 Zeina Dagher1
Zeina Dagher1 Shuying Xu1Paige E. Negoro1Nida S. Khan1,2
Shuying Xu1Paige E. Negoro1Nida S. Khan1,2 Michael B. Feldman3Jennifer L. Reedy1
Michael B. Feldman3Jennifer L. Reedy1 Jenny M. Tam1
Jenny M. Tam1 David B. Sykes4
David B. Sykes4 Michael K. Mansour1*
Michael K. Mansour1*
- 1Division of Infectious Disease, Massachusetts General Hospital, Harvard Medical School, Boston, MA, United States
- 2Biomedical Engineering and Biotechnology, University of Massachusetts Medical School, Worcester, MA, United States
- 3Division of Pulmonary and Critical Care, Massachusetts General Hospital, Boston, MA, United States
- 4Center for Regenerative Medicine, Massachusetts General Hospital, Boston, MA, United States
Macrophages play a critical role in the elimination of fungal pathogens. They are sensed via cell surface pattern-recognition receptors and are phagocytosed into newly formed organelles called phagosomes. Phagosomes mature through the recruitment of proteins and lysosomes, resulting in addition of proteolytic enzymes and acidification of the microenvironment. Our earlier studies demonstrated an essential role of Dectin-1-dependent activation of spleen tyrosine kinase (Syk) in the maturation of fungal containing phagosomes. The absence of Syk activity interrupted phago-lysosomal fusion resulting in arrest at an early phagosome stage. In this study, we sought to define the contribution of Syk to the control of phagocytosed live Candida glabrata in primary macrophages. To accurately measure intracellular yeast division, we designed a carboxyfluorescein succinimidyl ester (CFSE) yeast division assay in which bright fluorescent parent cells give rise to dim daughter cells. The CFSE-labeling of C. glabrata did not affect the growth rate of the yeast. Following incubation with macrophages, internalized CFSE-labeled C. glabrata were retrieved by cellular lysis, tagged using ConA-647, and the amount of residual CFSE fluorescence was assessed by flow cytometry. C. glabrata remained undivided (CFSE bright) for up to 18 h in co-culture with primary macrophages. Treatment of macrophages with R406, a specific Syk inhibitor, resulted in loss of intracellular control of C. glabrata with initiation of division within 4 h. Delayed Syk inhibition after 8 h was less effective indicating that Syk is critically required at early stages of macrophage–fungal interaction. In conclusion, we demonstrate a new method of tracking division of C. glabrata using CFSE labeling. Our results suggest that early Syk activation is essential for macrophage control of phagocytosed C. glabrata.
Introduction
The fungal pathogen Candida glabrata is a common cause of invasive bloodstream infections in immunocompromised patients (1). With the rise of organ transplantation, immunosuppressive chemotherapy, implanted medical devices, and antibacterial use, C. glabrata has emerged as a frequently isolated species, accounting for 20% of systemic bloodstream Candida-related infections in North America (2, 3).
In vitro, following interaction with human or murine macrophages, C. glabrata undergoes phagocytosis. Despite being internalized, C. glabrata survives and replicates inside host immune cells (4, 5). Intracellular killing of C. glabrata by macrophages is only about 10% effective (6). The capacity of this Candida species to evade the innate immune system complicates evaluation of intracellular pathogen control by simple phagocytosis assays. A more sensitive and accurate approach to quantifying C. glabrata intracellular proliferation is required to understand and identify the relevant pathogen–host interactions.
The carboxyfluorescein diacetate succinimidyl ester (CFSE) assay is a well-established method to fluorescently track lymphocyte cellular division (7, 8). The succinimidyl moiety of the dye covalently attaches to cellular aliphatic amine groups, forming stable amide bonds (9). This covalent-coupling dye allows for tracking of lymphocyte proliferation; each progressive cell division results in a twofold dilution of the fluorescent signal between daughter cells (10, 11). Unlike labeling techniques that rely on microscopic evaluation, CFSE can be visualized by both fluorescence microscopy and can be formally quantitated by flow cytometry (12). CFSE is highly fluorescent, has an excitation/emission profile at 491/518 nm, is stable in long-term tracking, and can be used in vitro and in vivo (10).
Although most published reports have used CFSE to monitor lymphocyte proliferation, recent studies have also used the dye to follow proliferation of other mammalian and non-mammalian cell types, such as bacterial cells and parasites (13, 14). CFSE also successfully labeled Paracoccidioides yeast in a flow cytometry based phagocytosis study, although the tracking of proliferation was not evaluated (15).
Here, we have applied the CFSE proliferation-tracking properties to C. glabrata, though several factors require special consideration. Unlike animal cells, fungi possess a cell wall composed of extracellular carbohydrate-rich polymers. CFSE brightly labels the cell wall, though does not permeate the yeast to label intracellular structures. Additionally, unlike mammalian cells where the cell membrane is shared symmetrically (in equal amounts) between daughter cells (16), the yeast cell wall is shared asymmetrically, with the budding yeast generating an almost-entirely new cell wall (17). This new (and CFSE-unlabeled) cell wall is synthesized at growing cell tips through the localization and activation of cell wall carbohydrate synthases (18, 19). Thus, unlike CFSE assays in lymphocytes, the CFSE labeling of yeast such as C. glabrata does not allow one to track beyond a single division.
Following infection, tissue macrophages sense fungi through the expression of pattern-recognition receptors (PRRs), such as Dectin-1 and Dectin-2, which recognize fungal cell wall carbohydrates (5, 20). Recognition through PRRs triggers phagocytosis, engulfing microorganisms within phagosomes, eventually fusing with additional subcellular compartments including lysosomes for acidification and killing (21). The process of acidification is termed phagosomal maturation, which in response to fungal cell wall ligands, is spleen tyrosine kinase (Syk)-dependent (22). Syk activation and signal transduction is central for immune activation in several immune cells (23). PRRs share the ability to associate with immunoreceptor tyrosine-based activation motifs (ITAM) in their cytoplasmic domains, leading to Syk recruitment and phosphorylation, resulting in downstream events including phagocytosis, and the production of reactive oxygen species (ROS) and proinflammatory cytokines (24–26). The central role of Syk in activation of several innate and adaptive immune cells has made this kinase a promising target for the development of anti-inflammatory therapeutics (27, 28).
