 Giorgio Santoni1*
Giorgio Santoni1* Maria Beatrice Morelli1,2
Maria Beatrice Morelli1,2 Consuelo Amantini3
Consuelo Amantini3 Matteo Santoni4
Matteo Santoni4 Massimo Nabissi1
Massimo Nabissi1 Oliviero Marinelli1,3
Oliviero Marinelli1,3 Angela Santoni2,5
Angela Santoni2,5
- 1Section of Experimental Medicine, School of Pharmacy, University of Camerino, Camerino, Italy
- 2Department of Molecular Medicine, Sapienza University, Rome, Italy
- 3School of Biosciences and Veterinary Medicine, University of Camerino, Camerino, Italy
- 4Clinical Oncology Unit, Macerata Hospital, Macerata, Italy
- 5Neuromed I.R.C.C.S. – Istituto Neurologico Mediterraneo, Pozzilli, Italy
Monocytes and macrophages play important roles in health and disease. They have a central role in protecting the host, as they clear pathogens and modulate other immune cell functions through the production of regulatory molecules. Their functions include immune surveillance, bacterial killing, tissue remodeling and repair, clearance of cell debris and more. Macrophages can have beneficial and detrimental effects on the outcome of several diseases depending on the microenvironment and the activation state of cells. Over the past few years, there has been an increasing interest in the expression and functions of ion channels, in particular of transient receptor potential (TRP) channel family in immune cells. The 30 members of mammalian TRP channels are subdivided into TRPC, TRPV, TRPM, TRPML, TRPP, and TRPA superfamily, and several members of TRP subfamily have been found to be functionally expressed in monocytes and macrophages. TRP are cation-selective channels that are weakly voltage-sensitive and diversely gated by temperature, mechanical force, electrophiles, ligands, and internal cues, such as membrane composition and pH, contributing to immune and inflammatory responses. The TRP channels play major roles in controlling several monocyte and macrophage functions such as phagocytosis, production of chemokines and cytokines, cell survival, polarization and so forth. In addition, they can also be potential therapeutic targets in a variety of inflammatory diseases. Thus, the goal of this review is to describe the role of TRP channels in the control of monocyte–macrophage functions in inflammatory and immune-mediated diseases.
Introduction
Macrophages play a crucial role in defense and disease by triggering immune surveillance, bacterial killing, tissue remodeling, and tissue repair (1–4). Macrophages show beneficial or detrimental effects in different diseases depending on their cell activation state and the microenvironment where they are present (5).
In the last years, there has been an increasing interest in the expression and functions of transient receptor potential (TRP) ion channel family in myeloid cells.
On the basis of amino acid sequence homology, TRP channels are grouped into different subfamily, called canonical (TRPC), melastatin (TRPM), vanilloid (TRPV), ankyrin (TRPA), mucolipin (TRPML), and polycystin (TRPP) subfamily (6, 7). Structurally they have six transmembrane spanning domains (S1–S6) with a pore domain between the fifth (S5) and sixth (S6) segment and intracellular C and N termini (8–10). TRP channels conduct cations, are weakly voltage-sensitive and non-selective for calcium, with a permeability ratio to Na (PCa/PNa) in a range between 0.3 and 10.
At present, TRP channel ligands are only partially known, although they function as multimodal signal integrators for exogenous ligands. The G protein-coupled receptors (Gq/11; linked to PLCβ) and tyrosine kinase receptors (linked to PLCγ) potentiate the signaling and function of most TRP channels (11). Elements of phosphatidylinositol signaling pathway, in particular, PIP2, can regulate TRP channels (12). In addition, intracellular Ca2+ increases TRP activity and modulates all TRP channels. For detailed description of TRP channels, there are many excellent reviews (13–16).
Several members of TRP subfamily are expressed in monocytes and macrophages (M/MΦ) (17). In these cells, they can recognize exogenous signals, including damage-associated molecular pattern molecules from the environment (heat, acidity, and chemicals) and endogenous danger signals released during trauma/tissue injury (ATP, mechanical, osmotic stress, and uric acid). In addition, they sensitize the pattern recognition receptors expressed in myeloid cells to respond to pathogen-associated molecular patterns (PAMPs) (18).
Aim of this review is to describe the cellular functions mediated by different members of TRP channels in M/MΦ.
Effects of TRP Channels on M/MΦ Survival and Proliferation
TRPM channels control the survival and proliferation of M/MΦ. In this regard, TRPM2 has been found to inhibit reactive oxygen species (ROS) generation in phagocytic cells and protect the mice from LPS-induced effects. LPS-treated TRPM2(−/−) mice show an increased inflammatory response and reduced cell viability with respect to wild-type mice. In addition, TRPM2 channels damp NADPH oxidase-stimulated ROS generation by phagocytes, through the induction of plasma membrane depolarization (19). The other TRP family member, TRPM4, controls M/MΦ survival in sepsis (20). The knockout of the TRPM4 gene increases the mortality in a murine model of LPS-induced sepsis. The lack of TRPM4 affects peritoneal macrophage infiltrate and increases the monocyte number, and the release of IL-1β and TNFα cytokines. Macrophages from TRPM4 knockout mice display reduced Ca2+ mobilization that inhibits the Akt pathway, and consequently macrophage survival, phagocytosis of bacteria (20).
