 Sylvain Latour1,2,3*
Sylvain Latour1,2,3* Sarah Winter1,2,3
Sarah Winter1,2,3
- 1Laboratory of Lymphocyte Activation and Susceptibility to EBV infection, INSERM UMR 1163, Paris, France
- 2Imagine Institute, Paris Descartes University, Sorbonne Paris Cité, Paris, France
- 3Equipe de Recherche Labéllisée, Ligue National contre le Cancer, Paris, France
Epstein–Barr Virus (EBV) is a gamma-herpes virus that infects 90% of humans without any symptoms in most cases, but has an oncogenic potential, especially in immunocompromised individuals. In the past 30 years, several primary immunodeficiencies (PIDs) associated with a high risk to develop EBV-associated lymphoproliferative disorders (LPDs), essentially consisting of virus-associated hemophagocytic syndrome, non-malignant and malignant B-cell LPDs including non-Hodgkin and Hodgkin’s types of B lymphomas have been characterized. Among them are SH2D1A (SAP), XIAP, ITK, MAGT1, CD27, CD70, CTPS1, RASGRP1, and CORO1A deficiencies. Penetrance of EBV infection ranges from 50 to 100% in those PIDs. Description of large cohorts and case reports has refined the specific phenotypes associated with these PIDs helping to the diagnosis. Specific pathways required for protective immunity to EBV have emerged from studies of these PIDs. SLAM-associated protein-dependent SLAM receptors and MAGT1-dependent NKG2D pathways are important for T and NK-cell cytotoxicity toward EBV-infected B-cells, while CD27–CD70 interactions are critical to drive the expansion of EBV-specific T-cells. CTPS1 and RASGRP1 deficiencies further strengthen that T-lymphocyte expansion is a key step in the immune response to EBV. These pathways appear to be also important for the anti-tumoral immune surveillance of abnormal B cells. Monogenic PIDs should be thus considered in case of any EBV-associated LPDs.
The Epstein–Barr virus (EBV) is a gamma-herpes family virus that infects most of humans with a marked tropism for B lymphocytes. EBV is an oncogenic virus known to be the causative agent of several types of neoplasms, including B, T, and NK cell lymphomas, nasopharyngeal and gastric carcinomas, and Epstein–Barr virus-associated smooth muscle tumors (EBV-SMT). 110,000–200,000 cancer cases per year are attributable to EBV worldwide (1).
The reservoir of EBV is strictly human. After the age of 35 years, the incidence of the infection in the general population is more than 90%. The primary infection occurs in the oropharynx where EBV virions infect epithelial cells and B lymphocytes via the CD21 molecule. During the primary infection, EBV drives the activation and the expansion of latently infected B lymphoblasts (2, 3). These proliferating B cells express EBV latent growth-transforming genes that establish EBV persistence (latency III program) and are mainly eliminated by specific CD8+ T cells that strongly expand during the immune response. Innate cytotoxic lymphocytes like NK cells, γδ T cells, and iNKT cells, specifically early differentiated KIR-negative NK cells and Vγ9Vδ2 T cells, are also thought to play an important role in the early phase of the primary infection by recognition of lytically and latently EBV-replicating cells, respectively (2, 4, 5). Some EBV-infected B cells escape to the immune response by downregulating latent genes expression (latency 0 program) and acquire a memory phenotype, becoming invisible to the immune system and establishing a reservoir for EBV. Subsequent stimulations of these EBV-containing “reservoir” memory B cells will lead to reactivation of EBV from latency into the lytic cycle, thus promoting infections of new B cells and their expansion. Ultimately, EBV-transformed lymphoblasts can lead to lymphoma. In some very rare cases, EBV can also infect T cells and NK cells. This peculiar profile of infection is rather observed in Asian and South American populations and is associated with a chronic viremia, infiltration of organs with by EBV-positive lymphocytes, and life-threatening lymphoproliferative disorders (LPDs) including hemophagocytic syndrome or/and EBV-positive T/NK cell lymphoma. The mechanisms underlying the pathogenesis of this infection are not clearly known, as well as its genetic determinants that are thought to be oligogenic or polygenic (6, 7). This unusual EBV infection will not be covered in this review.
The first encounter with EBV usually happens during infancy and adolescence by oral transmission and is largely asymptomatic. However, in some immunocompetent individuals particularly during adolescence, primary infection causes infectious mononucleosis (IM), a self-limiting lymphoproliferative disease clinically characterized by fever, sore throat, body aches, swollen lymph nodes, and general fatigue (3). The lymphoproliferation consists of a robust and sustained expansion of CD8+ T cells and infected B cells reflecting a strong immune response to the virus. Notably, CD8+ EBV-specific T cells can represent more than 40% of circulating T cells in some subjects (8).
In immunocompromised individuals, reactivations of EBV and persistence of proliferating latent growth-transforming EBV-infected B cells are associated with severe pathologies that can have fatal outcome. Those include hemophagocytic lymphohistiocytosis (HLH), also termed virus-associated hemophagocytic syndrome, non-malignant B-cell LPDs, and B-cell lymphomas including Hodgkin’s lymphomas and non-Hodgkin’s lymphomas such as Burkitt’s lymphoma and diffuse large B-cell lymphoma (DLBCL) (1). Such disorders defined as posttransplant lymproliferative disorders are often observed in patients with organ transplantation under immunosuppressive treatment. Similarly, HIV-infected patients with acquired immunodeficiency syndrome (AIDS) often experience lymphoproliferation disorders driven by EBV, that represent one of the most frequent cause of death in patients with AIDS (9). Those observations highlight that reactivations of EBV from latently EBV-infected B cells occur frequently in normal individuals throughout life and need to be tightly controlled by the adaptative immune response.
Beside acquired forms, several inherited combined immunodeficiencies (CIDs) leading to a particular susceptibility to EBV infection and to develop EBV-driven diseases have been identified over the last 20 years (10–12). Those genetic defects include mutations in SH2D1A, ITK, MAGT1, CTPS1, CD27, CD70, CORO1A, and RASGRP1 (Table 1). In these genetically determined forms, the penetrance of the EBV susceptibility is high with more than 50% patients having presented at least one severe episode of EBV-driven LPD including Hodgkin and non-Hodgkin lymphomas (Table 2). However, a number of carriers of these gene defects can also experience other severe viral infections caused by CMV, VZV, HSV, HHV-6, or HPV. This is particularly true for CTPS1 and CORO1A deficiencies since patients often presented VZV and HPV infections, respectively. Bacterial infections, in particular recurrent lung infections are noticed in a number of patients and can be the initial clinical presentation. This may be related to the hypogammaglobulinemia and/or dysgammaglobulinemia associated with low number of CD27+ memory B cells that are frequently observed in these defects. These phenotypes likely result or have been proven to result from abnormalities in T-cell help to B cells due to defects in T-helper cell maturation and/or activation. Intrinsic defects in B cell development and function may also contribute directly to the hypo/dysgammaglobulinemia, like in RASGRP1, CTPS1, CD27 deficiencies, for which there is clear evidence that these genes are directly involved in B-cell development and/or function. However, these B-cell defects do not to interfere with the ability to EBV to infect B cells and its oncogenic activity. CD4+ T cell lymphopenia and/or decrease naïve T cells is also a frequent phenotype, which can be considered as an immunological hallmark of a T-cell defect (13, 14). NK cells and iNKT cells are also often found to be diminished or absent. Finally, several of these deficiencies are associated with very unique features, independently of the high risk to develop EBV-driven lymphoproliferations, helping to the diagnosis, such as autoimmunity, inflammatory bowel disease (IBD), lung involvement, and neurological disorders that develop in RASGRP1, XIAP-, ITK-, and CORO1A-deficiency, respectively (Table 1).
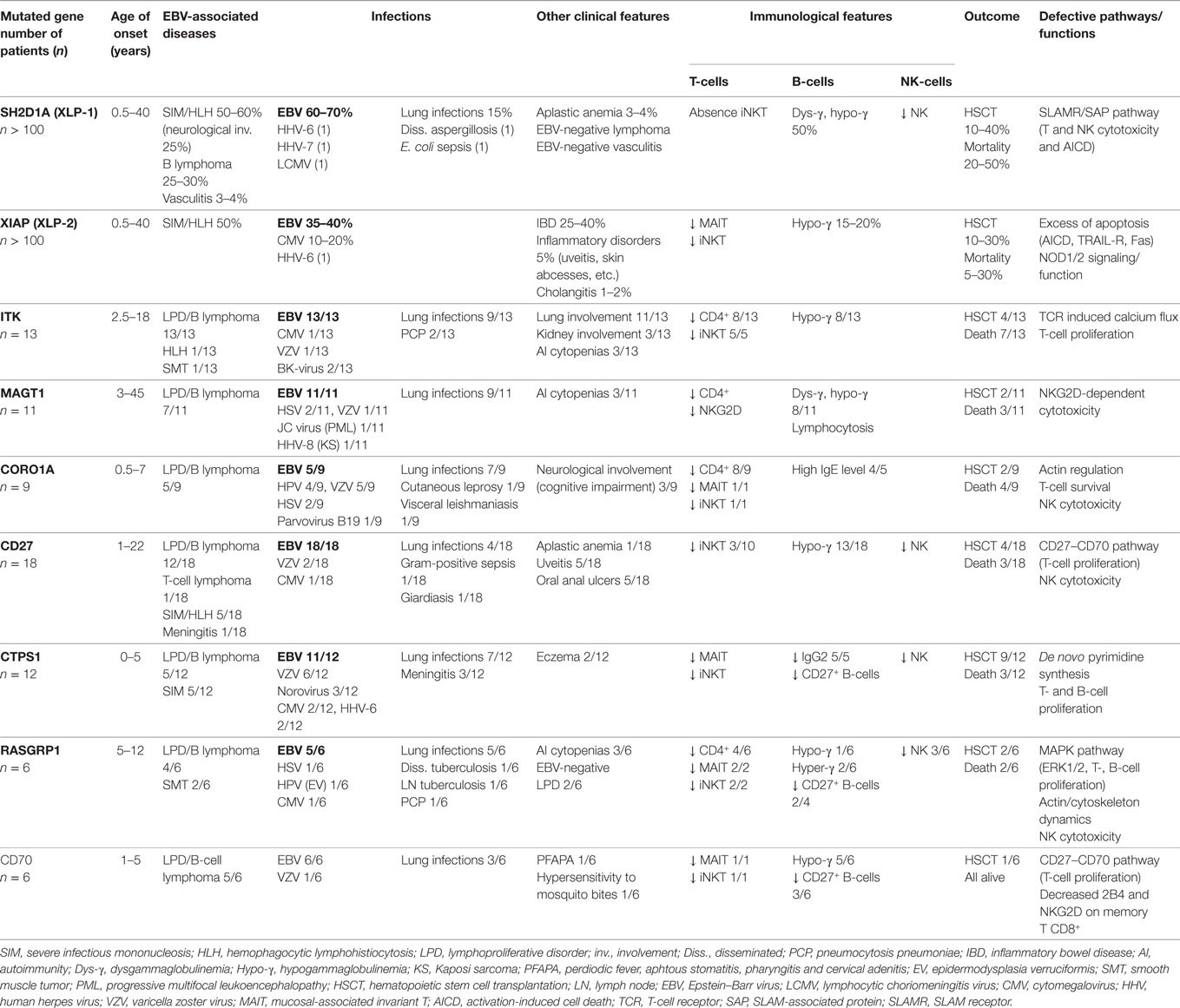
Table 1. Lymphoma subtypes in primary immunodeficiencies associated with high susceptibility to develop EBV-driven lymphoproliferative diseases.
