 Lipeng Tang
Lipeng Tang Xiaozhi Yang
Xiaozhi Yang Yongxin Liang
Yongxin Liang Hesong Xie
Hesong Xie Zhenhua Dai
Zhenhua Dai Guangjuan Zheng
Guangjuan Zheng- 1Department of Pharmacology of Traditional Chinese Medicine, The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, China
- 2School of Bioscience and Bio-Pharmaceutics, Guangdong Pharmaceutical University, Guangzhou, Guangdong, China
- 3Section of Immunology, The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, China
- 4Department of Pathology, The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, China
Psoriasis, which is a common chronic inflammatory skin disease, endangers human health and brings about a major economic burden worldwide. To date, treatments for psoriasis remain unsatisfied because of their clinical limitations and various side effects. Thus, developing a safer and more effective therapy for psoriasis is compelling. Previous studies have explicitly shown that psoriasis is an autoimmune disease that is predominantly mediated by T helper 17 (Th17) cells, which express high levels of interleukin-17 (IL-17) in response to interleukin-23 (IL-23). The discovery of the IL-23–Th17–IL-17 axis in the development of psoriasis has led to the paradigm shift of understanding pathogenesis of psoriasis. Although anti-IL-17 antibodies show marked clinical efficacy in treating psoriasis, compared with antibodies targeting IL-17A or IL-17R alone, targeting Th17 cells themselves may have a maximal benefit by affecting multiple proinflammatory cytokines, including IL-17A, IL-17F, IL-22, and granulocyte-macrophage colony-stimulating factor, which likely act synergistically to drive skin inflammation in psoriasis. In this review, we mainly focus on the critical role of Th17 cells in the pathogenesis of psoriasis. Especially, we explore the small molecules that target retinoid-related orphan receptor γt (RORγt), a vital transcription factor for Th17 cells. Given that RORγt is the lineage-defining transcription factor for Th17 cell differentiation, targeting RORγt via small molecular inverse agonists may be a promising strategy for the treatment of Th17-mediated psoriasis.
Introduction
Psoriasis is an autoimmune disease with chronic skin inflammation (1), affecting over 125 million people worldwide (up to 2–4% of the world’s population) (2). It is predominantly a skin disease, which can manifest itself as various phenotypes, including plaque-type psoriasis or psoriasis vulgaris, guttate psoriasis, pustular psoriasis such as palmoplantar pustulosis, and erythrodermic psoriasis.
Psoriasis vulgaris, a most common type of psoriasis, is characterized by well-defined areas of erythematous and plaques with overlying silvery scale. The main histopathological changes of psoriasis vulgaris include abnormal cell proliferation, parakeratosis, hyperkeratosis, angiogenesis, and inflammatory cell infiltration (1, 3).
Increasing evidence has shown that comorbid cardiovascular diseases are the leading causes of death among patients with psoriasis (4). In addition, a high prevalence of metabolic syndrome, psychosocial distress or psychiatric disorders, chronic kidney disease, and gastrointestinal disease has been demonstrated in individuals with psoriasis (5, 6). The global financial burden associated with the care of psoriatic patients is substantial and significant (7–10). It was reported that the annual costs for treating psoriasis in USA amounted to approximately $112 billion in 2013 (11). As for individuals, patients with psoriasis would incur a lifetime medical expense for relief of physical symptoms and emotional health (12).
Therapeutic Challenges for Psoriasis
Based on the immunological characteristics of psoriasis, researchers have developed topical treatments, including corticosteroids, vitamin D3 analogs and Victoria A acid, and systemic therapies, including methotrexate and cyclosporine, for psoriasis. In clinic, patients with mild-to-moderate plaque psoriasis are usually treated topically with corticosteroids and vitamin D3 analogs, whereas those with moderate-to-severe psoriasis are systemically treated with methotrexate and cyclosporine (13, 14). However, these treatments exhibit low efficacies, poor tolerability, and various adverse reactions (15) (Table 1).
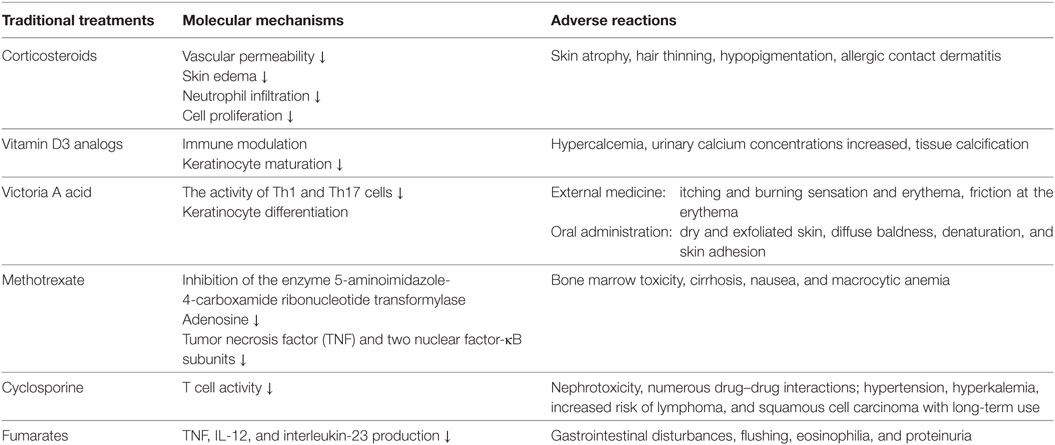
Table 1. Traditional treatment for psoriasis.
Although the introduction of biological treatments, including tumor necrosis factor (TNF)-α antagonists (Efalizumab), anti-TNF antibody (Adalimumab) (16), IL-12/interleukin-23 (IL-23) antagonists (Ustekinumab) (17), and interleukin-17 (IL-17) antagonists (Secukinumab, Ixekizumab, and Brodalumab) (18, 19), has revolutionized the short-term treatment of moderate-to-severe plaque psoriasis, the long-term use of biological therapies may cause loss of efficacy as well as severe adverse reactions, such as infection, cancer, and hepatic dysfunction (20, 21) (Table 2). These clinical side effects of existing treatments strongly suggest that it is still urgent to discover safer and more effective therapeutic drugs for psoriasis.