TRPC1 plays an important role in the protection from bacterial infection, through TLR4-TRPC1 activation of protein kinase (PK) Cα pathway (Figure 1) (21). Ca2+ entry, induced by TRPC1 channel, stimulates the production of pro-inflammatory cytokines in murine pneumocytes. The TLR4-dependent TRPC1 activation triggers Ca2+ depletion from endoplasmic reticulum (ER) store. After activation of PLC-γ, TRPC1 mediates Ca2+ entry and stimulates PKCα activity, which results in NF-κB/Jun kinase nuclear translocation and cytokine release leading to tissue destruction (21). The TRPC1(−/−) mice show reduced survival, lung tissue damage, and systemic infection. Moreover, bone-marrow macrophages from TRPC3(−/−) mice show reduction in basal Ca2+ influx, impaired TNFα-induced signal as compared to wild-type cells (22).
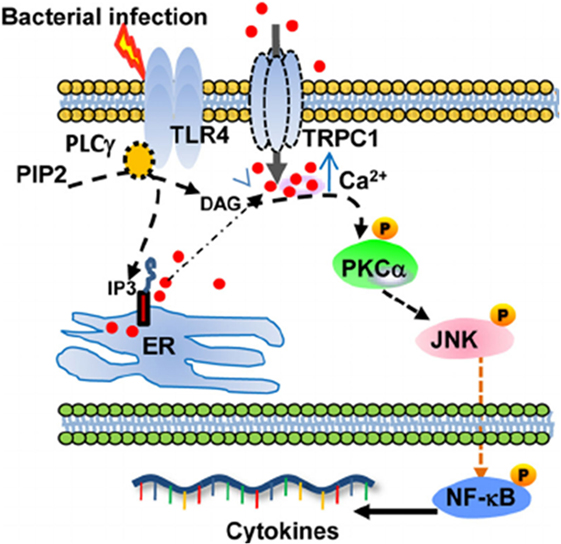
Figure 1. Schematic describing the proposed TRPC1/PKCα/JNK/NF-κB axis involved in the dysregulated pro-inflammatory response during bacterial infection. From Ref. (21) Copyright 2015 Molecular and Cellular Biology.
TRP Channels and Inflammasome Activation in M/MΦ
The inflammasomes are multiprotein platforms that mediate pro-caspase-1 cleavage and promote cytokine maturation (e.g., IL-1β and IL-18), in response to microbial and non-microbial stimuli, by canonical and non-canonical mechanisms. The activation of non-canonical inflammasome is mediated by caspase-11 that triggers IL-1β, IL-18, and IL-1α release in a caspase-1-dependent and -independent manner. Caspase-11 also promotes pyroptosis, a form of genetically programmed cell death (23). TRPC1 represents a substrate for caspase-11. Defects in TRPC1 expression enhance caspase-1-independent IL-1β release or macrophage death. Thus, intraperitoneal LPS injection in TRPC1(−/−) mice induces higher IL-1β secretion (24). Recently, in human U937 monocytes exposed to high glucose (HG) condition that induces the NLRP3-ASC inflammasome stimulation leading to caspase-1 activation and IL-1β and IL-18 secretion, TRPM2 regulates the thioredoxin-interacting protein-mediated triggering of NLRP3 inflammasome via interaction with the p47phox protein (25). In particular, TRPM2 activation and TRPM2-mediated Ca2+ influx represent the critical steps in NLRP3 activation. In response to HG, the reduction of TRPM2 expression reduces ROS generation and NADPH oxidase activity (25).
In phagocytes, the formation of crystals also induces oxidative stress that triggers NLRP3-mediated IL-1β secretion. Recently, Zhong et al. have demonstrated that liposomes are required for NLRP3 activation (26) and ROS-dependent TRPM2-mediated calcium influx (27). Infact, in macrophages from TRPM2 knockout mice, neither NLRP3 activation nor IL-1β production, is evidenced.
The NLRP3 inflammasome senses cell swelling and regulatory volume decrease (RVD), and the TRPV2 channel has been found to control volume regulation (18, 28). The reduction in extracellular osmolarity results in K(+)-dependent conformational change of the inactive NLRP3 inflammasome state followed by its activation, which is controlled by TRPV2 during RVD (28). Moreover, NLRP3-independent activation has been reported in human THP-1 macrophages (29). Apoptosis-associated speck-like protein containing a CARD domain (ASC) is required for the inflammatory processes. ASC bring NLRP proteins near to procaspase-1 into the inflammasome complex. Under hypotonic conditions, in TRPV2-dependent and independently by NLRP3, ASC forms specks that are unable to mediate pro-caspase-1 activation and pyroptosis. However, ASC speck formation leading to inflammasome and pro-caspase-1 cleavage is increased by interaction with NLRP3 (29).