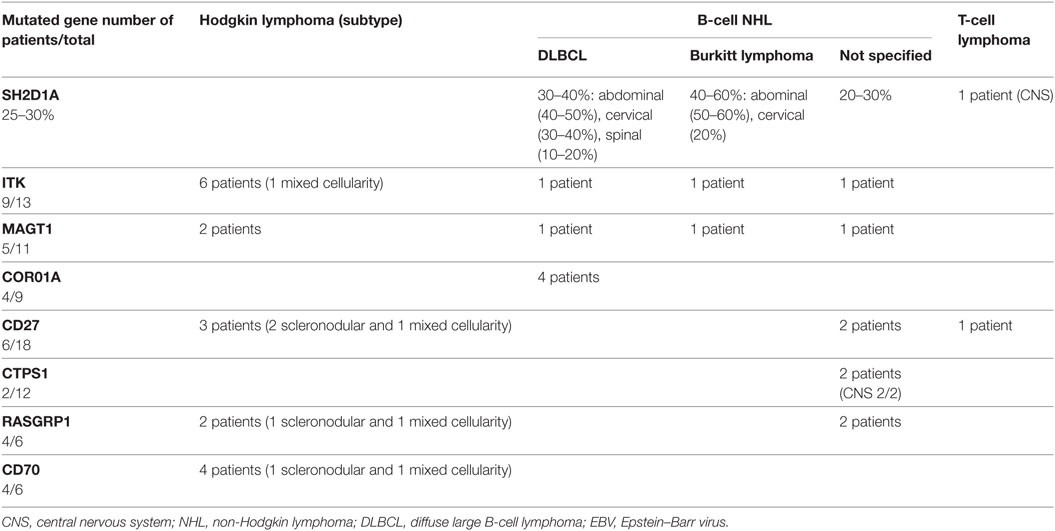
Table 2. Lymphoma subtypes in primary immunodeficiencies associated with high susceptibility to develop EBV-driven lymphoproliferative diseases.
Over the last two decades, these inherited defects have represented very unique in natura models to decipher the immune response to EBV. Molecular and genetic characterization of these disorders and their study have revealed several unexpected and novel pathways required for an efficient immunity to EBV, but also more generally involved in cancer immune surveillance of B lymphocytes, and adaptative immune responses.
SH2D1A/SLAM-Associated Protein (SAP) Deficiency
SH2D1A/SAP deficiency (also known as the X-linked lymphoproliferative syndrome type 1-XLP-1 or Purtilo syndrome) is caused by hemizygous mutations in the X-linked gene SH2D1A (15–19). More than 100 patients with hemizygous deleterious mutations in SH2D1A have been reported, but if considering the literature before the discovery of the gene, this is probably more than 200 cases (20–22). Clinical features of XLP-1 are EBV-driven fulminant or severe mononucleosis with all the clinical features of HLH including fulminant hepatitis, hepatic necrosis, bone marrow hypoplasia, neurological involvement in 20–30% of cases, hypogammaglobulinemia (50%), and B-cell lymphoma (25–30%) which often have abdominal localization. Few other rare phenotypes can be also observed such as vasculitis (2–5%), aplastic anemia (2–5%), and chronic gastritis (18, 23–26). All phenotypes can occur independently and some develop without any evidence of EBV infection (10–20%) or prior to EBV infection like HLH, hypogammaglobulinemia, lymphoma, aplastic anemia, and vasculitis (24–26). Some rare cases of T-cell lymphoma have also been reported (23, 27). However, a significant proportion of patients initially presented with combined variable immunodeficiency (CVID) associated with severe recurrent bacterial infections including disseminated aspergillosis and E. coli sepsis (28–31). Only five cases of other severe viral infections by HHV-6, HHV-7, and LCMV or HLH without EBV trigger have been reported in the recent literature (22, 26, 32, 33). This highlights that SAP has a very unique role in immunity to EBV, when it is not required for other viral infections except in very rare cases.
SH2D1A encodes a small adaptor protein of 128 amino acids named SAP, uniquely expressed in T and NK cells. SAP is only made of a unique SH2 domain that specifically binds to the intracytoplasmic domain of receptors of the SLAM family (SLAMF), which are self-ligands and are involved in homotypic cell–cell interactions with the exception of 2B4 (also known as CD244 or SLAMF4) that binds CD48 (34–36). On the one hand, SAP functions as a real adapter protein by its ability to recruit the tyrosine kinase FynT to SLAMF receptors, thus coupling those to downstream signaling pathways. On the other hand, SAP acts also as a blocker protein by inhibiting the recruitment of SH2-containing phosphatases to SLAMF (15, 35, 37–39). Early studies have documented NK-cell cytotoxicity defects toward EBV-infected B cells in XLP-1 patients (40, 41). Further investigations revealed that these defects involved two SLAM receptors, 2B4 and SLAMF6 (also known as NTB-A or Ly108) which have the capacity to trigger and activate NK- and CD8+ T-cell cytotoxicity toward EBV-infected B cells or transformed B cells when SAP is normally expressed (42–46). B-cell lymphomas and EBV-infected B cells are known to express high levels of ligands for SLAMF-R, including CD48 the ligand of 2B4 (47). This expression might signal abnormal “dangerous” proliferating B cells to NK cells and T lymphocytes. Following studies demonstrated that the stimulatory function of 2B4 and SLAMF6, which is dependent of FynT was not only lost in the absence of SAP but also shifted toward an inhibition of other stimulatory pathways of cell cytotoxicity in NK- and CD8+ T-cells (43, 48, 49). Additional findings in mice indicated that this inhibitory effect was independent of FynT, but depends of the blocking activity of SAP, and leads to decreased conjugate formation between CD8+ T or NK cells and SLAMF-expressing target cells like B cells (50, 51). In this context, other activation pathways of cytotoxicity such as NKG2D may not compensate for the defective function of 2B4 and SLAMF6. The inhibitory activity of 2B4 and SLAMF6 in the absence of SAP may have a trans-inhibitory effect on activating receptors (such as NKG2D and/or killer cell activating receptors) when co-engaged at the cytotoxic synapse.
Importantly, NK-cell and CD8+ T-cell cytotoxicity toward other APCs or target cells than B cells including non-hematopoietic cells or/and SLAMF-negative target cells was preserved, and even augmented for NK-cell cytotoxicity (47, 52, 53). Recent studies in mice indicate that this excessive NK-cell cytotoxicity is dependent of a role of SAP and SLAMF6 in NK cell licensing/education (52). In the absence of SAP, NK-cell responsiveness toward SLAM-negative cells is increased. In XLP-1 patients, increased NK cytotoxicity toward the SLAMF-negative target cells K562 was documented (54), associated with increased CD16- and NKp46-dependent cytotoxicity (52). Thus, extra-hematopoietic manifestations associated with HLH seen in XLP-1 patients, like extensive liver damage may be caused by this increased NK-cell cytotoxicity toward non-hematopoietic cells.
Beside its function in NK- and CD8+ T cell-cytotoxicity, SAP is also important to limit CD8+ T-cell expansion. SAP-deficient T cells both in mice and human exhibited increased survival due to impaired activation-induced cell death (AICD), an important pathway involves in the contraction of the pool of Ag-specific T-cells during immune responses (55, 56). The positive role of SAP on AICD is dependent of SLAMF6 that delivers signals for the expression of the pro-apoptotic molecules Bim and FasL (55). In concert, these distinct defects certainly contribute to the inability of EBV-specific T cells to eliminate EBV-infected B cells and to their uncontrolled expansion and activity during primary infection leading to fulminant mononucleosis or HLH. The fact that patients also frequently develop non-EBV B cell lymphoma argues for an important role of the SLAM–SAP pathway in the immune surveillance of B cells.
XIAP Deficiency
XIAP deficiency (also known as the X-linked lymphoproliferative syndrome type 2/XLP-2) is caused by hemizygous mutations in the XIAP gene coding the X-linked Inhibitor of Apoptosis Protein XIAP (54, 57). XIAP is an anti-apoptotic molecule member of the inhibitor of apoptosis protein family (IAP). XIAP is a potent inhibitor of program cell death, but it is also required for the function of (NOD)-like pattern recognition receptors (NLRs) NOD 1 and 2 and the regulation of the inflammasome activity of NLRP3 (58–61). More than 100 patients have been now reported with a XIAP deficiency. Initially reported in 2006 in a cohort of patients with a clinical phenotype close to that of SH2D1A-deficient patients (XLP-1), XIAP deficiency was further denominated XLP-2, since XIAP is located in the immediate vicinity of SH2D1A on the X chromosome (57). However, during the last 10 years, there were cumulative observations that the two diseases differ by many aspects, in particular, there is no evidence that they are functionally related. The main clinical phenotypes of the XIAP deficiency are the susceptibility to develop HLH in the context of EBV infection (36%), the recurrent splenomegaly corresponding to a minimal form of HLH (57%), and the IBD (26%) with features of Crohn’s disease (57, 62, 63). The HLH is often less severe than in the XLP-1 deficiency with very rare neurological involvement (24). Some patients also developed variable auto-inflammatory symptoms like uveitis, arthritis, skin abscesses, erythema nodosum, and nephritis (62, 63). Thus, today although the susceptibility to EBV remains one important and severe clinical manifestation of the XIAP deficiency, more than half of the patients never experienced a peculiar EBV susceptibility (57, 62). XIAP-deficient patients also never experience lymphoma in contrast to patients with SAP deficiency, likely related to the anti-apoptotic function of XIAP, which may protect patients from cancer, and XIAP is now considered as a promising therapeutic target for cancer treatment (64).
T lymphocytes from XLP-2 patients have been shown to exhibit increased AICD in response to T-cell receptor (TCR) activation and increased apoptosis to FAS/CD95 and TRAIL-R stimulations (54). On the other hand, monocytes from patients displayed impaired production of cytokines and chemokines (TNF-α, IL-10, IL-8, and MCP-1) to stimulation with NOD2 ligands (63, 65). Although the mechanisms of EBV-driven HLH in XIAP deficiency remains unclear, it is proposed that excessive AICD might compromise the expansion and proliferation of activated EBV-specific T cells like in CTPS1, CD27, or CD70 deficiencies (see below)(58). In this setting, accumulation of apoptotic cells and persistence of EBV-infected B cells could result in abnormal inflammation amplified by impaired inflammasome regulation and/or defective NOD1/2 activation of myeloid cell populations. In concert, T lymphocytes and myeloid defects may contribute to EBV-driven HLH in XIAP-deficient patients.
