 Pedro Santos e Sousa
Pedro Santos e Sousa Clare L. Bennett
Clare L. Bennett Ronjon Chakraverty
Ronjon Chakraverty- 1UCL Cancer Institute, University College London, London, United Kingdom
- 2UCL Institute of Immunity and Transplantation, University College London, London, United Kingdom
The skin is the most common target organ affected by graft-versus-host disease (GVHD), with severity and response to therapy representing important predictors of patient survival. Although many of the initiating events in GVHD pathogenesis have been defined, less is known about why treatment resistance occurs or why there is often a permanent failure to restore tissue homeostasis. Emerging data suggest that the unique immune microenvironment in the skin is responsible for defining location- and context-specific mechanisms of injury that are distinct from those involved in other target organs. In this review, we address recent advances in our understanding of GVHD biology in the skin and outline the new research themes that will ultimately enable design of precision therapies.
Introduction
Graft-versus-host disease (GVHD) is a severe immune-related complication of allogeneic hematopoietic stem cell transplantation (allo-HSCT), affecting 30–50% of transplanted patients and associated with both treatment resistance and excess mortality (1). Recognized by Billingham as a syndrome in which donor T cells recognize and attack host tissues in an immunocompromised recipient (2), GVHD has a wide range of clinical manifestations involving predominantly the skin, gastrointestinal tract, liver, and lungs. Although GVHD has been classically divided into acute or chronic subtypes depending on the time of its clinical onset, it is now accepted that the kinetics of acute and chronic GVHD are variable and that disease features frequently overlap (3). It is, therefore, not clear if acute and chronic GVHD are indeed sequential phases of the same disease or two independent disorders, with unique molecular and pathophysiological mechanisms (4–7). Although biomarkers have been identified that can predict acute and chronic GVHD onset and severity (8–14), it is unclear whether earlier therapeutic interventions in patients predicted to develop severe GVHD can change its natural history.
The skin is the most frequently affected organ both in acute and chronic GVHD (15, 16), and the cutaneous manifestations are often the presenting sign of the disease (17); the extent of involvement correlates with prognosis either by acting as a surrogate for GVHD severity at other sites (18), or more directly, by leading to loss of essential barrier functions and infections. In its acute form, the most prominent skin features include the sudden development of a symmetrical maculopapular rash, predominantly on the upper back and neck, the palms/soles, and face, which may progress to a diffuse erythroderma with the formation of bullae and epidermal necrosis (19). Histologically, cutaneous acute GVHD is characterized by an interface dermatitis, with extensive leukocyte infiltration of the superficial dermis and vacuolization of the epidermal basal layer (20). Apoptosis of individual keratinocytes along the basement membrane, including a subset of cytokeratin 15-expressing putative stem cells is a characteristic feature of acute injury (21). In contrast, cutaneous chronic GVHD may present a wide range of manifestations reflecting a spectrum of epidermal (lichenoid) and dermal (sclerodermatous) changes (22). Lichen planus-like eruptions and poikiloderma affecting the dorsal aspects of the hands, forearms, and trunk are the most typical features of early stage non-sclerotic cutaneous chronic GVHD, its histopathological hallmarks being hyperkeratosis, focal hypergranulosis, acanthosis, basal cell necrosis, vacuolar degeneration of the basal layer, and a distinctive superficial perivascular or band-like lymphoid cell infiltrate with perifollicular fibrosis (20). Sclerodermatous lesions usually develop progressively over the trunk, buttocks, hips, and thighs, with localized morphea-like features, diffuse sclerosis, or lichen sclerosus-like features, characterized by collagen homogenization of the dermis and/or subcutaneous tissues (17, 20). In this mini-review, we will address the pathophysiology of cutaneous GVHD as addressed mostly using models of experimental allo-HSCT in mice, highlighting recent advances in the field and discussing some of the key challenges that remain.
Overview of GVHD Pathogenesis
The pathogenesis of systemic acute and chronic GVHD has been extensively characterized in preclinical models and the reader is referred to several, excellent reviews (23–27). Briefly, initiation of acute GVHD is triggered by tissue injury as a result of the preparatory conditioning regimens leading to increased exposure to damage- or pathogen-associated molecules that activate host antigen-presenting cells (APC) (28). Host APC activation primes the proliferation of alloantigen-specific donor T cells and their migration toward target sites leading to immune-mediated injury through a broad array of cytotoxic and cytokine dependent mechanisms, a process that is amplified by the recruitment of additional effector populations. It is increasingly recognized that distortions to the local gut microbiome exacerbate injury by further reducing epithelial integrity at a time when repair mechanisms are also perturbed (28, 29). The mechanisms underlying chronic GVHD are multifaceted and include thymic injury and disordered T cell selection, loss of regulatory populations, disruption to B cell homeostasis, and aberrant tissue repair with fibrosis (30).
Although the skin is the major site of involvement, only a minority of experimental studies focus specifically upon distinct mechanisms that underpin injury at this location. Analysis of the kinetics of gene expression associated with the development of cutaneous GVHD in rodents has suggested complex spatial and temporal transcriptional signatures linked to inflammation, antigen presentation, effector cell recruitment and activation, apoptosis, and tissue repair, indicative of a sequential infiltration and activation of effector cells that correlate with in situ morphological alterations (31–33). Assembling this information into a comprehensive framework explaining the pathogenesis of skin GVHD and suggesting novel, targetable pathways remains a considerable challenge. Below, we review the existing literature as it relates to cutaneous GVHD and indicate those areas where we believe that critical information is still lacking (Figure 1).
