 Ilias Gkikas
Ilias Gkikas Konstantinos Palikaras
Konstantinos Palikaras Nektarios Tavernarakis
Nektarios Tavernarakis
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 05 June 2018
Sec. Immunological Tolerance and Regulation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01283
This article is part of the Research Topic Autophagy in Autoimmunity View all 13 articles
Mitochondria are cellular organelles essential for multiple biological processes, including energy production, metabolites biosynthesis, cell death, and immunological responses among others. Recent advances in the field of immunology research reveal the pivotal role of energy metabolism in innate immune cells fate and function. Therefore, the maintenance of mitochondrial network integrity and activity is a prerequisite for immune system homeostasis. Mitochondrial selective autophagy, known as mitophagy, surveils mitochondrial population eliminating superfluous and/or impaired organelles and mediating cellular survival and viability in response to injury/trauma and infection. Defective removal of damaged mitochondria leads to hyperactivation of inflammatory signaling pathways and subsequently to chronic systemic inflammation and development of inflammatory diseases. Here, we review the molecular mechanisms of mitophagy and highlight its critical role in the innate immune system homeostasis.
The immune system is an intricate network of distinct cell types, tissues, and organs acting synergistically to protect the entire organism against the invasion of various pathogens, including bacteria, fungi, parasites, and viruses among others. The regulation of immune system is a multistep and complex process that classically includes signaling pathways initiated in the surface of immune cells and transmitted to the nucleus through a cascade of phosphorylation events. In turn, epigenetic, transcriptional, posttranscriptional, translational, and posttranslational modifications take place and influence several aspects of innate and adaptive immunity (1, 2). These molecular mechanisms are tightly coordinated and define the onset, the duration, and the magnitude of immune responses neutralizing foreign pathogenic microorganisms and resolving injury (3–6).
Recent evidence underlines the pivotal role of energy metabolism in the regulation of immunity. Mitochondria are dynamic organelles that modify their function, distribution, and structure in response to metabolic state of the cell (7). Proper mitochondrial function not only provides the required energy but also is essential for the establishment and the maintenance of immune cells phenotype and activity (8). Mitochondrial defects, characterized by cytoplasmic calcium elevation, increased reactive oxygen species (ROS) levels, and pronounced release of pro-apoptotic factors and mitochondrial DNA (mtDNA), are key stimulators of inflammatory response pathways (9). Inflammation is a cytoprotective response preserving tissue homeostasis and ensuring viability upon infection or injury (10). Innate immune cells, such as neurotrophils and macrophages, detect harmful stimuli and initiate the inflammatory signaling pathways. However, persistent and unresolved inflammation in metabolic tissues, such as adipose, liver, pancreas, and muscle, leads to the development and progression of several inflammatory pathologies, including atherosclerosis, type-2 diabetes, metabolic syndrome, and inflammatory bowel disease among others (11). The actions of macrophages in these ailments are highly appreciated (12). Inflammasomes are innate immune system receptors and sensors initiating inflammatory responses (13). Excessive mitochondrial dysfunction mediates inflammasome overstimulation in response to noxious stimuli, such as pathogens and cellular debris. In turn, caspase-1 is activated resulting in the generation of pro-inflammatory cytokines and promoting inflammatory cell death (14). Accumulating evidence interconnects impaired energy metabolism and inflammasome hyperstimulation (14–20). Therefore, repairing of mitochondrial functional deficiency or removal of damaged organelles might be beneficial against the undesired chronic systemic inflammation.
In this review, we focus on the role of mitophagy in innate immune system. We first describe the molecular pathways that govern mitophagy as well as its complex interplay with microbe selective autophagy, known as xenophagy. Furthermore, we discuss the essential role of energy metabolism and mitophagy in macrophage homeostasis and inflammasome stimulation. Better understanding of mitochondrial degradation mechanisms is a key requirement for the development of novel therapeutic interventions to tackle numerous pathologies in humans, including inflammatory diseases.
Cellular homeostasis is often undermined by misfolded and aggregated proteins, damaged organelles, and invading microbes, among others. As a consequence, cells have developed sophisticated quality control mechanisms that remove superfluous and/or damaged cytoplasmic components. Autophagy serves as such a clearance mechanism that is highly responsive to the nature of the stimulus. Based on the response, three different types of autophagy have been described including microautophagy, chaperon-mediated autophagy, and macroautophagy (21, 22). For the purpose of this review, the prominent type of macroautophagy (hereby referred to as autophagy) will be described. General autophagy machinery comprises autophagosome formation and maturation via irreversible steps of double-membrane vesicle nucleation and elongation. Mature double-membrane autophagosomes followed by induction of autophagic adaptor proteins can recognize, sequester, and enclose cellular cargo. Ultimately, fusion of the mature autophagosome with the lysosome mediates cargo degradation and recycling of intracellular material (23).
In the immune system, proper mitochondrial function is a prerequisite for inflammatory responses and host defense (24). Accumulation of damaged mitochondria results in excessive ROS production, elevated cytoplasmic calcium levels, and mtDNA release to the cytosol, which in turn triggers inflammasome activation (25–27). Aberrant inflammatory responses have been associated with the development of several autoimmune diseases. Therefore, targeting damaged mitochondria for degradation could be a promising therapeutic strategy against progressive inflammatory pathologies. The removal of damaged mitochondria required the activation of a selective autophagic process, known as mitophagy. Although the crosstalk between mitophagy mechanisms and host defense has been established only recently, a growing body of evidence supports the importance of their coordination.
Following, recent evidence regarding the intricate role of mitophagy in inflammatory responses will be discussed in detail. The involvement of receptors and adaptors molecules is essential for mitophagy initiation and progression. Up to date, several mitochondrial proteins, located either in the outer (OMM) or the inner mitochondrial membrane (IMM), have been characterized as mitophagy receptors. Malfunctioning mitochondria are recognized by a microtubule-associated protein light chain 3 (LC3) in either ubiquitin-dependent or -independent manner (Figure 1). In turn, mitophagy receptors, which harbor an LC3-interacting region (LIR) motif, associate directly with LC3 and promote autophagosome formation (28).
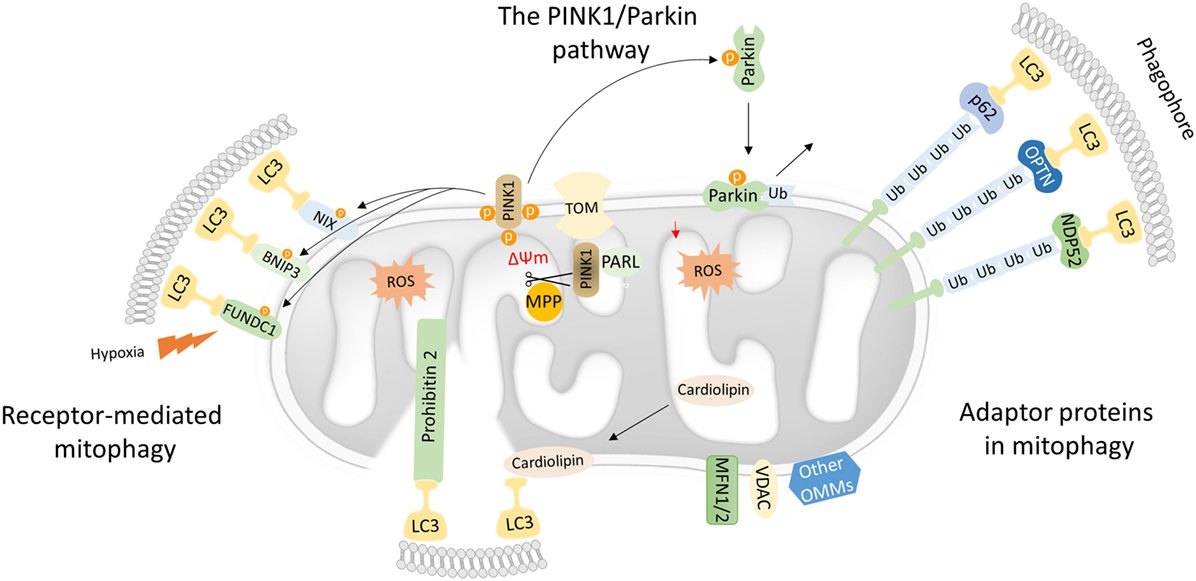
Figure 1. Mechanistic insights into mitophagy process. Dysfunctional mitochondria redirect PTEN-induced kinase 1 (PINK1) to the OMM while its proteolytic cleavage through mitochondrial processing peptidase (MPP) and presenilin-associated rhomboid-like (PARL) proteases is blocked. Concomitantly, PINK1 recruits Parkin through a series of modifications, such as phosphorylation of both Parkin and ubiquitin. In turn, Parkin triggers the polyubiquitination of various OMM proteins including voltage-dependent-anion-selective channel 1 (VDAC1) and MFN1/2. Polyubiquitinated proteins are recognized by several adaptor molecules, including p62, optineurin (OPTN), and NDP52, promoting their recognition by light chain 3 (LC3) and autophogosomal formation. Receptor-mediated mitophagy relies on various OMM proteins such as BNIP3, NIX, and FUN14 domain-containing protein 1 (FUNDC1). In addition, PHB2 and cardiolipin serve as inner mitochondrial membrane receptors in response to mitochondrial damage. Subsequently, PHB2 and cardiolipin are exposed to the cytosol mediating LC3 recruitment via their LIR motifs.
Mutations in the PINK1 and the E3-ubiquitin ligase (Parkin) were primary associated with Parkinson’s disease. Both PINK1 and Parkin are needed for proper mitochondrial function, although their role in mitochondrial turnover was appreciated only recently (29). Under physiological conditions, the transport of PINK1 preprotein into the IMM is followed by sequential proteolytic cleavage by the mitochondrial processing peptidase and presenilin-associated rhomboid-like protease (30–32). The remaining fragment of 52 kDa, which harbors the kinase domain of PINK1, is exposed to the cytosol until its final degradation by the proteasome. Under challenged conditions and loss of mitochondrial integrity, PINK1 fails to translocate to the IMM, and its proteolytic cleavage is blocked. Consequently, active PINK1 accumulates on the OMM though its interaction with the translocons of the outer mitochondrial membrane complex (TOM complex) (33). Then, PINK1 recruits Parkin through a circuit of modifications including phosphorylation of both Parkin and ubiquitin (34–38). Damaged mitochondria are tagged with active Parkin, which, in turn, mediates the polyubiqutination of several OMM proteins, including mitofusin 1 and 2 (MFN1/2), voltage-dependent-anion-selective channel 1, and mitochondrial import receptor subunit TOM20 homolog (TOMM20) among others (Figure 1) (39). In certain cases, Parkin-mediated polyubiquitination triggers the proteosomal degradation, as it has been documented for MFN1 and MFN2 (40, 41). As a consequence, mitochondrial fusion is prevented isolating damaged organelles form the healthy mitochondrial network. Thus, mitofusins degradation generates smaller mitochondria that can easily be sequestered by autophagosomal membranes.