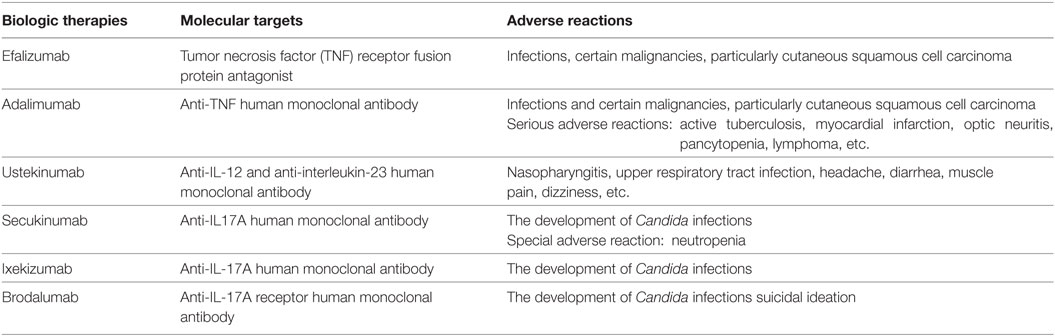
Table 2. Biologic therapies for psoriasis.
Pathogenesis of Autoimmune Psoriasis
To develop a better, safer, and more effective therapy for psoriasis, it is imperative to understand psoriatic pathogenesis. Previous studies have indicated that psoriasis is a skin disease mainly mediated by dendritic cells and T cells although macrophages, neutrophilic granulocytes, keratinocytes, vascular endothelial cells, and the cutaneous nervous system are involved in its pathogenesis (22, 23).
Epidermis-produced antimicrobial peptide LL-37 (cathelicidin), which acts as a dendritic cell activator, is upregulated in the initial phase of psoriasis (24). LL-37 stimulates dermal plasmacytoid dendritic cells to produce interferon-γ (IFN-γ), which in turn activates myeloid dendritic cells (mDCs) to secrete IL-12 and IL-23. IL-12 promotes the differentiation of Th1 cells, whereas IL-23 enhances T helper 17 (Th17) cell development. Th1 cells secrete more IFN-γ and TNF-α to further stimulate mDCs. In addition, Th17 cells secrete IL-17 to stimulate keratinocytes to over-proliferate, causing psoriasis-like lesions (25). Furthermore, the lesion cells secrete a series of chemokines, attracting more immune cells to inflamed tissue, while the damaged cells are digested by macrophages and produce LL-37, forming a positive feedback path that accelerates the development of psoriasis. Recently, LL-37 has been proved to be a T-cell-reactive autoantigen in psoriasis. LL37-specific CD4+ T cells can produce Th17-related cytokines (26). In summary, these results indicate that psoriasis is an autoimmune disease mediated by dendritic cells and T-cells (Figure 1).
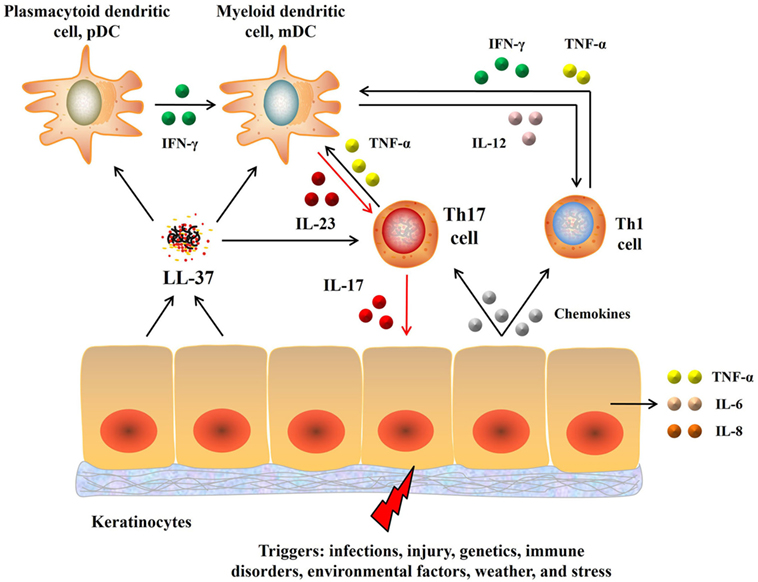
Figure 1. Pathogenesis of psoriasis. Upon activation, keratinocytes secrete LL-37 that in turn activates dendritic cells, which then produce IL-23 and IL-12. IL-23 induces differentiation of naive T cells into Th17 cells that then overproduce IL-17 and IL-22. IL-17 activates keratinocytes, promotes epidermal hyperplasia and recruits proinflammatory cells, resulting in a positive proinflammatory feedback that accelerates the development of psoriasis. Moreover, IL-12 produced by dendritic cells also promotes the differentiation of Th1 cells that in turn produce Th1 cytokines, including IFN-γ. Abbreviations: IL, interleukin; TNF, tumor necrosis factor; IFN-γ, interferon-γ; Th17, T helper 17; IL-23, interleukin-23; IL-17, interleukin-17.
The Main Role of Pathogenic Th17 Cells in Psoriasis
T helper 17 cells are a distinct subset of T helper cells that mainly produce IL-17A, IL-17F, and IL-22. Mounting evidence shows that there are two subsets of Th17 lineages. A non-pathogenic subset of Th17 cells induced by TGF-β1 and IL-6 has an important role in host defense against specific pathogens by producing IL-17 and IL-10 (27). The production of IL-10 by non-pathogenic Th17 cells restrains Th17 cell-mediated pathology so that they are incapable of promoting autoimmune inflammation. On the other hand, differentiation of highly pathogenic Th17 cells from naïve T cells occurs in the presence of IL-23, IL-6, and TGF-β1 (28, 29). More precisely, exposure to IL-23 diminishes the anti-inflammatory cytokine IL-10 in developing Th17 cells (27). In addition, IL-23 stabilizes and reinforces Th17 phenotypes by increasing expression of IL-23 receptor (30, 31) and endowing Th17 cells with pathogenic effector functions (32–34). These pathogenic Th17 cells contribute to various autoimmune diseases (35, 36).