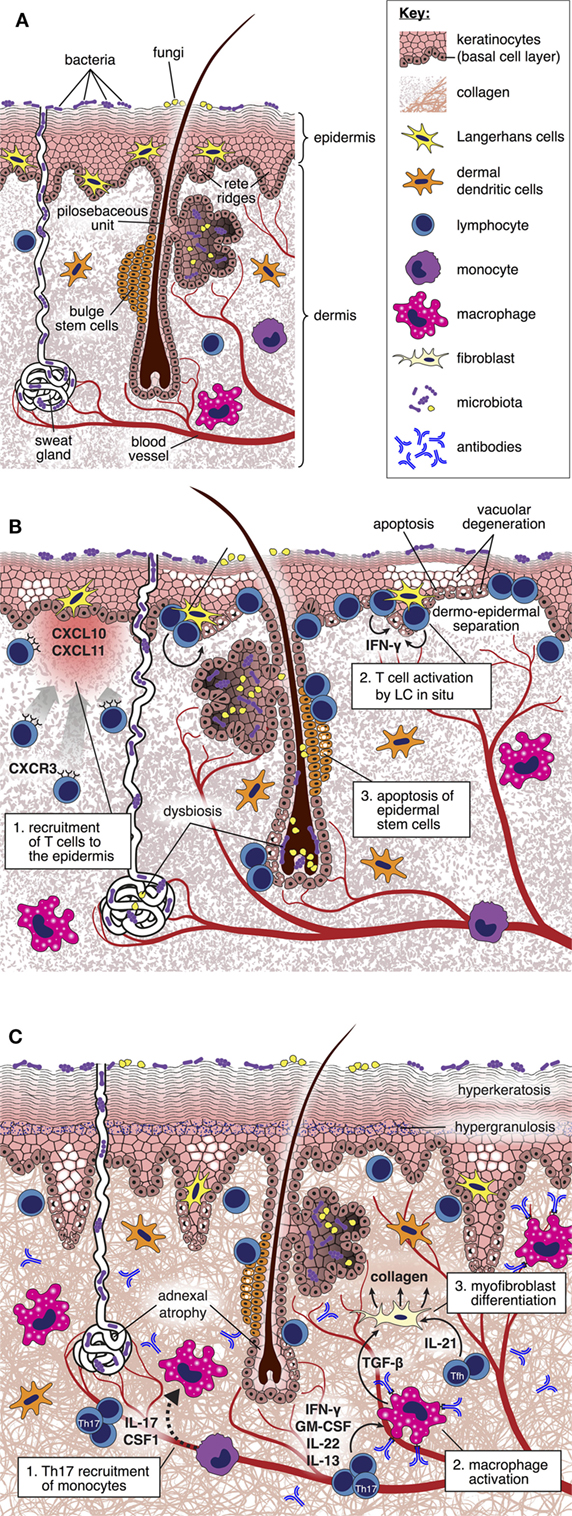
Figure 1. Pathogenesis of cutaneous graft-versus-host disease (GVHD). (A) Diagram depicting cellular and structural elements of normal skin. (B) During acute GVHD, the local production of IFN-γ inducible chemokines in response to tissue injury promotes the recruitment of CXCR3+ alloreactive T cells into the skin (34). Upon in situ interaction with host-type Langerhans cells, alloreactive T cells are reprogramed to differentiate into pathogenic effector cells characterized by enhanced survival and generation of IFN-γ (35). Pathogenic T cells kill Lgr5+ epidermal stem cells (36), thus disabling normal repair mechanisms and epidermopoiesis; these effects are manifested by vacuolar degeneration and apoptosis of the basal and suprabasal epidermal cells, focal dermo-epidermal separation, and necrosis of the epidermis with denudation. Distortion of the local microbiome as a consequence of the conditioning regimen and antibiotic therapy may also be an important factor driving skin inflammation (37). (C) During the evolution of chronic GVHD, donor T cell-derived IL-17 is required for recruitment of Ly6Clow monocytes to the skin, which then differentiate into macrophages (38). Generation of transforming growth factor (TGF)-β by activated macrophages induces fibroblasts to differentiate into myofibroblasts that promote collagen deposition through a heat shock protein 47-dependent mechanism (39). Binding of allo- or auto-antibodies to macrophage Fc receptors induces their polarization toward the M2 phenotype and production of TGF-β (27), thus amplifying the process of myofibroblast generation of collagen. IL-21 generation by follicular helper-like cells recruited to the skin may also have direct effects upon skin fibroblast differentiation (40). Collectively, these mechanisms lead to thickening and homogenization of collagen bundles throughout the dermis.
Pathogenic Mechanisms in Skin GVHD
The Role of T Cells
An extensive summary of the role of individual T cell subsets and cytokines in GVHD pathogenesis is beyond the scope of this discussion, but we refer readers to recent reviews by experts in the field (27, 41). In murine experimental systems, no single T cell differentiation pathway can explain the full spectrum of tissue injury at all sites; plasticity and heterogeneity of early T cell effector programs (35, 42), variation in housing conditions, clinical relevance of the mouse models employed (43), and the potential impact of immune suppressants that are used in the clinic (44) all complicate this issue further. In addition, experiments using genetic approaches to block differentiation down one pathway may distort the overall balance of differentiation across other subsets, making the results often difficult to interpret (45). Finally, cytokine profiling of human T cells derived from skin lesions from patients with acute or chronic GVHD reveal a greater level of heterogeneity than suggested by the animal models, cautioning against underestimating the complexity of this disorder (34, 46).
Proliferative, Th1 cell responses were long considered to represent the paradigm for T cell differentiation in acute GVHD (47, 48); in experimental BMT, donor T cell responsiveness to the Th1-related cytokine, IFN-γ, is in turn required for CXCR3 expression and recruitment to the skin, suggesting a feed-forward loop that drives injury (34). The requirement of Th1 differentiation in driving acute GVHD has been challenged by numerous studies, which have demonstrated that Th2 or Th17 polarized cells can also induce acute skin injury in mice (45, 49–51). For example, a skewing toward Th2/Tc2 differentiation in Stat4−/− splenocyte donors increases acute GVHD of the skin (52). In vitro polarized Th17 cells also induce greater acute GVHD of the skin when transferred to allo-HSCT recipient mice (50), akin to their pathogenic role in other autoimmune and skin inflammatory diseases, such as psoriasis (53), scleroderma (54), and lichen planus (55). In a murine model of acute GVHD where we tracked the evolution of effector T cell gene expression according to time and location, we recently showed that transcriptional profiles of CD8+ effector T cells in the epidermis were highly distinct from other GVHD sites (including the dermis) and did not readily conform to previously published Tc1 or Tc17 signatures (35). Taken together, these data suggest that there is substantial redundancy in the requirements for acute skin injury in GVHD and/or that the precise molecular mechanisms leading to pathogenicity do not segregate exclusively with the “classic” differentiation pathways. Reciprocal to enhancement of effector T cell expansion, acute GVHD is also associated with an early deficiency in Treg cells, a process that contributes to skin injury (56). In a human skin explant model that mimics acute GVHD, Treg cells block the priming of CD8+ effector T cells (57) and their subsequent homing to skin by reducing T cell expression of CXCR3 and skin-expression of CXCL10/CXCL11 (58).
Loss of Treg cells during experimental acute GVHD may also be permissive for the subsequent emergence of pathogenic autoreactive donor T cells capable of inducing chronic tissue damage (59, 60). Consistent with this concept, therapeutic use of low dose IL-2 in human patients with chronic GVHD enhances Treg expansion and is associated with significant improvements in skin disease (61). In human allo-HSCT patients, chronic lichenoid GVHD is characterized by a mixed Th1/Th17 signature, a profile that is distinct from that observed in early lesions where Th2 and IL-22-producing T cells were found to predominate (46). Interestingly, while recipient-derived IL-17 and IL-22 are protective against acute GVHD in mice, preventing gut dysbiosis and helping maintaining epithelial integrity in the gastrointestinal tract (62, 63), dysregulated production of the same cytokines by donor-derived T cells drive pathogenicity in chronic skin GVHD, leading to abnormal differentiation of keratinocytes and dermal fibrosis (44, 51). Skin-infiltration by ICOS+ follicular helper (Tfh)-like CD4+ T cells and ICOS+ Th1 cells may also be important in driving skin fibrosis in murine chronic GVHD, the latter process requiring the generation of the Tfh-related cytokine, IL-21 (40); of note, antibody-mediated depletion of ICOS-expressing T cells or IL-21 blockade was able to block collagen deposition. Like acute GVHD, the human and murine studies in chronic GVHD are indicative of substantial heterogeneity of T cells recruited to the skin; a priority, therefore, will be to define pathways that are both pathogenic and targetable.