Following, Parkin-mediated ubiqutination of mitochondrial substrates, several adaptors proteins have been described to bind ubiquitin chains on the OMM promoting LC3 recruitment (42). Similar to the canonical autophagy mechanism, LC3 recognizes and interacts with the adaptor molecules through LIR motifs initiating autophagosomal formation. Numerous autophagy adaptors have been identified so far, including p62/sequestosome-1 (SQSTM1), optineurin (OPTN), next to BRCA1 gene 1 (NBR1), nuclear domain 10 protein 52 (NDP52), and TAX1 binding protein 1 (TAX1BP1) (Figure 1) (43).
While the autophagy adaptor p62/SQSTM1 binds ubiquitin chains on depolarized mitochondria and is essential for mitochondrial clustering in a Parkin-dependent manner, the exact role of p62/SQSTM1 in mitophagy has not been verified yet (44–46). Despite the similar kinetics of NDP52, TAX1BP1, and OPTN to dysfunctional mitochondria, cells lacking these adaptors fail to induce mitophagy (47–49). Particularly, loss of OPTN results in most prominent inhibition of mitophagy. Studies in mammalian cells demonstrate that PINK1-mediated recruitment of OPTN and NDP52 autophagy adaptors albeit Parkin was dispensable for mitophagy induction (45). Recent findings suggest that both NDP52 and OPTN are phopshorylated by the Tank-binding kinase 1 (TBK1) and, thereby, enhancing their binding affinity (48, 50–52). Interestingly, TBK1 is activated and phosphorylates OPTN in response to mitochondrial damage. Then, OPTN is recruited on the OMM-promoting mitochondrial elimination (49).
Mitophagy receptors are commonly found on the outer and IMM. Certain OMM receptors of mitophagy have been identified, including BCL2 interacting protein 3 (BNIP3), Nip3-like protein X (NIX), and the FUN14 domain-containing protein 1 (FUNDC1) among others (43). Surprisingly, cardiolipin and prohibitin 2 (PHB2), which are located in IMM, have been also shown to serve as receptor proteins upon stress conditions (53–55). Mitophagy receptors contain LIR motifs that indicate their direct interaction with LC3 to promote the engulfment of defective mitochondria (Figure 1).
NIX contains a mitochondrial BH3 domain and interacts with LC3. NIX has been shown to mediate mitochondrial turnover during reticulocytes’ maturation (56). Specifically, NIX-mediated mitophagy relies on a specific motif within NIX cytoplasmic region, which acts as a signaling amplifier to launch additional mitophagic proteins (57). Under low oxygen levels, NIX transcriptional activity is regulated by hypoxia-inducible factor 1 (HIF1), while posttranslational phosphorylation at Ser81 drives mitochondrial clearance in ischemic stroke (58, 59). In addition, NIX phosphorylation at Ser34 and Ser35 residues surrounding LIR motif increases its binding affinity to LC3 (60). A recent study has also documented an alternative role of NIX-mediated mitochondrial quality control in human fibroblasts lacking PINK1 and Parkin (61, 62). This non-canonical regulation of mitophagy by NIX can give rise to novel therapeutic approaches for removal of malfunctioning mitochondria in Parkinson’s disease.
BNIP3 was also characterized as a BH3 protein on the OMM initially involved in cell death process (63). Despite its role in cell death, a potent role of BNIP3 in mitophagy has been reported. Specifically, its N-terminal LIR motif serves as a signaling platform for LC3-mediated mitochondrial sequestration through autophagosomes. Notably, sufficient LC3 binding is accompanied by BNIP3 phosphorylation at Ser17 and Ser24, proximal to the LIR motif (64–66). Surprisingly, BNIP3-deficient mammalian cells showed induction of PINK1 proteolysis and subsequently failed to promote mitophagy (67). Upon hypoxia, HIF1 triggers BNIP3 expression levels, which, in turn, inhibits cleavage of PINK1 proteolysis and promotes mitophagy (67). Depletion of DCT-1 the Caenorhabditis elegans homolog of both BNIP3 and NIX results in mitophagy inhibition suggesting a conserved role of autophagy receptors among species (68).
In mammalian cells, hypoxia promotes the binding of mitophagy receptor FUNDC1 to LC3 (69). Under normal oxygen levels, LC3 binding is perturbed due to phosphorylation of FUNDC1 at Tyr18 and Ser13 by Src and casein kinase II, respectively (70). In response to hypoxic conditions, phosphoglycerate mutase family member 5 phosphatase is activated and dephosphorylates FUNDC1 enabling its functional association with LC3 autophagosomal protein (71). Recently, it has been reported that FUNDC1 is a substrate of the serine/threonine-protein kinase unc-51-like kinase 1 (ULK1) (72). ULK1 translocates to damaged mitochondria and phosphorylates FUNDC1 at Ser17 triggering mitophagy in response to stress conditions (72). However, several homeostatic mechanisms have been evolved to regulate and fine-tune mitophagy during hypoxia (23, 73). FUNDC1-mediated mitophagy is block due to activation of receptor-interacting serine/threonine-protein kinase 3 followed by phosphorylation of FUNDC1 upon reperfusion injury (73). These results highlight the interplay between mitophagy and necroptosis to maintain cellular homeostasis during hypoxic conditions. Furthermore, mitochondrial E3-ubiquitin protein ligase 5 ubiquitinates FUNDC1 mediating its proteasomal degradation in response to hypoxia (23, 74).
Although the aforementioned receptors are located on the OMM, the possibility that an IMM protein could serve as a mitophagy receptor is not excluded. Toward this direction, PHB2 was recently characterized as an IMM mitophagy receptor (55). Particularly, it has been showed that Parkin-dependent loss of mitochondrial integrity and permeabilization of the OMM enhance the interaction between LC3 and PHB2, thereby promoting mitophagy. In addition, PHB2-mediated mitophagy is involved in selective clearance of paternal mitochondria in C. elegans embryos (55).
Similar to PHB2, cardiolipin belongs to the group of IMM mitophagy receptors. Biosynthesis of cardiolipin occurs in the IMM, where it is primary located. In response to mitochondrial stress, cardiolipin migrates to the OMM setting up a signaling platform for mitophagy and apoptosis initiation. Furthermore, migration of cardiolipin on the OMM is essential for its direct binding of with LC3 and mitophagy stimulation (53). A recent study in yeast showed that both the mitogen-activated protein kinase and the protein kinase C (PKC) are involved in cardiolipin-mediated mitophagy. Interestingly, activation of PKC was sufficient to reverse mitophagy defects phenotypes in cardiolipin-depleted cells (54). Taken together, detail mechanistic insights relative to the activation and function of IMM mitophagy receptors will provide novel therapeutics targets in numerous mitochondrial disorders.
The bacterial origin of mitochondria is a result of an endosymbiotic event that happened billions of years ago. Although the evolutionary changes, mitochondria retained several vestiges of their prokaryotic ancestors. First, mitochondria are semi-autonomous organelles that could expand or shrink their population through fission/fusion events independently of cell division (75). Mitochondria contain their own circular genome that displays evident bacterial characteristics such as decreased methylation events, lack of histones, polycistronic, and intron-less genetic loci (76). Furthermore, mitochondrial inner membrane is composed of cardiolipin, a specific phospholipid that exist uniquely in prokaryotic membranes (77). In addition, mitochondrial protein translation begins with N-formylmethionine, which is a derivative of methionine and a common feature of bacterial and organellar protein synthesis (78). Given the ancestral similarities between mitochondria and bacteria, it is worthwhile to investigate the common molecular mechanisms that regulate mitophagy and microbe selective autophagy, known as xenophagy.
The function of innate immunity is driven by the recognition of endogenous and exogenous signals by innate immune system receptors, such as toll-like receptors (TLRs), formyl peptide receptors, nucleotide oligomerization domain-like receptors (NLRs), retinoic acid-inducible gene 1 (RIG-1)-like receptors (RLRs), C-type lectin receptors (CLRs), and inflammasomes. Both pathogen-associated molecular pattern (PAMP) molecules, which are microbial derived stimulators (e.g., microbial nucleic acids, lipoproteins, and carbohydrates), and damage-associated molecular pattern (DAMPs) molecules, which are released by the cells of the host in response to injury or necrotic cell death (e.g., mtDNA, cardiolipin, ATP, and formyl peptides), are recognized by the immunity receptors mediating, in turn, inflammatory signaling pathways (10, 11).
Similar to mitophagy induction, PAMPs promote the recruitment of autophagic machinery through a series of ubiquitination events and the stimulation of several receptor and adaptor molecules in response to pathogen invasion. Following Mycobacterium tuberculosis infection in macrophages, bacterial DNA is recognized by cGMP-AMP synthase/stimulator of IFN genes mediating type 1 interferon generation and xenophagy initiation (51). Galectin-8 is a cytosolic PAMPs receptor that binds Salmonella typhimurium and prevents its proliferation. Interestingly, galectin-8 recruits NDP52 adaptor protein in a ubiquitin-dependent manner and promotes xenophagy (79). Moreover, several autophagy adaptor proteins, including p62, OPTN, and NBR1 with a well-established role in mitophagy, bind ubiquitinated bacteria and mediate autophagosome formation (Figure 2) (50, 80, 81). Thus, ubiquitination events have an important role in the recognition and the elimination of pathogenic microorganisms bearing a strong resemblance to mitophagy (82). Strikingly, the E3-ubiquitin ligase Parkin has been shown to mediate xenophagy (83). M. tuberculosis ubiqutination is abolished in murine and human Parkin-depleted macrophages resulting in defective bacterial elimination (83). Furthermore, Parkin-deficient nematodes, flies, and mice are more vulnerable to multiple pathogenic bacteria, such as M. tuberculosis, Mycobacterium marinum, S. typhimurium, Listeria monocytogenes, and Pseudomonas aeruginosa (83, 84). Congruently, several polymorphisms in the PARK2 genetic locus are correlated with enhanced susceptibility to Mycobacterium leprae and S. typhimurium in humans, highlighting the evolutionary conserved function of Parkin in innate immunity (85–87).
