 Li Bo
Li Bo Tan Guojun*
Tan Guojun*- Department of Neurology, The Second Hospital of Hebei Medical University, Shijiazhuang, China
Many neurologic diseases are related to autoimmune dysfunction and a variety of molecules or reaction pathways are involved in the regulation of immune function of the nervous system. Soluble CD83 (sCD83) is the soluble form of CD83, a specific marker of mature dendritic cell, which has recently been shown to have an immunomodulatory effect. Indoleamine 2,3-dioxygenase (IDO; corresponding enzyme intrahepatic, tryptophan 2,3-dioxygenase, TDO), a rate-limiting enzyme of extrahepatic tryptophan kynurenine pathway (KP) participates in the immunoregulation through a variety of mechanisms solely or with the synergy of sCD83, and the imbalances of metabolites of KP were associated with immune dysfunction. With the complement of sCD83 to IDO-KP, a previously known immunomodulatory axis, this review focused on an expanded neuroimmunomodulation axis: sCD83-IDO-KP and its involvement in nervous system diseases.
Introduction
sCD83 and Immunomodulation
CD83 is a type I transmembrane glycoprotein belonging to the immunoglobulin superfamily members (1), which is mainly expressed on membrane of mature dendritic cells (DCs) of human or mice (2), and considered as a specific marker of DCs. In addition to DCs, CD83 can be expressed on activated T cells (3), B cells, macrophages, and certain brain cells. There are two forms of CD83: membrane-bound CD83 (mCD83) is presented on the surface of the mature DCs membrane, soluble CD83 (sCD83) is released from the DCs membrane and dissolved into the body fluids.
sCD83 was found in healthy human serum and confirmed to possess immunosuppressive properties (4). Staab et al. established effective sCD83 expression and purification regimens with eukaryotic human embryonic kidney 293T cells (5). Guo et al. isolated and purified sCD83 molecules from Pichia pastoris (1). sCD83 inhibited the differentiation process of monocytes into DCs in vitro and during which there was a feedback regulatory mechanism (6). sCD83 also inhibited the maturation of T cells and immature DCs stimulated by mature DCs (7) and the expression and release of CD83 from mature DC membranes induced by lipopolysaccharide. Moreover, the production of autoreactive antibodies was confirmed to be regulated by sCD83 (8). The expression of indoleamine 2,3-dioxygenase (IDO), the rate-limiting enzyme in kynurenine pathway (KP) of tryptophan metabolism, was identified as the major molecular mechanism associated with the protective effects of sCD83 (9). Mediated by IDO and transforming growth factor-β (TGF-β), the immunomodulatory effects of sCD83 were associated with CD4+CD25+Foxp3+ regulatory T cells (Tregs). Studies in vitro had shown that sCD83 induced long-term expression of IDO in DCs through autocrine or paracrine of TGF-β, whereas the latter was an essential cytokine for IDO-dependent immune tolerance (10).
sCD83 was involved in the pathogenesis of immune-related diseases, such as multiple sclerosis and its animal models of experimental autoimmune encephalomyelitis (EAE) (11), systemic lupus erythematosus (SLE) (8), transplant rejection (12, 13) and would probably provide promising approaches for the treatment of autoimmune diseases.
IDO and Immunomodulation
Indoleamine-2,3-dioxygenase, an enzyme containing heme in the cytoplasm, is widely expressed in a variety of mammalian tissue cells, such as endothelial cells, macrophages, microglia, monocytes, DCs, fibroblasts, and certain cancer cells (14, 15). As the only extrahepatic rate-limiting enzyme that catalyzed the oxidative cleavage of indole ring structure in tryptophan (Trp) molecules along the KP (16), IDO was first identified in the intestinal tissue of rabbits (17). IDO expression is mainly distributed in the thymic medulla and the T cell regions of secondary lymphoid organs and dispersedly seen in immune tolerance or immunologically privileged sites such as the placenta, gastrointestinal mucosa, epididymis, anterior chamber, and brain tissue. By catabolizing Trp, cells expressing IDO induced the production of kynurenine metabolites, which orchestrated local and systemic responses to control inflammation, thus maintaining immune privilege (18).
Compared with normal condition, IDO expression in a variety of pathological processes increased significantly. IDO played an important metabolic immunoregulatory role through diverse mechanisms in tumor immune escape (19), maternal–fetal tolerance (20), chronic inflammatory diseases (21), autoimmune diseases (22), and transplantation tolerance (23). Trp is essential for T cell activation and hyperplasia. The induction of IDO by interferon-γ (IFN-γ) leaded to depletion of Trp in co-culture of monocytes and serum (24), which resulted in T cell proliferation arresting at Gl phase and reactivation disorder, thus bringing about the lack of effector T cells (25). With significant cytotoxic effect, l-kynurenine, picolinic acid (PIC), and other Trp catabolic products directly inhibited the proliferation of T cells and induced T cell apoptosis. IDO played immunoregulatory roles with the synergy of Tregs. IDO inhibited the activation of T cells by inducing proliferation of Tregs, resulting in the formation of local immune tolerance. Tregs promoted the production of IFN-γ, which increased the expression of IDO, and the latter strengthened the immune regulation of Tregs through feedback effects (26). Through the positive effects of DCs, Tregs directly promoted self-formation from helper T cells (27). With increased IDO expression upon IFN-γ stimulation in vitro and in vivo, microglial cells were able to block T cell responses by both Trp depletion and induction of Tregs (28). IDO was the mediator of communication between immunomodulation and oxidative stress (29). Activation of IDO-mediated tryptophan metabolism was strongly redox-sensitive and was involved in a variety of immunological diseases (30, 31). In pathological conditions, IDO mediates apoptosis through various mechanisms. By expressing abundant IDO, human umbilical cord-derived mesenchymal stem cells induced T lymphocyte apoptosis with significantly increased expression of caspase 3 (32). 1-Methyl-tryptophan (1-MT), an IDO inhibitor, promoted the apoptosis of hepatic stellate cells by increasing the expression of IFN-γRβ, IRF-1, and FAS (33). Blocking the IDO/aryl hydrocarbon receptor (AhR) metabolic circuitry resulted in enhanced repression of tumor growth by apoptosis of tumor-repopulating cells (34).
Agonists and Inhibitors of IDO
The immunoregulatory role of IDO in KP of tryptophan metabolism is under the tight control of a variety of factors (35) [Figure 1, Lee et al. (36)]. At the same time that IDO was separated and determined in 1967, researchers found that its native substrate, l-tryptophan, could inhibit the activity of IDO at high concentrations (37). Administration of a large dose of Trp could not significantly enhance IDO activity in rat intestine, which might be explained in part by a possible substrate inhibition mechanism (38). The inhibition of IDO activity with l-tryptophan loading exerted protective effects by suppressing the formation of neurotoxic substances and nitric oxide synthase in many central nervous system (CNS) disorders (39).
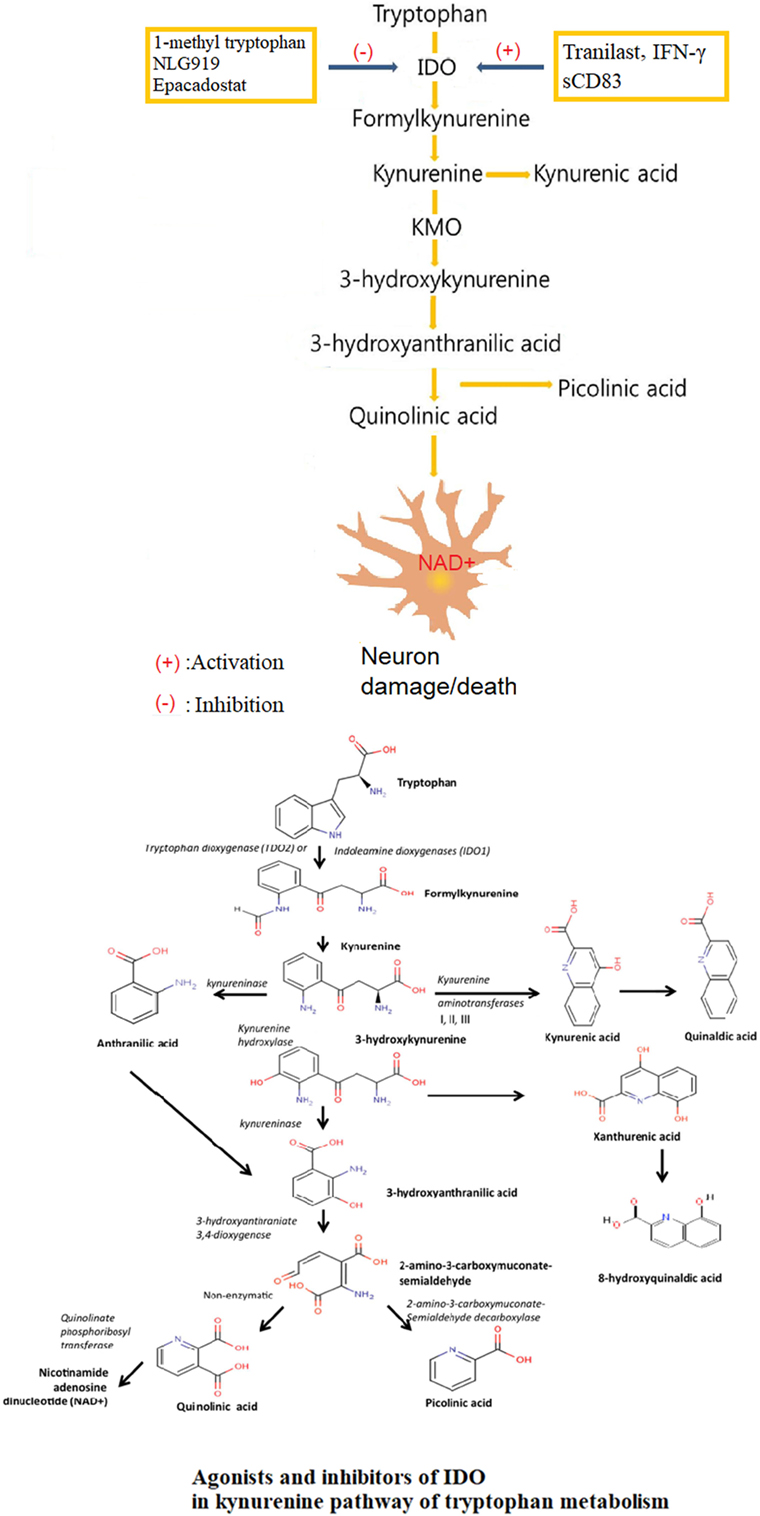
Figure 1. Agonists and inhibitors of indoleamine 2,3-dioxygenase (IDO) in kynurenine pathway (KP) of tryptophan metabolism. Tryptophan is finally converted to nicotinamide adenine dinucleotide (NAD+) through a series of biochemical steps along the KP with IDO as the rate-limiting enzyme and many neuroactive intermediates. The neuroprotectants include kynurenic acid and picolinic, and the neurotoxin, mainly QUIN. The role of IDO in KP is under tight control of a variety of factors, with tranilast, IFN-γ and sCD83 as agonists, and 1-methyl tryptophan (1-MT), NLG919, Epacadostat as inhibitors.
IDO is an enzyme that causes immunosuppression in tumors. In humans, elevated IDO expression in colon and ovarian cancer is associated with a poorer prognosis. Pharmacological, genetic, and immunological methods targeting IDO showed therapeutic benefits (40), and several pharmacological IDO inhibitors were undergoing clinical evaluation (41). Studies confirmed that 1-methyl tryptophan (1-MT) and other tryptophan structural analogs had certain IDO inhibitory activity (42). 1-MT was regarded as a specific inhibitor of IDO (43). NLG919, a newly developed IDO inhibitor, was effective in inhibiting IDO-induced T cell suppression (44). Studies in vitro confirmed that NLG919 promoted tumor tissue atrophy, which was further strengthened by d-1-methyl-tryptophan (D-1-MT) (45). With the significant pharmacological effects, NLG919 had entered phase I clinical trial but with a result of failure. Indoximod (d-1-methyl-tryptophan), a competitive inhibitor of IDO, can induce tumor responses in individuals with metastatic solid tumors (46). Indoximod functions as a tryptophan mimetic that suppresses the downstream effects of IDO activation on amino acid-sensing pathways and mammalian target of rapamycin (mTOR) signaling. Preclinical data support the ability of indoximod to reverse IDO-mediated immune suppression. D-1-MT treatment can reactivate mTOR suppressed by IDO1-mediated Trp depletion in vitro. Epacadostat (INCB024360), a potent and selective IDO enzymatic inhibitor, acted through affecting the downstream metabolites of KP, and a phase 1 dose-escalation study had been initiated in patients with advanced solid tumors (47).
There was an IFN-γ-IDO axis for IDO expression regulation and related immunoregulatory effects in different cell species. As a principal effector, IFN-γ most potently stimulated the expression of IDO (48). IFN-γ induced the gene expression and enhanced the enzyme activity of IDO (49).
