 Timo Jan Oberstein1
Timo Jan Oberstein1 Lava Taha1
Lava Taha1 Philipp Spitzer1
Philipp Spitzer1 Janina Hellstern1
Janina Hellstern1 Martin Herrmann2
Martin Herrmann2 Johannes Kornhuber1
Johannes Kornhuber1 Juan Manuel Maler1*
Juan Manuel Maler1*
- 1Department of Psychiatry and Psychotherapy, Friedrich-Alexander-University of Erlangen-Nuremberg, Erlangen, Germany
- 2Department of Internal Medicine 3 – Rheumatology and Immunology, Friedrich-Alexander-University of Erlangen-Nürnberg, Universitätsklinikum Erlangen, Erlangen, Germany
The neuropathological hallmarks of Alzheimer’s disease (AD), i.e., neuritic plaques and neurofibrillary tangles, consist of beta amyloid peptides (Aβ) and hyperphosphorylated Tau. These are accompanied by reactive microglia and astrocytes in the vicinity of the neuritic plaques and by changes to the peripheral immune system, e.g., an increase of the pro-inflammatory cytokines IL-1β, IL-6, and TNF-α in the peripheral blood. To address a potential involvement of peripheral T helper cell (Th) subsets in AD, we conducted a case control study with 54 individuals with AD dementia (n = 14), with mild cognitive impairment (MCI) due to AD (MCIAD, n = 14), with MCI unlikely due to AD (MCIother, n = 13), and controls without cognitive impairment (controls, n = 13). The proportions of CD3+CD8−IL-17A+IFNγ− Th17 cells, CD3+CD8−IL-17A−IFNγ+ Th1 cells, and CD4+CD127lowCD25+ regulatory T cells (Tregs) were assessed by flow cytometry. In addition, the correlations of the proportions of Th subsets to cerebrospinal fluid biomarkers were studied. CD3+CD8−IL-17A+IFNγ− Th17 cells were significantly increased in subjects with MCIAD compared to age- and sex-matched subjects with MCIother and controls (MCIAD mean = 1.13, SD = 0.77; MCIother mean = 0.58, SD = 0.28; and controls mean = 0.52, SD = 0.22; p = 0.008). The proportion of CD4+CD127lowCD25+ Tregs was not altered between the different groups, but it significantly positively related with the levels of total Tau and pTau181 (rTreg|totalTau = 0.43, p = 0.021, n = 28; rTreg|pTau181 = 0.46; p = 0.024, n = 28) in subjects with AD but not in nonAD controls (rTreg|totalTau = −0.51, p = 0.007, n = 26). The increase of circulating CD3+CD8−IL-17A+IFNγ− Th17 cells in the early stages of AD and the association of CD4+CD127lowCD25+ Tregs with neurodegeneration marker Tau may indicate that the adaptive immune system relates to neuropathological changes in AD.
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly. The neuritic plaques and neurofibrillary tangles are the neuropathological hallmarks of AD. They are accompanied by activated, cytokine-overexpressing microglia and astrocytes in the vicinity of the diffuse and neuritic plaques at early stages of progression in AD brain and by an increase of pro-inflammatory cytokines in the peripheral blood, such as IL-1β, IL-6, and TNF-α (1–6).
The blood–brain barrier (BBB) is impaired in patients with AD (7). This may facilitate the cytokine crosstalk between microglia and astrocytes with peripheral blood mononuclear cells (PBMCs) and the migration of PBMCs through the BBB. There is increasing evidence that blood or bone marrow-derived monocytes may either promote or inhibit chronic neuroinflammation in AD after crossing the BBB comparable with resident microglia (8, 9). Similarly, increased percentages of lymphocytes, both CD8+ cytotoxic T-cells and CD4+ T helper (Thelper) cells, have been observed in brain parenchyma of patients with AD (10).
Primarily based on murine models, various Th cell lineages, including Th1, Th17, and regulatory T cells (Tregs), have been described to be associated with AD (11–20). The Th1 lineage is promoted by IL-12, is characterized by the transcription factor T-bet and by the secretion of IFNγ (20–22). Th17 cells are elevated in various autoimmune diseases and diseases associated with chronic inflammation (23, 24). The differentiation of the Th17 lineage depends on IL-12p40/IL-23p19 and cytokines reportedly increased in AD, i.e., TGF-β, IL-1, and IL-6, and is inhibited among others by IFNγ and IL-4 (25, 26). Th17 cells are characterized by the secretion of IL-17 and typically express the transcription factor RORγt. IL-17A, which forms homodimers or heterodimers with IL-17F, acts synergistically with other pro-inflammatory cytokines and recruits neutrophils and monocytes to the sites of inflammation (25). High levels of TGF-β and IL-2, however, facilitate the differentiation of Th cells into regulatory T-cells (Tregs), which dampen pro-inflammatory responses. The influence of Tregs on neuroinflammation has been extensively studied in mouse models of experimental autoimmune encephalomyelitis (EAE), i.e., a commonly used model for multiple sclerosis (27). In this model, Treg cells are able to dampen deleterious pro-inflammatory effects, e.g., demyelination, and promote myelin regeneration directly via increased CCN3 expression and by immunomodulation among others via suppressing the Th1- and Th17 cell-mediated immune response (28–30). There is a great variety of CD4+ Tregs, usually identified by the expression of CD25+ and the intracellular transcription factor Foxp3 or the expression of CD25 combined with low levels of CD127 (31, 32).
In the revised criteria for AD by the diagnostic guidelines for AD of the National Institute on Aging-Alzheimer’s Association (NIA-AA) workgroups, the diagnosis of AD is no longer merely based on clinical examination, neuropsychiatric tests, and exclusion of other causes for dementia. In the revised criteria, the diagnostic procedures include the detection of amyloidopathy in terms of a decrease of amyloid beta (Aβ) 42 in cerebrospinal fluid (CSF) or a positive amyloid-PET and of neurodegeneration reflected by an increase of Tau and of hyperphosphorylated Tau (pTau) or glucose hypometabolism in FDG-PET (33, 34). The detection of the CSF biomarkers Tau and pTau does not only provide a higher sensitivity and specificity for the diagnosis of AD dementia but also indicate a higher rate of progression from mild cognitive impairment (MCI) to AD dementia (35, 36).