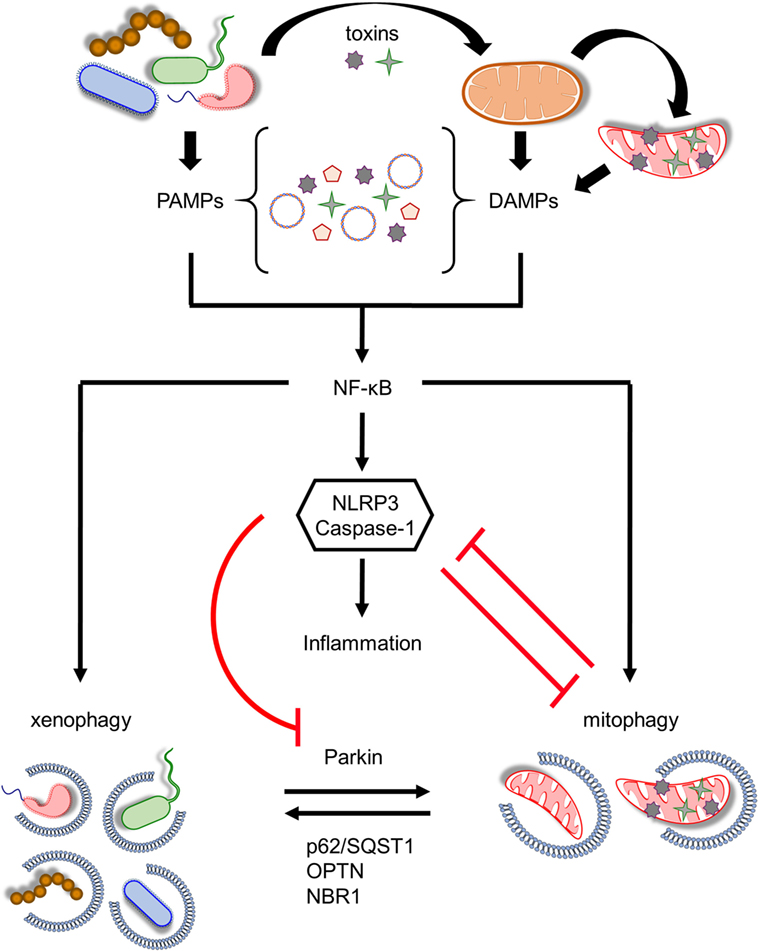
Figure 2. Coordination between mitophagy and xenophagy during infection. Pathogen-derived toxins and secreted proteins impair mitochondrial homeostasis leading to mitochondrial DNA and formyl peptides release and excessive generation of mitochondrial ROS. Consequently, pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) trigger nuclear factor-κB (NF-κB) transcription factor promoting immune responses through NLR family, pyrin domain-containing 3 (NLRP3) inflammasome activation and pro-inflammatory cytokines production. In parallel, NF-κB establishes a self-limiting program to prevent persistent and uncontrolled inflammation by augmenting mitophagy and pathogen removal via xenophagy. NLRP3 stimulation amplifies mitochondrial defects by inhibiting mitophagy through the direct caspase-1-mediated proteolytic cleavage of Parkin. Mitochondrial and bacterial autophagic processes share several common regulatory factors, including Parkin, p62/SQST1, optineurin (OPTN), and NBR1 among others, highlighting their tight communication. This intricate interplay between energy metabolism and innate immune responses upholds cellular and tissue homeostasis and survival during pathogen invasion.
The question then arises: How does bacterial infection stimulate the E3 ligase activity of Parkin? During mitochondrial removal, PINK1 is stabilized on the OMM mediating Parkin translocation and activation (74). Recently, PINK1 has emerged as a critical regulator of innate immunity, as it has been shown that loss of PINK1 enhances inflammation by attenuating the levels of pro- and anti-inflammatory cytokines leading subsequently to cell death (88). Moreover, PINK1-depleted nematodes are sensitive to P. aeruginosa infection (84). However, the role of PINK1 in xenophagy needs further to be elucidated. An alternative candidate of Parkin activation could be the serine/threonine kinase TBK1. Interestingly, TBK1 has an essential role in mitophagy regulation, as it has been found to phosphorylate several autophagy adaptor proteins including p62, OPTN, and NDP52, enhancing cargo recognition and autophagosomal engulfment (48, 50–52). Notably, TBK1 is required for efficient identification and removal of M. tuberculosis and S. typhimurium in mammals (50, 51). Hence, TBK1 kinase could mirror PINK1 activity during infection; however, further investigation of the functional association between TBK1 and Parkin is needed.
Considering the structural similarities between mitochondria and bacteria, an intricate question follows: Could mitochondria be misrecognized by the innate immunity as “invaders” promoting lethal inflammatory responses? Severe physical injury and/or trauma could lead to tissue disruption and cellular damage mediating the release of mitochondrial DAMPs molecules, such as formyl peptides and mtDNA, into the host bloodstream (89). Then, the immune system is alarmed resulting in the development of systemic inflammatory response syndrome (SIRS), which is characterized by fever, increased heart rate, low blood pressure, shortness of breath, multiple organ failure, and increased lethality rates. Although, the characteristic features of SIRS resemble sepsis, an inflammatory response to severe infection, pathogenic microorganisms are need not to be present. Thus, mitochondria lie in the heart of innate immunity initiating uncontrolled immune response upon noxious stimuli.
Several studies have shown the immunogenic capabilities of defective mitochondria (90). Impaired mitochondrial metabolism results in increased mitochondrial ROS (mtROS) levels and defective ion homeostasis. Compelling evidence has been accumulated suggesting that the cytoplasmic levels of mtDNA and mtROS signaling are critical factors in innate immunity via inflammasome activation (16–18). The NLR family, pyrin domain-containing 3 (NLRP3) inflammasome is one of the well-studied inflammasomes protecting the cell against pathogens invasion (13). However, dysregulation of NLRP3 activity leads to chronic inflammation and the development of several pathologies, such as neurodegeneration, metabolic disorders, and sepsis (91).
Infections impair mitochondrial homeostasis mediating mtDNA release, excessive mtROS production, and subsequently inflammasome stimulation (Figure 2). NLRP3 is triggered in response to mitochondrial damage promoting caspase-1 activation. Consequently, caspase-1 generates mature interleukin (IL)-1β and IL-18 promoting inflammatory cell death (14, 92). To this direction, there is evidence suggesting that LC3B-, ATG5-, ATG16L1-, and Beclin1-deficient macrophages display accrual of defective mitochondria, increased cytosolic levels of mtDNA, and mtROS in response to noxious stimuli, resulting in NLRP3 activation and IL-1β secretion (15, 17, 18). In addition to mtDNA and mtROS, NLRP3 is also activated by cardiolipin. Mitochondrial membrane depolarization triggers the translocation of cardiolipin from the IMM to OMM promoting its direct association with NLRP3 (93). NLRP3–cardiolipin interaction is pivotal for inflammasome stimulation indicating that mitochondria act as a central signaling platform for innate immune responses. Experimental evidence highlights the existence of a positive feedback loop between inflammasome and mitochondria, since caspase-1 amplifies mitochondrial dysfunction by impairing mitochondrial membrane potential, increasing mitochondrial membrane permeabilization, and promoting mitochondrial network fragmentation to enhance inflammatory responses (19). Notably, mitophagy is also inhibited upon inflammasome stimulation, since it is reported that Parkin is cleaved by caspase-1 preventing the degradation of damaged organelles (19, 94). Concurrently, accumulation of defective mitochondria results in enhanced mtROS production and hyperstimulation of NLRP3 (Figure 2).
Nuclear factor-κB (NF-κB) is the master coordinator of inflammatory signaling acting downstream of immune receptors (95, 96). In addition to the production of multiple inflammatory chemokines and cytokines, NF-κB also regulates inflammasome activation (97). The TLR9 innate immune receptor recognizes mtDNA, which is released from necrotic cells, resulting in NF-κB nuclearization and the induction of several pro-inflammatory cytokines, such as tumor necrosis factor α (TNFα) and IL-6 (98). A recent study uncovered a self-regulatory and anti-inflammatory pathway, whereby NF-κB restricts NLRP3 function through p62-dependent mitophagy (20). NF-κB enhances the expression of p62 adaptor molecule mediating the removal of damaged mitochondria. Moreover, p62-, Parkin-, and ATG7-depleted macrophages display pronounced NLRP3 activity, since they accumulate defective organelles releasing inflammasome-activating signals in response to harmful stimuli (20). Therefore, NF-κB establishes a self-limiting program to inhibit unresolved inflammation, whereby mitophagy has a central role preventing tissue damage through the maintenance of mitochondrial metabolism (Figure 2).
Mitochondrial antiviral signaling protein (MAVS) is an RLR immune receptor that is localized on the OMM (99). Elevated mtROS levels trigger oligomerization of MAVS, which subsequently activate NF-κB to regulate host defense and inflammation (100). Interestingly, MAVS recruits NLRP3 on the OMM during viral infection (Figure 3). Thereby, inflammasome assembly and activity is enhanced due to the close proximity of NLRP3 with the sites of mtROS generation (101, 102). MAVS signaling is negatively regulated by ubiquitination events mediated by the ubiquitin E3 ligases SMURF1, Gp78, and Mul1 (103–105). Notably, SMURF1, Gp78, and MUL1 are also involved in the regulation of mitochondrial removal indicating the immunosuppressive role of mitophagy in response to noxious stimuli (Figure 3) (106–108). Indeed, a very recent study revealed that anti-inflammatory cytokine IL-10 promotes mitophagy to restrain inflammasome activity and the uncontrolled inflammatory responses upon lipopolysaccharide (LPS) treatment (109).