N-(3,4-dimethoxycinnamonyl) anthranilic acid (3,4-DAA), known as tranilast in pharmacopeia, was a synthetic anthranilic acid derivative. With anthranilic acid as the core structure, Tranilast shared similar chemical structure with a variety of intermediate metabolites of tryptophan in KP mediated by IDO (50). Researches in vivo confirmed that tranilast upregulated the expression of IDO as an IDO-specific agonist (51). Tranilast had been approved in Japan and South Korea for the treatment of bronchial asthma, atopic dermatitis, allergic conjunctivitis, and scar hyperplasia (52, 53). Subsequent studies indicated that tranilast reversed paralysis in mice with EAE, offering a new strategy for treating TH1-mediated autoimmune disease such as multiple sclerosis (51).
Kynurenine Pathway
Trp is an essential amino acid that cannot be synthesized by the human body and must be obtained from the diet. Only Trp in free form could be transported across the blood–brain barrier (BBB) by non-specific L-type amino acid transporter (54). In the CNS, Trp acted as a precursor in several metabolic pathways, such as for the production of kynurenine (Kyn), serotonin, and melatonin (55), in addition to its role in protein synthesis. In human body, the vast majority of Trp, accounting for 95% of all, was metabolized directing to production of kynurenines (56), which was called KP (57) [Figure 1, Lee et al. (36)]. IDO was the rate-limiting enzyme of KP founded in various cells, including macrophages, microglia, neurons, and astrocytes. The KP presented in most cell types of the CNS, including astrocytes (58), neurons (59), infiltrating macrophages and microglia, oligodendrocytes (60), and endothelial cells (61). The extrahepatic KP became more significant under conditions of immune activation (62). The first and rate-limiting step of the KP was the oxidative cleavage of the 2,3-double bond of the indole ring of L-tryptophan, catalyzed by the iron dependent dioxygenases (63).
The biologically active metabolites of KP mainly included Kyn and its decomposition products such as neuroprotective kynurenic acid (KYNA) (64), neurotoxic 3-hydroxy-L-kynurenine (3-OH-KYN) and its downstream enzyme metabolites, 3-hydroxyanthranilic acid (3HAA) (65), the excitotoxin, and N-methyl-d-aspartate (NMDA) receptor agonist, quinolinic acid (QUIN) (66), PIC (67), and the final product nicotinamide adenine dinucleotide (phosphate) [NAD+ (P+)] or NAD(P)H (68, 69). KYNA was a NMDA receptor antagonist as well as a non-competitive antagonist of the nicotinic receptor α-A7 subtype (70). QUIN, 3-OH-L-KYN, and 3-OH-AA were responsible for the production of the extremely reactive free radical species. The central intermediate of the KP was L-kynurenine (L-KYN), where the metabolic pathway divided into two different branches. L-KYN was transformed to either KYNA or 3-OH-KYN, which was further metabolized in a sequence of enzymatic steps to yield finally NAD that played essential roles in several biological processes such as redox reactions essential for mitochondrial function (71).
QUIN was possibly the most important metabolite of KP in terms of neurobiological activity with a significant increase following inflammation and immune activation (72). QUIN disrupted the integrity of the BBB (73). Neurons within the neocortex, striatum, and hippocampus were sensitive to QUIN, but cerebellar and spinal cord neurons were insensitive (74). QUIN leaded to chronic dysfunction or death of human neuron in a concentration dependent manner through different mechanisms, including protein dysfunction (75), oxidative stress (76), glutamate excitotoxicity (77), mitochondrial dysfunction and energy depletion (78), neuroinflammation (79), autophagy (80), and apoptosis (81) [Figure 2, Lee et al. (36)].
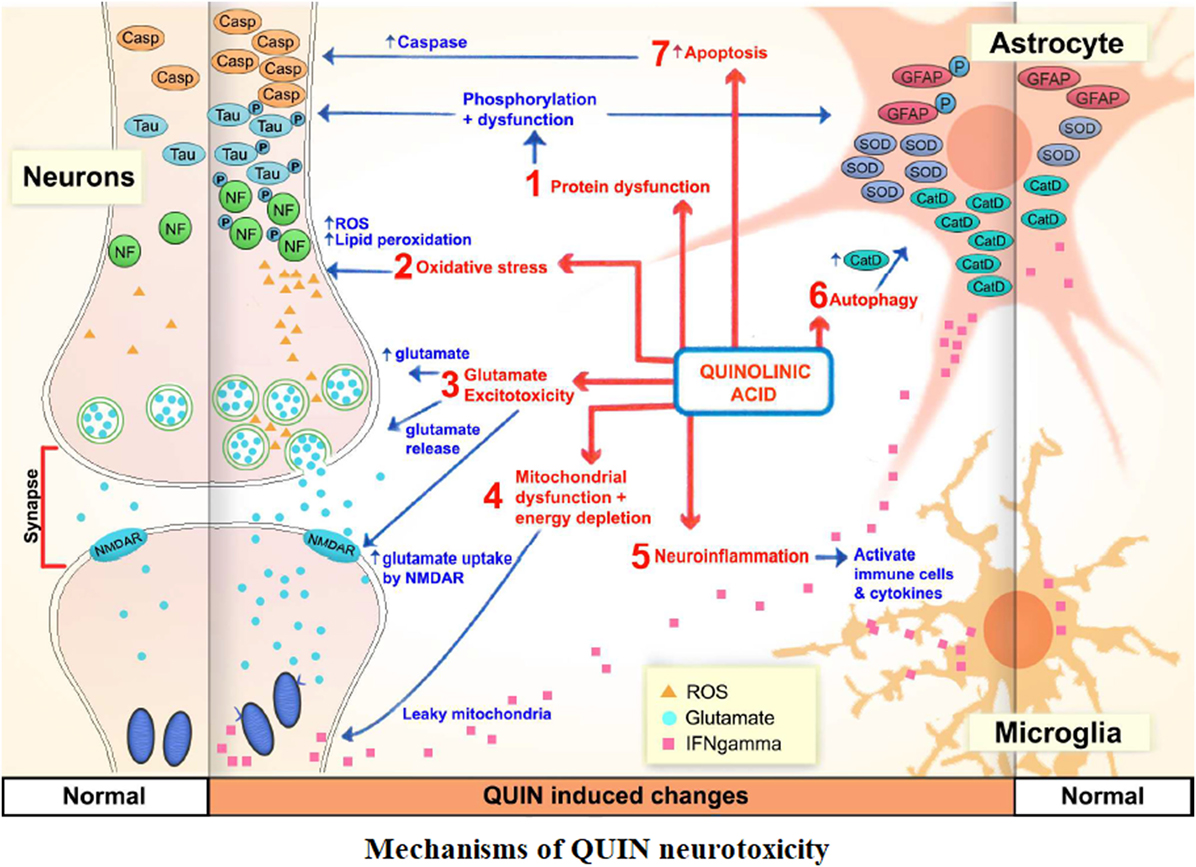
Figure 2. The mechanisms of QUIN neurotoxicity. Increased QUIN expression levels could increase blood–brain barrier permeability, initiate and/or exacerbate a myriad of neurotoxic processes, such as mitochondrial dysfunction, excitotoxicity, oxidative stress, protein phosphorylation, and autophagic processes.
Under pathological conditions, the concentrations of l-tryptophan, vasoactive l-kynurenine, neuroactive KYNA, QUIN, 3-OH-L-KYN, and enzymes responsible for their formation were significantly changed in blood, urine as well as in the brain (82). These Trp metabolites and related enzymes played short-term antimicrobial and long-term immunosuppressive roles (83). Certain Kyn productions from astrocytes played a protective role against neuronal excitotoxic-induced cell death (84). The KP contributed to immunosuppression mainly through the following four mechanisms: Trp depletion and the suppression of mammalian target of rapamycin 1 pathway, a process inhibiting effector T cell (T-eff) function and growth (85); the direct effect of Kyn on the AhR, inducing a predominant immune suppression through Tregs, especially in chronic inflammation (86); promoting the differentiation of CD4 T cells into Tregs expressing cytotoxic T lymphocyte antigen-4 (CTLA-4) and via phosphatase and tensin homolog encoded by a tumor suppressor gene (87); the Kyn-mediated inhibition of IL-2 signaling, which impaired memory CD4 T cell survival (88). Additionally, some of the kynurenines selectively targeted immune cells undergoing activation, resulting in the suppression of T cell proliferation (36). In conjunction with the adenosine/purinergic pathway and immune checkpoints such as CTLA-4 and programmed cell death-1 (89), KP contributed to immune privilege within organs.
With important neurological activity, the KP metabolites exerted neuroprotective or neurotoxic effects by affecting the brain glutamate, cholinergic and dopaminergic neurotransmitter delivery systems (90). Alterations in the KP had clinical and therapeutic implications, and targeting various KP enzymes was a possible strategy for addressing a variety of immune, cognitive, and neurodegenerative diseases (91, 92).
Previous studies have confirmed that both sCD83 and IDO can independently play immunoregulatory roles. Metabolites in KP of tryptophan metabolism also contribute to immunoregulation through cellular or molecular mechanisms in many neuroimmunological diseases. With the confirmation of IDO expression as the main molecular mechanism of the protective effects of CD83, and given the role of IDO as the rate-limiting enzyme in KP, an expanded immunomodulatory axis, sCD83-IDO-KP and its downstream metabolites, is formed, which may provide a more broader perspective for pathogenesis study and more target choices for drug intervention researches of immunological diseases.
KP in Neurologic Diseases
KP in Ischemic Stroke
Ischemic stroke is one of the major disabling and lethal diseases in adults. The pathogenesis of ischemic stroke is the decrease or complete disruption of local cerebral blood flow leading to ischemia and hypoxia, abnormal energy metabolism, cell dysfunction, and tissue necrosis.
Being enhanced both in animal models and patients with acute cerebral infarction, KP activity was significantly associated with severity and prognosis of cerebral infarction (93). Inflammation in cerebral infarction activated KP and induced the expression of IDO (94). Changes in kynurenine metabolites of KP had been confirmed to correlate with the infarct volume and the mortality of stroke patients (95). Earlier studies showed that elevated levels of QUIN after cerebral infarction were associated with reduced nerve cells in different regions of the brain. KYN expression in patients with blood stasis cerebral infarction was significantly higher (96). Studies about L-KYN in rodent stroke models showed conflicting results, the reason for which at present is unclear. l-kynurenine sulfate was protective when issued prior to induction of cerebral ischemia (97). In post-ischemic treatment, l-kynurenine sulfate worsened the histopathological outcome of a mouse stroke model. Deteriorated histopathological changes were observed in a focal ischemia/reperfusion model with distal middle cerebral artery occlusion treated with L-KYN, indicating that post-ischemic treatment with L-KYN might be harmful (98). As an endogenous ligand that mediated AhR activation in the brain after occlusion of the middle cerebral artery, L-KYN inhibited the combination of cyclic adenosine monophosphate response element with protein-dependent signaling that leading to acute brain injury after stroke (99). The inhibition of kynurenine 3-monooxygenase (KMO) activity reduced brain injury in experimental models of ischemic stroke. 3-hydroxyhydroanthranilic acid (3-HAA) and anthranilic acid (AA), downstream metabolites of KYN, were directly related to the volume of cerebral infarction. An synthetic KYNA analog, N-(2-N,N-dimethylaminoethyl)-4-oxo-1H-quinoline-2-carboxamide hydrochloride exerted neuroprotective effects in animal cerebral ischemia models because of the NMDA antagonism effects.
KP in Epilepsy
Epilepsy is a clinical syndrome that characterized by paroxysmal CNS dysfunction, caused by recurrently and suddenly abnormal discharge of brain neurons and manifested as paroxysmal motor sensory abnormalities, autonomic dysfunction, consciousness, and mental state changes.
Neuroactive metabolites of KP with amino acid structures played an important role in epileptic pathophysiology. The incidence of epilepsy was associated with decreased levels of KYN and KYNA in brains of the patients and increased production of KYNA relieved the symptoms of drug-related epilepsy. The changes of KP enzyme activity which resulted in the imbalance between glutamic acid (GLU)/gamma-aminobutyric acid (GABA) in epileptic foci were involved in the pathogenesis of epilepsy (100).