Although the role of Syk in intracellular signaling has been studied extensively, its involvement in the response to intracellular pathogens in innate immune cells, such as macrophages, is not delineated (23, 26). In this study, we introduce a CFSE-labeling assay that allows for intracellular tracking of yeast proliferation in macrophage phagosomes. We demonstrate that CFSE is a reliable method of evaluating C. glabrata division by fluorescence microscopy and quantitatively by flow cytometry. Employing the CFSE assay, we show that Syk inhibition in macrophages results in the loss of the ability to control C. glabrata proliferation. This loss of C. glabrata division control is independent of phagocytosis and is not a result of failure to activate the Dectin-1 and Dectin-2 pathways. These observations highlight CFSE live labeling as a useful and convenient tool for measuring yeast proliferation accurately and support further investigation into the role of Syk in antifungal immunity.
Materials and Methods
Reagents
R406 was purchased from Santa Cruz Biotechnology (Dallas, TX, USA), Nonidet P40 (NP40) and peptone from American Bioanalytical (Natick, MA, USA), CFSE dye, and all other chemicals from Sigma (St. Louis, MO, USA) unless otherwise stated. Wild type C. glabrata (ATCC2001) was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cell lysis buffer consisted of 4× NP40 containing 40 mM Tris–hydrochloric acid, 600 mM sodium chloride, 20 mM magnesium chloride, and 4% NP40 titrated to pH 7.5. Yeast culture media liquid YPD contained 1% yeast extract (BD Biosciences, San Jose, CA, USA), 2% peptone, and 2% dextrose. Cell culture media (RPMI-complete) was composed of RPMI 1640 (Corning, Tewksbury, MA, USA) with 2 mM l-glutamine, 10% heat-inactivated fetal bovine serum, and 1% penicillin–streptomycin (ThermoFisher Scientific, Waltham MA, USA).
Yeast Culture and CFSE Staining
Candida glabrata were grown overnight shaking in liquid YPD at 30°C, washed three times in phosphate buffered saline (PBS), counted using a Luna automated cell counter (Logos Biosystems, Annandale, VA, USA), and resuspended in PBS at the desired inoculum.
To stain yeast with CFSE, wild type C. glabrata were grown to log phase in liquid YPD, washed twice in 1 mL PBS, and 160 million yeast were stained with 25 µg/mL CFSE for 30 min in 4 mL PBS at 30°C on a vertical rotator. Yeasts were washed twice in 10 mL PBS containing 2% bovine serum albumin (BSA) to remove excess CFSE. Stained yeast cells were resuspended in 1 mL PBS and passed through a 25G 7/8-inch needle 10 times to dissociate clumps into single cell suspension.
Calcofluor White Staining
To stain all intracellular yeast cells, macrophages were fixed and permeabilized with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) for 20 min at room temperature in the dark. Cells were washed three times with PBS and incubated in 10% calcofluor white solution in PBS for 15 min and imaged using confocal microscopy with 405 nm laser excitation.
Collection of Murine Peritoneal Macrophages
Eight-week-old inbred C57BL/6 mice (Jackson Laboratory, ME, USA), Dectin-1−/− (B6 background, a gift from Dr. Gordon Brown, University of Aberdeen, UK) (29), or Dectin-2−/− (B6 background, a gift from Dr. Marcel Wuethrich, University of Wisconsin) (30) were housed in a specific pathogen-free facility at the Massachusetts General Hospital (MGH, Boston, MA, USA). All animal experiments were approved by the MGH Institutional Animal Care and Use Committee. Peritoneal macrophages were isolated from mice following intraperitoneally instillation of 2 mL thioglycollate (Northeast Laboratory, Waterville, ME, USA). After 3 days, the mice were euthanized and the peritoneum lavaged with 10 mL RPMI-complete to retrieve cells. Intraperitoneal cells were washed and plated at a density of 300,000 cells per well in 500 µL RPMI-complete in 24-well dishes and allowed to adhere overnight. Non-adherent contaminating cells were washed away after 24 h resulting in a >95% pure, viable macrophage population as determine by anti-Mac 1 staining using flow cytometry (data not shown).
Co-Culture of Peritoneal Macrophages and CFSE-C. glabrata
The media in each 24-well was reduced to 250 µL and wells were treated with R406 dissolved in DMSO or DMSO only either 10 min prior to C. glabrata inoculation or 2, 4, or 8 h after C. glabrata inoculation. A total of 200,000 CFSE-labeled C. glabrata were added for a final MOI of 0.6. Plates were centrifuged for 1 min at 700 × g to facilitate cell-ligand contacts and incubated at 37°C with 5% CO2. After the time indicated, the macrophages were lysed with 100 µL of 4× NP40 on ice for 5 min. Well contents were centrifuged for 7.5 min at 10,000 rpm. The pellets were then fixed with 4% formaldehyde for 10 min in the dark and washed with PBS/2% BSA. Concanavalin A conjugated to Alexa Fluor 647 (ThermoFisher, Waltham, MA, USA) at a final concentration 3 µg/mL was added for 20 min in the dark to stain C. glabrata. Yeast were washed and resuspended in PBS/2% BSA for flow cytometry.
Macrophage Phagocytosis of C. glabrata
To assess macrophage phagocytosis, CFSE-C. glabrata were co-incubated with macrophages in the presence or absence of inhibitor for the time indicated. Plates were transferred onto ice, wells washed with PBS, then macrophages mechanically lifted, and strained through a 40-μm filter and immediately subjected to flow cytometry gating on the macrophage population. The phagocytosis percent was measured as the number of CFSE positive macrophages over the total macrophage cell number.