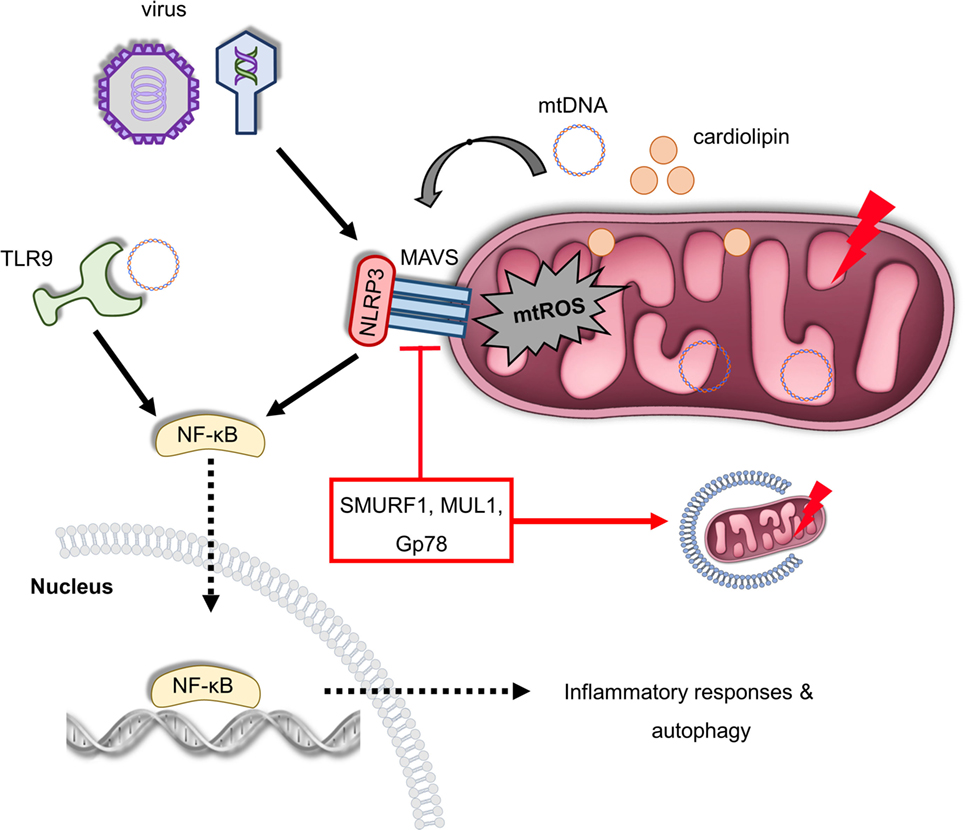
Figure 3. Mitochondria act as a signaling platform of innate immunity. Viral infection triggers mitochondrial antiviral signaling protein (MAVS) oligomerization in OMM, which promotes the expression of several inflammatory and immune genes via nuclear factor-κB (NF-κB) activation. Furthermore, MAVS can be activated by excessive mitochondrial ROS (mtROS) production recruiting NLRP3 on the OMM in response to infection and mitochondrial damage. In turn, NLRP3 assembly and activity is enhanced due to elevated reactive oxygen species levels. In addition, depolarized mitochondria release cardiolipin and mitochondrial DNA (mtDNA) in the cytosol-stimulating NLRP3, which generates pro-inflammatory cytokines and mediates inflammation. mtDNA can be also sensed by toll-like receptor (TLR) 9 immune receptors promoting activation of both NLRP3 and NF-κB. The immunosuppressive role of the ubiquitin E3 ligases SUMRF1, MUL1, and Gp78 is characterized both by the negative regulation of MAVS signaling and mitophagy stimulation. Thus, multiple innate immune signaling pathways, such as MAVS, NLRP3 inflammasome, and TLR9, depend on mitochondrial function to regulate immune responses.
Altogether, these results demonstrate the pivotal role of mitochondria in the innate immune signaling pathways and underline mitophagy as a key regulatory mechanism limiting excessive inflammation and preserving tissue homeostasis. Although the delineation of mitophagy–innate immunity interplay represents a milestone in the field of immunometabolism, several mechanistic questions still remain elusive, including how mitophagy and inflammasome activity are coordinated in response to infection, physical injury, and/or trauma and which already known mitophagy factors are involved during mitochondrial removal in immune cells.
Macrophages are indispensable phagocytic cells coordinating both pro-inflammatory and anti-inflammatory responses as wells as tissue homeostasis and repair during infection (Figure 4) (110). Halted macrophages can be stimulated and adapted to the host and pathogen nature while their activity undergoes dynamic changes (111). Following pathogen invasion and recognition through the innate immune receptors, such as TLRs and CLRs, immune cells produce inflammatory cytokines (112–114). Particularly, secretion of interferon γ (IFNγ) by helper T cells 1, triggers macrophages pro-inflammatory polarization, known as classically activated or M1 macrophages (115, 116). Similar to IFNγ production, LPS signaling also stimulates M1 macrophages through TLRs (117). The tumoricidal and microbicidal properties of M1 macrophages are highlighted by their ability to produce and release several pro-inflammatory cytokines, such as IL-1β and TNFα, and cellular byproducts including ROS and nitric oxide (NO) (118–120). On the contrary, polarization of macrophages toward an anti-inflammatory phenotype is known as alternatively activated or M2 macrophages. In response to specific stimulus, a large spectrum of mediators has been reported to activate M2 macrophages (121). Stimulation of M2 macrophages requires in part, secretion of IL-4 and IL-13 cytokines through Th2 cells (122, 123). Particularly, IL-4- and IL-13-induced M2 macrophages are important for wound healing, while IL-10-induced M2 macrophages regulating host immunity and tissue homeostasis (124). In addition, the secretion of immune complexes together with agonist of TLRs triggers M2 macrophages exerting an immunoresponsive function (125). Non-activated macrophages could be differentiated to M2 phenotype by the transforming growth factor-β activity (124, 126). Given the distinct modes of action, further classification of M2 macrophages has been proposed (57, 115, 127).
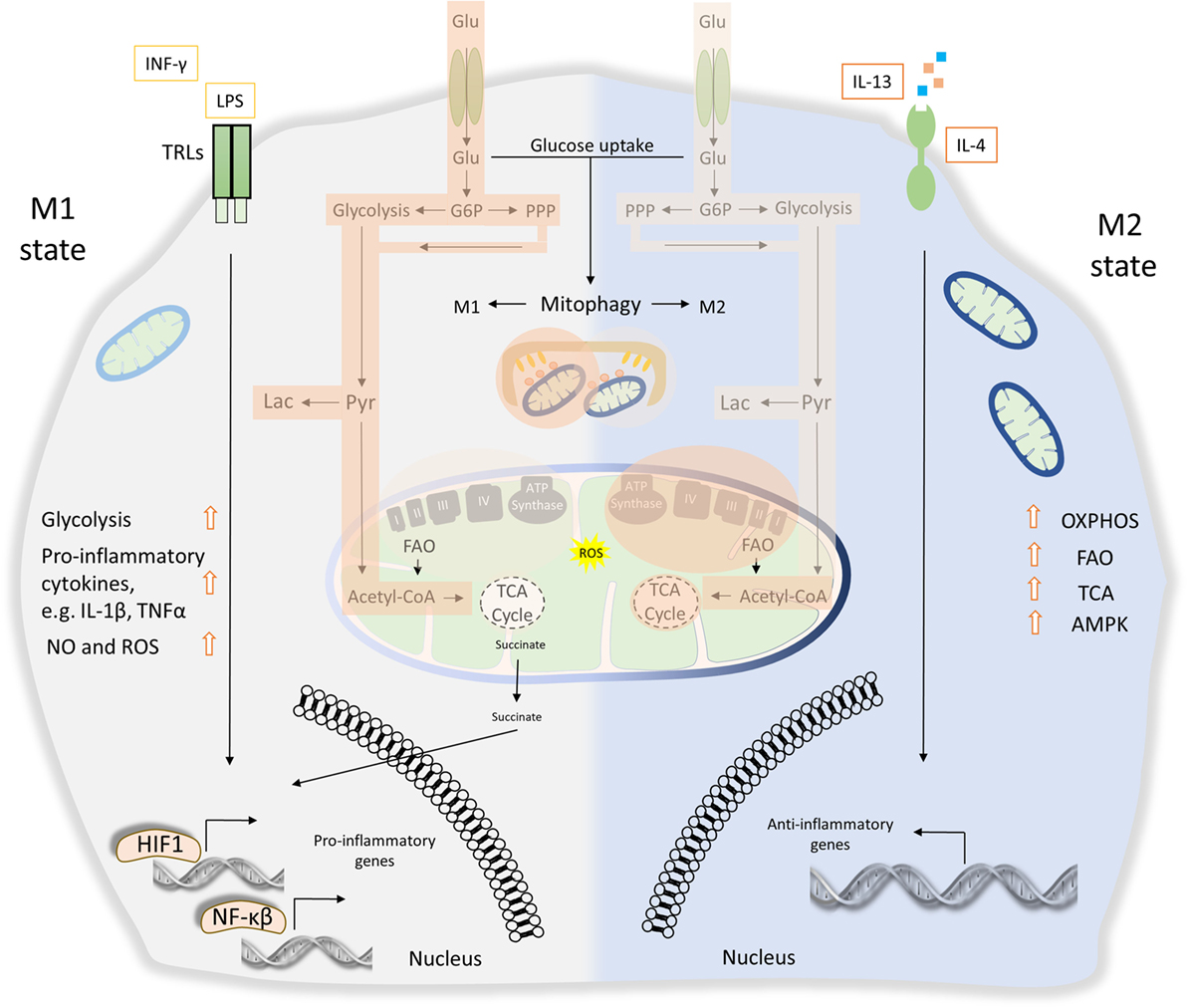
Figure 4. The role of energy homeostasis and mitophagy in metabolic signature of M1 and M2 polarization. M1 macrophages are characterized by secretion of pro-inflammatory cytokines, such as interleukin (IL)-1β and tumor necrosis factor α (TNFα). Thus, M1 macrophages display high tumoricidal and microbicidal properties. M1 macrophages rely heavily on glycolysis accompanied with increased glucose uptake and pentose phosphate pathway (PPP). Metabolic byproducts, such as reactive oxygen species (ROS) and nitric oxide (NO), are abundantly produced. During M1 macrophage polarization, succinate is released to the cytosol and promotes stabilization of hypoxia-inducible factor 1 (HIF1), which in turn drives the transcription of several genes involved in glycolysis. Translocation of both HIF1 and nuclear factor-κB (NF-κB) to the nucleus mediates the expression of numerous pro-inflammatory genes. On the other hand, anti-inflammatory function of M2 macrophages relies more on mitochondrial oxidative phosphorylation (OXPHOS), tricarboxylic cycle (TCA) fluxes, and fatty acid oxidation (FAO). AMP-activated protein kinase (AMPK) is activated and triggers FAO, which fuels OXPHOS. Mitophagy act as a key factor of M1/M2 differentiation. During M1 polarization, mitochondria clearance through mitophagy stimulation favors metabolic reprogramming to glycolysis. Activity of glycolysis, OXPHOS, TCA, and FAO is labeled with bright orange frames (less active state) and dark orange frames (more active state).