Based on the increase of pro-inflammatory cytokines and Th subsets found in AD brains, this study assesses the proportions of CD8+CD3−IL-17A+IFNγ− Th17 cells, CD8+CD3−IL-17A−IFNγ+ Th1 cells, and CD4+CD127lowCD25+ Tregs in cryopreserved PBMCs of subjects with Alzheimer’s dementia (AD dementia), MCI due to AD (MCIAD), MCI unlikely due to AD (MCIother), and subjects without cognitive impairment (controls). The associations between the percentages of the Th subsets and CSF biomarkers are investigated.
Materials and Methods
Study Population
The study protocol was approved by the clinical ethics committee of the University of Erlangen-Nuremberg. Patients and their authorized legal representatives provided written informed consent after receiving a complete description of the study. Fifty-four individuals with AD dementia (n = 14), with MCIAD (n = 14), MCIother (n = 13), and controls (n = 13) frequency matched for sex, age, and years of education were enrolled in this case control study.
Each participant was examined by a psychiatrist with advanced training in neuropsychiatry and dementia research according to the protocol of the German version of Consortium to Establish a Registry for AD (CERAD) clinical assessment battery. All participants with AD as well as 19 non-demented controls were assessed with the German version of the CERAD neuropsychological battery (CERAD-NB), trail making test part A and B (TMT-A and -B), and single letter Phonemic fluency (CERAD-NB+). Two subjects with AD dementia had severe cognitive deficits, therefore, only the MMSE was performed. TMT-A or TMT-B were dismissed, when the time limit had been exceeded. All subjects were assessed with SPECT and CSF dementia diagnostics [analysis of Aβ 42, Aβ 40, Aβ42/Aβ40 ratio, Tau, and phospho-Tau (pThr181, pTau)]. To exclude other potential causes for dementia, routine laboratory analyses (e.g., blood count, measurement of serum electrolytes, urea, creatinine, TSH, vitamin B12, folate, and CRP), routine CSF analyses, and MRI of the head were performed. Participants with focal brain lesions on MRI were excluded from this study. Classification into possible and probable AD dementia and MCIAD was performed by diagnostic guidelines for AD of the NIA-AA workgroups (33, 34). When the decrease of Aβ 42 was substituted by a decrease of Aβ42/Aβ40 ratio for the discrimination between possible and probable AD, all 13 subjects with AD dementia fulfilled the criteria for probable AD (37). MCI probable due to AD (MCIAD) was similarly defined by the decrease of Aβ42/Aβ40 ratio and increase of Tau/pTau181, while all other subjects with MCI (MCIother) had negative CSF biomarkers.
The severity of concomitant depressive symptoms was monitored by the Beck’s depression inventory II or the geriatric depression scale. The donors of the samples were enrolled in the study during the initial diagnosis, which is why none of them received treatment with acetylcholinesterase inhibitors or memantine at the time of sample acquisition.
CSF Biomarker Diagnostics
The concentrations of Aβ 40 and Aβ 42 in CSF were measured with commercially available ELISAs from IBL International (Hamburg, Germany). Total Tau and pTau181 in CSF were assessed with an ELISA from Fujire-bio Europe (Gent, Belgium).
Isolation of PBMCs
Whole blood was collected in EDTA containing S-Monovettes® (Sarstedt, Nümbrecht, Germany). PBMCs were isolated by density gradient centrifugation with Biocoll separatin solution (Merck, Darmstadt, Germany) and cryopreserved with Mr. Frosty™ Freezing Container (Thermo Fisher Scientific, Schwerte, Germany) in RPMI (Gibco, now Thermo Fisher Scientific) with 20% v/v FBS and 10% v/v DMSO (Roth, Karlsruhe, Germany) and stored in liquid nitrogen.
Flow Cytometry
The following fluorochrome-conjugated monoclonal antibodies were purchased from commercial vendors: PE/Dy647 conjugated anti-CD3 (clone: MEM-57, Immunotools, Friesoythe, Germany), APC conjugated anti-CD4 (clone: EDU-2, Immunotools), PE conjugated anti-CD127 APC (clone: eBioRDR5, eBioscience, now Thermo Fisher Scientific, Frankfurt, Germany), IL-17A (clone: eBio64DEC17, eBioscience), FITC conjugated anti-CD25 (clone: HI25a, Immunotools), and Alexa488 conjugated anti-IFNγ (clone: B27, BD Biosciences; Heidelberg, Germany). PE conjugated IgG1κ (eBioscience) and FITC conjugated IgG1 (clone: PPV-06, Immunotools) served as isotype controls.
Cryopreserved PMBCs were thawed, and the number of viable leukocytes was determined by the CASY® TT Cell Counter + Analyzer (OLS Omni Life Science, Bremen, Germany). Cells were resuspended in RPMI1640 with 10% v/v FBS superior (Merck Millipore, Darmstadt, Germany)/1% v/v penicillin (10,000 U/ml)/streptomycin (10 mg/ml) (S/P, Gibco)/20 µg/ml DNAse (Sigma-Aldrich, Taufkirchen, Germany), spun with 500 g for 5 min, and cultivated in RPMI1640/10% v/v FBS/1% v/v S/P overnight in an incubator at 37°C and 5% CO2. For surface staining, 0.2 × 106 cells were washed twice in PBS/0.1% v/v FBS and stained with combinations of APC conjugated anti-CD4, PE conjugated anti-CD127, FITC conjugated anti-CD25, and/or FITC conjugated IgG1 (1:200, each). For the staining of intracellular cytokines, 0.2 × 106 cells were stimulated with RPMI/10% FBS/1% S/P with 20 µg/ml DNAse, phorbol myristate acetate (PMA) 50 ng/ml (Sigma-Aldrich), and ionomycin 500 ng/ml (Sigma-Aldrich). After 1 h, 5 µg/ml Brefeldin (Sigma-Aldrich) and 5 µg/ml Monensin (BD Biosciences) were added, and the cells were incubated for an additional 5 h. Cells were washed twice in PBS/0.1% v/v FBS, stained with PE/Dy647 conjugated anti-CD3 and APC conjugated anti-CD8 for 20 min at room temperature in the dark. Permeabilization, fixation, and staining of intracellular cytokines with Alexa488 conjugated anti-IFNγ and PE conjugated anti-IL-17A or PE conjugated IgG1κ (1:200, each) were performed with Cytofix/Cytoperm® Plus Fixation/Permeabilization Kit (BD Biosciences) according to the manufacturer’s instructions. Th cells from PMA and ionomycin stimulated cultured PBMCs were defined by a combination of FSC and SSC characteristics and CD3/CD8 stain, as CD4 was extensively downregulated under PMA and ionomycin stimulation as previously reported (38–40).