Reactive Oxygen Species
Reactive oxygen species was measured as described (31), briefly macrophages were plated at 5 × 104 cells/well in white wall 96-well plate (Costar, Cambridge, MA, USA). Cells were placed on ice and washed three times with PBS. Lucigenin solution (0.9 mM CaCl2, 0.5 mM MgCl2, 20 mM dextrose, and 20 µM lucigenin) was added, and cells were incubated on ice for 10 min. Heat-killed C. glabrata for 10 min were added at an effector to target ratio of 25, and then the plate was centrifuged at 750 × g for 1 min to facilitate cell-ligand contacts. An initial reading was then taken immediately after centrifugation for baseline. The plate was then incubated at 37°C and read with SpectraMax i3x reader (Molecular Devices, Sunnyvale, CA, USA) for total luminescence every 10 min for 2 h.
Confocal Microscopy
2 × 104 peritoneal macrophages were plated onto eight-chambered coverslip slides (LabTek, Thermo Scientific, Rochester, NY, USA) in RPMI-complete. CFSE-labeled C. glabrata were added at MOI 1:1, spun at 700 g × 1 min for immediate co-culture and slides mounted on a Nikon Ti-E inverted microscope equipped with an EM-CCD camera (Hamamatsu Photonics K.K., Hamamatsu, Japan). The excitation source was an 89 North MultiLine LaserBank (89 North, Burlington, VT, USA) A piezo stage (Prior Instruments, Rockland, MA, USA) capable of X, Y, and Z movement was used for acquisition. A polarizer (Nikon, MEN 51941) and Wollaston prisms (Nikon, MBH76190) were used to acquire differential interference contrast (DIC) images. Emission light from the samples was collected after passage through the appropriate emission filters (Semrock, Rochester, NY, USA) (32). Images were acquired using MetaMorph software (Molecular Devices, Downingtown, PA, USA), and processed using Adobe Photoshop CS5 and assembled in Adobe Illustrator, version CS5 (Adobe Systems, San Jose, CA, USA).
Flow Cytometric Analysis of CFSE
Carboxyfluorescein succinimidyl ester-labeled C. glabrata that were fixed and stained with concanavalin A-Alexa Fluor 647 were flowed using FACS Calibur flow cytometer (Becton-Dickinson, San Jose, CA, USA) and CellQuest software (Becton-Dickinson). ConA-647 (FL4) was plotted on the y-axis and CFSE (FL1) on the x-axis and the percentage of CFSE bright undivided population was determined with FlowJo 10 (FlowJo, Ashland, OR, USA).
Statistics
Statistical calculations were performed using GraphPad Prism 7 software. Data were analyzed by two-tailed, unpaired t test and were considered significantly different when p ≤ 0.05.
Results
CFSE Labeling Tracks Division of C. glabrata
We evaluated CFSE staining of C. glabrata using fluorescence microscopy. C. glabrata were homogenously fluorescent throughout the cell wall, likely due to the high content of fungal cell wall esterases that permit CFSE activation and labeling. The CFSE-labeled C. glabrata retained normal cell shape and morphology (Figure 1). Following 6 h in culture, CFSE-labeled C. glabrata were counter-stained with calcofluor white for chitin to delineate all yeast cells and visualized using confocal microscopy. All yeast cells were evenly stained with calcofluor white. Parent yeast cells were brightly CFSE-fluorescent, while daughter cells exhibited diluted CFSE leading to reduced fluorescence intensity as identified by flow cytometry (Figure 2A).
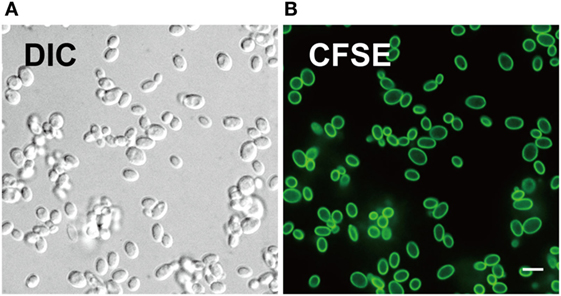
Figure 1. Carboxyfluorescein succinimidyl ester (CFSE) labels cell wall of Candida glabrata. (A) Differential interference contrast and (B) fluorescence confocal microscopy image of live C. glabrata stained with CFSE showing labeling of all yeast cells prior to cell division. Scale bar represents 5 µm.
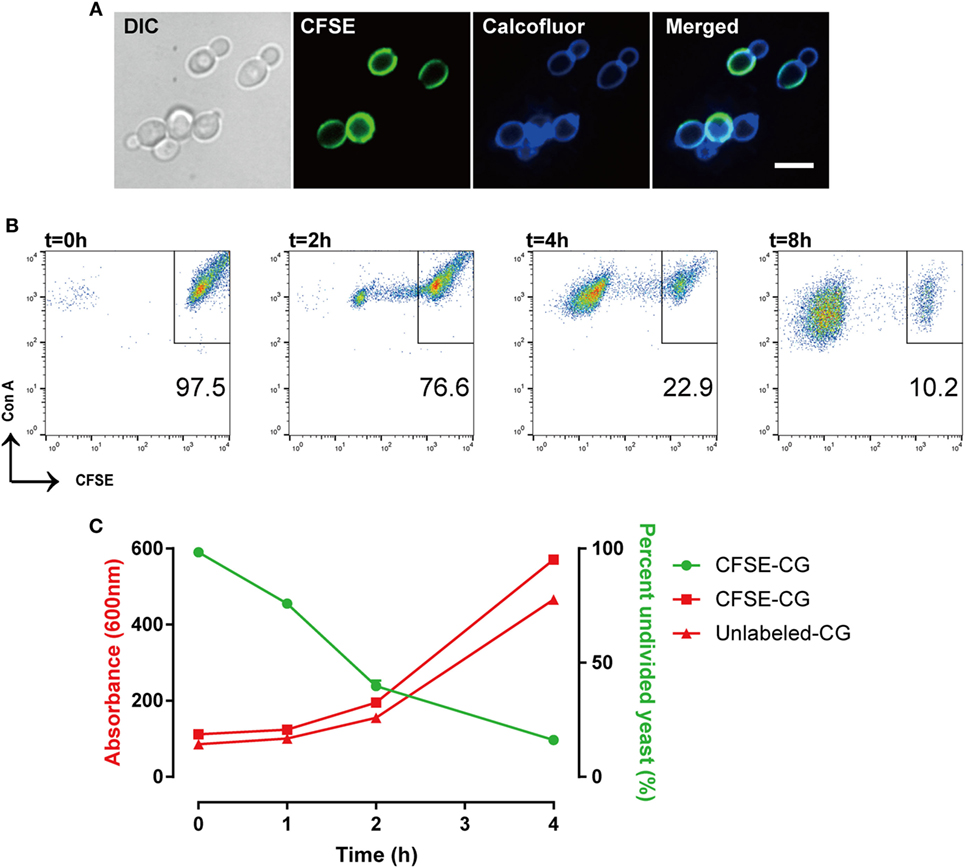
Figure 2. Carboxyfluorescein succinimidyl ester (CFSE)-labeling marks Candida glabrata yeast division. (A) Differential interference contrast and fluorescence microscopy image of CFSE-labeled C. glabrata that have undergone division and stained with calcofluor white. Parental cells exhibit full CFSE labeling, while daughter cells have diluted CFSE labeling leading to dimmer fluorescence signal. Scale bar represents 5 µm. (B) Scatter plot of Con A-AF647 and CFSE fluorescence. CFSE-bright parental yeast population in upper right quadrant gate. Percent (scatter plot inset) of undivided CFSE-bright population was determined over time indicated. (C) Correlation of percent CFSE-bright population to OD600 nm of proliferating yeast. Data represent a minimum of three independent triplicate experiments.