Psoriasis is primarily characterized as a Th1-driven disease because the levels of Th1 cytokines, such as IFN-γ, TNF-α, and interleukin (IL)-12, are markedly elevated in psoriatic lesions, while there is no such an increase in expression of Th2 cytokines (IL-4, IL-5, and IL-13) (37–39). With the characterization of a distinct subset of Th17 cells, the research field of psoriasis has experienced a major paradigm shift.
Indeed, previous results have confirmed that pathogenic Th17 cells play a central role in the development of psoriasis(40, 41). Pathological or immunohistochemical studies on psoriasis have shown that skin lesions are mainly infiltrated by Th17 cells. In addition, IL-23, which is produced by activated mDCs, drives naïve T cells to develop into pathogenic Th17 cells (42). IL-17, which is predominantly produced by pathogenic Th17 (43), is significantly elevated in patients with psoriasis compared with healthy subjects. Upregulated IL-17 has potent ability to recruit neutrophils (44, 45), to activate T cells, to stimulate fibroblasts (46), and to promote development of multiple lineages of macrophages (47, 48). Moreover, pathogenic Th17-secreted IL-17 induces proliferation of keratinocytes and secretion of antimicrobial peptides, cytokines, and chemokines, which in turn recruit more immune cells to inflamed tissue. This positive feedback loop between Th17 cells and keratinocytes has been proved to contribute to the chronic inflammatory phase of psoriasis (43, 49, 50). Other proinflammatory factors released by pathogenic Th17 cells, such as IL-22, TNF-α, and granulocyte-macrophage colony-stimulating factor (GM-CSF), stimulate keratinocytes to release chemokines, further sustaining the inflammatory cycle to promote the development of psoriasis (51, 52).
Retinoid-Related Orphan Receptor γt (RORγt): A Lineage-Defining Transcription Factor for Th17 Cells
The differentiation of Th17 cells, similar to that of Th1 and Th2 subsets (53, 54), relies on the action of a lineage-specific transcription factor, identified as the orphan nuclear receptor RORγt (55). RORγt, encoded by RORC2, is an isoform of RORγ that belongs to the NR1F subfamily of orphan receptors, including RORα and RORβ. Previous studies have indicated that RORγt is both necessary and sufficient for Th17 cell differentiation in mouse and human CD4+ T cells. Ivanov et al. reported that T cells lacking RORγt (Rorc−/−) failed to differentiate into Th17 cells even under Th17-polarizing culture conditions, while over-expression of Rorc in naïve CD4+ T cells was sufficient to accelerate the expression of Th17-related cytokines and chemokines, including IL-17A, IL-17F, IL-22, IL-26, CCR6, and CCL20. Moreover, mice lacking RORγt were much less susceptible to experimental autoimmune encephalomyelitis (EAE), and CD4+ splenocytes from those mice could not induce the disease (55). A similarly crucial role for RORγt in human Th17 cells was also demonstrated (56). IL-6 and IL-23 signals strongly phosphorylated and dimerized signal transducer and activator of transcription 3 (STAT3), resulting in enhanced expression and nuclear translocation of RORγt, which then promoted Th17 responses by activating Th17 gene promoters, including Il17a, Il17f, Il22, Il26, Il23r, Csf-2, Ccr6, and Ccl20. In addition, IL-23 signaling-induced transcription factor Blimp-1 enhanced pathogenic Th17 function by co-localizes RORγt and STAT-3 at Il17a, Il23r, and Csf-2 enhancer sites (34, 57, 58) (Figure 2). Interestingly, neither IL-23 nor IL-6 alone was sufficient to effectively generate Th17 cells (59). Nevertheless, either IL-23 or IL-6 induced IL-17 production by naïve precursors in the presence of IL-1β rather than TGF-β. T-bet + RORγt + Th17 cells were generated without TGF-β and were pathogenic in an EAE animal model, indicating an alternative pathway for Th17 differentiation (59).
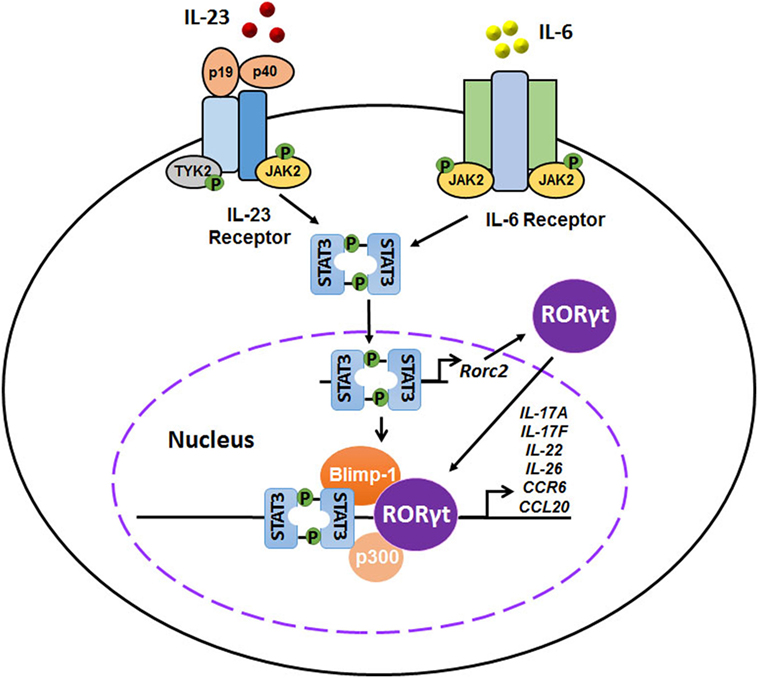
Figure 2. Interplays of interleukin-23 (IL-23), IL-6, signal transducer and activator of transcription 3 (STAT3), and retinoid-related orphan receptor γt (RORγt) in the differentiation of pathogenic T helper 17 (Th17) cells. IL-23 and IL-6 signals activate the JAK–STAT signaling pathway, inducing a strong phosphorylation and dimerization of STAT3. STAT3 homodimers induce the expression and nuclear translocation of RORγt, which in turn promotes Th17 responses by activating Th17 gene promoters, including Il17a, Il17f, Il22, Il26, Il23r, Csf-2, Ccr6, and Ccl20. In addition, IL-23 signaling-induced transcription factor Blimp-1 enhances pathogenic Th17 function by co-localizes RORγt and STAT-3 at Il17a, Il23r, and Csf-2 enhancer sites.