ITK Deficiency
ITK deficiency is an autosomal recessive disorder caused by bi-allelic mutations in ITK. Until now, 13 patients from 8 families have been reported in the literature (66–72). All experienced EBV-associated recurrent non-malignant LPD or malignant B-cell lymphoproliferations including Hodgkin lymphoma in five, and one develop an EBV-SMT (69). B-cell lymphoproliferations have often a pulmonary localization and more than 50% also develop pulmonary infections. ITK (IL-2-inducible tyrosine kinase) is a well-characterized protein tyrosine kinase of the TEC/BTK family specifically expressed in T lymphocytes and NK cells. ITK is involved in TCR signaling by its capacity to phosphorylate and activate the PLC-γ1, a key enzyme that stimulates Ca++ fluxes through the production of IP3, both being critical second messengers of T-cell activation (73, 74). ITK has been also involved in CXCR4 signaling (75). Mice studies demonstrated the importance of ITK in immunity particularly in CD4+ T-cell responses (73, 76). A recent report showed that ITK is also required for efficient CD8+ T-cell responses in mouse (77). In the absence of ITK, CD8+ T-cell expansion and maturation into cytolytic effector T cells is impaired leading to decreased CD8+ T-cell cytotoxic responses. Very few studies have been made to characterize ITK-deficient T cells from patients, albeit-derived T-cell lines from ITK-deficient patients have been obtained and studied showing decreased Ca++ mobilization (67). Defective T-cell proliferation in response to TCR engagement was documented in one patient (69). Although the exact mechanism(s) underlying EBV susceptibility in ITK deficiency need to be established, the recent study of CD8+ T cells from ITK-deficient mice (77), strongly supports that in T cells from ITK-deficient patients, TCR activation signals are impaired resulting in defective expansion and maturation of EBV-specific CD8+ T cells like in CD70 and CD27 deficiencies (see below).
MAGT1 Deficiency
MAGT1 deficiency also termed XMEN disease (for X-linked immunodeficiency, magnesium defect, EBV infection, and neoplasia syndrome) is caused by hemizygous mutations in MAGT1. To date, 11 male patients with MAGT1 deficiency have been identified and all developed susceptibility to EBV infection with chronic viremia and B-cell lymphomas including Hodgkin and DLBCL (78–82). In one patient, the initial clinical presentation was a HHV-8-associated Kaposi sarcoma (80). One particular trait of this disease is the B-cell lymphocytosis associated with a CD4 lymphopenia (83). MAGT1 encodes a ubiquitously expressed transmembrane Mg++ transporter involved in the maintenance of free basal intracellular Mg++ pools. However, MAGT1 also associates with the N-oligosaccharyl transferase complex, and therefore may have a role in protein N-glycosylation (84). Following the discovery of the MAGT1 immunodeficiency, MAGT1-dependent Mg++ influx was documented in T cells upon TCR engagement. Importantly, this Mg++ mobilization was shown to be involved in PLC-γ1 activation and subsequent dependent Ca++ influx (78). Notably, all these events were markedly impaired in activated MAGT1-deficient T cells in response to TCR, but T-cell proliferation has been considered to be normal or diminished (85). Based on these findings, it was proposed that MAGT1 might be necessary to activate PLC-γ1 possibly by acting on ITK. In a recent study, intriguingly, Ca++ mobilization was found to be only moderately decreased and delayed in T cells from a patient carrier of a deletion encompassing MAGT1 (80). Along these lines, an other recent report showed that MAGT1-deficient T cells from mice exhibited normal calcium flux upon TCR activation, while calcium flux was impaired in B cells in response to BCR stimulation (86). The discrepancy between these recent observations and earlier studies is not known. In any case, MAGT1 was further shown to be required for expression and function of NKG2D and its signaling adapter DAP10 (79). NKG2D is an activating NK-cell receptor expressed on NK cells, γδ T cells, and CD8+ T cells, that recognizes MHC class I-homologous proteins induced by cellular stress in response to infection or neoplasia. Importantly, ligands of NKG2D are upregulated on EBV-infected B cells and EBV-associated lymphoproliferations (87, 88). Consistent with defective NKG2D expression, MAGT1-deficient CD8+ T cells displayed impaired cytotoxic activity against autologous EBV-transformed B cells. Importantly, magnesium supplementation treatment in vivo and in vitro restored basal intracellular Mg++ concentration, NKG2D expression, cell cytotoxicity, and immunity to EBV in MAGT1-deficient patients (79). Defective N-glycosylation of NKG2D associated with increased ubiquitinylation leading to accelerated protein turnover might be considered as the main mechanism explaining the impaired NKG2D function. Thus, these observations highlight the essential role of the NKG2D pathway in immunity to EBV, albeit the exact role of MAGT1 in TCR signaling remains to be clarified.
CD27 Deficiency
This deficiency is caused by bi-allelic mutations in CD27 that encodes a protein belonging to the super family of TNF receptors (TNFSFR), also known as TNFSFR7. Until today, 18 patients have been reported and all developed EBV-associated LPDs including malignant B-cell proliferations, Hodgkin’s lymphoma, B lymphoma, and few patients developed HLH triggered by EBV (89–92). Some patients also experienced inflammatory symptoms such as uveitis and oral ulcers. CD27 binds to CD70 (also named TNFSF7), a member of the TNF superfamily ligands. CD27 is highly expressed by T cells including resting T cells and a small fraction of B cells corresponding to memory B cells (93–95). CD27 is a co-stimulatory molecule of T-cell activation and CD27–CD70 interactions in mice have been shown to enhance T-cell survival, effector functions, and memory T-cell expansion, in particular CD8+ T cells during anti-viral immune responses (96, 97). Insights to the mechanism underlying the high susceptibility to EBV in CD27 deficiency was recently given by the identification of CD70-deficient patients (see below).
CD70 Deficiency
Five patients with homozygous deleterious mutations in CD70 have been reported in 2017 and one in 2018 (91, 98, 99). Five out of six patients developed EBV-Hodgkin’s lymphoma and recurrent B-cell lymphoproliferations, and one initially presented clinical signs evoking a Behçet-like syndrome and had uncharacterized viral encephalitis. All patients had dysgammaglobulinemia and two patients developed other infections. By many aspects CD70-deficiency appears to be a phenocopy of the CD27 deficiency. In peripheral blood mononuclear cells, CD70 is only expressed by a very small fraction of B cells (91), but its expression is strongly upregulated on activated B cells and EBV-infected B cells during primary infection in tonsils of patients with IM (91). B-cell lymphomas and several other cancers such as solid carcinomas are also known to express CD70 (94). Izawa et al. demonstrated that CD70 expression on EBV-infected B cells drives the expansion of EBV-specific cytolytic T cells via a TCR-CD27-dependent co-stimulation. When CD70 is absent on EBV-infected B cells, or CD27 on T cells, EBV-specific T cells failed to expand leading to reduced cytotoxicity responses toward EBV-infected B cells (91). Decreased expression of 2B4 and NKG2D on memory CD8+ T cells of CD70-deficient patients was also noticed and may also contribute to the inability of T cells to eliminate EBV-infected B cells (98). These findings demonstrate that the CD70–CD27 axis represents a key component of the protective immunity to EBV. Furthermore, the implication of the CD27–CD70 axis in anti-tumoral immune surveillance of abnormal B cells is supported by the observations that CD70 is often found somatically mutated in B lymphomas, perhaps to escape to the immune surveillance (91).
CTPS1 Deficiency
CTPS1 deficiency is an autosomal recessive immunodeficiency caused by a unique homozygous deleterious mutation in CTPS1 with a founder effect in the population of the North West of England. Until now, 12 patients have been reported and all but 1 presented EBV susceptibility including severe infectious mononucleosis, LPD, and B-cell lymphoma, which was the initial clinical presentation in 40% of cases (100–102). Half of them also developed other viral infections including CMV, VZV, and HHV-6. Some also experienced recurrent bacterial infections with Haemophilus influenza, Streptococcus pneumonia, and/or Neisseria meningitis. CTPS1 codes for the CTP synthetase or synthase 1, a key enzyme of the de novo synthesis of the CTP nucleotide, which is the limiting nucleotide in cells (103). CTP is a critical precursor in the metabolism of nucleic acids. CTP is produced by two pathways, a salvage pathway and a de novo synthesis pathway. The salvage pathway utilizes cytidine, a degradation product from nucleic acids. The “de novo” CTP synthesis is dependent of two enzymes CTPS1 and CTPS2, which catalyze ATP-dependent amination of UTP to CTP with ammonia (-NH3) transfer from hydrolyzed glutamine. In normal tissues, CTPS activity is rather low, while it is high in proliferating cells like cancer cells including lymphoma. Importantly, CTPS1 is very low in resting T cells and is rapidly and strongly upregulated in response to TCR stimulation (100). In CTPS1-deficient patients, the proliferation of T cells in response to TCR engagement is markedly impaired, while other T-cell responses including cytokines production, AICD, and cytotoxicity are not affected, and T cells normally proliferate in response to IL-2. Addition of CTP or cytidine in the culture medium restored T-cell proliferation of activated T cells. CTPS1 expression is also upregulated in activated B cells. However, the role of CTPS1 in B cells may be less important than in T cells as patients developed rather infections associated with a T-cell defect and the absence of CTPS1 has no effect on proliferation of EBV-infected B cells and their transformation by EBV. The discovery of the CTPS1 deficiency emphasizes the importance of T-cell expansion during anti-viral responses, specifically in primary infection to EBV, since 40% T cells of circulating can be specific to EBV (8).
CORO1A Deficiency
Deficiency in the actin regulator CORO1A (Coronin-1A) has been identified in nine patients (104–108). Patients presented with severe infections, and five developed EBV-driven B cell lymphoma. Four patients had severe mucocutaneous-immunodeficiency manifestations including epidermodysplasia verruciformis-HPV (EV-HPV) (104, 108). Three patients also exhibited neurological abnormalities including autism-like symptoms. Patients with CORO1A deficiency are characterized by a profound T cell lymphopenia with strongly decreased or nearly absent naïve cells associated with defective thymic output. CORO1A-deficient T cells from patients showed increased spontaneous in vitro apoptosis, delayed ERK1/2 activation, increased filamentous actin, but normal or reduced T cell proliferation to mitogens and antigens and normal calcium flux and cytotoxicity. It is proposed that CORO1A deficiency is primarily a T-cell immunodeficiency caused by impaired thymic egress, migration, and survival of mature T cells, thereby affecting lymphocyte homeostasis, repertoire selection, and lineage commitment (109). CORO1A belongs to the family of Coronins that are evolutionarily conserved intracellular actin-binding proteins expressed at high level in most of leukocyte populations (110). In T cells, CORO1A has been shown to be a negative regulator of branched F-actin formation and be required for chemokine-mediated migration and lymphocyte survival given the fact that accumulation of F-actin is known to be toxic for cells (108, 111). However, CORO1A in T cells has been also involved in a variety of pathways including TCR and TGF-β signaling and immunologic synapse (IS) formation (112–115). Along these lines, defective NK cell degranulation was reported in one patient in association with increased density of F-actin at the cytotoxic synapse (113). Neurological abnormalities found in patients are likely explained by the role of coronin-1A in neurodevelopment that has been reported in mice (116). Finally, Coronin 1A was recently shown to be required for neutrophil trafficking. These observations suggest that the immune defects in patients may not only be restricted to T cells (117). While it is not clearly established why patients with CORO1A deficiency are susceptible to EBV, it is very likely that the poor T-cell survival may result in defective expansion of EBV-specific CD8+ T-cells leading to impaired control of EBV-infected B cells. Interestingly, four patients developed EV-HPV, which could bring closer CORO1A deficiency to primary immunodeficiencies (PIDs) associated with frequent and extensive HPV infections, especially those such as MST1/STK4 and DOCK8 deficiencies, that are characterized by CD4 lymphopenia, defective T cell migration, and/or abnormal F-actin polymerization and IS formation (118).
