 Fokhrul Hossain1,2
Fokhrul Hossain1,2 Samarpan Majumder1,2
Samarpan Majumder1,2 Deniz A. Ucar2Paulo C. Rodriguez3Todd E. Golde4
Deniz A. Ucar2Paulo C. Rodriguez3Todd E. Golde4 Lisa M. Minter5
Lisa M. Minter5 Barbara A. Osborne5
Barbara A. Osborne5 Lucio Miele1,2*
Lucio Miele1,2*
- 1Department of Genetics, Louisiana State University Health Sciences Center, New Orleans, LA, United States
- 2Stanley S. Scott Cancer Center, Louisiana State University Health Sciences Center, New Orleans, LA, United States
- 3H. Lee Moffitt Comprehensive Cancer Center, Tampa, FL, United States
- 4Department of Neurosciences, McKnight Brain Institute, University of Florida at Gainesville, Gainesville, FL, United States
- 5Department of Veterinary and Animal Sciences, University of Massachusetts, Amherst, MA, United States
Cancer immunotherapy, which stimulates or augments host immune responses to treat malignancies, is the latest development in the rapidly advancing field of cancer immunology. The basic principles of immunotherapies are either to enhance the functions of specific components of the immune system or to neutralize immune-suppressive signals produced by cancer cells or tumor microenvironment cells. When successful, these approaches translate into long-term survival for patients. However, durable responses are only seen in a subset of patients and so far, only in some cancer types. As for other cancer treatments, resistance to immunotherapy can also develop. Numerous research groups are trying to understand why immunotherapy is effective in some patients but not others and to develop strategies to enhance the effectiveness of immunotherapy. The Notch signaling pathway is involved in many aspects of tumor biology, from angiogenesis to cancer stem cell maintenance to tumor immunity. The role of Notch in the development and modulation of the immune response is complex, involving an intricate crosstalk between antigen-presenting cells, T-cell subpopulations, cancer cells, and other components of the tumor microenvironment. Elegant studies have shown that Notch is a central mediator of tumor-induced T-cell anergy and that activation of Notch1 in CD8 T-cells enhances cancer immunotherapy. Tumor-infiltrating myeloid cells, including myeloid-derived suppressor cells, altered dendritic cells, and tumor-associated macrophages along with regulatory T cells, are major obstacles to the development of successful cancer immunotherapies. In this article, we focus on the roles of Notch signaling in modulating tumor-infiltrating myeloid cells and discuss implications for therapeutic strategies that modulate Notch signaling to enhance cancer immunotherapy.
Introduction
Notch signaling, an evolutionarily conserved cell-fate-determination pathway, mediates close contact interactions between neighboring cells. Notch is involved in many aspects of tumor biology, from angiogenesis to cancer stem cells maintenance to tumor immunity (1–3). Mammals have four structurally related Notch receptors (Notch1–4) that bind transmembrane ligands of the Jagged (Jagged-1, Jagged-2) or the Delta-like (DLL1, DLL3, and DLL4) families (2, 4, 5). Binding of Notch receptors to ligands, or in some cases, ligand-independent receptor activation (6) triggers separation of the extracellular receptor subunit from the transmembrane subunit. The latter undergoes a multistep proteolytic process, which results in the release of a Notch intracellular domain (NICD) (7). NICD translocate into the nucleus and complexes with the CSL (CBF-1/Suppressor of Hairless/LAG-1, also known as RBP-J), and mastermind-like (MAML1-3) coactivator and other proteins to form the Notch transcriptional complex, which regulates the transcription of multiple genes (2, 4, 5, 7). In addition to canonical Notch signaling, several non-canonical (CSL-independent) Notch signals have been described in oncogenesis and inflammation (8–10). Context-dependent Notch signaling regulates many cell fate choices and Notch dysregulation contributes to the development of various malignancies (5). Notch signaling can produce different biological outcomes depending on the timing and the strength of the signals as well as the expression of different ligand/receptor pairs, post-translational modifications, or receptors and specific regulation at both the transcriptional and post-transcriptional level (11, 12). Hyperactivation of Notch has been considered as oncogenic in several cancers including breast cancer and lymphoid malignancies (T-cell acute lymphoblastic leukemia, T-ALL, B-chronic lymphocytic leukemia, and splenic marginal zone lymphoma). On the other hand, loss of function of individual Notch paralogs has revealed tumor-suppressive activities in other malignancies, as reviewed in Ref. (13, 14).
Myeloid cells are essential for the homeostasis of the innate and adaptive immune responses. Myeloid cells [granulocytes, macrophages, and dendritic cells (DCs)] develop from hematopoietic stem cells (HSCs) through sequential differentiation steps under normal physiological conditions. However, multiple soluble factors released by the tumor microenvironment (both tumor cells and tumor-associated stromal cells) perturb the normal myeloid development resulting in the accumulation of myeloid-derived suppressor cells (MDSCs), a heterogeneous group of immature myeloid cells with immune-suppressive properties. In addition, tumor-derived soluble factors induce defects in the differentiation of DCs and accumulation and polarization of tumor-associated macrophages (TAMs), as described in Ref. (15). Although the importance of Notch signaling in myeloid cells differentiation is well understood, the exact nature of Notch effects remains controversial. There is literature supporting a critical role of Notch in the maintenance of progenitor cells to delay the terminal differentiation of myeloid cells, while other data suggest that Notch signaling is required for differentiation of mature myeloid cells, as reviewed in Ref. (16). Overall, it is probably fair to say that the role of Notch signaling in myeloid cell differentiation is context dependent; it depends on the timing of Notch activation and the differentiation stages of myeloid cells.
T-cell-based cancer immunotherapy has shown effectiveness in some highly lethal malignancies and offers a great deal of promise for the treatment of others. Although the Food and Drug Administration (FDA) approved few T cell-based immunotherapy agents and several others are in phase I–II clinical trials, clinical outcomes have not been as universally positive as initially thought. The presence of a tolerogenic microenvironment that blocks the antitumor effector functions of T cells is a major factor limiting the clinical efficacy of T-cell-based immunotherapy (17). Tumor-infiltrating myeloid cells are central components of the tolerogenic tumor microenvironment, along with regulatory T cells (Tregs). Recently, Campese et al. described a role of Notch in immunoregulatory cells including Treg in the context of tumor microenvironment (18). In this review, we will discuss the role of Notch signaling in myeloid cells (MDSC, DC, and macrophages) as a modulator of tumor immune response.