Intravenous injection of QUIN or its direct injection into specific parts of the experimental animal brain could lead to epilepsy-like seizures in rats. Researches confirmed that particular anatomical site of the hippocampus was sensitive to QUIN excitotoxicity (101). In gliocytes proliferating in epileptic foci, the levels of QUIN synthetase, 3-hydroxyanthranilate oxidase, were relatively increased; the activity of quinolinate phosphoribosyltransferase (QPRTase), a QUIN degrading enzyme, decreased. PIC enhanced epilepsy activity and was essential for the pathogenesis of drug-resistant epilepsy (102). As naturally occurring vesicular glutamate transporters (VGLUTs) inhibitors, Kynurenine and xanthurenic acid inhibited the uptake of glutamate by presynaptic membrane into vesicles, thereby affecting synaptic function. VGLUTs dysfunction was involved in many neuropsychiatric disorders such as epilepsy and schizophrenia (103). KP metabolism of Trp provided a reasonable explanation for the correlation between epilepsy and depression. Neurobiochemistry and behavioral studies found that minocycline, an IDO inhibitor, could promote antiepileptic effects of valproate in a dose-dependent manner, and improve depression associated with epilepsy (104), Quercetin combined with levetiracetam had a similar effect (105).
KP in Alzheimer’s Disease (AD)
Alzheimer’s disease is one of the most prevalent progressive neurodegenerative diseases, the most common type of dementia with insidious onset mainly affecting the elderly (106). The characteristic clinical manifestations include the deterioration of memory function, progressive decline in daily living capacity, various neuropsychiatric symptoms and behavioral disorders. Extracellular amyloid-β accumulation and intracellular tau deposition are the significant features of the pathomechanism related to AD (107).
Neuroendocrine disorders served as one of the pathogenesis of AD (108). Glutamate excitotoxicity, neuroinflammation, cerebrovascular dysfunctions, and mitochondrial metabolic disorders were also factors known to contribute to the neurodegenerative process (109). The enhanced IDO expression and increased KYN/TRP ratio reflecting the activity of KP in tryptophan metabolism were observed in serum of AD patients, exhibiting an inverse correlation with the cognitive decline (110). Imbalances in the KP had been observed in many disorders with cognitive decline including AD, and influencing this delicate balance might be of therapeutic value (111).
Induced by upregulated generation of IDO mediated by interferon (IFN)-gamma and other proinflammatory cytokines, the production of various neurotoxic substances of KP involved in the pathogenesis of AD were increased (112, 113). QUIN was particularly associated with the pathogenesis of AD (59). Studies confirmed that QUIN immunoreactivity in cerebral hippocampus of AD patients was related to disease progression. Increased levels of QUIN were observed in both the cerebrospinal fluid (CSF) and serum of AD patients, causing significant neuronal damage either by direct activation of NMDA receptors or the release of endogenous glutamate (114). QUIN promoted the expression of IDO in small keratinocytes and astrocytes, which in turn affected KP. In brains of AD patients, QUIN accelerated cell damage by oxidative stress, aggravated the pathological changes as an inducer of β-amyloid peptide and promoted the release of extracellular glutamate which could directly cause neurotoxicity. QUIN was not only co-localized with hyperphosphorylated tau in the AD cortex, but also capable of inducing tau phosphorylation in primary neurone cultures (115). KYN-NAD was involved in AD since Aβ 1-42, a cleavage product of amyloid precursor protein, induced the production of QUIN with neurotoxic concentrations from macrophages and microglia (116).
Kynurenic acid levels were altered in the brain and CSF of AD patients with its significance differently described in various research conclusions (117). In brains of pathologically confirmed AD patients, decreased levels of L-KYN, 3-OH-KYN, and elevated activity of KYNA and kynurenine aminotransferase-I (KAT-I) had been detected in the striatum and caudate nucleus (118). Kling et al. showed that KYNA had a positive impact on AD by promoting the activity and gene expression of enkephalinaseand and facilitating Aβ degradation (119). KYNA exerted its neuroprotective effects by promoting amyloid degradation and increasing the neuronal cell survival through induction of the gene expression of neprilysin (120). Other mechanisms involved in the neuroprotective effects of KYNA and its derivatives included preferentially inhibiting of extrasynaptic NMDA and α7 nicotinic acetylcholine receptors while sparing the synaptic NMDA-mediated currents (121).
The level of 3-hydroxy-kynurenine (3-HK), a downstream metabolite of tryptophan in KP, increased in the brain and serum of AD patients (122). Through increasing β-amyloid protein accumulation, glial activation, oxidative stress and positive feedback regulation mechanism in KP, 3-HK exacerbated the neurodegenerative lesions of AD. 3-HK inhibited the activity of mitochondrial respiratory chain enzyme complex I, II, III, resulting in energy metabolism disorder in neurons and subsequent nerve tissue injuries.
Studies indicated that blocking KYN pathway could protect mouse models of AD. The application of KP enzyme inhibitors or its metabolite analogs had been shown to be effective in the treatment of AD in preclinical studies. The extracts of rhizoma coptidis demonstrated therapeutic effects on AD through inhibiting the activity of microglia and astrocytes and the expression of IDO, blocking the loss of neurons and reducing the formation of amyloid plaques in AβPP/PSI transgenic mice (123). Administered together with a transport inhibitor of KYNA known as probenecid, L-KYN prevented the morphological alteration and cellular damage caused by soluble Aβ which further resulted in a significant improvement of the spatial memory (124).
KP in Multiple Sclerosis
Multiple sclerosis (MS) is a demyelinating disease of the CNS, which is also regarded as a neurodegenerative disease triggered by inflammation. Multiple demyelinating plaques in the CNS white matter are the pathological characteristics of MS. Demyelination may partially or wholly involve the white matter of the lateral ventricles, optic nerve, spinal cord, cerebellum, and the brain stem, among which, periventricular lesions are especially typical. Demyelination founded in the cerebral cortex of MS patients might be the cause of cognitive impairment (125). The pathogeny of MS has not been elucidated. The particular effects of various KP metabolites on nervous system (126), imbalance between neuroprotective and neurotoxic intermediates of KP and immunomodulatory effects mediated by IDO were all involved in the pathogenesis of MS (127). Recent studies suggest that abnormalities in the KP may be associated with the switch from early mild stage MS to debilitating progressive forms of MS and that analysis of KP metabolites in MS patient serum may have application as MS disease biomarkers (128).
Associated with disease condition, there was activation of the KP in MS. Decreased levels of Trp in serum and CSF of MS patients suggested an increase in KP activity, the increased serum L-KYN levels in MS patients treated with IFN-β were associated with disease remission (129). KYNA is the only known endogenous ionotropic glutamate receptor antagonist that inhibits presynaptic α7 nicotinic acetylcholine receptors, regulates presynaptic glutamate release. Changed with patient’s condition, the fluctuation of KYNA levels in CSF demonstrated the probable neuroprotective effects on MS. QUIN was a potent agonist of NMDA receptors that could promote the onset of MS through inducing glutamate excitotoxicity, lipid peroxidation, and oxidative stress injury (130). Elevated levels of QUIN in the spinal cord of EAE rats were associated with an acute disease course (131). Multiple studies had consistently found structural changes in a variety of cellular proteins associated with QUIN that leading to the death of oligodendrocytes, neurons, and astrocytes. The QUIN/KYNA ratio has strong correlation with the disability and severity of MS patients (128). 3-HK, another potential neurotoxin, was found to be significantly higher in the serum of MS patients (128). QUIN and 3-HAA analogs selectively leaded to the apoptosis of Th1 cells, thereby further inducing immune tolerance. Increased levels of 3-HK in the spinal cord of EAE rats distinctly raised the production of free radicals.
Indoleamine 2,3-dioxygenase gene expression and activity were predictive of relapse in MS patients, indicating that KP orchestrated self-protective mechanisms that inhibited antigen-specific immune responses in the CNS (132). Studies confirmed that immunoregulatory effects of IDO were involved in the pathogenesis of EAE. Enhanced inflammatory responses of Th1/Th17 exacerbated EAE in IDO-deficient mice (22). IDO activation increased the level of KYNA, thereby inhibiting the proliferation of reactive T lymphocytes. The administration of 1-methyl-tryptophan (1-MT), a specific IDO inhibitor, deteriorated the condition of EAE (133).
Immunomodulatory effects mediated by KP and IDO were crucial to the pathogenesis and treatment of MS (134). Inhibitors of KP enzymes, NMDA antagonist KYNA, and its pharmacological derivatives provided new options for MS treatment (135). Enzymes and metabolites related to KP had become potential therapeutic targets for MS (16), such as endogenous tryptophan metabolites and their structural analogs (cinnabarinic acid, tranilast), IDO inhibitors (1-MT and berberine). The administration of 3HAA, a downstream KP metabolite, reduced EAE severity through increasing the proportion of Tregs and inhibiting Th1/Th17 response. Tranilast was an oral active synthetic Trp metabolite that inhibited myelin-specific T cell proliferation and the release of proinflammatory cytokines from Th1 cells, relieved inflammatory responses, modulated immunosuppressive effects, thereby reducing disease relapse frequency and the disease severity during relapse. Sundaram et al. (136) found that IDO inhibitors significantly reduced the levels of QUIN in cultured cells and eliminated oligodendrocyte apoptosis. As an endogenous Trp metabolite, cinnabarinic acid was a partial agonist of metabotropic glutamate receptor 4 (mGluR4), which has a protective effect on EAE. Treated mGlu4-deficient mice with cinnabarinic acid caused a turning of immune response to Tregs (137).
KP in Vascular Cognitive Impairment (VCI)
Vascular cognitive impairment is defined as a syndrome with evidence of clinical stroke or subclinical vascular brain injury and cognitive impairment affecting at least one cognitive domain, including attention, memory, language, visuospatial skills, and abstract reasoning (138, 139). Microvascular inflammation, cerebrovascular dysfunction, and neurodegeneration were all involved in the development of VCI as pathogenic mechanisms (2, 140).
Previous studies described an IFN-γ-IDO-KYN and inducible nitric oxide synthase (iNOS) pathway through which microvascular immune inflammation resulted in VCI. The impact of chronic inflammation on VCI was mediated by the unique ability of IFN-γ to transcriptionally induce the rate-limiting enzymes of tryptophan–kynurenine metabolic pathways with the synergistic action of tumor necrosis factor-α (TNF-α) (141). Triggered by upregulated production of IFN-γ in periphery (macrophages) and brain (glia), a merger of tryptophan–KP into inflammation cascade involved in VCI (142). IFN-γ-induced upregulation of IDO increased the levels of certain metabolites of KYN associated with cognitive impairment. The significantly increased levels of iNOS in CSF of patients with vascular dementia (VD) were involved in VCI by mediating excitatory neurotransmission and affecting synaptic plasticity. Under pathological conditions, excessive release of NO induced by positive feedback loop in inflammation that mediated by IDO leaded to neuronal oxidative stress injury which affected the learning and memory function of brain (16).
Indoleamine 2,3-dioxygenase and KYN metabolites increased markedly with deterioration of cognitive impairment accompanied with immune activation and nerve injury (8). The long-term activation of the KP resulted in an imbalance in critical neuroactive compounds involving the reduction of tryptophan and increase of tryptophan metabolites, which had definite implications in cognitive impairment (143). KP metabolites, some kynurenines (e.g., quinolinic and PICs) had also been implicated in VCI (55). Changes of kynurenine metabolites had also been suggested to correlate with the post-stroke cognitive impairment. Activation of IDO and increased production of kynurenine metabolites had been observed in post-stroke cognitive impairment patients. One study demonstrated an association between elevated kynurenine/tryptophan (K/T) ratios and the extent of cognitive impairment in patients with acute ischemic stroke, and what is more, the K/T ratio was described as an important biomarker of VCI (144). Kynurenine was also identified as candidate diagnostic biomarker for VCI in a metabolomics study, which provided a novel strategy for stratifying stroke patients with cognitive impairment using serum-based metabolite markers and might be of great importance in further investigating the pathological mechanisms of VCI (145).
VCI was also identified as age-associated neuroendocrine disorders. The transcriptional induction and activation of IDO shifted tryptophan metabolism from serotonin synthesis to formation of “kynurenines,” which might contribute to development of VCI via their apoptotic, neurotoxic, pro-oxidative effects, and the upregulation of iNOS, phospholipase A2, arachidonic acid, prostaglandin, 5-lipoxygenase, and leukotriene cascade (146).
KP in Depression
Depression is commonly characterized by anhedonia (loss of interest or pleasure) for 2 weeks or longer and the presence of at least four of the following persistent symptoms: weight loss or gain, sleep disturbances, psychomotor agitation or retardation, fatigue, worthlessness or inappropriate guilt, diminished concentration, or indecisiveness (147). As one of the most common mental disorders and social issues worldwide, depression causes a low quality of life or even suicide for many people (148). However, the underlying mechanisms of depression remain elusive and the effectiveness of the currently used antidepressants is still far from satisfactory (149).
The hypothesis of cytokine-induced sickness behavior, which was recognized within a few years of the cloning and expression of interferon-alpha, IL-1 and IL-2, may provide some meaningful references. The proinflammatory cytokines activated in the brain can induce common symptoms of sickness, such as loss of appetite, sleepiness, withdrawal from normal social activities, and fatigue. These cytokine actions may offer some of the first clues about the pathophysiology of certain mental health disorders, including depression.