Contribution of TRP Channels to MΦ Polarization
Similar to the Th1/Th2 nomenclature (30, 31), in response to different cytokines or PAMPs, there are specialized and polarized M1 and M2 macrophages. Activated M1 macrophages are induced by IFNγ alone or by microbial stimuli (e.g., LPS) or cytokines (e.g., TNF and GM-CSF). IL-4 and IL-13 other than to be inhibitors of macrophage activation, can induce the alternative M2 phenotype of macrophages (30). Activated M2 macrophages include cells exposed to IL-4 or IL-13, immune complexes, IL-10, glucocorticoids, or hormones (32). M1 cells secrete high levels of IL-12 and IL-23 and exhibit low IL-10 production; they generate NO and ROS and produce IL-1β, TNF, IL-6; they participate in Th1-polarized responses and mediate increased resistance against intracellular parasites and tumors. In contrast, M2 macrophages secrete low levels of IL-12 and IL-23 and high levels IL-10. Low expression of IL-1β and caspase-1 and high levels of IL-1ra, and decoy type II receptor were found in M2 cells (33). M1 and M2 cells also have distinct chemokine and chemokine receptor repertoire (31). M2 cells cooperate with Th2 cells in promoting the killing of parasites (34); they are present in some tumors and stimulate tissue repair (35). Moreover, recently, the analysis of transcriptomes in human macrophages stimulated with different stimuli has revealed the presence of distinct stimulus-specific macrophage polarization program and a broader spectrum of macrophage activation states, other that M1 and M2 (36).
A number of evidences indicate that the TRP channels regulate macrophage differentiation. Thus, gastric inflammation and reduced bacterial colonization were observed in Helicobacter pylori-infected TRPM2 knockout mice compared to controls (37). Loss of TRPM2 in H. pylori-infected macrophages triggers an increased production of inflammatory mediators and M1 polarization. Stimulation of TRPM2-deficient macrophages with H. pylori induces calcium overloading and increase of ERK1/2 and NADPH oxidase activities respect to wild type cells (37).
The expression and activity of TRPM7 are differentially regulated in bone-marrow derived murine M1 and M2 macrophages (38). Unlike M1 macrophages, in IL-4 stimulated M2 macrophages, higher TRPM7 current density (about 4.7-fold) was observed, whereas TRPM7 mRNA levels remain unchanged upon cell polarization. NS8593 and FTY720, two specific TRPM7 inhibitors, block IL-4- and M-CSF-induced macrophage proliferation and prevent M2 polarization. Inhibition of TRPM7 expression diminishes IL-4-induced arginase-1 mRNA levels and activity and completely inhibits the IL-4 or M-CSF mediated effects on TNF production in LPS-stimulated macrophages. In addition, TRPM7 inhibition decreases PI3K and ERK1/ERK2 phosphorylation levels and induces apoptosis in rat hepatic stellate cells (32, 39). In addition, adoptive transfer of macrophages from TRPM8-deficient mice, aggravates colitis, and IL-10 overexpression rescues M2 macrophage subpopulation. Thus, TNFα production in TRPM8-positive macrophages promotes the M1 macrophage phenotype and pro-inflammatory activity (40). Consequently, activation of TRPM8 channel in murine peritoneal macrophages triggers calcium transient currents in wild type but not TRPM8-deficient mice exhibiting defective phagocytosis and increased motility (40).
In addition, polarized macrophages from mice with specific TRPC3 deficiency show an increased in vitro phagocytic function (22). A crosstalk between TRP channels and unfolded protein response (UPR) system regulating macrophage polarization was also evidenced (41, 42). Thus, in Apoe(−/−) TRPC3(−/−) mice, M1 but not M2 macrophages show diminished ER stress-mediated apoptosis is reported. The reduced susceptibility of TRPC3-deficient M1 macrophages to apoptosis induced by ER stress is associated with impaired UPR and down-regulation of pro-apoptotic molecules as calmodulin-dependent PK II (Figure 2) (22, 42–44).
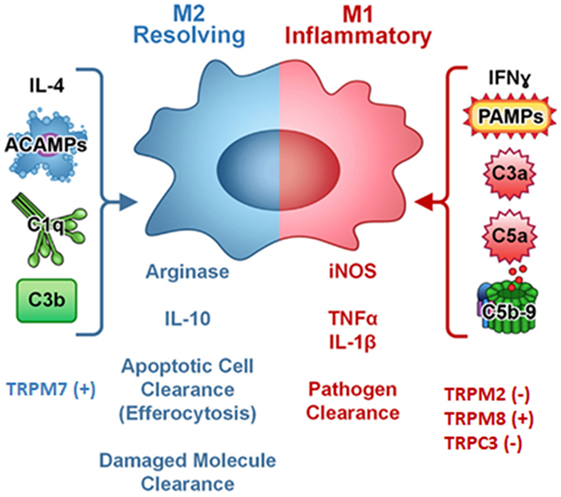
Figure 2. Complement components, cytokines, pathogen-associated molecular patterns (PAMPs), and TRP ion channels regulates macrophage polarization. Symbol (+) meaning stimulation, (−) meaning inhibition. Modified from (44) Copyright 2014 Front Immunol.
Role of TRP Channels in Adhesion and Migration in M/MΦ
Macrophage migration and infiltration is a multi-step process characterized by cell adhesion to different extracellular matrix (ECM) substrates, degradation of ECM proteins, topology and pericellular sense, intracellular transport, cell protrusion stabilization, and transmigration (45). In this regard, the organelles appointed to mediate these important functions are the podosomes. Recent studies have demonstrated that TRPV2 is localized in the podosome, and stimulation by fMLP further recruits TRPV2 to this compartment (46). Numerous signaling molecules including PI3K, Src, Cas, Pyk2, and Rho GTPases are associated with the podosome. TRPV2 may regulate Pyk2 activation, since TRPV2 knockdown inhibits the phospho-Pyk2 expression in macrophages. Activation of Pyk2 by ionomycin leads to breakdown of the podosome. On the contrary, increase of podosome numbers upon Pyk2 blocking by using a dominant negative variant of PyK2, PRNK, was observed. Gelsolin-assembled actin filaments and gelsolin activity are required for podosome assembly. It can be suggested that TRPV2, by activating gelsolin, promotes the formation of podosome. In murine macrophages, TRPV2 also contributes to fMLP-induced Ca2+ entry and migration (47). Notably, translocation of TRPV2 to the membrane induced by fMLP stimulation, is completely abrogated by PI3K inhibition or by Gi/0 trimeric G protein, suggesting that trafficking of TRPV2 channel is PI-(3,4,5)-P3 (PIP3)-dependent (46, 47).