Notch and MDSC
Myeloid-derived suppressor cells are major immune response regulators in cancer and other pathological conditions. MDSCs are a heterogeneous population of cells consisting of myeloid progenitor cells and immature myeloid cells that have immune-suppressive functions, as reviewed in Ref. (19). MDSCs adversely modulate the immune response to cancer and also facilitate tumor metastasis and angiogenesis (15, 19, 20). The immune-suppressive function of MDSC is mediated through the expression of arginase1 (ARG1), inducible NOS, formation of peroxynitrite, expression of TGF-β, IL10, and COX2, sequestration of cysteine, and induction of immunosuppressive Tregs, among others, as reviewed in Ref. (15, 21). In mice, MDSCs are defined by the co-expression of CD11b and Gr-1 markers and consist of two major subsets, the granulocytic polymorphonuclear (PMN)-MDSC (CD11b+Ly6G+Ly6Clo) and the M-MDSC (CD11b+Ly6G−Ly6Chi) (22). However, in humans, the situation appears to be more complex, and several different markers of MDSCs have been described (22).
Although the role of Notch signaling in myelopoiesis remains somewhat controversial, a number of studies have demonstrated that Notch signaling is important for the accumulation of MDSC (18, 23, 24). Transgenic mice that overexpress ADAM10 (responsible for the first proteolytic cleavage of Notch transmembrane subunits) resulted in abrogated B cell development, delayed T cell development in the thymus but systemic expansion of CD11b+Gr1+ MDSC (25). Gibb et al. (25) suggested that differential cleavage of Notch1 into S2 and S3 products modulated by ADAM10 is important to hematopoietic cell-fate determination. Notch was shown to induce myeloid differentiation of multipotent hemopoietic progenitor cells by upregulating the expression of the transcription factor PU.1, suggesting that Notch signaling functions as an extrinsic regulator of myeloid commitment (26).
Gabrilovich et al. reported that inhibition of Notch signaling in hematopoietic progenitor cells (HPCs), MDSCs, and DCs correlates with abnormal myeloid cell differentiation in cancer (23). The inhibition of Notch signaling in these cells is mediated by NICD phosphorylation by casein kinase 2, which disrupts the interaction between NICD and CSL. Another group (27) also reported that blockade of Notch signaling induced the generation of PMN-MDSC with lower immunosuppressive function, but inhibited the production of mononuclear-MDSC. They also showed that Notch-CSL signals modulate the differentiation process and immunosuppressive functions of MDSC. One possible mechanism whereby Notch signaling could regulate MDSC differentiation is through miR-223. Notch suppresses miR-223 expression in rheumatoid arthritis macrophages (28). In turn, miR-223 inhibits the differentiation of tumor-induced MDSC (29), regulating their number and immune-suppressive functions (30).
Myeloid-derived suppressor cells within the tumor microenvironment block the effects of adoptive T cell-based immunotherapy (ACT) by inhibiting several T cell functions, including T cell proliferation and the expression of various cytotoxic mediators. The success of ACT depends on differentiation of CD8+ T cells into cytolytic and cytokine-producing effector cells (31). However, limited exposure to MDSCs can paradoxically enhance the effectiveness of ACT. Acquisition of full effector function in vitro impairs the antitumor efficacy of CD8+ T cell-based ACT (32). In fact, transfer of activated stem cell memory T cells resulted in higher antitumor responses in mice than effector memory T cells (33). These results suggest that inhibition of CD8+ cell differentiation can enhance the antitumor activity of CD8+ T cells following ACT. Rodriguez et al. (34) reported that transient conditioning of CD8+ T cells with MDSC blocks their differentiations into effector T cells and significantly improves their antitumor activity following ACT. Their results indicated that conditioning of T cells with MDSC induces stress survival pathways through blunted mTOR signaling, which in turn modulated T cell differentiation and ACT efficacy. Thus, short-term conditioning T cells with MDSC could prove beneficial in ACT strategies for cancer immunotherapy.
An elegant study by Peng et al. (35) suggested that the presence of MDSC in tumors is correlated with the presence of cancer stem-like cells (CSCs) and both independently predict poor patient survival. These authors suggested that MDSC-derived IL-6 and nitric oxide (NO) may collaborate to activate STAT3 and Notch signaling and induce breast CSCs. Notch signaling has also been proposed to induce cancer metastasis by promoting the migration of MDSCs. Nakayama et al. reported that F-box protein FBXW7 has tumor-suppressive capacity and inhibits cancer metastasis (36). FBXW7 is an E3 ubiquitin protein ligase involved in the degradation of several oncoproteins including NICD. Deletion of Fbxw7 in murine bone marrow-derived stromal cells resulted in the accumulation of Notch1 and increased expression of CCL2. CCL2 in turn facilitated the recruitment of M-MDSC and macrophages, promoting metastatic tumor growth.
The role of Notch in T cell-mediated cancer immunity has been studied extensively (8, 37). Rodriguez et al. (38) reported that the tumor microenvironment suppresses Notch1 and Notch2 expression in CD8 T cells. Conditional expression of transgenic Notch1 intracellular domain (N1ICD) in activated antigen-specific CD8+ T cells induced cytotoxic responses and caused CD8+ T cells to become resistant to MDSC-mediated tolerogenic effects in tumor-bearing mice (38). MDSC blocked the expression of Notch in T cells via NO-dependent mechanisms. The authors suggested that transgenic expression of Notch1 or Notch2 NICD in CD8+ T cells or chimeric antigen receptor T (CAR-T) cells may overcome MDSC-mediated tolerogenic effects and prove therapeutically beneficial. However, the molecular mechanisms whereby MDSC-derived NO inhibits Notch signaling remain unclear.
Recently, the Rodriguez lab in collaboration with the Miele and Osborne labs showed that tumor MDSC, unlike circulating MDSC, upregulate expression of Notch ligand Jagged1, and to a lesser extent, Jagged2. This phenomenon is mediated by NF-κB (39). Treatment with an anti-Jagged1/2-blocking antibody had remarkable therapeutic activity in several mouse models (3LL lung carcinoma and EG-7, an ovalbumin-expressing form of EL-4 lymphoma), which depended upon enhanced CD8 responses (39). In EG-7 tumors, anti-Jagged antibodies enhanced the effect of anti-ovalbumin adoptive T-cell therapy (ACT). Interestingly, anti-Jagged therapy induces the appearance of potentially immune-stimulatory MDSC-like cells (MDSC-LC), which had lower expression of MDSC-suppressive mediators, iNOS and ARG1. It is unclear whether these MDSC-LC derive from the reprogramming of MDSC or from de novo differentiation from bone marrow myeloid precursors upon Jagged inhibition. It is also unclear how Jagged blockade produces this effect. It may allow DLL ligands to activate Notch with a different kinetics, or possibly relieve cis-inhibition of MDSC Notch receptors by Jagged ligands expressed on the same cells. Further mechanistic investigations are necessary to answer these questions. However, these findings provide a preclinical proof of concept for the use of anti-Jagged1/2 antibodies to reprogram MDSC-mediated T-cell suppression to enhance the efficacy of cancer immunotherapy.