The consistent and predictable decrease in fluorescence permits quantitation of C. glabrata intracellular division as a marker of control by macrophages. While calcofluor allows visualization of all yeast cells using 405 nm laser excitation under confocal microscopy, our FACSCalibur is fitted with a blue and red laser incapable of exciting calcofluor. For these reasons, the total number of yeast were identified by labeling with concanavalin A conjugated to AF647 for flow cytometry. Therefore, a C. glabrata parent cell population is double-positive for CFSE and ConA-647, while divided daughter cells are CFSE-dim, but remain ConA-647 (Figure 2B).
With each cell division, the percent of the parent cells decrease, as daughter cell numbers rise and are easily distinguished via CFSE intensity. In fact, the diminished CFSE-bright parent cells was inversely proportional to the increase in the optical density of the growing yeast, confirming that the percentage of CFSE-bright cells accurately reflects yeast proliferation (Figure 2C). Moreover, CFSE-labeled and unlabeled C. glabrata proliferated at the same rate (as measured by OD600) indicating that the CFSE-labeling process does not affect yeast proliferation.
Macrophages Control Intracellular C. glabrata Proliferation Using Syk
To evaluate the role of Syk in macrophages following exposure to C. glabrata, we used an in vitro co-culture system. Peritoneal macrophages were co-cultured with CFSE-labeled C. glabrata parent cells and monitored for evidence of intracellular proliferation. In wild type murine macrophages, yeast cells were phagocytosed by macrophages with high efficiency. There were rare numbers of extracellular yeast remaining as measured by microscopy (Figure 3A) and flow cytometry analysis of co-culture supernatant (data not shown). Following co-culture incubation, analysis of CFSE intensity of C. glabrata from lysed macrophages revealed that yeast did not divide for up to 16 h as measured by retained parent CFSE-bright (Figure 3B, top panel). However, in the presence of the reversible Syk inhibitor, R406, macrophages were unable to control C. glabrata division (Figure 3B bottom panel). Compared to untreated controls, R406-treated macrophages resulted in a diminished number of parent yeast and in a growing numbers of divided daughter population. While these yeast remained intracellular, they had escaped control and undergone division as noted by loss of CFSE (Figure 3C). To ensure that R406 does not alter growth kinetics of C. glabrata, division rates of C. glabrata in the presence of R406 and was not different from yeast control conditions (data not shown). These data suggest that loss of Syk activity significantly impaired the ability of macrophages to control C. glabrata intracellular division.
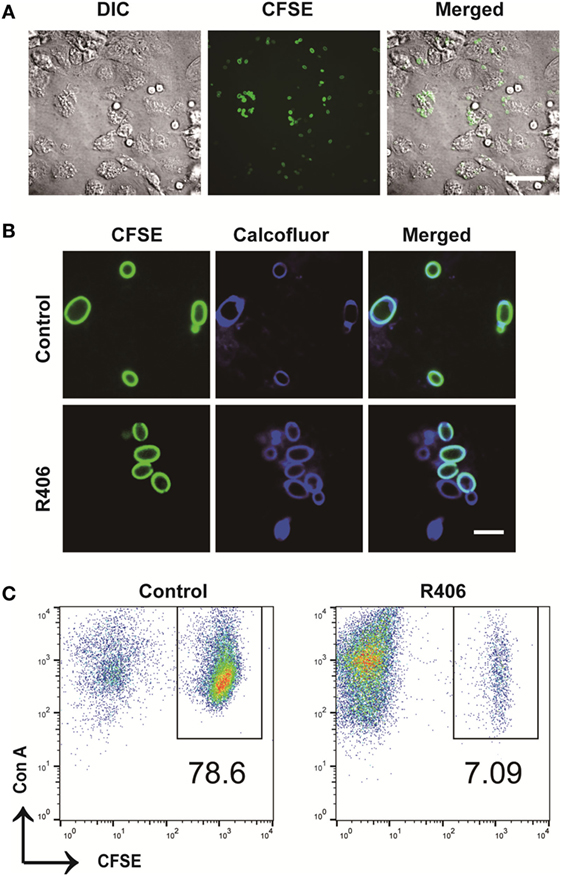
Figure 3. Spleen tyrosine kinase is required for peritoneal macrophage control of Candida glabrata. (A) Differential interference contrast (DIC) and fluorescence microscopy image of carboxyfluorescein succinimidyl ester (CFSE)-C. glabrata and macrophage after 1 h of coculture indicating complete uptake of the yeast by the macrophages. Scale bar represents 50 μm. (B) DIC and fluorescence microscopy image of CFSE C. glabrata infected macrophages 16 h post inoculation. Macrophages control C. glabrata division in CFSE-bright undivided state, while treatment with R406 impaired the macrophages’ ability in controlling C. glabrata. Scale bar represents 5 µm. (C) Flow cytometry scatter plots of Con A and CFSE showing macrophage controlling C. glabrata division and loss of control in R406-treated cells. Data represent three independent experiments.