Tryptophan degradation and its role in the availability of serotonin and the setting of inflammation had brought attention to the KP for potential mechanism and treatment studies of depression (150). Kynurenine metabolism was hypothesized to be a key mechanism that linked inflammation and depression. Increased inflammation and toxic KP activation, including excitotoxicity mediated by NMDA receptor, as well as decreased 5-HT levels were associated with pathophysiology of depression (151, 152). Bay-Richter et al. (153) revealed that levels of QUIN increased and KYNA decreased over time in suicidal depression patients versus healthy controls (153). The activation of KP as a marker of inflammation presented in a subgroup of individuals with major depressive disorder (MDD). The abnormalities in the concentrations of KP metabolites (KYNA and QUIN) were associated with reduced volume of striatum, which played the central role of in motivated behavior, reward processing, and anhedonia. The KYN/TRP ratio was inversely associated with striatal volumes in the MDD sample (154). Research revealed that relative neurotoxic shift in the balance of kynurenine metabolites with reduced KYNA/quinolinic acid (QA) was associated with sleep disturbance in the currently depressed patients. It was considered that altered kynurenine metabolism might also molecularly link sleep disturbance and depression (155).
Depression is predominately female disease but with unclear reason. With different disease forms such as premenstrual depression, postnatal depression, and perimenopausal depression, depression is obviously related to hormonal fluctuations. The successful treatment of certain common types of depression by estrogens provided further evidence. Meier et al. found that a reduction in KynA concentrations in women might constitute a vulnerability factor that partly explained the higher incidence of depression in females. Furthermore, the significant association between oral contraceptive (OC) use and reduced KynA could conceivably partially account for the epidemiological association between OC use and depression in females (156). Characterized by low tryptophan levels, increased breakdown toward kynurenine and a downstream shift toward the 3-OH-KYN arm, women in the first 2 months of the physiological postpartum period were at very high risk for the first onset of acute and severe depression (157). de Bie et al. found that progesterone attenuated interferon-γ-induced KP activity in macrophages with increased of neuroprotective KYNA levels and reduced the inflammatory marker neopterin, which could partly explain how hormones were involved in the pathogenesis of depression (158).
Patients with various chronic diseases of different systems might suffer from depression. Depression occurred more frequently in patients after stroke than in the general population (159), with a varied incidence from 18 to 61%. Several studies reported an increase activation of both tryptophan-2, 3-Dioxygenase (TDO)/IDO and also kynurenine aminotransferases, which leaded to an augmentation of 3-HK, QUIN, L-Kyn, and KYNA production, which finally induced ROS generation and alterations in glutamatergic neurotransmission in stroke patients (160). A reduction in 5-HT production and an increased KP catabolism was clearly observed in poststroke depression. Neuropsychiatric symptoms are among the earliest manifestations of SLE. Depression-like behavior was evident in the murine model of lupus. Increased levels of KP metabolites (kynurenine, 3-hydroxy-kynurenine, 3-hydroxynthranilic acid, and QUIN) in cortex and hippocampus of these murine models suggested that activation of the KP might among the potential pathophysiological mechanisms responsible for neuropsychiatric symptoms of SLE (NP-SLE) (161). Positive correlations were drawn between KYN, QUIN, KYNA, and specific pro-inflammatory immunological variables in the CSF, and depressive symptoms in patients with hepatitis on IFN-α treatment. In addition, a significant relationship was found between depression developed in patients with chronic viral hepatitis after IFN therapy and especially the type of IFN-α (162). With significantly higher kynurenine levels and KYN/TRP ratios post-chemotherapy, many women with breast cancer experienced more severe depression after chemotherapy treatment (163). Chronic inflammation was the likely common instigator between cardiovascular pathology and depression. Certain pro-inflammatory substances released by macrophages and microglia upregulated the rate-limiting enzymes in the KP, resulted in the formation of neurotoxic metabolites. Inflammation was closely associated with endothelial dysfunction in cardiovascular disease, a preamble to atherosclerosis and atherothrombosis, which had also been detected in depression (164).
KP in Parkinson’s Disease (PD)
Parkinson’s disease is a chronic progressive neurodegenerative disorder characterized by loss of dopaminergic neurons in the midbrain and presence of localized neuroinflammation and protein inclusions called Lewy bodies in the midbrain several years before the actual onset of symptoms (165, 166). PD is normally identified by motor symptoms (bradykinesia, rigidity, resting tremor, and gait disturbances), which are believed to be largely related to the loss of nigral dopamine neurons. Moreover, patients also frequently exhibit a range of non-motor disturbances including constipation, hyposmia, depression, and cognitive decline (167), being more debilitating than motor signs and worsening with disease progression (168).
The detailed pathogenesis of PD is unclear, but several mechanisms have been proposed including neuroinflammation, glutamatergic neurotoxicity, the dysregulation of the KP and alterations in serotoninergic and melatoninergic pathways (35). The KP of TRP catabolism is implicated in the inflammatory and neurotoxic events in Parkinsonism. Studies demonstrated that increased TRP catabolism positively correlated to the increases of inflammatory markers (IL-6, CRP, and MCP-1) in both the periphery and CSF of PD patients, which were associated with non-motor symptoms of PD such as fatigue, depression and cognitive impairment (169). Lewitt et al. assayed CSF of pathologically verified PD subjects using ultra-high-performance liquid and gas chromatography linked to mass spectrometry. The results showed that the mean 3-HK-KYN concentration was increased and the mean oxidized glutathione was decreased, providing direct support for the involvement of the KP and excitotoxicity in development of PD (170).
Several neuroactive compounds are produced through the KP, among which KYNA and QUIN are intensively studied in PD. KYNA possessed antioxidant properties and was a non-competitive antagonist of a7-nicotinic acetylcholine receptors at physiological levels regulating the levels of acetylcholine, dopamine, and glutamate in the CNS (171). QUIN could activate the NMDA receptor-signaling pathway leading to excitotoxicity and amplify the inflammatory response (164).
The increase in KYNA not only modulated glutamate release from cortical areas to striatum but also directly acted on the NMDA receptors as antagonist, thereby limiting glutamate excitotoxicity. Ogawa et al. reported for the first time that TRP/KYN and KYNA/TRP ratios were significantly increased in the frontal cortex, putamen of PD patients (172). There were also increased Kynurenine/Tryptophan (K/T) ratios in both serum and CSF of PD patients compared to controls (173, 174). By driving tryptophan down the KP, alterations in gut microbiota were widely accepted as relevant to the etiology, course, and treatment of many neuropsychiatric disorders, including PD (175). Investigators observed altered levels of kynurenine metabolites in PD patients with L-3, 4-Dihydroxyphenylalanine (L-DOPA)-induced dyskinesia. There were obvious increases of the 3-HK-MYN/KYNA ratio and 5-hydroxytryptophan (5-HTP) levels, but decrease of anthranilic acid levels in plasma and CSF of this patient group. Through affecting the KP of tryptophan metabolism, L-DOPA might play a role in the development of L-DOPA-induced dyskinesia as the most effective drug in the symptomatic treatment of Parkinson’s disease (176).
QUIN, a metabolite of the KP of tryptophan catabolism, plays a role in the oxidative stress associated with many neurological disorders including Parkinson’s disease. Intrastriatal administration of QUIN in rats was reported to be able to induce significant behavioral changes including involuntary movements (177). Through activating NMDA receptor, endogenous QUIN released by microglia played a crucial role in mediating the progressive loss of dopaminergic neurons in PD (178). Recent researches indicated that QUIN was directly involved in mood and behavioral changes in PD patients (179). The deficits in spatial reference memory had also been observed in rats following the development of QUIN lesions, suggesting that QUIN could induce cognitive deficits in PD patients. Apart from its excitotoxic effects, QUIN could also enhance lipid peroxidation in an iron (II) dependent manner (180).
The abnormalities of enzyme activity of KP are observed in PD. Diminished immunoreactivity of KAT-I, the KP enzyme, which leaded to KYNA formation in the pars compacta of the substania nigra, was found in mouse models of PD (181). The enzyme aminocarboxymuconate-semialdehyde-decarboxylase (ACMSD) located at a key branch-point of the KP, limited the production of the neurotoxin QUIN with inflammatory properties. The missense mutation in the ACMSD gene predicted to disrupt enzyme function in typical PD individual, suggesting that this enzyme might influence PD pathogenesis (167). Vilas et al. reported a novel ACMSD mutation resulting in the change of p.Glu298Lys amino-acid in a sporadic PD patient, which was not present in neurologically normal population, suggesting that not only common genetic variability but also rare variants in ACMSD alone might increase the risk of PD (182).
Using PD animal models, the modulation of the KP by enhancing endogenous KYNA and/or decreasing QUIN production was demonstrated to be a potential therapeutic strategy for PD (183). Administration of KYNA directly into the globus pallidus internus of parkinsonian non-primate protected against 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine hydrochloride (MPTP)-induced toxicity. In addition, systemic administration of exogenous KYNA showed very limited therapeutic efficacy because KYNA had poor permeability across the BBB and a very short biological half-life. KYNA injection into the brain could protect dopaminergic neurons against QUIN or NMDA-mediated excitotoxicity. Silva-Adaya et al. reported that L-kynurenine (L-KYN) pretreatment had a protective effect on locomotor asymmetry, striatal reactive gliosis, and neurodegeneration, and changes of dopamine levels in rodent PD models (184). Further study demonstrated that synthetic kynurenine analogs had neuroprotective effects on mice model of PD (185). Silva-Adaya et al. showed that co-administration of the main KYNA precursor, L-KYN and an inhibitor of organic anion transporter could increase KYNA and result in the reversal of glutamate-induced excitotoxicity in 6-hydroxydopamine (6-OHDA)-induced PD rats (184). Analogs of L-KYN and KYNA with long half-life such as 4-Cl-L-KYN, 7- Cl-KYNA and more recently 2-(2-N, N-dimethylaminoethylamine- 1-carboyl)-1H-quinolin-4-one hydrochloride had been designed to enhance their stability and pharmacological therapeutic properties (186–188). Several preclinical studies showed that activation of group II- metabotropic glutamate receptor (mGluR) and group III-mGluR were potentially important drug targets to provide both symptom relief and neuroprotection in PD implying that, apart from KYNA, these two KP metabolites might have therapeutic importance in PD (189). When administered intraventricularly with systemic L-KYN, kynureninase inhibitors elevated KYNA levels in brain and showed protective effects against QUIN-induced toxicity in PD rats. Recent work had highlighted the therapeutic potential of inhibiting two critical regulatory enzymes in KP, KMO, and TDO. In fly models of Parkinson’s disease, Breda et al. provided genetic evidence that inhibition of TDO or KMO improved locomotor performance and ameliorated shortened life span (190). Parasram revealed that phenolic compounds, a class of phytochemicals, including flavonoids and diarylheptanoids, had been shown to reduce striatal lesion size, reduce inflammation, and prevent lipid peroxidation caused by QUIN in PD and other oxidative stress related neurological disorders (191).
A growing body of evidence suggested that the KP and its metabolites of KP were involved in the pathogenesis of PD. Modification of altered KP represented important targets to prevent the progression of the underlying neurodegeneration in PD. Drugs targeting the KP are pivotal to further understanding of the therapeutic value of KP manipulation in patients with PD.
Conclusion
Dysfunction of neuroimmunomodulation and destruction of immune homeostasis are the pathogenesis of various neurological diseases in which a variety of immune molecules or reaction pathways were involved. With the determination of immunomodulatory property of sCD83 and related mechanisms, an expanded neuroimmunomodulation axis, sCD83-IDO-KP, was formed, which had been proved to have important impacts on the development of various neurological diseases, including ischemic stroke, epilepsy, AD, multiple sclerosis, PD, and depression. Further researches are still needed on doubts about the downstream effector molecules, other neuroactive effects of these cells and molecules besides immunoregulation and the specific processes of their interaction with immune effector cells. The answers to the above mysteries will also provide more new strategies for the prevention and treatment of neurological autoimmune diseases and other immune-related diseases.
Author Contributions
LB is responsible for literature search, data collection, and article writing. TG determines the structure of the article and content distribution. GL determines the content and is responsible for the article review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Natural Science Fund of Hebei Province: H2012206047.
Abbreviations
sCD83, soluble CD83; IDO, indoleamine 2, 3-dioxygenase; KP, kynurenine pathway; DCs, dendritic cells; Tregs, CD4+CD25+Foxp3+ regulatory T cells; 1-MT, 1-methyl-l-tryptophan; 3, 4-DAA, N-(3, 4-dimethoxycinnamonyl) anthranilic acid; KYN, kynurenine; KYNA, kynurenic acid; 3-OH-KYN, neurotoxic 3-hydroxy-L-kynurenine; 3HAA, 3-hydroxyanthranilic acid; NMDA, N-methyl-d-aspartate; LPS, lipopolysacchatide; EAE, experimental autoimmune encephalomyelitis; QUIN, quinolinic acid; PIC, picolinic acid; BBB, blood–brain barrier.
