 Masashi Satoh
Masashi Satoh Kazuya Iwabuchi
Kazuya Iwabuchi- Department of Immunology, Kitasato University School of Medicine, Sagamihara, Japan
Natural killer T (NKT) cells play important roles in adipose tissue inflammation, and thus influence the development of diet-induced obesity and insulin resistance. The interactions between cluster of differentiation (CD)1d and NKT T cell receptor are thought to be critical in this process, as demonstrated in two NKT cell-deficient mouse models—systemic CD1d gene knockout (KO) and prototypic Jα18 KO mice. The latter lacks some repertoires besides invariant (i)NKT cells due to manipulation of the Jα18 gene segment; therefore, the role of iNKT vs. variant NKT cells must be reinterpreted considering the availability of new Jα18 KO mice. NKT cells have varied roles in the development of obesity; indeed, studies have reported contradictory results depending on the mouse model, diet, and rearing conditions, all of which could affect the microbiome. In this mini-review, we discuss these points considering recent findings from our laboratory and others as well as the role of NKT cells in the development of obesity and insulin resistance based on data obtained from studies on conditional CD1d1 KO and new Jα18 KO mice generated through gene editing.
Introduction
Obesity as a Chronic Inflammatory Disorder
Inflammation in adipose tissue (AT) is induced by hypertrophy of adipocytes that secrete inflammatory cytokines and chemokines (1) and thus recruit various immune cells such as macrophages, T cells [αβ, γδ, regulatory T cells (Tregs), and natural killer T (NKT) cells], B cells, NK cells, and leukocytes that exist in a steady state in immune organs (2, 3). Fat accumulation is a major factor contributing to meta-inflammation and metabolic dysfunction (1, 4). Obesity alters the microenvironment in AT from an anti-inflammatory to a pro-inflammatory state, leading to impaired immune balance (5, 6). Visceral (V)AT in the lean state predominantly contains M2 macrophages, eosinophils, and Tregs that suppress inflammation and maintain insulin sensitivity (7, 8). By contrast, VAT in obese individuals has more M1 macrophages, cluster of differentiation (CD)8+ T cells, NK cells, B cells, and neutrophils that enhance inflammation and reduce insulin sensitivity (9–13). Notably, chronic low-grade inflammation accompanied by obesity is implicated in the etiology of lifestyle-related diseases such as atherosclerosis, type 2 diabetes, and various cancers (14).
NKT Cells
Natural killer T cells are a unique T cell subset that recognize lipid antigen presented by CD1d (15, 16). α-Galactosylceramide (α-GalCer) is a prototypical ligand recognized by invariant (i)NKT cells that harbors an invariant T cell receptor (TCR) α-chain (Vα14-Jα18 in mouse and Vα24-Jα18 in human) (17). Another type of NKT cell known as variant (v)NKT cells express diverse TCRs that are presumed to recognize various lipid antigens including sulfatide (18). Activated NKT cells secrete large amounts of cytokine that modulate immune balance, implying that they can either enhance or suppress inflammatory and immune responses. NKT cells have been reported to exacerbate, protect against, or have no role in the development of obesity through modulation of AT inflammation (19).
Here, we summarize the correlation between the CD1d/NKT cell axis and obesity with a focus on AT inflammation and discuss factors that may contribute to the discrepancies among reports considering recent progress.
Opposing Functions of NKT Cells in the Development of Obesity
Many studies have examined whether NKT cells play a role in diet-induced obesity (DIO) and have reported variable results.
NKT Cells as an Aggravator of DIO
Ohmura et al. first demonstrated that iNKT cells induce AT inflammation and glucose intolerance in β2-microglobulin (β2m) knockout (KO) mice fed a high-fat diet (HFD) and treated with the NKT cell stimulator α-GalCer (20). Since β2m KO mice also lack CD8+ T cells, the role of NKT cells in obesity has been examined using CD1d KO mice fed an HFD. However, two subsequent studies showed that NKT cell deficiency is insufficient to protect against or aggravate DIO (21) and that CD1d is important for the modulation of metabolic functions via a non-NKT cell-mediated mechanism (22). By contrast, we showed that CD1d KO mice lacking both iNKT and vNKT cells showed a reduced body weight (BW) gain along with improved AT inflammation and insulin resistance (23). Meanwhile, Jα18 KO mice lacking only iNKT cells demonstrated similar pathology to wild-type (WT) mice, suggesting that vNKT cells may contribute to DIO in the absence of iNKT cells (23). Wu et al. reported that iNKT cells responded to lipid excess and produced pro-inflammatory cytokines that promoted AT inflammation and insulin resistance (24).
iNKT vs. vNKT Cells
We investigated whether iNKT cells (24) or vNKT cells (23) contribute to the exacerbation of DIO, since distinct measures must be taken to control either subset. We speculated that vNKT cells contribute to the development of DIO in the absence of iNKT cells based on the aforementioned results (i.e., no difference in BW between WT and Jα18 KO mice on an HFD) and some additional observations (23): (1) the NK1.1+TCRβ+ population in AT was activated upon consumption of an HFD and contained more CD8+ but fewer CD4−CD8− subsets in Jα18 KO (referred hereafter as Jα281 KO) (25) mice, which differed from observations in either WT or CD1d KO mice; (2) WT mice harbored more non-iNKT (=vNKT) cells in AT; and (3) hepatic mononuclear cells from Jα281 KO mice [which are enriched in vNKT cells including CD1d+ antigen-presenting cells (APCs)] transferred insulin resistance to CD1d KO hosts.