Early Syk Activity Is Required for Intracellular C. glabrata Control
Given that Syk is essential for control of C. glabrata division, we next sought to define the temporal relationship between Syk activity and yeast control. Macrophages that had engulfed C. glabrata were exposed to R406 continuously or following a 2, 4, or 8-h delay from time of coincubation. Syk inhibition at the start, or 2- or 4-h resulted in a loss of macrophage control as evidenced by the significant decrease in the percent of undivided yeast (Figure 4). The delayed addition of R406 to 8 h, and beyond, did not have an influence C. glabrata division. These data suggest that it is the early Syk activity following macrophage phagocytosis of yeast that is required for intracellular C. glabrata control.
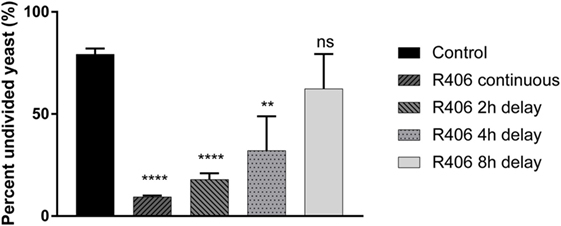
Figure 4. Spleen tyrosine kinase (Syk) is required at an early stage for macrophage control of Candida glabrata. Macrophages were treated with chemical Syk inhibitor, R406, prior to addition of C. glabrata for 2, 4, and 8 h after infection. All wells were lysed after 16 h of co-culture and the yeast retrieved and stained with ConA-AF647. Percent of parental CFSE-bright population was assessed via flow cytometry, with R406 treatments initiated at indicated time. **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. Data represent a minimum of three independent experiments.
Syk Inhibition Does Not Affect Macrophage Phagocytosis
In certain cell types, Syk has been shown to play a role in cytoskeletal rearrangement and endocytosis (33). If the process of macrophage phagocytosis was impaired by Syk inhibition (R406), this might leave extracellular C. glabrata in an actively dividing state. To determine if Syk inhibition impacts the process of phagocytosis of C. glabrata by peritoneal macrophages, we measured phagocytosis by flow cytometry in the presence and absence of R406. Cytochalasin D, an actin polymerization inhibitor, was used as a positive control inhibitor of phagocytosis.
We first determined the percent of macrophages that were associated with CFSE-bright C. glabrata. There was no difference in the ability of macrophages to phagocytose C. glabrata in the presence of R406 after 5 min (Figure 5). Macrophages that were treated with cytochalasin D showed dramatically reduced phagocytosis. The small percentage of yeast associated with cytochalasin D-treated macrophages as detected by flow possibly represents CFSE-bright C. glabrata attached extracellularly to the macrophage membrane or incomplete inhibition of phagocytosis by cytochalasin D. These findings indicate that Syk plays a minor role in the process of macrophage phagocytosis of C. glabrata.
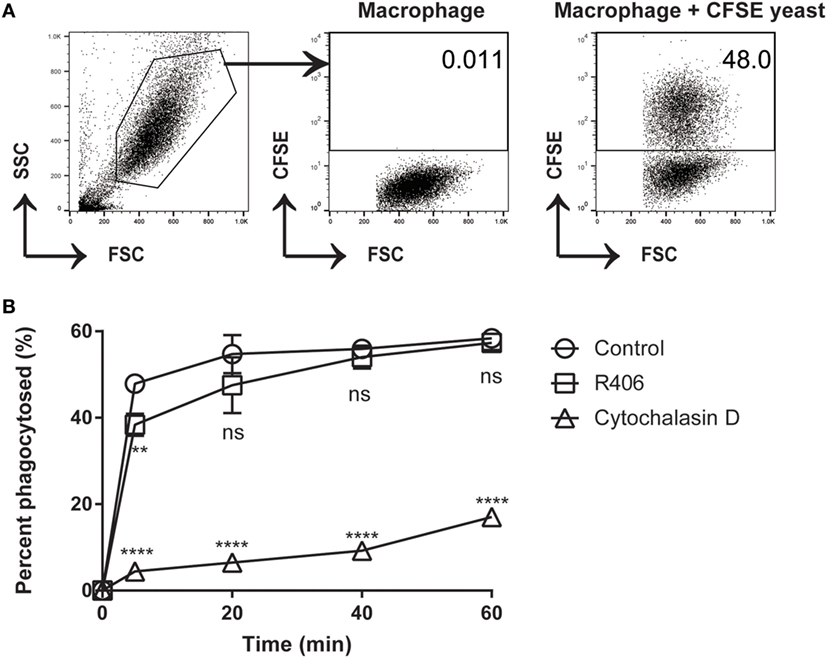
Figure 5. R406-induced spleen tyrosine kinase inhibition does not affect macrophage phagocytosis of C. glabrata. (A) Gating scheme for calculated percentage of macrophages that have phagocytosed CFSE-labeled C. glabrata by flow cytometry. (B) Percent phagocytosis of C. glabrata by macrophages over time incubated with control, R406, or cytochalasin D by flow cytometry. **p ≤ 0.01, ****p < 0.0001. Data represent three independent experiments.
R406 Inhibition of Syk Does Not Result in Permanent Macrophage Impairment
Given that Syk-mediated pathways are involved in multiple macrophage functions, we sought to determine if chemical Syk inhibition might result in a persistent hyporesponsive state. Given that ROS is a key mechanism in the response of macrophage to fungi, we evaluated ROS production in primary macrophages treated with R406 as compared to macrophages pulsed with R406 and then washed free of the inhibitor.