RASGRP1 Deficiency
RASGRP1 deficiency is caused by bi-allelic mutations in RASGRP1. So far, three different null homozygous mutations have been identified in four patients having developed severe EBV-driven LPDs including two Hodgkin lymphoma. One had also an EBV-SMT (119–121). RASGRP1 codes for a diacylglycerol-regulated guanidine exchange factor preferentially expressed in T and NK cells (122, 123), which acts as an activator of the small G protein RAS and the downstream RAF-MEK-ERK kinases cascade (also known as the MAP kinases pathway). In T lymphocytes, RASGRP1 is the main activator of the MAP kinases pathway (124, 125). RASGRP1-deficient T cells showed impaired ERK/MAPK activation and decreased T-cell proliferation in response to mitogens and antigens (119, 120). In the first report, RASGRP1-deficient T cells were also shown to have diminished cell cytotoxicity and migration capacity (120). NK cells also exhibited decreased cell cytotoxicity. A direct role of RASGRP1 in cytoskeletal dynamics during exocytosis of lytic granules in NK and T cells and in T-cell migration is suggested by its ability to interact with the dynein light chain DYNLL1 and to activate RhoA, respectively (120). This role may explain the impaired cytotoxic responses seen in RASGRP1-deficient T and NK cells. In a recent report by Winter et al., RASGRP1-deficient NK cells and CD8+ T cells were nonetheless found to have normal degranulation when stimulated (119). The discrepancy between these studies is not known. However, in the different reports, low numbers of NK cells were consistently noticed in RASGRP1-deficient patients that might contribute to the decreased NK cell cytotoxicity. Winter et al. further analyzed the possible mechanisms underlying the EBV susceptibility in RASGRP1 deficiency and showed that RASGRP1-deficient T cells failed to expand normally (119). In particular, RASGRP1-deficient T cells had impaired CD27-dependent proliferation toward CD70-expressing EBV-transformed B cells, a critical pathway to expand EBV-specific T cells (see above). Interestingly, the impaired proliferation of activated RASGRP1-deficient T cells correlated with their inability to upregulate CTPS1 protein expression, suggestive of a role of RASGRP1/MAPK pathway in CTPS1 expression, but also in other factors involved in T-cell proliferation, as CTP or cytidine failed to restore impaired proliferation of RASGRP1-deficient T cells (119).
Two RASGRP1 heterozygous compound mutations with no effect on RASGRP1 protein expression have been also recently identified in two siblings with multiple fungal, bacterial, and viral infections including EBV and CMV, and both also developed autoimmunity signs evoking autoimmune lymphoproliferative syndrome (126). Along these lines, RASGRP1-deficient mice developed autoimmune LPD resembling systemic lupus erythematous when getting older (127, 128) and RASGRP1 is considered as a risk locus for autoimmunity (129–131). T cells from the two patients showed defective TCR activation associated with impaired proliferation and AICD. However, complementation experiments with wild-type RASGRP1 were unsuccessful, leaving the possibility that other or additional genetic events contribute to this particular phenotype and/or these mutations do not behave as loss-of-function mutations. These observations could suggest a genotype–phenotype correlation.
Pathways in the Control of EBV Infection
Several key pathways required for an efficient immunity to EBV have emerged from studies of these genetic disorders (11). Interestingly, these pathways are involved in the cell–cell interaction and cross-talk between T and B cells and appear to play a critical role in the immune surveillance of B cells by T cells, which is consistent with B cells as the privileged target of EBV infection and the reservoir of EBV. These pathways implicate pairs of receptor–ligand expressed by T and B cells, respectively (Figure 1). The best known and in depth studied is the SLAMR–SAP pathway, which is defective in the SAP deficiency/XLP-1 syndrome and mainly involves two SLAM receptors, 2B4 and NTB-A; however, other SLAMR such as CD229 may be also implicated as suggested from mice and human studies (47, 49). These pathways appear to be important in the recognition of EBV-infected B cells by T cells and in the activation of the T- and NK-cell cytotoxicity responses toward EBV-infected B cells. Another important pathway is dependent of the NKG2D receptor, well known to activate T- and NK-cell cytotoxic responses. In the absence of MAGT1, NKG2D expression on CD8+ T cells and NK cells is impaired leading to defective killing of EBV-infected B cells (79). Nevertheless, it would be interesting to know whether EBV-specific T cells expand normally in MAGT1-deficient patients as NKG2D has been also involved in the survival and expansion of CD8+ T cells during viral infections. The pair CD27–CD70 molecules forms a critical axis required for survival and expansion of EBV-specific T cells (91). The key role of T-cell expansion/proliferation to control EBV-infected B cells is also highlighted by the CTPS1 and RASGRP1 deficiencies, in which the capacity of T lymphocytes to proliferate in response to antigenic stimulation is specifically impaired.
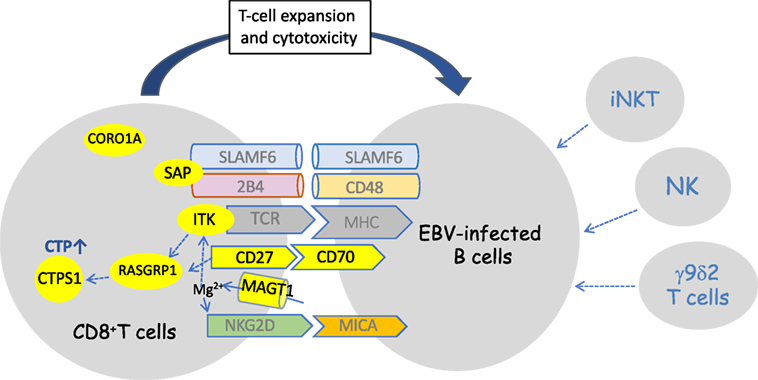
Figure 1. Schematic representation of defective identified pathways in immunodeficiencies predisposing to high susceptility to Epstein–Barr virus (EBV)-driven B-cell lymphoproliferative diseases. Defective components in the pathways are in yellow. Three non-redundant pathways have been shown to be impaired: the SLAMF6/2B4/SLAM-associated protein (SAP) pathway, the CD70/CD27 pathway, and the NKG2D/MICA/MAGT1 pathway. At some point, several of these pathways may converge toward distal effector molecules such as RASGRP1 and CTPS1 that are required for expansion of T cells. The connection of CORO1A to these pathways is not known. Besides the essential role of CD8+ T cells, NK and iNKT cells which are often decreased in these primary imunodeficiencies may play a role in the immune response against EBV, particularly during the early phase of the primary infection. Vγ9δ2 T cells are also thought to play an important role in the primary infection by recognition of latently EBV-replicating B cells.
Most of these deficiencies are characterized by a marked decreased or absence of invariant T cell populations iNKT and mucosal-associated invariant T (MAIT) cells, which raises the question of the role of these cells in immunity to EBV. iNKT cells are characterized by innate-like properties including prompt activation and production of large amounts of cytokines. These cells have been involved in a variety of immune responses including anti-viral immunity. Mice models have provided the evidence that SAP, ITK, and RASGRP1 are directly implicated in the development/homeostasis/function of iNKT cells (132–137). Analysis of XIAP-deficient patients also indicated that these cellular defects can be secondary to the EBV infection, leading to cell exhaustion in relation with the peculiar high sensitivity of iNKT and MAIT cells to AICD (138). There are only limited studies that assessed the role of iNKT cells in EBV infection, though it was shown that iNKT cells, in particular CD8+ iNKT cells have the capacity to directly lyse EBV-infected B cells expressing CD1d and to limit expansion of EBV-transformed B cells both in vitro and in vivo (139–141). However, RORC-deficient patients, who lack iNKT and MAIT cells are not particularly susceptible to EBV, indicating that these cells are not playing a critical role in immunity to EBV (142). Altered functions and decreased counts of NK cells are also often observed in a number of these immunodeficiencies. The role of NK cells might be particularly important during childhood, since KIR-negative early differentiated NK cells that expanded preferentially during IM and targeted lytically EBV-replicating B cells, progressively disappeared in the first decade of life (143). Recent data indicated that patients with severe combined immunodeficiency (SCID) who received bone marrow transplanted, although devoided of NK cells after immune reconstitution display an efficient immunity to EBV since none of them developed EBV-driven lymphoproliferation disorders, even 39 years after the transplantation (144). This suggests that NK cells are not essential for EBV immunity throughout the life. In any event, although they are not key components, accumulation of these different cellular defects might participate to the susceptibility to EBV in these genetic settings, particularly during childhood. CD4 T cell lymphopenia that is observed in several of these disease may also participate to EBV susceptibility, although the role of CD4+ T cells in the control of EBV infection is not clearly established (3). Arguing against a relevant role of CD4+ T cells, patients with MHC class II deficiency have a severe CD4 lymphopenia and do not develop EBV-driven LPDs and they have normal B cell counts. Although not really considered, some of these defects could also have intrinsic B-cell consequences that would further favor proliferation and/or lymphomagenesis of EBV-infected B cells in addition to the immune deficiency. In that respect, CD70 was shown to elicit reverse signaling involved in apoptosis of B cells and the recent study of MAGT1-deficient mice revealed a regulatory role of MAGT1 in B-cell development and proliferation that may explain the B lymphocytosis found in MAGT1-deficient patients (86, 145).
Differential Diagnosis
Given the importance of the CD8+ T-cell response in immunity to EBV, it is not surprising that EBV-driven LPDs are also found in other PIDs associated with T-cell defects, but with a lower frequency. Herpes virus, in particular EBV, are often the trigger of HLH in patients with familial haemophagocytic lymphohistiocytosis (FHL), a group of diseases associated with impaired cytolytic activity of CD8+ T cells and NK cells. These diseases are caused by gene defects in the perforin gene and in the components of lytic granule exocytosis machinery (146). Patients with CVID or CID, particularly those affecting T-cell survival, migration, and F-actin mobilization in T cells, can also develop EBV-associated disorders (12). Those include, among others, deficiencies in NFκB1 (147), MST1/STK4 (148, 149), WASP (150), DOCK8 (151), GATA2 (152), and gain-of-function mutations in PIK3CD known to cause activated PI3K-delta syndrome (153). Patients with hypomorphic mutations in genes involved in T-cell development such as RAG1/2, DCLRE1C (ARTEMIS), or ZAP-70 can also experience EBV susceptibility (154–156). It should be considered that patients with the most severe T-cell defects will never present EBV problems since they develop very early-onset severe infections (other than EBV), requiring rapid bone marrow transplantation before they encounter EBV. At last, some of these immunodeficiencies (SCID) are also associated with a severe block in B-cell development, a cellular context that in all likelihood does not allow EBV infection establishment and dissemination.
