
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 28 May 2018
Sec. Microbial Immunology
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01161
This article is part of the Research TopicUpdate on the Immune Mechanisms Against Respiratory PathogensView all 29 articles
 Marcia Campillo-Navarro1,2
Marcia Campillo-Navarro1,2 Kahiry Leyva-Paredes1
Kahiry Leyva-Paredes1 Luis Donis-Maturano3
Luis Donis-Maturano3 Gloria M. Rodríguez-López1
Gloria M. Rodríguez-López1 Rodolfo Soria-Castro1
Rodolfo Soria-Castro1 Blanca Estela García-Pérez1
Blanca Estela García-Pérez1 Nahum Puebla-Osorio4
Nahum Puebla-Osorio4 Stephen E. Ullrich5,6
Stephen E. Ullrich5,6 Julieta Luna-Herrera1Leopoldo Flores-Romo3Héctor Sumano-López2
Julieta Luna-Herrera1Leopoldo Flores-Romo3Héctor Sumano-López2 Sonia M. Pérez-Tapia1,7
Sonia M. Pérez-Tapia1,7 Sergio Estrada-Parra1
Sergio Estrada-Parra1 Iris Estrada-García1
Iris Estrada-García1 Rommel Chacón-Salinas1,7*
Rommel Chacón-Salinas1,7*
Tuberculosis is one of the leading causes of human morbidity and mortality. Mycobacterium tuberculosis (Mtb) employs different strategies to evade and counterattack immune responses persisting for years. Mast cells are crucial during innate immune responses and help clear infections via inflammation or by direct antibacterial activity through extracellular traps (MCETs). Whether Mtb induce MCETs production is unknown. In this study, we report that viable Mtb did not induce DNA release by mast cells, but heat-killed Mtb (HK-Mtb) did. DNA released by mast cells after stimulation with HK-Mtb was complexed with histone and tryptase. MCETs induced with PMA and HK-Mtb were unable to kill live Mtb bacilli. Mast cells stimulated with HK-Mtb induced hydrogen peroxide production, whereas cells stimulated with viable Mtb did not. Moreover, MCETs induction by HK-Mtb was dependent of NADPH oxidase activity, because its blockade resulted in a diminished DNA release by mast cells. Interestingly, catalase-deficient Mtb induced a significant production of hydrogen peroxide and DNA release by mast cells, indicating that catalase produced by Mtb prevents MCETs release by degrading hydrogen peroxide. Our findings show a new strategy employed by Mtb to overcome the immune response through inhibiting MCETs formation, which could be relevant during early stages of infection.
Mycobacterium tuberculosis (Mtb) is one of the most important pathogens affecting human health worldwide. The World Health Organization estimates that one-third of the human population is infected with this bacterium and approximately 5–10% of infected persons will develop a clinical manifestation of the infection (1).
Mycobacterium tuberculosis is an intracellular bacillus that has acquired different mechanisms to evade the immune response to survive and persist in the host. Mtb gains access to the host through the airways and reaches lung alveoli, where it interacts with different cells of the innate immune response (2). These cells recognize Mtb through different pattern-recognition receptors leading to the activation of different antimicrobial mechanisms (3). Phagocytosis is traditionally considered as one of the first mechanisms used by the host immune response. Macrophages, neutrophils, and dendritic cells have been identified as cells that phagocytose Mtb bacilli; however, elimination of the infection is usually not achieved (4). To this end, Mtb deploy different mechanisms to evade its killing in phagocytic cells, such as inhibiting phagosome maturation (5), interfering with phagosome acidification (6), and scavenging reactive oxygen and/or nitrogen species (7, 8).
Another strategy employed by phagocytic cells to clear infectious agents is through the production of extracellular traps (ETs), consisting of chromatin containing several proteins, commonly derived from intracellular compartments (9). Cells that release ETs following infection include neutrophils, macrophages, eosinophils, basophils, and mast cells (10). These structures have wide antimicrobial activities against many different pathogens including bacteria, protozoa, and fungi (11). Mycobacteria induce ETs formation by neutrophils and macrophages, but curiously, the ETs do not affect bacilli viability (12–14).
Mast cells are particularly abundant in human lungs and are able to detect and respond rapidly to different pathogens (15, 16). In this regard, several studies have shown the importance of mast cells during viral (17), bacterial (18, 19), fungal (20), and protozoan (21) infections. Recognition of bacteria by mast cells leads to release and de novo production of inflammatory mediators that recruit effector cells to control the infectious agent (22). However, mast cells also employ diverse mechanism to regulate bacterial growth, including phagocytosis (23), production of antimicrobial peptides (24), and by the production of ETs (MCETs) (25). In this regard, Mtb is able to activate mast cells in vitro activating degranulation, inducing the production of inflammatory cytokines, and internalizing bacteria through lipid rafts (26, 27). Moreover, mice treated with a potent inducer of mast cell degranulation C48/80 1 day before Mtb infection showed altered cytokine production and increased lung bacterial loads, suggesting the important protective role of mast cells early during Mtb infection (28).
Considering that mast cells are able to exert antimicrobial activity against both extracellular and intracellular bacteria via the release of MCETs (25, 29), here we evaluated whether Mtb induced such structures.
The bacteria employed in this work were Staphylococcus aureus (ATCC 6538), Mycobacterium tuberculosis H37Rv (Mtb), and the katG-deficient Mycobacterium tuberculosis Lehman and Neuman (Mtb KatG−) (ATCC 35822) (30). S. aureus was cultured in tryptic soy broth (Dibico, Mexico), while mycobacteria was growth in Middlebrook 7H9 broth (BD-Difco, USA) supplemented with 10% OADC (BD-Difco, USA) and incubated at 37°C in constant shaking at 150 rpm until exponential phase was reached. S. aureus inocula were prepared in tryptic soy broth and 10% glycerol, while mycobacteria inocula were done in RPMI-1640 Glutamax (Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA). Bacterial inoculums were adjusted to the McFarland nephelometer No.1 standard tube, corresponding to 3 × 108 bacteria/ml. Bacterial viability was determined after serial dilution and plated in tryptic soy agar (S. aureus) or Middlebrook agar (Mtb) and CFU was calculated. Heat-killed Mtb H37Rv (HK-Mtb) was done by incubating Mtb inoculum during 60 min in a water bath at 68°C and bacterial viability was confirmed by seeding in Middlebrook agar.