The Role of APCs
Although host-derived hematopoietic APC have been proposed to act as central players in acute GVHD initiation (64, 65), further studies using stringent conditional ablation of individual BM-derived APC subsets in mice have failed to show an absolute requirement of any single population for the induction of systemic disease (24); indeed, there appears to be considerable redundancy in the requirement for professional APC, especially, for CD4+ T cell-dependent models of GVHD where non-hematopoietic cells are sufficient to induce widespread tissue injury (66).
An area under active investigation is the role of tissue APC populations, particularly, within target organs affected by the disease. Through their functions as sensors capable of integrating complex environmental cues, resident, or recruited APC are well placed to influence effector T cell functions as they enter peripheral tissues. In the context of infection, cognate interactions with monocyte-derived CD11c+ dendritic cells recruited to inflamed tissues are necessary for amplification of local cytokine generation and proliferation of activated T cells in mice (67, 68). It is possible that this process becomes corrupted during the development of GVHD, a concept that is supported by the involvement of other CD11c+ APC populations recruited to the sites of T-cell mediated immunopathology in autoimmune (69–71) or infection-related (72) inflammation. In the skin, the role of epidermal Langerhans cells (LCs) has received significant attention because this population is radio-resistant and host-derived LC can persist long term following allo-HSCT in human patients especially following transplantation involving T cell depletion or reduced intensity conditioning (73). In an MHC-mismatched model of murine allo-HSCT, host LCs were shown to be capable of inducing acute skin GVHD (74), thus overriding their steady-state role in promoting tolerance (75). However, whether host LC populations are required for initiation of acute GVHD under conditions where other APC populations can efficiently present host antigens is controversial. In human patients, the persistence of recipient-derived LC does not predict clinical or histological skin GVHD (76), although this does not exclude a role of this population is initiating inflammation. Using models of inducible depletion in mice, we have found that host LC can “instruct” local T cell pathogenicity in an MHC-mismatched model of allo-HSCT where local GVHD was triggered by local application of a toll-like receptor agonist (77) and in several independent models of MHC-matched, minor antigen mismatched allo-HSCT (35). Host LCs were not required for activation of CD8+ T cells within the draining lymph node or subsequent homing of effector cells to the epidermis. Instead, epidermal LC triggered pathogenic differentiation of T cells in situ, an effect that was linked to the expression of a pro-inflammatory gene cluster that was also conserved in human patients at the onset of acute GVHD (35). In contrast, another study using Langerin.DTA BMT recipients (a model in which recipients lack LC through their lifespan) found that skin GVHD was unaffected in the absence of LC (78). However, these latter experimental findings are difficult to interpret because long-term absence of LC may be associated with abnormal baseline immunity and exaggerated T cell immunity (79). The finding that keratinocytes overexpressing a self-antigen can also activate naive CD8+ T cells directly and induce a local GVHD-like inflammation (80) suggests the possibility that allogeneic T cells entering the skin can potentially be regulated by multiple cell types via cognate interactions; in this case, the outcome of the interaction could be specific to the T cell clone or to the antigen involved.
Recruitment of other myeloid populations to the skin may also have the potential to influence local immunity. In models of acute systemic GVHD, early persistence of host-derived macrophages can limit GVHD through a process involving the CD47-dependent engulfment of allo-reactive T cells (81). Similarly, in collaboration with the Rubio group, we have found that granulocyte-colony stimulating factor (G-CSF)-mobilized peripheral blood stem cell grafts (which are used routinely in clinical practice) contain high frequencies of a variant, immunosuppressive CD34+ mature monocyte population that inhibits allogeneic T cell proliferation and correlates with a reduced risk of GVHD (82). A similar variant Ly6Chigh monocyte population can be found in the spleens of G-CSF-treated mice (82, 83); upon activation with IFN-γ, these cells produce NO that inhibits T cell function and also prevents GVHD. In contrast, emerging data support the concept that donor-derived monocytes and macrophages can also amplify GVHD. In human patients with GVHD, intermediate CD14++CD16+ monocytes can promote the induction of a subset of Th17 cells that are resistant to glucocorticoids (38). Furthermore, several clinical studies have reported a strong correlation between cutaneous macrophage infiltration and acute and chronic GVHD, in particular, in cases associated with steroid resistance and poor prognosis (84). In preclinical models of Th17-dependent chronic sclerodermatous GVHD, donor-derived M2-like macrophages infiltrate the skin in a macrophage colony-stimulating factor (CSF)-1 and CSF-1 receptor-dependent manner and contribute to dermal sclerosis via a mechanism that requires transforming growth factor (TGF)-β (85, 86). A recent study in mice has shown that TGF-β drives the differentiation of fibroblasts into collagen-producing myofibroblasts that express the collagen molecular chaperone, heat shock protein 47 (HSP47); of note, knock down of HSP47 can significantly block the process of collagen deposition in chronic GVHD (39). The immediate precursor population that traffics to the skin and gives rise to the macrophage population is not known, but appears to be distinct from Ly6Chigh inflammatory-type monocytes, which are usually thought to perform this role. Like LC, a sub-population of CD1a− CD14+ Factor XIIIa+-resident dermal macrophages are resistant to the conditioning therapy, retaining a mixed host–donor chimerism for over 1 year following allo-HSCT (87); ex vivo, this population is capable of regulating CD4+ T cell cytokine generation and promoting memory CD8+ T cell proliferation, suggesting their potential to sustain the alloreactive response (87).
The Role of B Cells and Antibodies
Profound disruption to B cell homeostasis is seen in chronic GVHD in human patients, with reduced reconstitution of memory B cells or IL-10 producing regulatory B cells and reciprocal increases in activated transitional B cells (88–90). B-cell supporting structures (e.g., the germinal center), cytokines (e.g., B-cell activating factor), and accessory populations (e.g., Tfh and regulatory populations) are also affected by chronic GVHD in murine studies. Although not required to initiate tissue injury, donor-derived B cell antibodies deposit in murine skin, where they promote cutaneous Th17 infiltration, leading to perpetuation of local chronic GVHD (91). In humans with chronic GVHD, auto-antibodies to platelet-derived growth factor induce accumulation of reactive oxygen species (ROS), and stimulate type 1 collagen gene expression through the Ha-Ras-ERK1/2-ROS signaling pathway (92); this process may be important in initiating the pathological fibrotic response. These findings provide the rationale for targeting B cells using Rituximab or using drugs that interfere with B-cell signaling to prevent or treat GVHD, the latter approach being associated with promising early results (93).