In summary, the Notch pathway can be considered a multifaceted modulator of MDSC biology. Notch signals modulate MDSC activity in different ways, depending on the receptors and ligands involved, microenvironmental clues (e.g., NF-κB activation by inflammatory cytokines), the stages of myeloid cells differentiation, as well as the subpopulation of cells. Targeting Jagged-family Notch ligands to inhibit MDSC is a promising strategy to overcome tumor tolerance.
Notch and DCs
Dendritic cells are professional antigen-presenting cells (APC) that recognize, acquire, process, and present antigens to resting T cells to activate antigen-specific immune responses. The engagement of DC in the induction of immune responses against a myriad of pathogens, tumor cells, and self-antigens is a cornerstone of adaptive immunity (40). DCs include distinct functional subsets including interferon-producing plasmacytoid DCs (pDCs) and classical DCs (myeloid) (41–43). Classical DCs are the dominant subset and differentiate along the myeloid lineage pathway. The mechanisms of differentiation of these two subsets are vastly different, although they converge on some pathways (41–43). Decreased DC function has been suggested as a major cause of the observed defect in cell-mediated immunity in patients with advanced breast cancer (44). DC differentiation from HPCs is controlled both by a network of soluble growth factors and cytokines produced by bone marrow stroma (BMS) and direct cell–cell contact with BMS via a complex network of soluble factors and cell-bound molecules. Several studies have implicated Notch signaling in DC differentiation and function (45–47).
There is both consensus and controversy surrounding the extent of Notch involvement in DC differentiation. Several groups have described a direct role of Notch in promoting DC differentiation. Expression of DLL1 in conjunction with GM-CSF induced differentiation of bone marrow cells to DCs at the expense of other lineages (48). In “emergency myelopoiesis,” DLL1 promoted DC differentiation while Jagged1 inhibited it. Both ligands activated Notch, but DLL1 also induced Wnt while Jagged suppressed it by inhibiting the expression of Wnt receptor Frizzled (49). Cheng et al. (50) showed that differentiation of DC was severely compromised in Notch1 antisense mice that have about half the physiological level of Notch1 in HPC. These findings were confirmed in an experimental model of DC differentiation from embryonic stem (ES) cells. Notch1−/− ES are unable to differentiate into DC. In this model, Notch signaling is necessary but not sufficient for DC differentiation (45). On the other hand, Radtke et al. (51) generated Notch1 conditional knockout mice using the Cre-Lox system and demonstrated that the number of thymic DCs, conventional DCs, and Langerhans cells were normal. Whether other Notch paralogs can compensate for Notch1 deficiency in this model is unclear. Conditional deletion of CSL (RBP-Jκ), which abrogates all canonical Notch signaling in BM cells and DCs resulted in substantial reduction in the presence of conventional DCs in spleens of the knockout mice (52). This decrease affected primarily the CD8− DC subset in the spleen marginal zone (52). Weijzen et al. (46) demonstrated that peptides from the DSL (Delta-Serrate-LAG1) receptor-binding region of Jagged1 promote the maturation of monocytes into myeloid DC. This effect may be mediated by direct activation of Notch receptors or relief of cis-inhibition by endogenous Jagged ligands. Lewis et al. (53) demonstrated that Notch2 is required for the functional differentiation of DCs in the spleen and intestine. De Smedt et al. (54) demonstrated an exquisite dose dependence of Notch signaling in the thymic microenvironment, with different levels of Notch signal intensity biasing cell fate decisions toward NK, B, DC, macrophage, or T cell lineages.
Similar contradictory data exist in the literature with respect to the role of Notch signaling in pDCs. It was reported that Notch signaling via DLL1 prevents the differentiation of pDC from early thymocyte precursors by decreasing expression of ETS transcription factor Spi-B. Conversely, Jagged1 did not suppress Spi-B expression. Stromal cells expressing DLL1 blocked pDC development (55). However, in a different study, Notch1−/− bone marrow precursors developed normally into thymic pDC, suggesting that thymocytes and pDC originate from different lineages and that Notch only modulates the thymocyte lineage (56).
There is emerging evidence of crosstalk between Notch and Wnt pathways in the regulation of DC differentiation (57). Inhibition of Notch signaling can lead to accelerated differentiation of HSCs in vitro and depletion of HSCs in vivo (57). Regulation of Notch signaling by the Wnt pathway also plays a vital role in differentiation of precursors along T or NK differentiation pathways (58). Table 1 summarizes some of the key findings reported on the role of Notch signaling in the differentiation and function of tumor-associated myeloid cells.
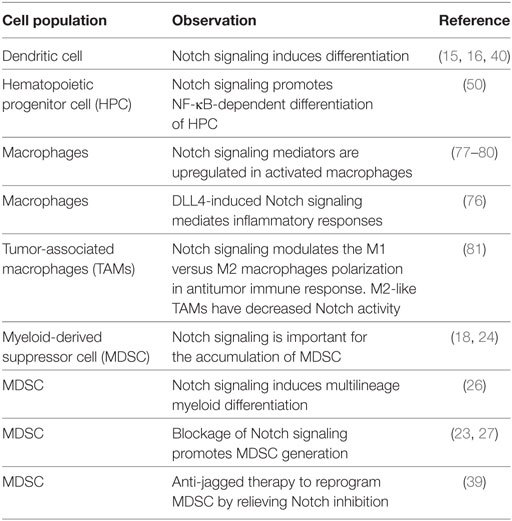
Table 1. Notch effects in the differentiation and function of tumor-associated myeloid cells.
These findings highlight two general features of Notch signaling, namely, its context dependence and dose dependence. Notch signals do not appear to operate as an on/off switch. Rather, in many systems, these signals appear to operate based on an intensity gradient that modulates and is modulated by other pathways. Complete blockade of Notch signals is not always necessary to change cellular phenotypes, and small variations in signal intensity or duration may have major phenotypic consequences. Figure 1 schematically depicts the current consensus on the role of Notch signaling in the differentiation and function of tumor microenvironment-associated myeloid cells.
