and pathogenesis. MBio (2016) 7(2):e1792. doi:10.1128/mBio.01792-15
39. Hackel M, Lascols C, Bouchillon S, Hilton B, Morgenstern D, Purdy J. Serotype prevalence and antibiotic resistance in Streptococcus pneumoniae clinical isolates among global populations. Vaccine (2013) 31(42):4881–7. doi:10.1016/j.vaccine.2013.07.054
40. Isaacman DJ, McIntosh ED, Reinert RR. Burden of invasive pneumococcal disease and serotype distribution among Streptococcus pneumoniae isolates in young children in Europe: impact of the 7-valent pneumococcal conjugate vaccine and considerations for future conjugate vaccines. Int J Infect Dis (2010) 14(3):e197–209. doi:10.1016/j.ijid.2009.05.010
41. Varon E. Epidemiology of Streptococcus pneumoniae. Med Mal Infect (2012) 42(8):361–5. doi:10.1016/j.medmal.2012.04.002
42. Chavanet P. Pneumococcus infections: is the burden still as heavy? Med Mal Infect (2012) 42(4):149–53. doi:10.1016/j.medmal.2012.02.002
43. Wroe PC, Finkelstein JA, Ray GT, Linder JA, Johnson KM, Rifas-Shiman S, et al. Aging population and future burden of pneumococcal pneumonia in the United States. J Infect Dis (2012) 205(10):1589–92. doi:10.1093/infdis/jis240
44. Torres A, Blasi F, Dartois N, Akova M. Which individuals are at increased risk of pneumococcal disease and why? Impact of COPD, asthma, smoking, diabetes, and/or chronic heart disease on community-acquired pneumonia and invasive pneumococcal disease. Thorax (2015) 70(10):984–9. doi:10.1136/thoraxjnl-2015-206780
45. Jung JA, Kita H, Yawn BP, Boyce TG, Yoo KH, McGree ME, et al. Increased risk of serious pneumococcal disease in patients with atopic conditions other than asthma. J Allergy Clin Immunol (2010) 125(1):217–21. doi:10.1016/j.jaci.2009.10.045
46. Zhao H, Kang C-I, Rouse MS, Patel R, Kita H, Juhn YJ. The role of IL-17 in the association between pneumococcal pneumonia and allergic sensitization. Int J Microbiol (2011) 2011:709509. doi:10.1155/2011/709509
47. Chao Y, Marks LR, Pettigrew MM, Hakansson AP. Streptococcus pneumoniae biofilm formation and dispersion during colonization and disease. Front Cell Infect Microbiol (2014) 4:194. doi:10.3389/fcimb.2014.00194
48. Adegbola RA, DeAntonio R, Hill PC, Roca A, Usuf E, Hoet B, et al. Carriage of Streptococcus pneumoniae and other respiratory bacterial pathogens in low and lower-middle income countries: a systematic review and meta-analysis. PLoS One (2014) 9(8):e103293. doi:10.1371/journal.pone.0103293
49. Korona-Glowniak I, Malm A. Characteristics of Streptococcus pneumoniae strains colonizing upper respiratory tract of healthy preschool children in Poland. ScientificWorldJournal (2012) 2012:732901. doi:10.1100/2012/732901
50. Marks LR, Parameswaran GI, Hakansson AP. Pneumococcal interactions with epithelial cells are crucial for optimal biofilm formation and colonization in vitro and in vivo. Infect Immun (2012) 80(8):2744–60. doi:10.1128/IAI.00488-12
51. Steel HC, Cockeran R, Anderson R, Feldman C. Overview of community-acquired pneumonia and the role of inflammatory mechanisms in the immunopathogenesis of severe pneumococcal disease. Mediators Inflamm (2013) 2013:490346. doi:10.1155/2013/490346
52. Mayo Clinic Staff. Meningitis. Mayo Foundation for Medical Education and Research (2016). Available from: https://www.mayoclinic.org/diseases-conditions/meningitis/symptoms-causes/syc-20350508 (Accessed: March 30, 2017).
53. Centers for Disease Control Prevention. Bacterial Meningitis. Atlanta, GA: Centers for Disease Control and Prevention (2017). Available from: https://www.cdc.gov/meningitis/bacterial.html (Accessed: March 30, 2017).
54. Mitchell AM, Mitchell TJ. Streptococcus pneumoniae: virulence factors and variation. Clin Microbiol Infect (2010) 16(5):411–8. doi:10.1111/j.1469-0691.2010.03183.x
55. Hyams C, Camberlein E, Cohen JM, Bax K, Brown JS. The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect Immun (2010) 78(2):704–15. doi:10.1128/IAI.00881-09
56. Rabes A, Suttorp N, Opitz B. Inflammasomes in pneumococcal infection: innate immune sensing and bacterial evasion strategies. Curr Top Microbiol Immunol (2016) 397:215–27. doi:10.1007/978-3-319-41171-2_11
57. Nelson AL, Roche AM, Gould JM, Chim K, Ratner AJ, Weiser JN. Capsule enhances pneumococcal colonization by limiting mucus-mediated clearance. Infect Immun (2007) 75(1):83–90. doi:10.1128/IAI.01475-06
58. Khan MN, Pichichero ME. The host immune dynamics of pneumococcal colonization: Implications for novel vaccine development. Hum Vaccin Immunother (2015) 10(12):3688–99. doi:10.4161/21645515.2014.979631
59. Zafar MA, Wang Y, Hamaguchi S, Weiser JN. Host-to-host transmission of Streptococcus pneumoniae is driven by its inflammatory toxin, pneumolysin. Cell Host Microbe (2017) 21(1):73–83. doi:10.1016/j.chom.2016.12.005
60. Keller LE, Bradshaw JL, Pipkins H, McDaniel LS. Surface proteins and pneumolysin of encapsulated and nonencapsulated Streptococcus pneumoniae mediate virulence in a chinchilla model of otitis media. Front Cell Infect Microbiol (2016) 6:55. doi:10.3389/fcimb.2016.00055
61. Hotomi M, Yuasa J, Briles DE, Yamanaka N. Pneumolysin plays a key role at the initial step of establishing pneumococcal nasal colonization. Folia Microbiol (Praha) (2016) 61(5):375–83. doi:10.1007/s12223-016-0445-z
62. Rubins JB, Janoff EN. Pneumolysin: a multifunctional pneumococcal virulence factor. J Lab Clin Med (1998) 131(1):21–7. doi:10.1016/S0022-2143(98)90073-7
63. Shak JR, Ludewick HP, Howery KE, Sakai F, Yi H, Harvey RM, et al. Novel role for the Streptococcus pneumoniae toxin pneumolysin in the assembly of biofilms. MBio (2013) 4(5):e00655–613. doi:10.1128/mBio.00655-13
64. Rai P, He F, Kwang J, Engelward BP, Chow VT. Pneumococcal pneumolysin induces DNA damage and cell cycle arrest. Sci Rep (2016) 6:22972. doi:10.1038/srep22972
65. Watson DA, Musher DM, Verhoef J. Pneumococcal virulence factors and host immune responses to them. Eur J Clin Microbiol Infect Dis (1995) 14(6):479–90. doi:10.1007/BF02113425
66. Paton JC, Lock RA, Hansman DJ. Effect of immunization with pneumolysin on survival time of mice challenged with Streptococcus pneumoniae. Infect Immun (1983) 40(2):548–52.
67. Kietzman CC, Gao G, Mann B, Myers L, Tuomanen EI. Dynamic capsule restructuring by the main pneumococcal autolysin LytA in response to the epithelium. Nat Commun (2016) 7:10859. doi:10.1038/ncomms10859
68. Shockman GD, Daneo-Moore L, Kariyama R, Massidda O. Bacterial walls, peptidoglycan hydrolases, autolysins, and autolysis. Microb Drug Resist (1996) 2(1):95–8. doi:10.1089/mdr.1996.2.95
69. Tomasz A. The role of autolysins in cell death. Ann N Y Acad Sci (1974) 235(1):439–47. doi:10.1111/j.1749-6632.1974.tb43282.x
71. Abeyta M, Hardy GG, Yother J. Genetic alteration of capsule type but not PspA type affects accessibility of surface-bound complement and surface antigens of Streptococcus pneumoniae. Infect Immun (2003) 71(1):218–25. doi:10.1128/IAI.71.1.218-225.2003
72. Bergmann S, Hammerschmidt S. Versatility of pneumococcal surface proteins. Microbiology (2006) 152(Pt 2):295–303. doi:10.1099/mic.0.28610-0
73. Tu AH, Fulgham RL, McCrory MA, Briles DE, Szalai AJ. Pneumococcal surface protein A inhibits complement activation by Streptococcus pneumoniae. Infect Immun (1999) 67(9):4720–4.
74. Rajem G, Anderton JM, Carlone GM, Sampson JS, Ades EW. Pneumococcal surface adhesin A (PsaA): a review. Crit Rev Microbiol (2008) 34:131–42. doi:10.1080/10408410802275352
75. Iannelli F, Chiavolini D, Ricci S, Oggioni MR, Pozzi G. Pneumococcal surface protein C contributes to sepsis caused by Streptococcus pneumoniae in mice. Infect Immun (2004) 72(5):3077–80. doi:10.1128/IAI.72.5.3077-3080.2004
76. Gosink KK, Mann ER, Guglielmo C, Tuomanen EI, Masure HR. Role of novel choline binding proteins in virulence of Streptococcus pneumoniae. Infect Immun (2000) 68(10):5690–5. doi:10.1128/IAI.68.10.5690-5695.2000
77. Maestro B, Sanz JM. Choline binding proteins from Streptococcus pneumoniae: a dual role as enzybiotics and targets for the design of new antimicrobials. Antibiotics (Basel) (2016) 5(2):E21. doi:10.3390/antibiotics5020021
78. Henderson B. Moonlighting Proteins: Novel Virulence Factors in Bacterial Infections. Hoboken, NJ: John Wiley & Sons (2017).
79. Terrasse R, Amoroso A, Vernet T, Di Guilmi AM. Streptococcus pneumoniae GAPDH is released by cell lysis and interacts with peptidoglycan. PLoS One (2015) 10(4):e0125377. doi:10.1371/journal.pone.0125377
80. Fulde M, Bergmann S. Impact of Streptococcal Enolase in Virulence. In: Henderson B, editor. Moonlighting Proteins: Novel Virulence Factors in Bacterial Infections. Hoboken, NJ: Wiley Blackwell (2017). p. 245–68.