Therapeutics
So far, the only curative treatment for these PIDs is hematopoietic stem cell transplantation (HSCT). The first studies on large cohorts of XLP-1 and XLP-2 patients reported poor survival with or without HSCT (23, 157). This pejorative prognosis was partly linked to the use of full conditioning regimens associated with a high toxicity in patients (XIAP) and/or the absence of HLH remission at the time of HSCT. In the last few years, the use of reduced intensity conditionings and the development of new therapies like alemtuzumab (anti-CD52 antibody) has strongly improved the management of these diseases (33, 158, 159). Furthermore, rituximab (anti-CD20 antibody) has now a major role in the management of LPDs associated with these PIDs with patients being in remission after having received this treatment.
Concluding Remarks
Molecular characterization of familial forms of EBV susceptibility has provided over the last 20 years, novel diagnostic tools for these disorders. Analysis of large cohorts and case reports have underlined non-EBV phenotypes associated with these PIDs such as IBD, pulmonary involvement, neurological disorders, or other typical infections (such as HPV), that can further help to the diagnosis. However, there are still a number of patients with a high susceptibility to EBV, in whom the molecular/genetic basis of their disease is not known and remains to be determined. In the light of the knowledge gained throughout the studies of the genetically determined forms of EBV susceptibility, we can speculate that these uncharacterized forms are caused by defects in molecules/components involved in T–B cell interactions and required for T-cell cytolytic responses and/or T-cell expansion.
Author Contributions
SL wrote the manuscript and made the Figure 1. SW compiled data for Tables 1 and 2 and did the tables and participated to the writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
SL is a senior scientist at the Centre National de la Recherche Scientifique-CNRS (France). SW is supported by Imagine Institut PhD program funded by the Fondation Bettencourt-Schueller. The work of the Laboratory of Lymphocyte Activation and Susceptibility to EBV headed by SL is supported by grants from Ligue Contre le Cancer-Equipe Labellisée (France), INSERM (France), the Agence Nationale de la Recherche, France (ANR-14-CE14-0028-01 (SL) and ANR-10-IAHU-01 (Imagine Institut)), the Rare Diseases Fondation (France), the Association François Aupetit (France), the Société Française de Lutte contre les Cancers et Leucémies de l’Enfant et de l’Adolescent (SFCE), AREMIG (France), Fédération Enfants et Santé (France), and the European Research Council (ERC-2009-AdG_20090506 no FP7-249816).
References
1. Cohen JI, Fauci AS, Varmus H, Nabel GJ. Epstein-Barr virus: an important vaccine target for cancer prevention. Sci Transl Med (2011) 3:107fs107. doi:10.1126/scitranslmed.3002878
2. Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu Rev Immunol (2007) 25:587–617. doi:10.1146/annurev.immunol.25.022106.141553
3. Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD. The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol (2015) 33:787–821. doi:10.1146/annurev-immunol-032414-112326
4. Munz C. Epstein-Barr virus-specific immune control by innate lymphocytes. Front Immunol (2017) 8:1658. doi:10.3389/fimmu.2017.01658
5. Djaoud Z, Guethlein LA, Horowitz A, Azzi T, Nemat-Gorgani N, Olive D, et al. Two alternate strategies for innate immunity to Epstein-Barr virus: one using NK cells and the other NK cells and gammadelta T cells. J Exp Med (2017) 214:1827–41. doi:10.1084/jem.20161017
6. Fujiwara S, Kimura H, Imadome K-I, Arai A, Kodama E, Morio T, et al. Current research on chronic active Epstein-Barr virus infection in Japan. Pediatr Int (2014) 56:159–66. doi:10.1111/ped.12314
7. Kimura H, Cohen JI. Chronic active Epstein-Barr virus disease. Front Immunol (2017) 8:1867. doi:10.3389/fimmu.2017.01867
8. Callan MF, Tan L, Annels N, Ogg GS, Wilson JD, O’Callaghan CA, et al. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus in vivo. J Exp Med (1998) 187:1395–402. doi:10.1084/jem.187.9.1395
9. Bibas M, Antinori A. EBV and HIV-related lymphoma. Mediterr J Hematol Infect Dis (2009) 1:e2009032. doi:10.4084/MJHID.2009.032
10. Veillette A, Perez-Quintero LA, Latour S. X-linked lymphoproliferative syndromes and related autosomal recessive disorders. Curr Opin Allergy Clin Immunol (2013) 13:614–22. doi:10.1097/ACI.0000000000000008
11. Tangye SG, Palendira U, Edwards ES. Human immunity against EBV-lessons from the clinic. J Exp Med (2017) 214:269–83. doi:10.1084/jem.20161846
12. Cohen JI. Primary immunodeficiencies associated with EBV disease. Curr Top Microbiol Immunol (2015) 390:241–65. doi:10.1007/978-3-319-22822-8_10
13. Fischer A. Recent advances in understanding the pathophysiology of primary T cell immunodeficiencies. Trends Mol Med (2015) 21:408–16. doi:10.1016/j.molmed.2015.04.002
14. Notarangelo LD. Combined immunodeficiencies with nonfunctional T lymphocytes. Adv Immunol (2014) 121:121–90. doi:10.1016/B978-0-12-800100-4.00004-0
15. Sayos J, Wu C, Morra M, Wang N, Zhang X, Allen D, et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature (1998) 395:462–9. doi:10.1038/26683
16. Coffey AJ, Brooksbank RA, Brandau O, Oohashi T, Howell GR, Bye JM, et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet (1998) 20:129–35. doi:10.1038/2424
17. Nichols KE, Harkin DP, Levitz S, Krainer M, Kolquist KA, Genovese C, et al. Inactivating mutations in an SH2 domain-encoding gene in X-linked lymphoproliferative syndrome. Proc Natl Acad Sci U S A (1998) 95:13765–70. doi:10.1073/pnas.95.23.13765
18. Tangye SG. XLP: clinical features and molecular etiology due to mutations in SH2D1A encoding SAP. J Clin Immunol (2014) 34:772–9. doi:10.1007/s10875-014-0083-7
19. Gaspar HB, Sharifi R, Gilmour KC, Thrasher AJ. X-linked lymphoproliferative disease: clinical, diagnostic and molecular perspective. Br J Haematol (2002) 119:585–95. doi:10.1046/j.1365-2141.2002.03851.x
20. Purtilo DT, Sakamoto K, Barnabei V, Seeley J, Bechtold T, Rogers G, et al. Epstein-Barr virus-induced diseases in boys with the X-linked lymphoproliferative syndrome (XLP): update on studies of the registry. Am J Med (1982) 73:49–56. doi:10.1016/0002-9343(82)90923-8
21. Sumegi J, Huang D, Lanyi A, Davis JD, Seemayer TA, Maeda A, et al. Correlation of mutations of the SH2D1A gene and Epstein-Barr virus infection with clinical phenotype and outcome in X-linked lymphoproliferative disease. Blood (2000) 96:3118–25.
22. Kanegane H, Yang X, Zhao M, Yamato K, Inoue M, Hamamoto K, et al. Clinical features and outcome of X-linked lymphoproliferative syndrome type 1 (SAP deficiency) in Japan identified by the combination of flow cytometric assay and genetic analysis. Pediatr Allergy Immunol (2012) 23:488–93. doi:10.1111/j.1399-3038.2012.01282.x
23. Booth C, Gilmour KC, Veys P, Gennery AR, Slatter MA, Chapel H, et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood (2011) 117:53–62. doi:10.1182/blood-2010-06-284935
24. Pachlopnik Schmid J, Canioni D, Moshous D, Touzot F, Mahlaoui N, Hauck F, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood (2011) 117:1522–9. doi:10.1182/blood-2010-07-298372
25. Talaat KR, Rothman JA, Cohen JI, Santi M, Choi JK, Guzman M, et al. Lymphocytic vasculitis involving the central nervous system occurs in patients with X-linked lymphoproliferative disease in the absence of Epstein-Barr virus infection. Pediatr Blood Cancer (2009) 53:1120–3. doi:10.1002/pbc.22185
26. Gray PE, O’Brien TA, Wagle M, Tangye SG, Palendira U, Roscioli T, et al. Cerebral vasculitis in X-linked lymphoproliferative disease cured by matched unrelated cord blood transplant. J Clin Immunol (2015) 35:604–9. doi:10.1007/s10875-015-0194-9
27. Seemayer TA, Gross TG, Egeler RM, Pirruccello SJ, Davis JR, Kelly CM, et al. X-linked lymphoproliferative disease: twenty-five years after the discovery. Pediatr Res (1995) 38:471–8. doi:10.1203/00006450-199510000-00001
28. Morra M, Silander O, Calpe S, Choi M, Oettgen H, Myers L, et al. Alterations of the X-linked lymphoproliferative disease gene SH2D1A in common variable immunodeficiency syndrome. Blood (2001) 98:1321–5. doi:10.1182/blood.V98.5.1321
29. Eastwood D, Gilmour KC, Nistala K, Meaney C, Chapel H, Sherrell Z, et al. Prevalence of SAP gene defects in male patients diagnosed with common variable immunodeficiency. Clin Exp Immunol (2004) 137:584–8. doi:10.1111/j.1365-2249.2004.02581.x
30. Nistala K, Gilmour KC, Cranston T, Davies EG, Goldblatt D, Gaspar HB, et al. X-linked lymphoproliferative disease: three atypical cases. Clin Exp Immunol (2001) 126:126–30. doi:10.1046/j.1365-2249.2001.01599.x
31. Soresina A, Lougaris V, Giliani S, Cardinale F, Armenio L, Cattalini M, et al. Mutations of the X-linked lymphoproliferative disease gene SH2D1A mimicking common variable immunodeficiency. Eur J Pediatr (2002) 161:656–9. doi:10.1007/s00431-002-1083-9
32. Liu J, Tian W, Wang F, Teng W, Zhang Y, Tong C, et al. Maternal onset de novo SH2D1A mutation and lymphocytic choriomeningitis virus infection in a patient with X-linked lymphoproliferative disease type 1: a case report. Mol Med Rep (2015) 11:3291–4. doi:10.3892/mmr.2015.3173
33. Marsh RA, Bleesing JJ, Chandrakasan S, Jordan MB, Davies SM, Filipovich AH. Reduced-intensity conditioning hematopoietic cell transplantation is an effective treatment for patients with SLAM-associated protein deficiency/X-linked lymphoproliferative disease type 1. Biol Blood Marrow Transplant (2014) 20:1641–5. doi:10.1016/j.bbmt.2014.06.003
34. Veillette A. Immune regulation by SLAM family receptors and SAP-related adaptors. Nat Rev Immunol (2006) 6:56–66. doi:10.1038/nri1761
35. Veillette A, Dong Z, Perez-Quintero LA, Zhong MC, Cruz-Munoz ME. Importance and mechanism of ’switch’ function of SAP family adapters. Immunol Rev (2009) 232:229–39. doi:10.1111/j.1600-065X.2009.00824.x
36. Cannons JL, Tangye SG, Schwartzberg PL. SLAM family receptors and SAP adaptors in immunity. Annu Rev Immunol (2011) 29:665–705. doi:10.1146/annurev-immunol-030409-101302
37. Latour S, Roncagalli R, Chen R, Bakinowski M, Shi X, Schwartzberg PL, et al. Binding of SAP SH2 domain to FynT SH3 domain reveals a novel mechanism of receptor signalling in immune regulation. Nat Cell Biol (2003) 5:149–54. doi:10.1038/ncb919
38. Latour S, Gish G, Helgason CD, Humphries RK, Pawson T, Veillette A. Regulation of SLAM-mediated signal transduction by SAP, the X-linked lymphoproliferative gene product. Nat Immunol (2001) 2:681–90. doi:10.1038/90615
39. Chan B, Lanyi A, Song HK, Griesbach J, Simarro-Grande M, Poy F, et al. SAP couples Fyn to SLAM immune receptors. Nat Cell Biol (2003) 5:155–60. doi:10.1038/ncb920
40. Harada S, Bechtold T, Seeley JK, Purtilo DT. Cell-mediated immunity to Epstein-Barr virus (EBV) and natural killer (NK)-cell activity in the X-linked lymphoproliferative syndrome. Int J Cancer (1982) 30:739–44. doi:10.1002/ijc.2910300610
41. Harada S, Sakamoto K, Seeley JK, Lindsten T, Bechtold T, Yetz J, et al. Immune deficiency in the X-linked lymphoproliferative syndrome. I. Epstein-Barr virus-specific defects. J Immunol (1982) 129:2532–5.