Analyses were performed using a Partec CyFlow® Space flow cytometer (Sysmex Europe, Norderstedt, Germany) and Flomax® operating sofware v2.9 (Sysmex Europe) and Kaluza Flow Cytometry Analysis Software v1.5.20207.16062 (Beckman Coulter Life Sciences, Krefeld, Germany). Viable lymphocytes were identified by their characteristics in forward scatter (FSC) and side scatter (SSC) and the percentage of viable lymphocytes in each preparation was determined by the CASY cell counting technology. The viable lymphocyte gate was based on Annexin V and propidium iodide stain. Representative dot plots are given in Figure S3 in Supplementary Material. Isotype control antibodies and single-stained samples were used periodically for color compensation and to check the settings.
Statistical Analysis
Scores in CERAD-NB+ were converted to z-scores. Shapiro–Wilk’s test was used to test for normality. Homogeneity of variance was assessed by Levene’s test. Group comparisons were performed using Pearson’s χ2 for categorical variables or the Mann–Whitney U test or Kruskal–Wallis test followed by Dunn’s multiple comparison test in case a significant effect was observed for ordinal or not normally distributed interval variables. The ANOVA and for groups with inhomogenous variances the Brown–Forsythe test were employed for normally distributed interval variables followed by Tukey B or Dunn–Bonferroni correction in case a significant effect was observed. Pearson correlation was used to examine the relationships between variables of interest. Data analysis was performed using the SPSS statistical package (version 20.0; SPSS, Chicago, IL, USA). Quartiles are indicated as follows: 1st quartile = Q1; 3rd quartile = Q3; significance levels are indicated as follows: ***p < 0.001; **p < 0.01; *p < 0.05; and ns, not significant.
Results
Description of the Study Population
The groups of subjects with AD dementia, MCI probably due to AD (MCIAD), MCI with negative CSF biomarkers (MCIother), and without cognitive impairment (controls) did not differ statistically in gender [χ2(3) = 1.261, p = 0.738, n = 54], age [F(3,54) = 2.338, p = 0.085, n = 54], or education (H = 2.718, d.f. = 3, p = 0.437, n = 47). A detailed listing of the levels of CSF biomarkers and z-scores of the CERAD-NB+ is given in Table 1.
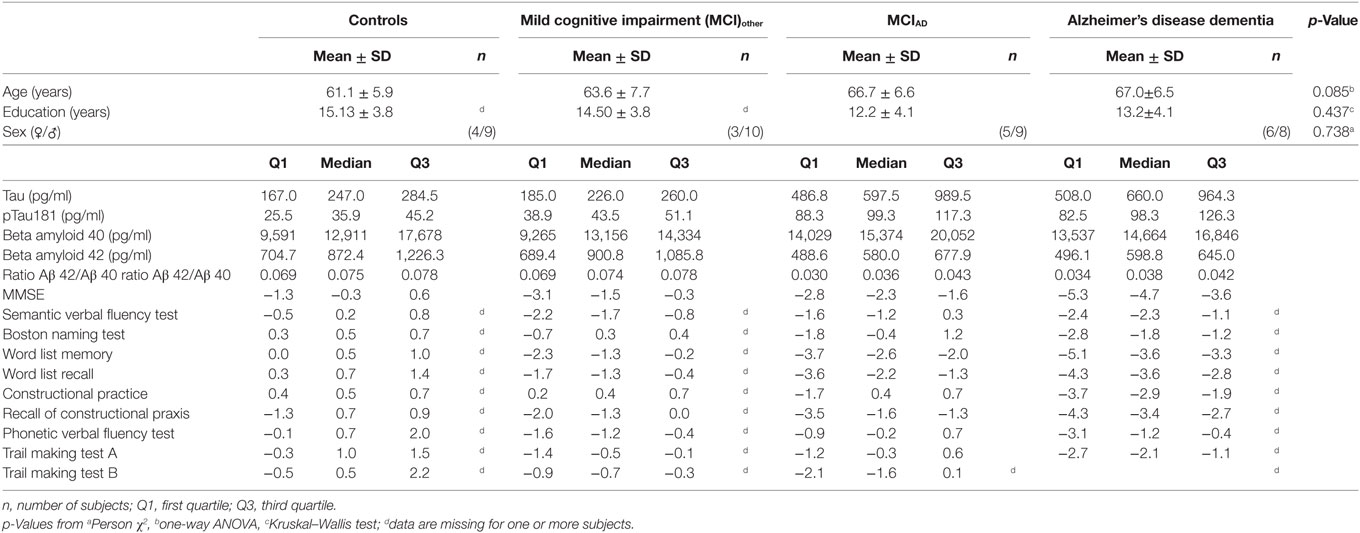
Table 1. Clinical data including cerebrospinal fluid biomarkers and z-scores of the CERAD-NB+ of the investigated groups.
Circulating CD3+CD8−IL-17A+IFNγ− Th17 Cells Were Increased in Participants With MCIAD Compared to Subjects Without Cognitive Impairment and With MCIother
The gating strategy for the analysis of CD3+CD8−IL-17A+IFNγ− Th17 cells, CD3+CD8−IL17A−IFNγ+ Th1, and CD4+CD127lowCD25+ Tregs from cultured cryopreserved PBMCs is depicted in Figure S1 in Supplementary Material. Doublet exclusion by FSC-H and FSC-A scatter did not relevantly alter the percentages of CD3+CD8−IL-17A+IFNγ− Th17 cells, CD3+CD8−IL17A−IFNγ+ Th1 and CD4+CD127lowCD25+ Tregs (Figure S3 in Supplementary Material). The proportion of peripheral PMA/ionomycin treated CD3+CD8−IL-17A+IFNγ− Th17 cells was significantly altered between subjects with AD dementia, with MCIAD, with MCIother and in controls [F*(d.f.1 = 3, d.f.2 = 24.339) = 5.314, p = 0.008, n = 54] (Figure 1). The highest abundance of CD3+CD8−IL-17A+IFNγ− Th17 cells in relation to CD3+CD8− cells was detected in the group with MCIAD (mean = 1.13, SD = 0.77), followed by AD dementia (mean = 0.79, SD = 0.33), MCIother (mean = 0.58, SD = 0.28), and controls (mean = 0.52, SD = 0.22). The subsequent Dunn–Bonferroni test showed that the proportion of CD3+CD8−IL-17A+IFNγ− Th17 cells in MCIAD was significantly elevated compared to controls (z = −0.61231, p = 0.007; dCohen = 1.07) and subjects with MCIother (z = −0.55007, p = 0.019; dCohen = 0.95), indicating that circulating CD3+CD8−IL-17A+IFNγ− Th17 cells are elevated especially in early stages of AD. Representative dotplots of CD3+CD8− and CD4+ lymphocytes are given in Figure S2 in Supplementary Material.