81. Barocchi MA, Ries J, Zogaj X, Hemsley C, Albiger B, Kanth A, et al. A pneumococcal pilus influences virulence and host inflammatory responses. Proc Natl Acad Sci U S A (2006) 103(8):2857–62. doi:10.1073/pnas.0511017103
82. Pancotto L, De Angelis G, Bizzarri E, Barocchi MA, Del Giudice G, Moschioni M, et al. Expression of the Streptococcus pneumoniae pilus-1 undergoes on and off switching during colonization in mice. Sci Rep (2013) 3:2040. doi:10.1038/srep02040
83. Janoff EN, Rubins JB, Fasching C, Charboneau D, Rahkola JT, Plaut AG, et al. Pneumococcal IgA1 protease subverts specific protection by human IgA1. Mucosal Immunol (2014) 7(2):249–56. doi:10.1038/mi.2013.41
84. Proctor M, Manning PJ. Production of immunoglobulin A protease by Streptococcus pneumoniae from animals. Infect Immun (1990) 58(9):2733–7.
85. Chi YC, Rahkola JT, Kendrick AA, Holliday MJ, Paukovich N, Roberts TS, et al. Streptococcus pneumoniae IgA1 protease: a metalloprotease that can catalyze in a split manner in vitro. Protein Sci (2017) 26(3):600–10. doi:10.1002/pro.3110
86. Wunderink RG, Waterer GW. Clinical practice. Community-acquired pneumonia. N Engl J Med (2014) 370(6):543–51. doi:10.1056/NEJMcp1214869
87. Heron MP. Deaths: Leading Causes for 2012. National Vital Statistics Reports. (2015) 64(10):1–93.
88. National Heart, Lung, and Blood Institute. Pneumonia. (2016). Available from: https://www.nhlbi.nih.gov/health-topics/pneumonia#Diagnosis (Accessed: March 20, 2017).
89. World Health Organization. World Pneumonia Day. (2017). Available from: http://www.who.int/life-course/news/events/2017-world-pneumonia-day/en/ (Accessed: May 20, 2018).
90. Centers for Disease Control Prevention. Pneumococcal Disease: Fast Facts. Washington, DC: Public Health Foundation (2017). Available from https://www.cdc.gov/pneumococcal/about/facts.html#ref1 (Accessed: May 20, 2018).
91. Bonten MJ, Huijts SM, Bolkenbaas M, Webber C, Patterson S, Gault S, et al. Polysaccharide conjugate vaccine against pneumococcal pneumonia in adults. N Engl J Med (2015) 372(12):1114–25. doi:10.1056/NEJMoa1408544
92. United Nations Children Fund. Pneumonia Claims the Lives of the World’s Most Vulnerable Children. (2018). Available from: https://data.unicef.org/topic/child-health/pneumonia/# (Accessed: May 20, 2018).
93. Simonetti AF, Viasus D, Garcia-Vidal C, Carratalà J. Management of community-acquired pneumonia in older adults. Ther Adv Infect Dis (2014) 2(1):3–16. doi:10.1177/2049936113518041
94. Stupka JE, Mortensen EM, Anzueto A, Restrepo MI. Community-acquired pneumonia in elderly patients. Aging Health (2009) 5(6):763–74. doi:10.2217/ahe.09.74
95. Wunderink RG, Waterer GW. Community-acquired pneumonia. N Engl J Med (2014) 370(19):1863. doi:10.1056/NEJMc1402692
96. World Health Organization. Fact Sheet: Children: Reducing Mortality. World Heath Organization (2016). Available from: http://www.who.int/news-room/fact-sheets/detail/children-reducing-mortality (Accessed: February 7, 2017).
97. World Health Organization. Pneumonia – Key Facts. World Heath Organization (2016). Available from: http://www.who.int/news-room/fact-sheets/detail/pneumonia (Accessed: May 20, 2018).
98. de Waroux OLP, Flasche S, Kucharski A, Langendorf C, Ndazima D, Mwanga-Amumpaire J, et al. Identifying Human Encounters that Shape the Transmission of Streptococcus pneumoniae and Other Respiratory Infections. Epidemics (2018) 116079. doi:10.1016/j.epidem.2018.05.008
99. Althouse B, Hammitt L, Grant L, Wagner B, Reid R, Larzelere-Hinton F, et al. Identifying transmission routes of Streptococcus pneumoniae and sources of acquisitions in high transmission communities. Epidemiol Infect (2017) 145(13):2750–8. doi:10.1017/S095026881700125X
100. Sá-Leão R, Nunes S, Brito-Avô A, Alves CR, Carriço JA, Saldanha J, et al. High rates of transmission of and colonization by Streptococcus pneumoniae and Haemophilus influenzae within a day care center revealed in a longitudinal study. J Clin Microbiol (2008) 46(1):225–34. doi:10.1128/JCM.01551-07
101. Lipsitch M, Abdullahi O, D’Amour A, Xie W, Weinberger DM, Tchetgen ET, et al. Estimating rates of carriage acquisition and clearance and competitive ability for pneumococcal serotypes in Kenya with a Markov transition model. Epidemiology (2012) 23(4):510. doi:10.1097/EDE.0b013e31824f2f32
102. Mosser JF, Grant LR, Millar EV, Weatherholtz RC, Jackson DM, Beall B, et al. Nasopharyngeal carriage and transmission of Streptococcus pneumoniae in American Indian households after a decade of pneumococcal conjugate vaccine use. PLoS One (2014) 9(1):e79578. doi:10.1371/journal.pone.0079578
103. Marks LR, Reddinger RM, Hakansson AP. Biofilm formation enhances fomite survival of Streptococcus pneumoniae and Streptococcus pyogenes. Infect Immun (2014) 82(3):1141–6. doi:10.1128/IAI.01310-13
104. Allan RN, Skipp P, Jefferies J, Clarke SC, Faust SN, Hall-Stoodley L, et al. Pronounced metabolic changes in adaptation to biofilm growth by Streptococcus pneumoniae. PLoS One (2014) 9(9):e107015. doi:10.1371/journal.pone.0107015
105. Walsh RL, Camilli A. Streptococcus pneumoniae is desiccation tolerant and infectious upon rehydration. MBio (2011) 2(3):e00092–11. doi:10.1128/mBio.00092-11
106. Davis BM, Aiello AE, Dawid S, Rohani P, Shrestha S, Foxman B. Influenza and community-acquired pneumonia interactions: the impact of order and time of infection on population patterns. Am J Epidemiol (2012) 175(5):363–7. doi:10.1093/aje/kwr402
107. Klein EY, Monteforte B, Gupta A, Jiang W, May L, Hsieh YH, et al. The frequency of influenza and bacterial coinfection: a systematic review and meta-analysis. Influenza Other Respi Viruses (2016) 10(5):394–403. doi:10.1111/irv.12398
108. Brundage JF, Shanks GD. Deaths from bacterial pneumonia during 1918–19 influenza pandemic. Emerg Infect Dis (2008) 14(8):1193. doi:10.3201/eid1408.071313
109. Chien Y-W, Klugman KP, Morens DM. Bacterial pathogens and death during the 1918 influenza pandemic. N Engl J Med (2009) 361(26):2582–3. doi:10.1056/NEJMc0908216
110. Bakaletz LO. Viral-bacterial co-infections in the respiratory tract. Curr Opin Microbiol (2017) 35(Suppl C):30–5. doi:10.1016/j.mib.2016.11.003
111. Cauley LS, Vella AT. Why is co-infection with influenza virus and bacteria so difficult to control? Discov Med (2015) 19(102):33–40.
112. Crotty MP, Meyers S, Hampton N, Bledsoe S, Ritchie DJ, Buller RS, et al. Epidemiology, co-infections, and outcomes of viral pneumonia in adults: an observational cohort study. Medicine (2015) 94(50):e2332. doi:10.1097/MD.0000000000002332
113. Hoffmann J, Machado D, Terrier O, Pouzol S, Messaoudi M, Basualdo W, et al. Viral and bacterial co-infection in severe pneumonia triggers innate immune responses and specifically enhances IP-10: a translational study. Sci Rep (2016) 6:38532. doi:10.1038/srep38532
114. Vu HT, Yoshida LM, Suzuki M, Nguyen HA, Nguyen CD, Nguyen AT, et al. Association between nasopharyngeal load of Streptococcus pneumoniae, viral coinfection, and radiologically confirmed pneumonia in Vietnamese children. Pediatr Infect Dis J (2011) 30(1):11–8. doi:10.1097/INF.0b013e3181f111a2
115. Khan MN, Xu Q, Pichichero ME. Protection against Streptococcus pneumoniae invasive pathogenesis by a protein-based vaccine is achieved by suppression of nasopharyngeal bacterial density during influenza A virus coinfection. Infect Immun (2017) 85(2):e530–516. doi:10.1128/IAI.00530-16
116. Brealey JC, Chappell KJ, Galbraith S, Fantino E, Gaydon J, Tozer S, et al. Streptococcus pneumoniae colonization of the nasopharynx is associated with increased severity during respiratory syncytial virus infection in young children. Respirology (2017) 23(2):220–7. doi:10.1111/resp.13179
117. Tettelin H, Chancey S, Mitchell T, Denapaite D, Schähle Y, Rieger M, et al. Chapter 5 – genomics, genetic variation, and regions of differences. In: Brown J, Hammerschmidt S, Orihuela C, editors. Streptococcus pneumoniae. Amsterdam: Academic Press (2015). p. 81–107.
118. Donati C, Hiller NL, Tettelin H, Muzzi A, Croucher NJ, Angiuoli SV, et al. Structure and dynamics of the pan-genome of Streptococcus pneumoniae and closely related species. Genome Biol (2010) 11(10):R107. doi:10.1186/gb-2010-11-10-r107
119. Hiller NL, Ahmed A, Powell E, Martin DP, Eutsey R, Earl J, et al. Generation of genic diversity among Streptococcus pneumoniae strains via horizontal gene transfer during a chronic polyclonal pediatric infection. PLoS Pathog (2010) 6(9):e1001108. doi:10.1371/journal.ppat.1001108
120. Weiser JN, Ferreira DM, Paton JC. Streptococcus pneumoniae: transmission, colonization and invasion. Nat Rev Microbiol (2018) 16:355–67. doi:10.1038/s41579-018-0001-8
121. Claverys J-P, Prudhomme M, Martin B. Induction of competence regulons as a general response to stress in Gram-positive bacteria. Annu Rev Microbiol (2006) 60:451–75. doi:10.1146/annurev.micro.60.080805.142139
122. Lees JA, Croucher NJ, Goldblatt D, Nosten F, Parkhill J, Turner C, et al. Genome-wide identification of lineage and locus specific variation associated with pneumococcal carriage duration. Elife (2017) 6:e26255. doi:10.7554/eLife.26255
123. Wartha F, Beiter K, Albiger B, Fernebro J, Zynchlinsky A, Normark S, et al. Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol (2007) 9(5):11162–71. doi:10.1111/j.1462-5822.2006.00857.x
124. Rodgers GL, Arguedas A, Cohen R, Dagan R. Global serotype distribution among Streptococcus pneumoniae isolates causing otitis media in children: potential implications for pneumococcal conjugate vaccines. Vaccine (2009) 27(29):3802–10. doi:10.1016/j.vaccine.2009.04.021
125. Wilson R, Cohen JM, Jose RJ, de Vogel C, Baxendale H, Brown JS. Protection against Streptococcus pneumoniae lung infection after nasopharyngeal colonization requires both humoral and cellular immune responses. Mucosal Immunol (2015) 8(3):627–39. doi:10.1038/mi.2014.95
126. Musher Daniel M. Pneumococcal Vaccination in Adults. (2015). Available from: http://www.uptodate.com/contents/pneumococcal-vaccination-in-adults (Accessed: February 7, 2017).