Apparently, numerous pathogen-derived molecules and biochemical signals orchestrate macrophage activation from pro-inflammatory M1 toward to anti-inflammatory M2 phenotype, including their intermediate responses (110). As a consequence of these extreme heterogeneities, M1 and M2 macrophages undergo broad transcriptional and metabolic alternations, beyond their energy demands (8). Metabolic signature of both M1 and M2 macrophages activation imposes a tight coordination between glycolysis, pentose phosphate pathway (PPP), fatty acid oxidation (FAO), mitochondrial oxidative phosphorylation (OXPHOS), and tricarboxylic cycle (TCA) fluxes (128). It has been shown that M1 macrophages activity is mostly affected by glycolysis and PPP while mitochondrial OXPHOS and TCA capacities are decreased (129, 130). Specifically, LPS-activated M1 macrophages promote the release of succinate dehydrogenase to the cytosol resulting in the stabilization of HIF1, which in turn regulates the expression of several pro-inflammatory genes such as IL-1β (Figure 4). Absence of HIF1 or inhibition of glycosis in bone marrow-derived macrophages (BMDMs) failed to induce LPS-mediated IL-1β expression (131). Metabolic rewiring from OXPHOS toward glycolysis followed by IL-1β induction requires the stimulation of pyruvate kinase M2 in LPS-activated M1 macrophages (132). On the other hand, M2 macrophages activity relies more on FAO, mitochondrial OXPHOS, and TCA, but less in glycolysis and PPP fluxes (133). Particularly, the stimulation of M2 macrophage requires AMP-activated protein kinase activation as well as induction of FAO to fuel OXPHOS (Figure 4) (134). Notably, byproducts of mitochondrial metabolism, such as mtROS, have also been involved in innate immune responses and macrophages activity. Production of mtROS has been shown to mediate inflammatory cytokine secretion (135). Accordingly, several studies suggest that augmented mtROS levels are required for the bactericidal activity of macrophages (119, 120).
Only recently, the metabolic signature underpinning macrophage activation has been associated with mitochondrial clearance through mitophagy. A latter report suggests that NIX-mediated mitophagy regulates metabolic shift during macrophage differentiation (136). Mitophagy is triggered during M1 macrophage polarization in response to LPS/IFNγ treatment favoring metabolic rewiring to glycolysis. Interestingly, NIX-depleted M1 macrophages present decreased levels of glycolytic enzymes and pro-inflammatory cytokines, indicating metabolic defects during their differentiation process (136). IFN-stimulated gene 15 (ISG15) has also been shown to regulate both cellular metabolism and mitophagy of BMDMs in response to vaccinia virus infection (137). In addition to impaired mitochondrial function, ISG15-deficient macrophages present reduced Parkin protein levels and inhibition of mitophagy upon IFNγ stimulation. Moreover, loss of ISG15 leads to defective macrophages polarization and subsequently to enhanced virus susceptibility (137).
As reported, IL-10-depleted murine BMDMs favors glucose uptake and glycolysis while inhibits OXPHOS in response to LPS treatment (109). IL-10-deficient macrophages display accumulation of damaged mitochondria due to inhibition of mitophagy. Interestingly, IL-10 regulates mitochondrial homeostasis through the inhibition of mTOR signaling (109). A recent study in mouse macrophages has also demonstrated that high glucose supplementation results in mitophagy defects and promotes M1 macrophages activation (138). Therefore, mitophagy regulation is indispensable for the proper determination of M1/M2 macrophage phenotypes (Figure 4). Recently, the essential role of fine-tuned mitochondrial metabolism and mitophagy was underlined in a mouse model of sepsis (139). Bone marrow-derived mesenchymal stem cells (BMSCs) promote survival and performance of various organs during septic shock. It is shown that the beneficial effects of BMSCs were mediated by mitophagy induction in cocultured BMDMs resulting in decreased mtROS levels and inflammasome restriction during cecal ligation and puncture-induced sepsis (139). Although the beneficial effect of mitophagy induction during pronounced inflammatory conditions, mitophagy hyperstimulation could also be detrimental for cellular physiology. Runaway mitophagy mediates mitochondrial content elimination conferring resistance to apoptosis in alveolar macrophages and subsequently leads to the development and progression of idiopathic pulmonary fibrosis (IPF) (140).
Taken together, mitochondrial homeostasis and mitophagy are crucial for the determination of macrophages functional behavior. However, the mechanistic details that orchestrate macrophage intracellular metabolism remain still elusive. A better understanding of the interconnection between mitophagy and macrophages fate and function in response to injury and/or infection could lead to unpreceded understanding of several immune disorders.
Infection triggers the activation of immune system promoting the production and release of several cytokines and chemokines into the host circulation. In turn, inflammatory responses are initiated mediating the signal throughout the body of the organism to confer protection against pathogens. However, persistent systemic inflammatory conditions, such as sepsis, impairs cellular metabolism leading to generalized shock, compromised function of multiple organs and eventually to death. Sepsis or septicemia is a life-threatening condition and a leading cause of morbidity and mortality worldwide (141–143). The pronounced mitochondrial defects, which are described in septic conditions, and the significant role of mitochondria in innate immune signaling indicate their involvement in the development and progression of sepsis (144).
Peripheral mononuclear blood cells, isolated form septic patients, present hyperactivated mitogen-activated protein (MAP) kinase kinase 3 (MKK3) (145). Notably, MKK3 stimulates p38 MAP kinase signaling to promote septic shock (146, 147). MKK3-depleted macrophages display improved energy metabolism, which is characterized by reduced mtROS production, larger and elongated mitochondria, elevated membrane potential and ATP generation during LPS challenge (148, 149). These results suggest that MKK3 alters mitochondrial function to further enhance inflammatory responses. Indeed, MKK3 depletion restricts NF-κB nuclearization and inflammasome stimulation conferring resistance to septic injury (147, 148). Recently, MKK3 has revealed as an essential factor of mitochondrial homeostasis, since MKK3 deficiency influences the modulation of several proteins, including sirtuin 1, PINK1, and Parkin among others, to promote both the induction of mitophagy and mitochondrial biogenesis (145).
Further supporting the immunosuppressive role of mitophagy in sepsis, a recent study showed that senstrin 2 (SESN2) restrains NLRP3 activity by promoting the elimination of damaged mitochondria in macrophages (150). Interestingly, SESN2 mediates the association between p62 and the ubiquitin chains on the OMM, thereby, promoting the perinuclear localization of dysfunctional organelles. In turn, SESN2 initiates autophagosomal formation and mitochondrial turnover by increasing the levels of the autophagy initiator protein ULK1 (150). It has been reported that NO prevents NLRP3 activation and protects against LPS-induced septic shock (151). Notably, NO, generated by nitric oxide synthase 2, upregulates SESN2 protein levels contributing to inflammasome suppression during LPS-induced sepsis (150). In addition, basal levels of NO could also promote mitochondrial translocation of Parkin mediating PINK1-independent mitophagy (152). On the other hand, under nitrosative stress conditions, the excessive NO generation induces S-nitrosylation of PINK1 inhibiting its kinase activity and preventing Parkin mitochondrial recruitment (153). Thus, further investigation is needed to delineate the role of NO activity in the regulation of mitophagy and energy metabolism in immune cells.
Mitophagy holds an essential role in the regulation of inflammatory responses. Several molecular mechanisms are coordinated to mediate mitophagy, preserving cellular and organismal survival in response to intracellular and environmental stimuli (68). Mitophagy deregulation leads to impaired mitochondrial metabolism and eventually to systemic unresolved inflammation and tissue collapse. While basal changes in mitochondrial number and function induce mitophagy, under severe mitochondrial damage excessive mitophagy leads to programmed cell death (154). Therefore, mitophagy impacts organismal health and disease in a context- and dose-dependent fashion (66, 140, 155, 156). To this direction, shortage of mitochondrial population due to induction or persistent mitophagy has also been reported (140, 157, 158). It is becoming evidence that mitophagy defects as well as excessive mitophagy events represent common features of several pathologies. In particular, runaway mitophagy lowers mitochondrial population in alveolar macrophages conferring resistance to apoptosis, which in turn, leads to IPF progression (140). As noted, growth of tumor cells upon hypoxia requires stimulation of glycolysis and lactate production accompanied by increased mitophagy (159, 160). Within this scope, mitochondrial localization of valosin-containing protein drives hyperactivation of mitophagy and leads to neurodegeneration in Huntington’s disease (161). Overall, the degree to which mitophagy contributes to these pathologies has not been elucidated yet. Moreover, it remains to be clarified whether pharmacological stimulation or inhibition of mitophagy represents a potent therapeutic strategy for several pathologies including immune disorders, cancer, and neurodegeneration among others. Although great progress has been already made in the field of mitophagy-inducing drugs, better understanding of the molecular mechanisms would ensure the identification of novel targets for maximum therapeutic efficiency.
Several synthetic and/or natural compounds have been shown to induce mitophagy maintaining cellular and organismal homeostasis (162). p62/SQST1-mediated mitophagy inducer (PMI) is a chemical compound that promotes mitochondrial removal through nuclear factor E2-related factor 2 (Nrf2) stimulation (163). Nrf2 is the master regulator of cellular homeostasis orchestrating the gene expression of multiple cytoprotective proteins, including antioxidant, anti-inflammatory, and detoxification enzymes among others, to enhance survival and viability during stress (164). Moreover, Nrf2 activity is pivotal for proper mitochondrial function and metabolism, since it regulates the expression levels of several mitochondrial related genes (165). PMI prevents the proteasomal degradation of Nrf2 by disrupting its association with Kelch-like ECH-associated protein 1 (163). In turn, Nrf2 is stabilized and enhances p62/SQST1 expression promoting PINK1/Parkin-independent mitophagy upon PMI supplementation (163). Notably, deregulation of Nrf2 function results in the development of autoimmune diseases and increased susceptibility to pathogens, since Nrf2 is implicated in several innate immune responses (166–169). Therefore, PMI administration could be beneficial against the aforementioned pathological conditions.