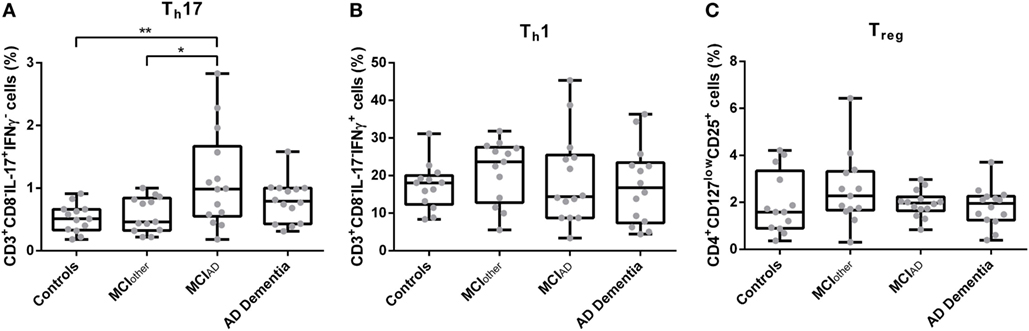
Figure 1. Percentages of CD3+CD8−IL-17+IFNγ− Th17 cells (A), CD3+CD8−IL-17−IFNγ+ Th1 cells (B), and CD4+CD127lowCD25+ Tregs (C) as assessed with flow cytometry from donor derived cryopreserved peripheral blood mononuclear cells were compared between controls, mild cognitive impairment (MCI)other, MCIAD, and Alzheimer’s disease (AD) dementia. Data are summarized as combined box- and scatter plots (p-values from post hoc tests are shown *p < 0.05, **p < 0.01).
The abundances of CD3+CD8−IFNγ+IL-17A− Th1 cells and CD4+CD127lowCD25+ Tregs from peripheral blood did not differ statistically significantly between the groups [F(3,50) = 0.498, p = 0.686, n = 54 and F*(3,50) = 1.106, d.f.1 = 3, d.f.2 = 34.213, p = 0.360, n = 54, respectively]. Similarly, the mean fluorescence intensities of CD3+CD8−IL-17A+IFNγ− Th17 cells, CD3+CD8−IFNγ+IL-17A− Th1 cells, and CD4+CD127lowCD25+ Tregs did not significantly vary between AD dementia, MCIAD, MCIother, and controls [F(3,50) = 0.274, p = 0.844, n = 54; H = 1.290, d.f. = 3, p = 0.731, n = 54; and H = 1.669, d.f. = 3, p = 0.644, n = 54, respectively]. Analyses of control parameters showed a significant difference in the serum cholesterol levels between the groups. The other control parameters, i.e., leukocyte count, levels of CRP, of TSH, of folate, of vitamin B12, of urea, of creatinine, and of fastening glucose did not differ statistically significantly in multivariate analyses as given in Table S1 in Supplementary Material.
The Proportion of Circulating CD4+CD127lowCD25+ Tregs Cells Was Positively Related to Total Tau, pTau181, and Aβ 40 in the AD but Not in the nonAD Group
Correlations of the proportions of CD3+CD8−IL-17A+IFNγ− Th17 cells, CD3+CD8−IFNγ+IL-17A− Th1 cells, and CD4+CD127lowCD25+ Tregs cells with total Tau, pTau181, Aβ 42, Aβ 40, and Aβ42/Aβ40 ratio were analyzed in subjects with probable AD and in subjects with negative AD biomarkers (nonAD group).
In the AD group, the proportions of peripheral CD4+CD127lowCD25+ Tregs were statistically significantly positively related to the amount of the neuronal injury marker total Tau in CSF (r = 0.433, p = 0.021, n = 28), to pTau (r = 0.462, p = 0.024, n = 28), and to Aβ 40 (r = 0.484, p = 0.009, n = 28) (Figure 2). In the nonAD group on the other hand, total Tau in CSF significantly negatively related to the level of CD4+CD127lowCD25+ Tregs (r = −0.513, p = 0.007, n = 26). In addition, the percentages of CD4+CD127lowCD25+ Tregs were only statistically significantly positively associated with the ratio of Aβ42/Aβ40 in the nonAD group (r = 0.450, p = 0.021, n = 26). Collectively, the percentage of CD4+CD127lowCD25+ Tregs in the AD group positively related to the level of neurodegeneration in means of increased levels of Tau/pTau181 and amyloidopathy in means of decrease of Aβ42/Aβ40 ratio, while in the nonAD group an inverse relation was observed.
