 Inmaculada Serrano
Inmaculada Serrano Ana Luque
Ana Luque Josep M. Aran
Josep M. Aran
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 30 April 2018
Sec. Immunological Tolerance and Regulation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00892
This article is part of the Research Topic Tolerogenic Antigen-Presenting Cells – Modulating Unwanted Immune Response at Their Core View all 23 articles
The acute phase response is generated by an overwhelming immune-inflammatory process against infection or tissue damage, and represents the initial response of the organism in an attempt to return to homeostasis. It is mediated by acute phase proteins (APPs), an assortment of highly conserved plasma reactants of seemingly different functions that, however, share a common protective role from injury. Recent studies have suggested a crosstalk between several APPs and the mononuclear phagocyte system (MPS) in the resolution of inflammation, to restore tissue integrity and function. In fact, monocyte-derived dendritic cells (Mo-DCs), an integral component of the MPS, play a fundamental role both in the regulation of antigen-specific adaptive responses and in the development of immunologic memory and tolerance, particularly in inflammatory settings. Due to their high plasticity, Mo-DCs can be modeled in vitro toward a tolerogenic phenotype for the treatment of aberrant immune-inflammatory conditions such as autoimmune diseases and allotransplantation, with the phenotypic outcome of these cells depending on the immunomodulatory agent employed. Yet, recent immunotherapy trials have emphasized the drawbacks and challenges facing tolerogenic Mo-DC generation for clinical use, such as reduced therapeutic efficacy and limited in vivo stability of the tolerogenic activity. In this review, we will underline the potential relevance and advantages of APPs for tolerogenic DC production with respect to currently employed immunomodulatory/immunosuppressant compounds. A further understanding of the mechanisms of action underlying the moonlighting immunomodulatory activities exhibited by several APPs over DCs could lead to more efficacious, safe, and stable protocols for precision tolerogenic immunotherapy.
In the superior organisms, inflammation is considered as an evolutionarily conserved, physiological response of the vascularized tissue against external physical, chemical, and biological insults, or internal threats such as metabolic stress. This complex, exquisitely fine-tuned and coordinated process is engaged with the final goal of restoring the homeostasis and repair/regenerate the damaged tissues in a relatively short-time window (1). Whether the insult persists, chronic undesirable inflammation ensues and is associated with a variety of pathologies such as autoimmune processes or vascular diseases. Innate immune cells with the capacity for antigen presentation, that is, specialized antigen-presenting cells (APCs) such as monocytes/macrophages and dendritic cells (DCs), are key players in all phases of inflammation (2). Thus, APCs are involved in the initial sensing of noxious agents through recognition of danger-associated molecular patterns (DAMPs), including pathogen-associated molecular patterns (PAMPs), in the amplification of the defense/protection by locally attracting other immune cells through the vasculature and, finally, are essential effector cells in the resolution of inflammation. All these events are orchestrated mainly by DCs, endowed with high plasticity to bridge innate and acquired immune responses within the inflammatory program (3, 4). Local DAMPs/PAMPs detection by pattern recognition molecules (PRMs), notably the toll-like receptor (TLR) family of proteins, in these cells initiates an adaptive immune process leading to the activation and expansion of antigen-specific effector T lymphocytes in the secondary lymphoid organs (5). Conversely, the absence of pro-inflammatory stimuli or engagement of particular immunoreceptors, such as co-inhibitory receptors (PD-L1, PD-L2, B7-H3, ILT3, etc.) or other tyrosine-based inhibitory motif-containing receptors by a variety of signals maintain DCs in an “immature-like” state. These, “immature” DCs are able to elicit generalized or antigen-specific unresponsiveness/tolerance in central lymphoid organs or in the periphery, promoting the further stimulation of T cells (Treg) able to regulate or suppress other T cells (6). Such actions are crucial to maintain or return to immune homeostasis and to prevent autoimmune responses.
Another inherent aspect of the innate immunity elicited in wounded hosts (particularly those severely injured by trauma or microbial infection), in parallel to the advent of the above-described cellular or acquired immune response, is the prompt occurrence of a prominent non-specific immune-inflammatory response involving systemic physiological and metabolic alterations and affecting tissues/organs distant to the injured site, namely, the acute phase response (7). Thus, immunological stress induces a pro-inflammatory cytokine “storm,” diffusing into the circulation and alerting the liver, which in turn reinforces a protective response through coordinated, cytokine-driven transcriptional changes in hepatocytes, leading to the secretion of a variety of molecules that limit tissue injury and participate in host defense, termed acute phase proteins (APPs), such as the prototypical C-reactive protein (CRP), serum amyloid P (SAP), and serum amyloid A (SAA). These proteins have been traditionally explored as diagnostic/prognostic biomarkers reflecting the presence and intensity of inflammation during infection or injury. Indeed, while most APPs have been traditionally viewed as having a pro-inflammatory function, for example, in immune cell recruitment for efficient pathogen clearance (8), more recent studies are suggesting that a variety of APPs, depending on the microenvironment and through molecular mechanisms not yet completely understood, are able to interact directly with mononuclear phagocytes inducing a regulatory phenotype to these cells.
Mirroring the recent success and increasing importance of cellular immunotherapy strategies for cancer, in the last years a substantial effort has been devoted to generate DCs from blood precursors with tolerogenic features for the treatment of autoimmune diseases, allergy, and transplantation. As the first phase I adoptive tolerogenic DC therapy clinical trials are being concluded, preliminary lessons learned include the overall safety of tolerogenic DC administration, although also highlight present limitations regarding its efficacy. Thus, important current challenges to overcome for a more effective therapeutic outcome include the achievement of antigen-specific tolerogenic responses and, particularly, the maintenance of a “stable” tolerogenic phenotype of the infused DCs regardless of the inflammatory microenvironment that they may confront. Therefore, more progress has to be achieved on the thorough characterization, using both in in vitro functional readouts and preclinical assays, of tolerogenic DCs generated through alternative immunomodulatory inducers able to increase their clinical performance in immune-inflammatory pathologies.
In this review, we will consider the potential of APPs as novel immunomodulators. We will overview the current knowledge regarding the interaction of relevant APPs with phagocytes, fundamentally monocytes, and monocyte-derived DCs (Mo-DCs), resulting in a bias toward immune tolerance. A better understanding of the crosstalk between the innate and the adaptive immune systems in homeostasis and inflammatory pathology, taking into account the unique roles of both APPs and DCs, may support therapeutic benefits of APP-induced tolerogenic DCs for transplantation and autoimmunity.
The immediate innate body defense against acute illnesses, that is, the acute phase response, features both, hepatic and extra-hepatic overproduction and release, typically within 24–48 h after the initial insult, of a variety of seemingly biochemically and functionally unrelated APPs into the circulation. In fact, phagocyte sentinels (macrophages, DCs, and neutrophils) sensing eminently damaged, stressed or infected cells, elicit a local pro-inflammatory response, and seek further help by secreting pro-inflammatory cytokines such as IL-6, IL-1, IL-8, TNF-α, and IFN-γ, and releasing a large assortment of “alarmins.” These key mediators travel through the circulation, induce neuroendocrine and behavioral changes (fever, hyponatremia, anorexia, somnolence, and lethargy), and reach the liver, whose most abundant cell type, the hepatocytes, hold also the capability to act as immunological agents and have a central role in the systemic innate immune response through the intravascular secretion of APPs (9). Indeed, APPs conform up to 40 different proteins whose serum concentration increase (positive APPs) or decrease (negative APPs) at least 25% in response to inflammation (10). Positive APPs include soluble PRMs [CRP, SAP, SAA, lipopolysaccharide binding protein, complement components, and α1 acid glycoprotein (AAG)], hemostasis factors (fibrinogen, plasminogen, prothrombin, and plasminogen activators), binding/transport proteins [haptoglobin (Hp), hemopexin, and ceruloplasmin], and antiproteases [α1-antichymotrypsin (AAC), antithrombin (AT), α1-antitrypsin (AAT), and α2-macroglobulin (α2M)]. These proteins participate in host defense (e.g., attracting inflammatory cells, inactivating proteolytic enzymes, activating complement, opsonizing, and clearing infectious agents) and limit tissue injury (scavenging free radicals and modulating the host’s immune response). Conversely, negative APPs comprise albumin, AT, transferrin, transthyretin, transcortin, and retinol-binding protein (8). It has been suggested that reduced albumin production enhances the amino acids “pool” available for positive APP production, and that decreased transferrin production could protect the host by starving microorganisms of the iron required for growth and virulence expression (11).
Based on their degree of response to inflammatory stimuli, APPs can be grouped as strong (more than 100-fold increase in blood levels; CRP, α2M, SAA), moderate (2–10-fold increase; haptoglobulin, fibrinogen, AAT), or weak (up to twofold increase; C3, ceruloplasmin). While strong APPs usually increase abruptly within the first 24–48 h after an acute inflammatory event, and further experience a quick decline related to their relatively short half-life, moderate to weak APPs are more likely present during chronic inflammatory processes. According to the differential regulation of their synthesis by cytokines, positive APPs can also be classified in type I and type II. Type I are induced by IL-1-like pro-inflammatory cytokines (SAA, CRP, C3, AAG, and SAP), and type II are induced by IL-6-like cytokines (fibrinogen, Hp, AAC, AAT, and α2M). In turn, the production of hepatic APPs may also be influenced by other cytokines and by hormones (insulin, dexamethasone, glucagon, and/or epinephrine) (12). Thus, at the level of the organism, the complex neuroendocrine-immunological axis seems to efficiently modulate the acute phase response through various feedback loops (13). For instance, cytokines released from monocytes/macrophages activated locally through noxious inflammatory agents stimulate the brain to release stress-response neuropeptides such as corticotropin (ACTH), which acts into the adrenal glands inducing glucocorticoid production. Glucocorticoids can downregulate pro-inflammatory cytokines (IL-1, TNF-α).
Due to their stability in the circulation compared with cytokines, which are cleared from the circulation within a few hours, several APPs have been extensively used as diagnostic/prognostic biomarkers because their increased/decreased levels reflect the presence and intensity of inflammation during infection or injury, remaining unchanged for 48 h or longer. Nevertheless, although presenting high sensitivity, the diagnostic value of APPs is being questioned due to their low specificity (14).