Natural occurring compounds able to induce mitochondrial turnover have attracted much attention in recent years. As such, spermidine and urolithin A have been shown to induce mitochondrial elimination promoting longevity and stress resistance in many organisms, including mice, flies, and nematodes (170–172). In addition to mitophagy, both spermidine and urolithin A present anti-inflammatory properties modulating mitochondrial metabolism and subsequently NF-κB activity (173–178).
The balanced interplay between the inflammatory and the immunosuppressive signaling pathways highlights that sustaining mitochondrial network integrity and energy metabolism safeguard tissue and organismal homeostasis in response to constant exposure to immunogenic signals. Hence, examination of already FDA-approved drugs and several pharmacological screenings are taking place to characterize novel molecules that can be used to enhance immune system homeostasis through the regulation of mitophagy. Although, experimental evidence underlines the cytoprotective effects of mitophagy modulators on animal disease models, the therapeutic potential and the levels of cytotoxicity on humans remain to be determined. Thus, interventional clinical studies need to be organized to monitor and validate the therapeutic capacity of mitophagy-inducing agents against immune system diseases.
IG and KP wrote the manuscript. NT organized and edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to those colleagues whose work could not be referenced owing to space limitations. KP is supported by an AXA Research Fund post-doctoral long-term fellowship. Work in the authors’ laboratory is funded by grants from the European Research Council (ERC—GA695190—MANNA, ERC—GA737599—NeuronAgeScreen), the European Commission Framework Programmes, and the Greek Ministry of Education.
1. Piccirillo CA, Bjur E, Topisirovic I, Sonenberg N, Larsson O. Translational control of immune responses: from transcripts to translatomes. Nat Immunol (2014) 15:503–11. doi:10.1038/ni.2891
2. Chen YG, Satpathy AT, Chang HY. Gene regulation in the immune system by long noncoding RNAs. Nat Immunol (2017) 18:962–72. doi:10.1038/ni.3771
3. Weinmann AS, Mitchell DM, Sanjabi S, Bradley MN, Hoffmann A, Liou HC, et al. Nucleosome remodeling at the IL-12 p40 promoter is a TLR-dependent, Rel-independent event. Nat Immunol (2001) 2:51–7. doi:10.1038/83168
4. Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature (2003) 425:516–21. doi:10.1038/nature01991
5. Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature (2007) 447:972–8. doi:10.1038/nature05836
6. Doxaki C, Kampranis SC, Eliopoulos AG, Spilianakis C, Tsatsanis C. Coordinated regulation of miR-155 and miR-146a genes during induction of endotoxin tolerance in macrophages. J Immunol (2015) 195:5750–61. doi:10.4049/jimmunol.1500615
7. Friedman JR, Nunnari J. Mitochondrial form and function. Nature (2014) 505:335–43. doi:10.1038/nature12985
8. Mehta MM, Weinberg SE, Chandel NS. Mitochondrial control of immunity: beyond ATP. Nat Rev Immunol (2017) 17:608–20. doi:10.1038/nri.2017.66
9. Mills EL, Kelly B, O’neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol (2017) 18:488–98. doi:10.1038/ni.3704
10. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454:428–35. doi:10.1038/nature07201
11. Netea MG, Balkwill F, Chonchol M, Cominelli F, Donath MY, Giamarellos-Bourboulis EJ, et al. A guiding map for inflammation. Nat Immunol (2017) 18:826–31. doi:10.1038/ni.3790
12. Cildir G, Akincilar SC, Tergaonkar V. Chronic adipose tissue inflammation: all immune cells on the stage. Trends Mol Med (2013) 19:487–500. doi:10.1016/j.molmed.2013.05.001
13. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21:677–87. doi:10.1038/nm.3893
14. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature (2012) 481:278–86. doi:10.1038/nature10759
15. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature (2008) 456:264–8. doi:10.1038/nature07383
16. Van Bruggen R, Koker MY, Jansen M, Van Houdt M, Roos D, Kuijpers TW, et al. Human NLRP3 inflammasome activation is Nox1-4 independent. Blood (2010) 115:5398–400. doi:10.1182/blood-2009-10-250803
17. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12:222–30. doi:10.1038/ni.1980
18. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi:10.1038/nature09663
19. Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, et al. Inflammasome activation leads to caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci U S A (2014) 111:15514–9. doi:10.1073/pnas.1414859111
20. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell (2016) 164:896–910. doi:10.1016/j.cell.2015.12.057
21. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol (2010) 221:3–12. doi:10.1002/path.2697
22. Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy (2017) 14(2):207–15. doi:10.1080/15548627.2017.1378838
23. Chen Z, Liu L, Cheng Q, Li Y, Wu H, Zhang W, et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep (2017) 18:495–509. doi:10.15252/embr.201643309
24. Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med (2011) 208:417–20. doi:10.1084/jem.20110367
25. Ichimura H, Parthasarathi K, Quadri S, Issekutz AC, Bhattacharya J. Mechano-oxidative coupling by mitochondria induces proinflammatory responses in lung venular capillaries. J Clin Invest (2003) 111:691–9. doi:10.1172/JCI17271
26. Maass DL, White J, Sanders B, Horton JW. Role of cytosolic vs. mitochondrial Ca2+ accumulation in burn injury-related myocardial inflammation and function. Am J Physiol Heart Circ Physiol (2005) 288:H744–51. doi:10.1152/ajpheart.00367.2004
27. Escames G, Lopez LC, Garcia JA, Garcia-Corzo L, Ortiz F, Acuna-Castroviejo D. Mitochondrial DNA and inflammatory diseases. Hum Genet (2012) 131:161–73. doi:10.1007/s00439-011-1057-y
28. Ploumi C, Daskalaki I, Tavernarakis N. Mitochondrial biogenesis and clearance: a balancing act. FEBS J (2017) 284:183–95. doi:10.1111/febs.13820
29. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol (2011) 12:9–14. doi:10.1038/nrm3028
30. Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol (2010) 191:933–42. doi:10.1083/jcb.201008084
31. Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet (2011) 20:867–79. doi:10.1093/hmg/ddq526
32. Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep (2012) 13:378–85. doi:10.1038/embor.2012.14
33. Pickles S, Vigie P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol (2018) 28:R170–85. doi:10.1016/j.cub.2018.01.004
34. Sha D, Chin LS, Li L. Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Hum Mol Genet (2010) 19:352–63. doi:10.1093/hmg/ddp501
35. Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol (2013) 200:163–72. doi:10.1083/jcb.201210111
36. Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol (2014) 205:143–53. doi:10.1083/jcb.201402104
37. Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature (2014) 510:162–6. doi:10.1038/nature13392
38. Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol (2014) 16:495–501. doi:10.1038/ncb2979
39. Palikaras K, Tavernarakis N. Mitophagy in neurodegeneration and aging. Front Genet (2012) 3:297. doi:10.3389/fgene.2012.00297
40. Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet (2010) 19:4861–70. doi:10.1093/hmg/ddq419
41. Chen Y, Dorn GW II. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science (2013) 340:471–5. doi:10.1126/science.1231031
42. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy (2011) 7:279–96. doi:10.4161/auto.7.3.14487
43. Lazarou M. Keeping the immune system in check: a role for mitophagy. Immunol Cell Biol (2015) 93:3–10. doi:10.1038/icb.2014.75
44. Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy (2010) 6:1090–106. doi:10.4161/auto.6.8.13426
45. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature (2015) 524:309–14. doi:10.1038/nature14893
46. Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet (2015) 24:4429–42. doi:10.1093/hmg/ddv179
47. Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A (2014) 111:E4439–48. doi:10.1073/pnas.1405752111
48. Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell (2015) 60:7–20. doi:10.1016/j.molcel.2015.08.016
49. Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A (2016) 113:E3349–58. doi:10.1073/pnas.1523810113
50. Wild P, Farhan H, Mcewan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science (2011) 333:228–33. doi:10.1126/science.1205405
51. Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell (2012) 150:803–15. doi:10.1016/j.cell.2012.06.040
52. Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A (2016) 113:4039–44. doi:10.1073/pnas.1523926113
53. Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol (2013) 15:1197–205. doi:10.1038/ncb2837
54. Shen Z, Li Y, Gasparski AN, Abeliovich H, Greenberg ML. Cardiolipin regulates mitophagy through the protein kinase C pathway. J Biol Chem (2017) 292:2916–23. doi:10.1074/jbc.M116.753574
55. Wei Y, Chiang WC, Sumpter R Jr, Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell (2017) 168:224–38.e10. doi:10.1016/j.cell.2016.11.042
56. Novak I, Kirkin V, Mcewan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep (2010) 11:45–51. doi:10.1038/embor.2009.256
57. Zhang J, Loyd MR, Randall MS, Waddell MB, Kriwacki RW, Ney PA. A short linear motif in BNIP3L (NIX) mediates mitochondrial clearance in reticulocytes. Autophagy (2012) 8:1325–32. doi:10.4161/auto.20764
58. Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res (2001) 61:6669–73.