127. Li Y, Weinberger DM, Thompson CM, Trzciński K, Lipsitch M. Surface charge of Streptococcus pneumoniae predicts serotype distribution. Infect Immun (2013) 81(12):4519–24. doi:10.1128/IAI.00724-13
128. Chang B, Nariai A, Sekizuka T, Akeda Y, Kuroda M, Oishi K, et al. Capsule switching and antimicrobial resistance acquired during repeated Streptococcus pneumoniae pneumonia episodes. J Clin Microbiol (2015) 53(10):3318–24. doi:10.1128/JCM.01222-15
129. Wyres KL, Lambertsen LM, Croucher NJ, McGee L, von Gottberg A, Liñares J, et al. Pneumococcal capsular switching: a historical perspective. J Infect Dis (2013) 207(3):439–49. doi:10.1093/infdis/jis703
130. Park IH, Kim K-H, Andrade AL, Briles DE, McDaniel LS, Nahm MH. Nontypeable pneumococci can be divided into multiple cps types, including one type expressing the novel gene pspK. MBio (2012) 3(3):e00035–12. doi:10.1128/mBio.00035-12
131. Gisch N, Peters K, Zähringer U, Vollmer W. Chapter 8 – the pneumococcal cell wall. In: Brown J, Hammerschmidt S, Orihuela C, editors. Streptococcus pneumoniae. Amsterdam: Academic Press (2015). p. 145–67.
132. Bui NK, Eberhardt A, Vollmer D, Kern T, Bougault C, Tomasz A, et al. Isolation and analysis of cell wall components from Streptococcus pneumoniae. Anal Biochem (2012) 421(2):657–66. doi:10.1016/j.ab.2011.11.026
133. Hirst RA, Kadioglu A, O’Callaghan C, Andrew PW. The role of pneumolysin in pneumococcal pneumonia and meningitis. Clin Exp Immunol (2004) 138(2):195–201. doi:10.1111/j.1365-2249.2004.02611.x
134. Paton JC, Andrew PW, Boulnois GJ, Mitchell TJ. Molecular analysis of the pathogenicity of Streptococcus pneumoniae: the role of pneumococcal proteins. Annu Rev Microbiol (1993) 47:89–115. doi:10.1146/annurev.mi.47.100193.000513
135. Marshall JE, Faraj BH, Gingras AR, Lonnen R, Sheikh MA, El-Mezgueldi M, et al. The crystal structure of pneumolysin at 2.0 a resolution reveals the molecular packing of the pre-pore complex. Sci Rep (2015) 5:13293. doi:10.1038/srep13293
136. Lemon JK, Weiser JN. Degradation products of the extracellular pathogen Streptococcus pneumoniae access the cytosol via its pore-forming toxin. MBio (2015) 6(1):e2110–4. doi:10.1128/mBio.02110-14
137. Best BP. Nuclear DNA damage as a direct cause of aging. Rejuvenation Res (2009) 12(3):199–208. doi:10.1089/rej.2009.0847
138. Neill DR, Mitchell TJ, Kadioglu A. Chapter 14 – pneumolysin. In: Brown J, Hammerschmidt S, Orihuela C, editors. Streptococcus pneumoniae. Amsterdam: Academic Press (2015). p. 257–75.
139. Jefferies JM, Johnston CH, Kirkham L-AS, Cowan GJ, Ross KS, Smith A, et al. Presence of nonhemolytic pneumolysin in serotypes of Streptococcus pneumoniae associated with disease outbreaks. J Infect Dis (2007) 196(6):936–44. doi:10.1086/520091
140. Lock RA, Zhang QY, Berry AM, Paton JC. Sequence variation in the Streptococcus pneumoniae pneumolysin gene affecting haemolytic activity and electrophoretic mobility of the toxin. Microb Pathog (1996) 21(2):71–83. doi:10.1006/mpat.1996.0044
141. Kirkham L-AS, Jefferies JM, Kerr AR, Jing Y, Clarke SC, Smith A, et al. Identification of invasive serotype 1 pneumococcal isolates that express nonhemolytic pneumolysin. J Clin Microbiol (2006) 44(1):151–9. doi:10.1128/JCM.44.1.151-159.2006
142. Morgan P, Andrew P, Mitchell T. Thiol-activated cytolysins. Rev Med Microbiol (1996) 7(4):221–30. doi:10.1097/00013542-199610000-00004
143. Saunders F, Mitchell T, Walker J, Andrew P, Boulnois G. Pneumolysin, the thiol-activated toxin of Streptococcus pneumoniae, does not require a thiol group for in vitro activity. Infect Immun (1989) 57(8):2547–52.
144. Rogers HJ, Forsberg CW. Role of autolysins in the killing of bacteria by some bactericidal antibiotics. J Bacteriol (1971) 108(3):1235–43.
145. Cundell DR, Pearce BJ, Sandros J, Naughton AM, Masure HR. Peptide permeases from Streptococcus pneumoniae affect adherence to eucaryotic cells. Infect Immun (1995) 63(7):2493–8.
146. Dintilhac A, Alloing G, Granadel C, Claverys JP. Competence and virulence of Streptococcus pneumoniae: Adc and PsaA mutants exhibit a requirement for Zn and Mn resulting from inactivation of putative ABC metal permeases. Mol Microbiol (1997) 25(4):727–39. doi:10.1046/j.1365-2958.1997.5111879.x
147. Rosenow C, Ryan P, Weiser JN, Johnson S, Fontan P, Ortqvist A, et al. Contribution of novel choline-binding proteins to adherence, colonization and immunogenicity of Streptococcus pneumoniae. Mol Microbiol (1997) 25(5):819–29. doi:10.1111/j.1365-2958.1997.mmi494.x
148. Pérez-Dorado I, Galan-Bartual S, Hermoso J. Pneumococcal surface proteins: when the whole is greater than the sum of its parts. Mol Oral Microbiol (2012) 27(4):221–45. doi:10.1111/j.2041-1014.2012.00655.x
149. Zomer A, Hermans PW, Bootsma HJ. Non-adhesive surface proteins of Streptococcus pneumoniae. Streptococcus pneumoniae. Elsevier (2015). p. 231–44.
150. Galán-Bartual S, Pérez-Dorado I, García P, Hermoso JA. Chapter 11 – structure and function of choline-binding proteins. In: Brown J, Hammerschmidt S, Orihuela C, editors. Streptococcus pneumoniae. Amsterdam: Academic Press (2015). p. 207–30.
151. Hammerschmidt S, Bethe G, Remane PH, Chhatwal GS. Identification of pneumococcal surface protein A as a lactoferrin-binding protein of Streptococcus pneumoniae. Infect Immun (1999) 67(4):1683–7.