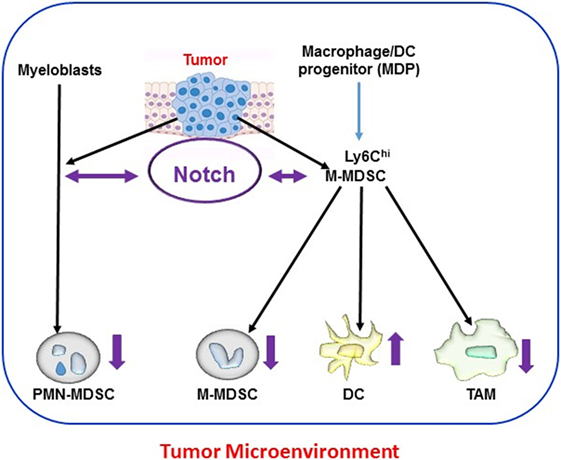
Figure 1. Notch and myeloid cells differentiation within tumor microenvironments. Myeloid cells [polymorphonuclear (PMN) cells, dendritic cells (DCs), and macrophages] derive from hematopoietic stem cells through common myeloid progenitors and the granulocyte-myeloid progenitors lineage. In tumor-bearing hosts, this differentiation process is altered by tumor-derived signals. Expansion of activated PMN-MDSC and M-MDSC occurs from myeloblasts and M-MDSC, respectively, during tumorigenesis. M-MDSC also differentiate into tumor-associated macrophages (TAMs) and DC at the tumor site. Notch signaling mediates bidirectional crosstalk at multiple steps of myeloid cells differentiation in the tumor microenvironment. Differential Notch expression and activity (as indicated by the direction of purple arrows) has been reported in different myeloid populations, with lower Notch expression in myeloid-derived suppressor cell (MDSC) and TAM and higher expression in DC.
Notch signals are involved not only in the maturation of DC but also in their effector functions. DCs express both Notch receptors and ligands as well as toll-like receptors (TLRs) (59). TLRs potently stimulate the expression of Notch ligands in DC (59). TLRs are being increasingly adopted in DC vaccine manufacturing protocols to stimulate DC maturation (60). DCs are composed of subsets that differ in their phenotype, localization, and function. DLL4 + DC promote CD4+ T cell effector response. Blocking DLL4 causes a dramatic reduction of inflammatory T cell responses (60). Gentle et al. (59) demonstrated that DC stimulated concurrently with both Notch and TLR ligands have a distinct cytokine profile and are more pro-inflammatory compared with DCs stimulated with either ligand alone. This effect appears to be mediated by non-canonical Notch signaling (61, 62). Non-canonical Notch signaling regulates various pathways in cancer and immune cells (59). In DC, PI3kinase stimulated by membrane-bound Notch modulates the response to pro-inflammatory signals (59).
In summary, Notch signals play important roles in DC maturation and activity. Canonical and non-canonical Notch signaling are involved. In most cases, Notch activity seems to promote DC maturation and function, but pDC may be an exception. Strategies leading to Notch activation in DC may enhance the effectiveness of DC-based cancer immunotherapy strategies.
Notch and TAMs
Macrophages are a multifunctional and heterogeneous cell population, which can originate from embryonic precursor cells within a tissue or derive from HSCs via the myelomonocytic lineage (63). They can function as phagocytes, APC, and modulators of innate and adaptive immune responses, tissue remodeling, and inflammation. Macrophages are phenotypically plastic, and at least in animal models two distinct polarization pathways have been identified: classic activation-M1 macrophages and alternative activation-M2 macrophages (64, 65). M1-macrophages are polarized and activated by interferon-γ and lipopolysaccharide. They are specialized in innate immune responses against intracellular pathogens. TLR receptors such as TLR4 in M1 macrophages trigger the activation of NF-κB, AP-1, and STAT1 and promote the release of pro-inflammatory cytokines such as IL1, TNFα, IL-12, IL-1, IL-6, IFNγ, and chemokines CCL2 and CXCL10 (66). M2 macrophages secrete anti-inflammatory cytokines such as IL-10 and TGF-β. These cells limit tissue damage caused by inflammation and promote tissue repair and remodeling. Their effects on the adaptive immune system are more complex, including activation and inhibition (67). Importantly, the M1 and M2 polarization states are not irreversible. They can be considered phenotypic manifestations of biological plasticity, and intermediate phenotypes are possible. Additional macrophage subpopulations are emerging (68) whose roles in cancer are still unclear.
Tumor-associated macrophages are important components of the tumor microenvironment (69). TAMs tend to acquire an M2-phenotype. Recent studies have shown that TAMs can originate either from resident tissue macrophages or from tumor-infiltrating monocytes (67). Studies in patient samples and animal models reveal that TAMs can promote tumor growth by modulating angiogenesis, remodeling the extracellular matrix, providing a niche for cancer stem cells, as well as directly enhancing invasion and metastasis (70–72). High numbers of TAMs are linked to poor prognosis in cancer and associated with increased angiogenesis, enhanced tumor cell invasion, and suppression of adaptive antitumor immunity (73, 74). In basal-like breast cancer, TAMs are associated with poor clinical outcomes (75).
Notch signals play important roles in the differentiation, polarization, and activation of macrophages. In general, Notch signaling mediators are upregulated in activated macrophages (76–80). Wang et al. reported that Notch signaling modulated the M1 or M2 polarization of macrophages in antitumor immune response (81). M2-like TAMs have decreased Notch activity. Activation of Notch signaling promoted an M1 phenotype, secretion of IL-12, and enhanced tumor immunity. These authors showed that canonical CSL/RBP-J-mediated Notch signaling modulates the M1 versus M2 polarization through SOCS3 (81). Xu et al. showed that Notch1 enhances the M1 polarization of inflammatory macrophages through canonical and mitochondrial signaling, whereby Notch1 NICD induces CSL-mediated expression of mitochondrial genes but also associates with mitochondria and modulates metabolic activity and mitochondrial genome expression (82).
An elegant study by the Reedijk group showed that Notch signaling in tumor cells regulates the expression of pro-inflammatory cytokines, IL1β and CCL2, and induced the recruitment of TAM (83). In addition, these authors found that Notch regulates TGFβ-mediated activation of tumor cells by TAMs, suggesting a paracrine loop between TAMs and cancer cells mediated by Notch signals. These authors found a strong association between Notch activation, IL1β and CCL2 production, macrophages infiltration in basal-like breast cancer (83). Zhang et al. analyzed patient samples of invasive micropapillary carcinoma of the breast and proposed that Jagged1-modulated TAM infiltration is associated with poor prognosis (84). Liu et al. found Jagged1 expression is associated with high stromal M2-like TAM and with reduced disease-free and overall survival in primary breast tumor tissues (85). Interestingly, they also found higher M2-like TAM infiltration in metastatic lesions than in primary tumor of patients with aromatase inhibitor resistant cancers. They concluded that Jagged1 promotes aromatase inhibitor resistance by inducing TAM differentiation in breast cancer patients (85). Tanase et al. proposed that TAM and Notch signaling cooperate in reprogramming the glioma stem cell niche, providing protection and support for glioma stem cells (86). Guo and Gonzalez-Perez described a novel crosstalk between Notch, IL-1, and leptin that induces angiogenesis in breast cancer (87). In their working model, leptin stimulates receptor and ligand expression in breast cancer cells. This phenomenon is dependent on IL-1 signaling. In turn, Notch contributes to the expression of VEGF/VEGFR2 and thus promotes angiogenesis. In this model, IL-1 produced by inflammatory cells such as TAM would enhance leptin-promoted Notch signaling. This crosstalk would be of particular importance in obesity-associated cancers, as leptin is increased in obese patients. Low-grade systemic chronic inflammation in obesity has been proposed to involve M1 macrophages (88). In this case, systemic production of pro-inflammatory cytokines such as IL-1 by M1 macrophages would promote tumor growth at least in part through Notch.