However, the Jα281 KO strain was shown to exhibit a marked reduction in TCR diversity, which could affect immune responses (26). Four novel Jα18 KO mouse strains were independently generated after the report (26) by deleting only the T-cell receptor alpha joining (Traj)18 locus and leaving the remaining Jα repertoire unperturbed using novel technologies [clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein-9 nuclease or transcription activator-like effector nuclease] (27–30). New Traj18 KO (referred hereafter as simply Traj18 KO) mice gained less weight and had heightened sensitivity to insulin compared with WT mice, suggesting that iNKT cells play a pathogenic role in DIO (30). In that study, the mice were fed the same HFD (HFD-32; CLEA Japan, Tokyo, Japan) as those in our experiments, and Jα281 KO mice fed this diet showed similar BW gain to WT mice. The interpretation of the results from Traj18 KO mice was that iNKT but not vNKT cells exacerbate the development of DIO. Experiments using Vα14-Jα18 transgenic mice lacking low-density lipoprotein receptor also demonstrated that the abundance of iNKT cells increased adiposity by inducing metabolic abnormalities and AT inflammation (31). The DIO results from Traj18 KO mice also imply that reduced TCR diversity or the lack of particular T cell subsets in Jα281 KO but not Traj18 KO mice account for the discrepancy among reports on the involvement of iNKT vs. vNKT cells. Mucosal-associated invariant T (MAIT) cells that utilize Jα33 may be lost in Jα281 KO mice and may thus affect the development of obesity, as was suggested in studies of human subjects (32, 33). However, the actual role of MAIT cells in obesity and their involvement (or that of other T cell subsets) in DIO in Jα281 KO mice require further investigation.
Protective Role of NKT Cells Against Obesity
Some studies have reported that NKT cells play a protective role against obesity. Regulatory cytokines such as interleukin (IL)-4 and -10 produced by AT iNKT cells prevented the development of DIO (34, 35) and insulin resistance even in mice fed a low-fat diet (36). IL-13-producing innate immune cells such as type 2 innate lymphoid cells (ILC2s), iNKT cells, and vNKT cells were shown to prevent DIO by suppressing inflammation in AT (37). AT-resident iNKT cells express transcriptional repressor E4-binding protein (E4BP) 4 (also known as nuclear factor, IL-3-regulated) but not promyelocytic leukemia zinc finger protein (PLZF), unlike iNKT cells in other tissues, reflecting their anti-inflammatory phenotype (38); moreover, IL-10-producing iNKT cells (NKT10) are enriched in subcutaneous white (W)AT (39). Interestingly, an F108Y substitution in TCRβ altered NKT cell development to an adipose-like phenotype (40) without affecting TCR activation nor its ability to bind CD1d–ligand complexes, suggesting that a hydrophobic patch created after TCRα–TCRβ pairing is essential for the development of a distinct NKT cell population (40). iNKT cells with TCRβ F108Y express E4BP4 but not PLZF, similar to AT-resident NKT cells (38). These results suggest that NKT cells in AT constitute a specialized subset and are not regular iNKT cells that localize there as passers-by.
Mechanism of Fat Reduction via Thermogenesis and Relationship With Protective NKT Cells
In the development of obesity, the inflammatory environment created by NKT cell activation leads to insulin resistance and impaired glucose tolerance, which further accelerates metabolic changes that promote weight gain through increased fat mass. Meanwhile, recent studies on the suppression of obesity have provided insight into how NKT cells prevent obesity other than by producing anti-inflammatory cytokines. Fat mass is actively reduced in brown (B)AT through thermogenensis (41). BAT contains thermogenic mitochondria that express uncoupling protein (UCP) 1 and contribute to energy expenditure, in contrast to WAT (42). UCP1-expressing adipocytes with thermogenic capacity—known as beige or brite cells—also develop in WAT in response to various stimuli (43). The relationship between iNKT cells and thermogenesis was demonstrated by the finding that activated iNKT cells enhanced fibroblast growth factor 21 production and increased the number of beige cells in WAT, which in turn increased thermogenesis and weight loss (44). Several recent studies have demonstrated that innate immune cells play an important role in the induction of beige cells. vNKT cells and ILC2s induced by IL-25 produce IL-13 and regulate glucose homeostasis to protect against obesity (37). ILC2s also sustain eosinophils that produce IL-4, which stimulates M2 macrophages in VAT (45). IL-4 further stimulates M2 macrophages to secrete catecholamines for the induction of thermogenic gene expression in BAT and lipolysis in WAT (46). IL-33 is also critical for the maintenance of ILC2s in the induction of beige cells in WAT and regulation of energy expenditure. ILC2s produce methionine–enkephalin peptides that can act directly on adipocytes to upregulate UCP1 expression and promote beiging (47). These findings indicate that the innate immune system—including iNKT cells, macrophages, and ILCs—in AT controls thermogenesis by inducing beige cells, which is an important mechanism for the regulation of obesity and insulin resistance in addition to the control of AT inflammation via production of anti-inflammatory cytokines.