The mast cell line HMC-1 was kindly donated by Dr. J. H. Butterfield, Mayo Clinic, Rochester, MN, USA (31). Cells were growth in RPMI-1640 Glutamax enriched with 10% FBS at 37°C and 5% CO2. The cell line was validated by STR DNA fingerprinting by the MD Anderson Cancer Center Characterized Cell Line Core using the AmpFLSTR identifier kit according to the manufacturer’s instructions (Applied Biosystems, Thermo Scientific, Rockford, IL, USA). The STR profiles were compared with known ATCC fingerprints,1 to the Cell Line Integrated Molecular Authentication database (CLIMA) version 0.1.200808,2 and to the MD Anderson fingerprint database. The STR profiles matched known DNA fingerprints or were unique.
Bone marrow-derived mast cells (BMMC) were obtained as previously described (32, 33). Briefly, bone marrow from the femurs and tibias of 6- to 10-week-old C57BL/6 mice were disaggregated and cultured at a concentration of 1 × 106 cells/ml of RPMI 1640 supplemented with 10% FBS and 10 ng/ml of murine recombinant IL-3 and SCF (Bio-Legend, USA). Non-adherent cells were transferred to fresh culture medium twice a week for 4–8 weeks. Mast cells purity was >90% according to CD117 and FcεRIα measured by flow cytometry (Figure S1 in Supplementary Material). The protocol for BMMC obtainment from mice was reviewed and approved by the Committee for Ethics in Research ENCB, IPN.
Extracellular DNA was stained as described (12). In brief, cells were seeded at a density of 5 × 105 cells/ml in RPMI-1640 Glutamax + 2% FBS and seeded on glass coverslips pretreated with 0.001% poly-l-lysine (Sigma Aldrich, USA). Cells were stimulated with 10 MOI of Mtb, KatG−, HK-Mtb, or 25 nM of Phorbol 12-Myristate 13-Acetate or PMA (Sigma Aldrich, USA). Afterward, cells were incubated for different times at 37°C and 5% CO2 and fixed with 4% paraformaldehyde (Sigma Aldrich, USA) for 20 min and DNA was stained with 5 µM SYTOX-Green (Molecular Probes, USA). Samples were mounted on a glass slide with Vectashield (Vector Laboratories, USA) and analyzed in a fluorescence microscope Nikon Eclipse E800.
Extracellular DNA was quantified as previously described (29). Briefly, MCETs were induced with the different stimuli in RPMI without phenol red + 2% FBS and incubated with 1 U DNase I (Invitrogen, USA) during 30 min. Cell supernatants were collected and 0.5 µM SYTOX-Green (Molecular Probes, USA) was added. Fluorescence was evaluated immediately in a Fluorometer (Fluoroskan Ascent FL, Thermo Scientific, USA) with a filter setting of 485 nm (excitation)/538 nm (emission). Data are presented as increase in the relative fluorescence units (RFU).
Cells were seeded on glass coverslips treated with 0.001% poly-l-lysine (Sigma Aldrich, USA) and left unstimulated or stimulated with HK-Mtb (MOI 10) or 25 nM PMA for different times. After incubation at 37°C and 5% CO2, cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 (Sigma Aldrich, USA) during 10 min and blocked with universal blocking reagent (BioGenex, USA). Cells were incubated with either anti-histone H3 (Lifespan Biosciences, USA) or anti-tryptase (Abcam, USA) and revealed with secondary antibody labeled with R-phycoerythrin (histone) (Invitrogen, USA) or Alexa Fluor 488 (tryptase) (Invitrogen USA). DNA was stained with 10 µg/ml 4′,6-Diamidino-2-Phenylindole or DAPI (Invitrogen, USA). Samples were mounted with Vectashield and images were obtained in an Axiovert 200 M confocal microscope (Carl Zeiss, Germany) with 63× oil immersion oil objective. Images were collected and processed with Zeiss LSM Image Pascal version 4.0.
To induce MCETs, mast cells were stimulated with 25 nM PMA or with HK-Mtb (MOI 10) for 4 h. They then were carefully washed with HBSS (Life Technologies, USA) and replaced with fresh media containing viable S. aureus or Mtb (MOI 1) and centrifuged at 400 × g for 10 min. The pellet was then incubated at 37°C for 30, 60, 90, 180, and 360 min. After incubation, the bacteria were suspended, the supernatant collected, and CFU was determined. Bacterial survival was determined as percentage in relation to bacteria incubated only in culture media, as previously described (12).
Intracellular hydrogen peroxide was evaluated using a commercial kit (Sigma, USA) following the manufacturer’s instructions. Briefly, 2.5 × 105/100 μl mast cells were plated in a 96-well plate. In some experiments, the cells were pretreated with 0.01 µM diphenyliodonium (DPI) (Sigma, USA) for 30 min to inhibit the NADPH oxidase activity. Cells were left unstimulated, stimulated with 10 MOI of Mtb, KatG−, HK-Mtb, or 25 nM PMA for different times. Data are presented as increase in the RFU.
This study was approved by the Bioethics Committee of Escuela Nacional de Ciencias Biológicas from the Instituto Politécnico Nacional (CEI-ENCB 006/2013).
Data represent the mean ± SD of three independent experiments. Statistical differences between controls and experimental groups were determined using one-way ANOVA followed by the Newman–Keuls method (GraphPad Prism Software V6, USA). A p-value <0.05 was considered statically significant.
Mast cells are activated by Mtb as evinced by their degranulation, the release of pro-inflammatory cytokines and the ability to internalize bacteria (26, 27). However, whether mast cells are able to release MCETs when activated with Mtb is unknown. To this end, we first evaluated whether Mtb was able to induce DNA release from mast cells at different times. We observed that mast cells, either HMC-1 cells or BMMC, stimulated with PMA released DNA 2-h post-activation (Figure 1A). However, when the mast cells were stimulated with viable Mtb, DNA release was not observed until 4 h of stimulation (Figure 1B). Because viable Mtb employ different strategies to avoid the host immune response, we stimulated mast cells with HK-Mtb to determine if this had any effect. Interestingly, mast cells were able to release DNA after 2 h of stimulation with HK-Mtb (Figures 1A,B), suggesting an active mechanism was employed by Mtb to inhibit the release of DNA. These results show that HK-Mtb, but not live Mtb, is able to induce DNA release by mast cells.