152. Shaper M, Hollingshead SK, Benjamin WH, Briles DE. PspA protects Streptococcus pneumoniae from killing by apolactoferrin, and antibody to PspA enhances killing of pneumococci by apolactoferrin. Infect Immun (2004) 72(9):5031–40. doi:10.1128/IAI.72.9.5031-5040.2004
153. Hammerschmidt S, Tillig MP, Wolff S, Vaerman JP, Chhatwal GS. Species-specific binding of human secretory component to SpsA protein of Streptococcus pneumoniae via a hexapeptide motif. Mol Microbiol (2000) 36(3):726–36. doi:10.1046/j.1365-2958.2000.01897.x
154. Dave S, Carmicle S, Hammerschmidt S, Pangburn MK, McDaniel LS. Dual roles of PspC, a surface protein of Streptococcus pneumoniae, in binding human secretory IgA and factor H. J Immunol (2004) 173(1):471–7. doi:10.4049/jimmunol.173.1.471
155. Kerr AR, Paterson GK, McCluskey J, Iannelli F, Oggioni MR, Pozzi G, et al. The contribution of PspC to pneumococcal virulence varies between strains and is accomplished by both complement evasion and complement-independent mechanisms. Infect Immun (2006) 74(9):5319–24. doi:10.1128/IAI.00543-06
156. Iannelli F, Oggioni MR, Pozzi G. Allelic variation in the highly polymorphic locus pspC of Streptococcus pneumoniae. Gene (2002) 284(1):63–71. doi:10.1016/S0378-1119(01)00896-4
157. Dieudonné-Vatran A, Krentz S, Blom AM, Meri S, Henriques-Normark B, Riesbeck K, et al. Clinical isolates of Streptococcus pneumoniae bind the complement inhibitor C4b-binding protein in a PspC allele-dependent fashion. J Immunol (2009) 182(12):7865. doi:10.4049/jimmunol.0802376
158. Martner A, Skovbjerg S, Paton JC, Wold AE. Streptococcus pneumoniae autolysis prevents phagocytosis and production of phagocyte-activating cytokines. Infect Immun (2009) 77(9):3826–37. doi:10.1128/IAI.00290-09
159. Kadioglu A, Andrew PW. The innate immune response to pneumococcal lung infection: the untold story. Trends Immunol (2004) 25(3):143–9. doi:10.1016/j.it.2003.12.006
160. Martner A, Dahlgren C, Paton JC, Wold AE. Pneumolysin released during Streptococcus pneumoniae autolysis is a potent activator of intracellular oxygen radical production in neutrophils. Infect Immun (2008) 76(9):4079–87. doi:10.1128/IAI.01747-07
161. Mellroth P, Daniels R, Eberhardt A, Ronnlund D, Blom H, Widengren J, et al. LytA, major autolysin of Streptococcus pneumoniae, requires access to nascent peptidoglycan. J Biol Chem (2012) 287(14):11018–29. doi:10.1074/jbc.M111.318584
162. Mellroth P, Sandalova T, Kikhney A, Vilaplana F, Hesek D, Lee M, et al. Structural and functional insights into peptidoglycan access for the lytic amidase LytA of Streptococcus pneumoniae. MBio (2014) 5(1):e01120–1113. doi:10.1128/mBio.01120-13
163. Ramos-Sevillano E, Urzainqui A, Campuzano S, Moscoso M, González-Camacho F, Domenech M, et al. Pleiotropic effects of cell wall amidase LytA on Streptococcus pneumoniae sensitivity to the host immune response. Infect Immun (2015) 83(2):591–603. doi:10.1128/IAI.02811-14
164. Bai X-H, Chen H-J, Jiang Y-L, Wen Z, Huang Y, Cheng W, et al. Structure of pneumococcal peptidoglycan hydrolase LytB reveals insights into the bacterial cell wall remodeling and pathogenesis. J Biol Chem (2014) 289(34):23403–16. doi:10.1074/jbc.M114.579714
165. Garcia P, Gonzalez MP, Garcia E, Lopez R, Garcia JL. LytB, a novel pneumococcal murein hydrolase essential for cell separation. Mol Microbiol (1999) 31(4):1275–81. doi:10.1046/j.1365-2958.1999.01238.x
166. Ramos-Sevillano E, Moscoso M, Garcia P, Garcia E, Yuste J. Nasopharyngeal colonization and invasive disease are enhanced by the cell wall hydrolases LytB and LytC of Streptococcus pneumoniae. PLoS One (2011) 6(8):e23626. doi:10.1371/journal.pone.0023626
167. Domenech M, Ruiz S, Moscoso M, García E. In vitro biofilm development of Streptococcus pneumoniae and formation of choline-binding protein-DNA complexes. Environ Microbiol Rep (2015) 7(5):715–27. doi:10.1111/1758-2229.12295
168. Pérez-Dorado I, González A, Morales M, Sanles R, Striker W, Vollmer W, et al. Insights into pneumococcal fratricide from the crystal structures of the modular killing factor LytC. Nat Struct Mol Biol (2010) 17(5):576. doi:10.1038/nsmb.1817
169. Eldholm V, Johnsborg O, Haugen K, Ohnstad HS, Håvarstein LS. Fratricide in Streptococcus pneumoniae: contributions and role of the cell wall hydrolases CbpD, LytA and LytC. Microbiology (2009) 155(7):2223–34. doi:10.1099/mic.0.026328-0
170. Molina R, González A, Stelter M, Pérez-Dorado I, Kahn R, Morales M, et al. Crystal structure of CbpF, a bifunctional choline-binding protein and autolysis regulator from Streptococcus pneumoniae. EMBO Rep (2009) 10(3):246–51. doi:10.1038/embor.2008.245
171. Guiral S, Mitchell TJ, Martin B, Claverys J-P. Competence-programmed predation of noncompetent cells in the human pathogen Streptococcus pneumoniae: genetic requirements. Proc Natl Acad Sci U S A (2005) 102(24):8710–5. doi:10.1073/pnas.0500879102
172. Brown JS, Gilliland SM, Ruiz-Albert J, Holden DW. Characterization of pit, a Streptococcus pneumoniae iron uptake ABC transporter. Infect Immun (2002) 70(8):4389–98. doi:10.1128/IAI.70.8.4389-4398.2002
173. Yang X-Y, Sun B, Zhang L, Li N, Han J, Zhang J, et al. Chemical interference with iron transport systems to suppress bacterial growth of Streptococcus pneumoniae. PLoS One (2014) 9(8):e105953. doi:10.1371/journal.pone.0105953
174. Deplazes E, Begg SL, Van Wonderen JH, Campbell R, Kobe B, Paton JC, et al. Characterizing the conformational dynamics of metal-free PsaA using molecular dynamics simulations and electron paramagnetic resonance spectroscopy. Biophys Chem (2015) 207:51–60. doi:10.1016/j.bpc.2015.08.004
175. Johnston JW, Myers LE, Ochs MM, Benjamin WH, Briles DE, Hollingshead SK. Lipoprotein PsaA in virulence of Streptococcus pneumoniae: surface accessibility and role in protection from superoxide. Infect Immun (2004) 72(10):5858–67. doi:10.1128/IAI.72.10.5858-5867.2004
176. Berry AM, Paton JC. Sequence heterogeneity of PsaA, a 37-kilodalton putative adhesin essential for virulence of Streptococcus pneumoniae. Infect Immun (1996) 64(12):5255–62.
177. Whalan RH, Funnell SG, Bowler LD, Hudson MJ, Robinson A, Dowson CG. Distribution and genetic diversity of the ABC transporter lipoproteins PiuA and PiaA within Streptococcus pneumoniae and related streptococci. J Bacteriol (2006) 188(3):1031–8. doi:10.1128/JB.188.3.1031-1038.2006
178. Whalan RH, Funnell SG, Bowler LD, Hudson MJ, Robinson A, Dowson CG. PiuA and PiaA, iron uptake lipoproteins of Streptococcus pneumoniae, elicit serotype independent antibody responses following human pneumococcal septicaemia. FEMS Immunol Med Microbiol (2005) 43(1):73–80. doi:10.1016/j.femsim.2004.07.010
179. Brown JS, Ogunniyi AD, Woodrow MC, Holden DW, Paton JC. Immunization with components of two iron uptake ABC transporters protects mice against systemic Streptococcus pneumoniae infection. Infect Immun (2001) 69(11):6702–6. doi:10.1128/IAI.69.11.6702-6706.2001
180. Brown JS, Gilliland SM, Holden DW. A Streptococcus pneumoniae pathogenicity island encoding an ABC transporter involved in iron uptake and virulence. Mol Microbiol (2001) 40(3):572–85. doi:10.1046/j.1365-2958.2001.02414.x
181. Honsa ES, Johnson MDL, Rosch JW. The roles of transition metals in the physiology and pathogenesis of Streptococcus pneumoniae. Front Cell Infect Microbiol (2013) 3:92. doi:10.3389/fcimb.2013.00092
182. Cheng W, Li Q, Jiang Y-L, Zhou C-Z, Chen Y. Structures of Streptococcus pneumoniae PiaA and its complex with ferrichrome reveal insights into the substrate binding and release of high affinity iron transporters. PLoS One (2013) 8(8):e71451. doi:10.1371/journal.pone.0071451
183. Johnson DI. Bacterial Pathogens and Their Virulence Factors. Cham, Switzerland: Springer (2017).
184. Zhang L, Li N, Cao K, Yang X-Y, Zeng G, Sun X, et al. Crucial residue Trp158 of lipoprotein PiaA stabilizes the ferrichrome-PiaA complex in Streptococcus pneumoniae. J Inorg Biochem (2017) 167(Suppl C):150–6. doi:10.1016/j.jinorgbio.2016.08.015
185. Tai SS, Lee CJ, Winter RE. Hemin utilization is related to virulence of Streptococcus pneumoniae. Infect Immun (1993) 61(12):5401–5.
186. Tai SS, Yu C, Lee JK. A solute binding protein of Streptococcus pneumoniae iron transport. FEMS Microbiol Lett (2003) 220(2):303–8. doi:10.1016/S0378-1097(03)00135-6
187. Yang X-Y, He K, Du G, Wu X, Yu G, Pan Y, et al. Integrated translatomics with proteomics to identify novel iron-transporting proteins in Streptococcus pneumoniae. Front Microbiol (2016) 7:78. doi:10.3389/fmicb.2016.00078
188. Boekhorst J, de Been MWHJ, Kleerebezem M, Siezen RJ. Genome-wide detection and analysis of cell wall-bound proteins with LPxTG-like sorting motifs. J Bacteriol (2005) 187(14):4928–34. doi:10.1128/JB.187.14.4928-4934.2005
189. Marraffini LA, DeDent AC, Schneewind O. Sortases and the art of anchoring proteins to the envelopes of Gram-positive bacteria. Microbiol Mol Biol Rev (2006) 70(1):192–221. doi:10.1128/MMBR.70.1.192-221.2006