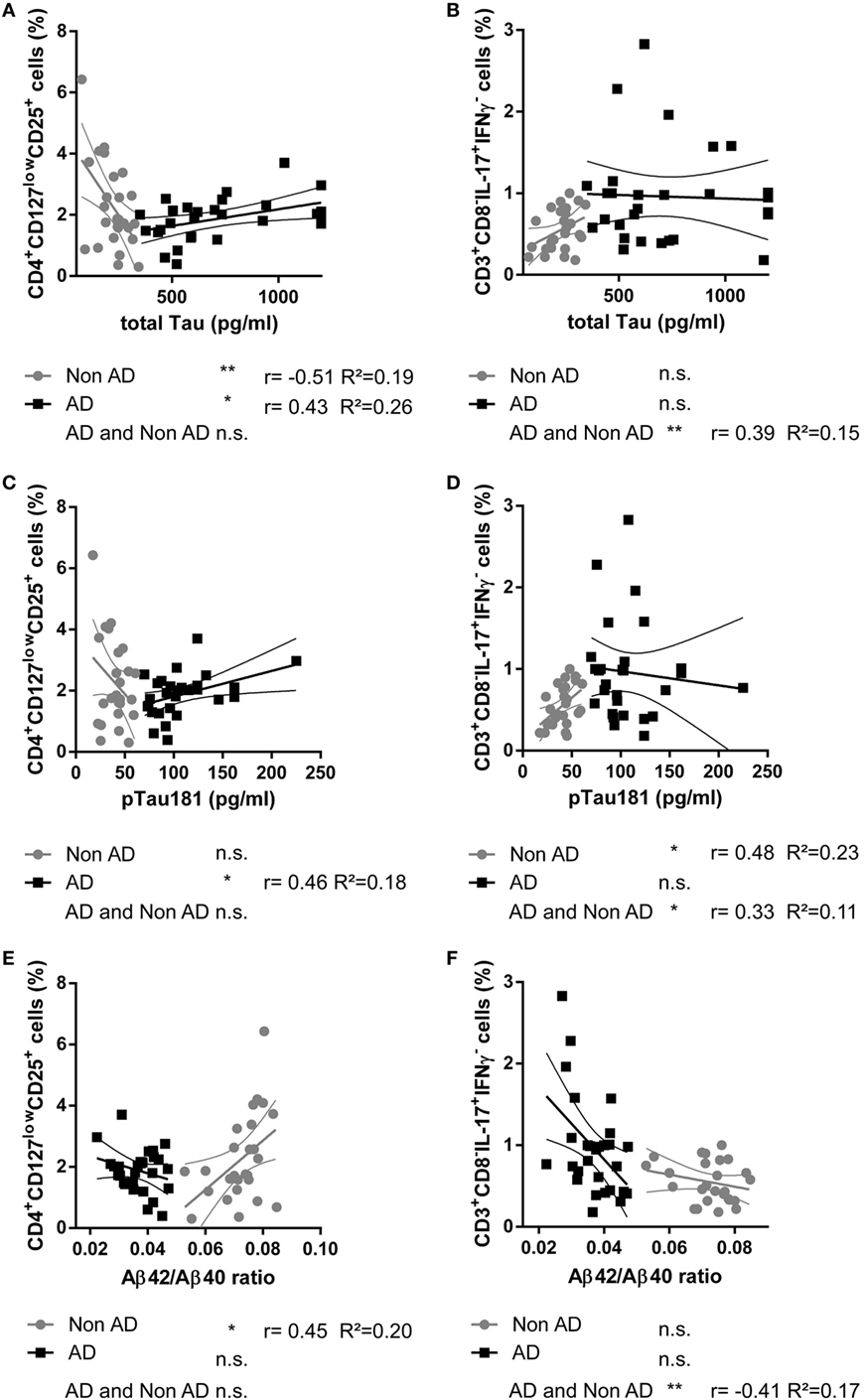
Figure 2. Scatter plots showing the relationship between the cerebrospinal fluid level of total Tau protein [pg/ml; (A,B)], pTau181 [pg/ml; (C,D)], Aβ42/Aβ40 ratio (E,F), and the percentage of CD4+CD127lowCD25+ Tregs (A,C,E) and CD3+CD8−IL-17+IFNγ− Th17 cells (B,D,F) in donors with Alzheimer’s disease (AD, black squares) and controls (nonAD, gray dots). The associations in AD group, nonAD group and all subjects combined were investigated with Spearman’s correlation. (Scatter plots with the linear regression lines for AD and nonAD groups and their 95% confidence intervals are shown; non-significant = n.s., *p < 0.05, **p < 0.01, **p < 0.001; r: Spearman’s correlation coefficient; R2: coefficient of determination.)
The percentages of CD4+CD127lowCD25+ Tregs did not correlate with age (rAD = 0.01, pAD = 0.98 and rnonAD = 0.02, pnonAD = 0.91) or education (rAD = 0.25, pAD = 0.21 and rnonAD = 0.19, pnonAD = 0.41). There was no significant difference in the proportion of CD4+CD127lowCD25+ Tregs between males and females [χ2(25)AD = 28.000, pAD = 0.411, nAD = 28 and χ2(25)nonAD = 27.000, pnonAD = 0.356, nnonAD = 27]. The percentage of CD3+CD8−IL-17A+IFNγ− Th17 was statistically significantly positively related to pTau181 in the nonAD group (r = 0.480, p = 0.013, n = 26).
Discussion
In this case control study, the proportion of peripheral CD3+CD8−IL-17A+IFNγ− Th17 cells was significantly elevated in subjects with MCIAD compared to controls and subjects with MCIother. Together with the observation, that the proportion of CD4+CD127lowCD25+ Tregs was significantly positively related with the level of neurodegeneration markers pTau181 and total Tau in patients with AD but not in controls, the results indicate that these Th cell lineages might be associated with the neurodegeneration in AD.
First indications, that the proportion of Th17 cells and RORγt+ cells, respectively, was elevated in Alzheimer’s dementia, were reported by Saresella et al. (16) and Agnes et al. (41). Both used the NINCDS ADRA criteria for the diagnosis of Alzheimer’s dementia without the detection of the CSF biomarkers. Saresella et al. similarly detected the highest proportions of RORγt+ cell in subjects with MCI, though no differentiation was made between subjects with MCI probably due to AD and MCI unlikely due to AD. An elevation of CCR6+ lymphocytes in 16 patients with AD has been described by Goldeck et al. (42). CCR6+ is a chemokine receptor found on Th17 cells, subsets of Tregs, and dendritic cells and among others is involved in the recruitment of dendritic cells and T cells to the site of inflammation (43). In this study, we were able to differentiate the proportions of Th17 cells between MCIAD and MCIother for the first time. In addition, due to the simultaneous detection of the CSF biomarkers used for the diagnosis of AD dementia and MCIAD, we were able to relate the levels of CSF biomarkers to the percentages of CD3+CD8−IL-17A+IFNγ− Th17 and CD4+CD127lowCD25+ Tregs. In our study, the level of Th17 cells statistically significantly related to amyloidopathy, i.e., the decrease of the ratio of Aβ42/Aβ40. Findings from previous studies indicate that Aβ can directly influence the cytokine expression of Th17 cells, as the disruption of the BBB by hippocampal application of Aβ 42 in rats was followed by an infiltration of Th17 cells into the brain parenchyma, increased expression of IL-17 and IL-22 in the hippocampus, and elevated concentrations of the two cytokines in both CSF and serum (20). The production of IL-17 from mice splenocytes was elevated after treatment with Aβ 42 and the level of IL-17A and of Th17 cells in human PBMCs was increased after the stimulation with Aβ 25–35 (13, 19).