59. Yuan Y, Zheng Y, Zhang X, Chen Y, Wu X, Wu J, et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy (2017) 13:1754–66. doi:10.1080/15548627.2017.1357792
60. Rogov VV, Suzuki H, Marinkovic M, Lang V, Kato R, Kawasaki M, et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep (2017) 7:1131. doi:10.1038/s41598-017-01258-6
61. Koentjoro B, Park JS, Sue CM. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci Rep (2017) 7:44373. doi:10.1038/srep44373
62. Park JS, Koentjoro B, Sue CM. Commentary: Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Front Mol Neurosci (2017) 10:297. doi:10.3389/fnmol.2017.00297
63. Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ (2009) 16:939–46. doi:10.1038/cdd.2009.16
64. Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem (2012) 287:19094–104. doi:10.1074/jbc.M111.322933
65. Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, et al. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem (2013) 288:1099–113. doi:10.1074/jbc.M112.399345
66. Yoo SM, Jung YK. A molecular approach to mitophagy and mitochondrial dynamics. Mol Cells (2018) 41:18–26. doi:10.14348/molcells.2018.2277
67. Zhang T, Xue L, Li L, Tang C, Wan Z, Wang R, et al. BNIP3 protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. J Biol Chem (2016) 291:21616–29. doi:10.1074/jbc.M116.733410
68. Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature (2015) 521:525–8. doi:10.1038/nature14300
69. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol (2012) 14:177–85. doi:10.1038/ncb2422
70. Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res (2014) 24:787–95. doi:10.1038/cr.2014.75
71. Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell (2014) 54:362–77. doi:10.1016/j.molcel.2014.02.034
72. Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep (2014) 15:566–75. doi:10.1002/embr.201438501
73. Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D, et al. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol (2017) 13:498–507. doi:10.1016/j.redox.2017.07.007
74. Palikaras K, Daskalaki I, Markaki M, Tavernarakis N. Mitophagy and age-related pathologies: development of new therapeutics by targeting mitochondrial turnover. Pharmacol Ther (2017) 178:157–74. doi:10.1016/j.pharmthera.2017.04.005
75. Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta (2008) 1777:1092–7. doi:10.1016/j.bbabio.2008.05.001
76. Archibald JM. Endosymbiosis and eukaryotic cell evolution. Curr Biol (2015) 25:R911–21. doi:10.1016/j.cub.2015.07.055
77. Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol (2011) 192:7–16. doi:10.1083/jcb.201006159
78. Dahlgren C, Gabl M, Holdfeldt A, Winther M, Forsman H. Basic characteristics of the neutrophil receptors that recognize formylated peptides, a danger-associated molecular pattern generated by bacteria and mitochondria. Biochem Pharmacol (2016) 114:22–39. doi:10.1016/j.bcp.2016.04.014
79. Thurston TL, Wandel MP, Von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature (2012) 482:414–8. doi:10.1038/nature10744
80. Cemma M, Kim PK, Brumell JH. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy (2011) 7:341–5. doi:10.4161/auto.7.3.14046
81. Von Muhlinen N, Akutsu M, Ravenhill BJ, Foeglein A, Bloor S, Rutherford TJ, et al. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol Cell (2012) 48:329–42. doi:10.1016/j.molcel.2012.08.024
82. Boyle KB, Randow F. The role of ‘eat-me’ signals and autophagy cargo receptors in innate immunity. Curr Opin Microbiol (2013) 16:339–48. doi:10.1016/j.mib.2013.03.010
83. Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature (2013) 501:512–6. doi:10.1038/nature12566
84. Kirienko NV, Ausubel FM, Ruvkun G. Mitophagy confers resistance to siderophore-mediated killing by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A (2015) 112:1821–6. doi:10.1073/pnas.1424954112
85. Mira MT, Alcais A, Nguyen VT, Moraes MO, Di Flumeri C, Vu HT, et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature (2004) 427:636–40. doi:10.1038/nature02326
86. Ali S, Vollaard AM, Widjaja S, Surjadi C, Van De Vosse E, Van Dissel JT. PARK2/PACRG polymorphisms and susceptibility to typhoid and paratyphoid fever. Clin Exp Immunol (2006) 144:425–31. doi:10.1111/j.1365-2249.2006.03087.x
87. Chopra R, Ali S, Srivastava AK, Aggarwal S, Kumar B, Manvati S, et al. Mapping of PARK2 and PACRG overlapping regulatory region reveals LD structure and functional variants in association with leprosy in unrelated Indian population groups. PLoS Genet (2013) 9:e1003578. doi:10.1371/journal.pgen.1003578
88. Sun L, Shen R, Agnihotri SK, Chen Y, Huang Z, Bueler H. Lack of PINK1 alters glia innate immune responses and enhances inflammation-induced, nitric oxide-mediated neuron death. Sci Rep (2018) 8:383. doi:10.1038/s41598-017-18786-w
89. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464:104–7. doi:10.1038/nature08780
90. Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol (2011) 32:157–64. doi:10.1016/j.it.2011.01.005
91. Kim MJ, Yoon JH, Ryu JH. Mitophagy: a balance regulator of NLRP3 inflammasome activation. BMB Rep (2016) 49:529–35. doi:10.5483/BMBRep.2016.49.10.115
92. Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature (1997) 386:619–23. doi:10.1038/386619a0
93. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity (2013) 39:311–23. doi:10.1016/j.immuni.2013.08.001
94. Kahns S, Kalai M, Jakobsen LD, Clark BF, Vandenabeele P, Jensen PH. Caspase-1 and caspase-8 cleave and inactivate cellular parkin. J Biol Chem (2003) 278:23376–80. doi:10.1074/jbc.M300495200
95. Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol (2009) 27:693–733. doi:10.1146/annurev.immunol.021908.132641
96. Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation-the negative regulation of NF-kappaB and the NLRP3 inflammasome. Nat Immunol (2017) 18:861–9. doi:10.1038/ni.3772
97. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140:821–32. doi:10.1016/j.cell.2010.01.040
98. Zhang JZ, Liu Z, Liu J, Ren JX, Sun TS. Mitochondrial DNA induces inflammation and increases TLR9/NF-kappaB expression in lung tissue. Int J Mol Med (2014) 33:817–24. doi:10.3892/ijmm.2014.1650
99. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell (2005) 122:669–82. doi:10.1016/j.cell.2005.08.012
100. Buskiewicz IA, Montgomery T, Yasewicz EC, Huber SA, Murphy MP, Hartley RC, et al. Reactive oxygen species induce virus-independent MAVS oligomerization in systemic lupus erythematosus. Sci Signal (2016) 9:ra115. doi:10.1126/scisignal.aaf1933
101. Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol (2013) 191:4358–66. doi:10.4049/jimmunol.1301170
102. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell (2013) 153:348–61. doi:10.1016/j.cell.2013.02.054
103. Wang Y, Tong X, Ye X. Ndfip1 negatively regulates RIG-I-dependent immune signaling by enhancing E3 ligase Smurf1-mediated MAVS degradation. J Immunol (2012) 189:5304–13. doi:10.4049/jimmunol.1201445
104. Jenkins K, Khoo JJ, Sadler A, Piganis R, Wang D, Borg NA, et al. Mitochondrially localised MUL1 is a novel modulator of antiviral signaling. Immunol Cell Biol (2013) 91:321–30. doi:10.1038/icb.2013.7
105. Jacobs JL, Zhu J, Sarkar SN, Coyne CB. Regulation of mitochondrial antiviral signaling (MAVS) expression and signaling by the mitochondria-associated endoplasmic reticulum membrane (MAM) protein Gp78. J Biol Chem (2014) 289:1604–16. doi:10.1074/jbc.M113.520254
106. Orvedahl A, Sumpter R Jr, Xiao G, Ng A, Zou Z, Tang Y, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature (2011) 480:113–7. doi:10.1038/nature10546
107. Lokireddy S, Wijesoma IW, Teng S, Bonala S, Gluckman PD, Mcfarlane C, et al. The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metab (2012) 16:613–24. doi:10.1016/j.cmet.2012.10.005
108. Fu M, St-Pierre P, Shankar J, Wang PT, Joshi B, Nabi IR. Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol Biol Cell (2013) 24:1153–62. doi:10.1091/mbc.E12-08-0607
109. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science (2017) 356:513–9. doi:10.1126/science.aal3535
110. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol (2017) 198:1006–14. doi:10.4049/jimmunol.1601515
111. Mills EL, O’Neill LA. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur J Immunol (2016) 46:13–21. doi:10.1002/eji.201445427
112. Kumagai Y, Akira S. Identification and functions of pattern-recognition receptors. J Allergy Clin Immunol (2010) 125:985–92. doi:10.1016/j.jaci.2010.01.058
114. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol (2014) 14:81–93. doi:10.1038/nri3600
115. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8:958–69. doi:10.1038/nri2448
116. Su X, Yu Y, Zhong Y, Giannopoulou EG, Hu X, Liu H, et al. Interferon-gamma regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat Immunol (2015) 16:838–49. doi:10.1038/ni.3205
117. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol (2009) 27:451–83. doi:10.1146/annurev.immunol.021908.132532
118. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25:677–86. doi:10.1016/j.it.2004.09.015
119. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature (2011) 472:476–80. doi:10.1038/nature09973
120. Hall CJ, Boyle RH, Astin JW, Flores MV, Oehlers SH, Sanderson LE, et al. Immunoresponsive gene 1 augments bactericidal activity of macrophage-lineage cells by regulating beta-oxidation-dependent mitochondrial ROS production. Cell Metab (2013) 18:265–78. doi:10.1016/j.cmet.2013.06.018
121. Roszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm (2015) 2015:816460. doi:10.1155/2015/816460
122. Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, Van Rooijen N, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science (2011) 332:1284–8. doi:10.1126/science.1204351
123. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol (2011) 11:723–37. doi:10.1038/nri3073
124. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi:10.1016/j.immuni.2014.06.008
125. Muraille E, Leo O, Moser M. TH1/TH2 paradigm extended: macrophage polarization as an unappreciated pathogen-driven escape mechanism? Front Immunol (2014) 5:603. doi:10.3389/fimmu.2014.00603
126. Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, et al. TGF-beta induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget (2016) 7:52294–306. doi:10.18632/oncotarget.10561