With constant R406, ROS production was completely suppressed (Figure 6). However, removal of R406 (following 2 h of exposure) restored ROS production to baseline levels. The observation that R406-pulsed macrophages were capable of generating robust ROS mirroring those of untreated control cells suggests that inhibition of Syk does not permanently alter the viability or biological activity of macrophages.
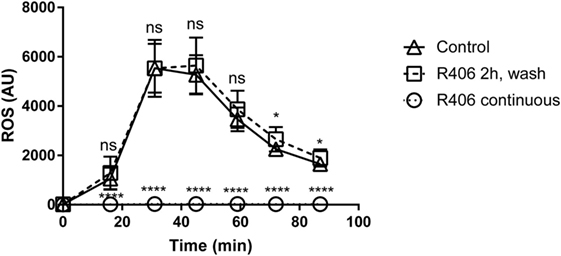
Figure 6. R406 does not result in permanent macrophage response defects to fungal stimuli as measured by reactive oxygen species (ROS) production. Plated macrophages were treated with R406 for 2 h, and then washed to remove the inhibitor. ROS production measured by lucigenin after addition of heat-killed Candida. Arbitrary light unites (AU) represents total luminescence corresponding with ROS production. *p ≤ 0.05, ****p ≤ 0.0001. Data represent a minimum of three independent experiments.
Dectin-1 and Dectin-2-Dependent Syk Activity Are Dispensable for Control of Intracellular C. glabrata Growth
Dectin-1 and Dectin-2 are upstream carbohydrate lectin PRRs that rely on Syk to regulate intracellular macrophage responses. To determine if the Syk activity required for intracellular C. glabrata control was derived through activation of Dectin-1 or Dectin-2, we compared peritoneal macrophages derived from wild type, to those derived from Dectin-1-deficient and Dectin-2-deficient mice. Following macrophage co-culture with CFSE-labeled C. glabrata, there was no difference in the control of intracellular C. glabrata growth between the macrophage genotypes signifying that the critical Syk activity required for yeast control is not related to individual Dectin-1 or Dectin-2 function (Figure 7).
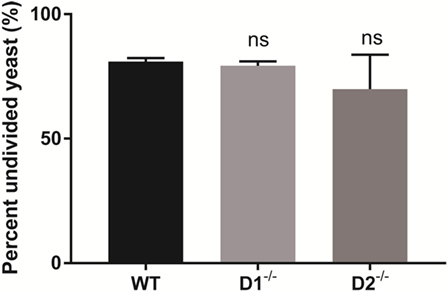
Figure 7. Spleen tyrosine kinase-mediated control of Candida glabrata is independent of Dectin-1 and Dectin-2. Percent undivided carboxyfluorescein succinimidyl ester-bright C. glabrata population from peritoneal macrophages-yeast co-cultures. Macrophages were obtained from wild type, Dectin-1−/−, and Dectin-2−/− mice. Comparisons between all groups are non-significant. Data represent a minimum of three independent experiments.
Discussion
Though the importance of the innate immune system in protecting against fungal pathogens has been articulated, defining the mechanisms responsible for the intracellular control of C. glabrata have yet to be defined. In this study, we define the role of Syk in the intracellular control of C. glabrata division in macrophages. Our approach outlines the CFSE-labeling technique as a new tool to track yeast division. We demonstrate that in macrophages, early Syk activity is essential for controlling C. glabrata division, and that this role is not solely dependent on the contribution of Dectin-1 or Dectin-2, both of which rely principally on Syk for intracellular signaling following ligand engagement.
We delineated a method of CFSE labeling that allows tracking of yeast multiplication. This process involved conjugating the cell wall of C. glabrata cells with CFSE, a fluorescent dye that links to amine groups through covalent bonds, and that can be visualized using confocal microscopy and flow cytometry. Microscopy demonstrated that CFSE does not change the morphology of C. glabrata cells. CFSE stained only the cell walls of parent yeasts, which permitted us to distinguish CFSE-bright parent populations and CFSE-dim daughter populations. The increase of CFSE-dim yeast cells, indicative of cell division and a growing daughter population, could be accurately measured by flow cytometry.
While CFSE assays have historically been used to follow the division of lymphocytes, our adaptation to track yeast division does face limitations. CFSE stains only the fungal cell wall rather than the contents of the cytoplasm, which allows us to track only a single generation of daughter cells. This observation is likely due to the presence of the esterase-rich fungal cell wall. Esterases are necessary for the activation of CFSE for covalent conjugation to amino groups on proteins. In mammalian cells, esterases are abundant within the cytoplasm, whereas in yeast, high levels of esterases are found within the fungal cell wall (34). This esterase distribution clarifies the reason for concentrated CFSE conjugation to the cell wall ultrastructure as opposed to homogenous cytoplasmic staining as seen in lymphocytes. Given the high concentration of CFSE in the cell wall, we confirmed that labeling did not alter the fungal cell wall or yeast division. By monitoring growth via optical density at 600 nm, we show that CFSE-labeled C. glabrata are capable of proliferating with normal kinetics as compared to unlabeled controls. While this method allows tracking of intracellular yeast division, it does not confirm killing of the yeast by macrophages.
Using CFSE-labeled yeast, we defined the role of Syk in the intracellular control of C. glabrata by macrophages. We hypothesized that Syk is indeed necessary for macrophages to maintain growth control of C. glabrata given its described contribution to phagolysosomal maturation (22) and autophagy (31). The downstream Syk-dependent molecular mechanisms critical for control of C. glabrata will require additional investigation and may involve sequestration of essential nutrients or disrupted fusion of lysosomal content. The C-type lectin receptors, Dectin-1 and Dectin-2, recognize fungi and are linked through Syk for intracellular signaling (35). We determined the contribution of each receptor to Syk activity in macrophages following Candida exposure. Data reveal that loss of either receptor does not impact the control of intracellular C. glabrata suggesting that Syk activity is stimulated through redundant pathways.