References
1. Guo Y, Li R, Song X, Zhong Y, Wang C, Jia H, et al. The expression and characterization of functionally active soluble CD83 by Pichia pastoris using high-density fermentation. PLoS One (2014) 9:e89264. doi:10.1371/journal.pone.0089264
2. Zhou LJ, Tedder TF. Human blood dendritic cells selectively express CD83, a member of the immunoglobulin superfamily. J Immunol (1995) 154:3821–35.
3. Kreiser S, Eckhardt J, Kuhnt C, Stein M, Krzyzak L, Seitz C, et al. Murine CD83-positive T cells mediate suppressor functions in vitro and in vivo. Immunobiology (2015) 220:270–9. doi:10.1016/j.imbio.2014.08.005
4. Fujimoto Y, Tu L, Miller AS, Bock C, Fujimoto M, Doyle C, et al. CD83 expression influences CD4+ T cell development in the thymus. Cell (2002) 108:755–67. doi:10.1016/S0092-8674(02)00673-6
5. Staab C, Mühl-Zürbes P, Steinkasserer A, Kummer M. Eukaryotic expression of functionally active recombinant soluble CD83 from HEK 293T cells. Immunobiology (2010) 215:849–54. doi:10.1016/j.imbio.2010.05.029
6. Lin H, Liang S, Zhong Z, Wen J, Li W, Wang L, et al. Soluble CD83 inhibits human monocyte differentiation into dendritic cells in vitro. Cell Immunol (2014) 292:25–31. doi:10.1016/j.cellimm.2014.08.003
7. Zinser E, Lechmann M, Golka A, Lutz MB, Steinkasserer A. Prevention and treatment of experimental autoimmune encephalomyelitis by soluble CD83. J Exp Med (2004) 200:345–51. doi:10.1084/jem.20030973
8. Starke C, Steinkasserer A, Voll RE, Zinser E. Soluble human CD83 ameliorates lupus in NZB/W F1 mice. Immunobiology (2013) 218:1411–5. doi:10.1016/j.imbio.2013.06.002
9. Eckhardt J, Kreiser S, Döbbeler M, Nicolette C, DeBenedette MA, Tcherepanova IY, et al. Soluble CD83 ameliorates experimental colitis in mice. Mucosal Immunol (2014) 7:1006–18. doi:10.1038/mi.2013.119
10. Bock F, Rössner S, Onderka J, Lechmann M, Pallotta MT, Fallarino F, et al. Topical application of soluble CD83 induces IDO-mediated immune modulation, increases Foxp3+ T cells, and prolongs allogeneic corneal graft survival. J Immunol (2013) 191:1965–75. doi:10.4049/jimmunol.1201531
11. Pashine A, Göpfert U, Chen J, Hoffmann E, Dietrich PS, Peng SL. Failed efficacy of soluble human CD83-Ig in allogeneic mixed lymphocyte reactions and experimental autoimmune encephalomyelitis: implications for a lack of therapeutic potential. Immunol Lett (2008) 115:9–15. doi:10.1016/j.imlet.2007.10.015
12. Lan Z, Lian D, Liu W, Arp J, Charlton B, Ge W, et al. Prevention of chronic renal allograft rejection by soluble CD83. Transplantation (2010) 90:1278–85. doi:10.1097/TP.0b013e318200005c
13. Xu JF, Huang BJ, Yin H, Xiong P, Feng W, Xu Y, et al. A limited course of soluble CD83 delays acute cellular rejection of MHC-mismatched mouse skin allografts. Transpl Int (2007) 20:266–76. doi:10.1111/j.1432-2277.2006.00426.x
14. Currier AR, Ziegler MH, Riley MM, Babcock TA, Telbis VP, Carlin JM. Tumor necrosis factor-alpha and lipopolysaccharide enhance interferon-induced antichlamydial indoleamine dioxygenase activity independently. J Interferon Cytokine Res (2000) 20:369–76. doi:10.1089/107999000312306
15. Mándi Y, Vécsei L. The kynurenine system and immunoregulation. J Neural Transm (Vienna) (2012) 119:197–209. doi:10.1007/s00702-011-0681-y
16. Rajda C, Majláth Z, Pukoli D, Vécsei L. Kynurenines and multiple sclerosis: the dialogue between the immune system and the central nervous system. Int J Mol Sci (2015) 16:18270–82. doi:10.3390/ijms160818270
18. Routy JP, Routy B, Graziani GM, Mehraj V. The kynurenine pathway is a double-edged sword in immune-privileged sites and in cancer: implications for immunotherapy. Int J Tryptophan Res (2016) 9:67–77. doi:10.4137/IJTR.S38355
19. Théate I, van Baren N, Pilotte L, Moulin P, Larrieu P, Renauld JC, et al. Extensive profiling of the expression of the indoleamine 2, 3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol Res (2015) 3:161–72. doi:10.1158/2326-6066.CIR-14-0137
20. Bianchi PKFDC, Leandro RM, Poscai AN, Yoshinaga T, Gonçalez PO, Kfoury Junior JR. Progesterone decreases in vitro indoleamine 2, 3-dioxygenase expression in dendritic and CD4+ cells from maternal-fetal interface of rats. Immunol Invest (2017) 46:447–59. doi:10.1080/08820139.2017.1296856
21. Ott M, Litzenburger UM, Rauschenbach KJ, Bunse L, Ochs K, Sahm F, et al. Suppression of TDO-mediated tryptophan catabolism in glioblastoma cells by a steroid-responsive FKBP52-dependent pathway. Glia (2015) 63:78–90. doi:10.1002/glia.22734
22. Yan Y, Zhang GX, Gran B, Fallarino F, Yu S, Li H, et al. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J Immunol (2010) 185:5953–61. doi:10.4049/jimmunol.1001628
23. Lu Y, Giver CR, Sharma A, Li JM, Darlak KA, Owens LM, et al. IFN-gamma and indoleamine 2, 3-dioxygenase signaling between donor dendritic cells and T cells regulates graft versus host and graft versus leukemia activity. Blood (2012) 119:1075–85. doi:10.1182/blood-2010-12-322891
24. Fuchs D, Forsman A, Hagberg L, Larsson M, Norkrans G, Reibnegger G, et al. Immune activation and decreased tryptophan in patients with HIV-1 infection. J Interferon Res (1990) 10:599–603. doi:10.1089/jir.1990.10.599
25. Rueda CM, Jackson CM, Chougnet CA. Regulatory T-cell-mediated suppression of conventional T-cells and dendritic cells by different cAMP intracellular pathways. Front Immunol (2016) 7:216. doi:10.3389/fimmu.2016.00216
26. Harden JL, Egilmez NK. Indoleamine 2, 3-dioxygenase and dendritic cell tolerogenicity. Immunol Invest (2012) 41:738–64. doi:10.3109/08820139.2012.676122
27. Julliard W, Fechner JH, Mezrich JD. The aryl hydrocarbon receptor meets immunology: friend or foe? A tittle of roth. Front Immunol (2014) 5:458. doi:10.3389/fimmu.2014.00458
28. Kwidzinski E, Bunse J, Kovac AD, Ullrich O, Zipp F, Nitsch R, et al. IDO (indolamine 2,3-dioxygenase) expression and function in the CNS. Adv Exp Med Biol (2003) 527:113–8. doi:10.1007/978-1-4615-0135-0_13
29. Böttcher M, Hofmann AD, Bruns H, Haibach M, Loschinski R, Saul D, et al. Mesenchymal stromal cells disrupt mTOR-signaling and aerobic glycolysis during T-cell activation. Stem Cells (2016) 34:516–21. doi:10.1002/stem.2234
30. Gostner JM, Becker K, Ueberall F, Fuchs D. The good and bad of antioxidant foods: an immunological perspective. Food Chem Toxicol (2015) 80:72–9. doi:10.1016/j.fct.2015.02.012
31. Abuelezz SA, Hendawy N, Magdy Y. Targeting oxidative stress, cytokines and serotonin interactions via indoleamine 2, 3-dioxygenase by coenzyme Q10: role in suppressing depressive like behavior in rats. J Neuroimmune Pharmacol (2017) 12:277–91. doi:10.1007/s11481-016-9712-7
32. Li X, Xu Z, Bai J, Yang S, Zhao S, Zhang Y, et al. Umbilical cord tissue-derived mesenchymal stem cells induce T lymphocyte apoptosis and cell cycle arrest by expression of indoleamine 2, 3-dioxygenase. Stem Cells Int (2016) 2016:7495135. doi:10.1155/2016/7495135
33. Oh JE, Shim KY, Lee JI, Choi SI, Baik SK, Eom YW. 1-Methyl-L-tryptophan promotes the apoptosis of hepatic stellate cells arrested by interferon-γ by increasing the expression of IFN-γRβ, IRF-1 and FAS. Int J Mol Med (2017) 40:576–82. doi:10.3892/ijmm.2017.3043
34. Liu Y, Liang X, Yin X, Lv J, Tang K, Ma J, et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-γ-induced immunologic dormancy of tumor-repopulating cells. Nat Commun (2017) 8:15207. doi:10.1038/ncomms15207
35. Campbell BM, Charych E, Lee AW, Möller T. Kynurenines in CNS disease: regulation by inflammatory cytokines. Front Neurosci (2014) 8:12. doi:10.3389/fnins.2014.00012
36. Lee JM, Tan V, Lovejoy D, Braidy N, Rowe DB, Brew BJ, et al. Involvement of quinolinic acid in the neuropathogenesis of amyotrophic lateral sclerosis. Neuropharmacology (2017) 112:346–64. doi:10.1016/j.neuropharm.2016.05.011
37. Weber B, Nickel E, Horn M, Nienhaus K, Nienhaus GU. Substrate inhibition in human indoleamine 2, 3-dioxygenase. J Phys Chem Lett (2014) 5:756–61. doi:10.1021/jz500220k
38. Cook JS, Pogson CI, Smith SA. Indoleamine 2, 3-dioxygenase. A new, rapid, sensitive radiometric assay and its application to the study of the enzyme in rat tissues. Biochem J (1980) 189:461–6. doi:10.1042/bj1890461
39. Chiarugi A, Dello Sbarba P, Paccagnini A, Donnini S, Filippi S, Moroni F. Combined inhibition of indoleamine 2, 3-dioxygenase and nitric oxide synthase modulates neurotoxin release by interferon-gamma-activated macrophages. J Leukoc Biol (2000) 68:260–6. doi:10.1189/jlb.68.2.260
40. Vacchelli E, Aranda F, Eggermont A, Sautès-Fridman C, Tartour E, Kennedy EP, et al. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology (2014) 3:e957994. doi:10.4161/21624011.2014.957994
41. Buqué A, Bloy N, Aranda F, Cremer I, Eggermont A, Fridman WH, et al. Trial Watch-Small molecules targeting the immunological tumor microenvironment for cancer therapy. Oncoimmunology (2016) 5:e1149674. doi:10.1080/2162402X.2016.1149674
42. Tourino MC, de Oliveira EM, Bellé LP, Knebel FH, Albuquerque RC, Dörr FA, et al. Tryptamine and dimethyltryptamine inhibit indoleamine 2, 3-dioxygenase and increase the tumor-reactive effect of peripheral blood mononuclear cells. Cell Biochem Funct (2013) 31:361–4. doi:10.1002/cbf.2980
43. Löb S, Königsrainer A, Rammensee HG, Opelz G, Terness P. Inhibitors of indoleamine 2, 3-dioxygenase for cancer therapy: can we see the wood for the trees? Nat Rev Cancer (2009) 9:445–52. doi:10.1038/nrc2639
44. Mario M, Sanjeev K, Jesse W, et al. Fused imidazole derivatives useful as IDO inhibitors [P]. (2016) Patent WO2012142237, 2012-20-10.