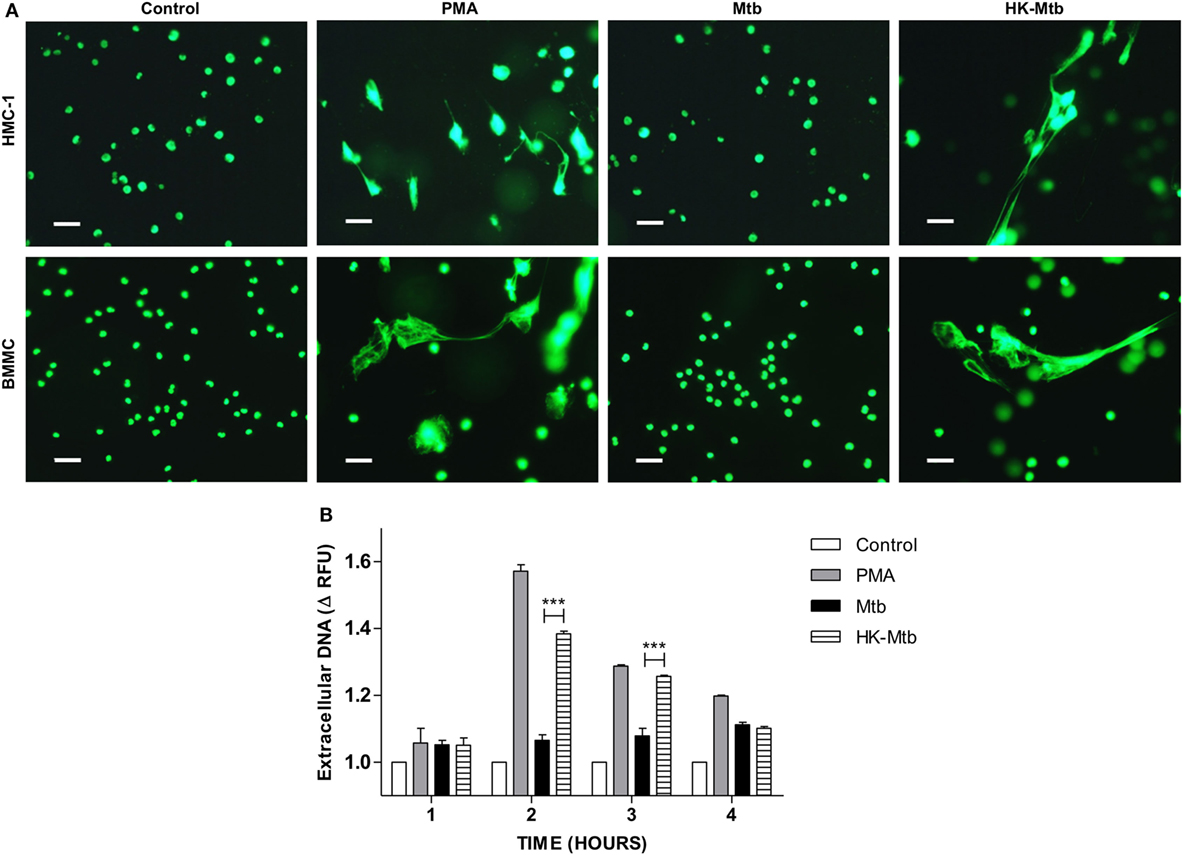
Figure 1. Mast cells release DNA in response to heat-killed but not live Mycobacterium tuberculosis (Mtb). (A) Representative micrographs of HMC-1 cells or bone marrow-derived mast cells (BMMC) unstimulated (control) or stimulated during 2 h with PMA, live Mtb, or heat-killed Mtb (HK-Mtb) at a MOI of 10. DNA was visualized after staining with SYTOX-Green. Scale bar 20 µm (magnification 400×). (B) Released DNA was quantified in unstimulated HMC-1 cells (control) or stimulated at indicated times with PMA, live Mtb, or HK-Mtb at a MOI of 10. Released DNA was partially digested with DNase I and quantified in supernatants with SYTOX Green I in a fluorometer. The graph represents the change in fluorescence ± SD of stimulated cells compared to control. ***p < 0.001 as indicated.
MCETs formation imply the combination of granular proteins of mast cells with nuclear DNA and then its release to the extracellular environment (25). When we analyzed by confocal microscopy the structures released by HK-Mtb-treated mast cells, we observed that extracellular DNA contained both tryptase and histone (Figure 2), indicating that the DNA released by HK-Mtb have components classically found in MCETs.
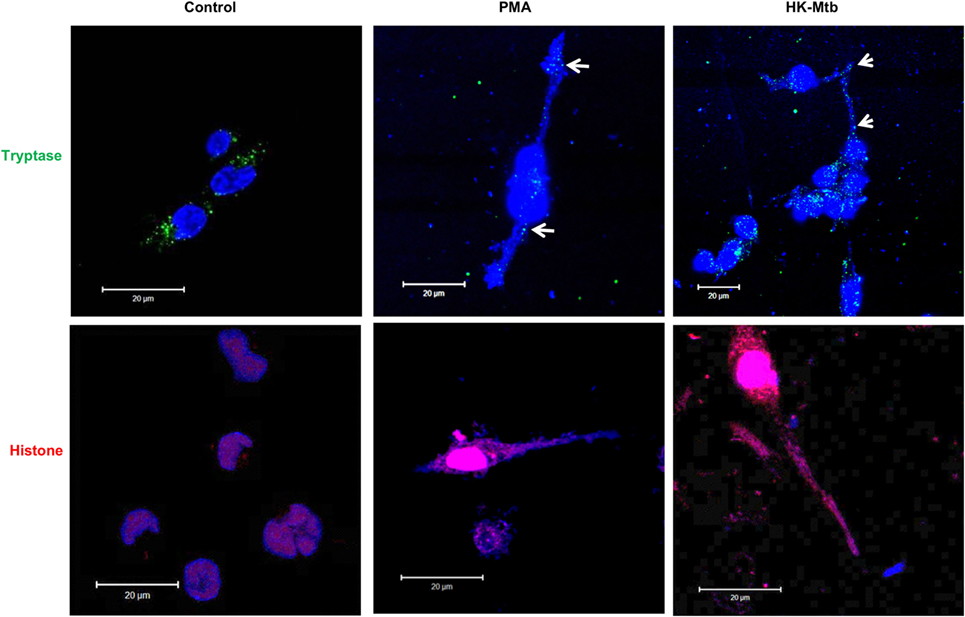
Figure 2. MCETs induced by heat-killed Mtb (HK-Mtb) contain both tryptase and histone. Representative micrographs of unstimulated HMC-1 mast cells (control) or stimulated for 2 h with PMA or HK-Mtb at a MOI of 10. Mast cell tryptase is shown in green, while histone is shown in red and DNA in blue. White arrows indicate DNA zones that showed co-localization with mast cell tryptase. Scale bar 20 µm.