127. Leopold Wager CM, Wormley FL Jr. Classical versus alternative macrophage activation: the Ying and the Yang in host defense against pulmonary fungal infections. Mucosal Immunol (2014) 7:1023–35. doi:10.1038/mi.2014.65
128. Tur J, Vico T, Lloberas J, Zorzano A, Celada A. Macrophages and mitochondria: a critical interplay between metabolism, signaling, and the functional activity. Adv Immunol (2017) 133:1–36. doi:10.1016/bs.ai.2016.12.001
129. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab (2012) 15:813–26. doi:10.1016/j.cmet.2012.04.023
130. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity (2015) 42:406–17. doi:10.1016/j.immuni.2015.02.002
131. Tannahill GM, Curtis AM, Adamik J, Palsson-Mcdermott EM, Mcgettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496:238–42. doi:10.1038/nature11986
132. Alves-Filho JC, Palsson-McDermott EM. Pyruvate kinase M2: a potential target for regulating inflammation. Front Immunol (2016) 7:145. doi:10.3389/fimmu.2016.00145
133. Van Den Bossche J, Baardman J, Otto NA, Van Der Velden S, Neele AE, Van Den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep (2016) 17:684–96. doi:10.1016/j.celrep.2016.09.008
134. Mounier R, Theret M, Arnold L, Cuvellier S, Bultot L, Goransson O, et al. AMPKalpha1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab (2013) 18:251–64. doi:10.1016/j.cmet.2013.06.017
135. Chandel NS, Schumacker PT, Arch RH. Reactive oxygen species are downstream products of TRAF-mediated signal transduction. J Biol Chem (2001) 276:42728–36. doi:10.1074/jbc.M103074200
136. Esteban-Martinez L, Sierra-Filardi E, Mcgreal RS, Salazar-Roa M, Marino G, Seco E, et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J (2017) 36:1688–706. doi:10.15252/embj.201695916
137. Baldanta S, Fernandez-Escobar M, Acin-Perez R, Albert M, Camafeita E, Jorge I, et al. ISG15 governs mitochondrial function in macrophages following vaccinia virus infection. PLoS Pathog (2017) 13:e1006651. doi:10.1371/journal.ppat.1006651
138. Zhao Y, Guo Y, Jiang Y, Zhu X, Liu Y, Zhang X. Mitophagy regulates macrophage phenotype in diabetic nephropathy rats. Biochem Biophys Res Commun (2017) 494:42–50. doi:10.1016/j.bbrc.2017.10.088
139. Li S, Wu H, Han D, Ma S, Fan W, Wang Y, et al. A novel mechanism of mesenchymal stromal cell-mediated protection against sepsis: restricting inflammasome activation in macrophages by increasing mitophagy and decreasing mitochondrial ROS. Oxid Med Cell Longev (2018) 2018:15. doi:10.1155/2018/3537609
140. Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity (2016) 44:582–96. doi:10.1016/j.immuni.2016.01.001
141. Slade E, Tamber PS, Vincent JL. The surviving sepsis campaign: raising awareness to reduce mortality. Crit Care (2003) 7:1–2. doi:10.1186/cc2147
142. Moss M. Epidemiology of sepsis: race, sex, and chronic alcohol abuse. Clin Infect Dis (2005) 41(Suppl 7):S490–7. doi:10.1086/432003
143. Camacho-Gonzalez A, Spearman PW, Stoll BJ. Neonatal infectious diseases: evaluation of neonatal sepsis. Pediatr Clin North Am (2013) 60:367–89. doi:10.1016/j.pcl.2012.12.003
144. Arulkumaran N, Deutschman CS, Pinsky MR, Zuckerbraun B, Schumacker PT, Gomez H, et al. Mitochondrial function in sepsis. Shock (2016) 45:271–81. doi:10.1097/SHK.0000000000000463
145. Mannam P, Shinn AS, Srivastava A, Neamu RF, Walker WE, Bohanon M, et al. MKK3 regulates mitochondrial biogenesis and mitophagy in sepsis-induced lung injury. Am J Physiol Lung Cell Mol Physiol (2014) 306:L604–19. doi:10.1152/ajplung.00272.2013
146. Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, et al. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science (1995) 267:682–5. doi:10.1126/science.7839144
147. Mannam P, Zhang X, Shan P, Zhang Y, Shinn AS, Zhang Y, et al. Endothelial MKK3 is a critical mediator of lethal murine endotoxemia and acute lung injury. J Immunol (2013) 190:1264–75. doi:10.4049/jimmunol.1202012
148. Srivastava A, Mcginniss J, Wong Y, Shinn AS, Lam TT, Lee PJ, et al. MKK3 deletion improves mitochondrial quality. Free Radic Biol Med (2015) 87:373–84. doi:10.1016/j.freeradbiomed.2015.06.024
149. Srivastava A, Shinn AS, Lee PJ, Mannam P. MKK3 mediates inflammatory response through modulation of mitochondrial function. Free Radic Biol Med (2015) 83:139–48. doi:10.1016/j.freeradbiomed.2015.01.035
150. Kim MJ, Bae SH, Ryu JC, Kwon Y, Oh JH, Kwon J, et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy (2016) 12:1272–91. doi:10.1080/15548627.2016.1183081
151. Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res (2013) 23:201–12. doi:10.1038/cr.2013.6
152. Han JY, Kang MJ, Kim KH, Han PL, Kim HS, Ha JY, et al. Nitric oxide induction of Parkin translocation in PTEN-induced putative kinase 1 (PINK1) deficiency: functional role of neuronal nitric oxide synthase during mitophagy. J Biol Chem (2015) 290:10325–35. doi:10.1074/jbc.M114.624767
153. Oh CK, Sultan A, Platzer J, Dolatabadi N, Soldner F, Mcclatchy DB, et al. S-nitrosylation of PINK1 attenuates PINK1/Parkin-dependent mitophagy in hiPSC-based Parkinson’s disease models. Cell Rep (2017) 21:2171–82. doi:10.1016/j.celrep.2017.10.068
154. Kim EH, Sohn S, Kwon HJ, Kim SU, Kim MJ, Lee SJ, et al. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res (2007) 67:6314–24. doi:10.1158/0008-5472.CAN-06-4217
155. Tasdemir E, Maiuri MC, Tajeddine N, Vitale I, Criollo A, Vicencio JM, et al. Cell cycle-dependent induction of autophagy, mitophagy and reticulophagy. Cell Cycle (2007) 6:2263–7. doi:10.4161/cc.6.18.4681
156. Cloonan SM, Choi AM. Mitochondria in lung disease. J Clin Invest (2016) 126:809–20. doi:10.1172/JCI81113
157. Chakrabarti L, Eng J, Ivanov N, Garden GA, La Spada AR. Autophagy activation and enhanced mitophagy characterize the Purkinje cells of pcd mice prior to neuronal death. Mol Brain (2009) 2:24. doi:10.1186/1756-6606-2-24
158. Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol (2012) 8:831–8. doi:10.1038/nchembio.1059
159. Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: a simple solution to the autophagy paradox. Cell Cycle (2010) 9:4297–306. doi:10.4161/cc.9.21.13817
160. Carito V, Bonuccelli G, Martinez-Outschoorn UE, Whitaker-Menezes D, Caroleo MC, Cione E, et al. Metabolic remodeling of the tumor microenvironment: migration stimulating factor (MSF) reprograms myofibroblasts toward lactate production, fueling anabolic tumor growth. Cell Cycle (2012) 11:3403–14. doi:10.4161/cc.21701
161. Guo X, Sun X, Hu D, Wang YJ, Fujioka H, Vyas R, et al. VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease. Nat Commun (2016) 7:12646. doi:10.1038/ncomms12646
162. Georgakopoulos ND, Wells G, Campanella M. The pharmacological regulation of cellular mitophagy. Nat Chem Biol (2017) 13:136–46. doi:10.1038/nchembio.2287
163. East DA, Fagiani F, Crosby J, Georgakopoulos ND, Bertrand H, Schaap M, et al. PMI: a DeltaPsim independent pharmacological regulator of mitophagy. Chem Biol (2014) 21:1585–96. doi:10.1016/j.chembiol.2014.09.019
164. Vomund S, Schafer A, Parnham MJ, Brune B, Von Knethen A. Nrf2, the master regulator of anti-oxidative responses. Int J Mol Sci (2017) 18:E2772. doi:10.3390/ijms18122772
165. Holmstrom KM, Kostov RV, Dinkova-Kostova AT. The multifaceted role of Nrf2 in mitochondrial function. Curr Opin Toxicol (2016) 1:80–91. doi:10.1016/j.cotox.2016.10.002
166. Braun S, Hanselmann C, Gassmann MG, Auf Dem Keller U, Born-Berclaz C, Chan K, et al. Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol Cell Biol (2002) 22:5492–505. doi:10.1128/MCB.22.15.5492-5505.2002
167. Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest (2006) 116:984–95. doi:10.1172/JCI25790
168. Schaedler S, Krause J, Himmelsbach K, Carvajal-Yepes M, Lieder F, Klingel K, et al. Hepatitis B virus induces expression of antioxidant response element-regulated genes by activation of Nrf2. J Biol Chem (2010) 285:41074–86. doi:10.1074/jbc.M110.145862
169. Wruck CJ, Fragoulis A, Gurzynski A, Brandenburg LO, Kan YW, Chan K, et al. Role of oxidative stress in rheumatoid arthritis: insights from the Nrf2-knockout mice. Ann Rheum Dis (2011) 70:844–50. doi:10.1136/ard.2010.132720
170. Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol (2009) 11:1305–14. doi:10.1038/ncb1975
171. Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med (2016) 22:1428–38. doi:10.1038/nm.4222
172. Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Felix AA, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med (2016) 22:879–88. doi:10.1038/nm.4132
173. Lagishetty CV, Naik SR. Polyamines: potential anti-inflammatory agents and their possible mechanism of action. Indian J Pharmacol (2008) 40:121–5. doi:10.4103/0253-7613.42305
174. Gonzalez-Sarrias A, Larrosa M, Tomas-Barberan FA, Dolara P, Espin JC. NF-kappaB-dependent anti-inflammatory activity of urolithins, gut microbiota ellagic acid-derived metabolites, in human colonic fibroblasts. Br J Nutr (2010) 104:503–12. doi:10.1017/S0007114510000826
175. Larrosa M, Gonzalez-Sarrias A, Yanez-Gascon MJ, Selma MV, Azorin-Ortuno M, Toti S, et al. Anti-inflammatory properties of a pomegranate extract and its metabolite urolithin-A in a colitis rat model and the effect of colon inflammation on phenolic metabolism. J Nutr Biochem (2010) 21:717–25. doi:10.1016/j.jnutbio.2009.04.012
176. Ishimoto H, Shibata M, Myojin Y, Ito H, Sugimoto Y, Tai A, et al. In vivo anti-inflammatory and antioxidant properties of ellagitannin metabolite urolithin A. Bioorg Med Chem Lett (2011) 21:5901–4. doi:10.1016/j.bmcl.2011.07.086
177. Choi YH, Park HY. Anti-inflammatory effects of spermidine in lipopolysaccharide-stimulated BV2 microglial cells. J Biomed Sci (2012) 19:31. doi:10.1186/1423-0127-19-31
Keywords: autophagy, energy homeostasis, immunity, inflammation, metabolism, macrophages, mitochondria, mitophagy
Citation: Gkikas I, Palikaras K and Tavernarakis N (2018) The Role of Mitophagy in Innate Immunity. Front. Immunol. 9:1283. doi: 10.3389/fimmu.2018.01283
Received: 08 March 2018; Accepted: 22 May 2018;
Published: 05 June 2018
Edited by:
Panayotis Verginis, Biomedical Research Foundation of the Academy of Athens, GreeceReviewed by:
Agustina Alaimo, Universidad de Buenos Aires, ArgentinaCopyright: © 2018 Gkikas, Palikaras and Tavernarakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nektarios Tavernarakis, dGF2ZXJuYXJha2lzQGltYmIuZm9ydGguZ3I=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.