The Role of Repair Mechanisms
Ongoing inflammation and/or damage to normal repair processes lead to a profound failure to restore normal skin tissue homeostasis in untreated GVHD. In models of acute GVHD, early tissue responses including new blood vessel (94) and lymphatic formation (95) are abnormal even before the influx of T cells. Furthermore, the normal restoration of epithelial integrity through proliferation and differentiation of adjacent epidermal stem cells within the hair follicle or inter-follicular epithelium does not occur (96). Instead, the epidermal proliferative response is uncoupled from differentiation (97) leading to dysregulation of epidermopoiesis. This abnormal differentiation program leads to the induction of genes that are not expressed by keratinocytes in the normal skin and the abnormal overexpression of differentiation markers (31), in a similar manner to that described in other inflammatory skin disorders, such as psoriasis and atopic dermatitis (98, 99). Experimental and human studies in acute GVHD have shown that apoptotic cells in the epidermis are spatially restricted to sites that harbor putative epidermal stem cells in the hair follicle bulge or human rete ridges, or rete-like prominences (RLP) found in the murine dorsal tongue (100–103). A recent study by the Teshima group has demonstrated that leucine-rich repeat-containing G-protein coupled receptor (Lgr)5+ epidermal stem cells in the RLP of the murine dorsal tongue and bulge region of hair follicles are targeted early following the initiation of GVHD (36). Loss of epidermal stem cells in GVHD led to both impairment in the normal progression to hair follicle growth (anagen) and a reduced capacity for wound healing (36). Of note, use of topical corticosteroids compounded loss of Lgr5+ cells in the epithelium, whereas inhibition of JAK1/2 with Ruxolitinib was effective at blocking T cell infiltration and restoring Lgr5+ cell populations. It is, therefore, likely that the disordered attempt at repair in GVHD relates significantly to the targeting of stem cells necessary to restore epithelial integrity. The role of other factors relevant to disordered repair requires further investigation but could include examining the impact of the inflammatory microenvironment upon basement membrane integrity, the constitution of the extracellular matrix, the availability of local differentiation signals, and the effect upon accessory populations.
The Role of the Skin Microbiome
A large number of recent studies have detailed the changes in the gut microbiome during the development of acute GVHD in human patients (29), where intestinal injury is associated with loss of diversity and increases in pathogenic bacteria. In murine models, intestinal dysbiosis in GVHD can disrupt epithelial integrity by interfering with the formation of mucus or the intraluminal generation of short chain fatty acids that fortify gap junctions (104–107). Other changes in the human gut virome during GVHD are also emerging (108). To date, the impact of GVHD upon the skin microbiota and its potential role in promoting injury has not been defined. In the steady state, the human skin microbiome is highly stable (with Propionobacterium, Corynebacterium, Staphylococcus, and Melassezia being the most prominent genera) and possesses a significantly smaller biomass than the gut (109). Relative acidity and the presence of antimicrobial molecules, such as free fatty acids and antimicrobial peptides that inhibit microbial colonization, make conditions unfavorable for the growth of certain microorganisms (109). Conditioning, the use of antibiotics, inflammation, and relative immune deficiency all have the potential to distort the local microbiota. Skin-resident microorganisms have been shown to control the expression of different antimicrobial peptides, such as cathelicidins and β-defensins (110), as well as components of the complement system (111) while enhancing the induction and/or activation of lymphocytes both in the steady state and during infection (112, 113). In a murine model of atopic dermatitis, for instance, it has been found that dysbiosis is an important pathological factor driving skin inflammation, with the emergence of Staphylococcus aureus and Corynebacterium bovis as dominant populations being correlated with the induction of dermatitis and the enhancement of Th2 responses, respectively, in a process mediated by LC (114). The association between alterations in the diversity and composition of the skin microbiota has now been reported in a variety of other inflammatory skin conditions; however, for most of them, it is not yet clear whether these are the cause or a consequence of the disease (37). Studies in gut GVHD have provided an experimental framework for testing the role of specific changes to the microbiota in the skin and testing the feasibility of its manipulation therapeutically.
Concluding Remarks
Precision-based therapies for skin GVHD will require an in depth understanding of tissue-specific processes that initiate or amplify disease. There remains a significant information gap in relation to the specific pathogenesis of skin GVHD because only a minority of preclinical studies focus primarily on injury to this site. The distinct immune microenvironment of the skin and the likely context-specific interactions between multiple cell types cautions against extrapolation of mechanisms pertinent to other sites involved by GVHD. Future research should also focus upon pathological immune responses as measured in tissues rather than secondary lymphoid organs, and refine models to include components that reflect the complexity of the clinical setting (for example, with the use of prophylactic immune suppression or antibiotics). Corroborative evidence that mechanisms identified in preclinical models are conserved in human patients will be essential in selection of potential targetable pathways. Ultimately, the next step change will derive from unbiased, systems-level approaches interrogating whole tissues to identify candidate mechanisms that drive inflammation.
Author Contributions
PS, CB, and RC formulated opinions and concepts for the mini-review and wrote it together.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the following research grants: RC, CB (Bloodwise, 12006); CB (Biotechnology and Biological Sciences Research Council, BB/L001608/1); PS (Fundação para a Ciência e a Tecnologia, SFRH/BD/76516/2011).
References
1. Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet (2009) 373(9674):1550–61. doi:10.1016/S0140-6736(09)60237-3
3. Filipovich AH, Weisdorf D, Pavletic S, Socie G, Wingard JR, Lee SJ, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant (2005) 11(12):945–56. doi:10.1016/j.bbmt.2005.09.004
4. Shulman HM, Sullivan KM, Weiden PL, McDonald GB, Striker GE, Sale GE, et al. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med (1980) 69(2):204–17. doi:10.1016/0002-9343(80)90380-0
5. Hamilton BL, Parkman R. Acute and chronic graft-versus-host disease induced by minor histocompatibility antigens in mice. Transplantation (1983) 36(2):150–5. doi:10.1097/00007890-198308000-00008
6. Snover DC. Acute and chronic graft versus host disease: histopathological evidence for two distinct pathogenetic mechanisms. Hum Pathol (1984) 15(3):202–5. doi:10.1016/S0046-8177(84)80180-X
7. Toubai T, Sun Y, Reddy P. GVHD pathophysiology: is acute different from chronic? Best Pract Res Clin Haematol (2008) 21(2):101–17. doi:10.1016/j.beha.2008.02.005
8. Vander Lugt MT, Braun TM, Hanash S, Ritz J, Ho VT, Antin JH, et al. ST2 as a marker for risk of therapy-resistant graft-versus-host disease and death. N Engl J Med (2013) 369(6):529–39. doi:10.1056/NEJMoa1213299
9. Ahmed SS, Wang XN, Norden J, Pearce K, El-Gezawy E, Atarod S, et al. Identification and validation of biomarkers associated with acute and chronic graft versus host disease. Bone Marrow Transplant (2015) 50(12):1563–71. doi:10.1038/bmt.2015.191
10. Yu J, Storer BE, Kushekhar K, Abu Zaid M, Zhang Q, Gafken PR, et al. Biomarker panel for chronic graft-versus-host disease. J Clin Oncol (2016) 34(22):2583–90. doi:10.1200/JCO.2015.65.9615
11. Abu Zaid M, Wu J, Wu C, Logan BR, Yu J, Cutler C, et al. Plasma biomarkers of risk for death in a multicenter phase 3 trial with uniform transplant characteristics post-allogeneic HCT. Blood (2017) 129(2):162–70. doi:10.1182/blood-2016-08-735324
12. Hartwell MJ, Ozbek U, Holler E, Renteria AS, Major-Monfried H, Reddy P, et al. An early-biomarker algorithm predicts lethal graft-versus-host disease and survival. JCI Insight (2017) 2(3):e89798. doi:10.1172/jci.insight.89798
13. Weissinger EM, Human C, Metzger J, Hambach L, Wolf D, Greinix HT, et al. The proteome pattern cGvHD_MS14 allows early and accurate prediction of chronic GvHD after allogeneic stem cell transplantation. Leukemia (2017) 31(3):654–62. doi:10.1038/leu.2016.259
14. Major-Monfried H, Renteria AS, Pawarode A, Reddy P, Ayuk F, Holler E, et al. MAGIC biomarkers predict long term outcomes for steroid-resistant acute GVHD. Blood (2018). doi:10.1182/blood-2018-01-822957
15. Ratanatharathorn V, Nash RA, Przepiorka D, Devine SM, Klein JL, Weisdorf D, et al. Phase III study comparing methotrexate and tacrolimus (prograf, FK506) with methotrexate and cyclosporine for graft-versus-host disease prophylaxis after HLA-identical sibling bone marrow transplantation. Blood (1998) 92(7):2303–14.