APC for NKT Cells in AT
Natural killer T cells in DIO act as NKT1 or NKT2 (or AT-resident NKT) cells through interactions with CD1d-expressing cells in AT. Many cell types in AT express CD1d including macrophages, dendritic cells, adipocytes, and possibly others. Recent studies have shown that adipocytes can activate T cells and NKT cells through antigen presentation (48, 49). CD1d expressed on the surface of adipocytes can induce helper T cell (Th)1 and Th2 cytokine release by iNKT cells depending on the co-expression of microsomal triglyceride transfer protein and CCAAT/enhancer-binding protein-β and -δ even in the absence of exogenous ligands (48), suggesting that adipocytes express ligands that are recognized by NKT cells. To determine whether interaction between NKT cells and adipocytes influence DIO, we analyzed mice with adipocyte-specific CD1d1 deletion (adipoqcre-CD1d1fl/fl) and found that they gained less weight than control mice fed an HFD (50), consistent with our findings from conventional CD1d KO mice (24). A decrease in IFN-γ and concomitant increase in adiponectin was observed following disruption of the NKT cell/adipocyte interaction, which ameliorated AT inflammation and insulin resistance. On the contrary, another study showed that adipocyte-specific CD1d1 deletion reduced IL-4 expression in adipose iNKT cells and increased AT inflammation and insulin resistance (51), in accordance with an earlier report (49). The fact that these studies reported opposite results using the same conditional (c)KO mice provides strong evidence that adipocytes are the APCs for NKT cells in modulating AT inflammation, and that different HFDs can explain the discrepancy in the reported roles of NKT cells in the development of obesity (50, 51).
CD1d2-Restricted NKT Cells
The fact that opposite results were obtained using the same cKO mice is critical, because it excludes the possibility that the results simply reflect the use of either pro-aggravating [CD1d1 KO; (52)] or pro-ameliorating [CD1d1/d2 KO; (53, 54)] mice (Table 1). Although it was reported that CD1d2 does not specify a specific NKT cell population (55), CD1d2 may affect the development of obesity in CD1d1 KO mice. Indeed, it was recently reported that CD1d2 can present distinct species of glycosylceramide (GlyCer) and affect the differentiation of NKT cells (56). Thus, the possible contribution of CD1d2-restricted NKT cells to the development of obesity remains to be determined, although contradictory results were obtained regarding DIO using the same cKO mice that express neither CD1d1 nor CD1d2 on adipocytes (50, 51).
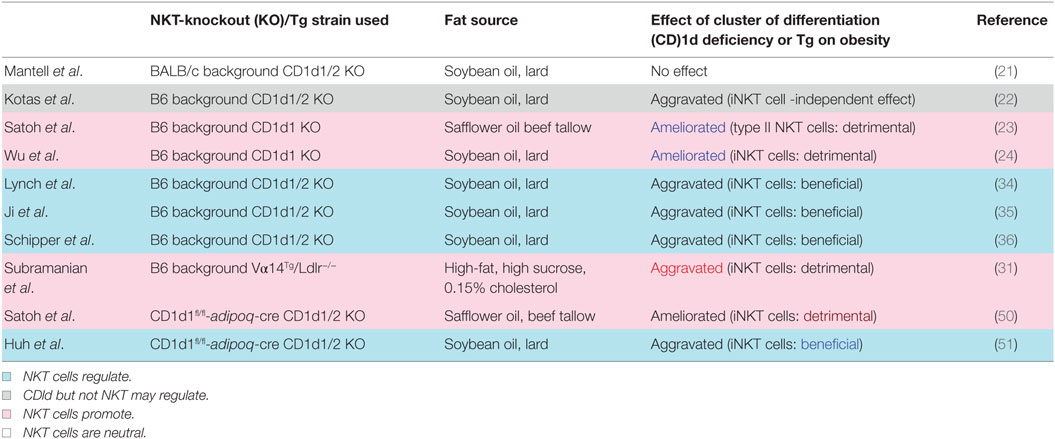
Table 1. Roles of natural killer T (NKT) cells in the development of diet-induced obesity.
In addition to studies of genetically engineered mice, other factors affecting the development of obesity have been investigated, including microbiota—especially those in the gut—and fat composition, both of which are related to diet and influence the presentation of ligands to NKT cells.
Other Factors that affect the Development of DIO
Microbiota
The findings that gut microbiota composition is a critical factor in the development of obesity come from studies using germ-free (GF) animals. Conventionally raised mice have higher total body fat than GF mice, although the latter consume more food (57). When the two types of mice are fed a sugar-rich HFD, GF mice are protected from DIO owing to increased fatty acid (FA) oxidation and AMP-activated protein kinase activity (58). On the other hand, pathogenic alterations in gut microbiome profiles (i.e., dysbiosis) in obesity affect energy metabolism (59). In fact, the abundance of Firmicutes is increased whereas that of Bacteroidetes is decreased in ob/ob mice with a mutation in the gene encoding leptin; on the contrary, lean ob/+ mice fed a polysaccharide-rich diet predominantly harbor Bacteroidetes (60). Similar differences in gut microbiota composition are also observed between obese and lean human subjects (61). Furthermore, GF mice inoculated with microbiota from obese twin donors developed increased adiposity when compared with those receiving transplants from lean twin donors and did not develop increased adiposity when they were cohoused with the latter mice (62). It is unclear whether microbiota or diet (calorie excess) is responsible for obesity.