The main function of MCETs is to ensnare and induce the killing of the entrapped pathogen. Because different signals on mast cells have an impact on the presence of different microbicidal molecules in MCETs (34), we evaluated the ability of induced MCETs with PMA and HK-Mtb to exert antimicrobial activity against viable Mtb. As a control we employed S. aureus, because it was previously observed that MCETs showed antimicrobial activity against this bacterium (35). We found that MCETs were able to kill S. aureus, but not Mtb, independently if MCETs were induced with PMA or HK-Mtb (Figures 3A,B). These findings indicate that MCETs induced with different stimuli are unable to exert antimicrobial activity against this bacterium.
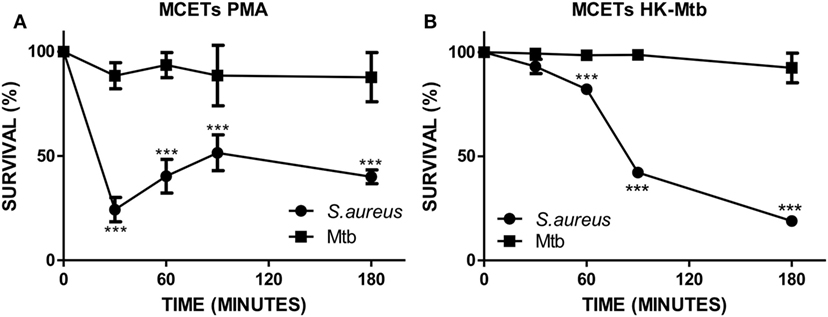
Figure 3. Mast cell extracellular traps do not kill Mycobacterium tuberculosis (Mtb). (A) HMC-1 cells were activated with PMA or (B) heat-killed Mtb (HK-Mtb) to induce MCETs, then Mtb or Staphylococcus aureus were added at a MOI of 1 for 3 h Bacteria survival was evaluated by CFU. ***p < 0.001 compared with the initial time.
Reactive oxygen species (ROS) generated by NADPH oxidase is a crucial step in the induction of MCETs. In particular, the generation of hydrogen peroxide (H2O2) is considered as a trigger for the release of MCETs (25). Because we previously observed that only HK-Mtb was able to induce MCETs, we evaluated the intracellular production of H2O2 by live Mtb and HK-Mtb. We noticed that mast cells stimulated with HK-Mtb induced a significant production of intracellular H2O2 90 min after stimulation, comparable to that induced by PMA (Figure 4). However, live Mtb was unable to induce a significant amount of H2O2 at this time (Figure 4). To evaluate whether H2O2 produced by NADPH oxidase was necessary for the production of MCETs by HK-Mtb we blocked the enzyme activity with DPI. Inhibition of NADPH oxidase activity with DPI significantly diminished H2O2 production by HK-Mtb and PMA (Figure 5A), and also diminished DNA release by mast cells when compared to control cells stimulated in the absence of DPI (Figure 5B). These results show that H2O2 produced during the respiratory burst, initiated by NADPH oxidase, is critical for the production of MCETs by HK-Mtb. Moreover, our data suggest that the reduced production of H2O2 is associated with a poor induction of MCETs.
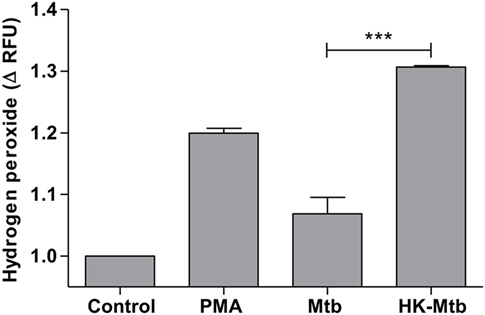
Figure 4. Mast cells stimulated with Mycobacterium tuberculosis (Mtb) shows low levels of hydrogen peroxide. HMC-1 cells were left unstimulated (control) or stimulated with PMA, live Mtb, or heat-killed Mtb (HK-Mtb) for 90 min and hydrogen peroxide was evaluated. The graph represents the change in fluorescence ± SD of stimulated cells compared to unstimulated cells. ***p < 0.001 as indicated.