42. Tangye SG, Phillips JH, Lanier LL, Nichols KE. Functional requirement for SAP in 2B4-mediated activation of human natural killer cells as revealed by the X-linked lymphoproliferative syndrome. J Immunol (2000) 165:2932–6. doi:10.4049/jimmunol.165.6.2932
43. Parolini S, Bottino C, Falco M, Augugliaro R, Giliani S, Franceschini R, et al. X-linked lymphoproliferative disease. 2B4 molecules displaying inhibitory rather than activating function are responsible for the inability of natural killer cells to kill Epstein-Barr virus-infected cells. J Exp Med (2000) 192:337–46. doi:10.1084/jem.192.3.337
44. Bottino C, Falco M, Parolini S, Marcenaro E, Augugliaro R, Sivori S, et al. NTB-A [correction of GNTB-A], a novel SH2D1A-associated surface molecule contributing to the inability of natural killer cells to kill Epstein-Barr virus-infected B cells in X-linked lymphoproliferative disease. J Exp Med (2001) 194:235–46. doi:10.1084/jem.194.3.235
45. Dupre L, Andolfi G, Tangye SG, Clementi R, Locatelli F, Arico M, et al. SAP controls the cytolytic activity of CD8+ T cells against EBV-infected cells. Blood (2005) 105:4383–9. doi:10.1182/blood-2004-08-3269
46. Sharifi R, Sinclair JC, Gilmour KC, Arkwright PD, Kinnon C, Thrasher AJ, et al. SAP mediates specific cytotoxic T-cell functions in X-linked lymphoproliferative disease. Blood (2004) 103:3821–7. doi:10.1182/blood-2003-09-3359
47. Hislop AD, Palendira U, Leese AM, Arkwright PD, Rohrlich PS, Tangye SG, et al. Impaired Epstein-Barr virus-specific CD8+ T-cell function in X-linked lymphoproliferative disease is restricted to SLAM family-positive B-cell targets. Blood (2010) 116:3249–57. doi:10.1182/blood-2009-09-238832
48. Bloch-Queyrat C, Fondaneche MC, Chen R, Yin L, Relouzat F, Veillette A, et al. Regulation of natural cytotoxicity by the adaptor SAP and the Src-related kinase Fyn. J Exp Med (2005) 202:181–92. doi:10.1084/jem.20050449
49. Dong Z, Cruz-Munoz ME, Zhong MC, Chen R, Latour S, Veillette A. Essential function for SAP family adaptors in the surveillance of hematopoietic cells by natural killer cells. Nat Immunol (2009) 10:973–80. doi:10.1038/ni.1763
50. Dong Z, Davidson D, Perez-Quintero LA, Kurosaki T, Swat W, Veillette A. The adaptor SAP controls NK cell activation by regulating the enzymes Vav-1 and SHIP-1 and by enhancing conjugates with target cells. Immunity (2012) 36:974–85. doi:10.1016/j.immuni.2012.03.023
51. Zhao F, Cannons JL, Dutta M, Griffiths GM, Schwartzberg PL. Positive and negative signaling through SLAM receptors regulate synapse organization and thresholds of cytolysis. Immunity (2012) 36:1003–16. doi:10.1016/j.immuni.2012.05.017
52. Wu N, Zhong MC, Roncagalli R, Perez-Quintero LA, Guo H, Zhang Z, et al. A hematopoietic cell-driven mechanism involving SLAMF6 receptor, SAP adaptors and SHP-1 phosphatase regulates NK cell education. Nat Immunol (2016) 17:387–96. doi:10.1038/ni.3369
53. Palendira U, Low C, Chan A, Hislop AD, Ho E, Phan TG, et al. Molecular pathogenesis of EBV susceptibility in XLP as revealed by analysis of female carriers with heterozygous expression of SAP. PLoS Biol (2011) 9:e1001187. doi:10.1371/journal.pbio.1001187
54. Rigaud S, Fondaneche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature (2006) 444:110–4. doi:10.1038/nature05257
55. Snow AL, Marsh RA, Krummey SM, Roehrs P, Young LR, Zhang K, et al. Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. J Clin Invest (2009) 119:2976–89. doi:10.1172/JCI39518
56. Chen G, Tai AK, Lin M, Chang F, Terhorst C, Huber BT. Increased proliferation of CD8+ T cells in SAP-deficient mice is associated with impaired activation-induced cell death. Eur J Immunol (2007) 37:663–74. doi:10.1002/eji.200636417
57. Aguilar C, Latour S. X-linked inhibitor of apoptosis protein deficiency: more than an X-linked lymphoproliferative syndrome. J Clin Immunol (2015) 35:331–8. doi:10.1007/s10875-015-0141-9
58. Latour S, Aguilar C. XIAP deficiency syndrome in humans. Semin Cell Dev Biol (2015) 39:115–23. doi:10.1016/j.semcdb.2015.01.015
59. Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O’Reilly L, et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity (2012) 36:215–27. doi:10.1016/j.immuni.2012.01.012
60. Damgaard RB, Fiil BK, Speckmann C, Yabal M, zur Stadt U, Bekker-Jensen S, et al. Disease-causing mutations in the XIAP BIR2 domain impair NOD2-dependent immune signalling. EMBO Mol Med (2013) 5:1278–95. doi:10.1002/emmm.201303090
61. Krieg A, Correa RG, Garrison JB, Le Negrate G, Welsh K, Huang Z, et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci U S A (2009) 106:14524–9. doi:10.1073/pnas.0907131106
62. Speckmann C, Lehmberg K, Albert MH, Damgaard RB, Fritsch M, Gyrd-Hansen M, et al. X-linked inhibitor of apoptosis (XIAP) deficiency: the spectrum of presenting manifestations beyond hemophagocytic lymphohistiocytosis. Clin Immunol (2013) 149:133–41. doi:10.1016/j.clim.2013.07.004
63. Aguilar C, Lenoir C, Lambert N, Begue B, Brousse N, Canioni D, et al. Characterization of Crohn disease in X-linked inhibitor of apoptosis-deficient male patients and female symptomatic carriers. J Allergy Clin Immunol (2014) 134:1131–41.e9. doi:10.1016/j.jaci.2014.04.031
64. Gu L, Zhang H, Liu T, Zhou S, Du Y, Xiong J, et al. Discovery of dual inhibitors of MDM2 and XIAP for cancer treatment. Cancer Cell (2016) 30:623–36. doi:10.1016/j.ccell.2016.08.015
65. Ammann S, Elling R, Gyrd-Hansen M, Duckers G, Bredius R, Burns SO, et al. A new functional assay for the diagnosis of X-linked inhibitor of apoptosis (XIAP) deficiency. Clin Exp Immunol (2014) 176:394–400. doi:10.1111/cei.12306
66. Huck K, Feyen O, Niehues T, Ruschendorf F, Hubner N, Laws HJ, et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest (2009) 119:1350–8. doi:10.1172/JCI37901
67. Linka RM, Risse SL, Bienemann K, Werner M, Linka Y, Krux F, et al. Loss-of-function mutations within the IL-2 inducible kinase ITK in patients with EBV-associated lymphoproliferative diseases. Leukemia (2012) 26:963–71. doi:10.1038/leu.2011.371
68. Mansouri D, Mahdaviani SA, Khalilzadeh S, Mohajerani SA, Hasanzad M, Sadr S, et al. IL-2-inducible T-cell kinase deficiency with pulmonary manifestations due to disseminated Epstein-Barr virus infection. Int Arch Allergy Immunol (2012) 158:418–22. doi:10.1159/000333472
69. Serwas NK, Cagdas D, Ban SA, Bienemann K, Salzer E, Tezcan I, et al. Identification of ITK deficiency as a novel genetic cause of idiopathic CD4+ T-cell lymphopenia. Blood (2014) 124:655–7. doi:10.1182/blood-2014-03-564930
70. Cipe FE, Aydogmus C, Serwas NK, Tugcu D, Demirkaya M, Bicici FA, et al. ITK deficiency: how can EBV be treated before lymphoma? Pediatr Blood Cancer (2015) 62:2247–8. doi:10.1002/pbc.25648
71. Cagdas D, Erman B, Hanoglu D, Tavil B, Kuskonmaz B, Aydin B, et al. Course of IL-2-inducible T-cell kinase deficiency in a family: lymphomatoid granulomatosis, lymphoma and allogeneic bone marrow transplantation in one sibling; and death in the other. Bone Marrow Transplant (2017) 52:126–9. doi:10.1038/bmt.2016.185
72. Stepensky P, Weintraub M, Yanir A, Revel-Vilk S, Krux F, Huck K, et al. IL-2-inducible T-cell kinase deficiency: clinical presentation and therapeutic approach. Haematologica (2011) 96:472–6. doi:10.3324/haematol.2010.033910
73. Andreotti AH, Schwartzberg PL, Joseph RE, Berg LJ. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb Perspect Biol (2010) 2:a002287. doi:10.1101/cshperspect.a002287
74. Liu KQ, Bunnell SC, Gurniak CB, Berg LJ. T cell receptor-initiated calcium release is uncoupled from capacitative calcium entry in Itk-deficient T cells. J Exp Med (1998) 187:1721–7. doi:10.1084/jem.187.10.1721
75. Fischer AM, Mercer JC, Iyer A, Ragin MJ, August A. Regulation of CXC chemokine receptor 4-mediated migration by the Tec family tyrosine kinase ITK. J Biol Chem (2004) 279:29816–20. doi:10.1074/jbc.M312848200
76. Schaeffer EM, Debnath J, Yap G, McVicar D, Liao XC, Littman DR, et al. Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science (1999) 284:638–41. doi:10.1126/science.284.5414.638
77. Kapnick SM, Stinchcombe JC, Griffiths GM, Schwartzberg PL. Inducible T cell kinase regulates the acquisition of cytolytic capacity and degranulation in CD8(+) CTLs. J Immunol (2017) 198:2699–711. doi:10.4049/jimmunol.1601202
78. Li FY, Chaigne-Delalande B, Kanellopoulou C, Davis JC, Matthews HF, Douek DC, et al. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature (2011) 475:471–6. doi:10.1038/nature10246
79. Chaigne-Delalande B, Li FY, O’Connor GM, Lukacs MJ, Jiang P, Zheng L, et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science (2013) 341:186–91. doi:10.1126/science.1240094
80. Brigida I, Chiriaco M, Di Cesare S, Cittaro D, Di Matteo G, Giannelli S, et al. Large deletion of MAGT1 gene in a patient with classic kaposi sarcoma, CD4 lymphopenia, and EBV infection. J Clin Immunol (2017) 37:32–5. doi:10.1007/s10875-016-0341-y