Although gut microbiotas are transmissible and can be altered by diet, they may have the ability to directly alter systemic energy metabolism and thereby control weight gain. Several studies have demonstrated that NKT cells play a central role in maintaining homeostasis at mucosal surfaces (63, 64). CD1d KO mice exhibit altered gut microbiome profiles, which exacerbate intestinal inflammation induced by dextran sodium sulfate treatment and even in the steady state. Compared to non-littermate B6 mice, these mice have a higher abundance of segmented filamentous bacteria that can induce Th17 cells but reduced levels of Akkermansia, which may protect mice from developing colitis (65). A recent study using CD1dfl/flCD11cCre cKO mice also showed that CD1d expression on CD11c+ cells contributes to the maintenance of intestinal homeostasis by regulation of the immunoglobulin A repertoire and induction of Tregs in the gut (66). Bacteroides fragilis, a prominent gut bacterial species, produces the glycosphingolipid α-GalCerBf, which is structurally related to the prototypic ligand α-GalCer or KRN7000 (67). α-GalCerBf stimulates iNKT cells in the context of CD1d, suggesting that alterations in the abundance of B. fragilis caused by obesity can affect NKT cell homeostasis. Disruption of the NKT cell/CD1d interaction—which is required to maintain intestinal homeostasis—may affect energy consumption and fat storage by modulating gut microbiota composition.
Fat Composition
α-Galactosylceramide is a potent activator of NKT cells, and various analogs have been synthesized that elicit distinct cytokine responses (68). For instance, α-C-GalCer and RCAI-56 promote Th1-biased responses (69, 70), whereas OCH and 20:2 promote Th2-biased responses (71). Natural ligands for CD1d have also been identified. Several mammalian glycosphingolipids such as isoglobotrihexosylceramide and β-glucosylceramide (β-GlcCer) were shown to act as self-lipid antigens (72). However, a recent study showed that a small quantity of stimulatory α-GlyCer was present in β-GlcCer preparations (73). Accordingly, pure β-GlcCer may not activate iNKT cells, which can respond to a minor fraction of α-GlyCer. Phospholipids (PHLs) that are a major component of mammalian cell membranes including phosphatidylinositol (PI), phosphatidylethanolamine (PE), and phosphatidylglycerol (PG) are natural antigens recognized by NKT cells (74). Lysophosphatidylcholine is also a natural ligand that is recognized not only by human iNKT cell clones (75) but also by Vα24−/Vβ11− vNKT cells (76). Characteristic lipid abnormalities observed during the course of obesity include an increase in triacylglycerol and cholesterol levels in the low-density lipoprotein fraction, with a corresponding decrease in high-density lipoprotein cholesterol. In addition, obesity-related changes of serum lipids such as FAs, PHLs, and their oxidation products as well as oxylipins, sphingolipids, and their metabolites contribute to the health status and risk of comorbidities in obese patients (77). A lipidomics analysis demonstrated that changes in PHL concentrations may contribute to the development of insulin resistance and metabolic syndrome (78). Elevated circulating levels of phosphatidylcholine, PI, PE, and PG have been detected in subjects with when compared with those without non-alcoholic steatohepatitis (79). Although the molecular basis for the correlation between NKT cell activation and altered PHL levels in obese subjects remains unclear, some PHLs may affect NKT cell biology, based on the observations that the concentration of Cer species (C18:0, C20:0, and C24:1) and total Cer level was higher in type 2 diabetes; insulin sensitivity was inversely correlated with C18:0, C20:0, C24:1, C24:0, and total Cer levels; and increased tumor necrosis factor (TNF)-α concentration was correlated with the levels of C18:1 and C18:0 ceramide (80).
The mechanism of insulin resistance in obese patients with an elevated Cer concentration may involve inflammation induced by NKT cell activation, since certain Cer species stimulate NKT cells. FAs are the major components of fat and mediate immune responses. In AT, free FAs secreted by adipocytes—especially saturated FAs (SAFAs) such as palmitate and laurate—activate macrophages via toll-like receptor 4 to induce TNF-α expression, whereas polyunsaturated (PU)FAs such as linolenate and eicosapentaenoic acid do not have this effect (81). SAFAs, but not PUFAs, stimulate the expression of inflammatory cytokines such as IL-6 and TNF-α in adipocytes (82) that further promote metabolic syndrome. The composition and concentration of FA in sera that are altered and elevated in obese subjects are determined based on endogenous synthesis rates and dietary fat characteristics (83, 84). Thus, dietary fats may affect AT inflammation by modulating the functions of immune cells and adipocytes, suggesting that HFDs with different compositions of FA species presumably affect NKT cell response to either promote or suppress AT inflammation and obesity.
Conclusion
Obesity-associated inflammation in AT contributes to metabolic syndrome and is controlled by adipocytes and NKT cells with other immune cells, as discussed in this review. NKT cells appear to respond to lipid antigens on adipocytes and modulate inflammation (either by ameliorating or by aggravating this process) depending on the input—i.e., dietary lipids and ligands derived from the microbiome (Figure 1). Although the critical factors that give rise to the distinct outcomes of NKT cells remain elusive, future investigations should focus on two mutually interactive topics: (1) gut microorganisms that regulate energy consumption and modulation/maintenance by NKT cells and (2) diet/fat composition that can alter gut microbiota, the balance of lipid species, and the synthesis of endogenous lipid antigens that affect NKT cell activation. Furthermore, elucidating the mechanism of BAT maintenance and WAT beiging by NKT cells can provide a basis for the development of strategies to reverse metabolic dysregulation and reduce fat mass.