Taken together, previous studies have confirmed an essential role of RORγt in the differentiation of pathogenic Th17 cells. Given that pathogenic Th17 cells play such a pivotal role in the pathogenesis of psoriasis, targeting Th17 cells, especially via blocking RORγt, may be a good option for treating psoriasis. In addition, RORγt might be a uniquely tractable drug target by virtue of being a nuclear receptor. Therefore, RORγt can be an attractive pharmacologic target for the treatment of Th17-mediated autoimmune diseases, including psoriasis.
Small Molecules Targeting RORγt
Retinoid-related orphan receptor γt contains identical DNA-binding domain and ligand-binding domain (LBD). Like other nuclear receptors, the binding of ligands to the region LBD causes a conformational change, which results in recruiting transcriptional co-activators as well as activating transcriptional activity.
Since RORγt receptor was initially identified as an orphan receptor, its endogenous ligands attracted more attention at first. Previous studies have revealed that several oxysterols are endogenous modulators of RORγt activity with high-affinity. For example, 7-oxygenated sterols function as high-affinity ligands for RORγt via directly binding their LBDs, modulating co-activator binding, and suppressing the transcriptional activity of the receptors (60). In addition, 24S-hydroxycholestrol acts as an inverse agonist that suppresses the transcriptional activity of RORγt (61).
To develop potent synthetic RORγt ligands that selectively suppress pathogenic effector functions of Th17 cells, researchers have used many strategies to screen for potentially regulatory drug candidates, as described below.
Digoxin, the cardiotonic glycoside extracted mainly from Digitalis lanata, has been identified as a specific inhibitor of RORγ transcriptional activity without affecting other nuclear hormone receptors, including human androgen receptor (AR) and liver X receptor α (62). It specifically inhibits IL-17 production by Th17 cells. Moreover, it is effective in attenuating EAE in mice and decreasing the disease severity in a rat model of arthritis (62–64). However, it is toxic for human cells at high doses and may cause some adverse reactions, including arrhythmia, nausea, vomiting, blurred vision, diarrhea, depression, and even lethargy. Further studies have shown that derivatives of digoxin, such as Dig(dhd) 20,22-dihydrodigoxin-21,23-diol, and Dig(sal) digoxin-21-salicylidene, specifically inhibit the differentiation of Th17 cells in human CD4+ T cells without significant toxicity (62), indicating that nontoxic derivatives of digoxin may be utilized as chemical templates for the development of RORγt negative regulators.
SR1001, a derivative of liver X receptor agonist, is capable of suppressing the transcriptional activity of RORα and RORγ (65). It is a high-affinity synthetic ligand that can bind the LBD of RORα and RORγ, resulting in inhibition of murine Th17 cell differentiation and IL-17 expression by inducing conformational changes that in turn suppress the receptors’ transcriptional activity. Thence, SR1001 might be an attractive lead compound for drug development to treat Th17-mediated autoimmune diseases, such as psoriasis as well as RORα- and RORγ-mediated metabolic diseases (66, 67).
SR2211, a derivative of SR1001, only binds the LBD of RORγ and inhibits the transcriptional activity of RORγ without affecting RORα function (68). In addition, SR2211 suppresses the intracellular expression of IL-17 and has potential utility for the treatment of inflammatory diseases, such as experimental arthritis (69, 70). SR2211 has been shown to diminish genome-wide AR binding, H3K27ac abundance and expression of the AR target gene networks, and it could serve as a potential drug for the treatment of castration-resistant prostate cancer (71).
Ursolic acid (UA), a small molecule present in medicinal herbs such as Prunella vulgaris L., effectively inhibits the function of RORγt, resulting in greatly reduced IL-17 expression in both developing and differentiated Th17 cells (72, 73). However, UA also has other cellular targets, including the liver kinase B1–AMP-activated protein kinase (74), the NFE2-related factor 2 (75), nuclear factor-κB (76), and STAT3 pathway (77, 78), suggesting that it is not RORγt-specific in vivo.
TMP920, which can displace RORγt from its target loci, suppresses Th17 cell differentiation and Th17 signature gene expression (79). Based on TMP920, additional inverse agonists are developed, including TMP778, which exhibits an increase in potency and specificity. It predominantly affects RORγt transcription without removing DNA binding (79). Interestingly, the diastereomer of TMP778 or TMP776 displays no inverse agonist activity against RORγt. In experiments in vivo, TMP778 suppresses imiquimod-induced cutaneous inflammation and attenuates EAE. Furthermore, TMP778 also reduces expression of Th17-signature genes in cells isolated from the blood and skin of psoriatic patients (80).
Other RORγt inverse agonists have also been discovered. Using a scaffold hybridization strategy, a series of carbazole carboxamides are found to be potent RORγt inverse agonists (81). In addition, MG 2778, a cyclopenta[a]phenanthrene derivative, is identified as a lead compound for developing synthetic steroidal inverse agonists of RORγt (82). Furthermore, TAK-828F, a potent and selective RORγt inverse agonist, strongly inhibits Tc17 and Th17 cell differentiation from naive T cells and memory CD4+ T cells without affecting Th1 cell differentiation (83). In another study, Barbay et al. have identified 6-substituted quinolines as modulators of RORγt using a RORγt-driven cell-based reporter assay. They have further elucidated the interaction between 6-substituted quinolones and RORγt in an X-ray crystal structure (84). Moreover, A213, a potent and selective antagonist of RORγt, is found to inhibit Th17 cell differentiation in vitro. It also attenuates psoriatic skin lesion in two different mouse models by suppressing IL-17 production (85).