190. Kharat AS, Tomasz A. Inactivation of the srtA gene affects localization of surface proteins and decreases adhesion of Streptococcus pneumoniae to human pharyngeal cells in vitro. Infect Immun (2003) 71(5):2758–65. doi:10.1128/IAI.71.5.2758-2765.2003
191. Noske N, Kämmerer U, Rohde M, Hammerschmidt S. Pneumococcal interaction with human dendritic cells: phagocytosis, survival, and induced adaptive immune response are manipulated by PavA. J Immunol (2009) 183(3):1952–63. doi:10.4049/jimmunol.0804383
192. Kolberg J, Aase A, Bergmann S, Herstad TK, Rødal G, Frank R, et al. Streptococcus pneumoniae enolase is important for plasminogen binding despite low abundance of enolase protein on the bacterial cell surface. Microbiology (2006) 152(5):1307–17. doi:10.1099/mic.0.28747-0
193. Díaz-Ramos À, Roig-Borrellas A, García-Melero A, López-Alemany R. α-Enolase, a multifunctional protein: its role on pathophysiological situations. J Biomed Biotechnol (2012) 2012:156795. doi:10.1155/2012/156795
194. Agarwal V, Hammerschmidt S, Malm S, Bergmann S, Riesbeck K, Blom AM. Enolase of Streptococcus pneumoniae binds human complement inhibitor C4b-binding protein and contributes to complement evasion. J Immunol (2012) 189(7):3575–84. doi:10.4049/jimmunol.1102934
195. Mori Y, Yamaguchi M, Terao Y, Hamada S, Ooshima T, Kawabata S. α-Enolase of Streptococcus pneumoniae induces formation of neutrophil extracellular traps. J Biol Chem (2012) 287(13):10472–81. doi:10.1074/jbc.M111.280321
196. Bergmann S, Rohde M, Hammerschmidt S. Glyceraldehyde-3-phosphate dehydrogenase of Streptococcus pneumoniae is a surface-displayed plasminogen-binding protein. Infect Immun (2004) 72(4):2416–9. doi:10.1128/IAI.72.4.2416-2419.2004
197. De Angelis G, Moschioni M, Muzzi A, Pezzicoli A, Censini S, Delany I, et al. The Streptococcus pneumoniae pilus-1 displays a biphasic expression pattern. PLoS One (2011) 6(6):e21269. doi:10.1371/journal.pone.0021269
198. Bagnoli F, Moschioni M, Donati C, Dimitrovska V, Ferlenghi I, Facciotti C, et al. A second pilus type in Streptococcus pneumoniae is prevalent in emerging serotypes and mediates adhesion to host cells. J Bacteriol (2008) 190(15):5480–92. doi:10.1128/JB.00384-08
199. Muschiol S, Erlendsson S, Aschtgen M-S, Oliveira V, Schmieder P, de Lichtenberg C, et al. Structure of the competence pilus major pilin ComGC in Streptococcus pneumoniae. J Biol Chem (2017) 292(34):14134–46. doi:10.1074/jbc.M117.787671
200. Laurenceau R, Péhau-Arnaudet G, Baconnais S, Gault J, Malosse C, Dujeancourt A, et al. A type IV pilus mediates DNA binding during natural transformation in Streptococcus pneumoniae. PLoS Pathog (2013) 9(6):e1003473. doi:10.1371/journal.ppat.1003473
201. Woof J, Russell M. Structure and function relationships in IgA. Mucosal Immunol (2011) 4(6):590–7. doi:10.1038/mi.2011.39
202. Rai P, Parrish M, Tay IJJ, Li N, Ackerman S, He F, et al. Streptococcus pneumoniae secretes hydrogen peroxide leading to DNA damage and apoptosis in lung cells. Proc Natl Acad Sci U S A (2015) 112(26):E3421–30. doi:10.1073/pnas.1424144112
203. Pericone CD, Overweg K, Hermans PWM, Weiser JN. Inhibitory and bactericidal effects of hydrogen peroxide production by Streptococcus pneumoniae on other inhabitants of the upper respiratory tract. Infect Immun (2000) 68(7):3990–7. doi:10.1128/IAI.68.7.3990-3997.2000
204. Loose M, Hudel M, Zimmer K-P, Garcia E, Hammerschmidt S, Lucas R, et al. Pneumococcal hydrogen peroxide-induced stress signaling regulates inflammatory genes. J Infect Dis (2015) 211(2):306–16. doi:10.1093/infdis/jiu428
205. Pesakhov S, Benisty R, Sikron N, Cohen Z, Gomelsky P, Khozin-Goldberg I, et al. Effect of hydrogen peroxide production and the Fenton reaction on membrane composition of Streptococcus pneumoniae. Biochim Biophys Acta (2007) 1768(3):590–7. doi:10.1016/j.bbamem.2006.12.016
206. Yesilkaya H, Andisi VF, Andrew PW, Bijlsma JJ. Streptococcus pneumoniae and reactive oxygen species: an unusual approach to living with radicals. Trends Microbiol (2013) 21(4):187–95. doi:10.1016/j.tim.2013.01.004
207. Pericone CD, Park S, Imlay JA, Weiser JN. Factors contributing to hydrogen peroxide resistance in Streptococcus pneumoniae include pyruvate oxidase (SpxB) and avoidance of the toxic effects of the Fenton reaction. J Bacteriol (2003) 185(23):6815–25. doi:10.1128/JB.185.23.6815-6825.2003
208. Dobrindt U, Hochhut B, Hentschel U, Hacker J. Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol (2004) 2(5):414. doi:10.1038/nrmicro884
209. Schmidt H, Hensel M. Pathogenicity islands in bacterial pathogenesis. Clin Microbiol Rev (2004) 17(1):14–56. doi:10.1128/CMR.17.1.14-56.2004
210. Shivshankar P, Sanchez C, Rose LF, Orihuela CJ. The Streptococcus pneumoniae adhesin PsrP binds to keratin 10 on lung cells. Mol Microbiol (2009) 73(4):663–79. doi:10.1111/j.1365-2958.2009.06796.x
211. Lizcano A, Akula Suresh Babu R, Shenoy AT, Saville AM, Kumar N, D’Mello A, et al. Transcriptional organization of pneumococcal psrP-secY2A2 and impact of GtfA and GtfB deletion on PsrP-associated virulence properties. Microbes Infect (2017) 19(6):323–33. doi:10.1016/j.micinf.2017.04.001
212. Donlan RM. Biofilms: microbial life on surfaces. Emerg Infect Dis (2002) 8(9):881–90. doi:10.3201/eid0809.020063
213. Oliver MB, Swords WE. Chapter 16 – pneumococcal biofilms and bacterial persistence during otitis media infections. In: Brown J, Hammerschmidt S, Orihuela C, editors. Streptococcus pneumoniae. Amsterdam: Academic Press (2015). p. 293–308.
214. Marks LR, Reddinger RM, Hakansson AP. High levels of genetic recombination during nasopharyngeal carriage and biofilm formation in Streptococcus pneumoniae. MBio (2012) 3(5):e00200–12. doi:10.1128/mBio.00200-12
215. de la Fuente-Núñez C, Reffuveille F, Fernández L, Hancock REW. Bacterial biofilm development as a multicellular adaptation: antibiotic resistance and new therapeutic strategies. Curr Opin Microbiol (2013) 16(5):580–9. doi:10.1016/j.mib.2013.06.013
216. Sanchez CJ, Shivshankar P, Stol K, Trakhtenbroit S, Sullam PM, Sauer K, et al. The pneumococcal serine-rich repeat protein is an intra-species bacterial adhesin that promotes bacterial aggregation in vivo and in biofilms. PLoS Pathog (2010) 6(8):e1001044. doi:10.1371/journal.ppat.1001044
217. Wu H, Moser C, Wang H-Z, Høiby N, Song Z-J. Strategies for combating bacterial biofilm infections. Int J Oral Sci (2015) 7(1):1–7. doi:10.1038/ijos.2014.65
218. Fliegauf M, Sonnen AFP, Kremer B, Henneke P. Mucociliary clearance defects in a murine in vitro model of pneumococcal airway infection. PLoS One (2013) 8(3):e59925. doi:10.1371/journal.pone.0059925
219. Murphy K, Weaver C. Innate immunity: the first line of defense. 9th ed. Janeway’s Immunobiology. Garland Science (2017). p. 37–76.
220. Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol (2015) 16(1):27–35. doi:10.1038/ni.3045
221. Antunes M, Cohen N. Mucociliary clearance – a critical upper airway host defense mechanism and methods of assessment. Curr Opin Allergy Clin Immunol (2007) 7(1):5–10. doi:10.1097/ACI.0b013e3280114eef
222. André GO, Politano WR, Mirza S, Converso TR, Ferraz LFC, Leite LCC, et al. Combined effects of lactoferrin and lysozyme on Streptococcus pneumoniae killing. Microb Pathog (2015) 89:7–17. doi:10.1016/j.micpath.2015.08.008
223. Nawrocki KL, Crispell EK, McBride SM. Antimicrobial peptide resistance mechanisms of Gram-positive bacteria. Antibiotics (Basel) (2014) 3(4):461–92. doi:10.3390/antibiotics3040461
224. Kovács M, Halfmann A, Fedtke I, Heintz M, Peschel A, Vollmer W, et al. A functional dlt operon, encoding proteins required for incorporation of d-alanine in teichoic acids in Gram-positive bacteria, confers resistance to cationic antimicrobial peptides in Streptococcus pneumoniae. J Bacteriol (2006) 188(16):5797–805. doi:10.1128/JB.00336-06
225. Dockrell DH, Brown JS. Chapter 21 – Streptococcus pneumoniae interactions with macrophages and mechanisms of immune evasion. In: Brown J, Hammerschmidt S, Orihuela C, editors. Streptococcus pneumoniae. Amsterdam: Academic Press (2015). p. 401–22.
226. Shelhamer J, Marom Z, Logun C, Kaliner M. Human respiratory mucous glycoproteins. Exp Lung Res (1984) 7(2):149–62. doi:10.3109/01902148409069675
227. Yesilkaya H, Manco S, Kadioglu A, Terra VS, Andrew PW. The ability to utilize mucin affects the regulation of virulence gene expression in Streptococcus pneumoniae. FEMS Microbiol Lett (2007) 278(2):231–5. doi:10.1111/j.1574-6968.2007.01003.x
228. Davis KM, Akinbi HT, Standish AJ, Weiser JN. Resistance to mucosal lysozyme compensates for the fitness deficit of peptidoglycan modifications by Streptococcus pneumoniae. PLoS Pathog (2008) 4(12):e1000241. doi:10.1371/journal.ppat.1000241
229. Weiser JN, Austrian R, Sreenivasan PK, Masure HR. Phase variation in pneumococcal opacity: relationship between colonial morphology and nasopharyngeal colonization. Infect Immun (1994) 62(6):2582–9.