A recent study demonstrated that miR-148a-3p acts downstream of Notch to promote the differentiation of monocytes into macrophages (89). Following Notch activation, miR-148a-3p promoted M1 but inhibited M2 polarization of macrophages. In a transgenic mouse model, conditional overexpression of NICD had no effect of TAM differentiation, but abrogated TAM functions (90). The same study identified miR-125a as a novel downstream mediator of Notch signaling. A miR-125a mimetic increased the phagocytic activity of macrophages and suppressed tumor growth by remodeling tumor microenvironment (90).
In conclusion, Notch signaling participates in the polarization of macrophages and modulates their activity. Furthermore, cytokines produced by macrophages stimulate Notch in cancer cells, and paracrine loops between macrophages and cancer cells can promote tumor survival.
Concluding Remarks
After decades of preclinical studies with only anecdotal clinical successes, cancer immunotherapy has entered a new phase. Immune checkpoint blockade therapy is one of the most radical innovations in clinical oncology in recent years (91). The FDA approval of CAR-T cell therapy in 2017 was another momentous development (92). However, despite the power of these approaches, there remain plenty of challenges to their clinical application on a large scale. For instance, cancers with low mutational burden are less likely to respond to immunotherapy, perhaps due to their limited antigen repertoire (93). The identification of patients and tumors most likely to respond to immunotherapy through precision medicine approaches is one of the most promising strategies to enhance the impact of cancer immunotherapy. In 2017, in a landmark development, the U.S. FDA granted accelerated approval of an anti-PD-1 antibody to treat patients whose cancers show microsatellite instability or somatic defects in DNA mismatch repair. This was the first FDA approval of an anti-neoplastic agent based not on anatomical cancer location or tumor type but on genomic biomarkers.
Immune suppression by TME myeloid cells is one of the main challenges to the large scale application of cancer immunotherapy. The intricate crosstalk between systemic inflammation, myeloid cells in tumor microenvironment, the cancer cell themselves, and multiple lymphocyte subpopulations modulates tumor immunity. Notch signaling plays multiple roles in this crosstalk (Figure 2), and potential therapeutic applications of Notch modulation in immunotherapy have shown significant promise. Among these, the inhibition of MDSC functions by Jagged antibodies and the enhancement of CD8 resistance to MDSC by CD8 T cell-selective Notch activation appear particularly attractive. Another attractive target is DLL4. Tumor-infiltrating myeloid cells activate Dll4/Notch/TGF-β signaling to drive malignant progression (94). A human DLL4 monoclonal antibody by Oncomed Pharmaceuticals is presently in a phase Ib clinical trial in combination with anti PD-1. Combination cancer immunotherapy, particularly targeting the interaction between myeloid cells and T cells in the tumor microenvironment, is a potentially attractive strategy for Notch-targeted drugs and biologics.
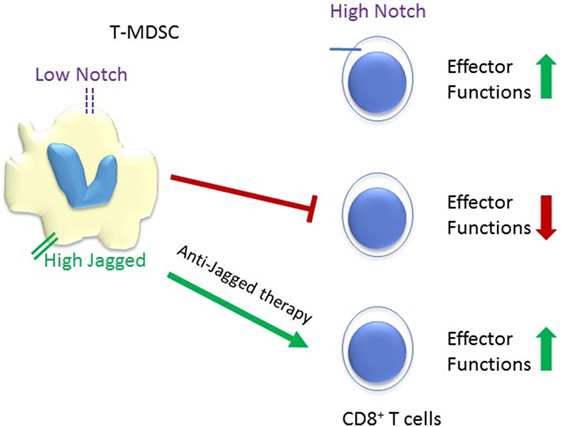
Figure 2. Schematic representation of the reciprocal responses of tumor-infiltrating MDSC (T-MDSC) and CD8+ T cells to Notch signaling. High Notch signaling promotes CD8+ T cells effector functions, while low Notch signaling spurs T-MDSC. Tumor microenvironments upregulate the expression of Notch ligand Jagged on T-MDSC and anti-Jagged therapy overcome tumor-induced T cells tolerance (36). It is unclear whether Jagged expressed in myeloid-derived suppressor cell (MDSC) competes with DLL ligands for Notch1 and Notch2 in CD8 T-cells, or potentially with TCR-induced ligand-independent activation. However, blockade of Jagged1 and 2 in MDSC restores CD8 effector functions.
Author Contributions
FH, SM, and DU wrote different sections of this manuscript. PR, BO, TG, and LMM provided critical input. LM reviewed and edited the draft, and wrote the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
FH, TG, LMM, BO, and LM are supported by P01CA166009 National Cancer Institute. FH and LM are also supported by U54 GM104940 Louisiana Clinical and Translational Sciences Center.