Taken together, previous studies have implicated a potential therapeutic application of RORγt antagonist for the treatment of Th17-mediated diseases, including psoriasis. Especially, targeting RORγt for the treatment of cutaneous inflammatory disorders may afford additional therapeutic benefits over existing modalities, in which only one Th17 cytokine such as IL-17A is targeted. However, the small molecules targeting RORγt could generate unwanted or unexpected results given that they may exert off-target effects in vivo. Those molecules must undergo rigorous clinical trials prior to a clinical application to carefully evaluate their potential side effects. In addition, other types of immune cells, including type 3 innate lymphoid cells, CD8+ IL-17-producing (Tc17) cells, γδT, and even Treg cells, may also express RORγt. Target RORγt could affect these cells as well. Thus, strategies targeting RORγt in Th17 cells are preferred so that we can attenuate Th17-mediated inflammation while limiting potential side effects.
Summary and Outlook
Since there are many limitations of traditional and biological treatments for psoriasis, it is important to develop more effective and safer therapies of psoriasis. The finding of RORγt/Th17/IL-17 signaling pathway has provided further insights into the pathogenesis of psoriasis. Compared with antibodies targeting IL-17A or IL-17R alone, targeting Th17 cells themselves might benefit psoriatic patients to a greatest extent by impacting multiple proinflammatory cytokines (IL-17A, IL-17F, IL-22, and GM-CSF) that are likely to act synergistically to drive psoriatic inflammation. Hence, targeting RORγt via small molecule inverse agonists is a promising strategy for treating psoriasis via suppressing Th17 cell differentiation. Furthermore, small molecules disrupting RORγt are also expected to be safer than global immunosuppressive agents, such as cyclosporine. However, there are several challenges that need to be overcome. Researchers should generate safer and more potent compounds. Moreover, rigorous clinical studies are needed to assess their actual clinical efficacy and side effects since they could generate off-target effects. In conclusion, given the importance of Th17 cells and their proinflammatory cytokines in the pathogenesis of psoriasis, targeting RORγt seems to be a promising approach to treating psoriasis effectively and perhaps safely.
Ethics Statement
The epidemiological data were cited without any commercial or financial uses.
Author Contributions
LT and XY wrote the manuscript; YL and HX searched the literature; ZD and GZ edited the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81602748); Natural Science Foundation of Guangdong Province (No. 2016A030310291, No. 2017A030313639); the Specific research fund for TCM Science and Technology of Guangdong Provincial Hospital of Chinese Medicine (YN2015QN03, YN2016YX03, YN2016ZD01); and Medical Scientific Research Foundation of Guangdong Province (No. A2017277).
References
1. Boehncke W, Schön MP. Psoriasis. Lancet (2015) 386(9997):983–94. doi:10.1016/S0140-6736(14)61909-7
2. Kaufman BP, Alexis AF. Psoriasis in skin of color: insights into the epidemiology, clinical presentation, genetics, quality-of-life impact, and treatment of psoriasis in non-white racial/ethnic groups. Am J Clin Dermatol (2017) 19(3):405–23. doi:10.1007/s40257-017-0332-7
3. Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet (2007) 370(9583):263–71. doi:10.1016/S0140-6736(07)61128-3
4. Puig L. Cardiometabolic comorbidities in psoriasis and psoriatic arthritis. Int J Mol Sci (2018) 19:58. doi:10.3390/ijms19010058
5. Oliveira MDFS, Rocha BDO, Duarte GV. Psoriasis: classical and emerging comorbidities. An Bras Dermatol (2015) 90:9–20. doi:10.1590/abd1806-4841.20153038
6. Dominguez PL, Han J, Li T, Ascherio A, Qureshi AA. Depression and the risk of psoriasis in US women. J Eur Acad Dermatol Venereol (2013) 27:1163–7. doi:10.1111/j.1468-3083.2012.04703.x
7. Al SS, Foster SA, Goldblum OM, Malatestinic WN, Zhu B, Shi N, et al. Healthcare costs in psoriasis and psoriasis sub-groups over time following psoriasis diagnosis. J Med Econ (2017) 20:982–90. doi:10.1080/13696998.2017.1345749
8. Hay RJ, Johns NE, Williams HC, Bolliger IW, Dellavalle RP, Margolis DJ, et al. The global burden of skin disease in 2010: an analysis of the prevalence and impact of skin conditions. J Invest Dermatol (2014) 134:1527–34. doi:10.1038/jid.2013.446
9. Brezinski EA, Dhillon JS, Armstrong AW. Economic burden of psoriasis in the United States: a systematic review. JAMA Dermatol (2015) 151:651–8. doi:10.1001/jamadermatol.2014.3593
10. Goff KL, Karimkhani C, Boyers LN, Weinstock MA, Lott JP, Hay RJ, et al. The global burden of psoriatic skin disease. Br J Dermatol (2015) 172:1665–8. doi:10.1111/bjd.13715
11. Feldman SR, Tian H, Gilloteau I, Mollon P, Shu M. Economic burden of comorbidities in psoriasis patients in the United States: results from a retrospective U.S. database. BMC Health Serv Res (2017) 17(1):337. doi:10.1186/s12913-017-2278-0