230. Mook-Kanamori BB, Geldhoff M, van der Poll T, van de Beek D. Pathogenesis and pathophysiology of pneumococcal meningitis. Clin Microbiol Rev (2011) 24(3):557–91. doi:10.1128/CMR.00008-11
231. Wanner A, Salathé M, O’Riordan TG. Mucociliary clearance in the airways. Am J Respir Crit Care Med (1996) 154(6):1868–902. doi:10.1164/ajrccm.154.6.8970383
232. Grubb BR, Livraghi-Butrico A, Rogers TD, Yin W, Button B, Ostrowski LE. Reduced mucociliary clearance in old mice is associated with a decrease in Muc5b mucin. Am J Physiol Lung Cell Mol Physiol (2016) 310(9):L860–7. doi:10.1152/ajplung.00015.2016
233. Craig A, Mai J, Cai S, Jeyaseelan S. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect Immun (2009) 77(2):568–75. doi:10.1128/IAI.00832-08
234. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13(3):159–75. doi:10.1038/nri3399
235. Falloon J, Gallin JI. Neutrophil granules in health and disease. J Allergy Clin Immunol (1986) 77(5):653–62. doi:10.1016/0091-6749(86)90404-5
236. Gardiner EE, Andrews RK. Neutrophil extracellular traps (NETs) and infection-related vascular dysfunction. Blood Rev (2012) 26(6):255–9. doi:10.1016/j.blre.2012.09.001
237. Simon AK, Hollander GA, McMichael A. Evolution of the immune system in humans from infancy to old age. Proc Biol Sci (2015) 282(1821):20143085. doi:10.1098/rspb.2014.3085
238. Nussbaum C, Gloning A, Pruenster M, Frommhold D, Bierschenk S, Genzel-Boroviczény O, et al. Neutrophil and endothelial adhesive function during human fetal ontogeny. J Leukoc Biol (2013) 93(2):175–84. doi:10.1189/jlb.0912468
239. Filias A, Theodorou GL, Mouzopoulou S, Varvarigou AA, Mantagos S, Karakantza M. Phagocytic ability of neutrophils and monocytes in neonates. BMC Pediatr (2011) 11:29. doi:10.1186/1471-2431-11-29
240. Sapey E, Greenwood H, Walton G, Mann E, Love A, Aaronson N, et al. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood (2014) 123(2):239–48. doi:10.1182/blood-2013-08-519520
241. Simell B, Vuorela A, Ekström N, Palmu A, Reunanen A, Meri S, et al. Aging reduces the functionality of anti-pneumococcal antibodies and the killing of Streptococcus pneumoniae by neutrophil phagocytosis. Vaccine (2011) 29(10):1929–34. doi:10.1016/j.vaccine.2010.12.121
242. Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol (2014) 5:491. doi:10.3389/fimmu.2014.00491
243. Gregory CD, Devitt A. The macrophage and the apoptotic cell: an innate immune interaction viewed simplistically? Immunology (2004) 113(1):1–14. doi:10.1111/j.1365-2567.2004.01959.x
244. Kraal G, van der Laan LJ, Elomaa O, Tryggvason K. The macrophage receptor MARCO. Microbes Infect (2000) 2(3):313–6. doi:10.1016/S1286-4579(00)00296-3
245. Förster-Waldl E, Sadeghi K, Tamandl D, Gerhold B, Hallwirth U, Rohrmeister K, et al. Monocyte toll-like receptor 4 expression and LPS-induced cytokine production increase during gestational aging. Pediatr Res (2005) 58(1):121. doi:10.1203/01.PDR.0000163397.53466.0F
246. Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol (2005) 17(1):1–14. doi:10.1093/intimm/dxh186
247. Anas A, van der Poll T, de Vos AF. Role of CD14 in lung inflammation and infection. Crit Care (2010) 14(2):209. doi:10.1186/cc8850
248. Fitzner N, Clauberg S, Essmann F, Liebmann J, Kolb-Bachofen V. Human skin endothelial cells can express all 10 TLR genes and respond to respective ligands. Clin Vaccine Immunol (2008) 15(1):138–46. doi:10.1128/CVI.00257-07
249. O’Neill LA, Bryant CE, Doyle SL. Therapeutic targeting of toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol Rev (2009) 61(2):177–97. doi:10.1124/pr.109.001073
250. Dessing MC, Schouten M, Draing C, Levi M, von Aulock S, van der Poll T. Role played by toll-like receptors 2 and 4 in lipoteichoic acid-induced lung inflammation and coagulation. J Infect Dis (2008) 197(2):245–52. doi:10.1086/524873
251. Kohler S, Voss F, Gomez Mejia A, Brown JS, Hammerschmidt S. Pneumococcal lipoproteins involved in bacterial fitness, virulence, and immune evasion. FEBS Lett (2016) 590(21):3820–39. doi:10.1002/1873-3468.12352
252. Tomlinson G, Chimalapati S, Pollard T, Lapp T, Cohen J, Camberlein E, et al. TLR-mediated inflammatory responses to Streptococcus pneumoniae are highly dependent on surface expression of bacterial lipoproteins. J Immunol (2014) 193(7):3736–45. doi:10.4049/jimmunol.1401413
253. Richard AL, Siegel SJ, Erikson J, Weiser JN. TLR2 signaling decreases transmission of Streptococcus pneumoniae by limiting bacterial shedding in an infant mouse influenza A co-infection model. PLoS Pathog (2014) 10(8):e1004339. doi:10.1371/journal.ppat.1004339
254. Srivastava A, Henneke P, Visintin A, Morse SC, Martin V, Watkins C, et al. The apoptotic response to pneumolysin is toll-like receptor 4 dependent and protects against pneumococcal disease. Infect Immun (2005) 73(10):6479–87. doi:10.1128/IAI.73.10.6479-6487.2005
255. Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol (2009) 21(4):317–37. doi:10.1093/intimm/dxp017
256. Godowski PJ. A smooth operator for LPS responses. Nat Immunol (2005) 6(6):544–6. doi:10.1038/ni0605-544
257. Warner N, Núñez G. MyD88: a critical adaptor protein in innate immunity signal transduction. J Immunol (2013) 190(1):3–4. doi:10.4049/jimmunol.1203103
258. Rahman AH, Taylor DK, Turka LA. The contribution of direct TLR signaling to T cell responses. Immunol Res (2009) 45(1):25–36. doi:10.1007/s12026-009-8113-x
259. Simpson JL, McDonald VM, Baines KJ, Oreo KM, Wang F, Hansbro PM, et al. Influence of age, past smoking, and disease severity on TLR2, neutrophilic inflammation, and MMP-9 levels in COPD. Mediators Inflamm (2013) 2013:462934. doi:10.1155/2013/462934
260. Ramirez A, Rathinam V, Fitzgerald KA, Golenbock DT, Mathew A. Defective pro-IL-1β responses in macrophages from aged mice. Immun Ageing (2012) 9(1):27. doi:10.1186/1742-4933-9-27
261. Cho SJ, Rooney K, Choi AM, Stout-Delgado HW. NLRP3 inflammasome activation in aged macrophages is diminished during Streptococcus pneumoniae infection. Am J Physiol Lung Cell Mol Physiol (2017) 314(3):L372–87. doi:10.1152/ajplung.00393.2017
262. Boyd AR, Shivshankar P, Jiang S, Berton MT, Orihuela CJ. Age-related defects in TLR2 signaling diminish the cytokine response by alveolar macrophages during murine pneumococcal pneumonia. Exp Gerontol (2012) 47(7):507–18. doi:10.1016/j.exger.2012.04.004
263. Murray PJ. Beyond peptidoglycan for Nod2. Nat Immunol (2009) 10(10):1053–4. doi:10.1038/ni1009-1053
264. Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev (2009) 227(1):106–28. doi:10.1111/j.1600-065X.2008.00734.x
265. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem (2003) 278(11):8869–72. doi:10.1074/jbc.C200651200
266. Davis KM, Nakamura S, Weiser JN. Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J Clin Invest (2011) 121(9):3666–76. doi:10.1172/JCI57761
267. Dessing MC, Knapp S, Florquin S, de Vos AF, van der Poll T. CD14 facilitates invasive respiratory tract infection by Streptococcus pneumoniae. Am J Respir Crit Care Med (2007) 175(6):604–11. doi:10.1164/rccm.200606-824OC
268. Murphy K, Weaver C. In: Toledo M, Bochicchio A, Acevedo-Quinones C, Zayetz E, Divakaran D, editors. Janeway’s Immunobiology. 9th ed. New York: Garland Science (2016). p. 1–855.
269. Zhang L, Li Z, Wan Z, Kilby A, Kilby JM, Jiang W. Humoral immune responses to Streptococcus pneumoniae in the setting of HIV-1 infection. Vaccine (2015) 33(36):4430–6. doi:10.1016/j.vaccine.2015.06.077
270. Weiser JN, Bae D, Fasching C, Scamurra RW, Ratner AJ, Janoff EN. Antibody-enhanced pneumococcal adherence requires IgA1 protease. Proc Natl Acad Sci U S A (2003) 100(7):4215–20. doi:10.1073/pnas.0637469100
271. Romagnani S. Th1/Th2 cells. Inflamm Bowel Dis (1999) 5(4):285–94. doi:10.1097/00054725-199911000-00009
272. Malley R, Trzcinski K, Srivastava A, Thompson CM, Anderson PW, Lipsitch M. CD4(+) T cells mediate antibody-independent acquired immunity to pneumococcal colonization. Proc Natl Acad Sci U S A (2005) 102(13):4848–53. doi:10.1073/pnas.0501254102
273. Hoe E, Anderson J, Nathanielsz J, Toh ZQ, Marimla R, Balloch A, et al. The contrasting role of Th17 immunity in human health and disease. Microbiol Immunol (2017) 61(2):49–56. doi:10.1111/1348-0421.12471
274. Azarian T, Grant LR, Georgieva M, Hammitt LL, Reid R, Bentley SD, et al. Association of pneumococcal protein antigen serology with age and antigenic profile of colonizing isolates. J Infect Dis (2017) 215(5):713–22. doi:10.1093/infdis/jiw628
275. AlonsoDeVelasco E, Verheul AF, Verhoef J, Snippe H. Streptococcus pneumoniae: virulence factors, pathogenesis, and vaccines. Microbiol Rev (1995) 59(4):591–603.
276. Gray BM, Converse GM III, Dillon HC Jr. Epidemiologic studies of Streptococcus pneumoniae in infants: acquisition, carriage, and infection during the first 24 months of life. J Infect Dis (1980) 142(6):923–33. doi:10.1093/infdis/142.6.923
277. Gray BM, Dillon HC Jr. Epidemiological studies of Streptococcus pneumoniae in infants: antibody to types 3, 6, 14, and 23 in the first two years of life. J Infect Dis (1988) 158(5):948–55. doi:10.1093/infdis/158.5.948
278. Coder B, Su D-M. Thymic involution beyond T-cell insufficiency. Oncotarget (2015) 6(26):21777. doi:10.18632/oncotarget.4970
279. van der Geest KS, Abdulahad WH, Tete SM, Lorencetti PG, Horst G, Bos NA, et al. Aging disturbs the balance between effector and regulatory CD4+ T cells. Exp Gerontol (2014) 60:190–6. doi:10.1016/j.exger.2014.11.005
280. Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol (2014) 15(8):738–48. doi:10.1038/ni.2919
281. McNeela EA, Burke Á, Neill DR, Baxter C, Fernandes VE, Ferreira D, et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog (2010) 6(11):e1001191. doi:10.1371/journal.ppat.1001191
282. Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol (2011) 187(1):434–40. doi:10.4049/jimmunol.1003143
283. Brown JS, Hussell T, Gilliland SM, Holden DW, Paton JC, Ehrenstein MR, et al. The classical pathway is the dominant complement pathway required for innate immunity to Streptococcus pneumoniae infection in mice. Proc Natl Acad Sci U S A (2002) 99(26):16969–74. doi:10.1073/pnas.012669199
284. Ali YM, Lynch NJ, Haleem KS, Fujita T, Endo Y, Hansen S, et al. The lectin pathway of complement activation is a critical component of the innate immune response to pneumococcal infection. PLoS Pathog (2012) 8(7):e1002793. doi:10.1371/journal.ppat.1002793
285. Kerr AR, Paterson GK, Riboldi-Tunnicliffe A, Mitchell TJ. Innate immune defense against pneumococcal pneumonia requires pulmonary complement component C3. Infect Immun (2005) 73(7):4245–52. doi:10.1128/IAI.73.7.4245-4252.2005
286. Davis CA, Vallota EH, Forristal J. Serum complement levels in infancy: age related changes. Pediatr Res (1979) 13(9):1043–6. doi:10.1203/00006450-197909000-00019
287. Grumach A, Ceccon M, Rutz R, Fertig A, Kirschfink M. Complement profile in neonates of different gestational ages. Scand J Immunol (2014) 79(4):276–81. doi:10.1111/sji.12154
288. MacGregor RR, Shalit M. Neutrophil function in healthy elderly subjects. J Gerontol (1990) 45(2):M55–60. doi:10.1093/geronj/45.2.M55