In addition to Syk being important for C. glabrata control, we also demonstrates the importance of timing of Syk activity. Early Syk activation, within the first 4 h after macrophage phagocytosis C. glabrata, is the essential window for effective control of C. glabrata division. Macrophages in which Syk was inhibited 2 or 4 h after addition of C. glabrata enabled the yeast to divide unchecked. Syk may have less relevance when inhibited 8 h after co-infection because the macrophages retain their ability to control yeast division in similar fashion to untreated macrophages. These observations may be due to time-dependent phagosomal mechanisms, which have no further consequence on C. glabrata division at later stages following phagocytosis. In addition, C. glabrata-specific immune evasion pathways may be upregulated at later time points, and these may bypass Syk-related control mechanisms. In this study, we have only evaluated chemical Syk inhibition, and have yet to test Syk knockout models.
Spleen tyrosine kinase has an established role in cytoskeletal changes, which could potentially impact phagocytosis. To control for this possibility, we measured uptake of C. glabrata by macrophages and found no difference in phagocytosis between control and R406-treated macrophages. This observation is in line with our previous observation using tyrosine mutations in the hemi-ITAM of Dectin-1, which result in complete loss of Syk phosphorylation, yet maintains near normal rates of phagocytosis (22). In addition, to address the likelihood of a lasting effect triggered through Syk inhibition, we utilized ROS activity as a surrogate marker of macrophage activity. After washout of R406, ROS activity is fully restored, indicating that transient Syk inhibition does not result in a long-lasting inhibition of macrophages, and does not account for loss of intracellular C. glabrata activity.
In conclusion, our results demonstrate that macrophages control the division of C. glabrata in a Syk-dependent manner. A limitation of this observation is the use of a single ATCC strain and further investigation is required to determine if Syk-dependent control is generalizable to other Candida species. These observations were verified by a CFSE-labeling technique that accurately distinguished CFSE-bright parent and CFSE-dim daughter cell populations through flow cytometric analysis or microscopy. We show that Syk in macrophages is essential in the early stages of C. glabrata infection and that Dectin-1 or Dectin-2 are not individual contributors to Syk activity. Further investigations are required to define the precise downstream mechanisms of Syk activity in the intracellular control of fungal pathogens.
Ethics Statement
This study was carried out in accordance with the recommendations of Massachusetts General Hospital Institutional Animal Care and Use Committee. The protocol was approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee.
Author Contributions
ZD, SX, PN, MM, and DS contributed to experimental design. ZD, SX, PN, JT, and MM executed the experiments. ZD, SX, PN, NK, MF, JR, JT, DS, and MM performed analysis and interpretation of experiments. ZD, SX, PN, and MM contributed in writing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported, in whole or in part, by NIH NIAID T32 AI007061 (to JR), NIH NIAID K08 AI110655 (to MM), and K08 CA201640 (to DS).
References
1. Sardi JC, Scorzoni L, Bernardi T, Fusco-Almeida AM, Mendes Giannini MJ. Candida species: current epidemiology, pathogenicity, biofilm formation, natural antifungal products and new therapeutic options. J Med Microbiol (2013) 62(Pt 1):10–24. doi:10.1099/jmm.0.045054-0
2. Rodrigues CF, Silva S, Henriques M. Candida glabrata: a review of its features and resistance. Eur J Clin Microbiol Infect Dis (2014) 33(5):673–88. doi:10.1007/s10096-013-2009-3
3. Silva S, Henriques M, Hayes A, Oliveira R, Azeredo J, Williams DW. Candida glabrata and Candida albicans co-infection of an in vitro oral epithelium. J Oral Pathol Med (2011) 40(5):421–7. doi:10.1111/j.1600-0714.2010.00981.x
4. Kaur R, Ma B, Cormack BP. A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc Natl Acad Sci U S A (2007) 104(18):7628–33. doi:10.1073/pnas.0611195104
5. Kasper L, Seider K, Hube B. Intracellular survival of Candida glabrata in macrophages: immune evasion and persistence. FEMS Yeast Res (2015) 15(5):fov042. doi:10.1093/femsyr/fov042
6. Ifrim DC, Bain JM, Reid DM, Oosting M, Verschueren I, Gow NA, et al. Role of Dectin-2 for host defense against systemic infection with Candida glabrata. Infect Immun (2014) 82(3):1064–73. doi:10.1128/IAI.01189-13
7. Filby A, Begum J, Jalal M, Day W. Appraising the suitability of succinimidyl and lipophilic fluorescent dyes to track proliferation in non-quiescent cells by dye dilution. Methods (2015) 82:29–37. doi:10.1016/j.ymeth.2015.02.016
8. Tario JD Jr, Humphrey K, Bantly AD, Muirhead KA, Moore JS, Wallace PK. Optimized staining and proliferation modeling methods for cell division monitoring using cell tracking dyes. J Vis Exp (2012) (70):e4287. doi:10.3791/4287
9. Weston SA, Parish CR. New fluorescent dyes for lymphocyte migration studies. Analysis by flow cytometry and fluorescence microscopy. J Immunol Methods (1990) 133(1):87–97. doi:10.1016/0022-1759(90)90322-M
10. Hawkins ED, Hommel M, Turner ML, Battye FL, Markham JF, Hodgkin PD. Measuring lymphocyte proliferation, survival and differentiation using CFSE time-series data. Nat Protoc (2007) 2(9):2057–67. doi:10.1038/nprot.2007.297