16. Flowers ME, Parker PM, Johnston LJ, Matos AV, Storer B, Bensinger WI, et al. Comparison of chronic graft-versus-host disease after transplantation of peripheral blood stem cells versus bone marrow in allogeneic recipients: long-term follow-up of a randomized trial. Blood (2002) 100(2):415–9. doi:10.1182/blood-2002-01-0011
17. Strong Rodrigues K, Oliveira-Ribeiro C, de Abreu Fiuza Gomes S, Knobler R. Cutaneous graft-versus-host disease: diagnosis and treatment. Am J Clin Dermatol (2018) 19(1):33–50. doi:10.1007/s40257-017-0306-9
18. Palmer J, Chai X, Pidala J, Inamoto Y, Martin PJ, Storer B, et al. Predictors of survival, nonrelapse mortality, and failure-free survival in patients treated for chronic graft-versus-host disease. Blood (2016) 127(1):160–6. doi:10.1182/blood-2015-08-662874
19. Aractingi S, Carosella ED, Gluckman E. [Cutaneous manifestations of graft versus host reaction]. Ann Dermatol Venereol (1998) 125(9):620–7.
20. Vargas-Diez E, Garcia-Diez A, Marin A, Fernandez-Herrera J. Life-threatening graft-vs-host disease. Clin Dermatol (2005) 23(3):285–300. doi:10.1016/j.clindermatol.2004.06.005
21. Zhan Q, Korngold R, Lezcano C, McKeon F, Murphy GF. Graft-versus-host disease-related cytokine-driven apoptosis depends on p73 in cytokeratin 15-positive target cells. Biol Blood Marrow Transplant (2012) 18(6):841–51. doi:10.1016/j.bbmt.2012.02.004
22. Shulman HM, Sale GE, Lerner KG, Barker EA, Weiden PL, Sullivan K, et al. Chronic cutaneous graft-versus-host disease in man. Am J Pathol (1978) 91(3):545–70.
23. Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol (2012) 12(6):443–58. doi:10.1038/nri3212
24. Markey KA, MacDonald KP, Hill GR. The biology of graft-versus-host disease: experimental systems instructing clinical practice. Blood (2014) 124(3):354–62. doi:10.1182/blood-2014-02-514745
25. Teshima T, Reddy P, Zeiser R. Acute graft-versus-host disease: novel biological insights. Biol Blood Marrow Transplant (2016) 22(1):11–6. doi:10.1016/j.bbmt.2015.10.001
26. Cooke KR, Luznik L, Sarantopoulos S, Hakim FT, Jagasia M, Fowler DH, et al. The biology of chronic graft-versus-host disease: a task force report from the national institutes of health consensus development project on criteria for clinical trials in chronic graft-versus-host disease. Biol Blood Marrow Transplant (2017) 23(2):211–34. doi:10.1016/j.bbmt.2016.09.023
27. MacDonald KPA, Hill GR, Blazar BR. Chronic graft-versus-host disease: biological insights from preclinical and clinical studies. Blood (2017) 129(1):13–21. doi:10.1182/blood-2016-06-686618
28. Penack O, Holler E, van den Brink MR. Graft-versus-host disease: regulation by microbe-associated molecules and innate immune receptors. Blood (2010) 115(10):1865–72. doi:10.1182/blood-2009-09-242784
29. Staffas A, Burgos da Silva M, van den Brink MR. The intestinal microbiota in allogeneic hematopoietic cell transplant and graft-versus-host disease. Blood (2017) 129(8):927–33. doi:10.1182/blood-2016-09-691394
30. Socie G, Ritz J. Current issues in chronic graft-versus-host disease. Blood (2014) 124(3):374–84. doi:10.1182/blood-2014-01-514752
31. Sugerman PB, Faber SB, Willis LM, Petrovic A, Murphy GF, Pappo J, et al. Kinetics of gene expression in murine cutaneous graft-versus-host disease. Am J Pathol (2004) 164(6):2189–202. doi:10.1016/S0002-9440(10)63776-5
32. Zhou L, Askew D, Wu C, Gilliam AC. Cutaneous gene expression by DNA microarray in murine sclerodermatous graft-versus-host disease, a model for human scleroderma. J Invest Dermatol (2007) 127(2):281–92. doi:10.1038/sj.jid.5700517
33. Novota P, Zinocker S, Norden J, Wang XN, Sviland L, Opitz L, et al. Expression profiling of major histocompatibility and natural killer complex genes reveals candidates for controlling risk of graft versus host disease. PLoS One (2011) 6(1):e16582. doi:10.1371/journal.pone.0016582
34. Choi J, Ziga ED, Ritchey J, Collins L, Prior JL, Cooper ML, et al. IFNgammaR signaling mediates alloreactive T-cell trafficking and GVHD. Blood (2012) 120(19):4093–103. doi:10.1182/blood-2012-01-403196
35. Santos E Sousa P, Cire S, Conlan T, Jardine L, Tkacz C, Ferrer IR, et al. Peripheral tissues reprogram CD8+ T cells for pathogenicity during graft-versus-host disease. JCI Insight (2018) 3(5):e97011. doi:10.1172/jci.insight.97011
36. Takahashi S, Hashimoto D, Hayase E, Ogasawara R, Ohigashi H, Ara T, et al. Ruxolitinib protects skin stem cells and maintains skin homeostasis in murine graft-versus-host disease. Blood (2018). doi:10.1182/blood-2017-06-792614
37. Rodrigues Hoffmann A. The cutaneous ecosystem: the roles of the skin microbiome in health and its association with inflammatory skin conditions in humans and animals. Vet Dermatol (2017) 28(1):60–e15. doi:10.1111/vde.12408
38. Reinhardt-Heller K, Hirschberg I, Lang P, Vogl T, Handgretinger R, Bethge WA, et al. Increase of intermediate monocytes in graft-versus-host disease: correlation with MDR1(+)Th17.1 levels and the effect of prednisolone and 1alpha,25-dihydroxyvitamin D3. Biol Blood Marrow Transplant (2017) 23(12):2057–64. doi:10.1016/j.bbmt.2017.08.008
39. Yamakawa T, Ohigashi H, Hashimoto D, Hayase E, Takahashi S, Miyazaki M, et al. Vitamin A-coupled liposomes containing siRNA against HSP47 ameliorate skin fibrosis in chronic graft-versus-host disease. Blood (2018) 131(13):1476–85. doi:10.1182/blood-2017-04-779934
40. Taylor DK, Mittereder N, Kuta E, Delaney T, Burwell T, Dacosta K, et al. T follicular helper-like cells contribute to skin fibrosis. Sci Transl Med (2018) 10(431):eaaf5307. doi:10.1126/scitranslmed.aaf5307