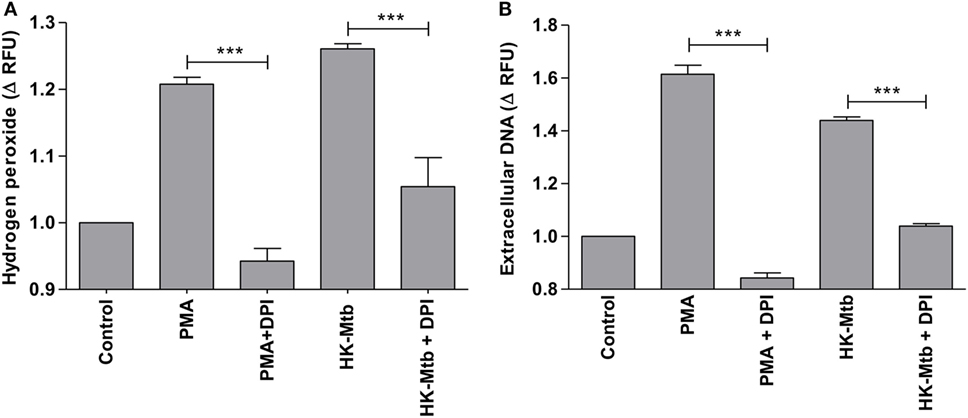
Figure 5. Induction of MCETs by heat-killed Mtb (HK-Mtb) depends on NADPH oxidase. (A) Hydrogen peroxide was evaluated in HMC-1 mast cells that were left unstimulated (control) or stimulated for 90 min with PMA or HK-Mtb. A group of cells was incubated with the NADPH oxidase inhibitor, diphenyliodonium (DPI), previous to activation. The graph represents the change in fluorescence ± SD of stimulated cells compared to unstimulated cells. ***p < 0.001 as indicated. (B) Extracellular DNA was evaluated in HMC-1 mast cells after 2 h of stimulation with PMA or HK-Mtb in the presence or absence of DPI. The graph represents the change in fluorescence ± SD of stimulated cells compared to the control. ***p < 0.001 as indicated.
Mycobacterium tuberculosis overrides ROS produced by cells of the immune system through several ways, one of which implies the presence of genes that code for proteins with catalase activity. It is well known that Mtb strains that develop resistance to isoniazid have deletions in katG gene that codes for an enzyme with catalase activity (36). Therefore, we evaluated whether a katG-deficient Mtb strain (Mtb katG−) was able to induce a significant generation of H2O2 in mast cells. We observed that mast cells stimulated with Mtb katG− had increased levels of H2O2, comparable to that induced by HK-Mtb and PMA (Figure 6A), and significantly higher than that produced by katG+ live Mtb strain (p < 0.001). Next, we evaluated the ability of Mtb katG− to induce MCETs production. We observed that mast cells stimulated with Mtb katG− induced a significant release of DNA, similar to that induced by HK-Mtb or PMA, but significantly higher than that promoted by katG+ live Mtb (Figures 6B,C). Taken as a whole, our results indicate that Mtb inhibits the release of MCETs through the production of mycobacterial catalase that activates the decomposition of hydrogen peroxide.
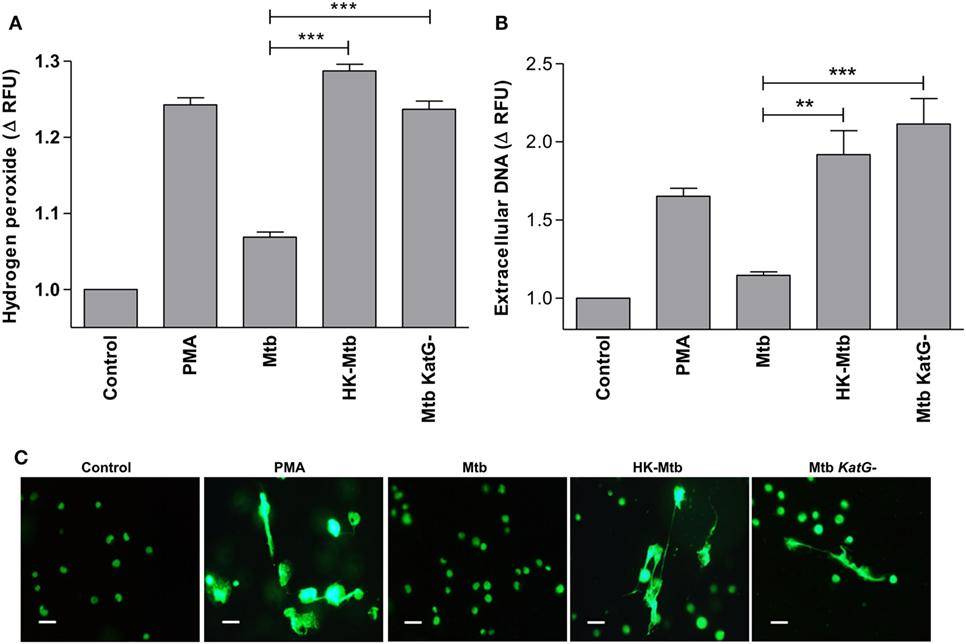
Figure 6. Catalase deficient Mycobacterium tuberculosis (Mtb KatG-) induce release of MCETs. (A) HMC-1 cells were left unstimulated (Control) or stimulated with PMA, live Mtb, heat-killed Mtb (HK-Mtb), or with a Mtb katG-deleted strain (Mtb KatG−) for 90 min and H2O2 was evaluated. The graph represents the change in fluorescence ± SD of stimulated cells compared to unstimulated cells ***p < 0.001 as indicated. (B) Extracellular DNA was evaluated in HMC-1 mast cells after 2 h of stimulation with PMA, live Mtb, HK-Mtb, or Mtb katG−. The graph represents the change in fluorescence ± SD of stimulated cells compared to the control. ***p < 0.001, **p < 0.01 as indicated. (C) Representative micrographs of HMC-1 cells unstimulated (Control) or stimulated during 2 h with PMA, Mtb, HK-Mtb or Mtb KatG− at a MOI of 10. DNA was visualized after staining with SYTOX-Green. Scale bar 20 µm (magnification 400×).
Mast cells develop different functions during an immune response. They play a role in induction of immune regulation, allergy, and can both positively and negatively affect cancer survival, depending upon the type of cancer (16, 32, 33, 37). Of course, one of the main functions of mast cells is to initiate an immune response to different pathogens that surpass the epithelial barrier (22). In this regard, several mechanisms are employed by mast cells to control pathogens, including the induction of a rapid inflammatory response and the development of different antimicrobial activities, such as the production of MCETs. Several pathogens have been identified to induce these structures, including extracellular and intracellular bacteria, fungi, and protozoa (25, 29, 38–40). In this work, we show that Mtb inhibits the release of MCETs in relation to that induced by PMA and other intracellular bacteria, such as Listeria monocytogenes (29). Our findings indicate that mycobacterial catalase encoded by katG plays an essential role in the ability of this organism to evade the immune response through the decomposition of hydrogen peroxide that is an essential trigger for MCETs induction (25). To the best of our knowledge, this represents a novel, so far unrecognized strategy employed by pathogens to override ETs.