The role of Tregs in the pathogenesis of AD is a matter of recent debate. In accordance with previous studies, we did not detect a significant difference between CD4+CD127lowCD25+ Tregs in subjects with AD dementia or MCIAD compared to age-matched controls or MCIother (15, 17). However, Saresella et al. reported an increase of CD4+Foxp3+ Tregs in Alzheimer’s dementia especially in MCI diagnosed by NINCDS ADRA criteria, Le Page et al. reported an increase of CD4+Foxp3+CD25high cells in amnestic MCI but not in mild Alzheimer’s dementia, and Larbi et al. reported an overall decreased frequency of CD4+CD25high T cells in subjects with mild AD dementia when compared with age-matched controls (17, 44, 45).
Our findings suggest that in AD but not in controls the number of CD4+CD127lowCD25+ Treg cells was rather related to the level of Tau and pTau181, which predict the rate of cognitive decline in the different stages of AD and correlated with neurofibrillary tangle pathology in the neocortex in AD brains (35, 36, 46, 47). A possible pathomechanism might be that the number of peripheral Tregs was elevated in case of increased neurodegeneration in AD to prevent further demyelination and axonal loss similar to the findings in the models of EAE. Observations in a transgenic mouse model of AD (APPPS1 mice) likewise indicated that Tregs are able to slow the disease progression and restore cognitive function (48). In addition, an increased suppressive activity of Treg cells in AD and Parkinson’s disease has been described (15). However, in 5XFAD mice carrying five mutations associated with early onset familial AD, the depletion of Foxp3+ Tregs was accompanied with increased Aβ clearance and reversal of cognitive decline and in a model for neuronal injury in BALB/c/OLA mice, the transfer of CD4+CD25+ Tregs worsened the outcome, indicating a deleterious effect of Tregs in these models (49, 50). Taken together, the function or loss of function of Tregs in AD has still to be determined and further studies are required to identify causal relationship and to provide evidence that the relations to the CSF biomarkers in this study are not due to a common response variable.
Complementing but further complicating the immunological changes in AD, the profile of interleukins supports our study’s findings, such as IL-6 and TNF-α, which are commonly elevated in serum in AD, are induced by IL-17 from the Th17 cells (5, 25). Furthermore, the inhibition of the IL-12/IL-23 pathway, which supports the differentiation of Th17 cells, attenuated AD pathology and cognitive deficits in an AD mouse model (51). IL-17 has been reported to be increased in AD by several reports; however, Doecke et al. detected on the contrary a significant decrease of IL-17 in plasma from AD patients in a larger study with 151 subjects (52–55). For the level of TGF-β, studies regularly reported increased as well decreased serum levels in AD (56–58). In low doses, TGF-β promotes the differentiation of Th17 cells, whereas high levels promote Tregs, which dampen the pro-inflammatory response of Th17 cells. Interestingly, low levels of TGF-β were associated with a faster progression from MCI to AD dementia, as it was observed for high levels of total Tau and pTau181 (35, 36, 59).
Nevertheless, this study has limitations, which include the lack of a healthy control sample because of the difficulties associated with collecting CSF from healthy subjects. Instead, patients without cognitive impairment were enrolled as control. Furthermore, the small sample size and a potentially skewed population due to the fact that subjects were enrolled from our memory unit or patients hospitalized in our psychiatric ward may have some bearing on our results. A larger population study will be informative to further elaborate our initial findings.
Conclusion
Our clinical data showed an increase of circulating CD3+CD8−IL-17A+IFNγ− Th17 cells in subjects with MCIAD and an association of CD4+CD25+CD127low Tregs with the neurodegeneration markers phospho and total Tau, which complements the observations from fundamental research, that the adaptive immune system seems to be involved in the pathogenesis of AD and its neuropathological changes especially in the early stages of the disease. Further studies might provide more insight into disease progression and the interplay between the neuropathological hallmarks of AD and the peripheral immune system.
Datasets are Available on Request
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The study protocol was approved by the clinical ethics committee of the University of Erlangen-Nuremberg (project no. 3987). Patients and their authorized legal representatives provided written informed consent after receiving a complete description of the study.
Author Contributions
TO designed the study, performed experiments, analyzed the data, and together with JM drafted the manuscript. LT and JH performed experiments and contributed to revision of the manuscript. JK provided reagents and contributed to the interpretation of findings and revision of the manuscript. PS and MH contributed to the interpretation of findings and revision of the manuscript. All the authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The author declares that he has no relevant or material financial interests that relate to the research described in this paper. This work was partly supported by the Interdisciplinary Center for Clinical Research (IZKF) Erlangen.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01213/full#supplementary-material.