81. Dhalla F, Murray S, Sadler R, Chaigne-Delalande B, Sadaoka T, Soilleux E, et al. Identification of a novel mutation in MAGT1 and progressive multifocal leucoencephalopathy in a 58-year-old man with XMEN disease. J Clin Immunol (2015) 35:112–8. doi:10.1007/s10875-014-0116-2
82. Patiroglu T, Haluk Akar H, Gilmour K, Unal E, Akif Ozdemir M, Bibi S, et al. A case of XMEN syndrome presented with severe auto-immune disorders mimicking autoimmune lymphoproliferative disease. Clin Immunol (2015) 159:58–62. doi:10.1016/j.clim.2015.04.015
83. Li FY, Chaigne-Delalande B, Su H, Uzel G, Matthews H, Lenardo MJ. XMEN disease: a new primary immunodeficiency affecting Mg2+ regulation of immunity against Epstein-Barr virus. Blood (2014) 123:2148–52. doi:10.1182/blood-2013-11-538686
84. Cherepanova N, Shrimal S, Gilmore R. N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr Opin Cell Biol (2016) 41:57–65. doi:10.1016/j.ceb.2016.03.021
85. Li FY, Lenardo MJ, Chaigne-Delalande B. Loss of MAGT1 abrogates the Mg2+ flux required for T cell signaling and leads to a novel human primary immunodeficiency. Magnes Res (2011) 24:S109–14. doi:10.1684/mrh.2011.0286
86. Gotru SK, Gil-Pulido J, Beyersdorf N, Diefenbach A, Becker IC, Vogtle T, et al. Cutting edge: imbalanced cation homeostasis in MAGT1-deficient B Cells dysregulates B cell development and signaling in mice. J Immunol (2018) 200(8):2529–34. doi:10.4049/jimmunol.1701467
87. Pappworth IY, Wang EC, Rowe M. The switch from latent to productive infection in Epstein-Barr virus-infected B cells is associated with sensitization to NK cell killing. J Virol (2007) 81:474–82. doi:10.1128/JVI.01777-06
88. Obeidy P, Sharland AF. NKG2D and its ligands. Int J Biochem Cell Biol (2009) 41:2364–7. doi:10.1016/j.biocel.2009.07.005
89. van Montfrans JM, Hoepelman AI, Otto S, van Gijn M, van de Corput L, de Weger RA, et al. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J Allergy Clin Immunol (2012) 129:787–93.e6. doi:10.1016/j.jaci.2011.11.013
90. Alkhairy OK, Perez-Becker R, Driessen GJ, Abolhassani H, van Montfrans J, Borte S, et al. Novel mutations in TNFRSF7/CD27: clinical, immunologic, and genetic characterization of human CD27 deficiency. J Allergy Clin Immunol (2015) 136:703–12.e10. doi:10.1016/j.jaci.2015.02.022
91. Izawa K, Martin E, Soudais C, Bruneau J, Boutboul D, Rodriguez R, et al. Inherited CD70 deficiency in humans reveals a critical role for the CD70-CD27 pathway in immunity to Epstein-Barr virus infection. J Exp Med (2017) 214:73–89. doi:10.1084/jem.20160784
92. Salzer E, Daschkey S, Choo S, Gombert M, Santos-Valente E, Ginzel S, et al. Combined immunodeficiency with life-threatening EBV-associated lymphoproliferative disorder in patients lacking functional CD27. Haematologica (2013) 98:473–8. doi:10.3324/haematol.2012.068791
93. Klein U, Rajewsky K, Kuppers R. Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med (1998) 188:1679–89. doi:10.1084/jem.188.9.1679
94. Jacobs J, Deschoolmeester V, Zwaenepoel K, Rolfo C, Silence K, Rottey S, et al. CD70: an emerging target in cancer immunotherapy. Pharmacol Ther (2015) 155:1–10. doi:10.1016/j.pharmthera.2015.07.007
95. Tangye SG, Liu YJ, Aversa G, Phillips JH, de Vries JE. Identification of functional human splenic memory B cells by expression of CD148 and CD27. J Exp Med (1998) 188:1691–703. doi:10.1084/jem.188.9.1691
96. Hendriks J, Gravestein LA, Tesselaar K, van Lier RA, Schumacher TN, Borst J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol (2000) 1:433–40. doi:10.1038/80877
97. Borst J, Hendriks J, Xiao Y. CD27 and CD70 in T cell and B cell activation. Curr Opin Immunol (2005) 17:275–81. doi:10.1016/j.coi.2005.04.004
98. Abolhassani H, Edwards ES, Ikinciogullari A, Jing H, Borte S, Buggert M, et al. Combined immunodeficiency and Epstein-Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. J Exp Med (2017) 214:91–106. doi:10.1084/jem.20160849
99. Caorsi R, Rusmini M, Volpi S, Chiesa S, Pastorino C, Sementa AR, et al. CD70 deficiency due to a novel mutation in a patient with severe chronic EBV infection presenting as a periodic fever. Front Immunol (2017) 8:2015. doi:10.3389/fimmu.2017.02015
100. Martin E, Palmic N, Sanquer S, Lenoir C, Hauck F, Mongellaz C, et al. CTP synthase 1 deficiency in humans reveals its central role in lymphocyte proliferation. Nature (2014) 510:288–92. doi:10.1038/nature13386
101. Kucuk ZY, Zhang K, Filipovich L, Bleesing JJ. CTP synthase 1 deficiency in successfully transplanted siblings with combined immune deficiency and chronic active EBV infection. J Clin Immunol (2016) 36:750–3. doi:10.1007/s10875-016-0332-z
102. Truck J, Kelly DF, Taylor JM, Kienzler AK, Lester T, Seller A, et al. Variable phenotype and discrete alterations of immune phenotypes in CTP synthase 1 deficiency: report of 2 siblings. J Allergy Clin Immunol (2016) 138:1722–5.e6. doi:10.1016/j.jaci.2016.04.059
103. Evans DR, Guy HI. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J Biol Chem (2004) 279:33035–8. doi:10.1074/jbc.R400007200
104. Stray-Pedersen A, Jouanguy E, Crequer A, Bertuch AA, Brown BS, Jhangiani SN, et al. Compound heterozygous CORO1A mutations in siblings with a mucocutaneous-immunodeficiency syndrome of epidermodysplasia verruciformis-HPV, molluscum contagiosum and granulomatous tuberculoid leprosy. J Clin Immunol (2014) 34:871–90. doi:10.1007/s10875-014-0074-8
105. Shiow LR, Roadcap DW, Paris K, Watson SR, Grigorova IL, Lebet T, et al. The actin regulator coronin 1A is mutant in a thymic egress-deficient mouse strain and in a patient with severe combined immunodeficiency. Nat Immunol (2008) 9:1307–15. doi:10.1038/ni.1662
106. Moshous D, Martin E, Carpentier W, Lim A, Callebaut I, Canioni D, et al. Whole-exome sequencing identifies coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. J Allergy Clin Immunol (2013) 131:1594–603. doi:10.1016/j.jaci.2013.01.042
107. Punwani D, Pelz B, Yu J, Arva NC, Schafernak K, Kondratowicz K, et al. Coronin-1A: immune deficiency in humans and mice. J Clin Immunol (2015) 35:100–7. doi:10.1007/s10875-015-0130-z
108. Yee CS, Massaad MJ, Bainter W, Ohsumi TK, Foger N, Chan AC, et al. Recurrent viral infections associated with a homozygous CORO1A mutation that disrupts oligomerization and cytoskeletal association. J Allergy Clin Immunol (2016) 137:879–88.e2. doi:10.1016/j.jaci.2015.08.020
109. Moshous D, de Villartay JP. The expanding spectrum of human coronin 1A deficiency. Curr Allergy Asthma Rep (2014) 14:481. doi:10.1007/s11882-014-0481-1
110. Pieters J, Muller P, Jayachandran R. On guard: coronin proteins in innate and adaptive immunity. Nat Rev Immunol (2013) 13:510–8. doi:10.1038/nri3465
111. Foger N, Rangell L, Danilenko DM, Chan AC. Requirement for coronin 1 in T lymphocyte trafficking and cellular homeostasis. Science (2006) 313:839–42. doi:10.1126/science.1130563
112. Mueller P, Massner J, Jayachandran R, Combaluzier B, Albrecht I, Gatfield J, et al. Regulation of T cell survival through coronin-1-mediated generation of inositol-1,4,5-trisphosphate and calcium mobilization after T cell receptor triggering. Nat Immunol (2008) 9:424–31. doi:10.1038/ni1570
113. Mace EM, Orange JS. Lytic immune synapse function requires filamentous actin deconstruction by coronin 1A. Proc Natl Acad Sci U S A (2014) 111:6708–13. doi:10.1073/pnas.1314975111
114. Kaminski S, Hermann-Kleiter N, Meisel M, Thuille N, Cronin S, Hara H, et al. Coronin 1A is an essential regulator of the TGFbeta receptor/SMAD3 signaling pathway in Th17 CD4(+) T cells. J Autoimmun (2011) 37:198–208. doi:10.1016/j.jaut.2011.05.018
115. Siegmund K, Thuille N, Posch N, Fresser F, Baier G. Novel protein kinase C theta: coronin 1A complex in T lymphocytes. Cell Commun Signal (2015) 13:22. doi:10.1186/s12964-015-0100-3
116. Jayachandran R, Liu X, Bosedasgupta S, Muller P, Zhang CL, Moshous D, et al. Coronin 1 regulates cognition and behavior through modulation of cAMP/protein kinase A signaling. PLoS Biol (2014) 12:e1001820. doi:10.1371/journal.pbio.1001820
117. Pick R, Begandt D, Stocker TJ, Salvermoser M, Thome S, Bottcher RT, et al. Coronin 1A, a novel player in integrin biology, controls neutrophil trafficking in innate immunity. Blood (2017) 130:847–58. doi:10.1182/blood-2016-11-749622
118. Leiding JW, Holland SM. Warts and all: human papillomavirus in primary immunodeficiencies. J Allergy Clin Immunol (2012) 130:1030–48. doi:10.1016/j.jaci.2012.07.049
119. Winter S, Martin E, Boutboul D, Lenoir C, Boudjemaa S, Petit A, et al. Loss of RASGRP1 in humans impairs T-cell expansion leading to Epstein-Barr virus susceptibility. EMBO Mol Med (2018) 10(2):188–99. doi:10.15252/emmm.201708292
120. Salzer E, Cagdas D, Hons M, Mace EM, Garncarz W, Petronczki OY, et al. RASGRP1 deficiency causes immunodeficiency with impaired cytoskeletal dynamics. Nat Immunol (2016) 17:1352–60. doi:10.1038/ni.3575