One of the classical mechanisms identified to evade ETs is by the presence of pathogen-derived nucleases, which dismantle the DNA backbone of ETs. This phenomenon was observed during the infection with S. aureus, whose nuclease is able to degrade neutrophil extracellular traps (NETs), generating deoxyadenosine that exert toxic effects in infiltrating macrophages (41). A different identified mechanism is by interfering with the activation of the immune cells that produce ETs. For instance, Group A Streptococcus expresses a high-molecular weight hyaluronan that engages Siglec-9 in human neutrophils that blocks the cell signaling that leads to ROS generation and NETs formation (42). On the other hand, Acinetobacter baumannii inhibits NET formation by altering neutrophil adhesion by reducing CD11a expression on neutrophils (43), while the capsular polysaccharides glucoronoxylomanan from Cryptococcus neoformans inhibit NET production by blocking ROS production (44).
Hydrogen peroxide is an essential molecule in the formation of MCETs (25), and several pathogens promote the degradation of this molecule through the presence of genes that code for enzymes with catalase activity (45). In this work, we observed that HK-Mtb, but not viable Mtb, was able to induce the release of MCETs. Moreover, we noticed that HK-Mtb induced significant levels of hydrogen peroxide, that were diminished in mast cells stimulated with live Mtb, indicating that live Mtb targeted production of this molecule. We decided to evaluate if mycobacterial katG was involved in this blockade because of two reasons: (1) it codes for an enzyme with the ability to decompose hydrogen peroxide into water and molecular oxygen and (2) the deletion of this gene usually occur in Mtb strains that develop resistance to isoniazid, but are less virulent in guinea pig model of infection (36). Our findings indicate that mast cells stimulated with live Mtb, in which katG was deleted, showed an increased accumulation of hydrogen peroxide and were able to induce the release of MCETs. These results show that katG helps mycobacteria to override MCETs production by depressing mast cell production of hydrogen peroxide, thus suppressing a mechanism that triggers MCETs formation.
Because Mtb inhibited MCETs formation it seems logical that this is one mechanism by which Mtb avoids the antibacterial activity of such structures. To test this hypothesis, we induced MCETs with PMA or HK-Mtb and incubated them with viable Mtb to evaluate their antibacterial activity. We noticed that although such MCETs were able to exert antimicrobial activity against S. aureus, they did not affect mycobacterial viability. The resistance of Mtb to ETs is also observed with those released by neutrophils and macrophages (12, 13). This is an interesting observation because although the main effect associated with antimicrobial activity in ETs is related to the presence of antimicrobial peptides, the presence of specific granule enzymes also contributes to antimicrobial activity (29), and Mtb is resistant to the antimicrobial activity of the different components present in the different ETs of macrophages, neutrophils, and mast cells. This is probably due to the particular characteristics of the cell wall of Mtb, although the components that are implied in this process need to be clarified. In this regard, resistance to the antimicrobial activity is described in other pathogens such as Streptococcus pneumoniae that alters lipoteichoic acid by the incorporation of d-alanine rendering resistance to the bactericidal activity of NETs, while Cryptococcus gattii produce extracellular fibrils that render resistance to NETs (46, 47).
A second possibility is that Mtb blocks MCETs formation to avoid the death of mast cells and that these cells exert an inflammatory process that favors infection. It is well known that the inflammatory process during the early phase of mycobacterial infection is crucial for the recruitment of macrophages that are susceptible to the infection and promote the spread to other tissues (48). On the other hand, mast cells release preformed inflammatory mediators and induce the production of pro-inflammatory cytokines when stimulated with Mtb (26). However, further work is needed to clarify the role of the blockade of MCETs production during Mtb infection.
Finally, one limitation of our work is that although we used two different sources of mast cells with different levels of maturity, we did not employ a completely mature mast cell that represent the phenotype of lung mast cells. It is well known that mast cells have a great heterogeneity in their phenotype and function depending on the tissue they reside (49), so further work is needed to clarify this issue.
In conclusion, our work unravels a previously unrecognized mechanism employed by Mtb to override the immune response through the inhibition of MCETs production, employing katG to decompose hydrogen peroxide. We hypothesize that MCETs blockade by this bacterium could be relevant during the early phase of the infection.
This study was approved by the Bioethics Committee of Escuela Nacional de Ciencias Biológicas from the Instituto Politécnico Nacional (CEI-ENCB 006/2013).
MC-N, KL-P, LD-M, GR-L, and RS-C performed experiments and analyzed data. MC-N, KL-P, LD-M, BG-P, LF-R, SE-P, IE-G, and RC-S analyzed and interpreted data. MC-N, NP-O, SU, JL-H, LF-R, HS-L, SP-T, and RC-S interpreted data, drafted the manuscript, and contributed with intellectual content. RC-S designed, supervised the study, and obtained funding. All the authors critically revised and approved the final version of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by grants from Consejo Nacional de Ciencia y Tecnología SEP-CONACYT 157100 to RC-S, and from Secretaría de Investigación y Posgrado, Instituto Politécnico Nacional (SIP-IPN), México.
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01161/full#supplementary-material.
Figure S1. Bone marrow-derived mast cell purity. (A) Representative flow cytometry density plot of bone marrow cells after 6 weeks of culture with IL-3 and SCF. The percentage of FcεRI+CD117+ cells is shown. (B) BMMC were stained with toluidine blue and analyzed by light microscopy (magnification 1,000×).
2. Campillo-Navarro M, Wong-Baeza I, Serafín-López J, Hernández-Pando R, Estrada-Parra S, Estrada-García I, et al. Regulation of the immune response by Mycobacterium tuberculosis Beijing genotype. In: Ribón W, editor. Tuberculosis – Expanding Knowledge. London, UK: InTech (2015). p. 65–88.