Overexpression of mouse TRPM7 channel results in focal adhesion (FA) formation, spreading, and adhesion by increasing Ca2+ levels. The transformation of FA into podosomes depends by a kinase-dependent TRPM7-mediated activation (48). Non-activated TRPM7 channel is not associated with the actomyosin protein in the cytoskeleton. Triggering with PLC agonists induces TRPM7-mediated Ca2+ influx and TRPM7 kinase activity. Autophosphorylation of TRPM7 protein promotes a conformational change in the channel structure that allows Ca2+-dependent myosin IIA association, myosin IIA heavy chain phosphorylation leading to myosin dissociation and cytoskeletal remodeling. Finally, silencing of TRPC6 by siRNA or treatment with SKF-96365, a TRP blocker induce cytoskeleton disruption in murine podocytes (49).
Cellular migration and contractility are regulated by cytoskeleton rearrangements, FA turnover and changes in Ca2+ flux. In this regard, a role for TRPM4 as regulator of FA/cytoskeleton dynamics, mechanotransduction, and adhesome has been reported (50). The mouse TRPM4 channel localizes at FAs, where it contributes to FA turnover and disassembly of lamellipodial actin cytoskeleton components. Moreover, TRPM4 by regulating FAK and Rac GTPase activities modulates cellular contractility and migration in M/MΦ (51).
TRPM2 is involved in chemokine production from M/MΦ (52). The expression of TRPML2 is negligible in resting macrophages, but its levels increase in response to TLR4, TRL7, and TLR8 stimulation. In activated macrophages, TRPML2 facilitates the fusion of recycling endosomes or plasma membrane, thus promoting secretion of specific chemokines and cytokines. Recent data (53) demonstrated that CCL2, CCL3, and CCL5 chemokines are reduced in TRPML2(−/−) mice. Furthermore, TRPML2 knockout mice display impaired recruitment of peripheral macrophages in response to intraperitoneal injection of either LPS or live bacteria (53). In human U937 monocyte cell line, CXCL8 production depends on TRPM2-mediated Ca2+ influx. Monocytes from TRPM2 knockout mice exhibit reduced hydrogen peroxide-stimulated CXCL2 production (52). Activation of TRPM2 in human monocytes increases LPS-induced TNFα, IL-6, IL-8, and IL-10 production and phagocytosis in vitro (54).
The expression of TRPA1 mRNA in macrophages is upregulated in inflammatory bowel disease patients (55). In colitis, human TRPA1 channel activation exerts a mucosal protective role by reducing the expression of pro-inflammatory neuropeptides (SP, NKA, NKB, and VIP), cytokines (IL-1β, IFN, and TNF α/β), and of MCP-1 chemokine (55). Blocking of TRPA1 increases IL-10 levels and decreases TNF α secretion and TRPA1 siRNA normalizes monocyte IL-10 secretion (56).
Contribution of TRP Channels in M/MΦ Phagocytosis
Macrophage phagocytosis of pathogens is essential function of innate immune responses and depends on a large repertoire of receptors capable to recognize different targets. Phagosome maturation requires endosomal pathway regulators, including the phosphoinositide lipids. Both, PtdIns(3,5)P2 and PIP3, are required for phagosome maturation. Inhibition of the lipid kinase that generates PtdIns(3,5)P2, PIKfyve, and phosphatidylinositol-5-phosphate [PtdIns(5)P] blocks phagosome-lysosome fusion and abrogates the phagosome degradative capability in RAW264.7 macrophages. PIKfyve inactivation disrupts membrane recycling by causing lysosome swelling and blocks phagosome and endosome maturation (57). In this regard, TRPML1 regulates phagosome biogenesis; both particle ingestion and lysosomal exocytosis are inhibited by TRPML1 blockers (56, 58). Instead, TRPML1 overexpression and TRPML1 agonist stimulation trigger lysosomal exocytosis and particle uptake. The particle binding stimulates lysosomal PI(3,5)P2 increase that triggers TRPML1-dependent lysosomal Ca2+ release, rapidly delivering TRPML1 translocation from lysosomal membranes to Lamp1+ nascent phagosomes (59, 60). PIKfyve and PtdIns(3,5)P2 trigger the TRPML1 channel to mediate phagosome–lysosome fusion. Genetic deletion of TRPML1 gene hinders the acquisition of lysosomal markers in the phagosomes and reduces their bactericidal activity. Finally, cytosolic Ca2+ level increases during the TRPML1- and PIKfyve-dependent phagocytosis (57).