45. Nayak A, Hao Z, Sadek R, Vahanian N, Ramsey WJ, Kennedy E, et al. A phase I study of NLG919 for adult patients with recurrent advanced solid tumors. J Immunother Cancer (2014) 2:250. doi:10.1186/2051-1426-2-S3-P250
46. Soliman H, Ringash J, Jiang H, Singh K, Kim J, Dinniwell R, et al. Phase II trial of palliative radiotherapy for hepatocellular carcinoma and liver metastases. J Clin Oncol (2013) 31:3980–6. doi:10.1200/JCO.2013.49.9202
47. Beatty GL, O’Dwyer PJ, Clark J, Shi JG, Bowman KJ, Scherle PA, et al. First-in-human phase I study of the oral inhibitor of indoleamine 2,3-dioxygenase-1 epacadostat (INCB024360) in patients with advanced solid malignancies. Clin Cancer Res (2017) 23:3269–76. doi:10.1158/1078-0432.CCR-16-2272
48. Jones SP, Franco NF, Varney B, Sundaram G, Brown DA, de Bie J, et al. Expression of the kynurenine pathway in human peripheral blood mononuclear cells: implications for inflammatory and neurodegenerative disease. PLoS One (2015) 10:e0131389. doi:10.1371/journal.pone.0131389
49. Li Q, Harden JL, Anderson CD, Egilmez NK. Tolerogenic phenotype of IFN-γ-induced IDO+ dendritic cells is maintained via an autocrine IDO-kynurenine/AhR-IDO loop. J Immunol (2016) 197:962–70. doi:10.4049/jimmunol.1502615
50. Cole JE, Astola N, Cribbs AP, Goddard ME, Park I, Green P, et al. Indoleamine 2, 3-dioxygenase-1 is protective in atherosclerosis and its metabolites provide new opportunities for drug development. Proc Natl Acad Sci U S A (2015) 112:13033–8. doi:10.1073/pnas.1517820112
51. Platten M, Ho PP, Youssef S, Fontoura P, Garren H, Hur EM, et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science (2005) 310:850–5. doi:10.1126/science.1117634
52. Ammendola M, Leporini C, Marech I, Gadaleta CD, Scognamillo G, Sacco R, et al. Targeting mast cells tryptase in tumor microenvironment: a potential antiangiogenetic strategy. Biomed Res Int (2014) 2014:154702. doi:10.1155/2014/154702
53. Azuma H, Banno K, Yoshimura T. Pharmacological properties of N-(3′,4′-dimethoxycinnamoyl) anthranilic acid (N-5′), a new anti-atopic agent. Br J Pharmacol (1976) 58:483–8. doi:10.1111/j.1476-5381.1976.tb08614.x
54. Hargreaves KM, Pardridge WM. Neutral amino acid transport at the human blood-brain barrier. J Biol Chem (1988) 263:19392–7.
55. Oxenkrug GF. Genetic and hormonal regulation of tryptophan kynurenine metabolism: implications for vascular cognitive impairment, major depressive disorder, and aging. Ann N Y Acad Sci (2007) 1122:35–49. doi:10.1196/annals.1403.003
56. Moffett JR, Namboodiri MA. Tryptophan and the immune response. Immunol Cell Biol (2003) 81:247–65. doi:10.1046/j.1440-1711.2003.t01-1-01177.x
57. Leklem JE. Quantitative aspects of tryptophan metabolism in humans and other species: a review. Am J Clin Nutr (1971) 24:659–72. doi:10.1093/ajcn/24.6.659
58. Guillemin GJ, Smith DG, Kerr SJ, Smythe GA, Kapoor V, Armati PJ, et al. Characterisation of kynurenine pathway metabolism in human astrocytes and implications in neuropathogenesis. Redox Rep (2000) 5:108–11. doi:10.1179/135100000101535375
59. Guillemin GJ, Cullen KM, Lim CK, Smythe GA, Garner B, Kapoor V, et al. Characterization of the kynurenine pathway in human neurons. J Neurosci (2007) 27:12884–92. doi:10.1523/JNEUROSCI.4101-07.2007
60. Lim CK, Smythe GA, Stocker R, Brew BJ, Guillemin GJ. Characterization of the kynurenine pathway in human oligodendrocytes. Int Congr Ser (2007) 1304:213–7. doi:10.1016/j.ics.2007.07.011
61. Owe-Young R, Webster NL, Mukhtar M, Pomerantz RJ, Smythe G, Walker D, et al. Kynurenine pathway metabolism in human blood-brain-barrier cells: implications for immune tolerance and neurotoxicity. J Neurochem (2008) 105:1346–57. doi:10.1111/j.1471-4159.2008.05241.x
62. Badawy AA. Tryptophan metabolism, disposition and utilization in pregnancy. Biosci Rep (2015) 35:e00261. doi:10.1042/BSR20150197
63. Aitken JB, Austin CJ, Hunt NH, Ball HJ, Lay PA. The Fe-heme structure of metindoleamine 2, 3-dioxygenase-2 determined by X-ray absorption fine structure. Biochem Biophys Res Commun (2014) 450:25–9. doi:10.1016/j.bbrc.2014.05.054
64. Reyes Ocampo J, Lugo Huitrón R, González-Esquivel D, Ugalde-Muñiz P, Jiménez-Anguiano A, Pineda B, et al. Kynurenines with neuroactive and redox properties: relevance to aging and brain diseases. Oxid Med Cell Longev (2014) 2014:646909. doi:10.1155/2014/646909
65. Goldstein LE, Leopold MC, Huang X, Atwood CS, Saunders AJ, Hartshorn M, et al. 3-Hydroxykynurenine and 3-hydroxyanthranilic acid generate hydrogen peroxide and promote alpha-crystallin cross-linking by metal ion reduction. Biochemistry (2000) 39:7266–75. doi:10.1021/bi992997s
66. Lugo-Huitrón R, Ugalde Muñiz P, Pineda B, Pedraza-Chaverrí J, Ríos C, Pérez-de la Cruz V. Quinolinic acid: an endogenous neurotoxin with multiple targets. Oxid Med Cell Longev (2013) 2013:104024. doi:10.1155/2013/104024
67. Jhamandas K, Boegman RJ, Beninger RJ, Bialik M. Quinolinate-induced cortical cholinergic damage: modulation by tryptophan metabolites. Brain Res (1990) 529:185–91. doi:10.1016/0006-8993(90)90826-W
68. Ball HJ, Jusof FF, Bakmiwewa SM, Hunt NH, Yuasa HJ. Tryptophan catabolizing enzymes-party of three. Front Immunol (2014) 5:485. doi:10.3389/fimmu.2014.00485
69. Jiang T, Sun Y, Yin Z, Feng S, Sun L, Li Z. Research progress of indoleamine 2, 3-dioxygenase inhibitors. Future Med Chem (2015) 7:185–201. doi:10.4155/fmc.14.151
70. Hilmas C, Pereira EF, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci (2001) 21:7463–73. doi:10.1523/JNEUROSCI.21-19-07463.2001
71. Badawy AA. Pellagra and alcoholism: a biochemical perspective. Alcohol Alcohol (2014) 49:238–50. doi:10.1093/alcalc/agu010
72. Chen Y, Guillemin GJ. Kynurenine pathway metabolites in humans: disease and healthy states. Int J Tryptophan Res (2009) 2:1–19. doi:10.4137/IJTR.S2097
73. Steiner J, Bogerts B, Sarnyai Z, Walter M, Gos T, Bernstein HG, et al. Bridging the gap between the immune and glutamate hypotheses of schizophrenia and major depression: potential role of glial NMDA receptor modulators and impaired blood-brain barrier integrity. World J Biol Psychiatry (2012) 13:482–92. doi:10.3109/15622975.2011.583941
74. Kumar U. Characterization of striatal cultures with the effect of QUIN and NMDA. Neurosci Res (2004) 49:29–38. doi:10.1016/j.neures.2004.01.011
75. Pierozan P, Zamoner A, Soska AK, Silvestrin RB, Loureiro SO, Heimfarth L, et al. Acute intrastriatal administration of quinolinic acid provokes hyperphosphorylation of cytoskeletal intermediate filament proteins in astrocytes and neurons of rats. Exp Neurol (2010) 224:188–96. doi:10.1016/j.expneurol.2010.03.009
76. Vandresen-Filho S, Martins WC, Bertoldo DB, Mancini G, De Bem AF, Tasca CI. Cerebral cortex, hippocampus, striatum and cerebellum show differential susceptibility to quinolinic acid-induced oxidative stress. Neurol Sci (2015) 36:1449–56. doi:10.1007/s10072-015-2180-7
77. Ting KK, Brew BJ, Guillemin GJ. Effect of quinolinic acid on human astrocytes morphology and functions: implications in Alzheimer’s disease. J Neuroinflammation (2009) 6:36. doi:10.1186/1742-2094-6-36
78. Pérez-De La Cruz V, Carrillo-Mora P, Santamaría A. Quinolinic Acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. Int J Tryptophan Res (2012) 5:1–8. doi:10.4137/IJTR.S8158
79. Chen Y, Brew BJ, Guillemin GJ. Characterization of the kynurenine pathway in NSC-34 cell line: implications for amyotrophic lateral sclerosis. J Neurochem (2011) 118:816–25. doi:10.1111/j.1471-4159.2010.07159.x
80. Braidy N, Brew BJ, Inestrosa NC, Chung R, Sachdev P, Guillemin GJ. Changes in Cathepsin D and Beclin-1 mRNA and protein expression by the excitotoxin quinolinic acid in human astrocytes and neurons. Metab Brain Dis (2014) 29:873–83. doi:10.1007/s11011-014-9557-9
81. Guillemin GJ, Wang L, Brew BJ. Quinolinic acid selectively induces apoptosis of human astrocytes: potential role in AIDS dementia complex. J Neuroinflammation (2005) 2:16. doi:10.1186/1742-2094-2-16
82. Bohár Z, Toldi J, Fülöp F, Vécsei L. Changing the face of kynurenines and neurotoxicity: therapeutic considerations. Int J Mol Sci (2015) 16:9772–93. doi:10.3390/ijms16059772
83. Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature (2014) 511:184–90. doi:10.1038/nature13323
84. Guillemin GJ, Smythe G, Takikawa O, Brew BJ. Expression of indoleamine 2, 3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia (2005) 49:15–23. doi:10.1002/glia.20090
85. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol (2006) 176:6752–61. doi:10.4049/jimmunol.176.11.6752
86. Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature (2015) 523:221–5. doi:10.1038/nature14452
87. Sharma MD, Shinde R, McGaha TL, Huang L, Holmgaard RB, Wolchok JD, et al. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci Adv (2015) 1:e1500845. doi:10.1126/sciadv.1500845
88. Dagenais-Lussier X, Aounallah M, Mehraj V, El-Far M, Tremblay C, Sekaly RP, et al. Kynurenine reduces memory CD4 T-cell survival by interfering with interleukin-2 signaling early during HIV-1 infection. J Virol (2016) 90:7967–79. doi:10.1128/JVI.00994-16
89. Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity (2016) 44:1255–69. doi:10.1016/j.immuni.2016.06.001
90. Lee YE, An J, Lee KH, Kim SS, Song HJ, Pyeon H, et al. Correction: the synergistic local immunosuppressive effects of neural stem cells expressing indoleamine 2, 3-Dioxygenase (IDO) in an experimental autoimmune encephalomyelitis (EAE) animal model. PLoS One (2016) 11:e0148720. doi:10.1371/journal.pone.0148720
91. Jayawickrama GS, Sadig RR, Sun G, Nematollahi A, Nadvi NA, Hanrahan JR, et al. Kynurenine aminotransferases and the prospects of inhibitors for the treatment of schizophrenia. Curr Med Chem (2015) 22:2902–18. doi:10.2174/0929867322666150608094054
92. Stone TW, Darlington LG. The kynurenine pathway as a therapeutic target in cognitive and neurodegenerative disorders. Br J Pharmacol (2013) 169:1211–27. doi:10.1111/bph.12230
93. Brouns R, Verkerk R, Aerts T, De Surgeloose D, Wauters A, Scharpé S, et al. The role of tryptophan catabolism along the kynurenine pathway in acute ischemic stroke. Neurochem Res (2010) 35:1315–22. doi:10.1007/s11064-010-0187-2
94. Jackman KA, Brait VH, Wang Y, Maghzal GJ, Ball HJ, McKenzie G, et al. Vascular expression, activity and function of indoleamine 2,3-dioxygenase-1 following cerebral ischaemia-reperfusion in mice. Naunyn Schmiedebergs Arch Pharmacol (2011) 383:471–81. doi:10.1007/s00210-011-0611-4
95. Darlington LG, Mackay GM, Forrest CM, Stoy N, George C, Stone TW. Altered kynurenine metabolism correlates with infarct volume in stroke. Eur J Neurosci (2007) 26:2211–21. doi:10.1111/j.1460-9568.2007.05838.x
96. Cha MH, Kim MJ, Jung J, Kim JH, Lee MS, Kim MS. Metabolomic analysis of clinical plasma from cerebral infarction patients presenting with blood stasis. Evid Based Complement Alternat Med (2015) 2015:453423. doi:10.1155/2015/453423
97. Gigler G, Szénási G, Simó A, Lévay G, Hársing LG Jr, Sas K, et al. Neuroprotective effect of L-kynurenine sulfate administered before focal cerebral ischemia in mice and global cerebral ischemia in gerbils. Eur J Pharmacol (2007) 564:116–22. doi:10.1016/j.ejphar.2007.02.029
98. Gellért L, Knapp L, Németh K, Herédi J, Varga D, Oláh G, et al. Post-ischemic treatment with L-kynurenine sulfate exacerbates neuronal damage after transient middle cerebral artery occlusion. Neuroscience (2013) 247:95–101. doi:10.1016/j.neuroscience.2013.04.063
99. Cuartero MI, Ballesteros I, de la Parra J, Harkin AL, Abautret-Daly A, Sherwin E, et al. L-kynurenine/aryl hydrocarbon receptor pathway mediates brain damage after experimental stroke. Circulation (2014) 130:2040–51. doi:10.1161/CIRCULATIONAHA.114.011394
100. Majewski M, Kozlowska A, Thoene M, Lepiarczyk E, Grzegorzewski WJ. Overview of the role of vitamins and minerals on the kynurenine pathway in health and disease. J Physiol Pharmacol (2016) 67:3–19.