41. Henden AS, Hill GR. Cytokines in graft-versus-host disease. J Immunol (2015) 194(10):4604–12. doi:10.4049/jimmunol.1500117
42. Gartlan KH, Markey KA, Varelias A, Bunting MD, Koyama M, Kuns RD, et al. Tc17 cells are a proinflammatory, plastic lineage of pathogenic CD8+ T cells that induce GVHD without antileukemic effects. Blood (2015) 126(13):1609–20. doi:10.1182/blood-2015-01-622662
43. Zeiser R, Blazar BR. Preclinical models of acute and chronic graft-versus-host disease: how predictive are they for a successful clinical translation? Blood (2016) 127(25):3117–26. doi:10.1182/blood-2016-02-699082
44. Gartlan KH, Bommiasamy H, Paz K, Wilkinson AN, Owen M, Reichenbach DK, et al. A critical role for donor-derived IL-22 in cutaneous chronic GVHD. Am J Transplant (2018) 18(4):810–20. doi:10.1111/ajt.14513
45. Yi T, Chen Y, Wang L, Du G, Huang D, Zhao D, et al. Reciprocal differentiation and tissue-specific pathogenesis of Th1, Th2, and Th17 cells in graft-versus-host disease. Blood (2009) 114(14):3101–12. doi:10.1182/blood-2009-05-219402
46. Bruggen MC, Klein I, Greinix H, Bauer W, Kuzmina Z, Rabitsch W, et al. Diverse T-cell responses characterize the different manifestations of cutaneous graft-versus-host disease. Blood (2014) 123(2):290–9. doi:10.1182/blood-2013-07-514372
47. Krenger W, Ferrara JL. Graft-versus-host disease and the Th1/Th2 paradigm. Immunol Res (1996) 15(1):50–73. doi:10.1007/BF02918284
48. Reddy P, Ferrara JL. Immunobiology of acute graft-versus-host disease. Blood Rev (2003) 17(4):187–94. doi:10.1016/S0268-960X(03)00009-2
49. Tawara I, Maeda Y, Sun Y, Lowler KP, Liu C, Toubai T, et al. Combined Th2 cytokine deficiency in donor T cells aggravates experimental acute graft-vs-host disease. Exp Hematol (2008) 36(8):988–96. doi:10.1016/j.exphem.2008.02.010
50. Carlson MJ, West ML, Coghill JM, Panoskaltsis-Mortari A, Blazar BR, Serody JS. In vitro-differentiated TH17 cells mediate lethal acute graft-versus-host disease with severe cutaneous and pulmonary pathologic manifestations. Blood (2009) 113(6):1365–74. doi:10.1182/blood-2008-06-162420
51. Hill GR, Olver SD, Kuns RD, Varelias A, Raffelt NC, Don AL, et al. Stem cell mobilization with G-CSF induces type 17 differentiation and promotes scleroderma. Blood (2010) 116(5):819–28. doi:10.1182/blood-2009-11-256495
52. Nikolic B, Lee S, Bronson RT, Grusby MJ, Sykes M. Th1 and Th2 mediate acute graft-versus-host disease, each with distinct end-organ targets. J Clin Invest (2000) 105(9):1289–98. doi:10.1172/JCI7894
53. Cai Y, Fleming C, Yan J. New insights of T cells in the pathogenesis of psoriasis. Cell Mol Immunol (2012) 9(4):302–9. doi:10.1038/cmi.2012.15
54. Yang X, Yang J, Xing X, Wan L, Li M. Increased frequency of Th17 cells in systemic sclerosis is related to disease activity and collagen overproduction. Arthritis Res Ther (2014) 16(1):R4. doi:10.1186/ar4430
55. Speeckaert R, Lambert J, Grine L, Van Gele M, De Schepper S, van Geel N. The many faces of interleukin-17 in inflammatory skin diseases. Br J Dermatol (2016) 175(5):892–901. doi:10.1111/bjd.14703
56. Schneidawind D, Pierini A, Negrin RS. Regulatory T cells and natural killer T cells for modulation of GVHD following allogeneic hematopoietic cell transplantation. Blood (2013) 122(18):3116–21. doi:10.1182/blood-2013-08-453126
57. Wang XN, Haniffa MA, Holtick U, Collin MP, Jackson G, Hilkens CM, et al. Regulatory T-cell suppression of CD8+ T-cell-mediated graft-versus-host reaction requires their presence during priming. Transplantation (2009) 88(2):188–97. doi:10.1097/TP.0b013e3181ac14ce
58. Mavin E, Ahmed SS, O’Boyle G, Turner B, Douglass S, Norden J, et al. Regulatory T cells inhibit CD8(+) T-cell tissue invasion in human skin graft-versus-host reactions. Transplantation (2012) 94(5):456–64. doi:10.1097/TP.0b013e31826205d6
59. Chen BJ, Deoliveira D, Cui X, Le NT, Son J, Whitesides JF, et al. Inability of memory T cells to induce graft-versus-host disease is a result of an abortive alloresponse. Blood (2007) 109(7):3115–23. doi:10.1182/blood-2006-04-016410
60. Chen X, Vodanovic-Jankovic S, Johnson B, Keller M, Komorowski R, Drobyski WR. Absence of regulatory T-cell control of TH1 and TH17 cells is responsible for the autoimmune-mediated pathology in chronic graft-versus-host disease. Blood (2007) 110(10):3804–13. doi:10.1182/blood-2007-05-091074
61. Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med (2013) 5(179):179ra143. doi:10.1126/scitranslmed.3005265
62. Hanash AM, Dudakov JA, Hua G, O’Connor MH, Young LF, Singer NV, et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity (2012) 37(2):339–50. doi:10.1016/j.immuni.2012.05.028
63. Varelias A, Ormerod KL, Bunting MD, Koyama M, Gartlan KH, Kuns RD, et al. Acute graft-versus-host disease is regulated by an IL-17-sensitive microbiome. Blood (2017) 129(15):2172–85. doi:10.1182/blood-2016-08-732628
64. Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, et al. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science (1999) 285(5426):412–5. doi:10.1126/science.285.5426.412
65. Matte CC, Liu J, Cormier J, Anderson BE, Athanasiadis I, Jain D, et al. Donor APCs are required for maximal GVHD but not for GVL. Nat Med (2004) 10(9):987–92. doi:10.1038/nm1089
66. Koyama M, Kuns RD, Olver SD, Raffelt NC, Wilson YA, Don AL, et al. Recipient nonhematopoietic antigen-presenting cells are sufficient to induce lethal acute graft-versus-host disease. Nat Med (2011) 18(1):135–42. doi:10.1038/nm.2597