References
1. Farnie G, Clarke RB. Mammary stem cells and breast cancer – role of Notch signalling. Stem Cell Rev (2007) 3:169–75. doi:10.1007/s12015-007-0023-5
2. Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet (2012) 13:654–66. doi:10.1038/nrg3272
3. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol (2015) 12:445–64. doi:10.1038/nrclinonc.2015.61
4. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science (1999) 284:770–6. doi:10.1126/science.284.5415.770
5. Aster JC. In brief: Notch signalling in health and disease. J Pathol (2014) 232:1–3. doi:10.1002/path.4291
6. Palmer WH, Deng WM. Ligand-independent mechanisms of Notch activity. Trends Cell Biol (2015) 25:697–707. doi:10.1016/j.tcb.2015.07.010
7. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell (2009) 137:216–33. doi:10.1016/j.cell.2009.03.045
8. Ayaz F, Osborne BA. Non-canonical Notch signaling in cancer and immunity. Front Oncol (2014) 4:345. doi:10.3389/fonc.2014.00345
9. Lee KS, Wu Z, Song Y, Mitra SS, Feroze AH, Cheshier SH, et al. Roles of PINK1, mTORC2, and mitochondria in preserving brain tumor-forming stem cells in a noncanonical Notch signaling pathway. Genes Dev (2013) 27:2642–7. doi:10.1101/gad.225169.113
10. Minter LM, Osborne BA. Canonical and non-canonical Notch signaling in CD4(+) T cells. Curr Top Microbiol Immunol (2012) 360:99–114. doi:10.1007/82_2012_233
11. Palermo R, Checquolo S, Bellavia D, Talora C, Screpanti I. The molecular basis of Notch signaling regulation: a complex simplicity. Curr Mol Med (2014) 14:34–44. doi:10.2174/1566524013666131118105216
12. Wang H, Zang C, Liu XS, Aster JC. The role of Notch receptors in transcriptional regulation. J Cell Physiol (2015) 230:982–8. doi:10.1002/jcp.24872
13. Lobry C, Oh P, Mansour MR, Look AT, Aifantis I. Notch signaling: switching an oncogene to a tumor suppressor. Blood (2014) 123:2451–9. doi:10.1182/blood-2013-08-355818
14. Aster JC, Pear WS, Blacklow SC. The varied roles of Notch in cancer. Annu Rev Pathol (2017) 12:245–75. doi:10.1146/annurev-pathol-052016-100127
15. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12:253–68. doi:10.1038/nri3175
16. Cheng P, Zhou J, Gabrilovich D. Regulation of dendritic cell differentiation and function by Notch and Wnt pathways. Immunol Rev (2010) 234:105–19. doi:10.1111/j.0105-2896.2009.00871.x
17. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med (2004) 10:909–15. doi:10.1038/nm1100
18. Grazioli P, Felli MP, Screpanti I, Campese AF. The mazy case of Notch and immunoregulatory cells. J Leukoc Biol (2017) 102:361–8. doi:10.1189/jlb.1VMR1216-505R
19. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
20. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol (2009) 182:4499–506. doi:10.4049/jimmunol.0802740
21. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest (2015) 125:3356–64. doi:10.1172/JCI80005
22. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi:10.1038/ncomms12150
23. Cheng P, Kumar V, Liu H, Youn JI, Fishman M, Sherman S, et al. Effects of Notch signaling on regulation of myeloid cell differentiation in cancer. Cancer Res (2014) 74:141–52. doi:10.1158/0008-5472.CAN-13-1686
24. Saleem SJ, Conrad DH. Hematopoietic cytokine-induced transcriptional regulation and Notch signaling as modulators of MDSC expansion. Int Immunopharmacol (2011) 11:808–15. doi:10.1016/j.intimp.2011.03.010
25. Gibb DR, Saleem SJ, Kang DJ, Subler MA, Conrad DH. ADAM10 overexpression shifts lympho- and myelopoiesis by dysregulating site 2/site 3 cleavage products of Notch. J Immunol (2011) 186:4244–52. doi:10.4049/jimmunol.1003318
26. Schroeder T, Kohlhof H, Rieber N, Just U. Notch signaling induces multilineage myeloid differentiation and up-regulates PU.1 expression. J Immunol (2003) 170:5538–48. doi:10.4049/jimmunol.170.11.5538
27. Wang SH, Lu QY, Guo YH, Song YY, Liu PJ, Wang YC. The blockage of Notch signalling promoted the generation of polymorphonuclear myeloid-derived suppressor cells with lower immunosuppression. Eur J Cancer (2016) 68:90–105. doi:10.1016/j.ejca.2016.08.019
28. Ogando J, Tardaguila M, Diaz-Alderete A, Usategui A, Miranda-Ramos V, Martinez-Herrera DJ, et al. Notch-regulated miR-223 targets the aryl hydrocarbon receptor pathway and increases cytokine production in macrophages from rheumatoid arthritis patients. Sci Rep (2016) 6:20223. doi:10.1038/srep20223
29. Liu Q, Zhang M, Jiang X, Zhang Z, Dai L, Min S, et al. miR-223 suppresses differentiation of tumor-induced CD11b(+) Gr1(+) myeloid-derived suppressor cells from bone marrow cells. Int J Cancer (2011) 129:2662–73. doi:10.1002/ijc.25921
30. Cantoni C, Cignarella F, Ghezzi L, Mikesell B, Bollman B, Berrien-Elliott MM, et al. miR-223 regulates the number and function of myeloid-derived suppressor cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol (2017) 133:61–77. doi:10.1007/s00401-016-1621-6
31. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348:62–8. doi:10.1126/science.aaa4967
32. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest (2005) 115:1616–26. doi:10.1172/JCI24480
33. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med (2011) 17:1290–7. doi:10.1038/nm.2446
34. Raber PL, Sierra RA, Thevenot PT, Shuzhong Z, Wyczechowska DD, Kumai T, et al. T cells conditioned with MDSC show an increased anti-tumor activity after adoptive T cell based immunotherapy. Oncotarget (2016) 7:17565–78. doi:10.18632/oncotarget.8197
35. Peng D, Tanikawa T, Li W, Zhao L, Vatan L, Szeliga W, et al. Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/Notch cross-talk signaling. Cancer Res (2016) 76:3156–65. doi:10.1158/0008-5472.CAN-15-2528
36. Yumimoto K, Akiyoshi S, Ueo H, Sagara Y, Onoyama I, Ueo H, et al. F-box protein FBXW7 inhibits cancer metastasis in a non-cell-autonomous manner. J Clin Invest (2015) 125:621–35. doi:10.1172/JCI78782
37. Tsukumo SI, Yasutomo K. Regulation of CD8(+) T cells and antitumor immunity by Notch signaling. Front Immunol (2018) 9:101. doi:10.3389/fimmu.2018.00101
38. Sierra RA, Thevenot P, Raber PL, Cui Y, Parsons C, Ochoa AC, et al. Rescue of Notch-1 signaling in antigen-specific CD8+ T cells overcomes tumor-induced T-cell suppression and enhances immunotherapy in cancer. Cancer Immunol Res (2014) 2:800–11. doi:10.1158/2326-6066.CIR-14-0021
39. Sierra RA, Trillo-Tinoco J, Mohamed E, Yu L, Achyut BR, Arbab A, et al. Anti-Jagged immunotherapy inhibits MDSCs and overcomes tumor-induced tolerance. Cancer Res (2017) 77:5628–38. doi:10.1158/0008-5472.CAN-17-0357
40. Cheng P, Gabrilovich D. Notch signaling in differentiation and function of dendritic cells. Immunol Res (2008) 41:1–14. doi:10.1007/s12026-007-8011-z
41. Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med (1996) 2:1096–103. doi:10.1038/nm1096-1096
42. Gabrilovich DI, Nadaf S, Corak J, Berzofsky JA, Carbone DP. Dendritic cells in antitumor immune responses. II. Dendritic cells grown from bone marrow precursors, but not mature DC from tumor-bearing mice, are effective antigen carriers in the therapy of established tumors. Cell Immunol (1996) 170:111–9. doi:10.1006/cimm.1996.0140
43. Toi M, Kondo S, Suzuki H, Yamamoto Y, Inada K, Imazawa T, et al. Quantitative analysis of vascular endothelial growth factor in primary breast cancer. Cancer (1996) 77:1101–6. doi:10.1002/(SICI)1097-0142(19960315)77:6<1101::AID-CNCR15>3.0.CO;2-5
44. Gabrilovich DI, Corak J, Ciernik IF, Kavanaugh D, Carbone DP. Decreased antigen presentation by dendritic cells in patients with breast cancer. Clin Cancer Res (1997) 3:483–90.