11. Lastovicka J, Rataj M, Bartunkova J. Assessment of lymphocyte proliferation for diagnostic purpose: comparison of CFSE staining, Ki-67 expression and (3)H-thymidine incorporation. Hum Immunol (2016) 77(12):1215–22. doi:10.1016/j.humimm.2016.08.012
12. Parish CR. Fluorescent dyes for lymphocyte migration and proliferation studies. Immunol Cell Biol (1999) 77(6):499–508. doi:10.1046/j.1440-1711.1999.00877.x
13. Ludington JG, Ward HD. The Cryptosporidium parvum C-type lectin CpClec mediates infection of intestinal epithelial cells via interactions with sulfated proteoglycans. Infect Immun (2016) 84(5):1593–602. doi:10.1128/IAI.01410-15
14. Ueckert JE, Nebe von-Caron G, Bos AP, ter Steeg PF. Flow cytometric analysis of Lactobacillus plantarum to monitor lag times, cell division and injury. Lett Appl Microbiol (1997) 25(4):295–9. doi:10.1046/j.1472-765X.1997.00225.x
15. Batista VG, Toledo MS, Straus AH, Mendes-Giannini MJ, Duarte AJ, Takahashi HK, et al. Glycolipid sensing and innate immunity in paracoccidioidomycosis. Mycopathologia (2014) 178(3–4):153–62. doi:10.1007/s11046-014-9783-z
16. Bocharov G, Luzyanina T, Cupovic J, Ludewig B. Asymmetry of cell division in CFSE-based lymphocyte proliferation analysis. Front Immunol (2013) 4:264. doi:10.3389/fimmu.2013.00264
17. Seider K, Brunke S, Schild L, Jablonowski N, Wilson D, Majer O, et al. The facultative intracellular pathogen Candida glabrata subverts macrophage cytokine production and phagolysosome maturation. J Immunol (2011) 187(6):3072–86. doi:10.4049/jimmunol.1003730
18. Davi V, Minc N. Mechanics and morphogenesis of fission yeast cells. Curr Opin Microbiol (2015) 28:36–45. doi:10.1016/j.mib.2015.07.010
19. Piel M, Tran PT. Cell shape and cell division in fission yeast. Curr Biol (2009) 19(17):R823–7. doi:10.1016/j.cub.2009.08.012
20. Hohl TM, Rivera A, Pamer EG. Immunity to fungi. Curr Opin Immunol (2006) 18(4):465–72. doi:10.1016/j.coi.2006.05.003
21. Collette JR, Lorenz MC. Mechanisms of immune evasion in fungal pathogens. Curr Opin Microbiol (2011) 14(6):668–75. doi:10.1016/j.mib.2011.09.007
22. Mansour MK, Tam JM, Khan NS, Seward M, Davids PJ, Puranam S, et al. Dectin-1 activation controls maturation of beta-1,3-glucan-containing phagosomes. J Biol Chem (2013) 288(22):16043–54. doi:10.1074/jbc.M113.473223
23. Turner M, Schweighoffer E, Colucci F, Di Santo JP, Tybulewicz VL. Tyrosine kinase SYK: essential functions for immunoreceptor signalling. Immunol Today (2000) 21(3):148–54. doi:10.1016/S0167-5699(99)01574-1
24. Miller YI, Choi SH, Wiesner P, Bae YS. The SYK side of TLR4: signalling mechanisms in response to LPS and minimally oxidized LDL. Br J Pharmacol (2012) 167(5):990–9. doi:10.1111/j.1476-5381.2012.02097.x
25. Underhill DM, Rossnagle E, Lowell CA, Simmons RM. Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood (2005) 106(7):2543–50. doi:10.1182/blood-2005-03-1239
26. Yi YS, Son YJ, Ryou C, Sung GH, Kim JH, Cho JY. Functional roles of Syk in macrophage-mediated inflammatory responses. Mediators Inflamm (2014) 2014:270302. doi:10.1155/2014/270302
27. Geahlen RL. Getting Syk: spleen tyrosine kinase as a therapeutic target. Trends Pharmacol Sci (2014) 35(8):414–22. doi:10.1016/j.tips.2014.05.007
28. Ruzza P, Biondi B, Calderan A. Therapeutic prospect of Syk inhibitors. Expert Opin Ther Pat (2009) 19(10):1361–76. doi:10.1517/13543770903207039
29. Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol (2007) 8(1):31–8. doi:10.1038/ni1408
30. Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, et al. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity (2010) 32(5):681–91. doi:10.1016/j.immuni.2010.05.001
31. Tam JM, Mansour MK, Khan NS, Seward M, Puranam S, Tanne A, et al. Dectin-1-dependent LC3 recruitment to phagosomes enhances fungicidal activity in macrophages. J Infect Dis (2014) 210(11):1844–54. doi:10.1093/infdis/jiu290
32. Reedy JL, Negoro PE, Feliu M, Lord AK, Khan NS, Lukason DP, et al. The carbohydrate lectin receptor dectin-1 mediates the immune response to Exserohilum rostratum. Infect Immun (2017) 85(3):e903–16. doi:10.1128/IAI.00903-16
33. Le Roux D, Lankar D, Yuseff MI, Vascotto F, Yokozeki T, Faure-Andre G, et al. Syk-dependent actin dynamics regulate endocytic trafficking and processing of antigens internalized through the B-cell receptor. Mol Biol Cell (2007) 18(9):3451–62. doi:10.1091/mbc.E06-12-1114
34. Peter J, Armstrong D, Lyman CA, Walsh TJ. Use of fluorescent probes to determine MICs of amphotericin B and caspofungin against Candida spp. and Aspergillus spp. J Clin Microbiol (2005) 43(8):3788–92. doi:10.1128/JCM.43.8.3788-3792.2005
Keywords: Candida, Candida glabrata, macrophages, spleen tyrosine kinase, phagosome, carboxyfluorescein succinimidyl ester
Citation: Dagher Z, Xu S, Negoro PE, Khan NS, Feldman MB, Reedy JL, Tam JM, Sykes DB and Mansour MK (2018) Fluorescent Tracking of Yeast Division Clarifies the Essential Role of Spleen Tyrosine Kinase in the Intracellular Control of Candida glabrata in Macrophages. Front. Immunol. 9:1058. doi: 10.3389/fimmu.2018.01058
Received: 06 March 2018; Accepted: 27 April 2018;
Published: 16 May 2018
Edited by:
Steven Templeton, Indiana University School of Medicine-Terre Haute, United StatesReviewed by:
Jeniel E. Nett, University of Wisconsin-Madison, United StatesAttila Gacser, University of Szeged, Hungary
Copyright: © 2018 Dagher, Xu, Negoro, Khan, Feldman, Reedy, Tam, Sykes and Mansour. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael K. Mansour, bWttYW5zb3VyQG1naC5oYXJ2YXJkLmVkdQ==