Relevant features of the acute phase response are an increase in the number of peripheral leukocytes and the dilation and leakage of the vasculature through the release of inflammatory mediators such as reactive oxygen species, arachidonate metabolites, and pro-inflammatory cytokines and chemokines (15). Pro-inflammatory cytokines activate and mobilize blood cell precursors in both bone marrow and peripheral blood (16–18). Moreover, stimulated endothelial cells allow the extravasation and migration of circulating leukocytes. Among these, Mo-DCs have been appealing due to: (1) their influence on adaptive immune function and rapid accumulation in the inflammatory focus and (2) their easy ex vivo isolation, amplification, and manipulation. Mo-DCs arise from monocyte precursors both in vitro and in vivo (19, 20). Monocytes are recruited to sites of inflammation, having a major role in the protective immune response of the host (21). For instance, local differentiation of monocytes into inflammatory macrophages and DCs is induced in response to natural killer cell-produced IFN-γ (22). In fact, by depletion of tissue-resident cell populations it has been shown that circulating monocyte precursors in the blood can replenish functionally specialized macrophages and DCs (23), which reinforces the concept of blood monocytes as reservoirs that can be utilized on demand, particularly in inflammatory processes where monocyte recruitment is strongly increased. Accordingly, monocytes have been shown to migrate to inflammatory sites and differentiate into DCs in various murine models of inflammation (24, 25). Sequential trafficking and/or differentiation of the different monocyte subsets to the sites of inflammation is likely modulated by diverse mechanisms (26–28). Following tissue damage, classical monocytes (human: CD14++CD16−; mouse: Ly6C+CCR2highCX3CR1low) appear to be recruited within the first few hours, after their egression from the bone marrow being modulated by the CCR2–CCL2/CCL7 axis (29). Once in the inflammatory milieu, they differentiate into DCs and macrophages and exert a potent pro-inflammatory immune response through high-level production of IL-1β and TNF-α, among other protective functions (30–32). When the progression of the immune-inflammatory response is not halted, the prolonged action of classical inflammatory monocytes may result in tissue damage and drive autoimmunity (33). Several days after the initial damaging insult, acute inflammation enters in a resolution phase where the classical monocyte levels are reduced and progressively replaced by intermediate [CD14+(+)CD16+] and non-classical (human: CD14+CD16++; mouse: Ly6C−CCR2lowCX3CR1high) monocytes, which relay on the CX3CR1–CX3CL1 axis to accumulate in the damaged tissue and, after DC/macrophage differentiation, secrete anti-inflammatory cytokines (IL-10, TGF-β) that counteract tissue injury and promote wound healing (34). Certainly, it has been suggested that, in response to inflammatory stimuli, patrolling non-classical CD16-expressing monocytes could leave the blood vessels and function as DC precursors (35). Thus, these inflammatory Mo-DCs seem to hold unique features influenced by the microenvironmental status of the inflamed tissue, boosting more potent immune responses DCs derived from classical monocytes, and better immune tolerance DCs generated from non-classical monocytes (36).
Monocytes from human or mouse peripheral blood or bone marrow are widely utilized to generate in vitro large amounts of Mo-DCs upon differentiation, typically with IL-4 and GM-CSF (37), allowing comprehensive mechanistic studies regarding their key role in the immune-inflammatory processes at the molecular level and to initiate DC therapy approaches in the clinic. In fact, a comparative transcriptional profiling has revealed that human DCs isolated from inflammatory fluids are the in vivo counterpart of in vitro-generated Mo-DCs from CD14+ monocytes (38), in the same way that murine inflammatory DCs share equivalent developmental and functional features to in vitro GM-CSF/IL-4-induced BM-DCs (39).
Monocyte-derived cells have been deemed essential for inducing protective Th1 cell-mediated immunity following both pathogen infection and non-infectious conditions (40, 41), and may acquire DC-specific functions such as cross-presentation (41, 42).
Conversely, DCs play a key role in tolerance, whether participating in the negative selection of autoreactive T cells in the thymus (central tolerance) (43), or limiting effector T cells through deletion or anergy and, instead, promoting Treg differentiation (peripheral tolerance). A variety of mechanisms are orchestrated by DCs to induce tolerance and suppress inflammatory responses against innocuous stimuli, including the overexpression of inhibitory immunoreceptors (e.g., PD-L1, B7H, and CD80/86), the ligand-activated transcription factor aryl hydrocarbon receptor, the pore-forming cytolytic protein perforin, and the release and/or control of several immunomodulatory mediators, such as anti-inflammatory cytokines (IL-10, IL-27, and TGF-β), indoleamine 2,3-dioxygenase (IDO) metabolites, retinoic acid, vitamins A and D, ATP, and adenosine [see Ref. (44, 45), and references therein]. The regulatory function of DCs is determined by their maturation/activation status (46). Hence, tolerogenic DCs hold an “immature” or “semi-mature” state.
A myriad of recent studies has reported the in vitro generation of monocyte-derived “permissive,” “tolerogenic,” “regulatory,” “alternatively activated,” or “maturation-resistant” cell types (47), although most attention has been focused on Mo-DCs. This is being achieved by incubation with a variety of different biological or pharmacological agents such as cytokines (IL-10, TNF-α, IFN-γ, TGF-β, IL-21, and thymic stromal lymphopoietin), immunosuppressant drugs (dexamethasone, tacrolimus, and mycophenolate), organic molecules (vitamin D3, salycilate, vasoactive intestinal peptide, intravenous immunoglobulin, and hepatocyte growth factor), other agents (pathogen products, mesenchymal stem cells), or their combinations, or by genetic engineering (48–50). Mimicking the in vivo circumstances, the resulting tolerogenic Mo-DCs are characterized essentially by reduced surface expression of co-stimulatory molecules (CD80, CD86, and CD40) and maturation markers (CD83), increased expression of inhibitory receptors (ILT3, PD-L1, and PD-L2), reduced or null production of pro-inflammatory cytokines (IL-12, TNF-α, IFN-γ, and IL-8) and, conversely, increased production of anti-inflammatory cytokines (IL-10, TGF-β), even in the presence of inflammation (51–54). Thus, the main features of these cells would be to present a state of unresponsiveness through hampering key activation/maturation pathways such as the pro-inflammatory NF-κB pathway, and to support the differentiation and maintenance of different types of Treg cells.
There are clear evidences showing that the acute phase response can directly influence the differentiation of DCs toward a tolerogenic state. In sepsis, an overwhelming systemic inflammatory response syndrome, an expansion of intermediate monocytes has been detected in the circulation (55). Monocytes from sepsis patients preferentially differentiated into alternative CD1a− DCs, holding increased capacity to induce Foxp3+ Treg cells, when compared with monocytes from healthy individuals in which classical monocytes predominated (56). On the other hand, the hepatic APPs SAA and Cxcl1/KC cooperatively promoted myeloid-derived suppressor cell (MDSC) mobilization, accumulation and survival, reversed dysregulated inflammation, and restored survival of mice deficient for gp130 (the signaling receptor shared by IL-6 family cytokines) undergoing polymicrobial sepsis (57). Thus, hepatocytes may also modulate innate immune cells through the acute phase response, for example, by recruitment and promotion of MDSC function.
Accordingly, it is not unreasonable to consider a number of APPs, acting either systemically or locally in a restricted time window coinciding with a parallel increase of monocyte recruitment toward the inflammatory focus, as a part of a protective network to restrain the harmful consequences of continued overinflammation. That is, APPs could directly exert a feedback loop redirecting the differentiation of these inflammatory monocytes to regulatory or tolerogenic DCs, in an attempt to regain homeostasis and maintain tissue integrity through the resolution of the immune-inflammatory response (Figure 1). We will now focus in representative APPs and APP-related proteins that are able to induce tolerogenesis through modulation of Mo-DC differentiation and/or maturation.
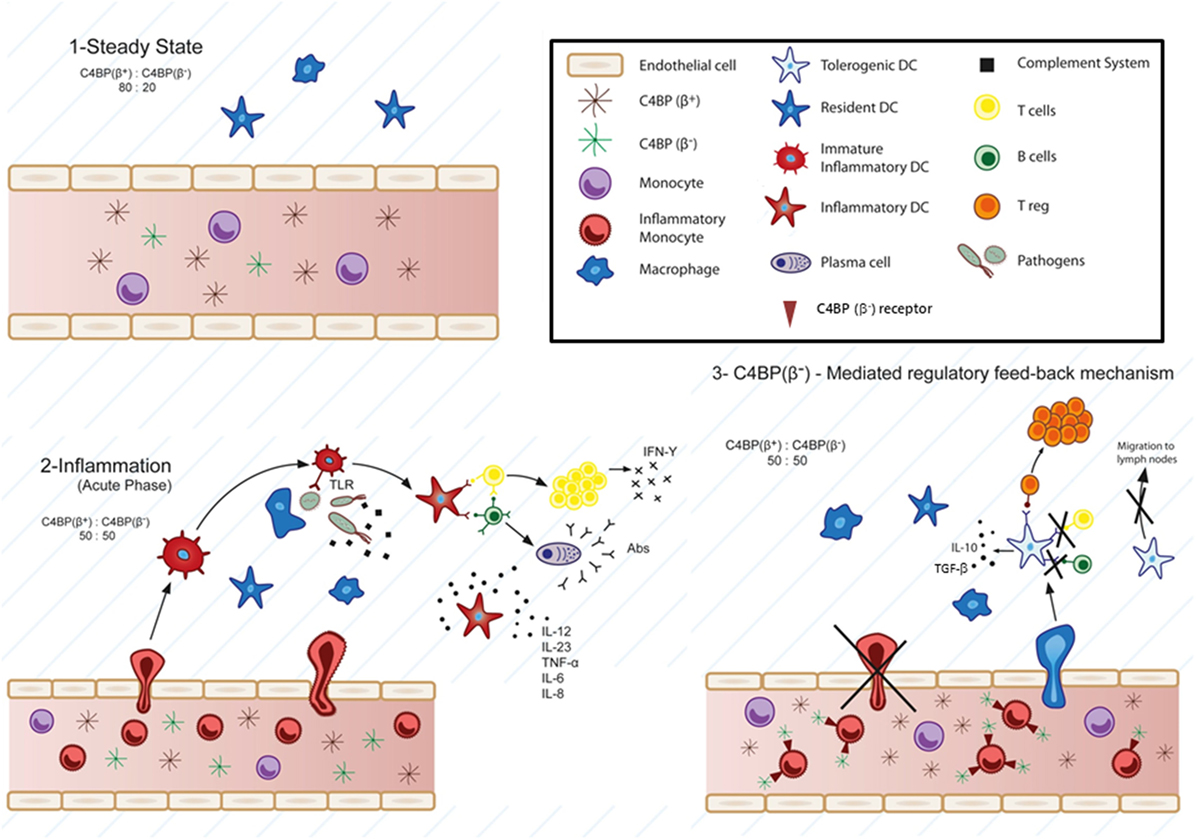
Figure 1. Physiological involvement of C4b-binding protein (C4BP)(β−) in cell-mediated immune-inflammatory responses. According to this model, at the steady-state, the classical and lectin pathways complement inhibitor C4BP circulates in two main isoforms: C4BP(β+) (80%) and C4BP(β−) (20%) (1). Strong pro-inflammatory conditions (acute phase) induce a modification of the C4BP(β+):C4BP(β−) ratio to 50:50, through increased levels of the acute phase protein C4BP(β−) (2). Several evidences support a specific action of overexpressed C4BP(β−) over inflammatory monocytes and monocyte-derived dendritic cells (DCs). Strong inflammatory stimuli (infection, lupus nephritis, etc.) trigger the presence of inflammatory monocytes in the blood, which are actively recruited to inflamed tissues, and differentiated to inflammatory DCs, having the ability to stimulate naïve T cells. Under these conditions, besides its function as complement inhibitor, the increased presence of C4BP(β−) in the blood would act in one or both ways upon engaging one or some, as yet unknown, cell surface receptor(s): 1) reducing transendothelial migration and accumulation of the inflammatory monocytes into the inflamed tissue and 2) inducing a tolerogenic phenotype in the recruited inflammatory DCs, which would led to: (a) inhibition of T cell proliferation and differentiation into Th1, Th2, and/or Th17 cells depending on the inflammatory microenvironment, (b) decreased pro-inflammatory cytokine secretion (IL-12, TNF-α, IFN-γ, etc.), (c) reduced migration to the lymph nodes, and conversely, to: induction of anti-inflammatory cytokine release (IL-10, TGF-β, etc.), and (d) Treg generation within the inflamed tissues (3).
Soluble PRMs are a heterogeneous group of molecules (collectins, ficolins, pentraxins, and other complement components) belonging to the humoral arm of innate immunity that have been proposed to represent the functional ancestor of antibodies (58). They share basic functions with the membrane-bound PRMs from DCs, such as the recognition of “non-self” and “modified self” and, additionally, play an important role in opsonization and complement activation. In the last years, several studies have evidenced that APPs, particularly soluble PRMs, acting directly in the early stages of monocyte differentiation mediated by GM-CSF/IL-4 (a faithful in vitro model for the generation of inflammatory DCs), are able to confer a tolerogenic phenotype and function to the ensuing Mo-DCs, although the detailed molecular mechanisms of APPs action over DCs are still not known for most of them.