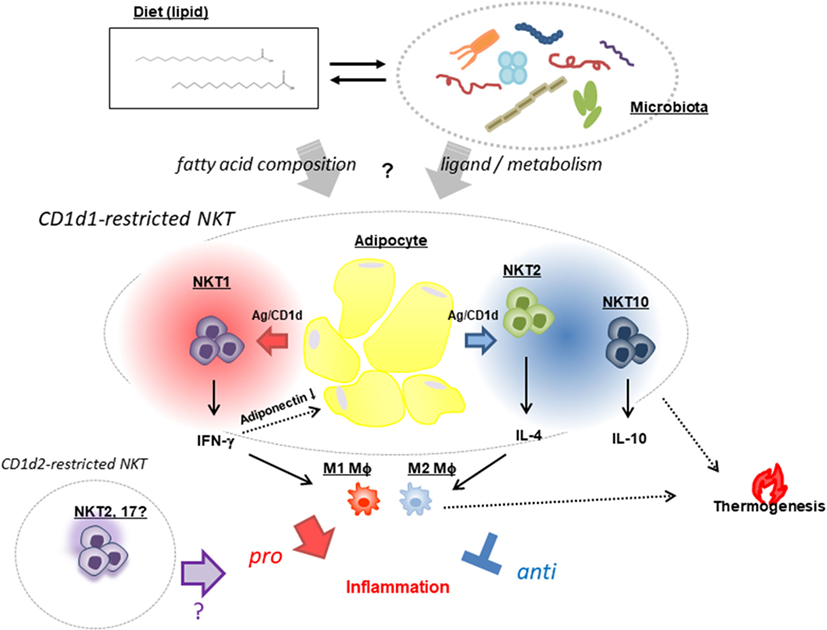
Figure 1. Natural killer T (NKT) cell-based modulation of AT inflammation. NKT cells exhibit opposing functions: a pro-inflammatory response that promotes AT inflammation and insulin resistance through release of IFN-γ (NKT1) that reduces the pro-ameliorating adipokine adiponectin, and a regulatory response that suppresses inflammation via production of IL-4 (NKT2) and -10 (NKT10) and increased thermogenesis leading to energy expenditure. These NKT [mostly cluster of differentiation (CD)1d1-restricted and very few, if any, CD1d2-restricted] cell functions are presumed to be affected by two mutually interacting factors—namely, dietary fat composition and a microbiome in which the Bacteroidetes and Firmicutes phyla predominate.
Author Contributions
MS wrote the first draft. MS and KI edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by the Japan Society for the Promotion of Science (Grant-in-Aid for Young Scientists [B] [no. 16K21347 to MS] and Grant-in-Aid for Scientific Research [C] [no. 17K08791 to KI and MS]); by an A-MED grant (no. 16ek0109076s0202 to KI); and by MEXT (Grant-in-Aid for Research Branding Project; Agromedicine at Kitasato University to KI and MS).
References
1. Hotamisligil GS. Inflammation and metabolic disorders. Nature (2006) 444:860–7. doi:10.1038/nature05485
2. Caspar-Bauguil S, Cousin B, Galinier A, Segafredo C, Nibbelink M, Andre M, et al. Adipose tissues as an ancestral immune organ: site-specific change in obesity. FEBS Lett (2005) 579:3487–92. doi:10.1016/j.febslet.2005.05.031
3. Exley MA, Hand L, O’Shea D, Lynch L. Interplay between the immune system and adipose tissue in obesity. J Endocrinol (2014) 223:R41–8. doi:10.1530/JOE-13-0516
4. Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell (2001) 104:531–43. doi:10.1016/S0092-8674(01)00240-9
5. Hotamisligil GS. Foundations of immunometabolism and implications for metabolic health and disease. Immunity (2017) 47:406–20. doi:10.1016/j.immuni.2017.08.009
6. Bansal A, Henao-Mejia J, Simmons RA. Immune system: an emerging player in mediating effects of endocrine disruptors on metabolic health. Endocrinology (2018) 159:32–45. doi:10.1210/en.2017-00882
7. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med (2009) 15:930–9. doi:10.1038/nm.2002
8. Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science (2011) 332:243–7. doi:10.1126/science.1201475
9. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112:1796–808. doi:10.1172/JCI200319246
10. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med (2009) 15:914–20. doi:10.1038/nm.1964
11. Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med (2011) 17:610–7. doi:10.1038/nm.2353
12. Wensveen FM, Jelenčić V, Valentić S, Šestan M, Wensveen TT, Theurich S, et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol (2015) 16:376–85. doi:10.1038/ni.3120
13. Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med (2012) 18:1407–12. doi:10.1038/nm.2885
14. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest (2011) 121:2111–7. doi:10.1172/JCI57132
15. Van Kaer L. NKT cells: T lymphocytes with innate effector functions. Curr Opin Immunol (2007) 19:354–64. doi:10.1016/j.coi.2007.03.001
16. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol (2007) 25:297–336. doi:10.1146/annurev.immunol.25.022106.141711
17. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science (1997) 278:1626–9. doi:10.1126/science.278.5343.1626
18. Halder RC, Aguilera C, Maricic I, Kumar V. Type II NKT cell-mediated anergy induction in type I NKT cells prevents inflammatory liver disease. J Clin Invest (2007) 117:2302–12. doi:10.1172/JCI31602
19. Wu L, Van Kaer L. Contribution of lipid-reactive natural killer T cells to obesity-associated inflammation and insulin resistance. Adipocyte (2013) 2:12–6. doi:10.4161/adip.22296
20. Ohmura K, Ishimori N, Ohmura Y, Tokuhara S, Nozawa A, Horii S, et al. Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice. Arterioscler Thromb Vasc Biol (2010) 30:193–9. doi:10.1161/ATVBAHA.109.198614
21. Mantell BS, Stefanovic-Racic M, Yang X, Dedousis N, Sipula IJ, O’Doherty RM. Mice lacking NKT cells but with a complete complement of CD8+ T-cells are not protected against the metabolic abnormalities of diet-induced obesity. PLoS One (2011) 6:e19831. doi:10.1371/journal.pone.0019831