3. Mishra A, Akhtar S, Jagannath C, Khan A. Pattern recognition receptors and coordinated cellular pathways involved in tuberculosis immunopathogenesis: emerging concepts and perspectives. Mol Immunol (2017) 87:240–8. doi:10.1016/j.molimm.2017.05.001
4. Cadena AM, Flynn JL, Fortune SM. The importance of first impressions: early events in Mycobacterium tuberculosis infection influence outcome. MBio (2016) 7(2):e342–316. doi:10.1128/mBio.00342-16
5. Armstrong JA, Hart PD. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J Exp Med (1971) 134(3 Pt 1):713–40. doi:10.1084/jem.134.3.713
6. Sturgill-Koszycki S, Schlesinger PH, Chakraborty P, Haddix PL, Collins HL, Fok AK, et al. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science (1994) 263(5147):678–81. doi:10.1126/science.8303277
7. Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science (2003) 302(5652):1963–6. doi:10.1126/science.1091176
8. Ng VH, Cox JS, Sousa AO, MacMicking JD, McKinney JD. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol Microbiol (2004) 52(5):1291–302. doi:10.1111/j.1365-2958.2004.04078.x
9. Stoiber W, Obermayer A, Steinbacher P, Krautgartner WD. The role of reactive oxygen species (ROS) in the formation of extracellular traps (ETs) in humans. Biomolecules (2015) 5(2):702–23. doi:10.3390/biom5020702
10. Mollerherm H, von Kockritz-Blickwede M, Branitzki-Heinemann K. Antimicrobial activity of mast cells: role and relevance of extracellular DNA traps. Front Immunol (2016) 7:265. doi:10.3389/fimmu.2016.00265
11. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol (2017) 18(2):134–47. doi:10.1038/nri.2017.105
12. Ramos-Kichik V, Mondragon-Flores R, Mondragon-Castelan M, Gonzalez-Pozos S, Muniz-Hernandez S, Rojas-Espinosa O, et al. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (Edinb) (2009) 89(1):29–37. doi:10.1016/j.tube.2008.09.009
13. Je S, Quan H, Yoon Y, Na Y, Kim BJ, Seok SH. Mycobacterium massiliense induces macrophage extracellular traps with facilitating bacterial growth. PLoS One (2016) 11(5):e0155685. doi:10.1371/journal.pone.0155685
14. Wong KW, Jacobs WR Jr. Mycobacterium tuberculosis exploits human interferon gamma to stimulate macrophage extracellular trap formation and necrosis. J Infect Dis (2013) 208(1):109–19. doi:10.1093/infdis/jit097
15. Andersson CK, Mori M, Bjermer L, Lofdahl CG, Erjefalt JS. Novel site-specific mast cell subpopulations in the human lung. Thorax (2009) 64(4):297–305. doi:10.1136/thx.2008.101683
16. Campillo-Navarro M, Chavez-Blanco AD, Wong-Baeza I, Serafin-Lopez J, Flores-Mejia R, Estrada-Parra S, et al. Mast cells in lung homeostasis: beyond type I hypersensitivity. Curr Respir Med Rev (2014) 10(2):115–23. doi:10.2174/1573398X10666141024220151
17. Graham AC, Hilmer KM, Zickovich JM, Obar JJ. Inflammatory response of mast cells during influenza A virus infection is mediated by active infection and RIG-I signaling. J Immunol (2013) 190(9):4676–84. doi:10.4049/jimmunol.1202096
18. Malaviya R, Ikeda T, Ross E, Abraham SN. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-alpha. Nature (1996) 381(6577):77–80. doi:10.1038/381077a0
19. Chiba N, Shimada K, Chen S, Jones HD, Alsabeh R, Slepenkin AV, et al. Mast cells play an important role in Chlamydia pneumoniae lung infection by facilitating immune cell recruitment into the airway. J Immunol (2015) 194(8):3840–51. doi:10.4049/jimmunol.1402685
20. Nieto-Patlan A, Campillo-Navarro M, Rodriguez-Cortes O, Munoz-Cruz S, Wong-Baeza I, Estrada-Parra S, et al. Recognition of Candida albicans by Dectin-1 induces mast cell activation. Immunobiology (2015) 220(9):1093–100. doi:10.1016/j.imbio.2015.05.005
21. Cruz A, Mendes EA, de Andrade MV, do Nascimento VC, Cartelle CT, Arantes RM, et al. Mast cells are crucial in the resistance against Toxoplasma gondii oral infection. Eur J Immunol (2014) 44(10):2949–54. doi:10.1002/eji.201344185
22. St John AL, Abraham SN. Innate immunity and its regulation by mast cells. J Immunol (2013) 190(9):4458–63. doi:10.4049/jimmunol.1203420
23. Malaviya R, Ross EA, MacGregor JI, Ikeda T, Little JR, Jakschik BA, et al. Mast cell phagocytosis of FimH-expressing enterobacteria. J Immunol (1994) 152(4):1907–14.
24. Di Nardo A, Yamasaki K, Dorschner RA, Lai Y, Gallo RL. Mast cell cathelicidin antimicrobial peptide prevents invasive group A Streptococcus infection of the skin. J Immunol (2008) 180(11):7565–73. doi:10.4049/jimmunol.180.11.7565
25. von Kockritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, et al. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood (2008) 111(6):3070–80. doi:10.1182/blood-2007-07-104018
26. Munoz S, Hernandez-Pando R, Abraham SN, Enciso JA. Mast cell activation by Mycobacterium tuberculosis: mediator release and role of CD48. J Immunol (2003) 170(11):5590–6. doi:10.4049/jimmunol.170.11.5590
27. Munoz S, Rivas-Santiago B, Enciso JA. Mycobacterium tuberculosis entry into mast cells through cholesterol-rich membrane microdomains. Scand J Immunol (2009) 70(3):256–63. doi:10.1111/j.1365-3083.2009.02295.x
28. Carlos D, de Souza Junior DA, de Paula L, Jamur MC, Oliver C, Ramos SG, et al. Mast cells modulate pulmonary acute inflammation and host defense in a murine model of tuberculosis. J Infect Dis (2007) 196(9):1361–8. doi:10.1086/521830
29. Campillo-Navarro M, Leyva-Paredes K, Donis-Maturano L, Gonzalez-Jimenez M, Paredes-Vivas Y, Cerbulo-Vazquez A, et al. Listeria monocytogenes induces mast cell extracellular traps. Immunobiology (2017) 222(2):432–9. doi:10.1016/j.imbio.2016.08.006
30. Rouse DA, Morris SL. Molecular mechanisms of isoniazid resistance in Mycobacterium tuberculosis and Mycobacterium bovis. Infect Immun (1995) 63(4):1427–33.