In the next paragraphs, we will address the state of understanding and arguments regarding APP-mediated tolerogenic DC generation and functional outcome, according to common features currently defining tolerogenic DCs.
Pentraxins constitute a superfamily of evolutionarily conserved multimeric and multifunctional proteins sharing an 8-amino acid “pentraxin domain” (HxCxS/TWxS, where “x” is any amino acid) in their carboxy terminus. Based on the primary structure of the promoter, pentraxins are divided into short pentraxins (CRP and SAP) and long pentraxins (PTX3) (58). Both CRP and SAP are homooligomeric proteins arranged in a ~25 kDa subunit pentameric radial symmetry and hold 51% amino acid sequence identity. They constitute the main APPs in human and mouse, respectively, are produced by hepatocytes and have wide capacity for pathogen recognition, phagocytosis, and cytokine secretion through interaction with Fcγ receptors (59). Moreover, CRP and SAP are able to regulate the activation of the complement system by interaction with C1q, ficolins, C4b-binding protein (C4BP) and factor H, favoring efferocytosis and preventing the onset of autoimmune diseases (60, 61).
C-reactive protein has been shown to transform biological functions of Mo-DCs toward a tolerogenic phenotype. Interestingly, when CRP was added at the early stage of Mo-DC differentiation from CD14+ monocytes, it downregulated surface expression of DC-SIGN and the antigen uptake molecules CD205 and CD206, resulting in reduced endocytosis capacity (62, 63). Moreover, LPS-mediated Mo-DC maturation was also impaired, through downregulation of co-stimulatory molecules CD80 and CD86, and of the maturation marker CD83, inhibition of allogeneic T cell proliferation and decreased production of pro-inflammatory cytokines (IL-12, IL-8, IL-6, TNF-α, MIP-1α, MIP-1β, and MCP-1). These effects seemed to be mediated through the immunoreceptor FcγRII/CD32, which is downregulated during differentiation into Mo-DCs. Conversely, another study reported just the opposite, that is, CRP was able to activate Mo-DCs through upregulation of DC activation markers (CD40, CD80, CD83, and CCR7) and induced allogeneic T cell proliferation and IFN-γ production (64). Nevertheless, in that case the pulsation of Mo-DCs was started at day 6 of culture, once the Mo-DCs were fully differentiated. These results evidence the restricted tolerogenic activity window characterizing CRP, at the initial steps of Mo-DC differentiation. Analogously, human SAP has been reported to bind strongly to monocytes but weakly to differentiated Mo-DCs (65). SAP also inhibits neutrophil recruitment and monocyte to fibrocyte differentiation, in part, by binding to the FcγRs (66, 67), and polarizes macrophages toward an immunoregulatory phenotype through PI3K/Akt-ERK signaling (68). Thus, SAP regulates key components of the innate immune system and inflammation.
Pentraxins is a multimeric 340 kDa glycoprotein with a complex quaternary structure (elongated, with a large and a small domain interconnected by a stalk region) composed of two tetramers linked by interchain bridges to form an octamer. PTX3 expression is induced in a variety of cell types (particularly in phagocytes) by inflammatory cytokines, TLR agonists or pathogens, binds to a wide range of microorganisms, and plays a relevant role in host defense and inflammation (69), for example, by regulating leukocyte recruitment (70). Moreover, analogously to CRP and SAP, PTX3 is also able to modulate the activation of the complement system by binding C1q, ficolins, mannose-binding lectin (MBL), and the complement regulators C4BP and FH, and increases phagocytosis in an FcγRII-dependent manner. Hence, PTX3 binds to apoptotic cells and recruits C4BP, limiting complement activation and an exacerbated inflammatory response (71). In this context, PTX3 reduces the release of TNF-α and IL-10 by LPS-challenged Mo-DCs, and consistently inhibits the upregulation of membrane molecules (CD86, HLA-ABC, HLA-DR) on an inflammatory cell surface induced by LPS. Moreover, PTX3 also induces macrophages to secrete anti-inflammatory cytokines such as TGF-β and IL-10 (72), modulates LPS-induced inflammatory response and attenuates liver injury (73).
The evolutionarily conserved complement system, in addition to its crucial function in the innate defense against common pathogens, holds also a key regulatory non-immunogenic role in the “silent” clearance of immune complexes from the circulation and apoptotic cells from damaged tissues, in close crosstalk with the mononuclear phagocyte system (74). We have recently discussed the “non-canonical” activities of a variety of complement effectors and modulators able to transform DCs toward a tolerogenic phenotype (75). Thus, we will instead focus here on the functional outcome of a few representative complement components directly interacting with Mo-DCs.
In addition to their central role as complement cascade initiators for microbial phagocytosis and killing, it is becoming evident that both, complement cascade initiators such as mannose-binding lectin (MBL) and soluble complement inhibitors such as C4BP, are able to promote an immunomodulatory and anti-inflammatory environment by direct interaction with DCs and other immune cells.
MBL, the prototypic initiator of the lectin pathway of complement activation, belongs to the collectin family and, through its carbohydrate-recognition domains, is able to bind to oligosaccharides (mannose, N-acetyl-glucosamine) on the pathogen surface (76). DCs from MBL-deficient individuals showed increased IL-6 production and poor allogeneic T cell responses, features of pathogen-stimulated DCs, which could be reversed by in vitro addition of MBL (77). In fact MBL, at supraphysiological concentrations, influences the phenotype and function of DCs by attenuating LPS binding to immature DCs and their further maturation and pro-inflammatory cytokine production (IL-12, TNF-α), while preventing allogeneic T lymphocyte proliferation (78). Moreover, MBL not only attenuates LPS-induced Mo-DC maturation, but also affects early Mo-DC differentiation from CD14+ monocytes, yielding Mo-DCs with tolerogenic features (low MHC-II, CD80 and CD40 expression, increased IL-10 and IL-6 secretion, and reduced T cell alloproliferation), and being possibly mediated by members of the STAT family (79).
Among the complement inhibitors, the regulator of the classical and lectin pathways of complement activation C4BP has a complex oligomeric structure. The major C4BP isoform, C4BP(β+), has an heterooligomeric radial structure (570 kDa). It is composed by seven identical 70 kDa modular α-chains (responsible for the complement inhibitory activity, and for pentraxin, heparin, DNA, and pathogen binding, among others), and a single 40 kDa β-chain (high-affinity binding site for anticoagulant vitamin K-dependent Protein S, allowing a strong interaction with apoptotic/necrotic cells) (80, 81). The minor C4BP isoform, C4BP α7β0 or C4BP(β−), holds the same oligomeric structure and complement inhibitory function than C4BP(β+), but lacks the β-chain. Under acute phase conditions (poly-traumatisms, sepsis) the levels of circulating C4BP(β−) isoform increase significantly as a consequence of the differential hepatic regulation of the α- and β-chains by pro-inflammatory cytokines (82). Thus, C4BP(β−) is a genuine APP. We have shown that the C4BP(β−) isoform, but not the C4BP(β+) isoform, by direct interaction with Mo-DCs through as yet unknown receptor(s), only in the early stages of monocyte to Mo-DC differentiation, is able to confer an anti-inflammatory, tolerogenic phenotype to these cells, retaining a high-endocytic activity, and morphological features of immaturity. Upon LPS priming, these C4BP(β−)-treated Mo-DCs featured low-surface expression of CD83, CD80, and CD86, inhibition of pro-inflammatory IL-12, TNF-α, IFN-γ, IL-6, and IL-8 production and, instead, increased expression of anti-inflammatory IL-10 and TGF-β, reduced CCR7 expression and chemotaxis, and promoted Treg expansion. Moreover, C4BP(β−) induced tolerogenic DCs with increased viability and yield when compared with the immunomodulator vitamin D3, and similarly prevented T cell alloproliferation (83).
Although perhaps not a bona fide APP, C1q, the recognition unit from the classical pathway of complement activation and major component of the C1 complex, binds to various APPs including CRP, SAP, and PTX3, thereby regulating the classical complement pathway. C1q has also been recognized to modulate cellular functions within the adaptive immune response (84). Certainly, C1q has even been proposed as a tolerogenic DC marker because relevant immunomodulatory agents such as dexamethasone, IL-10, or vitamin D3 are able to induce at least a 10-fold overexpression of C1q at both mRNA and protein levels in Mo-DCs (85). The regulatory effects of C1q on monocyte/DC precursors could be mediated by gC1qR, occurring within a narrow timeframe of monocyte to Mo-DC transition and being influenced by the microenvironment. Accordingly, while in the presence of danger signals C1q would recognize and bind antigens through its globular head domains, leading to activation of a pro-inflammatory immune response in immature Mo-DCs, in the absence of danger signals C1q would maintain immature Mo-DCs in a tolerance state through gC1qR (86). Certainly, gC1qR ligation on the surface of Mo-DCs suppresses TLR4-induced IL-12 production through PI3K pathway activation (87). Furthermore, an alternative mechanism of C1q-mediated immunomodulation involves high-affinity binding between C1q and the inhibitory immunoreceptor LAIR-1, which inhibits monocyte-to-Mo-DC differentiation (88). More recently, this interaction has been refined through the characterization of a tri-molecular engagement encompassing C1q-CD33/LAIR-1 crosslinking (89).
Essential cellular processes, such as energy generation, DNA replication, oxygen transport, and protection from oxidative stress are dependent on iron. Since bacterial pathogens also require iron for replication and infection, iron sequestration strategies from vertebrates constitutes a significant form of nutritional innate immunity (90). Thus, in homeostatic healthy conditions iron is largely intracellular and sequestered within ferritin. Conversely, acute inflammatory processes such as infection, include the release of lactoferrin from secondary granules contained within polymorphonuclear leukocytes. Furthermore, hemoglobin released by physiological and pathological hemolysis is captured by Haptoglobin (Hp). All together, these proteins ensure a virtually free iron environment in vertebrate tissues.
Ferritin is a major tissue iron-binding protein with a molecular weight of 500 kDa, whose main function is to store iron in a soluble non-toxic form, protecting the cell from iron-mediated redox reactions. The levels of this APP remain elevated in many chronic inflammatory diseases such as periodontitis (91). Ferritin is composed of 24 subunits consisting of heavy (H) and light (L) chains, and may heterooligomerize forming isoferritins depending on the proportions of H and L chains (92). The immunosuppressive effects of cancer cell supernatants, such as melanoma supernatants, correlated with their content of H-ferritin. Accordingly, H-ferritin has been shown to inhibit anti-CD3-stimulated lymphocyte proliferation, probably mediated by increased IL-10 production (93). Importantly, H-ferritin is also able to induce a semi-mature, tolerogenic phenotype on Mo-DCs featuring increased expression of CD86 (B7-2) and B7-H1, and the activation of IL-10-producing Treg cells (94).