67. McLachlan JB, Catron DM, Moon JJ, Jenkins MK. Dendritic cell antigen presentation drives simultaneous cytokine production by effector and regulatory T cells in inflamed skin. Immunity (2009) 30(2):277–88. doi:10.1016/j.immuni.2008.11.013
68. Iijima N, Mattei LM, Iwasaki A. Recruited inflammatory monocytes stimulate antiviral Th1 immunity in infected tissue. Proc Natl Acad Sci U S A (2011) 108(1):284–9. doi:10.1073/pnas.1005201108
69. Bartholomaus I, Kawakami N, Odoardi F, Schlager C, Miljkovic D, Ellwart JW, et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature (2009) 462(7269):94–8. doi:10.1038/nature08478
70. Friedman RS, Lindsay RS, Lilly JK, Nguyen V, Sorensen CM, Jacobelli J, et al. An evolving autoimmune microenvironment regulates the quality of effector T cell restimulation and function. Proc Natl Acad Sci U S A (2014) 111(25):9223–8. doi:10.1073/pnas.1322193111
71. Teichmann LL, Cullen JL, Kashgarian M, Dong C, Craft J, Shlomchik MJ. Local triggering of the ICOS coreceptor by CD11c(+) myeloid cells drives organ inflammation in lupus. Immunity (2015) 42(3):552–65. doi:10.1016/j.immuni.2015.02.015
72. Hufford MM, Kim TS, Sun J, Braciale TJ. Antiviral CD8+ T cell effector activities in situ are regulated by target cell type. J Exp Med (2011) 208(1):167–80. doi:10.1084/jem.20101850
73. Collin MP, Hart DN, Jackson GH, Cook G, Cavet J, Mackinnon S, et al. The fate of human Langerhans cells in hematopoietic stem cell transplantation. J Exp Med (2006) 203(1):27–33. doi:10.1084/jem.20051787
74. Merad M, Hoffmann P, Ranheim E, Slaymaker S, Manz MG, Lira SA, et al. Depletion of host Langerhans cells before transplantation of donor alloreactive T cells prevents skin graft-versus-host disease. Nat Med (2004) 10(5):510–7. doi:10.1038/nm1038
75. Malissen B, Tamoutounour S, Henri S. The origins and functions of dendritic cells and macrophages in the skin. Nat Rev Immunol (2014) 14(6):417–28. doi:10.1038/nri3683
76. Mielcarek M, Kirkorian AY, Hackman RC, Price J, Storer BE, Wood BL, et al. Langerhans cell homeostasis and turnover after nonmyeloablative and myeloablative allogeneic hematopoietic cell transplantation. Transplantation (2014) 98(5):563–8. doi:10.1097/TP.0000000000000097
77. Bennett CL, Fallah-Arani F, Conlan T, Trouillet C, Goold H, Chorro L, et al. Langerhans cells regulate cutaneous injury by licensing CD8 effector cells recruited to the skin. Blood (2011) 117(26):7063–9. doi:10.1182/blood-2011-01-329185
78. Li H, Kaplan DH, Matte-Martone C, Tan HS, Venkatesan S, Johnson K, et al. Langerhans cells are not required for graft-versus-host disease. Blood (2011) 117(2):697–707. doi:10.1182/blood-2010-07-299073
79. Kaplan DH. Ontogeny and function of murine epidermal Langerhans cells. Nat Immunol (2017) 18(10):1068–75. doi:10.1038/ni.3815
80. Kim BS, Miyagawa F, Cho YH, Bennett CL, Clausen BE, Katz SI. Keratinocytes function as accessory cells for presentation of endogenous antigen expressed in the epidermis. J Invest Dermatol (2009) 129(12):2805–17. doi:10.1038/jid.2009.176
81. Hashimoto D, Chow A, Greter M, Saenger Y, Kwan WH, Leboeuf M, et al. Pretransplant CSF-1 therapy expands recipient macrophages and amelio-rates GVHD after allogeneic hematopoietic cell transplantation. J Exp Med (2011) 208(5):1069–82. doi:10.1084/jem.20101709
82. D’Aveni M, Rossignol J, Coman T, Sivakumaran S, Henderson S, Manzo T, et al. G-CSF mobilizes CD34+ regulatory monocytes that inhibit graft-versus-host disease. Sci Transl Med (2015) 7(281):281ra242. doi:10.1126/scitranslmed.3010435
83. Sivakumaran S, Henderson S, Ward S, Santos E Sousa P, Manzo T, Zhang L, et al. Depletion of CD11c(+) cells in the CD11c.DTR model drives expansion of unique CD64(+) Ly6C(+) monocytes that are poised to release TNF-alpha. Eur J Immunol (2016) 46(1):192–203. doi:10.1002/eji.201545789
84. Nishiwaki S, Terakura S, Ito M, Goto T, Seto A, Watanabe K, et al. Impact of macrophage infiltration of skin lesions on survival after allogeneic stem cell transplantation: a clue to refractory graft-versus-host disease. Blood (2009) 114(14):3113–6. doi:10.1182/blood-2009-03-209635
85. Alexander KA, Flynn R, Lineburg KE, Kuns RD, Teal BE, Olver SD, et al. CSF-1-dependant donor-derived macrophages mediate chronic graft-versus-host disease. J Clin Invest (2014) 124(10):4266–80. doi:10.1172/JCI75935
86. Du J, Paz K, Flynn R, Vulic A, Robinson TM, Lineburg KE, et al. Pirfenidone ameliorates murine chronic GVHD through inhibition of macrophage infiltration and TGF-beta production. Blood (2017) 129(18):2570–80. doi:10.1182/blood-2017-01-758854
87. Haniffa M, Ginhoux F, Wang XN, Bigley V, Abel M, Dimmick I, et al. Differential rates of replacement of human dermal dendritic cells and macrophages during hematopoietic stem cell transplantation. J Exp Med (2009) 206(2):371–85. doi:10.1084/jem.20081633
88. Greinix HT, Pohlreich D, Kouba M, Kormoczi U, Lohmann I, Feldmann K, et al. Elevated numbers of immature/transitional CD21- B lymphocytes and deficiency of memory CD27+ B cells identify patients with active chronic graft-versus-host disease. Biol Blood Marrow Transplant (2008) 14(2):208–19. doi:10.1016/j.bbmt.2007.10.009
89. Corre E, Carmagnat M, Busson M, de Latour RP, Robin M, Ribaud P, et al. Long-term immune deficiency after allogeneic stem cell transplantation: B-cell deficiency is associated with late infections. Haematologica (2010) 95(6):1025–9. doi:10.3324/haematol.2009.018853
90. Allen JL, Fore MS, Wooten J, Roehrs PA, Bhuiya NS, Hoffert T, et al. B cells from patients with chronic GVHD are activated and primed for survival via BAFF-mediated pathways. Blood (2012) 120(12):2529–36. doi:10.1182/blood-2012-06-438911