22. Kotas ME, Lee H-Y, Gillum MP, Annicelli C, Guigni BA, Shulman GI, et al. Impact of CD1d deficiency on metabolism. PLoS One (2011) 6:e25478. doi:10.1371/journal.pone.0025478
23. Satoh M, Andoh Y, Clingan CS, Ogura H, Fujii S, Eshima K, et al. Type II NKT cells stimulate diet-induced obesity by mediating adipose tissue inflammation, steatohepatitis and insulin resistance. PLoS One (2012) 7:e30568. doi:10.1371/journal.pone.0030568
24. Wu L, Parekh VV, Gabriel CL, Bracy DP, Marks-Shulman PA, Tamboli RA, et al. Activation of invariant natural killer T cells by lipid excess promotes tissue inflammation, insulin resistance, and hepatic steatosis in obese mice. Proc Natl Acad Sci U S A (2012) 109:E1143–52. doi:10.1073/pnas.1200498109
25. Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, et al. Requirement for Va14NKT cells in IL-12-mediated rejection of tumors. Science (1997) 278:1623–6. doi:10.1126/science.278.5343.1623
26. Bedel R, Matsuda JL, Brigl M, White J, Kappler J, Marrack P, et al. Lower TCR repertoire diversity in Traj18-deficient mice. Nat Immunol (2012) 13:705–6. doi:10.1038/ni.2347
27. Chandra S, Zhao M, Budelsky A, de Mingo Pulido A, Day J, Fu Z, et al. A new mouse strain for the analysis of invariant NKT cell function. Nat Immunol (2015) 16:799–800. doi:10.1038/ni.3203
28. Dashtsoodol N, Shigeura T, Ozawa R, Harada M, Kojo S, Watanabe T, et al. Generation of novel Traj18-deficient mice lacking Vα14 natural killer T cells with an undisturbed T cell receptor α-chain repertoire. PLoS One (2016) 11:e0153347. doi:10.1371/journal.pone.0153347
29. Zhang J, Bedel R, Krovi SH, Tuttle KD, Zhang B, Gross J, et al. Mutation of the Traj18 gene segment using TALENs to generate natural killer T cell deficient mice. Sci Rep (2016) 6:27375. doi:10.1038/srep27375
30. Ren Y, Sekine-Kondo E, Shibata R, Kato-Itoh M, Umino A, Yanagida A, et al. A novel mouse model of iNKT cell-deficiency generated by CRISPR/Cas9 reveals a pathogenic role of iNKT cells in metabolic disease. Sci Rep (2017) 7:12765. doi:10.1038/s41598-017-12475-4
31. Subramanian S, Turner MS, Ding Y, Goodspeed L, Wang S, Buckner JH, et al. Increased levels of invariant natural killer T lymphocytes worsen metabolic abnormalities and atherosclerosis in obese mice. J Lipid Res (2013) 54:2831–41. doi:10.1194/jlr.M041020
32. Carolan E, Tobin LM, Mangan BA, Corrigan M, Gaoatswe G, Byrne G, et al. Altered distribution and increased IL-17 production by mucosal-associated invariant T cells in adult and childhood obesity. J Immunol (2015) 194:5775–80. doi:10.4049/jimmunol.1402945
33. Magalhaes I, Pingris K, Poitou C, Bessoles S, Venteclef N, Kiaf B, et al. Mucosal-associated invariant T cell alterations in obese and type 2 diabetic patients. J Clin Invest (2015) 125:1752–62. doi:10.1172/JCI78941
34. Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V, et al. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity (2012) 37:574–87. doi:10.1016/j.immuni.2012.06.016
35. Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KS, et al. Activation of natural killer T cells promotes M2 macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4 (IL-4)/STAT6 protein signaling axis in obesity. J Biol Chem (2012) 287:13561–71. doi:10.1074/jbc.M112.350066
36. Schipper HS, Rakhshandehroo M, van de Graaf SF, Venken K, Koppen A, Stienstra R, et al. Natural killer T cells in adipose tissue prevent insulin resistance. J Clin Invest (2012) 122:3343–54. doi:10.1172/JCI62739
37. Hams E, Locksley RM, McKenzie AN, Fallon PG. Cutting edge: IL-25 elicits innate lymphoid type 2 and type II NKT cells that regulate obesity in mice. J Immunol (2013) 191:5349–53. doi:10.4049/jimmunol.1301176
38. Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat Immunol (2015) 16:85–95. doi:10.1038/ni.3047
39. Sag D, Krause P, Hedrick CC, Kronenberg M, Wingender G. IL-10-producing NKT10 cells are a distinct regulatory invariant NKT cell subset. J Clin Invest (2014) 124:3725–40. doi:10.1172/JCI72308
40. Vieth JA, Das J, Ranaivoson FM, Comoletti D, Denzin LK, Sant’Angelo DB. TCRα-TCRβ pairing controls recognition of CD1d and directs the development of adipose NKT cells. Nat Immunol (2017) 18:36–44. doi:10.1038/ni.3622
41. Kajimura S, Saito M. A new era in brown adipose tissue biology: molecular control of brown fat development and energy homeostasis. Annu Rev Physiol (2014) 76:225–49. doi:10.1146/annurev-physiol-021113-170252