Lactoferrin, also known as lactotransferrin, is produced in a number of tissues and is frequently found in mucosal secretions and neutrophil secretory granules (95). This 80 kDa iron-binding glycoprotein is an important component of innate immunity, and holds a key role in the protection of mucosal surfaces from microbial infections (96). Lactoferrin also modulates innate and adaptive immune-inflammatory responses, including cytokine production, promotion of T and B cell maturation, and enhancement of delayed-type hypersensitivity against defined antigens. Moreover, it has been suggested that lactoferrin might exert adjuvant activity, enhancing DC function to promote generation of antigen-specific T cells (97). In contrast, bovine lactoferrin (bLF) seems to play an opposite role. Thus, Mo-DCs differentiated in the presence of bLF showed a fully tolerogenic or immunomodulatory behavior [potent anti-inflammatory activity, high-endocytic capacity, increased expression of molecules with negative immunoregulatory functions (ILT3, PD-L1, IDO, and SOCS3), CCL1 production, and impaired capacity to undergo activation and to promote Th1 responses]. bLF is internalized and seems to reach the nucleus, although the molecular details mediating the bLF-mediated transcriptional regulation of Mo-DC differentiation are still unknown (98, 99).
Hp is the major hemoglobin-binding protein in plasma. This APP, whose hepatic expression is induced by inflammatory mediators such as IL-6-type cytokines, interacts with free hemoglobin neutralizing and restricting its oxidative damage to various organs (100). Hp has been suggested to exert immunomodulatory effects constituent with suppression of lymphocyte function (101). During physiological and pathological hemolysis, the Hp-CD163-heme oxygenase (HO-1) pathway efficiently scavenges and circumvents hemoglobin/heme-induced toxicity. This pathway plays an anti-inflammatory role in phagocytes, and the resulting heme metabolites, such as bilirubin, reinforce its cytoprotective and anti-inflammatory efficacy (102). Hp seems also to prevent epidermal Langerhans cells from spontaneously undergoing functional maturation in the skin, inhibiting their capacity to activate autologous T cells in vitro (103).
Serum amyloid A is an APP produced mainly by the hepatocytes, but also by other cell types such as macrophages, smooth muscle cells, chondrocytes, epithelial cells, and adipocytes, under pro-inflammatory stimuli (58). SAA interacts with Gram-negative bacteria and, through its opsonic activity, increases their phagocytosis and the production of TNF-α and IL-10 by phagocytes (104). SAA has also been recently shown to be involved in the expression of the “alarmin” IL-33 by monocytes and macrophages (105). Notably, it has been recently shown that SAA-stimulated monocytes (HLA-DRhi HVEMlo) most resemble immature Mo-DCs, and are able to drive Treg proliferation (106). SAA is also a chemoattractant for immature Mo-DCs through formyl peptide receptor like 1/formyl peptide receptor 2 (107). Furthermore, mice lacking SAA3, an acutely expressed isoform found in non-primate mammals, develop metabolic dysfunction, and exacerbated pro-inflammatory responses from innate immune cells. Particularly, bone marrow-derived DCs from SAA3(−/−) mice produce increased levels of IL-1β, IL-6, IL-23, and TNF-α in response to LPS compared with cells from wild-type mice (108). Thus, endogenous SAA3 likely modulates metabolic and immune homeostasis.
α1-Antitrypsin, a member of the SERPIN superfamily of protease inhibitors, is a major inhibitor of the neutrophil-derived serine proteases [neutrophil elastase (NE), cathepsin G, and proteinase 3]. It has a primary anti-inflammatory role by irreversible binding and inactivation of NE, protecting the lung against the destructive effects of NE released by degranulating neutrophils during inflammation (109). AAT is predominantly produced by the liver, and its secretion, increased under acute phase conditions, is mediated by pro-inflammatory cytokines (IL-6, IL-1β, and TNF-α) (12). Recent studies have reported tolerogenic activities of AAT that are difficult to explain solely by serine-protease inhibition or by its anti-inflammatory actions (110). Circulating AAT is bound to lipoprotein particles (LDL and HDL) and docks onto lipid-rafts. Thus, TLR2 and TLR4 contained in lipid-rafts from macrophages and DCs are downregulated by AAT (111). Moreover, AAT induces a tolerogenic phenotype on DCs characterized by low levels of CD40, CD86, and MHC class II, increased production of IL-10 and enhanced generation of Tregs through a so far unknown mechanism. These tolerogenic DCs maintain, nevertheless, the inflammation-driven cell migration capacity (112). In fact, AAT monotherapy has been shown to induce tolerance in islet allograft and kidney transplantation (113, 114), graft-versus-host disease (115), improved islet function in type I diabetes (116), and attenuated lupus nephritis (117).
Fibrinogen is synthesized mainly by hepatocytes, and its level increases substantially during infections and inflammatory conditions. This 340 kDa glycoprotein, made up of two identical subunits joined together by disulfide bonds, functions as a blood coagulation factor, supporting platelet aggregation, and fibrin cloth formation at the site of vessel injury. Fibrinogen had an Mo-DC maturation effect comparable with poly I:C, TNF-α, and PGE2, but it failed to induce IL-12 production (118). On the other hand, it has been recently reported that fibrinogen cleavage products generated by protease allergens, through induction of IL-13 production by mast cells, increased the number of TH2-favorable (PD-L2+) DCs in allergic asthma (119). Interestingly, another member of the fibrinogen-related protein superfamily, soluble fibroleukin or fibrinogen-like protein 2 (sFGL2), highly inducible by IFN-γ and with features of APP, has a 50 kDa weight and is highly expressed in cytotoxic T cells and Tregs upon activation (120). sFGL2 seems to act as an immunosuppressor, repressing the proliferation of alloreactive T lymphocytes and the maturation of DCs (121, 122). Thus, by binding to FcγRIIB and FcγRIII, sFGL2 can adjust the antigen presentation ability of APCs. Accordingly, the levels of Th2 cytokines and the activity of DCs have found to be increased in FGL2-deficient mice (123).
Pharmacological immunosuppression has gone mainstream of past and, still, current therapeutic strategies to prevent transplant rejection and to restore autoantigen tolerance in autoimmune disorders. Yet, the downside of the immunosuppressive regimens is the appearance of numerous and often severe side effects and increased risk of infection as a consequence of the general suppression of the host immune system (124–126). Thus, the attractive concept of using DCs, central orchestrators of other immune cells, with the aim to modulate immune-inflammatory responses that have gone awry while leaving protective immunity intact is becoming gradually a reality in the clinical setting. In fact, in addition to being explored in experimental animal models of autoimmune diseases such as collagen-induced arthritis (127, 128), diabetes (129, 130), and experimental autoimmune encephalomyelitis (EAE) (131), along with experimental graft rejection after transplantation (132, 133), tolerogenic DCs have recently been, and are currently being tested in phase I clinical trials for alloimmune (transplantation, graft-versus-host disease) and autoimmune processes (type I diabetes, rheumatoid arthritis, multiple sclerosis, and Crohn’s disease), and allergy, and there are ongoing collaborative efforts to harmonize/standardize tolerance-inducing therapies for upcoming trials (134–136).
The identification, characterization and, most notably, isolation and amplification of genuine regulatory or tolerogenic DCs populating a given healthy or diseased tissue, much like MDSCs, has proven a daunting task and a real limitation when planning to adoptively transfer them for therapeutic benefit. Therefore, due to the high plasticity of mononuclear myeloid cells such as monocytes, well-established ex vivo protocols of monocyte to Mo-DC expansion and differentiation, relying in the use of inflammatory cytokines (GM-CSF and IL-4) have become instrumental to adopt Mo-DCs as therapeutic cell products for clinical use (137).
A central aspect for the successful clinical application of tolerogenic Mo-DC relates to their development and manufacture. As previously stated, a variety of agents have been employed in vitro to skew Mo-DCs toward a tolerogenic or regulatory phenotype (notably vitamin D3, immunosuppressive drugs-like dexamethasone, or NF-κB inhibitors), opposing their “natural” tendency to be activated in vivo in a pro-inflammatory environment. Yet limited efficacy has been reported in terms of disease outcome, although most trials have noted an increase in Treg levels in the recipient’s blood during tolerogenic Mo-DC administration (138–140). Clearly, maintaining tolerogenic Mo-DCs in an activation- or maturation-resistant state is a fundamental requirement for a successful tolerogenic Mo-DC therapy, because unstable tolerogenic Mo-DCs able to reverse back in vivo to an immunogenic phenotype in contact with a pro-inflammatory microenvironment could aggravate the pathology. Indeed, semi-mature DCs, considered tolerogenic in in vitro assays, may become immunogenic when administered in vivo (141, 142). Furthermore, it has been recently reported that continuous treatment of DCs during their differentiation from bone marrow cells (10-day treatment) with the histone deacetylase inhibitor suberoxylanilide hydroxamic acid generated tolerogenic DCs that, however, were not stable and, therefore, inefficacious when administered in mice with EAE (143). Hence, the possibility to anticipate and modulate the stability of tolerogenic Mo-DCs in vivo, particularly in the pro-inflammatory allo- or autoimmune environments in which these cells are applied, would enhance their therapeutic efficacy. As yet, there is not enough mechanistic knowledge to ascertain which stimuli guarantee the induction of stable tolerogenic Mo-DCs adapted to particular in vivo situations. Still, in tolerogenic Mo-DC conditioning protocols, establishing the appropriate timing and intensity of the tolerogenic reagent treatment, its toxicity, as well as the migration capacity of the resulting conditioned cells are crucial aspects to take into account for a successful Mo-DC-based immunotherapy (134). For example, both rapamycin- and dexamethasone-conditioned cell cultures have been shown to markedly reduce DC recovery (144, 145). Importantly, none of the APPs proved cytotoxic in the Mo-DC cultures, most likely because of the wide range of physiological concentrations that these proteins are able to reach in serum, fluctuating between homeostatic and acute phase conditions. Thus, APP-derived tolerogenic Mo-DCs might overcome some of the limitations of the current tolerogenic Mo-DCs employed for immunotherapy approaches regarding consistency, safety, and efficacy (124, 146).
On the other hand, the common tolerogenic moonlighting activity of APPs over Mo-DCs seems surprising, given the variety of different physiological functions ascribed to these proteins. Remarkably, nearly all of them share a complex, oligomeric, and multi-modular structure (Table 1), providing flexibility in their capacity for binding, with a different grade of specificity, a variety of humoral and/or cellular determinants, including different receptors in the surface of Mo-DCs. This feature might constitute an advantage for the fine-tuning of the desired tolerogenic phenotype on Mo-DCs.