289. Abbas AK, Lichtman AH, Pillai S. Cellular and Molecular Immunology. Philadelphia: Elsevier Health Sciences (2015).
290. Mukerji R, Mirza S, Roche AM, Widener RW, Croney CM, Rhee D-K, et al. Pneumococcal surface protein A inhibits complement deposition on the pneumococcal surface by competing with the binding of C-reactive protein to cell-surface phosphocholine. J Immunol (2012) 189(11):5327–35. doi:10.4049/jimmunol.1201967
291. Eisen DP, Dean MM, Boermeester MA, Fidler KJ, Gordon AC, Kronborg G, et al. Low serum mannose-binding lectin level increases the risk of death due to pneumococcal infection. Clin Infect Dis (2008) 47(4):510–6. doi:10.1086/590006
292. Bradley JS, Byington CL, Shah SS, Alverson B, Carter ER, Harrison C, et al. The management of community-acquired pneumonia in infants and children older than 3 months of age: clinical practice guidelines by the pediatric infectious diseases society and the Infectious Diseases Society of America. Clin Infect Dis (2011) 53(7):e25–76. doi:10.1093/cid/cir531
293. Niederman MS, Mandell LA, Anzueto A, Bass JB, Broughton WA, Campbell GD, et al. Guidelines for the management of adults with community-acquired pneumonia. Diagnosis, assessment of severity, antimicrobial therapy, and prevention. Am J Respir Crit Care Med (2001) 163(7):1730–54. doi:10.1164/ajrccm.163.7.at1010
294. Grossman LK, Caplan SE. Clinical, laboratory, and radiological information in the diagnosis of pneumonia in children. Ann Emerg Med (1988) 17(1):43–6. doi:10.1016/S0196-0644(88)80502-X
295. Mulholland K. Problems with the WHO guidelines for management of childhood pneumonia. Lancet Glob Health (2018) 6(1):e8–9. doi:10.1016/S2214-109X(17)30468-0
296. Werno AM, Murdoch DR. Medical microbiology: laboratory diagnosis of invasive pneumococcal disease. Clin Infect Dis (2008) 46(6):926–32. doi:10.1086/528798
297. Gardner JG, Bhamidipati DR, Rueda AM, Nguyen DTM, Graviss EA, Musher DM. White blood cell counts, alcoholism, and cirrhosis in pneumococcal pneumonia. Open Forum Infect Dis (2017) 4(2):ofx034. doi:10.1093/ofid/ofx034
298. Harris AM, Beekmann SE, Polgreen PM, Moore MR. Rapid urine antigen testing for Streptococcus pneumoniae in adults with community-acquired pneumonia: clinical use and barriers. Diagn Microbiol Infect Dis (2014) 79(4):454–7. doi:10.1016/j.diagmicrobio.2014.05.008
299. D’Amato M, Rea G, Carnevale V, Grimaldi MA, Saponara AR, Rosenthal E, et al. Assessment of thoracic ultrasound in complementary diagnosis and in follow up of community-acquired pneumonia (cap). BMC Med Imaging (2017) 17(1):52. doi:10.1186/s12880-017-0225-5
300. Man SC, Fufezan O, Sas V, Schnell C. Performance of lung ultrasonography for the diagnosis of communityacquired pneumonia in hospitalized children. Med Ultrason (2017) 19(3):276–81. doi:10.11152/mu-1027
301. Jun S, Park B, Seo JB, Lee S, Kim N. Development of a computer-aided differential diagnosis system to distinguish between usual interstitial pneumonia and non-specific interstitial pneumonia using texture- and shape-based hierarchical classifiers on HRCT images. J Digit Imaging (2017) 31(2):235–44. doi:10.1007/s10278-017-0018-y
302. Alcoba G, Keitel K, Maspoli V, Lacroix L, Manzano S, Gehri M, et al. A three-step diagnosis of pediatric pneumonia at the emergency department using clinical predictors, C-reactive protein, and pneumococcal PCR. Eur J Pediatr (2017) 176(6):815–24. doi:10.1007/s00431-017-2913-0
303. Song JY, Eun BW, Nahm MH. Diagnosis of pneumococcal pneumonia: current pitfalls and the way forward. Infect Chemother (2013) 45(4):351–66. doi:10.3947/ic.2013.45.4.351
304. Azzari C, Moriondo M, Indolfi G, Cortimiglia M, Canessa C, Becciolini L, et al. Realtime PCR is more sensitive than multiplex PCR for diagnosis and serotyping in children with culture negative pneumococcal invasive disease. PLoS One (2010) 5(2):e9282. doi:10.1371/journal.pone.0009282
305. Kim L, McGee L, Tomczyk S, Beall B. Biological and epidemiological features of antibiotic-resistant Streptococcus pneumoniae in pre-and post-conjugate vaccine eras: a United States perspective. Clin Microbiol Rev (2016) 29(3):525–52. doi:10.1128/CMR.00058-15
306. Tan SY, Tatsumura Y. Alexander Fleming (1881–1955): discoverer of penicillin. Singapore Med J (2015) 56(7):366. doi:10.11622/smedj.2015105
307. Centers for Disease Control Prevention. Antibiotic Resistance Threats in the United States, 2013. Atlanta, GA: Centers for Disease Control and Prevention (2014). Available from: https://www.cdc.gov/drugresistance/threat-report-2013/index.html (Accessed: March 23, 2017).
308. McGee L, Pletz MW, Fobiwe JP, Klugman KP. Chapter 2 – antibiotic resistance of pneumococci. In: Brown J, Hammerschmidt S, Orihuela C, editors. Streptococcus pneumoniae. Amsterdam: Academic Press (2015). p. 21–40.
309. King DT, Sobhanifar S, Strynadka NC. The mechanisms of resistance to β-lactam antibiotics. In: Gotte M, Berghuis A, Matlashewski G, Wainberg M, Sheppard D, editors. Handbook of Antimicrobial Resistance. New York, NY: Springer (2017). p. 177–201.
310. Cherazard R, Epstein M, Doan TL, Salim T, Bharti S, Smith MA. Antimicrobial resistant Streptococcus pneumoniae: prevalence, mechanisms, and clinical implications. Am J Ther (2017) 24(3):e361–9. doi:10.1097/MJT.0000000000000551
311. Cornick JE, Bentley SD. Streptococcus pneumoniae: the evolution of antimicrobial resistance to beta-lactams, fluoroquinolones and macrolides. Microbes Infect (2012) 14(7–8):573–83. doi:10.1016/j.micinf.2012.01.012
312. Reinert R. The antimicrobial resistance profile of Streptococcus pneumoniae. Clin Microbiol Infect (2009) 15(s3):7–11. doi:10.1111/j.1469-0691.2009.02724.x
313. Butler JC, Breiman RF, Campbell JF, Lipman HB, Broome CV, Facklam RR. Pneumococcal polysaccharide vaccine efficacy. An evaluation of current recommendations. JAMA (1993) 270(15):1826–31. doi:10.1001/jama.1993.03510150060030
314. Smit P, Oberholzer D, Hayden-Smith S, Koornhof HJ, Hilleman MR. Protective efficacy of pneumococcal polysaccharide vaccines. JAMA (1977) 238(24):2613–6. doi:10.1001/jama.1977.03280250039019
315. Centers for Disease Control and Prevention. Recommended immunization schedules for persons aged 0 through 18 years. MMWR Morb Mortal Wkly Rep (2012) 61(5):1–4.
316. US Food and Drug Administration. Pneumovax 23 Prescribing Information. Whitehouse Station, NJ: Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. (2014). Available from: https://www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM257088.pdf (Accessed: March 27, 2017).
317. Pollard AJ, Perrett KP, Beverley PC. Maintaining protection against invasive bacteria with protein-polysaccharide conjugate vaccines. Nat Rev Immunol (2009) 9(3):213–20. doi:10.1038/nri2494
318. Westerink MA, Schroeder HW Jr, Nahm MH. Immune responses to pneumococcal vaccines in children and adults: rationale for age-specific vaccination. Aging Dis (2012) 3(1):51–67.
319. US Food and Drug Administration. Prevnar 13 Prescribing Information. (2016). Available from: https://www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM201669.pdf (Accessed: March 27, 2017).
320. van den Biggelaar AH, Pomat W, Bosco A, Phuanukoonnon S, Devitt CJ, Nadal-Sims MA, et al. Pneumococcal conjugate vaccination at birth in a high-risk setting: no evidence for neonatal T-cell tolerance. Vaccine (2011) 29(33):5414–20. doi:10.1016/j.vaccine.2011.05.065
321. Andrews NJ, Waight PA, Burbidge P, Pearce E, Roalfe L, Zancolli M, et al. Serotype-specific effectiveness and correlates of protection for the 13-valent pneumococcal conjugate vaccine: a postlicensure indirect cohort study. Lancet Infect Dis (2014) 14(9):839–46. doi:10.1016/S1473-3099(14)70822-9
322. Woodland DL. Jump-starting the immune system: prime-boosting comes of age. Trends Immunol (2004) 25(2):98–104. doi:10.1016/j.it.2003.11.009
323. Musher DM, Rueda AM, Nahm MH, Graviss EA, Rodriguez-Barradas MC. Response to pneumococcal polysaccharide and protein-conjugate vaccines singly or sequentially in adults who have recovered from pneumococcal pneumonia. J Infect Dis (2008) 198(7):1019–27. doi:10.1086/591629
324. Ali A, Milad A, Taleb M, Al Ahwel Y, Shehnaz R, Olson D. Use of PCV13 and PPSV23 in adult population: are we following the advisory committee on immunization practices recommendations? Chest (2016) 150(4 Suppl):604A. doi:10.1016/j.chest.2016.08.696
325. Lu S. Heterologous prime-boost vaccination. Curr Opin Immunol (2009) 21(3):346–51. doi:10.1016/j.coi.2009.05.016
326. Centers for Disease Control Prevention. Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine among adults aged ≥65 years: recommendations of the advisory committee on immunization practices (ACIP). MMWR Morb Mortal Wkly Rep (2014) 63(37):822–5.