45. Cheng P, Nefedova Y, Miele L, Osborne BA, Gabrilovich D. Notch signaling is necessary but not sufficient for differentiation of dendritic cells. Blood (2003) 102:3980–8. doi:10.1182/blood-2003-04-1034
46. Weijzen S, Velders MP, Elmishad AG, Bacon PE, Panella JR, Nickoloff BJ, et al. The Notch ligand Jagged-1 is able to induce maturation of monocyte-derived human dendritic cells. J Immunol (2002) 169:4273–8. doi:10.4049/jimmunol.169.8.4273
47. Ohishi K, Varnum-Finney B, Serda RE, Anasetti C, Bernstein ID. The Notch ligand, Delta-1, inhibits the differentiation of monocytes into macrophages but permits their differentiation into dendritic cells. Blood (2001) 98:1402–7. doi:10.1182/blood.V98.5.1402
48. Mizutani K, Matsubayashi T, Iwase S, Doi TS, Kasai K, Yazaki M, et al. Murine Delta homologue, mDelta1, expressed on feeder cells controls cellular differentiation. Cell Struct Funct (2000) 25:21–31. doi:10.1247/csf.25.21
49. Liu H, Zhou J, Cheng P, Ramachandran I, Nefedova Y, Gabrilovich DI. Regulation of dendritic cell differentiation in bone marrow during emergency myelopoiesis. J Immunol (2013) 191:1916–26. doi:10.4049/jimmunol.1300714
50. Cheng P, Zlobin A, Volgina V, Gottipati S, Osborne B, Simel EJ, et al. Notch-1 regulates NF-kappaB activity in hemopoietic progenitor cells. J Immunol (2001) 167:4458–67. doi:10.4049/jimmunol.167.8.4458
51. Radtke F, Ferrero I, Wilson A, Lees R, Aguet M, MacDonald HR. Notch1 deficiency dissociates the intrathymic development of dendritic cells and T cells. J Exp Med (2000) 191:1085–94. doi:10.1084/jem.191.7.1085
52. Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med (2007) 204:1653–64. doi:10.1084/jem.20062648
53. Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, et al. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity (2011) 35:780–91. doi:10.1016/j.immuni.2011.08.013
54. De Smedt M, Hoebeke I, Reynvoet K, Leclercq G, Plum J. Different thresholds of Notch signaling bias human precursor cells toward B-, NK-, monocytic/dendritic-, or T-cell lineage in thymus microenvironment. Blood (2005) 106:3498–506. doi:10.1182/blood-2005-02-0496
55. Dontje W, Schotte R, Cupedo T, Nagasawa M, Scheeren F, Gimeno R, et al. Delta-like1-induced Notch1 signaling regulates the human plasmacytoid dendritic cell versus T-cell lineage decision through control of GATA-3 and Spi-B. Blood (2006) 107:2446–52. doi:10.1182/blood-2005-05-2090
56. Ferrero I, Held W, Wilson A, Tacchini-Cottier F, Radtke F, MacDonald HR. Mouse CD11c(+) B220(+) Gr1(+) plasmacytoid dendritic cells develop independently of the T-cell lineage. Blood (2002) 100:2852–7. doi:10.1182/blood-2002-01-0214
57. Zhou J, Cheng P, Youn JI, Cotter MJ, Gabrilovich DI. Notch and wingless signaling cooperate in regulation of dendritic cell differentiation. Immunity (2009) 30:845–59. doi:10.1016/j.immuni.2009.03.021
58. Aoyama K, Delaney C, Varnum-Finney B, Kohn AD, Moon RT, Bernstein ID. The interaction of the Wnt and Notch pathways modulates natural killer versus T cell differentiation. Stem Cells (2007) 25:2488–97. doi:10.1634/stemcells.2007-0102
59. Gentle ME, Rose A, Bugeon L, Dallman MJ. Noncanonical Notch signaling modulates cytokine responses of dendritic cells to inflammatory stimuli. J Immunol (2012) 189:1274–84. doi:10.4049/jimmunol.1103102
60. Meng L, Hu S, Wang J, He S, Zhang Y. DLL4(+) dendritic cells: key regulators of Notch signaling in effector T cell responses. Pharmacol Res (2016) 113:449–57. doi:10.1016/j.phrs.2016.09.001
61. Berechid BE, Kitzmann M, Foltz DR, Roach AH, Seiffert D, Thompson LA, et al. Identification and characterization of presenilin-independent Notch signaling. J Biol Chem (2002) 277:8154–65. doi:10.1074/jbc.M108238200
62. Martinez Arias A, Zecchini V, Brennan K. CSL-independent Notch signalling: a checkpoint in cell fate decisions during development? Curr Opin Genet Dev (2002) 12:524–33. doi:10.1016/S0959-437X(02)00336-2
63. Franklin RA, Li MO. Ontogeny of tumor-associated macrophages and its implication in cancer regulation. Trends Cancer (2016) 2:20–34. doi:10.1016/j.trecan.2015.11.004
64. Baonza A, Garcia-Bellido A. Notch signaling directly controls cell proliferation in the Drosophila wing disc. Proc Natl Acad Sci U S A (2000) 97:2609–14. doi:10.1073/pnas.040576497
65. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018). doi:10.1002/jcp.26429
66. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8:958–69. doi:10.1038/nri2448
67. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi:10.12703/P6-13
68. Chavez-Galan L, Olleros ML, Vesin D, Garcia I. Much more than M1 and M2 macrophages, there are also CD169(+) and TCR(+) macrophages. Front Immunol (2015) 6:263. doi:10.3389/fimmu.2015.00263
69. Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer (2017) 117:1583–91. doi:10.1038/bjc.2017.356
70. Guo C, Buranych A, Sarkar D, Fisher PB, Wang XY. The role of tumor-associated macrophages in tumor vascularization. Vasc Cell (2013) 5:20. doi:10.1186/2045-824X-5-20
71. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol (2017) 14:399–416. doi:10.1038/nrclinonc.2016.217