A role of TRPV2 in early phagocytosis was also demonstrated (61). The chemoattractant-elicited mobility, zymosan or complement-mediated particle binding, and phagocytosis are impaired in macrophages from TRPV2 knockout mice. The TRPV2 recruitment to the nascent phagosome and plasma membrane depolarization increases PIP2 synthesis that triggers actin depolymerization indispensable for phagocytic receptor clustering (61). Moreover, recently recruitment of TRPV2 at cell surface, preferential localization in lipid rafts, and calcium influx upon P. aeruginosa infection have been reported. Furthermore, deregulated TRPV2-signaling in macrophages from cystic fibrosis is responsible for their defective phagocytosis and consequently chronic infection (62). Moreover, in RAW 264.7 macrophages, the TRPM8 activator icilin stimulates cation currents that result in macrophage membrane depolarization. It is intriguing to hypothesize that TRPM8 alters macrophage efferocytosis by inducing actin depolymerization and indirectly influences Ca2+-dependent macrophage survival or apoptosis by reducing the driving force for Ca2+-mediated positive feedback on other Ca2+ permeable channels (63).
Finally, Riazanski et al. have demonstrated that TRPC6 channel translocation into phagosomal membrane increases phagosomal functions. TRPC6 channel restores microbicidal function in compromised alveolar macrophages from cystic fibrosis patients (64).
Collectively, these findings indicate that TRP expression sensitizes M/MΦ to recognize phagocyte bacteria, and defective TRP channel expression and function lead to inefficient bacterial killing. Thus, TRPV4 mediates LPS-stimulated murine macrophage phagocytosis of Escherichia coli in vitro and opsonized particles in vitro and in vivo in mice model (65). Intracellular Ca2+ is a second messenger in TLR4-dependent recognition and signaling (65). In this regard, Ca2+-depletion in TRPV2-deficient mice challenged with Listeria monocytogenes induces accelerated mortality and greater bacterial organ load (61).
TRPM2 is required for bacterial clearance in E. coli sepsis. Thus, during polymicrobial sepsis, macrophages from TRPM2 knockout mice show inefficient bacterial killing and increased infection and death. Disruption of TRPM2 affects phagolysosomal acidification, impairs the phagosome-lysosome fusion, impedes the phagosome maturation, and increases intracellular Ca2+-facilitated phagosome maturation in TRPM2(−/−) macrophages (66). TRPM2(−/−) mice are also extremely susceptible to Listeria monocytogenes infection and exhibit a defective innate immune response (19, 67). Similarly, the catalase from Francinella tularensis restricts ROS generation by hindering TRPM2-dependent Ca2+ entry in murine macrophages (68). In addition, TRPM2 disruption reduces heme oxygenase-1 expression and increases bacterial-induced macrophage infiltration. Pretreatment of macrophages from TRPM2 knockout mice, with heme oxygenase-1 inducer, reduces bacterial burden (69). Finally, suppression of macrophage activation through inhibition of the TRPC1 activity has been evidenced in parasites-(helminths) induced diseases (70).
Conclusion
Several evidences suggest the involvement of ion channels, in particular of TRP cation channel superfamily, in the pathogenesis of immune-mediated chronic inflammatory diseases. In this regard, the study of TRP channel functional expression in the M1/M2 macrophage polarization is an interesting research field to better understand how ion channels might participate in the generation of endogenous signaling capable of modifying macrophage polarization and differentiation, in the view to maintain health or to induce diseases. Crosstalk between inflammatory receptors and ion channels belonging to the TRP channel superfamily and the specific signaling pathway activated upon protein to protein interaction have been only partially elucidated and the contribution of a single TRP channel in the inflammatory response is still lacking. Further studies, both in vitro and in vivo aimed at uncovering the direct impact of different members of TRP subfamily in inflammatory processes are required. Thus, there is the need in the next future to explore and fully characterize the monocyte and macrophage expression of specific pattern of TRP channels and their signaling pathways activated in different immune-mediated diseases in order to identify new molecular targets for therapy of these inflammatory conditions.
Author Contributions
GS supervised the work and wrote the manuscript. CA and MM contributed to the preparation of the subchapters about TRP channels and macrophage phagocytosis and migration. MN and OM cooperated in the preparation of the subchapters about TRP channels and macrophage survival and polarization. MS collaborated in the drafting of the introduction and conclusion. AS provided critical revision of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
AIRC Investigator Grant IG-2014.