327. Centers for Disease Control Prevention. Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine among children aged 6-18 years with immunocompromising conditions: recommendations of the advisory committee on immunization practices (ACIP). MMWR Morb Mortal Wkly Rep (2013) 62(25):521–4.
328. Sadlier C, O’Dea S, Bennett K, Dunne J, Conlon N, Bergin C. Immunological efficacy of pneumococcal vaccine strategies in HIV-infected adults: a randomized clinical trial. Sci Rep (2016) 6:32076. doi:10.1038/srep32076
329. Kobayashi M, Bennett NM, Gierke R, Almendares O, Moore MR, Whitney CG, et al. Intervals between PCV13 and PPSV23 vaccines: recommendations of the advisory committee on immunization practices (ACIP). MMWR Morb Mortal Wkly Rep (2015) 64(34):944–7. doi:10.15585/mmwr.mm6434a4
330. Weinberger DM, Harboe ZB, Shapiro ED. Developing better pneumococcal vaccines for adults. JAMA Int Med (2017) 177(3):303–4. doi:10.1001/jamainternmed.2016.8289
331. Centers for Disease Control Prevention. Updated recommendations for prevention of invasive pneumococcal disease among adults using the 23-valent pneumococcal polysaccharide vaccine (PPSV23). MMWR Morb Mortal Wkly Rep (2010) 59(34):1102–6.
332. Shiri T, Datta S, Madan J, Tsertsvadze A, Royle P, Keeling MJ, et al. Indirect effects of childhood pneumococcal conjugate vaccination on invasive pneumococcal disease: a systematic review and meta-analysis. Lancet Glob Health (2017) 5(1):e51–9. doi:10.1016/S2214-109X(16)30306-0
333. Suzuki M, Dhoubhadel BG, Ishifuji T, Yasunami M, Yaegashi M, Asoh N, et al. Serotype-specific effectiveness of 23-valent pneumococcal polysaccharide vaccine against pneumococcal pneumonia in adults aged 65 years or older: a multicentre, prospective, test-negative design study. Lancet Infect Dis (2017) 17(3):313–21. doi:10.1016/S1473-3099(17)30049-X
334. Lu CY, Huang LM. Is pneumococcal serotype replacement impending? Pediatr Neonatol (2016) 57(5):363–4. doi:10.1016/j.pedneo.2015.08.012
335. Mulholland K, Satzke C. Serotype replacement after pneumococcal vaccination. Lancet (2012) 379(9824):1387; author reply 8–9. doi:10.1016/S0140-6736(12)60588-1
336. McFetridge R, Sobanjo-ter Meulen A, Folkerth SD, Hoekstra JA, Dallas M, Hoover PA, et al. Safety, tolerability, and immunogenicity of 15-valent pneumococcal conjugate vaccine in healthy adults. Vaccine (2015) 33(24):2793–9. doi:10.1016/j.vaccine.2015.04.025
337. Pilishvili T, Lexau C, Farley MM, Hadler J, Harrison LH, Bennett NM, et al. Sustained reductions in invasive pneumococcal disease in the era of conjugate vaccine. J Infect Dis (2010) 201(1):32–41. doi:10.1086/648593
338. Granton J, Grossman R. Community-acquired pneumonia in the elderly patient. Clinical features, epidemiology, and treatment. Clin Chest Med (1993) 14(3):537.
339. Hayden LP, Hobbs BD, Cohen RT, Wise RA, Checkley W, Crapo JD, et al. Childhood pneumonia increases risk for chronic obstructive pulmonary disease: the COPDGene study. Respir Res (2015) 16:115. doi:10.1186/s12931-015-0273-8
340. Tiewsoh K, Lodha R, Pandey RM, Broor S, Kalaivani M, Kabra SK. Factors determining the outcome of children hospitalized with severe pneumonia. BMC Pediatr (2009) 9(1):15. doi:10.1186/1471-2431-9-15
341. Palmu AA, Jokinen J, Nieminen H, Rinta-Kokko H, Ruokokoski E, Puumalainen T, et al. Vaccine-preventable disease incidence of pneumococcal conjugate vaccine in the Finnish invasive pneumococcal disease vaccine trial. Vaccine (2018) 36(14):1816–22. doi:10.1016/j.vaccine.2018.02.088
342. Bourcier JE, Paquet J, Seinger M, Gallard E, Redonnet JP, Cheddadi F, et al. Performance comparison of lung ultrasound and chest x-ray for the diagnosis of pneumonia in the ED. Am J Emerg Med (2014) 32(2):115–8. doi:10.1016/j.ajem.2013.10.003
343. Chavez MA, Shams N, Ellington LE, Naithani N, Gilman RH, Steinhoff MC, et al. Lung ultrasound for the diagnosis of pneumonia in adults: a systematic review and meta-analysis. Respir Res (2014) 15(1):50. doi:10.1186/1465-9921-15-50
344. Long L, Zhao H-T, Zhang Z-Y, Wang G-Y, Zhao H-L. Lung ultrasound for the diagnosis of pneumonia in adults: a meta-analysis. Medicine (2017) 96(3):e5713. doi:10.1097/MD.0000000000005713
345. Al-Tarawneh SK, Border MB, Dibble CF, Bencharit S. Defining salivary biomarkers using mass spectrometry-based proteomics: a systematic review. OMICS (2011) 15(6):353–61. doi:10.1089/omi.2010.0134
346. Schnabel R, Fijten R, Smolinska A, Dallinga J, Boumans ML, Stobberingh E, et al. Analysis of volatile organic compounds in exhaled breath to diagnose ventilator-associated pneumonia. Sci Rep (2015) 5:17179. doi:10.1038/srep17179
347. Leeming JP, Cartwright K, Morris R, Martin SA, Smith MD; South-West Pneumococcus Study Group. Diagnosis of invasive pneumococcal infection by serotype-specific urinary antigen detection. J Clin Microbiol (2005) 43(10):4972–6. doi:10.1128/JCM.43.10.4972-4976.2005
348. Safdar A, Shelburne SA, Evans SE, Dickey BF. Inhaled therapeutics for prevention and treatment of pneumonia. Expert Opin Drug Saf (2009) 8(4):435–49. doi:10.1517/14740330903036083
349. Berical AC, Harris D, Dela Cruz CS, Possick JD. Pneumococcal vaccination strategies. An update and perspective. Ann Am Thorac Soc (2016) 13(6):933–44. doi:10.1513/AnnalsATS.201511-778FR
350. Centers for Disease Control Prevention. Vaccine Price List. Atlanta, GA: Centers for Disease Control and Prevention (2017). Available from: https://www.cdc.gov/vaccines/programs/vfc/awardees/vaccine-management/price-list/index.html (Accessed: March 23, 2017).
351. Lee LH, Gu X-X, Nahm MH. Towards new broader spectrum pneumococcal vaccines: the future of pneumococcal disease prevention. Vaccines (Basel) (2014) 2(1):112–28. doi:10.3390/vaccines2010112
352. Moffitt K, Malley R. Rationale and prospects for novel pneumococcal vaccines. Hum Vaccin Immunother (2016) 12(2):383–92. doi:10.1080/21645515.2015.1087625
353. Rodgers GL, Klugman KP. The future of pneumococcal disease prevention. Vaccine (2011) 29:C43–8. doi:10.1016/j.vaccine.2011.07.047
354. Daniels CC, Rogers PD, Shelton CM. A review of pneumococcal vaccines: current polysaccharide vaccine recommendations and future protein antigens. J Pediatr Pharmacol Ther (2016) 21(1):27–35. doi:10.5863/1551-6776-21.1.27
355. Frey SE, Lottenbach KR, Hill H, Blevins TP, Yu Y, Zhang Y, et al. A phase I, dose-escalation trial in adults of three recombinant attenuated Salmonella Typhi vaccine vectors producing Streptococcus pneumoniae surface protein antigen PspA. Vaccine (2013) 31(42):4874–80. doi:10.1016/j.vaccine.2013.07.049
356. Kamtchoua T, Bologa M, Hopfer R, Neveu D, Hu B, Sheng X, et al. Safety and immunogenicity of the pneumococcal pneumolysin derivative PlyD1 in a single-antigen protein vaccine candidate in adults. Vaccine (2013) 31(2):327–33. doi:10.1016/j.vaccine.2012.11.005
357. Khan F, Vidal JE, Khan MA, Ahmed N, Khan MI, Bashir H, et al. Molecular Characterization of Pneumococcal Surface Protein A (PspA), Serotype Distribution and Antibiotic Susceptibility of Streptococcus pneumoniae Strains Isolated from Pakistan. Infect Dis Ther (2018) 7(2):227–89. doi:10.1007/s40121-018-0195-0
358. Ginsburg AS, Nahm MH, Khambaty FM, Alderson MR. Issues and challenges in the development of pneumococcal protein vaccines. Expert Rev Vaccines (2012) 11(3):279–85. doi:10.1586/erv.12.5
359. HogenEsch H, Dunham A, Hansen B, Anderson K, Maisonneuve J-F, Hem SL. Formulation of a killed whole cell pneumococcus vaccine-effect of aluminum adjuvants on the antibody and IL-17 response. J Immune Based Ther Vaccines (2011) 9(1):5. doi:10.1186/1476-8518-9-5
360. Pilishvili T, Bennett NM. Pneumococcal disease prevention among adults: strategies for the use of pneumococcal vaccines. Vaccine (2015) 33:D60–5. doi:10.1016/j.vaccine.2015.05.102
361. Zhang X, Cui J, Wu Y, Wang H, Wang J, Qiu Y, et al. Streptococcus pneumoniae attenuated strain SPY1 with an artificial mineral shell induces humoral and Th17 cellular immunity and protects mice against pneumococcal infection. Front Immunol (2017) 8:1983. doi:10.3389/fimmu.2017.01983
Keywords: pneumococcal, pathogenesis, respiratory, immunology, virulence
Citation: Brooks LRK and Mias GI (2018) Streptococcus pneumoniae’s Virulence and Host Immunity: Aging, Diagnostics, and Prevention. Front. Immunol. 9:1366. doi: 10.3389/fimmu.2018.01366
Received: 20 March 2018; Accepted: 01 June 2018;
Published: 22 June 2018
Edited by:
Amy Rasley, United States Department of Energy (DOE), United StatesReviewed by:
Jessica Lynn Humann, Florida A&M University, United StatesMuhammad Zubair Shabbir, University of Veterinary and Animal Sciences, Pakistan
Copyright: © 2018 Brooks and Mias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George I. Mias, gmias@msu.edu