References
1. Delacourte A, Defossez A. Alzheimer’s disease: tau proteins, the promoting factors of microtubule assembly, are major components of paired helical filaments. J Neurol Sci (1986) 76:173–86. doi:10.1016/0022-510X(86)90167-X
2. Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun (1984) 120:885–90. doi:10.1016/S0006-291X(84)80190-4
3. Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A (1986) 83:4913–7. doi:10.1073/pnas.83.13.4913
4. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A (1985) 82:4245–9. doi:10.1073/pnas.82.12.4245
5. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging (2000) 21:383–421. doi:10.1016/S0197-4580(00)00124-X
6. Thal DR, Capetillo-Zarate E, Del Tredici K, Braak H. The development of amyloid beta protein deposits in the aged brain. Sci Aging Knowledge Environ (2006) 2006:re1. doi:10.1126/sageke.2006.6.re1
7. Kalaria RN. The blood-brain barrier and cerebrovascular pathology in Alzheimer’s disease. Ann N Y Acad Sci (1999) 893:113–25. doi:10.1111/j.1749-6632.1999.tb07821.x
8. Minogue AM. Role of infiltrating monocytes/macrophages in acute and chronic neuroinflammation: effects on cognition, learning and affective behaviour. Prog Neuropsychopharmacol Biol Psychiatry (2017) 79(Pt A):15–8. doi:10.1016/j.pnpbp.2017.02.008
9. Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron (2006) 49:489–502. doi:10.1016/j.neuron.2006.01.022
10. Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M, et al. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J Neuroimmunol (2002) 124:83–92. doi:10.1016/S0165-5728(01)00496-9
11. Browne TC, McQuillan K, McManus RM, O’Reilly JA, Mills KH, Lynch MA. IFN-gamma production by amyloid beta-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J Immunol (2013) 190:2241–51. doi:10.4049/jimmunol.1200947
12. Fisher Y, Nemirovsky A, Baron R, Monsonego A. T cells specifically targeted to amyloid plaques enhance plaque clearance in a mouse model of Alzheimer’s disease. PLoS One (2010) 5:e10830. doi:10.1371/journal.pone.0010830
13. Lambracht-Washington D, Qu BX, Fu M, Anderson LD Jr, Stuve O, Eagar TN, et al. DNA immunization against amyloid beta 42 has high potential as safe therapy for Alzheimer’s disease as it diminishes antigen-specific Th1 and Th17 cell proliferation. Cell Mol Neurobiol (2011) 31:867–74. doi:10.1007/s10571-011-9680-7
14. Pellicano M, Larbi A, Goldeck D, Colonna-Romano G, Buffa S, Bulati M, et al. Immune profiling of Alzheimer patients. J Neuroimmunol (2012) 242:52–9. doi:10.1016/j.jneuroim.2011.11.005
15. Rosenkranz D, Weyer S, Tolosa E, Gaenslen A, Berg D, Leyhe T, et al. Higher frequency of regulatory T cells in the elderly and increased suppressive activity in neurodegeneration. J Neuroimmunol (2007) 188:117–27. doi:10.1016/j.jneuroim.2007.05.011
16. Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Alberoni M, et al. Increased activity of Th-17 and Th-9 lymphocytes and a skewing of the post-thymic differentiation pathway are seen in Alzheimer’s disease. Brain Behav Immun (2011) 25:539–47. doi:10.1016/j.bbi.2010.12.004
17. Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Calvo MG, et al. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis (2010) 21:927–38. doi:10.3233/JAD-2010-091696
18. Villa C, Fenoglio C, De Riz M, Clerici F, Marcone A, Benussi L, et al. Role of hnRNP-A1 and miR-590-3p in neuronal death: genetics and expression analysis in patients with Alzheimer disease and frontotemporal lobar degeneration. Rejuvenation Res (2011) 14:275–81. doi:10.1089/rej.2010.1123
19. Yin Y, Wen S, Li G, Wang D. Hypoxia enhances stimulating effect of amyloid beta peptide (25–35) for interleukin 17 and T helper lymphocyte subtype 17 upregulation in cultured peripheral blood mononuclear cells. Microbiol Immunol (2009) 53:281–6. doi:10.1111/j.1348-0421.2009.00120.x
20. Zhang J, Ke KF, Liu Z, Qiu YH, Peng YP. Th17 cell-mediated neuroinflammation is involved in neurodegeneration of abeta1-42-induced Alzheimer’s disease model rats. PLoS One (2013) 8:e75786. doi:10.1371/journal.pone.0075786
21. Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol (1989) 7:145–73. doi:10.1146/annurev.iy.07.040189.001045
22. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell (2000) 100:655–69. doi:10.1016/S0092-8674(00)80702-3
23. Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature (2007) 448:484–7. doi:10.1038/nature05970
24. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201:233–40. doi:10.1084/jem.20041257
25. Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med (2009) 361:888–98. doi:10.1056/NEJMra0707449
26. Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity (2000) 13:715–25. doi:10.1016/S1074-7613(00)00070-4
27. O’Connor RA, Anderton SM. Foxp3+ regulatory T cells in the control of experimental CNS autoimmune disease. J Neuroimmunol (2008) 193:1–11. doi:10.1016/j.jneuroim.2007.11.016
28. Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, O’Farrelly C, et al. CD39+Foxp3+ regulatory T cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol (2009) 183:7602–10. doi:10.4049/jimmunol.0901881
29. Frisullo G, Nociti V, Iorio R, Patanella AK, Caggiula M, Marti A, et al. Regulatory T cells fail to suppress CD4T+-bet+ T cells in relapsing multiple sclerosis patients. Immunology (2009) 127:418–28. doi:10.1111/j.1365-2567.2008.02963.x
30. Dombrowski Y, O’Hagan T, Dittmer M, Penalva R, Mayoral SR, Bankhead P, et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat Neurosci (2017) 20:674–80. doi:10.1038/nn.4528
31. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med (2006) 203:1701–11. doi:10.1084/jem.20060772
32. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med (2006) 203:1693–700. doi:10.1084/jem.20060468
33. Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement (2011) 7:270–9. doi:10.1016/j.jalz.2011.03.008
34. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement (2011) 7:263–9. doi:10.1016/j.jalz.2011.03.005
35. Buerger K, Ewers M, Andreasen N, Zinkowski R, Ishiguro K, Vanmechelen E, et al. Phosphorylated tau predicts rate of cognitive decline in MCI subjects: a comparative CSF study. Neurology (2005) 65:1502–3. doi:10.1212/01.wnl.0000183284.92920.f2