101. St’astný F, Skultétyová I, Pliss L, Jezová D. Quinolinic acid enhances permeability of rat brain microvessels to plasma albumin. Brain Res Bull (2000) 53:415–20. doi:10.1016/S0361-9230(00)00368-3
102. Cioczek-Czuczwar A, Czuczwar P, Turski WA, Parada-Turska J. Influence of picolinic acid on seizure susceptibility in mice. Pharmacol Rep (2017) 69:77–80. doi:10.1016/j.pharep.2016.10.009
103. Neale SA, Copeland CS, Salt TE. Effect of VGLUT inhibitors on glutamatergic synaptic transmission in the rodent hippocampus and prefrontal cortex. Neurochem Int (2014) 73:159–65. doi:10.1016/j.neuint.2013.10.001
104. Singh T, Goel RK. Adjuvant indoleamine 2, 3-dioxygenase enzyme inhibition for comprehensive management of epilepsy and comorbid depression. Eur J Pharmacol (2016) 784:111–20. doi:10.1016/j.ejphar.2016.05.019
105. Singh T, Kaur T, Goel RK. Adjuvant quercetin therapy for combined treatment of epilepsy and comorbid depression. Neurochem Int (2017) 104:27–33. doi:10.1016/j.neuint.2016.12.023
106. Nestor PJ, Scheltens P, Hodges JR. Advances in the early detection of Alzheimer’s disease. Nat Med (2004) 10:S34–41. doi:10.1038/nrn1433
107. Majláth Z, Toldi J, Vécsei L. The potential role of kynurenines in Alzheimer’s disease: pathomechanism and therapeutic possibilities by influencing the glutamate receptors. J Neural Transm (Vienna) (2014) 121:881–9. doi:10.1007/s00702-013-1135-5
108. Wu W, Nicolazzo JA, Wen L, Chung R, Stankovic R, Bao SS, et al. Expression of tryptophan 2, 3-dioxygenase and production of kynurenine pathway metabolites in triple transgenic mice and human Alzheimer’s disease brain. PLoS One (2013) 8:e59749. doi:10.1371/journal.pone.0059749
109. Zádori D, Klivényi P, Szalárdy L, Fülöp F, Toldi J, Vécsei L. Mitochondrial disturbances, excitotoxicity, neuroinflammation and kynurenines: novel therapeutic strategies for neurodegenerative disorders. J Neurol Sci (2012) 322:187–91. doi:10.1016/j.jns.2012.06.004
110. Widner B, Leblhuber F, Walli J, Tilz GP, Demel U, Fuchs D. Tryptophan degradation and immune activation in Alzheimer’s disease. J Neural Transm (Vienna) (2000) 107:343–53. doi:10.1007/s007020050029
111. Majláth Z, Tajti J, Vécsei L. Kynurenines and other novel therapeutic strategies in the treatment of dementia. Ther Adv Neurol Disord (2013) 6:386–97. doi:10.1177/1756285613494989
112. Lovelace MD, Varney B, Sundaram G, Lennon MJ, Lim CK, Jacobs K, et al. Recent evidence for an expanded role of the kynurenine pathway of tryptophan metabolism in neurological diseases. Neuropharmacology (2017) 112:373–88. doi:10.1016/j.neuropharm.2016.03.024
113. Souza LC, Jesse CR, Del Fabbro L, de Gomes MG, Goes ATR, Filho CB, et al. Swimming exercise prevents behavioural disturbances induced by an intracerebroventricular injection of amyloid-beta1-42 peptide through modulation of cytokine/NF-kappaB pathway and indoleamine-2, 3-dioxygenase in mouse brain. Behav Brain Res (2017) 331:1–13. doi:10.1016/j.bbr.2017.05.024
114. Connick JH, Stone TW. The effect of kainic, quinolinic and beta-kainic acids on the release of endogenous amino acids from rat brain slices. Biochem Pharmacol (1986) 35:3631–5. doi:10.1016/0006-2952(86)90636-2
115. Rahman A, Ting K, Cullen KM, Braidy N, Brew BJ, Guillemin GJ. The excitotoxin quinolinic acid induces tau phosphorylation in human neurons. PLoS One (2009) 4:e6344. doi:10.1371/journal.pone.0006344
116. Kincses ZT, Toldi J, Vécsei L. Kynurenines, neurodegeneration and Alzheimer’s disease. J Cell Mol Med (2010) 14:2045–54. doi:10.1111/j.1582-4934.2010.01123.x
117. Wennström M, Nielsen HM, Orhan F, Londos E, Minthon L, Erhardt S. Kynurenic Acid levels in cerebrospinal fluid from patients with Alzheimer’s disease or dementia with Lewy bodies. Int J Tryptophan Res (2014) 2014:1–7. doi:10.4137/IJTR.S13958
118. Baran H, Jellinger K, Deecke L. Kynurenine metabolism in Alzheimer’s disease. J Neural Transm (Vienna) (1999) 106:165–81. doi:10.1007/s007020050149
119. Kling MA, Trojanowski JQ, Wolk DA, Lee VM, Arnold SE. Vascular disease and dementias: paradigm shifts to drive research in new directions. Alzheimers Dement (2013) 9:76–92. doi:10.1016/j.jalz.2012.02.007
120. Klein C, Patte-Mensah C, Taleb O, Bourguignon JJ, Schmitt M, Bihel F, et al. The neuroprotector kynurenic acid increases neuronal cell survival through neprilysin induction. Neuropharmacology (2013) 70:254–60. doi:10.1016/j.neuropharm.2013.02.006
121. Demeter I, Nagy K, Farkas T, Kis Z, Kocsis K, Knapp L, et al. Paradox effects of kynurenines on LTP induction in the Wistar rat. An in vivo study. Neurosci Lett (2013) 553:138–41. doi:10.1016/j.neulet.2013.08.028
122. Schwarz MJ, Guillemin GJ, Teipel SJ, Buerger K, Hampel H. Increased 3-hydroxykynurenine serum concentrations differentiate Alzheimer’s disease patients from controls. Eur Arch Psychiatry Clin Neurosci (2013) 263:345–52. doi:10.1007/s00406-012-0384-x
123. Yu D, Tao BB, Yang YY, Du LS, Yang SS, He XJ, et al. The IDO inhibitor coptisine ameliorates cognitive impairment in a mouse model of Alzheimer’s disease. J Alzheimers Dis (2015) 43:291–302. doi:10.3233/JAD-140414
124. Carrillo-Mora P, Méndez-Cuesta LA, Pérez-De La Cruz V, Fortoul-van Der Goes TI, Santamaría A. Protective effect of systemic L-kynurenine and probenecid administration on behavioural and morphological alterations induced by toxic soluble amyloid beta (25-35) in rat hippocampus. Behav Brain Res (2010) 210:240–50. doi:10.1016/j.bbr.2010.02.041
125. Nielsen AS, Kinkel RP, Madigan N, Tinelli E, Benner T, Mainero C. Contribution of cortical lesion subtypes at 7T MRI to physical and cognitive performance in MS. Neurology (2013) 81:641–9. doi:10.1212/WNL.0b013e3182a08ce8
126. Mancuso R, Hernis A, Agostini S, Rovaris M, Caputo D, Fuchs D, et al. Indoleamine 2, 3-dioxygenase (IDO) expression and activity in relapsing-remitting multiple sclerosis. PLoS One (2015) 10:e0130715. doi:10.1371/journal.pone.0130715
127. Vécsei L. Nature reviews drug discovery: editorial article of neuroscientists from Szeged about kynurenine. Ideggyogy Sz (2014) 67:70.
128. Lim CK, Bilgin A, Lovejoy DB, Tan V, Bustamante S, Taylor BV, et al. Kynurenine pathway metabolomics predicts and provides mechanistic insight into multiple sclerosis progression. Sci Rep (2017) 7:41473. doi:10.1038/srep41473
129. Sadowska-Bartosz I, Adamczyk-Sowa M, Gajewska A, Bartosz G. Oxidative modification of blood serum proteins in multiple sclerosis after interferon or mitoxantrone treatment. J Neuroimmunol (2014) 266:67–74. doi:10.1016/j.jneuroim.2013.11.005
130. Lim CK, Brew BJ, Sundaram G, Guillemin GJ. Understanding the roles of the kynurenine pathway in multiple sclerosis progression. Int J Tryptophan Res (2010) 3:157–67. doi:10.4137/IJTR.S4294
131. Flanagan EM, Erickson JB, Viveros OH, Chang SY, Reinhard JF Jr. Neurotoxin quinolinic acid is selectively elevated in spinal cords of rats with experimental allergic encephalomyelitis. J Neurochem (1995) 64:1192–6. doi:10.1046/j.1471-4159.1995.64031192.x
132. Lovelace MD, Varney B, Sundaram G, Franco NF, Ng ML, Pai S, et al. Current evidence for a role of the kynurenine pathway of tryptophan metabolism in multiple sclerosis. Front Immunol (2016) 7:246. doi:10.3389/fimmu.2016.00246
133. Kwidzinski E, Bunse J, Aktas O, Richter D, Mutlu L, Zipp F, et al. Indolamine 2, 3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. FASEB J (2005) 19:1347–9. doi:10.1096/fj.04-3228fje
134. Vécsei L, Szalárdy L, Fülöp F, Toldi J. Kynurenines in the CNS: recent advances and new questions. Nat Rev Drug Discov (2013) 12:64–82. doi:10.1038/nrd3793
135. Dounay AB, Tuttle JB, Verhoest PR. Challenges and opportunities in the discovery of new therapeutics targeting the kynurenine pathway. J Med Chem (2015) 58:8762–82. doi:10.1021/acs.jmedchem.5b00461
136. Sundaram G, Brew BJ, Jones SP, Adams S, Lim CK, Guillemin GJ. Quinolinic acid toxicity on oligodendroglial cells: relevance for multiple sclerosis and therapeutic strategies. J Neuroinflammation (2014) 11:204. doi:10.1186/s12974-014-0204-5
137. Fazio F, Zappulla C, Notartomaso S, Busceti C, Bessede A, Scarselli P, et al. Cinnabarinic acid, an endogenous agonist of type-4 metabotropic glutamate receptor, suppresses experimental autoimmune encephalomyelitis in mice. Neuropharmacology (2014) 81:237–43. doi:10.1016/j.neuropharm.2014.02.011
138. Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, et al. American Heart Association Stroke Council, council on epidemiology and prevention, council on cardiovascular nursing, council on cardiovascular radiology and intervention, and council on cardiovascular surgery and anesthesia. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke (2011) 42:2672–713. doi:10.1161/STR.0b013e3182299496
139. Cumming TB, Marshall RS, Lazar RM. Stroke, cognitive deficits, and rehabilitation: still an incomplete picture. Int J Stroke (2013) 8:38–45. doi:10.1111/j.1747-4949.2012.00972.x
140. Iadecola C. The pathobiology of vascular dementia. Neuron (2013) 80:844–66. doi:10.1016/j.neuron.2013.10.008
141. Robinson CM, Hale PT, Carlin JM. The role of IFN-gamma and TNF-alpha-responsive regulatory elements in the synergistic induction of indoleamine dioxygenase. J Interferon Cytokine Res (2005) 25:20–30. doi:10.1089/jir.2005.25.20
142. Oxenkrug GF. Interferon-gamma-inducible kynurenines/pteridines inflammation cascade: implications for aging and aging-associated psychiatric and medical disorders. J Neural Transm (Vienna) (2011) 118:75–85. doi:10.1007/s00702-010-0475-7
143. Allison DJ, Ditor DS. The common inflammatory etiology of depression and cognitive impairment: a therapeutic target. J Neuroinflammation (2014) 11:1–12. doi:10.1186/s12974-014-0151-1
144. Gold AB, Herrmann N, Swardfager W, Black SE, Aviv RI, Tennen G, et al. The relationship between indoleamine 2, 3-dioxygenase activity and post-stroke cognitive impairment. J Neuroinflammation (2011) 8:17–34. doi:10.1186/1742-2094-8-17
145. Liu M, Zhou K, Li H, Dong X, Tan G, Chai Y, et al. Potential of serum metabolites for diagnosing post-stroke cognitive impairment. Mol Biosyst (2015) 11:3287–96. doi:10.1039/c5mb00470e
146. Oxenkrug GF. Metabolic syndrome, age-associated neuroendocrine disorders, and dysregulation of tryptophan-kynurenine metabolism. Ann N Y Acad Sci (2010) 1199:1–14. doi:10.1111/j.1749-6632.2009.05356.x
147. Battle DE. Diagnostic and statistical manual of mental disorders (DSM). CODAS (2013) 25:191–2. doi:10.1590/S2317-17822013000200017
148. Ferrari AJ, Charlson FJ, Norman RE, Patten SB, Freedman G, Murray CJ, et al. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med (2013) 10:e1001547. doi:10.1371/journal.pmed.1001547