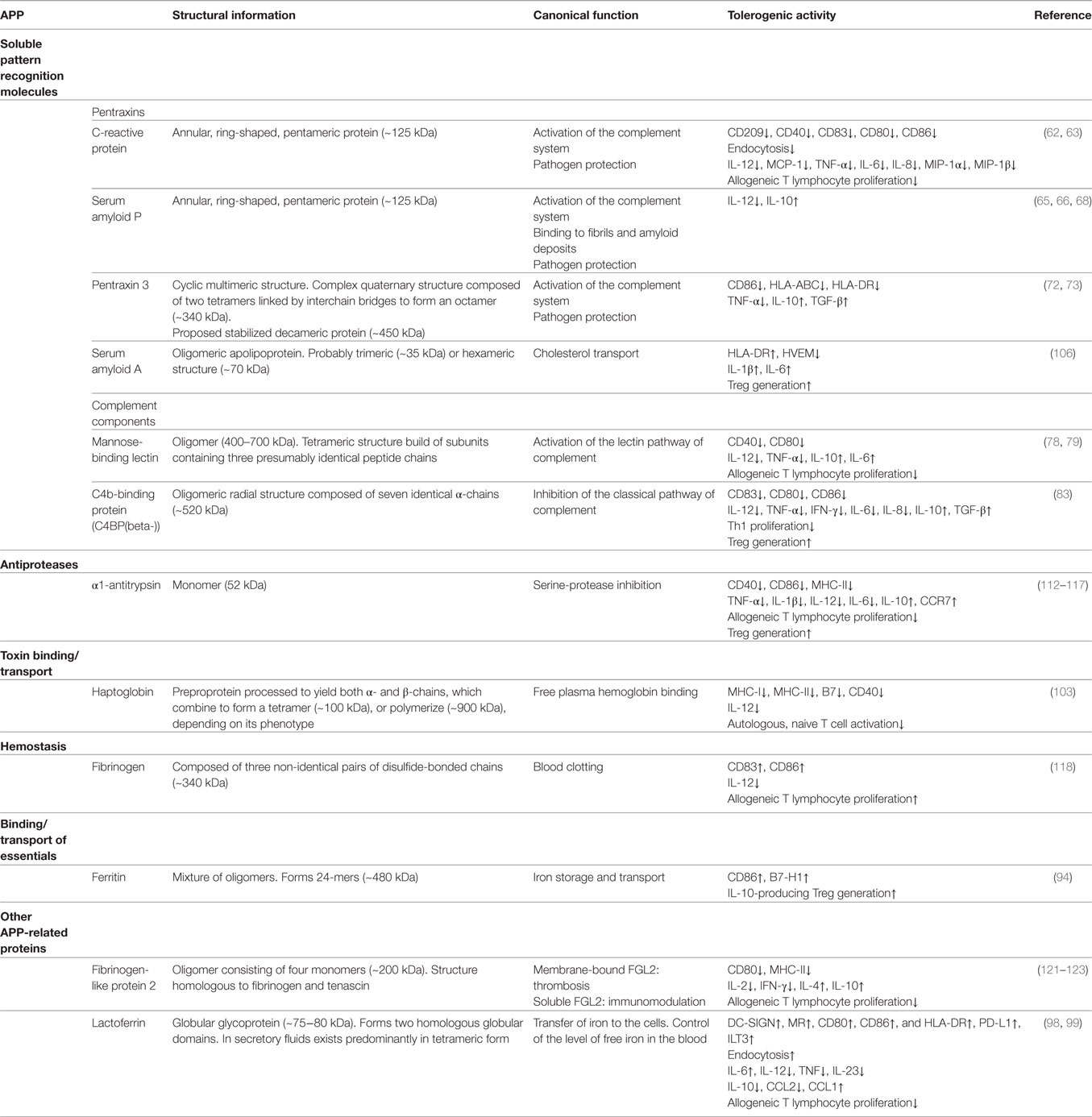
Table 1. Immunomodulatory actions of acute phase proteins (APPs) on dendritic cells (DCs).
Another key aspect of APP action, as outlined in previous sections, relates to the simultaneous presence and increased levels of both APPs and Mo-DCs under overwhelming immune-inflammatory conditions such as the acute phase response, which incites a physiological crosstalk between these humoral (APPs) and cellular (Mo-DCs) systems with the common goal of providing protection and progress toward the resolution of inflammation. In this regard, APPs probably contribute in vivo to mononuclear phagocyte switching toward an anti-inflammatory mode aimed at restoration of tissue integrity and function. Consequently, the APP interaction with Mo-DCs will be safer and effective over a wide range of concentrations, according to the significantly increased blood levels reached by APPs under acute phase conditions. On the other hand, most APPs have been shown to act over a narrow window within the Mo-DC differentiation and/or maturation program, limiting also their hypothetical toxicity, if any, and increasing their specificity compared with some of the current immunosuppressive/immunomodulatory agents, which need to be present over the full differentiation/maturation program to induce a tolerogenic outcome into Mo-DCs. Furthermore, several APPs and, particularly, all soluble PRMs, operate only in the early stages of monocyte to Mo-DC differentiation, while IL-10, for example, is active on Mo-DCs up to their terminal differentiation, when Mo-DCs downregulate the IL-10 receptor (147). This restricted activity of APPs at the beginning of the Mo-DC differentiation program may induce a more permanent and stable modification of the Mo-DC tolerogenic phenotype than that achieved by immunomodulators/immunossupressors influencing Mo-DCs late in their differentiation process, or by agents affecting only their maturation/activation status. These last agents may be more prone to be influenced by the pro-inflammatory microenvironment that the tolerogenic Mo-DCs face upon clinical administration. Comparing the performance and outcome of in vitro assays, APPs seem to hold a tolerogenic activity over Mo-DCs (low expression of co-stimulatory molecules, low production of pro-inflammatory cytokines, increased release of anti-inflammatory cytokines, low T cell alloproliferation and, instead, increased Treg generation, etc.) at least as efficient as the agents (rapamycin, dexamethasone and/or vitamin D3, NF-κB inhibitors, etc.) currently employed to generate clinical-grade tolerogenic Mo-DCs for the induction or restoration of immune tolerance in autoimmune pathologies and transplantation (148). Furthermore, given the broadened presence that APPs can reach in serum under acute phase conditions and the present hurdles facing adoptive DC-based immunotherapy (time-consuming, expensive, and arduous to implement in the current regulatory environment), the direct in vivo administration of APPs, either naked or complexed with nanoparticles, may become a useful and efficacious alternative in inflammatory pathologies. In fact, nanoparticle formulations for DC-specific receptor targeting (DEC205, DC-SIGN, CD40, CD11c, etc.) are being used in preclinical assays and phase I clinical trials as vaccines for onco-immunotherapy (149–151).
Nevertheless, presently the most important drawback for the use of APPs as tolerogenic agents lies in the fact that the detailed molecular mechanisms of action of APP-mediated transformation of Mo-DCs toward a tolerogenic phenotype are not known for most of these proteins. Thus, current efforts employing high-throughput genomics and proteomics approaches will certainly dissect cell surface-interacting partner(s) and relevant signaling and metabolic pathways underlying APP-mediated programming and distinctive functional outcome of the ensuing tolerogenic Mo-DCs (152, 153).
For a successful tolerogenic immunotherapy, Mo-DC conditioning must regulate antigen-specific immune responses in the intrinsically complex pro-inflammatory environments evolving in autoimmune disorders and transplantation, sustaining the development of immunological memory toward tolerance. Thus, it is critically important to thoroughly test the performance of novel tolerance-inducing agents regarding the potency and durability of the ensuing tolerogenic Mo-DC phenotype.
Besides being proposed as useful biomarkers for a variety of inflammatory pathologies, recent studies have proposed that APPs play important roles in tissue homeostasis and repair following overwhelming immune-inflammatory processes, probably in close interaction with inflammatory monocytes and DCs. In fact, APPs are able to generate tolerogenic Mo-DCs in vitro with the desired regulatory features (increased expression of immunomodulatory molecules, enhanced production of anti-inflammatory cytokines, and Treg generation) and low immunogenicity (Table 1), comparable with the currently used clinical tolerogenic Mo-DC-inducing immunomodulatory/immunosuppressive agents. Although the precise mechanism of action of tolerogenic Mo-DC skewing induced by most APPs is still unknown, these proteins may prove useful alternatives to overcome the present limitations for a more efficacious, safe, and stable Mo-DC-based tolerogenic immunotherapy. In this regard, attractive attributes of APPs include a physiological basis regarding their interaction with Mo-DCs in the context of the acute phase response, and a wide range of action due to the own intrinsic features of APPs, which would ensure reduced toxicity at the cellular level and increased safety upon in vivo administration. Moreover, the narrow activity window in the early stages of monocyte to Mo-DC differentiation shown by several APPs, notably soluble PRMs, should increase specificity and, more importantly, may contribute to a more stable tolerogenic phenotype. APPs targeting differentiating Mo-DCs could turn these cells unresponsive to the in vivo pro-inflammatory microenvironment present in autoimmune or alloimmune conditions and, therefore, refractory to Mo-DC maturation. Furthermore, taking into account novel findings, such as the proteomic characterization of tissue-/disease-specific posttranslational modifications of APPs (14), or the influence of the clinical status of the Mo-DC recipient (154) may fine-tune the tolerogenic potential of APP-treated Mo-DCs, e.g., their ability to modulate T cell responses. Nevertheless, additional research should help clarify whether some APPs, particularly pentraxins, or complement activators, are able to maintain their induced tolerogenic DCs in a stable and functional state upon administration in complex pathological tissue contexts, because of the dual protective and pro-inflammatory role played by these multifaceted molecules in physiology.
Definitely, although further work is warranted to establish which method, or perhaps combination of methods, is most suitable to generate tolerogenic Mo-DCs in the clinical setting, APPs may contribute, either on their own, combined with currently employed immunomodulators/immunosuppressants (155), and/or with recently proposed tolerogenic DC boosters such as minocycline (156), or reinforcing the tolerogenic properties of iPSC-derived CD141+ DCs holding enhanced capacity for antigen cross-presentation (157), to the design of tailored protocols to induce or re-establish immunological tolerance in different clinical settings including allogeneic transplantation and autoimmune diseases.
JA wrote the manuscript. IS and AL added additional insights. The final version was proofread and edited by all authors.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflict of interest.
JA received support from Ministerio de Economía y Competitividad (Madrid, Spain) [grants FIS-ISCIII PI13/01490 and PI16/00377, co-funded by FEDER funds/European Regional Development Fund (ERDF)-a way to build Europe-], from “La Marató de TV3” Foundation (grant 12/1210), and from Generalitat de Catalunya (grant 2014SGR541). JA is sponsored by the “Researchers Consolidation Program” from the SNS-Dpt. Salut Generalitat de Catalunya (Exp. CES06/012).
1. Ortega-Gómez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med (2003) 5(5):661–74. doi:10.1002/emmm.201202382
2. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol (2017) 17(6):349–62. doi:10.1038/nri.2017.28
3. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392(6673):245–52. doi:10.1038/32588
4. Bancherau J, Paczesny S, Blanco P, Bennet L, Pascual V, Fay J, et al. Dendritic cells: controllers of the immune system and a new promise for immunotherapy. Ann N Y Acad Sci (2003) 987:180–7. doi:10.1111/j.1749-6632.2003.tb06047.x
5. Macagno A, Napolitani G, Lanzavecchia A, Sallusto F. Duration, combination and timing: the signal integration model of dendritic cell activation. Trends Immunol (2007) 28(5):227–33. doi:10.1016/j.it.2007.03.008
6. Gregori S. Dendritic cells in networks of immunological tolerance. Tissue Antigens (2011) 77(4):89–99. doi:10.1111/j.1399-0039.2010.01615.x
7. Whicher JT, Westacott CI. The acute phase response. In: Whicher JT, Evans SW, editors. Biochemistry of Inflammation Boston: Kluwer Academic Publishers (1992). vol. 18, p. 243–69.
8. Ebersole JL, Cappelli D. Acute-phase reactants in infections and inflammatory diseases. Periodontol 2000 (2000) 23(1):19–49. doi:10.1034/j.1600-0757.2000.2230103.x
9. Cripse IN. Hepatocytes as immunological agents. J Immunol (2016) 196(1):17–21. doi:10.4049/jimmunol.1501668
10. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med (1999) 340(6):448–54. doi:10.1056/NEJM199902113400607
11. Parrow NL, Fleming RE, Minnick MF. Sequestration and scavenging of iron in infection. Infect Immun (2013) 81(10):3503–14. doi:10.1128/IAI.00602-13
12. Baumann H, Gauldie J. The acute phase response. Immunol Today (1994) 15(2):74–80. doi:10.1016/0167-5699(94)90137-6
13. Chrousos GP. The hypothalamic-pituitary-adrenal axis and the immune-mediated inflammation. N Engl J Med (1995) 332(20):1351–62. doi:10.1056/NEJM199505183322008
14. Schrödl W, Büchler R, Wendler S, Reinhold P, Muckova P, Reindl J, et al. Acute phase proteins as promising biomarkers: perspectives and limitation for human veterinary medicine. Proteomics Clin Appl (2016) 10(11):1077–92. doi:10.1002/prca.201600028
15. Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol (2009) 78(6):539–52. doi:10.1016/j.bcp.2009.04.029
16. Dinarello CA. Interleukin-1 and its biologically related cytokines. Adv Immunol (1989) 44:153–205. doi:10.1016/S0065-2776(08)60642-2
17. Heinrich PC, Castell JV, Andus T. Interleukin-6 and the acute phase response. Biochem J (1990) 265(3):621–36. doi:10.1042/bj2650621
18. Sehgal PB, Greininger G, Tosato G. Acute phase and immune responses: interleukin-6. Ann N Y Acad Sci (1989) 557:1–583.
19. Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med (1994) 179(4):1109–18. doi:10.1084/jem.179.4.1109
20. Randolph GJ, Inaba K, Robbiani DF, Steinman RM, Muller WA. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity (1999) 11(6):753–61. doi:10.1016/S1074-7613(00)80149-1
21. Segura E, Amigorena S. Inflammatory dendritic cells in mice and humans. Trends Immunol (2013) 34(9):440–5. doi:10.1016/j.it.2013.06.001
22. Askenase MH, Han SJ, Byrd AL, Morais da Fonseca D, Bouladoux N, Wilhelm C, et al. Bone-marrow-resident NK cells prime monocytes for regulatory function during infection. Immunity (2015) 42(6):1130–42. doi:10.1016/j.immuni.2015.05.011
23. Tamoutounour S, Guilliams M, Sanchis F, Liu H, Terhost D, Malosse C, et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity (2013) 39(5):925–38. doi:10.1016/j.immuni.2013.10.004
24. Campbell IK, van Nieuwenhuijze A, Segura E, O’Donnell K, Coghill E, Hommel M, et al. Differentiation of inflammatory dendritic cells is mediated by NF-kB1-dependent GM-CSF production in CD4 T cells. J Immunol (2011) 186(9):5468–77. doi:10.4049/jimmunol.1002923
25. Schmid MA, Harris E. Monocyte recruitment to the dermis and differentiation to dendritic cells increases the targets for dengue virus replication. PLoS Pathog (2014) 10(12):e1004541. doi:10.1371/journal.ppat.1004541
26. Van der Laan AM, Ter Horst EN, Delewi R, Begieneman MP, Krijnen PA, Hirsh A, et al. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur Heart J (2014) 35(6):376–85. doi:10.1093/eurheartj/eht331
27. Clements M, Gershenovich M, Chaber C, Campos-Rivera J, Du P, Zhang M, et al. Differential Ly6C expression after renal ischemia-reperfusion identifies unique macrophage populations. J Am Soc Nephrol (2016) 27(1):159–70. doi:10.1681/ASN.2014111138
28. Liaskou E, Zimmermann HW, Li KK, Oo YH, Suresh S, Stamataki Z, et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology (2013) 57(1):385–98. doi:10.1002/hep.26016
29. Jung H, Mithal DS, Park JE, Miller RJ. Localized CCR2 activation in the bone marrow niche mobilizes monocytes by desensitizing CXCR4. PLoS One (2015) 10(6):e0128387. doi:10.1371/journal.pone.0128387
30. Thiesen S, Janciauskiene S, Uronen-Hansson H, Agace W, Högerkorp CM, Spee P, et al. CD14(hi)HLA-DR(dim) macrophages, with a resemblance to classical blood monocytes, dominate inflamed mucosa in Crohn’s disease. J Leukoc Biol (2014) 95(3):531–41. doi:10.1189/jlb.0113021
31. Serti E, Werner JM, Chattergoon M, Cox AL, Lohmann V, Rehermann B. Monocytes activate natural killer cells via inflammasome-induced interleukin 18 in response to hepatitis C virus replication. Gastroenterology (2014) 147(1):209–20.e3. doi:10.1053/j.gastro.2014.03.046
32. Chow KV, Sutherland RM, Zhan Y, Lew AM. Heterogeneity, functional specialization and differentiation of monocyte-derived dendritic cells. Immunol Cell Biol (2017) 95(3):244–51. doi:10.1038/icb.2016.104
33. Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity (2015) 43(3):502–14. doi:10.1016/j.immuni.2015.08.010
34. Morias Y, Abels C, Laoui D, Van Overmeire E, Guilliams M, Schouppe E, et al. Ly6C-monocytes regulate parasite-induced liver inflammation by inducing the differentiation of pathogenic Ly6C+ monocytes into macrophages. PLoS Pathog (2015) 11(5):e1004873. doi:10.1371/journal.ppat.1004873
35. Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK, et al. Nonclassical Ly6C(-) monocytes drive the development of inflammatory arthritis in mice. Cell Rep (2014) 9(2):591–604. doi:10.1016/j.celrep.2014.09.032
36. Peng Y, Latchman Y, Elkon KB. Ly6C (low) monocytes differentiate into dendritic cells and cross-tolerize T cells through PDL-1. J Immunol (2009) 182(5):2777–85. doi:10.4049/jimmunol.0803172
37. Syme R, Glück S. Generation of dendritic cells: role of cytokines and potential clinical application. Transfus Apher Sci (2001) 24(2):117–24. doi:10.1016/S1473-0502(01)00005-2
38. Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity (2013) 38(2):336–48. doi:10.1016/j.immuni.2012.10.018
39. Xu Y, Zhan Y, Lew AM, Naik SH, Kershaw MH. Differential development of murine dendritic cells by GM-CSF versus Flt3 ligand has implications for inflammation and trafficking. J Immunol (2007) 179(11):7577–84. doi:10.4049/jimmunol.179.11.7577
40. León B, López-Bravo M, Ardavín C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity (2007) 26(4):519–31. doi:10.1016/j.immuni.2007.01.017
41. Nakano H, Lin KL, Yanagita M, Charbonneau C, Cook DN, Kakiuchi T, et al. Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat Immunol (2009) 10(4):394–402. doi:10.1038/ni.1707
42. Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell (2010) 143(3):416–29. doi:10.1016/j.cell.2010.09.039
43. Bonasio R, Scimone ML, Schaerli P, Grabie N, Lichtman AH, von Andrian UH. Clonal deletion of thymocytes by circulating dendritic cells homing to the thymus. Nat Immunol (2006) 7(10):1092–100. doi:10.1038/ni1106-1234b
44. Takenaka MC, Quintana FJ. Tolerogenic dendritic cells. Semin Immunopathol (2017) 39(2):113–20. doi:10.1007/s00281-016-0587-8
45. Iberg CA, Jones A, Hawiger D. Dendritic cells as inducers of peripheral tolerance. Trends Immunol (2017) 38(11):793–804. doi:10.1016/j.it.2017.07.007
46. Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med (2001) 194(6):769–79. doi:10.1084/jem.194.6.769
47. Riquelme P, Geissler EK, Hutchinson JA. Alternative approaches to myeloid suppressor cell therapy in transplantation: comparing regulatory macrophages to tolerogenic DCs and MDSCs. Transplant Res (2012) 1(17):1–14. doi:10.1186/2047-1440-1-17
48. Maldonado RA, von Andrian UH. How tolerogeneic dendritic cells induce regulatory T cell. Adv Immunol (2010) 108:111–65. doi:10.1016/B978-0-12-380995-7.00004-5
49. Sim WJ, Ahl PJ, Connolly JE. Metabolism is central to tolerogenic dendritic cell function. Mediators Inflamm (2016) 2016:2636701. doi:10.1155/2016/2636701
50. García-González P, Ubilla-Olquín G, Catalán D, Schinnerling K, Aguillón JC. Tolerogenic dendritic cells for reprogramming of lymphocyte responses in autoinmune diseases. Autoimmun Rev (2016) 15(11):1071–80. doi:10.1016/j.autrev.2016.07.032
51. Steinbrink K, Wölfl M, Jonuleit H, Knop J, Enk AH. Induction of tolerance by IL-10-treated dendritic cells. J Immunol (1997) 159(10):4772–80.
52. Anderson AE, Sayers BL, Haniffa MA, Swan DJ, Diboll J, Wang XN, et al. Differential regulation of naïve and memory CD4+ T cells by alternatively activated dendritic cells. J Leukoc Biol (2008) 84(1):124–33. doi:10.1189/jlb.1107744
53. Adorini L, Giarratana N, Penna G. Pharmacological induction of tolerogenic dendritic cells and regulatory T cells. Semin Immunol (2004) 16(2):127–34. doi:10.1016/j.smim.2003.12.008
54. Anderson AE, Swan DJ, Wong OY, Buck M, Eltherington O, Harry RA, et al. Tolerogenic dendritic cells generated with dexamethasone and vitamin D3 regulate rheumatoid arthritis CD4+ T cells partly via transforming growth factor-β1. Clin Exp Immunol (2017) 187(1):113–23. doi:10.1111/cei.12870
55. Wong KL, Yeap WH, Tai JJ, Ong SM, Dang TM, Wong SC. The three human monocyte subsets: implications for health and disease. Immunol Res (2012) 53(1–3):41–57. doi:10.1007/s12026-012-8297-3
56. Faivre V, Lukaszewicz AC, Alves A, Charron D, Payen D, Haziot A. Human monocytes differentiate into dendritic cells subsets that induce anergic and regulatory T cells in sepsis. PLoS One (2012) 7(10):e47209. doi:10.1371/journal.pone.0047209
57. Sander LE, Sackett SD, Dierssen U, Beraza N, Linke RP, Müller M, et al. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J Exp Med (2010) 207(7):1453–64. doi:10.1084/jem.20091474
58. Bottazzi B, Doni A, Garlanda C, Matovani A. An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu Rev Immunol (2010) 28:157–83. doi:10.1146/annurev-immunol-030409-101305
59. Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, Sun PD. Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature (2008) 456(7224):989–92. doi:10.1038/nature07468
60. Gershov D, Kim S, Brot N, Elkon KB. C-reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. J Exp Med (2000) 192(9):1353–64. doi:10.1084/jem.192.9.1353
61. Familian A, Zwart B, Huisman HG, Rensink I, Roem D, Hordijk PL, et al. Chromatin-independent binding of serum amyloid P component to apoptotic cells. J Immunol (2001) 167(2):647–54. doi:10.4049/jimmunol.167.2.647