36. Blom ES, Giedraitis V, Zetterberg H, Fukumoto H, Blennow K, Hyman BT, et al. Rapid progression from mild cognitive impairment to Alzheimer’s disease in subjects with elevated levels of tau in cerebrospinal fluid and the APOE epsilon4/epsilon4 genotype. Dement Geriatr Cogn Disord (2009) 27:458–64. doi:10.1159/000216841
37. Lewczuk P, Matzen A, Blennow K, Parnetti L, Molinuevo JL, Eusebi P, et al. Cerebrospinal fluid Abeta42/40 corresponds better than Abeta42 to amyloid PET in Alzheimer’s disease. J Alzheimers Dis (2017) 55:813–22. doi:10.3233/JAD-160722
38. Wang W, Shao S, Jiao Z, Guo M, Xu H, Wang S. The Th17/Treg imbalance and cytokine environment in peripheral blood of patients with rheumatoid arthritis. Rheumatol Int (2012) 32:887–93. doi:10.1007/s00296-010-1710-0
39. Baran J, Kowalczyk D, Ozog M, Zembala M. Three-color flow cytometry detection of intracellular cytokines in peripheral blood mononuclear cells: comparative analysis of phorbol myristate acetate-ionomycin and phytohemagglutinin stimulation. Clin Diagn Lab Immunol (2001) 8:303–13. doi:10.1128/CDLI.8.2.303-313.2001
40. Petersen CM, Christensen EI, Andresen BS, Moller BK. Internalization, lysosomal degradation and new synthesis of surface membrane CD4 in phorbol ester-activated T-lymphocytes and U-937 cells. Exp Cell Res (1992) 201:160–73. doi:10.1016/0014-4827(92)90360-K
41. Agnes P-K, Christiane S, Peter D-B. T-cells show increased production of cytokines and activation markers in Alzheimer’s disease. Brain Disord Ther (2013) 3(1):3–112. doi:10.4172/2168-975X.1000112
42. Goldeck D, Larbi A, Pellicano M, Alam I, Zerr I, Schmidt C, et al. Enhanced chemokine receptor expression on leukocytes of patients with Alzheimer’s disease. PLoS One (2013) 8:e66664. doi:10.1371/journal.pone.0066664
43. Lee AY, Phan TK, Hulett MD, Korner H. The relationship between CCR6 and its binding partners: does the CCR6-CCL20 axis have to be extended? Cytokine (2015) 72:97–101. doi:10.1016/j.cyto.2014.11.029
44. Le Page A, Garneau H, Dupuis G, Frost EH, Larbi A, Witkowski JM, et al. Differential phenotypes of myeloid-derived suppressor and T regulatory cells and cytokine levels in amnestic mild cognitive impairment subjects compared to mild Alzheimer diseased patients. Front Immunol (2017) 8:783. doi:10.3389/fimmu.2017.00783
45. Larbi A, Pawelec G, Witkowski JM, Schipper HM, Derhovanessian E, Goldeck D, et al. Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J Alzheimers Dis (2009) 17:91–103. doi:10.3233/JAD-2009-1015
46. Buerger K, Ewers M, Pirttila T, Zinkowski R, Alafuzoff I, Teipel SJ, et al. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain (2006) 129:3035–41. doi:10.1093/brain/awl269
47. Tapiola T, Overmyer M, Lehtovirta M, Helisalmi S, Ramberg J, Alafuzoff I, et al. The level of cerebrospinal fluid tau correlates with neurofibrillary tangles in Alzheimer’s disease. Neuroreport (1997) 8:3961–3. doi:10.1097/00001756-199712220-00022
48. Dansokho C, Ait Ahmed D, Aid S, Toly-Ndour C, Chaigneau T, Calle V, et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain (2016) 139:1237–51. doi:10.1093/brain/awv408
49. Kipnis J, Avidan H, Caspi RR, Schwartz M. Dual effect of CD4+CD25+ regulatory T cells in neurodegeneration: a dialogue with microglia. Proc Natl Acad Sci U S A (2004) 101(Suppl 2):14663–9. doi:10.1073/pnas.0404842101
50. Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, et al. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer’s disease pathology. Nat Commun (2015) 6:7967. doi:10.1038/ncomms8967
51. Vom Berg J, Prokop S, Miller KR, Obst J, Kalin RE, Lopategui-Cabezas I, et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat Med (2012) 18:1812–9. doi:10.1038/nm.2965
52. Doecke JD, Laws SM, Faux NG, Wilson W, Burnham SC, Lam CP, et al. Blood-based protein biomarkers for diagnosis of Alzheimer disease. Arch Neurol (2012) 69:1318–25. doi:10.1001/archneurol.2012.1282
53. Chen JM, Jiang GX, Li QW, Zhou ZM, Cheng Q. Increased serum levels of interleukin-18, -23 and -17 in Chinese patients with Alzheimer’s disease. Dement Geriatr Cogn Disord (2014) 38:321–9. doi:10.1159/000360606
54. Behairi N, Belkhelfa M, Mesbah-Amroun H, Rafa H, Belarbi S, Tazir M, et al. All-trans-retinoic acid modulates nitric oxide and interleukin-17A production by peripheral blood mononuclear cells from patients with Alzheimer’s disease. Neuroimmunomodulation (2015) 22:385–93. doi:10.1159/000435885
55. D’Anna L, Abu-Rumeileh S, Fabris M, Pistis C, Baldi A, Sanvilli N, et al. Serum interleukin-10 levels correlate with cerebrospinal fluid amyloid beta deposition in Alzheimer disease patients. Neurodegener Dis (2017) 17:227–34. doi:10.1159/000474940
56. Chao CC, Hu S, Frey WH II, Ala TA, Tourtellotte WW, Peterson PK. Transforming growth factor beta in Alzheimer’s disease. Clin Diagn Lab Immunol (1994) 1:109–10.
57. Rodriguez-Rodriguez E, Sanchez-Juan P, Mateo I, Llorca J, Infante J, Garcia-Gorostiaga I, et al. Serum levels and genetic variation of TGF-beta1 are not associated with Alzheimer’s disease. Acta Neurol Scand (2007) 116:409–12. doi:10.1111/j.1600-0404.2007.00892.x
58. Huang L, Jia J, Liu R. Decreased serum levels of the angiogenic factors VEGF and TGF-beta1 in Alzheimer’s disease and amnestic mild cognitive impairment. Neurosci Lett (2013) 550:60–3. doi:10.1016/j.neulet.2013.06.031
Keywords: Th17, regulatory T cell, Th1, Alzheimer’s disease, mild cognitive impairment, Tau, amyloid beta
Citation: Oberstein TJ, Taha L, Spitzer P, Hellstern J, Herrmann M, Kornhuber J and Maler JM (2018) Imbalance of Circulating Th17 and Regulatory T Cells in Alzheimer’s Disease: A Case Control Study. Front. Immunol. 9:1213. doi: 10.3389/fimmu.2018.01213
Received: 26 February 2018; Accepted: 15 May 2018;
Published: 04 June 2018
Edited by:
Martin Hadamitzky, Universitätsklinikum Essen, GermanyReviewed by:
Carlos Spuch, Galicia Sur Health Research Institute, CIBERSAM, SpainNiels Hellings, University of Hasselt, Belgium
Copyright: © 2018 Oberstein, Taha, Spitzer, Hellstern, Herrmann, Kornhuber and Maler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Manuel Maler, bWFudWVsLm1hbGVyQHVrLWVybGFuZ2VuLmRl