149. Pytka K, Dziubina A, Młyniec K, Dziedziczak A, Żmudzka E, Furgała A, et al. The role of glutamatergic, GABA-ergic, and cholinergic receptors in depression and antidepressant-like effect. Pharmacol Rep (2016) 68:443–50. doi:10.1016/j.pharep.2015.10.006
150. Réus GZ, Jansen K, Titus S, Carvalho AF, Gabbay V, Quevedo J. Kynurenine pathway dysfunction in the pathophysiology and treatment of depression: evidences from animal and human studies. J Psychiatr Res (2015) 68:316–28. doi:10.1016/j.jpsychires.2015.05.007
151. Myint AM, Schwarz MJ, Müller N. The role of the kynurenine metabolism in major depression. J Neural Transm (Vienna) (2012) 119:245–51. doi:10.1007/s00702-011-0741-3
152. Myint AM. Kynurenines: from the perspective of major psychiatric disorders. FEBS J (2012) 279:1375–85. doi:10.1111/j.1742-4658.2012.08551.x
153. Bay-Richter C, Linderholm KR, Lim CK, Samuelsson M, Träskman-Bendz L, Guillemin GJ, et al. A role for inflammatory metabolites as modulators of the glutamate N-methyl-D-aspartate receptor in depression and suicidality. Brain Behav Immun (2015) 43:110–7. doi:10.1016/j.bbi.2014.07.012
154. Savitz J, Dantzer R, Meier TB, Wurfel BE, Victor TA, McIntosh SA, et al. Activation of the kynurenine pathway is associated with striatal volume in major depressive disorder. Psychoneuroendocrinology (2015) 62:54–8. doi:10.1016/j.psyneuen.2015.07.609
155. Cho HJ, Savitz J, Dantzer R, Teague TK, Drevets WC, Irwin MR. Sleep disturbance and kynurenine metabolism in depression. J Psychosom Res (2017) 99:1–7. doi:10.1016/j.jpsychores.2017.05.016
156. Meier TB, Drevets WC, Teague TK, Wurfel BE, Mueller SC, Bodurka J, et al. Kynurenic acid is reduced in females and oral contraceptive users: implications for depression. Brain Behav Immun (2018) 67:59–64. doi:10.1016/j.bbi.2017.08.024
157. Veen C, Myint AM, Burgerhout KM, Schwarz MJ, Schütze G, Kushner SA, et al. Tryptophan pathway alterations in the postpartum period and in acute postpartum psychosis and depression. J Affect Disord (2016) 189:298–305. doi:10.1016/j.jad.2015.09.064
158. de Bie J, Lim CK, Guillemin GJ. Progesterone alters kynurenine pathway activation in IFN-γ-activated macrophages – relevance for neuroinflammatory diseases. Int J Tryptophan Res (2016) 9:89–93. doi:10.4137/IJTR.S40332
159. Ayerbe L, Ayis S, Wolfe CD, Rudd AG. Natural history, predictors and outcomes of depression after stroke: systematic review and meta-analysis. Br J Psychiatry (2013) 202:14–21. doi:10.1192/bjp.bp.111.107664
160. Braidy N, Grant R, Adams S, Guillemin GJ. Neuroprotective effects of naturally occurring polyphenols on quinolinic acid-induced excitotoxicity in human neurons. FEBS J (2010) 277:368–82. doi:10.1111/j.1742-4658.2009.07487.x
161. Li Y, Eskelund AR, Zhou H, Budac DP, Sánchez C, Gulinello M. Behavioral deficits are accompanied by immunological and neurochemical changes in a mouse model for neuropsychiatric lupus (NP-SLE). Int J Mol Sci (2015) 16:15150–71. doi:10.3390/ijms160715150
162. Cicek IE, Cicek E, Kayhan F, Uguz F, Erayman I, Kurban S, et al. The roles of BDNF, S100B, and oxidative stress in interferon-induced depression and the effect of antidepressant treatment in patients with chronic viral hepatitis: a prospective study. J Psychosom Res (2014) 76:227–32. doi:10.1016/j.jpsychores.2014.01.003
163. Lyon DE, Starkweather A, Yao Y, Garrett T, Kelly DL, Menzies V, et al. Pilot study of metabolomics and psychoneurological symptoms in women with early stage breast cancer. Biol Res Nurs (2018) 20:227–36. doi:10.1177/1099800417747411
164. Halaris A. Inflammation-associated co-morbidity between depression and cardiovascular disease. Curr Top Behav Neurosci (2017) 31:45–70. doi:10.1007/7854_2016_28
165. Zinger A, Barcia C, Herrero MT, Guillemin GJ. The involvement of neuroinflammation and kynurenine pathway in Parkinson’s disease. Parkinsons Dis (2011) 2011:716859. doi:10.4061/2011/716859
166. Lim CK, Fernández-Gomez FJ, Braidy N, Estrada C, Costa C, Costa S, et al. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog Neurobiol (2017) 155:76–95. doi:10.1016/j.pneurobio.2015.12.009
167. Thirtamara-Rajamani K, Li P, Escobar Galvis ML, Labrie V, Brundin P, Brundin L. Is the enzyme ACMSD a novel therapeutic target in Parkinson’s disease? J Parkinsons Dis (2017) 7:577–87. doi:10.3233/JPD-171240
168. Berg D, Postuma RB, Bloem B, Chan P, Dubois B, Gasser T, et al. Time to redefine PD? Introductory statement of the MDS task force on the definition of Parkinson’s disease. Mov Disord (2014) 29:454–62. doi:10.1002/mds.25844
169. Lindqvist D, Kaufman E, Brundin L, Hall S, Surova Y, Hansson O. Non-motor symptoms in patients with Parkinson’s disease-correlations with inflammatory cytokines in serum. PLoS One (2012) 7:e47387. doi:10.1371/journal.pone.0047387
170. Lewitt PA, Li J, Lu M, Beach TG, Adler CH, Guo L, et al. 3-Hydroxykynurenine and other Parkinson’s disease biomarkers discovered by metabolomic analysis. Mov Disord (2013) 28:1653–60. doi:10.1002/mds.25555
171. Lugo-Huitrón R, Blanco-Ayala T, Ugalde-Muñiz P, Carrillo-Mora P, Pedraza-Chaverrí J, Silva-Adaya D, et al. On the antioxidant properties of kynurenic acid: free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol Teratol (2011) 33:538–47. doi:10.1016/j.ntt.2011.07.002
172. Ogawa T, Matson WR, Beal MF, Myers RH, Bird ED, Milbury P, et al. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology (1992) 42:1702–6. doi:10.1212/WNL.42.9.1702
173. Widner B, Laich A, Sperner-Unterweger B, Ledochowski M, Fuchs D. Neopterin production, tryptophan degradation, and mental depression—what is the link? Brain Behav Immun (2002) 16:590–5. doi:10.1016/S0889-1591(02)00006-5
174. Widner B, Leblhuber F, Fuchs D. Increased neopterin production and tryptophan degradation in advanced Parkinson’s disease. J Neural Transm (Vienna) (2002) 109:181–9. doi:10.1007/s007020200014
175. Anderson G, Seo M, Berk M, Carvalho AF, Maes M. Gut permeability and microbiota in Parkinson’s disease: role of depression, tryptophan catabolites, oxidative and nitrosative stress and melatonergic pathways. Curr Pharm Des (2016) 22:6142–51. doi:10.2174/1381612822666160906161513
176. Havelund JF, Andersen AD, Binzer M, Blaabjerg M, Heegaard NHH, Stenager E, et al. Changes in kynurenine pathway metabolism in Parkinson patients with L-DOPA-induced dyskinesia. J Neurochem (2017) 142:756–66. doi:10.1111/jnc.14104
177. Foster AC, Collins JF, Schwarcz R. On the excitotoxic properties of quinolinic acid, 2,3-piperidine dicarboxylic acids and structurally related compounds. Neuropharmacology (1983) 22:1331–42. doi:10.1016/0028-3908(83)90221-6
178. McNally L, Bhagwagar Z, Hannestad J. Inflammation, glutamate, and glia in depression: a literature review. CNS Spectr (2008) 13:501–10. doi:10.1017/S1092852900016734
179. Erhardt S, Lim CK, Linderholm KR, Janelidze S, Lindqvist D, Samuelsson M, et al. Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology (2013) 38:743–52. doi:10.1038/npp.2012.248
180. Kubicova L, Hadacek F, Chobot V. Quinolinic acid: neurotoxin or oxidative stress modulator? Int J Mol Sci (2013) 14:21328–38. doi:10.3390/ijms141121328
181. Knyihár-Csillik E, Csillik B, Pákáski M, Krisztin-Péva B, Dobó E, Okuno E, et al. Decreased expression of kynurenine aminotransferase-I (KAT-I) in the substantia nigra of mice after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treatment. Neuroscience (2004) 126:899–914. doi:10.1016/j.neuroscience.2004.04.043
182. Vilas D, Fernández-Santiago R, Sanchez E, Azcona LJ, Santos-Montes M, Casquero P, et al. A novel p.Glu298Lys mutation in the ACMSD gene in sporadic Parkinson’s disease. J Parkinsons Dis (2017) 7:459–63. doi:10.3233/JPD-171146
183. Németh H, Toldi J, Vécsei L. Kynurenines, Parkinson’s disease and other neurodegenerative disorders: preclinical and clinical studies. J Neural Transm Suppl (2006) 70:285–304. doi:10.1007/978-3-211-45295-0_45
184. Silva-Adaya D, Pérez-De La Cruz V, Villeda-Hernández J, Carrillo-Mora P, González-Herrera IG, García E, et al. Protective effect of L-kynurenine and probenecid on 6-hydroxydopamine-induced striatal toxicity in rats: implications of modulating kynurenate as a protective strategy. Neurotoxicol Teratol (2011) 33:303–12. doi:10.1016/j.ntt.2010.10.002
185. Acuña-Castroviejo D, Tapias V, López LC, Doerrier C, Camacho E, Carrión MD, et al. Protective effects of synthetic kynurenines on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in mice. Brain Res Bull (2011) 85:133–40. doi:10.1016/j.brainresbull.2011.03.008
186. Demeter I, Nagy K, Gellért L, Vécsei L, Fülöp F, Toldi J. A novel kynurenic acid analog (SZR104) inhibits pentylenetetrazole-induced epileptiform seizures. An electrophysiological study: special issue related to kynurenine. J Neural Transm (Vienna) (2012) 119:151–4. doi:10.1007/s00702-011-0755-x
187. Gellért L, Fuzik J, Göblös A, Sárközi K, Marosi M, Kis Z, et al. Neuroprotection with a new kynurenic acid analog in the four-vessel occlusion model of ischemia. Eur J Pharmacol (2011) 667:182–7. doi:10.1016/j.ejphar.2011.05.069
188. Zádori D, Ilisz I, Klivényi P, Szatmári I, Fülöp F, Toldi J, et al. Time-course of kynurenic acid concentration in mouse serum following the administration of a novel kynurenic acid analog. J Pharm Biomed Anal (2011) 55:540–3. doi:10.1016/j.jpba.2011.02.014
189. Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, Schoepp DD, et al. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology (2011) 60:1017–41. doi:10.1016/j.neuropharm.2010.10.022
190. Breda C, Sathyasaikumar KV, Sograte Idrissi S, Notarangelo FM, Estranero JG, Moore GG, et al. Tryptophan-2, 3-dioxygenase (TDO) inhibition ameliorates neurodegeneration by modulation of kynurenine pathway metabolites. Proc Natl Acad Sci U S A (2016) 113:5435–40. doi:10.1073/pnas.1604453113
Keywords: soluble CD83, indoleamine 2 3-dioxygenase, kynurenine pathway, neuroimmunomodulation, neurologic disease
Citation: Bo L, Guojun T and Li G (2018) An Expanded Neuroimmunomodulation Axis: sCD83-Indoleamine 2,3-Dioxygenase—Kynurenine Pathway and Updates of Kynurenine Pathway in Neurologic Diseases. Front. Immunol. 9:1363. doi: 10.3389/fimmu.2018.01363
Received: 13 February 2018; Accepted: 01 June 2018;
Published: 15 June 2018
Edited by:
Fabienne Brilot, University of Sydney, AustraliaReviewed by:
Chai K. Lim, Macquarie University, AustraliaAlexander Steinkasserer, Universitätsklinikum Erlangen, Germany
Copyright: © 2018 Bo, Guojun and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tan Guojun, dHRhbmdqdW4mI3gwMDA0MDtob3RtYWlsLmNvbQ==;
Guo Li, Z3VvbGk2JiN4MDAwNDA7MTYzLmNvbQ==