91. Jin H, Ni X, Deng R, Song Q, Young J, Cassady K, et al. Antibodies from donor B cells perpetuate cutaneous chronic graft-versus-host disease in mice. Blood (2016) 127(18):2249–60. doi:10.1182/blood-2015-09-668145
92. Svegliati S, Olivieri A, Campelli N, Luchetti M, Poloni A, Trappolini S, et al. Stimulatory autoantibodies to PDGF receptor in patients with extensive chronic graft-versus-host disease. Blood (2007) 110(1):237–41. doi:10.1182/blood-2007-01-071043
93. Miklos D, Cutler CS, Arora M, Waller EK, Jagasia M, Pusic I, et al. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood (2017) 130(21):2243–50. doi:10.1182/blood-2017-07-793786
94. Riesner K, Shi Y, Jacobi A, Krater M, Kalupa M, McGearey A, et al. Initiation of acute graft-versus-host disease by angiogenesis. Blood (2017) 129(14):2021–32. doi:10.1182/blood-2016-08-736314
95. Mertlitz S, Shi Y, Kalupa M, Grotzinger C, Mengwasser J, Riesner K, et al. Lymphangiogenesis is a feature of acute GVHD, and VEGFR-3 inhibition protects against experimental GVHD. Blood (2017) 129(13):1865–75. doi:10.1182/blood-2016-08-734210
96. Gonzales KAU, Fuchs E. Skin and its regenerative powers: an alliance between stem cells and their niche. Dev Cell (2017) 43(4):387–401. doi:10.1016/j.devcel.2017.10.001
97. Favre A, Cerri A, Bacigalupo A, Lanino E, Berti E, Grossi CE. Immunohistochemical study of skin lesions in acute and chronic graft versus host disease following bone marrow transplantation. Am J Surg Pathol (1997) 21(1):23–34. doi:10.1097/00000478-199701000-00003
98. Nomura I, Gao B, Boguniewicz M, Darst MA, Travers JB, Leung DY. Distinct patterns of gene expression in the skin lesions of atopic dermatitis and psoriasis: a gene microarray analysis. J Allergy Clin Immunol (2003) 112(6):1195–202. doi:10.1016/j.jaci.2003.08.049
99. de Jongh GJ, Zeeuwen PL, Kucharekova M, Pfundt R, van der Valk PG, Blokx W, et al. High expression levels of keratinocyte antimicrobial proteins in psoriasis compared with atopic dermatitis. J Invest Dermatol (2005) 125(6):1163–73. doi:10.1111/j.0022-202X.2005.23935.x
100. Sale GE, Shulman HM, Gallucci BB, Thomas ED. Young rete ridge keratinocytes are preferred targets in cutaneous graft-versus-host disease. Am J Pathol (1985) 118(2):278–87.
101. Sale GE, Beauchamp M. Parafollicular bulge, but not papillary bulb keratinocytes, are attacked in graft-versus-host disease (Gvhd) of human skin. Lab Invest (1994) 70(1):A49–49.
102. Whitaker-Menezes D, Jones SC, Friedman TM, Korngold R, Murphy GF. An epithelial target site in experimental graft-versus-host disease and cytokine-mediated cytotoxicity is defined by cytokeratin 15 expression. Biol Blood Marrow Transplant (2003) 9(9):559–70. doi:10.1016/S1083-8791(03)00288-X
103. Murphy GF, Korngold R. Significance of selectively targeted apoptotic rete cells in graft-versus-host disease. Biol Blood Marrow Transplant (2004) 10(6):357–65. doi:10.1016/j.bbmt.2004.02.005
104. Shono Y, Docampo MD, Smith OM, Gupta J, Liberman SR, Taur Y, et al. Treatment with antibiotics containing activity against obligate anaerobes worsens GVHD survival in mice and humans after allogeneic BMT. Biol Blood Marrow Transplant (2015) 21(2):S27–8. doi:10.1016/j.bbmt.2014.11.015
105. Wang W, Xu S, Ren Z, Jiang J, Zheng S. Gut microbiota and allogeneic transplantation. J Transl Med (2015) 13:275. doi:10.1186/s12967-015-0640-8
106. Mathewson ND, Jenq R, Mathew AV, Koenigsknecht M, Hanash A, Toubai T, et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol (2016) 17(5):505–13. doi:10.1038/ni.3400
107. Zama D, Biagi E, Masetti R, Gasperini P, Prete A, Candela M, et al. Gut microbiota and hematopoietic stem cell transplantation: where do we stand? Bone Marrow Transplant (2017) 52(1):7–14. doi:10.1038/bmt.2016.173
108. Legoff J, Resche-Rigon M, Bouquet J, Robin M, Naccache SN, Mercier-Delarue S, et al. The eukaryotic gut virome in hematopoietic stem cell transplantation: new clues in enteric graft-versus-host disease. Nat Med (2017) 23(9):1080–5. doi:10.1038/nm.4380
109. Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol (2018) 16(3):143–55. doi:10.1038/nrmicro.2017.157
110. Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, et al. Compartmentalized control of skin immunity by resident commensals. Science (2012) 337(6098):1115–9. doi:10.1126/science.1225152
111. Chehoud C, Rafail S, Tyldsley AS, Seykora JT, Lambris JD, Grice EA. Complement modulates the cutaneous microbiome and inflammatory milieu. Proc Natl Acad Sci U S A (2013) 110(37):15061–6. doi:10.1073/pnas.1307855110
112. Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol (2015) 16(4):343–53. doi:10.1038/ni.3123
113. Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature (2015) 520(7545):104–8. doi:10.1038/nature14052
Keywords: cutaneous graft-versus-host disease, pathophysiology, T cells, antigen-presenting cells, B cells, repair mechanisms, microbiome
Citation: Santos e Sousa P, Bennett CL and Chakraverty R (2018) Unraveling the Mechanisms of Cutaneous Graft-Versus-Host Disease. Front. Immunol. 9:963. doi: 10.3389/fimmu.2018.00963
Received: 29 January 2018; Accepted: 18 April 2018;
Published: 02 May 2018
Edited by:
Chiara Bonini, San Raffaele Hospital (IRCCS), ItalyReviewed by:
Robert Zeiser, Albert Ludwigs Universität Freiburg, GermanyAnne Mary Dickinson, Newcastle University, United Kingdom
Copyright: © 2018 Santos e Sousa, Bennett and Chakraverty. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pedro Santos e Sousa, cGVkcm8uc291c2EuMTFAdWNsLmFjLnVr;
Ronjon Chakraverty, ci5jaGFrcmF2ZXJ0eUB1Y2wuYWMudWs=