62. Zhang R, Becnel L, Li M, Chen C, Yao Q. C-reactive protein impairs human CD14+ monocyte-derived dendritic cell differentiation, maturation and function. Eur J Immnol (2006) 36(11):2993–3006. doi:10.1002/eji.200635207
63. Jimenez RV, Wright TT, Jones NR, Wu J, Gibson AW, Szalai AJ. C-reactive protein impairs dendritic cell development, maturation, and function: implications for peripheral tolerance. Front Immunol (2018) 9:372. doi:10.3389/fimmu.2018.00372
64. Van Vré EA, Bult H, Hoymans VY, Van Tendeloo VF, Vrints CJ, Bosmans JM. Human C-reactive protein activates monocyte-derived dendritic cells and induces dendritic cell-mediated T-cell activation. Arterioscler Thromb Vasc Biol (2008) 28(3):511–8. doi:10.1161/ATVBAHA.107.157016
65. MacDonald SL, Kilpatrick DC. Human serum amyloid P component binds to peripheral blood monocytes. Scand J Immunol (2006) 64(1):48–52. doi:10.1111/j.1365-3083.2006.01774.x
66. Castaño AP, Lin SL, Surowy T, Nowlin BT, Turlapati SA, Patel T, et al. Serum amyloid P inhibits fibrosis through Fc gamma R-dependent monocyte-macrophage regulation in vivo. Sci Transl Med (2009) 1(5):5ra13. doi:10.1126/scitranslmed.3000111
67. Crawford JR, Pilling D, Gomer RH. FcγRI mediates serum amyloid P inhibition of fibrocyte differentation. J Leukoc Biol (2012) 92(4):699–711. doi:10.1189/jlb.0112033
68. Zhang W, Xu W, Xiong S. Macrophage differentation and polarization via phosphatidylinositol 3-kinase/Akt-ERK signaling pathway conferred by serum amyloid P component. J Immunol (2011) 187(4):1764–77. doi:10.4049/jimmunol.1002315
69. Moalli F, Doni A, Deban L, Zelante T, Zagarella S, Bottazzi B, et al. Role of complement and Fcγ receptors in the protective activity of the long pentraxin PTX3 against Aspergillus fumigatus. Blood (2010) 116(24):5170–80. doi:10.1182/blood-2009-12-258376
70. Deban L, Russo RC, Sironi M, Moalli F, Scanziani M, Zambelli V, et al. Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol (2010) 11(4):328–34. doi:10.1038/ni.1854
71. Braunschweig A, Józsi M. Human pentraxin 3 binds to the complement regulator c4b-binding protein. PLoS One (2011) 6(8):e23991. doi:10.1371/journal.pone.0023991
72. Ortega-Hernandez OD, Bassi N, Shoenfeld Y, Anaya JM. The long pentraxin 3 and its role in autoimmunity. Semin Arthritis Rheum (2009) 39(1):38–54. doi:10.1016/j.semarthrit.2008.03.006
73. Perea L, Coll M, Sanjurjo L, Blaya D, Taghdouini AE, Rodrigo-Torres D, et al. Pentraxin-3 modulates lipopolysaccharide-induced inflammatory response and attenuates liver injury. Hepatology (2017) 66(3):953–68. doi:10.1002/hep.29215
74. Freely S, Kemper C, Le Friec G. The “ins and outs” of complement-drive immune responses. Immunol Rev (2016) 274(1):16–32. doi:10.1111/imr.12472
75. Luque A, Serrano I, Aran JM. Complement components as promoters of immunological tolerance in dendritic cells. Semin Cell Dev Biol (2017) S1084-9521(17):30131–3. doi:10.1016/j.semcdb.2017.11.022
76. Ip WK, Takahashi K, Ezekowith RA, Stuart LM. Mannose-binding lectin and innate immunity. Immunol Rev (2009) 230(1):9–21. doi:10.1111/j.1600-065X.2009.00789.x
77. Dean MM, Flower RL, Eisen DP, Minchinton RM, Hart DN, Vuckovic S. Mannose-binding lectin deficiency influences innate and antigen-presenting functions of blood myeloid dendritic cells. Immunology (2011) 132(2):296–305. doi:10.1111/j.1365-2567.2010.03365.x
78. Wang M, Zhang Y, Chen Y, Zhang L, Lu X, Chen Z. Mannan-binding lectin regulates dendritic cell maturation and cytokine production induced by lipopolysaccharide. BMC Immunol (2011) 12:1. doi:10.1186/1471-2172-12-1
79. Xu XY, Li HJ, Zhang LY, Lu X, Zuo DM, Shan GQ, et al. Mannan-binding lectin at supraphysiological concentrations inhibits differentiation of dendritic cells from human CD14+ monocytes. Microbiol Immunol (2015) 59(12):724–34. doi:10.1111/1348-0421.12337
80. Blom AM, Villoutreix BO, Dahlbäck B. Complement inhibitor C4b-binding protein-friend or foe in the innate immune system? Mol Immunol (2004) 40(18):1333–46. doi:10.1016/j.molimm.2003.12.002
81. Ermert D, Blom AM. C4b-binding protein: the good, the bad and the deadly. Novel functions of an old friend. Immunol Lett (2016) 169:82–92. doi:10.1016/j.imlet.2015.11.014
82. Criado-García O, Sánchez-Corral P, Rodríguez de Córdoba S. Isoforms of human C4b-binding protein II. Differential modulation of the C4BPA and C4BPB genes by acute phase cytokines. J Immunol (1995) 155(8):4037–43.
83. Olivar R, Luque A, Naranjo-Gómez M, Quer J, García de Frutos P, Borràs FE, et al. The α7β0 isoform of the complement regulator C4b-binding protein induces a semimature, anti-inflamatory state in dendritic cells. J Immunol (2013) 190(6):2857–72. doi:10.4049/jimmunol.1200503
84. Ghebrehiwt B, Hosszu KH, Peerschke EI. C1q as an autocrine and paracrine regulator of cellular functions. Mol Immunol (2017) 84:26–33. doi:10.1016/j.molimm.2016.11.003
85. Zimmer A, Bouley J, Le Mignon M, Pliquet E, Horiot S, Turfkruyer M, et al. A regulatory dendritic cell signature correlates with the clinical efficacy of allergen-specific sublingual immunotherapy. J Allergy Clin Immunol (2012) 129(4):1020–30. doi:10.1016/j.jaci.2012.02.014
86. Hosszu KK, Santiago-Schwarz F, Peershke EI, Ghebrehiwet B. Evidence that a C1q/C1qR system regulates monocyte-derived dendritic cell differentiation at the interface of innate and acquired immunity. Innate Immun (2010) 16(2):115–27. doi:10.1177/1753425909339815
87. Waggoner SN, Cruise MW, Kassel R, Hahn YS. gC1q receptor ligation selectively down-regulates human IL-12 production through activation of the phosphoinositide 3-kinase pathway. J Immunol (2005) 175(7):4706–14. doi:10.4049/jimmunol.175.7.4706
88. Son M, Santiago-Schwarz F, Al-Abed Y, Diamond B. C1q limits dendritic cell differentiation and activation by engaging LAIR-1. Proc Natl Acad Sci U S A (2012) 109(46):E3160–7. doi:10.1073/pnas.1212753109
89. Son M, Diamond B, Volpe BT, Aranow CB, Mackay MC, Santiago-Schwarz F. Evidence for C1q-mediated crosslinking of CD33/LAIR-1 inhibitory immunoreceptors and biological control of CD33/LAIR-1 expression. Sci Rep (2017) 7(270):1–13. doi:10.1038/s41598-017-00290-w
90. Weinberg ED. Iron availability and infection. Biochim Biophys Acta (2009) 1790(7):600–5. doi:10.1016/j.bbagen.2008.07.002
91. Chakraborty S, Tewari S, Sharma RK, Narula SC. Effect of non-surgical periodontal therapy on serum ferritin levels: an interventional study. J Periodontol (2014) 85(5):688–96. doi:10.1902/jop.2013.130107
92. Theil EC. Ferritin: structure, gene regulation, and cellular function in animals, plants, and microorganisms. Annu Rev Biochem (1987) 56:289–315. doi:10.1146/annurev.bi.56.070187.001445
93. Gray CP, Franco AV, Arosio P, Hersey P. Immunosuppressive effects of melanoma-derived heavy-chain ferritin are dependent on stimulation of IL-10 production. Int J Cancer (2001) 92(6):843–50. doi:10.1002/ijc.1269
94. Gray CP, Arosio P, Hersey P. Heavy chain ferritin activates regulatory T cells by induction of changes in dendritic cells. Blood (2002) 99(9):3326–34. doi:10.1182/blood.V99.9.3326
95. Brock JH. The physiology of lactoferrin. Biochem Cell Biol (2002) 80(1):1–6. doi:10.1139/o01-212
96. Arslan SY, Leung KP, Wu CD. The effect of lactoferrin on oral bacterial attachment. Oral Microbiol Immunol (2009) 24(5):411–6. doi:10.1111/j.1399-302X.2009.00537.x
97. Hwang SA, Actor JK. Lactoferrin modulation of BCG-infected dendritic cell functions. Int Immunol (2009) 21(10):1185–97. doi:10.1093/intimm/dxp084
98. Puddu P, Latorre D, Carollo M, Catizone A, Ricci G, Valenti P, et al. Bovine lactoferrin counteracts toll-like receptor mediated activation signals in antigen presenting cells. PLoS One (2011) 6(7):e22504. doi:10.1371/journal.pone.0022504
99. Latorre D, Pulvirenti N, Covino DA, Varano B, Purificato C, Rainaldi G, et al. Bovine lactoferrin-induced CCL1 expression involves distinct receptors in monocyte-derived dendritic cells and their monocyte precursors. Toxins (Basel) (2015) 7(12):5472–83. doi:10.3390/toxins7124897
100. Dobryszycka W. Biological functions of haptoglobin-new pieces to an old puzzle. Eur J Clin Chem Clin Biochem (1997) 35(9):647–54.