12. Hawro T, Zalewska A, Hawro M, Kaszuba A, Królikowska M, Maurer M. Impact of psoriasis severity on family income and quality of life. J Eur Acad Dermatol Venereol (2015) 29:438–43. doi:10.1111/jdv.12572
13. Menter A, Korman NJ, Elmets CA, Feldman SR, Gelfand JM, Gordon KB, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. Guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol (2009) 61:451–85. doi:10.1016/j.jaad.2009.03.027
14. Greb JE, Goldminz AM, Elder JT, Lebwohl MG, Gladman DD, Wu JJ, et al. Psoriasis. Nat Rev Dis Primers (2016) 2:16082. doi:10.1038/nrdp.2016.82
15. Descalzo MA, Carretero G, Ferrandiz C, Rivera R, Dauden E, Gomez-Garcia FJ, et al. Change over time in the rates of adverse events in patients receiving systemic therapy for psoriasis: a cohort study. J Am Acad Dermatol (2018) 78(4):798–800. doi:10.1016/j.jaad.2017.10.051
16. Lee EB, Amin M, Egeberg A, Wu JJ. Treatment changes in patients with psoriasis on etanercept or adalimumab: a retrospective chart review. J Dermatolog Treat (2018) 6:1–2. doi:10.1080/09546634.2018.1443198
17. Mugheddu C, Atzori L, Del Piano M, Lappi A, Pau M, Murgia S, et al. Successful ustekinumab treatment of noninfectious uveitis and concomitant severe psoriatic arthritis and plaque psoriasis. Dermatol Ther (2017) 30:e12527. doi:10.1111/dth.12527
18. Sekhon S, Jeon C, Nakamura M, Yan D, Afifi L, Bhutani T, et al. Clinical utility of ixekizumab in the treatment of moderate-to-severe plaque psoriasis. Psoriasis (2017) 7:65–72. doi:10.2147/PTT.S129792
19. Rothstein B, Gottlieb A. Secukinumab for treating plaque psoriasis. Expert Opin Biol Ther (2016) 16:119–28. doi:10.1517/14712598.2016.1121986
20. Belinchón I, Ramos JM, Carretero G, Ferrándiz C, Rivera R, Daudén E, et al. Adverse events associated with discontinuation of the biologics/classic systemic treatments for moderate-to-severe plaque psoriasis: data from the Spanish Biologics Registry, Biobadaderm. J Eur Acad Dermatol Venereol (2017) 31:1700–8. doi:10.1111/jdv.14314
21. Lockwood SJ, Prens LM, Kimball AB. Adverse reactions to biologics in psoriasis. Curr Probl Dermatol (2018) 53:1–14. doi:10.1159/000478072
22. Diani M, Altomare G, Reali E. T cell responses in psoriasis and psoriatic arthritis. Autoimmun Rev (2015) 14:286–92. doi:10.1016/j.autrev.2014.11.012
23. Kim T, Kim DS, Kim H, Lee M. The pathophysiological role of dendritic cell subsets in psoriasis. BMB Rep (2014) 47:60–8. doi:10.5483/BMBRep.2014.47.2.014
24. Frohm M, Agerberth B, Ahangari G, Stâhle-Bäckdahl M, Lidén S, Wigzell H, et al. The expression of the gene coding for the antibacterial peptide LL-37 is induced in human keratinocytes during inflammatory disorders. J Biol Chem (1997) 272:15258–63. doi:10.1074/jbc.272.24.15258
25. Ogawa E, Sato Y, Minagawa A, Okuyama R. Pathogenesis of psoriasis and development of treatment. J Dermatol (2018) 45(3):264–72. doi:10.1111/1346-8138.14139
26. Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, Conrad C, et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun (2014) 5:5621. doi:10.1038/ncomms6621
27. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol (2007) 8:1390–7. doi:10.1038/ni1539
28. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol (2012) 13:991–9. doi:10.1038/ni.2416
29. Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature (2013) 496:513–7. doi:10.1038/nature11984
30. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201:233–40. doi:10.1084/jem.20041257
31. Chen Z, Tato CM, Muul L, Laurence A, O’Shea JJ. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum (2007) 56:2936–46. doi:10.1002/art.22866
32. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6:1133–41. doi:10.1038/ni1261
33. Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature (2007) 445(7128):648–51. doi:10.1038/nature05505
34. Jain R, Chen Y, Kanno Y, Joyce-Shaikh B, Vahedi G, Hirahara K, et al. Interleukin-23-induced transcription factor Blimp-1 promotes pathogenicity of T helper 17 cells. Immunity (2016) 44:131–42. doi:10.1016/j.immuni.2015.11.009
35. Puwipirom H, Hirankarn N, Sodsai P, Avihingsanon Y, Wongpiyabovorn J, Palaga T. Increased interleukin-23 receptor(+) T cells in peripheral blood mononuclear cells of patients with systemic lupus erythematosus. Arthritis Res Ther (2010) 12:R215. doi:10.1186/ar3194
36. Martin BN, Wang C, Zhang C, Kang Z, Gulen MF, Zepp JA, et al. T cell-intrinsic ASC critically promotes TH17-mediated experimental autoimmune encephalomyelitis. Nat Immunol (2016) 17:583–92. doi:10.1038/ni.3389
37. Nestle FO, Turka LA, Nickoloff BJ. Characterization of dermal dendritic cells in psoriasis. Autostimulation of T lymphocytes and induction of Th1 type cytokines. J Clin Invest (1994) 94:202–9. doi:10.1172/JCI117308
38. Schlaak JF, Buslau M, Jochum W, Hermann E, Girndt M, Gallati H, et al. T cells involved in psoriasis vulgaris belong to the Th1 subset. J Invest Dermatol (1994) 102:145–9. doi:10.1111/1523-1747.ep12371752
39. Austin LM, Ozawa M, Kikuchi T, Walters IB, Krueger JG. The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon-γ, interleukin-2, and tumor necrosis factor-α, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol (1999) 113:752–9.