121. Platt CD, Fried AJ, Hoyos-Bachiloglu R, Usmani GN, Schmidt B, Whangbo J, et al. Combined immunodeficiency with EBV positive B cell lymphoma and epidermodysplasia verruciformis due to a novel homozygous mutation in RASGRP1. Clin Immunol (2017) 183:142–4. doi:10.1016/j.clim.2017.08.007
122. Kortum RL, Rouquette-Jazdanian AK, Samelson LE. Ras and extracellular signal-regulated kinase signaling in thymocytes and T cells. Trends Immunol (2013) 34:259–68. doi:10.1016/j.it.2013.02.004
123. Hogquist K. RasGRP: the missing link for Ras activation in thymocytes. Trends Immunol (2001) 22:69. doi:10.1016/S1471-4906(00)01845-7
124. Roose JP, Mollenauer M, Gupta VA, Stone J, Weiss A. A diacylglycerol-protein kinase C-RasGRP1 pathway directs Ras activation upon antigen receptor stimulation of T cells. Mol Cell Biol (2005) 25:4426–41. doi:10.1128/MCB.25.11.4426-4441.2005
125. Warnecke N, Poltorak M, Kowtharapu BS, Arndt B, Stone JC, Schraven B, et al. TCR-mediated Erk activation does not depend on Sos and Grb2 in peripheral human T cells. EMBO Rep (2012) 13:386–91. doi:10.1038/embor.2012.17
126. Mao H, Yang W, Latour S, Yang J, Winter S, Zheng J, et al. RASGRP1 mutation in autoimmune lymphoproliferative syndrome-like disease. J Allergy Clin Immunol (2017). doi:10.1016/j.jaci.2017.10.026
127. Bartlett A, Buhlmann JE, Stone J, Lim B, Barrington RA. Multiple checkpoint breach of B cell tolerance in Rasgrp1-deficient mice. J Immunol (2013) 191:3605–13. doi:10.4049/jimmunol.1202892
128. Daley SR, Coakley KM, Hu DY, Randall KL, Jenne CN, Limnander A, et al. Rasgrp1 mutation increases naive T-cell CD44 expression and drives mTOR-dependent accumulation of Helios(+) T cells and autoantibodies. Elife (2013) 2:e01020. doi:10.7554/eLife.01020
129. Sun C, Molineros JE, Looger LL, Zhou XJ, Kim K, Okada Y, et al. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat Genet (2016) 48:323–30. doi:10.1038/ng.3496
130. Zhou XJ, Nath SK, Qi YY, Sun C, Hou P, Zhang YM, et al. Novel identified associations of RGS1 and RASGRP1 variants in IgA nephropathy. Sci Rep (2016) 6:35781. doi:10.1038/srep35781
131. Qu HQ, Grant SF, Bradfield JP, Kim C, Frackelton E, Hakonarson H, et al. Association of RASGRP1 with type 1 diabetes is revealed by combined follow-up of two genome-wide studies. J Med Genet (2009) 46:553–4. doi:10.1136/jmg.2009.067140
132. Pasquier B, Yin L, Fondaneche MC, Relouzat F, Bloch-Queyrat C, Lambert N, et al. Defective NKT cell development in mice and humans lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product. J Exp Med (2005) 201:695–701. doi:10.1084/jem.20042432
133. Gadue P, Stein PL. NK T cell precursors exhibit differential cytokine regulation and require Itk for efficient maturation. J Immunol (2002) 169:2397–406. doi:10.4049/jimmunol.169.5.2397
134. Felices M, Berg LJ. The Tec kinases Itk and Rlk regulate NKT cell maturation, cytokine production, and survival. J Immunol (2008) 180:3007–18. doi:10.4049/jimmunol.180.5.3007
135. Shen S, Chen Y, Gorentla BK, Lu J, Stone JC, Zhong XP. Critical roles of RasGRP1 for invariant NKT cell development. J Immunol (2011) 187:4467–73. doi:10.4049/jimmunol.1003798
136. Nichols KE, Hom J, Gong SY, Ganguly A, Ma CS, Cannons JL, et al. Regulation of NKT cell development by SAP, the protein defective in XLP. Nat Med (2005) 11:340–5. doi:10.1038/nm1189
137. Das R, Bassiri H, Guan P, Wiener S, Banerjee PP, Zhong MC, et al. The adaptor molecule SAP plays essential roles during invariant NKT cell cytotoxicity and lytic synapse formation. Blood (2013) 121:3386–95. doi:10.1182/blood-2012-11-468868
138. Gerart S, Siberil S, Martin E, Lenoir C, Aguilar C, Picard C, et al. Human iNKT and MAIT cells exhibit a PLZF-dependent proapoptotic propensity that is counterbalanced by XIAP. Blood (2013) 121:614–23. doi:10.1182/blood-2012-09-456095
139. Chung BK, Tsai K, Allan LL, Zheng DJ, Nie JC, Biggs CM, et al. Innate immune control of EBV-infected B cells by invariant natural killer T cells. Blood (2013) 122:2600–8. doi:10.1182/blood-2013-01-480665
140. Yuling H, Ruijing X, Li L, Xiang J, Rui Z, Yujuan W, et al. EBV-induced human CD8+ NKT cells suppress tumorigenesis by EBV-associated malignancies. Cancer Res (2009) 69:7935–44. doi:10.1158/0008-5472.CAN-09-0828
141. Xiao W, Li L, Zhou R, Xiao R, Wang Y, Ji X, et al. EBV-induced human CD8(+) NKT cells synergise CD4(+) NKT cells suppressing EBV-associated tumours upon induction of Th1-bias. Cell Mol Immunol (2009) 6:367–79. doi:10.1038/cmi.2009.48
142. Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, et al. IMMUNODEFICIENCIES. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science (2015) 349:606–13. doi:10.1126/science.aaa4282
143. Azzi T, Lunemann A, Murer A, Ueda S, Beziat V, Malmberg KJ, et al. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood (2014) 124:2533–43. doi:10.1182/blood-2014-01-553024
144. Vely F, Barlogis V, Vallentin B, Neven B, Piperoglou C, Ebbo M, et al. Evidence of innate lymphoid cell redundancy in humans. Nat Immunol (2016) 17:1291–9. doi:10.1038/ni.3553
145. Park GB, Kim YS, Lee HK, Song H, Cho DH, Lee WJ, et al. Endoplasmic reticulum stress-mediated apoptosis of EBV-transformed B cells by cross-linking of CD70 is dependent upon generation of reactive oxygen species and activation of p38 MAPK and JNK pathway. J Immunol (2010) 185:7274–84. doi:10.4049/jimmunol.1001547
146. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol (2017) 49:20–6. doi:10.1016/j.coi.2017.08.004
147. Boztug H, Hirschmugl T, Holter W, Lakatos K, Kager L, Trapin D, et al. NF-kappaB1 haploinsufficiency causing Immunodeficiency and EBV-driven lymphoproliferation. J Clin Immunol (2016) 36:533–40. doi:10.1007/s10875-016-0306-1
148. Nehme NT, Schmid JP, Debeurme F, Andre-Schmutz I, Lim A, Nitschke P, et al. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood (2012) 119:3458–68. doi:10.1182/blood-2011-09-378364
149. Abdollahpour H, Appaswamy G, Kotlarz D, Diestelhorst J, Beier R, Schaffer AA, et al. The phenotype of human STK4 deficiency. Blood (2012) 119:3450–7. doi:10.1182/blood-2011-09-378158
150. Du S, Scuderi R, Malicki DM, Willert J, Bastian J, Weidner N. Hodgkin’s and non-Hodgkin’s lymphomas occurring in two brothers with Wiskott-Aldrich syndrome and review of the literature. Pediatr Dev Pathol (2011) 14:64–70. doi:10.2350/10-01-0787-CR.1
151. Aydin SE, Kilic SS, Aytekin C, Kumar A, Porras O, Kainulainen L, et al. DOCK8 deficiency: clinical and immunological phenotype and treatment options – a review of 136 patients. J Clin Immunol (2015) 35:189–98. doi:10.1007/s10875-014-0126-0
152. Cohen JI. GATA2 deficiency and Epstein-Barr virus disease. Front Immunol (2017) 8:1869. doi:10.3389/fimmu.2017.01869
153. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: a large patient cohort study. J Allergy Clin Immunol (2016) 139(2):597–606.e4. doi:10.1016/j.jaci.2016.06.021
154. Newell A, Dadi H, Goldberg R, Ngan BY, Grunebaum E, Roifman CM. Diffuse large B-cell lymphoma as presenting feature of Zap-70 deficiency. J Allergy Clin Immunol (2011) 127:517–20. doi:10.1016/j.jaci.2010.09.016
155. Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med (2008) 358:2030–8. doi:10.1056/NEJMoa073966
156. Felgentreff K, Lee YN, Frugoni F, Du L, van der Burg M, Giliani S, et al. Functional analysis of naturally occurring DCLRE1C mutations and correlation with the clinical phenotype of ARTEMIS deficiency. J Allergy Clin Immunol (2015) 136:140–150.e7. doi:10.1016/j.jaci.2015.03.005
157. Marsh RA, Rao K, Satwani P, Lehmberg K, Muller I, Li D, et al. Allogeneic hematopoietic cell transplantation for XIAP deficiency: an international survey reveals poor outcomes. Blood (2013) 121:877–83. doi:10.1182/blood-2012-06-432500
158. Worth AJ, Nikolajeva O, Chiesa R, Rao K, Veys P, Amrolia PJ. Successful stem cell transplant with antibody-based conditioning for XIAP deficiency with refractory hemophagocytic lymphohistiocytosis. Blood (2013) 121:4966–8. doi:10.1182/blood-2013-01-478735
Keywords: immunodeficiencies, Epstein–Barr virus, lymphoproliferative disorders, genetic predisposition to disease, T lymphocytes, T lymphocyte activation
Citation: Latour S and Winter S (2018) Inherited Immunodeficiencies With High Predisposition to Epstein–Barr Virus-Driven Lymphoproliferative Diseases. Front. Immunol. 9:1103. doi: 10.3389/fimmu.2018.01103
Received: 15 February 2018; Accepted: 02 May 2018;
Published: 04 June 2018
Edited by:
Isabelle Meyts, KU Leuven, BelgiumReviewed by:
Jeffrey I. Cohen, National Institutes of Health (NIH), United StatesArndt Borkhardt, Heinrich Heine Universität Düsseldorf, Germany
Stuart G. Tangye, Garvan Institute of Medical Research, Australia
Copyright: © 2018 Latour and Winter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvain Latour, c3lsdmFpbi5sYXRvdXJAaW5zZXJtLmZy