42. Enerbäck S, Jacobsson A, Simpson EM, Guerra C, Yamashita H, Harper ME, et al. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature (1997) 387:90–4. doi:10.1038/387090a0
43. Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med (2013) 19:1252–63. doi:10.1038/nm.3361
44. Lynch L, Hogan AE, Duquette D, Lester C, Banks A, LeClair K, et al. iNKT cells induce FGF21 for thermogenesis and are required for maximal weight loss in GLP1. Therapy Cell Metab (2016) 24:510–9. doi:10.1016/j.cmet.2016.08.003
45. Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med (2013) 210:535–49. doi:10.1084/jem.20121964
46. Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature (2011) 480:104–8. doi:10.1038/nature10653
47. Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature (2015) 519:242–6. doi:10.1038/nature14115
48. Rakhshandehroo M, Gijzel SM, Siersbaek R, Broekema MF, de Haar C, Schipper HS, et al. CD1d-mediated presentation of endogenous lipid antigens by adipocytes requires microsomal triglyceride transfer protein. J Biol Chem (2014) 289:22128–39. doi:10.1074/jbc.M114.551242
49. Huh JY, Kim JI, Park YJ, Hwang IJ, Lee YS, Sohn JH, et al. A novel function of adipocytes in lipid antigen presentation to iNKT cells. Mol Cell Biol (2013) 33:328–39. doi:10.1128/MCB.00552-12
50. Satoh M, Hoshino M, Fujita K, Iizuka M, Fujii S, Clingan CS, et al. Adipocyte-specific CD1d-deficiency mitigates diet-induced obesity and insulin resistance in mice. Sci Rep (2016) 6:28473. doi:10.1038/srep28473
51. Huh JY, Park J, Kim JI, Park YJ, Lee YK, Kim JB. Deletion of CD1d in adipocytes aggravates adipose tissue inflammation and insulin resistance in obesity. Diabetes (2017) 66:835–47. doi:10.2337/db16-1122
52. Mendiratta SK, Martin WD, Hong S, Boesteanu A, Joyce S, Van Kaer L. CD1d1 mutant mice are deficient in natural T cells that promptly produce IL-4. Immunity (1997) 6:469–77. doi:10.1016/S1074-7613(00)80290-3
53. Chen YH, Chiu NM, Mandal M, Wang N, Wang CR. Impaired NK1+ T cell development and early IL-4 production in CD1-deficient mice. Immunity (1997) 6:459–67. doi:10.1016/S1074-7613(00)80289-7
54. Smiley ST, Kaplan MH, Grusby MJ. Immunoglobulin E production in the absence of interleukin-4-secreting CD1-dependent cells. Science (1997) 275:977–9. doi:10.1126/science.275.5302.977
55. Chen YH, Wang B, Chun T, Zhao L, Cardell S, Behar SM, et al. Expression of CD1d2 on thymocytes is not sufficient for the development of NK T cells in CD1d1-deficient mice. J Immunol (1999) 162:4560–6.
56. Sundararaj S, Zhang J, Krovi SH, Bedel R, Tuttle KD, Veerapen N, et al. Differing roles of CD1d2 and CD1d1 proteins in type I natural killer T cell development and function. Proc Natl Acad Sci U S A (2018) 115:E1204–13. doi:10.1073/pnas.1716669115
57. Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A (2004) 101:15718–23. doi:10.1073/pnas.0407076101
58. Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A (2007) 104:979–84. doi:10.1073/pnas.0605374104
59. Sonnenburg JL, Bäckhed F. Diet-microbiota interactions as moderators of human metabolism. Nature (2016) 535:56–64. doi:10.1038/nature18846
60. Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A (2005) 102:11070–5. doi:10.1073/pnas.0504978102
61. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature (2006) 444:1022–3. doi:10.1038/4441022a
62. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science (2013) 341:1241214. doi:10.1126/science.1241214
63. Nieuwenhuis EE, Matsumoto T, Lindenbergh D, Willemsen R, Kaser A, Simons-Oosterhuis Y, et al. Cd1d-dependent regulation of bacterial colonization in the intestine of mice. J Clin Invest (2009) 119:1241–50. doi:10.1172/JCI36509
64. Olszak T, Neves JF, Dowds CM, Baker K, Glickman J, Davidson NO, et al. Protective mucosal immunity mediated by epithelial CD1d and IL-10. Nature (2014) 509:497–502. doi:10.1038/nature13150
65. Selvanantham T, Lin Q, Guo CX, Surendra A, Fieve S, Escalante NK, et al. NKT cell-deficient mice harbor an altered microbiota that fuels intestinal inflammation during chemically induced colitis. J Immunol (2016) 197:4464–72. doi:10.4049/jimmunol.1601410
66. de Guinoa JS, Jimeno R, Gaya M, Kipling D, Garzón MJ, Dunn-Walters D, et al. CD1d-mediated lipid presentation by CD11c+ cells regulates intestinal homeostasis. EMBO J (2018) 37:e97537. doi:10.15252/embj.201797537