40. Harden JL, Krueger JG, Bowcock AM. The immunogenetics of psoriasis: a comprehensive review. J Autoimmun (2015) 64:66–73. doi:10.1016/j.jaut.2015.07.008
41. Marinoni B, Ceribelli A, Massarotti MS, Selmi C. The Th17 axis in psoriatic disease: pathogenetic and therapeutic implications. Auto Immun Highlights (2014) 5:9–19. doi:10.1007/s13317-013-0057-4
42. Boutet M, Nerviani A, Gallo Afflitto G, Pitzalis C. Role of the IL-23/IL-17 axis in psoriasis and psoriatic arthritis: the clinical importance of its divergence in skin and joints. Int J Mol Sci (2018) 19:530. doi:10.3390/ijms19020530
43. Couderc E, Morel F, Levillain P, Buffière-Morgado A, Camus M, Paquier C, et al. Interleukin-17A-induced production of acute serum amyloid A by keratinocytes contributes to psoriasis pathogenesis. PLoS One (2017) 12:e181486. doi:10.1371/journal.pone.0181486
44. Reich K, Papp KA, Matheson RT, Tu JH, Bissonnette R, Bourcier M, et al. Evidence that a neutrophil-keratinocyte crosstalk is an early target of IL-17A inhibition in psoriasis. Exp Dermatol (2015) 24:529–35. doi:10.1111/exd.12710
45. Keijsers RR, Hendriks AGM, Van Erp PEJ, Van Cranenbroek B, Van De Kerkhof PCM, Koenen HJPM, et al. In vivo induction of cutaneous inflammation results in the accumulation of extracellular trap-forming neutrophils expressing RORγt and IL-17. J Invest Dermatol (2014) 134:1276––1284. doi:10.1038/jid.2013.526
46. Schirmer C, Klein C, von Bergen M, Simon JC, Saalbach A. Human fibroblasts support the expansion of IL-17-producing T cells via up-regulation of IL-23 production by dendritic cells. Blood (2010) 116:1715–25. doi:10.1182/blood-2010-01-263509
47. Zhang J, Lin Y, Li C, Zhang X, Cheng L, Dai L, et al. IL-35 decelerates the inflammatory process by regulating inflammatory cytokine secretion and M1/M2 macrophage ratio in psoriasis. J Immunol (2016) 197:2131–44. doi:10.4049/jimmunol.1600446
48. Lorthois I, Asselineau D, Seyler N, Pouliot R. Contribution of in vivo and organotypic 3D models to understanding the role of macrophages and neutrophils in the pathogenesis of psoriasis. Mediators Inflamm (2017) 2017:7215072. doi:10.1155/2017/7215072
49. Senra L, Stalder R, Alvarez MD, Chizzolini C, Boehncke WH, Brembilla NC. Keratinocyte-derived IL-17E contributes to inflammation in psoriasis. J Invest Dermatol (2016) 136:1970–80. doi:10.1016/j.jid.2016.06.009
50. Pfaff CM, Marquardt Y, Fietkau K, Baron JM, Lüscher B. The psoriasis-associated IL-17A induces and cooperates with IL-36 cytokines to control keratinocyte differentiation and function. Sci Rep (2017) 7(1):15631. doi:10.1038/s41598-017-15892-7
51. Schäkel K, Schön MP, Ghoreschi K. Pathogenesis of psoriasis. Hautarzt (2016) 67:422–31. doi:10.1007/s00105-016-3800-8
52. Eberle FC, Bruck J, Holstein J, Hirahara K, Ghoreschi K. Recent advances in understanding psoriasis. F1000Res (2016) 5. doi:10.12688/f1000research.7927.1
53. Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell (1997) 89:587–96. doi:10.1016/S0092-8674(00)80240-8
54. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell (2000) 100:655–69. doi:10.1016/S0092-8674(00)80702-3
55. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi:10.1016/j.cell.2006.07.035
56. Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, et al. Phenotypic and functional features of human Th17 cells. J Exp Med (2007) 204:1849–61. doi:10.1084/jem.20070663
57. Calautti E, Avalle L, Poli V. Psoriasis: a STAT3-centric view. Int J Mol Sci (2018) 19:171. doi:10.3390/ijms19010171
58. McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol (2009) 10:314–24. doi:10.1038/ni.1698
59. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signaling. Nature (2010) 467:967–71. doi:10.1038/nature09447
60. Wang Y, Kumar N, Solt LA, Richardson TI, Helvering LM, Crumbley C, et al. Modulation of retinoic acid receptor-related orphan receptor α and γ activity by 7-oxygenated sterol ligands. J Biol Chem (2010) 285:5013–25. doi:10.1074/jbc.M109.080614
61. Wang Y, Kumar N, Crumbley C, Griffin PR, Burris TP. A second class of nuclear receptors for oxysterols: regulation of RORα and RORγ activity by 24S-hydroxycholesterol (cerebrosterol). Biochim Biophys Acta (2010) 1801:917–23. doi:10.1016/j.bbalip.2010.02.012
62. Huh JR, Leung MWL, Huang P, Ryan DA, Krout MR, Malapaka RRV, et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature (2011) 472:486–90. doi:10.1038/nature09978
63. Lee J, Baek S, Lee J, Lee J, Lee D, Park M, et al. Digoxin ameliorates auto-immune arthritis via suppression of Th17 differentiation. Int Immunopharmacol (2015) 26:103–11. doi:10.1016/j.intimp.2015.03.017
64. Fujita-Sato S, Ito S, Isobe T, Ohyama T, Wakabayashi K, Morishita K, et al. Structural basis of digoxin that antagonizes RORγt receptor activity and suppresses Th17 cell differentiation and interleukin (IL)-17 production. J Biol Chem (2011) 286:31409–17. doi:10.1074/jbc.M111.254003
65. Solt LA, Kumar N, Nuhant P, Wang Y, Lauer JL, Liu J, et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature (2011) 472:491–4. doi:10.1038/nature10075