References
1. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol (2005) 5(12):953–64. doi:10.1038/nri1733
2. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol (2009) 27:451–83. doi:10.1146/annurev.immunol.021908.132532
3. Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol (2009) 9(4):259–70. doi:10.1038/nri2528
4. Wynn TA, Chawla A, Pollard JW. Origins and hallmarks of macrophages: development, homeostasis, and disease. Nature (2013) 496(7446):445–55. doi:10.1038/nature12034
5. Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol (2015) 33:291–353. doi:10.1146/annurev-immunol-032414-112212
6. Clapham DE, Julius D, Montell C, Schultz G. International union of pharmacology. XLIX. Nomenclature and structure-function relationships o transient receptor potential channels. Pharmacol Rev (2005) 57(4):427–50. doi:10.1124/pr.57.4.6
7. Wu LJ, Sweet TB, Clapham DE. International union of basic and clinical pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol Rev (2010) 62:381–404. doi:10.1124/pr.110.002725
8. Yu FH, Yarov-Yarovoy V, Gutman GA, Catterall WA. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol Rev (2005) 57(4):387–95. doi:10.1124/pr.57.4.13
9. Clapham DE. TRP channels as cellular sensors. Nature (2003) 426(6966):517–24. doi:10.1038/nature02196
10. Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol (2006) 68:619–47. doi:10.1146/annurev.physiol.68.040204.100431
11. Trebak M, Lemonnier L, Smyth JT, Vazquez G, Putney JW Jr. Phospholipase C-coupled receptors and activation of TRPC channels. Handb Exp Pharmacol (2007) 179:593–614. doi:10.1007/978-3-540-34891-7_35
12. Voets T, Nilius B. Modulation of TRPs by PIPs. J Physiol (2007) 582(Pt 3):939–44. doi:10.1113/jphysiol.2007.132522
13. Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem (2007) 76:387–417. doi:10.1146/annurev.biochem.75.103004.142819
14. Talavera K, Nilius B, Voets T. Neuronal TRP channels: thermometers, pathfinders and life-savers. Trends Neurosci (2008) 31(6):287–95. doi:10.1016/j.tins.2008.03.002
15. Latorre R, Zaelzer C, Brauchi S. Structure-functional intimacies of transient receptor potential channels. Q Rev Biophys (2009) 42(3):201–46. doi:10.1017/S0033583509990072
16. Nilius B, Owsianik G. Transient receptor potential channelopathies. Pflugers Arch (2010) 460(2):437–50. doi:10.1007/s00424-010-0788-2
17. Parenti A, De Logu F, Geppetti P, Benemei S. What is the evidence for the role of TRP channels in inflammatory and immune cells? Br J Pharmacol (2016) 173(6):953–69. doi:10.1111/bph.13392
18. Santoni G, Cardinali C, Morelli MB, Santoni M, Nabissi M, Amantini C. Danger- and pathogen-associated molecular patterns recognition by pattern-recognition receptors and ion channels of the transient receptor potential family triggers the inflammasome activation in immune cells and sensory neurons. J Neuroinflammation (2015) 12:21. doi:10.1186/s12974-015-0239-2
19. Di A, Gao XP, Qian F, Kawamura T, Han J, Hecquet C, et al. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat Immunol (2011) 13(1):29–34. doi:10.1038/ni.2171
20. Serafini N, Dahdah A, Barbet G, Demion M, Attout T, Gautier G, et al. The TRPM4 channel controls monocyte and macrophage, but not neutrophil, function for survival in sepsis. J Immunol (2012) 189:3689–99. doi:10.4049/jimmunol.1102969
21. Zhou X, Ye Y, Sun Y, Li X, Wang W, Privratsky B, et al. Transient receptor potential channel 1 deficiency impairs host defense and proinflammatory responses to bacterial infection by regulating protein kinase Cα signaling. Mol Cell Biol (2015) 35:2729–39. doi:10.1128/MCB.00256-15
22. Tano JY, Solanki S, Lee RH, Smedlund K, Birnbaumer L, Vazquez G. Bone marrow deficiency of TRPC3 channel reduces early lesion burden and necrotic core of advanced plaques in a mouse model of atherosclerosis. Cardiovasc Res (2014) 101(1):138–44. doi:10.1093/cvr/cvt231
23. Viganò E, Mortellaro A. Caspase-11: the driving factor for non-canonical inflammasomes. Eur J Immunol (2013) 43:2240–5. doi:10.1002/eji.201343800
24. Py BF, Jin M, Desai BN, Penumaka A, Zhu H, Kober M, et al. Caspase-11 controls interleukin-1β release through degradation of TRPC1. Cell Rep (2014) 6:1122–8. doi:10.1016/j.celrep.2014.02.015
25. Tseng HH, Vong CT, Kwan YW, Lee SM, Hoi MP. TRPM2 regulates TXNIP-mediated NLRP3 inflammasome activation via interaction with p47 phox under high glucose in human monocytic cells. Sci Rep (2016) 6:35016. doi:10.1038/srep35016
26. Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, et al. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun (2013) 4:1611. doi:10.1038/ncomms2608
27. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38:1142–53. doi:10.1016/j.immuni.2013.05.016
28. Compan V, Baroja-Mazo A, López-Castejón G, Gomez AI, Martínez CM, Angosto D, et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity (2012) 37:487–500. doi:10.1016/j.immuni.2012.06.013