72. Zarif JC, Taichman RS, Pienta KJ. TAM macrophages promote growth and metastasis within the cancer ecosystem. Oncoimmunology (2014) 3:e941734. doi:10.4161/21624011.2014.941734
73. Chen JJ, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT, et al. Tumor-associated macrophages: the double-edged sword in cancer progression. J Clin Oncol (2005) 23:953–64. doi:10.1200/JCO.2005.12.172
74. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141:39–51. doi:10.1016/j.cell.2010.03.014
75. Mahmoud SM, Lee AH, Paish EC, Macmillan RD, Ellis IO, Green AR. Tumour-infiltrating macrophages and clinical outcome in breast cancer. J Clin Pathol (2012) 65:159–63. doi:10.1136/jclinpath-2011-200355
76. Fung E, Tang SM, Canner JP, Morishige K, Arboleda-Velasquez JF, Cardoso AA, et al. Delta-like 4 induces Notch signaling in macrophages: implications for inflammation. Circulation (2007) 115:2948–56. doi:10.1161/CIRCULATIONAHA.106.675462
77. Goh F, Irvine KM, Lovelace E, Donnelly S, Jones MK, Brion K, et al. Selective induction of the Notch ligand Jagged-1 in macrophages by soluble egg antigen from Schistosoma mansoni involves ERK signalling. Immunology (2009) 127:326–37. doi:10.1111/j.1365-2567.2008.02979.x
78. Hu X, Chung AY, Wu I, Foldi J, Chen J, Ji JD, et al. Integrated regulation of toll-like receptor responses by Notch and interferon-gamma pathways. Immunity (2008) 29:691–703. doi:10.1016/j.immuni.2008.08.016
79. Monsalve E, Perez MA, Rubio A, Ruiz-Hidalgo MJ, Baladron V, Garcia-Ramirez JJ, et al. Notch-1 up-regulation and signaling following macrophage activation modulates gene expression patterns known to affect antigen-presenting capacity and cytotoxic activity. J Immunol (2006) 176:5362–73. doi:10.4049/jimmunol.176.9.5362
80. Palaga T, Buranaruk C, Rengpipat S, Fauq AH, Golde TE, Kaufmann SH, et al. Notch signaling is activated by TLR stimulation and regulates macrophage functions. Eur J Immunol (2008) 38:174–83. doi:10.1002/eji.200636999
81. Wang YC, He F, Feng F, Liu XW, Dong GY, Qin HY, et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res (2010) 70:4840–9. doi:10.1158/0008-5472.CAN-10-0269
82. Xu J, Chi F, Guo T, Punj V, Lee WN, French SW, et al. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J Clin Invest (2015) 125:1579–90. doi:10.1172/JCI76468
83. Shen Q, Cohen B, Zheng W, Rahbar R, Martin B, Murakami K, et al. Notch shapes the innate immunophenotype in breast cancer. Cancer Discov (2017) 7:1320–35. doi:10.1158/2159-8290.CD-17-0037
84. Liu H, Wang J, Liu Z, Wang L, Liu S, Zhang Q. Jagged1 modulated tumor-associated macrophage differentiation predicts poor prognosis in patients with invasive micropapillary carcinoma of the breast. Medicine (2017) 96:e6663. doi:10.1097/MD.0000000000006663
85. Liu H, Wang J, Zhang M, Xuan Q, Wang Z, Lian X, et al. Jagged1 promotes aromatase inhibitor resistance by modulating tumor-associated macrophage differentiation in breast cancer patients. Breast Cancer Res Treat (2017) 166:95–107. doi:10.1007/s10549-017-4394-2
86. Codrici E, Enciu AM, Popescu ID, Mihai S, Tanase C. Glioma stem cells and their microenvironments: providers of challenging therapeutic targets. Stem Cells Int (2016) 2016:5728438. doi:10.1155/2016/5728438
87. Guo S, Gonzalez-Perez RR. Notch, IL-1 and leptin crosstalk outcome (NILCO) is critical for leptin-induced proliferation, migration and VEGF/VEGFR-2 expression in breast cancer. PLoS One (2011) 6:e21467. doi:10.1371/journal.pone.0021467
88. Catalan V, Gomez-Ambrosi J, Rodriguez A, Fruhbeck G. Adipose tissue immunity and cancer. Front Physiol (2013) 4:275. doi:10.3389/fphys.2013.00275
89. Huang F, Zhao JL, Wang L, Gao CC, Liang SQ, An DJ, et al. miR-148a-3p mediates Notch signaling to promote the differentiation and M1 activation of macrophages. Front Immunol (2017) 8:1327. doi:10.3389/fimmu.2017.01327
90. Zhao JL, Huang F, He F, Gao CC, Liang SQ, Ma PF, et al. Forced activation of Notch in macrophages represses tumor growth by upregulating miR-125a and disabling tumor-associated macrophages. Cancer Res (2016) 76:1403–15. doi:10.1158/0008-5472.CAN-15-2019
91. Bolos V, Blanco M, Medina V, Aparicio G, Diaz-Prado S, Grande E. Notch signalling in cancer stem cells. Clin Transl Oncol (2009) 11:11–9. doi:10.1007/s12094-009-0305-2
92. Miller JF, Sadelain M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell (2015) 27:439–49. doi:10.1016/j.ccell.2015.03.007
93. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348:56–61. doi:10.1126/science.aaa8172
Keywords: Notch, cancer, immunity, cellular, inflammation, myeloid cells
Citation: Hossain F, Majumder S, Ucar DA, Rodriguez PC, Golde TE, Minter LM, Osborne BA and Miele L (2018) Notch Signaling in Myeloid Cells as a Regulator of Tumor Immune Responses. Front. Immunol. 9:1288. doi: 10.3389/fimmu.2018.01288
Received: 31 March 2018; Accepted: 22 May 2018;
Published: 04 June 2018
Edited by:
Giovanna Schiavoni, Istituto Superiore di Sanità, ItalyReviewed by:
Valeria Tosello, Istituto Oncologico Veneto (IRCCS), ItalySantos Mañes, Consejo Superior de Investigaciones Científicas (CSIC), Spain
Copyright: © 2018 Hossain, Majumder, Ucar, Rodriguez, Golde, Minter, Osborne and Miele. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucio Miele, bG1pZWxlQGxzdWhzYy5lZHU=