31. Butterfield JH, Weiler DA, Hunt LW, Wynn SR, Roche PC. Purification of tryptase from a human mast cell line. J Leukoc Biol (1990) 47(5):409–19. doi:10.1002/jlb.47.5.409
32. Chacon-Salinas R, Chen L, Chavez-Blanco AD, Limon-Flores AY, Ma Y, Ullrich SE. An essential role for platelet-activating factor in activating mast cell migration following ultraviolet irradiation. J Leukoc Biol (2014) 95(1):139–48. doi:10.1189/jlb.0811409
33. Chacon-Salinas R, Limon-Flores AY, Chavez-Blanco AD, Gonzalez-Estrada A, Ullrich SE. Mast cell-derived IL-10 suppresses germinal center formation by affecting T follicular helper cell function. J Immunol (2011) 186(1):25–31. doi:10.4049/jimmunol.1001657
34. Garcia-Rodriguez KM, Goenka A, Alonso-Rasgado MT, Hernandez-Pando R, Bulfone-Paus S. The role of mast cells in tuberculosis: orchestrating innate immune crosstalk? Front Immunol (2017) 8:1290. doi:10.3389/fimmu.2017.01290
35. Abel J, Goldmann O, Ziegler C, Holtje C, Smeltzer MS, Cheung AL, et al. Staphylococcus aureus evades the extracellular antimicrobial activity of mast cells by promoting its own uptake. J Innate Immun (2011) 3(5):495–507. doi:10.1159/000327714
36. Zhang Y, Heym B, Allen B, Young D, Cole S. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature (1992) 358(6387):591–3. doi:10.1038/358591a0
37. Limon-Flores AY, Chacon-Salinas R, Ramos G, Ullrich SE. Mast cells mediate the immune suppression induced by dermal exposure to JP-8 jet fuel. Toxicol Sci (2009) 112(1):144–52. doi:10.1093/toxsci/kfp181
38. Scheb-Wetzel M, Rohde M, Bravo A, Goldmann O. New insights into the antimicrobial effect of mast cells against Enterococcus faecalis. Infect Immun (2014) 82(11):4496–507. doi:10.1128/IAI.02114-14
39. Lopes JP, Stylianou M, Nilsson G, Urban CF. Opportunistic pathogen Candida albicans elicits a temporal response in primary human mast cells. Sci Rep (2015) 5:12287. doi:10.1038/srep12287
40. Naqvi N, Ahuja K, Selvapandiyan A, Dey R, Nakhasi H, Puri N. Role of mast cells in clearance of Leishmania through extracellular trap formation. Sci Rep (2017) 7(1):13240. doi:10.1038/s41598-017-12753-1
41. Thammavongsa V, Missiakas DM, Schneewind O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science (2013) 342(6160):863–6. doi:10.1126/science.1242255
42. Secundino I, Lizcano A, Roupe KM, Wang X, Cole JN, Olson J, et al. Host and pathogen hyaluronan signal through human siglec-9 to suppress neutrophil activation. J Mol Med (Berl) (2016) 94(2):219–33. doi:10.1007/s00109-015-1341-8
43. Kamoshida G, Kikuchi-Ueda T, Nishida S, Tansho-Nagakawa S, Ubagai T, Ono Y. Pathogenic bacterium Acinetobacter baumannii inhibits the formation of neutrophil extracellular traps by suppressing neutrophil adhesion. Front Immunol (2018) 9:178. doi:10.3389/fimmu.2018.00178
44. Rocha JD, Nascimento MT, Decote-Ricardo D, Corte-Real S, Morrot A, Heise N, et al. Capsular polysaccharides from Cryptococcus neoformans modulate production of neutrophil extracellular traps (NETs) by human neutrophils. Sci Rep (2015) 5:8008. doi:10.1038/srep08008
45. Eason MM, Fan X. The role and regulation of catalase in respiratory tract opportunistic bacterial pathogens. Microb Pathog (2014) 74:50–8. doi:10.1016/j.micpath.2014.07.002
46. Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A, Normark S, et al. Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol (2007) 9(5):1162–71. doi:10.1111/j.1462-5822.2006.00857.x
47. Springer DJ, Ren P, Raina R, Dong Y, Behr MJ, McEwen BF, et al. Extracellular fibrils of pathogenic yeast Cryptococcus gattii are important for ecological niche, murine virulence and human neutrophil interactions. PLoS One (2010) 5(6):e10978. doi:10.1371/journal.pone.0010978
48. Davis JM, Ramakrishnan L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell (2009) 136(1):37–49. doi:10.1016/j.cell.2008.11.014
Keywords: tuberculosis, Mycobacterium tuberculosis, mast cell, mast cell extracellular trap, catalase
Citation: Campillo-Navarro M, Leyva-Paredes K, Donis-Maturano L, Rodríguez-López GM, Soria-Castro R, García-Pérez BE, Puebla-Osorio N, Ullrich SE, Luna-Herrera J, Flores-Romo L, Sumano-López H, Pérez-Tapia SM, Estrada-Parra S, Estrada-García I and Chacón-Salinas R (2018) Mycobacterium tuberculosis Catalase Inhibits the Formation of Mast Cell Extracellular Traps. Front. Immunol. 9:1161. doi: 10.3389/fimmu.2018.01161
Received: 10 February 2018; Accepted: 09 May 2018;
Published: 28 May 2018
Edited by:
Jesús Gonzalo-Asensio, Universidad de Zaragoza, SpainReviewed by:
Javier Rangel-Moreno, University of Rochester, United StatesCopyright: © 2018 Campillo-Navarro, Leyva-Paredes, Donis-Maturano, Rodríguez-López, Soria-Castro, García-Pérez, Puebla-Osorio, Ullrich, Luna-Herrera, Flores-Romo, Sumano-López, Pérez-Tapia, Estrada-Parra, Estrada-García and Chacón-Salinas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rommel Chacón-Salinas, cm9tbWVsY2hhY29uc0B5YWhvby5jb20ubXg=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.