66. Beurel E, Harrington LE, Jope RS. Inflammatory T helper 17 cells promote depression-like behavior in mice. Biol Psychiatry (2013) 73:622–30. doi:10.1016/j.biopsych.2012.09.021
67. Solt LA, Banerjee S, Campbell S, Kamenecka TM, Burris TP. ROR inverse agonist suppresses insulitis and prevents hyperglycemia in a mouse model of type 1 diabetes. Endocrinology (2015) 156:869–81. doi:10.1210/en.2014-1677
68. Kumar N, Lyda B, Chang MR, Lauer JL, Solt LA, Burris TP, et al. Identification of SR2211: a potent synthetic RORγ-selective modulator. ACS Chem Biol (2012) 7:672–7. doi:10.1021/cb200496y
69. Chang MR, Lyda B, Kamenecka TM, Griffin PR. Pharmacologic repression of retinoic acid receptor-related orphan nuclear receptor γ is therapeutic in the collagen-induced arthritis experimental model. Arthritis Rheumatol (2014) 66:579–88. doi:10.1002/art.38272
70. Lin H, Song P, Zhao Y, Xue L, Liu Y, Chu C. Targeting Th17 cells with small molecules and small interference RNA. Mediators Inflamm (2015) 2015:290657. doi:10.1155/2015/290657
71. Wang J, Zou JX, Xue X, Cai D, Zhang Y, Duan Z, et al. ROR-γ drives androgen receptor expression and represents a therapeutic target in castration-resistant prostate cancer. Nat Med (2016) 22:488–96. doi:10.1038/nm0616-692b
72. Baek S, Lee J, Lee D, Park M, Lee J, Kwok S, et al. Ursolic acid ameliorates autoimmune arthritis via suppression of Th17 and B cell differentiation. Acta Pharmacol Sin (2014) 35:1177–87. doi:10.1038/aps.2014.58
73. Xu T, Wang X, Zhong B, Nurieva RI, Ding S, Dong C. Ursolic acid suppresses interleukin-17 (IL-17) production by selectively antagonizing the function of RORγt protein. J Biol Chem (2011) 286:22707–10. doi:10.1074/jbc.C111.250407
74. He Y, Li Y, Zhao T, Wang Y, Sun C. Ursolic acid inhibits adipogenesis in 3T3-L1 adipocytes through LKB1/AMPK pathway. PLoS One (2013) 8:e70135. doi:10.1371/journal.pone.0070135
75. Li L, Zhang X, Cui L, Wang L, Liu H, Ji H, et al. Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res (2013) 1497:32–9. doi:10.1016/j.brainres.2012.12.032
76. You HJ, Choi CY, Kim JY, Park SJ, Hahm KS, Jeong HG. Ursolic acid enhances nitric oxide and tumor necrosis factor-alpha production via nuclear factor-kappaB activation in the resting macrophages. FEBS Lett (2001) 509:156–60. doi:10.1016/S0014-5793(01)03161-1
77. Lin J, Chen Y, Wei L, Shen A, Sferra TJ, Hong Z, et al. Ursolic acid promotes colorectal cancer cell apoptosis and inhibits cell proliferation via modulation of multiple signaling pathways. Int J Oncol (2013) 43:1235–43. doi:10.3892/ijo.2013.2040
78. Pathak AK, Bhutani M, Nair AS, Ahn KS, Chakraborty A, Kadara H, et al. Ursolic acid inhibits STAT3 activation pathway leading to suppression of proliferation and chemosensitization of human multiple myeloma cells. Mol Cancer Res (2007) 5:943–55. doi:10.1158/1541-7786.MCR-06-0348
79. Xiao S, Yosef N, Yang J, Wang Y, Zhou L, Zhu C, et al. Small-molecule RORγ antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity (2014) 40:477–89. doi:10.1016/j.immuni.2014.04.004
80. Skepner J, Ramesh R, Trocha M, Schmidt D, Baloglu E, Lobera M, et al. Pharmacologic inhibition of RORγ regulates Th17 signature gene expression and suppresses cutaneous inflammation in vivo. J Immunol (2014) 192:2564–75. doi:10.4049/jimmunol.1302190
81. Huang Y, Yu M, Sun N, Tang T, Yu F, Song X, et al. Discovery of carbazole carboxamides as novel RORγt inverse agonists. Eur J Med Chem (2018) 148:465–76. doi:10.1016/j.ejmech.2018.02.050
82. Dal Prà M, Carta D, Szabadkai G, Suman M, Frión-Herrera Y, Paccagnella N, et al. Targeting RORs nuclear receptors by novel synthetic steroidal inverse agonists for autoimmune disorders. Bioorg Med Chem (2018) 26(8):1686–704. doi:10.1016/j.bmc.2018.02.018
83. Shibata A, Uga K, Sato T, Sagara M, Igaki K, Nakamura Y, et al. Pharmacological inhibitory profile of TAK-828F, a potent and selective orally available RORγt inverse agonist. Biochem Pharmacol (2018) 150:35–45. doi:10.1016/j.bcp.2018.01.023
84. Barbay JK, Cummings MD, Abad M, Castro G, Kreutter KD, Kummer DA, et al. 6-Substituted quinolines as RORgammat inverse agonists. Bioorg Med Chem Lett (2017) 27(23):5277–83. doi:10.1016/j.bmcl.2017.10.027
Keywords: autoimmune disorder, psoriasis, T helper 17 cells, retinoid-related orphan receptor γt nuclear receptor, retinoid-related orphan receptor γt inverse agonist
Citation: Tang L, Yang X, Liang Y, Xie H, Dai Z and Zheng G (2018) Transcription Factor Retinoid-Related Orphan Receptor γt: A Promising Target for the Treatment of Psoriasis. Front. Immunol. 9:1210. doi: 10.3389/fimmu.2018.01210
Received: 20 March 2018; Accepted: 15 May 2018;
Published: 30 May 2018
Edited by:
Nicolò Costantino Brembilla, Université de Genève, SwitzerlandReviewed by:
Raffaele De Palma, Università degli Studi della Campania “Luigi Vanvitelli” Caserta, ItalyGiuseppe Sciumè, Sapienza Università di Roma, Italy
Copyright: © 2018 Tang, Yang, Liang, Xie, Dai and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenhua Dai, emRhaTIwMDlAb3V0bG9vay5jb20=;
Guangjuan Zheng, emhlbmdndWFuZ2p1YW5AZ3p1Y20uZWR1LmNu
†These authors have contributed equally to this work.