67. Wieland Brown LC, Penaranda C, Kashyap PC, Williams BB, Clardy J, Kronenberg M, et al. Production of α-galactosylceramide by a prominent member of the human gut microbiota. PLoS Biol (2013) 11:e1001610. doi:10.1371/journal.pbio.1001610
68. Wun KS, Cameron G, Patel O, Pang SS, Pellicci DG, Sullivan LC, et al. A molecular basis for the exquisite CD1d-restricted antigen specificity and functional responses of natural killer T cells. Immunity (2011) 34:327–39. doi:10.1016/j.immuni.2011.02.001
69. Schmieg J, Yang G, Franck RW, Tsuji M. Superior protection against malaria and melanoma metastases by a C-glycoside analogue of the natural killer T cell ligand alpha-galactosylceramide. J Exp Med (2003) 198:1631–41. doi:10.1084/jem.20031192
70. Tashiro T, Nakagawa R, Hirokawa T, Inoue S, Watarai H, Taniguchi M, et al. RCAI-37, 56, 59, 60, 92, 101, and 102, cyclitol and carbasugar analogs of KRN7000: their synthesis and bioactivity for mouse lymphocytes to produce Th1-biased cytokines. Bioorg Med Chem (2009) 17:6360–73. doi:10.1016/j.bmc.2009.07.025
71. Yu KO, Im JS, Molano A, Dutronc Y, Illarionov PA, Forestier C, et al. Modulation of CD1d-restricted NKT cell responses by using N-acyl variants of α-galactosylceramides. Proc Natl Acad Sci U S A (2005) 102:3383–8. doi:10.1073/pnas.0407488102
72. Macho-Fernandez E, Brigl M. The extended family of CD1d-restricted NKT cells: sifting through a mixed bag of TCRs, antigens, and functions. Front Immunol (2015) 6:362. doi:10.3389/fimmu.2015.00362
73. Kain L, Webb B, Anderson BL, Deng S, Holt M, Costanzo A, et al. The identification of the endogenous ligands of natural killer T cells reveals the presence of mammalian α-linked glycosylceramides. Immunity (2014) 41:543–54. doi:10.1016/j.immuni.2014.08.017
74. Gumperz JE, Roy C, Makowska A, Lum D, Sugita M, Podrebarac T, et al. Murine CD1d-restricted T cell recognition of cellular lipids. Immunity (2000) 12:211–21. doi:10.1016/S1074-7613(00)80174-0
75. Fox LM, Cox DG, Lockridge JL, Wang X, Chen X, Scharf L, et al. Recognition of lyso-phospholipids by human natural killer T lymphocytes. PLoS Biol (2009) 7:e1000228. doi:10.1371/journal.pbio.1000228
76. Chang DH, Deng H, Matthews P, Krasovsky J, Ragupathi G, Spisek R, et al. Inflammation-associated lysophospholipids as ligands for CD1d-restricted T cells in human cancer. Blood (2008) 112:1308–16. doi:10.1182/blood-2008-04-149831
77. Mika A, Sledzinski T. Alterations of specific lipid groups in serum of obese humans: a review. Obes Rev (2017) 18:247–72. doi:10.1111/obr.12475
78. Goto-Inoue N, Yamada K, Inagaki A, Furuichi Y, Ogino S, Manabe Y, et al. Lipidomics analysis revealed the phospholipid compositional changes in muscle by chronic exercise and high-fat diet. Sci Rep (2013) 3:3267. doi:10.1038/srep03267
79. Anjani K, Lhomme M, Sokolovska N, Poitou C, Aron-Wisnewsky J, Bouillot JL, et al. Circulating phospholipid profiling identifies portal contribution to NASH signature in obesity. J Hepatol (2015) 62:905–12. doi:10.1016/j.jhep.2014.11.002
80. Haus JM, Kashyap SR, Kasumov T, Zhang R, Kelly KR, Defronzo RA, et al. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes (2009) 58:337–43. doi:10.2337/db08-1228
81. Suganami T, Tanimoto-Koyama K, Nishida J, Itoh M, Yuan X, Mizuarai S, et al. Role of the Toll-like receptor 4/NF-κB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler Thromb Vasc Biol (2007) 27:84–91. doi:10.1161/01.ATV.0000251608.09329.9a
82. Bradley RL, Fisher FF, Maratos-Flier E. Dietary fatty acids differentially regulate production of TNF-alpha and IL-10 by murine 3T3-L1 adipocytes. Obesity (Silver Spring) (2008) 16:938–44. doi:10.1038/oby.2008.39
83. Warensjö E, Risérus U, Vessby B. Fatty acid composition of serum lipids predicts the development of the metabolic syndrome in men. Diabetologia (2005) 48:1999–2005. doi:10.1007/s00125-005-1897-x
Keywords: natural killer T cell, cluster of differentiation 1d, adipocyte, lipid, obesity, insulin resistance, adipose tissue inflammation
Citation: Satoh M and Iwabuchi K (2018) Role of Natural Killer T Cells in the Development of Obesity and Insulin Resistance: Insights From Recent Progress. Front. Immunol. 9:1314. doi: 10.3389/fimmu.2018.01314
Received: 03 April 2018; Accepted: 28 May 2018;
Published: 11 June 2018
Edited by:
Yun-Cai Liu, Tsinghua University, ChinaCopyright: © 2018 Satoh and Iwabuchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuya Iwabuchi, YWtpbWFyaUBraXRhc2F0by11LmFjLmpw