29. Compan V, Martín-Sánchez F, Baroja-Mazo A, López-Castejón G, Gomez AI, Verkhratsky A, et al. Apoptosis-associated speck-like protein containing a CARD forms specks but does not activate caspase-1 in the absence of NLRP3 during macrophage swelling. J Immunol (2015) 194(3):1261–73. doi:10.4049/jimmunol.1301676
30. Gordon S. Alternative activation of macrophages. Nat Rev Immunol (2003) 3:23–35. doi:10.1038/nri978
31. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25(12):677–86. doi:10.1016/j.it.2004.09.015
32. Fang L, Zhan S, Huang C, Cheng X, Lv X, Si H, et al. TRPM7 channel regulates PDGF-BB-induced proliferation of hepatic stellate cells via PI3K and ERK pathways. Toxicol Appl Pharmacol (2013) 272:713–25. doi:10.1016/j.taap.2013.08.009
33. Dinarello CA. Blocking IL-1 in systemic inflammation. J Exp Med (2005) 201(9):1355–9. doi:10.1084/jem.20050640
34. Noël W, Raes G, Hassanzadeh Ghassabeh G, De Baetselier P, Beschin A. Alternatively activated macrophages during parasite infections. Trends Parasitol (2004) 20(3):126–33. doi:10.1016/j.pt.2004.01.004
35. Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol (2004) 4:583–94. doi:10.1038/nri1412
36. Xue J, Schimdt SV, Sander J, Draffenhn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity (2014) 40(2):274–88. doi:10.1016/j.immuni.2014.01.006
37. Beceiro S, Radin JN, Chatuvedi R, Piazuelo MB, Horvarth DJ, Cortado H, et al. TRPM2 ion channels regulate macrophage polarization and gastric inflammation during Helicobacter pylori infection. Mucosal Immunol (2017) 10(2):493–507. doi:10.1038/mi.2016.6
38. Schilling T, Miralles F, Eder C. TRPM7 regulates proliferation and polarisation of macrophages. J Cell Sci (2014) 127:4561–6. doi:10.1242/jcs.151068
39. Zhu Y, Men R, Wen M, Hu X, Liu X, Yang L. Blockage of TRPM7 channel induces hepatic stellate death through endoplasmic reticulum stress-mediated apoptosis. Life Sci (2014) 94(1):37–44. doi:10.1016/j.lfs.2013.10.030
40. Khalil M, Babes A, Lakra R, Försch S, Reeh PW, Wirtz S, et al. Transient receptor potential melastatin 8 ion channel in macrophages modulates colitis through a balance-shift in TNF-alpha and interleukin-10 production. Mucosal Immunol (2016) 9(6):1500–13. doi:10.1038/mi.2016.16
41. Solanki S, Dube PR, Tano JY, Birnbaumer L, Vazquez G. Reduced endoplasmic reticulum stress-induced apoptosis and impaired unfolded protein response in TRPC3-deficient M1 macrophages. Am J Physiol Cell Physiol (2014) 307:C521–31. doi:10.1152/ajpcell.00369.2013
42. Amantini C, Farfariello V, Cardinali C, Morelli MB, Marinelli O, Nabissi M, et al. The TRPV1 ion channel regulates thymocyte differentiation by modulating autophagy and proteasome activity. Oncotarget (2017) 8(53):90766–80. doi:10.18632/oncotarget.21798
43. Vazquez G, Solanki S, Dube P, Smedlund K, Ampem P. On the roles of the transient receptor potential canonical 3 (TRPC3) channel in endothelium and macrophages: implications in atherosclerosis. Adv Exp Med Biol (2016) 898:185–99. doi:10.1007/978-3-319-26974-0_9
44. Bohlson SS, O’Conner SD, Hulsebus HJ, Ho MM, Fraser DA. Complement, C1q, and C1q-related molecules regulate macrophage polarization. Front Immunol (2014) 5:402. doi:10.3389/fimmu.2014.00402
45. Linder S, Wiesner C. Tools of the trade: podosomes as multipurpose organelles of monocytic cells. Cell Mol Life Sci (2015) 72(1):121–35. doi:10.1007/s00018-014-1731-z
46. Nagasawa M, Kojima I. Translocation of calcium-permeable TRPV2 channel to the podosome: its role in the regulation of podosome assembly. Cell Calcium (2012) 51(2):186–93. doi:10.1016/j.ceca.2011.12.012
47. Nagasawa M, Nakagawa Y, Tanaka S, Kojima I. Chemotactic peptide fMetLeuPhe induces translocation of the TRPV2 channel in macrophages. J Cell Physiol (2007) 210:692–702. doi:10.1002/jcp.20883
48. Clark K, Langeslag M, van Leeuwen B, Ran L, Ryazanov AG, Figdor CG, et al. TRPM7, a novel regulator of actomyosin contractility and cell adhesion. EMBO J (2006) 25(2):290–301. doi:10.1038/sj.emboj.7600931
49. Liu Z, Yang J, Zhang X, Xu P, Zhang T, Yang Z. Developmental changes in the expression and function of TRPC6 channels related the F-actin organization during differentiation in podocytes. Cell Calcium (2015) 58(6):541–8. doi:10.1016/j.ceca.2015.09.001
50. Barbet G, Demion M, Moura IC, Serafini N, Léger T, Vrtovsnik F, et al. The calcium-activated nonselective cation channel TRPM4 is essential for the migration but not the maturation of dendritic cells. Nat Immunol (2008) 9(10):1148–56. doi:10.1038/ni.1648
51. Cáceres M, Ortiz L, Recabarren T, Romero A, Colombo A, Leiva-Salcedo E, et al. TRPM4 is a novel component of the adhesome required for focal adhesion disassembly, migration and contractility. PLoS One (2015) 10(6):e0130540. doi:10.1371/journal.pone.0130540
52. Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med (2008) 14:738–47. doi:10.1038/nm1758
53. Sun L, Hua Y, Vergarajauregui S, Diab HI, Puertollano R. Novel role of TRPML2 in the regulation of the innate immune response. J Immunol (2015) 195:4922–32. doi:10.4049/jimmunol.1500163
54. Wehrhahn J, Kraft R, Harteneck C, Hauschildt S. Transient receptor potential melastatin 2 is required for lipopolysaccharide-induced cytokine production in human monocytes. J Immunol (2010) 184(5):2386–93. doi:10.4049/jimmunol.0902474
55. Kun J, Szitter I, Kemény A, Perkecz A, Kereskai L, Pohóczky K, et al. Upregulation of the transient receptor potential ankyrin 1 ion channel in the inflamed human and mouse colon and its protective roles. PLoS One (2014) 9(9):e108164. doi:10.1371/journal.pone.0108164
56. Billeter AT, Galbraith N, Walker S, Lawson C, Gardner SA, Sarojini H, et al. TRPA1 mediates the effects of hypothermia on the monocyte inflammatory response. Surgery (2015) 158(3):646–51. doi:10.1016/j.surg.2015.03.065
57. Dayam RM, Saric A, Shilliday RE, Botelho RJ. The phosphoinositide-gated lysosomal Ca(2+) channel, TRPML1, is required for phagosome maturation. Traffic (2015) 16(9):1010–26. doi:10.1111/tra.12303
58. Samie M, Wang X, Zhang X, Goschka A, Li X, Cheng X, et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev Cell (2013) 26:511–24. doi:10.1016/j.devcel.2013.08.003
59. Thompson EG, Schaheen L, Dang H, Fares H. Lysosomal trafficking functions of mucolipin-1 in murine macrophages. BMC Cell Biol (2007) 8:54. doi:10.1186/1471-2121-8-54
60. Kim GH, Dayam RM, Prashar A, Terebiznik M, Botelho RJ. PIKfyve inhibition interferes with phagosome and endosome maturation in macrophages. Traffic (2014) 15(10):1143–63. doi:10.1111/tra.12199
61. Link TM, Park U, Vonakis BM, Raben DM, Soloski MJ, Caterina MJ. TRPV2 has a pivotal role in macrophage particle binding and phagocytosis. Nat Immunol (2010) 11:232–9. doi:10.1038/ni.1842
62. Lévêque M, Penna A, Le Trionnaire S, Belleguic C, Desrues B, Brinchault G, et al. Phagocytosis depends on TRPV2-mediated calcium influx and requires TRPV2 in lipids rafts: alteration in macrophages from patients with cystic fibrosis. Sci Rep (2018) 8(1):4310. doi:10.1038/s41598-018-22558-5
63. Wu SN, Wu PY, Tsai ML. Characterization of TRPM8-like channels activated by the cooling agent icilin in the macrophage cell line RAW 264.7. J Membr Biol (2011) 241:11–20. doi:10.1007/s00232-011-9358-6
64. Riazanski V, Gabdoulkhakova AG, Boynton LS, Eguchi RR, Deriy LV, Hogarth DK, et al. TRPC6 channel translocation into phagosomal membrane augments phagosomal function. Proc Natl Acad Sci U S A (2015) 112(47):E6486–95. doi:10.1073/pnas.1518966112
65. Scheraga RG, Abraham S, Niese KA, Southern BD, Grove LM, Hite RD, et al. TRPV4 mechanosensitive ion channel regulates lipopolysaccharide-stimulated macrophage phagocytosis. J Immunol (2016) 196:428–36. doi:10.4049/jimmunol.1501688
66. Zhang ZQ, Cui P, Zhang K, Chen QP, Fang XM. Transient receptor potential melastatin 2 regulates phagosome maturation and is required for bacterial clearance in Escherichia coli sepsis. Anesthesiology (2017) 126(1):128–39. doi:10.1097/ALN.0000000000001430
67. Knowles H, Heizer JW, Li Y, Chapman K, Ogden CA, Andreasen K, et al. Transient receptor potential melastatin 2 (TRPM2) ion channel is required for innate immunity against Listeria monocytogenes. Proc Natl Acad Sci U S A (2011) 108:11578–83. doi:10.1073/pnas.1010678108
68. Shakerley NL, Chandrasekaran A, Trebak M, Miller BA, Melendez JA. Francisella tularensis catalase restricts immune function by impairing TRPM2 channel activity. J Biol Chem (2016) 291(8):3871–81. doi:10.1074/jbc.M115.706879
69. Qian X, Numata T, Zhang K, Li C, Hou J, Mori Y, et al. Transient receptor potential melastatin 2 protects mice against polymicrobial sepsis by enhancing bacterial clearance. Anesthesiology (2014) 121:336–51. doi:10.1097/ALN.0000000000000275
Keywords: macrophages, transient receptor potential, macrophage polarization, migration, phagocytosis
Citation: Santoni G, Morelli MB, Amantini C, Santoni M, Nabissi M, Marinelli O and Santoni A (2018) “Immuno-Transient Receptor Potential Ion Channels”: The Role in Monocyte- and Macrophage-Mediated Inflammatory Responses. Front. Immunol. 9:1273. doi: 10.3389/fimmu.2018.01273
Received: 20 February 2018; Accepted: 22 May 2018;
Published: 06 June 2018
Edited by:
Uday Kishore, Brunel University London, United KingdomReviewed by:
Francesco Borriello, Harvard University, United StatesMarco A. Cassatella, University of Verona, Italy
Copyright: © 2018 Santoni, Morelli, Amantini, Santoni, Nabissi, Marinelli and Santoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giorgio Santoni, Z2lvcmdpby5zYW50b25pQHVuaWNhbS5pdA==