101. Huntoon KM, Wang Y, Eppolito CA, Barbour KW, Berger FG, Shrikant PA, et al. The acute phase protein haptoglobin regulates host immunity. J Leukoc Biol (2008) 84(1):170–81. doi:10.1189/jlb.0208100
102. Thomsen JH, Etzerodt A, Svendsen P, Moestrup SK. The haptoglobin-CD163-heme oxygenase-1 pathway for hemoglobin scavenging. Oxid Med Cell Longev (2013) 2013:523652. doi:10.1155/2013/523652
103. Xie Y, Li Y, Zhang Q, Stiller MJ, Wang CL, Streilein JW. Haptoglobin is a natural regulator of Langerhans cell function in the skin. J Dermatol Sci (2000) 24(1):25–37. doi:10.1016/S0923-1811(00)00078-5
104. Shah C, Hari-Dass R, Raynes JG. Serum amyloid A is an innate immune opsonin for Gram-negative bacteria. Blood (2006) 108(5):1751–7. doi:10.1182/blood-2005-11-011932
105. Sun L, Zhu Z, Cheng N, Yan Q, Ye RD. Serum amyloid A induces interleukin-33 expression through an IRF7-dependent pathway. Eur J Immunol (2014) 44(7):2153–64. doi:10.1002/eji.201344310
106. Nguyen KD, Macaubas C, Truong P, Wang N, Hou T, Yoon T, et al. Serum amyloid A induces mitogenic signals in regulatory T cells via monocyte activation. Mol Immunol (2014) 59(2):172–9. doi:10.1016/j.molimm.2014.02.011
107. Gouwy M, De Buck M, Pörtner N, Opdenakker G, Proost P, Struyf S, et al. Serum amyloid A chemoattracts immature dendritic cells and indirectly provokes monocyte chemotaxis by induction of cooperating CC and CXC chemokines. Eur J Immunol (2015) 45(1):101–12. doi:10.1002/eji.201444818
108. Ather JL, Poynter ME. Serum amyloid A3 is required for normal weight and immunometabolic function in mice. PLoS One (2018) 13(2):e0192352. doi:10.1371/journal.pone.0192352
109. Korkmaz B, Poutrain P, Hazouard E, de Monte M, Attucci S, Gauthier FL. Competition between elastase and related proteases from human neutrophil for binding to alpha1-protease inhibitor. Am J Respir Cell Mol Biol (2005) 32(6):553–9. doi:10.1165/rcmb.2004-0374OC
110. Janciauskiene S, Larsson S, Larsson P, Virtala R, Jansson L, Stevens T. Inhibition of lipopolysaccharide-mediated human monocyte activaction, in vitro, by alpha1-antitrypsin. Biochem Biophys Res Commun (2004) 321(3):592–600. doi:10.1016/j.bbrc.2004.06.123
111. Nita IM, Serapinas D, Janciauskiene SM. α1-Antitrypsin regulates CD14 expression and soluble CD14 levels in human monocytes in vitro. Int J Biochem Cell Biol (2007) 39(6):1165–76. doi:10.1016/j.biocel.2007.02.017
112. Ozeri E, Mizrahi M, Shahaf G, Lewis EC. A-1 antitrypsin promotes semimature, IL-10-producing and readily migrating tolerogenic dendritic cells. J Immunol (2012) 189(1):146–53. doi:10.4049/jimmunol.1101340
113. Lewis EC, Mizrahi M, Toledano M, Defelice N, Wright JL, Churg A, et al. α1-Antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc Natl Acad Sci U S A (2008) 105(42):16236–41. doi:10.1073/pnas.0807627105
114. Chen G, Li J, Chen L, Lai X, Qiu J. α1-Antitrypsin-primed tolerogenic dendritic cells prolong allograft kidney transplant survival in rats. Int Immunopharmacol (2016) 31:216–21. doi:10.1016/j.intimp.2015.12.038
115. Tawara I, Sun Y, Lewis EC, Toubai T, Evers R, Nieves E, et al. Alpha-1-antitrypsin monotherapy reduces graft-versus-host disease after experimental allogeneic bone marrow transplantation. Proc Natl Acad Sci U S A (2012) 109(2):564–9. doi:10.1073/pnas.1117665109
116. Gottlieb PA, Alkanani AK, Michels AW, Lewis EC, Shapiro L, Dinarello CA, et al. α1-Antitrypsin therapy downregulates toll-like receptor-induced IL-1β responses in monocytes and myeloid dendritic cells and may improve islet function in recently diagnosed patients with type 1 diabetes. J Clin Endocrinol Metab (2014) 99(8):E1418–26. doi:10.1210/jc.2013-3864
117. Elshikha AS, Lu Y, Chen MJ, Akbar M, Zeumer L, Ritter A, et al. Alpha 1 antitrypsin inhibits dendritic cell activation and attenuates nephritis in a mouse model of lupus. PLoS One (2016) 11(5):e0156583. doi:10.1371/journal.pone.0156583
118. Tobiásová-Czetoová Z, Palmborg A, Lundgvist A, Karlsson G, Adamson L, Bartunková J, et al. Effects of human plasma proteins on maturation of monocyte-derived dendritic cells. Immunol Lett (2005) 100(2):113–9. doi:10.1016/j.imlet.2005.03.009
119. Cho M, Lee JE, Lim H, Shin HW, Khalmuratova R, Choi G, et al. Fibrinogen cleavage products and toll-like receptor 4 promote the generation of programmed cell death 1 ligand 2-positive dendritic cells in allergic asthma. J Allergy Clin Immunol (2017) S0091-6749(17):31582–8. doi:10.1016/j.jaci.2017.09.019
120. Shalev I, Wong KM, Foerster K, Zhu Y, Chan C, Maknojia A, et al. The novel CD4+CD25+ regulatory T cell effector molecule fibrinogen-like protein 2 contributes to the outcome of murine fulminant viral hepatitis. Hepatology (2009) 49(2):387–97. doi:10.1002/hep.22684
121. Chan CW, Kay LS, Khadaroo RG, Chan MW, Lakatoo S, Young KJ, et al. Soluble fibrinogen-like protein 2/fibroleukin exhibits immunosuppressive properties: suppressing T cell proliferation and inhibiting maturation of bone marrow-derived dendritic cells. J Immunol (2003) 170(8):4036–44. doi:10.4049/jimmunol.170.8.4036
122. Liu H, Shalev I, Manuel J, He W, Leung E, Crookshank J, et al. the FGL2-FcγRIIB pathway: a novel mechanism leading to immunosuppression. Eur J Immunol (2008) 38(11):3114–26. doi:10.1002/eji.200838338
123. Shalev I, Liu H, Koscik C, Bartczak A, Javadi M, Wong KM, et al. Targeted deletion of fgl2 leads to impaired regulatory T cell activity and development of autoimmune glomerulonephritis. J Immunol (2008) 180(1):249–60. doi:10.4049/jimmunol.180.1.249
124. Hilkens CM, Isaacs JD. Tolerogenic dendritic cell therapy for rheumatoid arthritis: where we are now? Clin Exp Immunol (2013) 172(2):148–57. doi:10.1111/cei.12038
125. Bays AM, Gardner G. Pharmacologic therapies for rheumatologic and autoimmune conditions. Med Clin North Am (2016) 100(4):719–31. doi:10.1016/j.mcna.2016.03.001
126. Katabathina V, Menias CO, Pickhardt P, Lubner M, Prasad SR. Complication of immunosuppressive therapy in solid organ transplantation. Radiol Clin North Am (2016) 54(2):303–19. doi:10.1016/j.rcl.2015.09.009
127. Popov I, Li M, Zheng X, San H, Zhang X, Ichim TE, et al. Preventing autoimmune arthritis using antigen-specific immature dendritic cells: a novel tolerogenic vaccine. Arthirits Res Ther (2006) 8(5):R141. doi:10.1186/ar2031
128. van Duivenvoorde LM, Han WG, Bakker AM, Louis-Plence P, Charbonnier LM, Apparailly F, et al. Immunomodulatory dendritic cells inhibit Th1 responses and arthritis via different mechanisms. J Immunol (2007) 179(3):1506–15. doi:10.4049/jimmunol.179.3.1506
129. Phillips BE, Giannoukakis N, Trucco M. Dendritic cell mediated therapy for immunoregulation of type 1 diabetes mellitus. Pediatr Endocrinol Rev (2008) 5(4):873–9.
130. Tai N, Yasuda H, Xiang Y, Zhang L, Rodríguez-Pinto D, Yokono K, et al. IL-10-conditioned dendritic cells prevent autoimmune diabetes in NOD and humanized HLA-DQ8/RIP-B7.1 mice. Clin Immunol (2011) 139(3):336–49. doi:10.1016/j.clim.2011.03.003
131. Chorny A, Gonzalez-Rey E, Fernandez-Martin A, Pozo D, Ganea D, Delgado M. Vasoactive intestinal peptide induces regulatory dendritic cells with therapeutic effects on autoimmune disorders. Proc Natl Acad Sci U S A (2005) 102(38):13562–7. doi:10.1073/pnas.0504484102
132. Horibe EK, Sacks J, Unadkat J, Raimondi G, Wang Z, Ikeguchi R, et al. Rapamycin-conditioned, alloantigen-pulsed dendritic cells promote indefinite survival of vascularized skin allografts in association with T regulatory cell expansion. Transpl Immunol (2008) 18(4):307–18. doi:10.1016/j.trim.2007.10.007
133. Raimondi G, Sumpter TL, Matta BM, Pillai M, Corbitt N, Vodovotz Y, et al. Mammalian target of rapamycin inhibition and alloantigen-specific regulatory T cells synergize to promote long-term graft survival in immunocompetent recipients. J Immunol (2010) 184(2):624–36. doi:10.4049/jimmunol.0900936
134. Phillips BE, Garciafigueroa Y, Trucco M, Giannoukakis N. Clinical tolerogenic dendritic cells: exploring therapeutic impact on human autoimmune disease. Front Immunol (2017) 8:1279. doi:10.3389/fimmu.2017.01279
135. Lord P, Spiering R, Aquillon JC, Anderson AE, Appel S, Benitez-Ribas D, et al. Minimum information about tolerogenic antigen-presenting cells (MITAP): a first step towards reproducibility and standardization of cellular therapies. PeerJ (2016) 4:e2300. doi:10.7717/peerj.2300
136. Ten Brinke A, Hilkens CM, Cools N, Geissler EK, Hutchinson JA, Lombardi G, et al. Clinical use of tolerogenic dendritic cells-harmonization approach in European collaborative effort. Mediators Inflamm (2015) 2015:471719. doi:10.1155/2015/471719
137. Palucka K, Bancherau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer (2012) 12(4):265–77. doi:10.1038/nrc3258
138. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care (2011) 34(9):2026–32. doi:10.2337/dc11-0472
139. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Transl Med (2015) 7(290):290ra87. doi:10.1126/scitranslmed.aaa9301
140. Jauregui-Amezaga A, Cabezón R, Ramírez-Morros A, España C, Rimola J, Bru C, et al. Intraperitoneal administration of autologous tolerogenic dendritic cells for refractory Crohn’s disease: a phase I study. J Crohns Colitis (2015) 9(12):1071–8. doi:10.1093/ecco-jcc/jjv144
141. Voigtländer C, Rössner S, Cierpka E, Theiner G, Wiethe C, Menges M, et al. Dendritic cells matured with TNF can be further activated in vitro and after subcutaneous injection in vivo which converts their tolerogenicity into immunogenicity. J Immunother (2006) 29(4):407–15. doi:10.1097/01.cji.0000210081.60178.b4
142. Lim DS, Kang MS, Jeong JA, Bae YS. Semi-mature DC are immunogenic and not tolerogenic then inoculated at a high dose in collagen-induced arthritis mice. Eur J Immunol (2009) 39(5):1334–43. doi:10.1002/eji.200838987
143. Theweissen K, Broux B, Hendriks JJ, Vanhees M, Stinssen P, Slaets H, et al. Tolerogenic dendritic cells generated by in vitro treatment with SAHA are not stable in vivo. Cell Transplant (2016) 25(6):1207–18. doi:10.3727/096368915X690305
144. Turnguist HR, Raimondi G, Zahorchak AF, Fischer RT, Wang Z, Thomson AW. Rapamycin-conditioned dendritic cells are poor stimulators of allogeneic CD4+ T cells, but enrich for antigen-specific Foxp3+ T regulatory cells and promote organ transplant tolerance. J Immunol (2007) 178(11):7018–31. doi:10.4049/jimmunol.178.11.7018
145. Xia CQ, Peng R, Beato F, Clare-Salzler MJ. Dexamethasone induces IL-10-producing monocyte-derived dendritic cells with durable immaturity. Scand J Immunol (2005) 62(1):45–54. doi:10.1111/j.1365-3083.2005.01640.x
146. Horton C, Shanmugarajah K, Fairchild PJ. Harnessing the properties of dendritic cells in the pursuit of immunological tolerance. Biomed J (2017) 40(2):80–93. doi:10.1016/j.bj.2017.01.002
147. Kajino K, Nakamura I, Bamba H, Sawai T, Ogasawara K. Involvement of IL-10 in exhaustion of myeloid dendritic cells and rescue by CD40 stimulation. Immunology (2007) 120(1):28–37. doi:10.1111/j.1365-2567.2006.02474.x
148. Gordon JR, Ma Y, Churchman L, Gordon SA, Dawicki W. Regulatory dendritic cells for immunotherapy in immunologic diseases. Front Immunol (2014) 5:7. doi:10.3389/fimmu.2014.00007
149. Dhodapkar MV, Sznol M, Zhao B, Wang D, Carvajal RD, Keohan ML, et al. Induction of antigen-specific immunity with a vaccine targetinc NY-ESO-1 to the dendritic cell receptor DEC-205. Sci Transl Med (2014) 6(232):232ra51. doi:10.1126/scitranslmed.3008068
150. Dominguez AL, Lustgarten J. Targeting the tumor microenvirontment with anti-neu/anti-CD40 conjugated nanoparticles for the induction of antitumor immune responses. Vaccine (2010) 28(5):1383–90. doi:10.1016/j.vaccine.2009.10.153
151. Cruz LJ, Rosalia RA, Kleinovink JW, Rueda F, Löwik CW, Ossendorp F. Targeting nanoparticles to CD40, DEC-205 or CD11c molecules on dendritic cells for efficient CD8(+) T cell response: a comparative study. J Control Release (2014) 192:209–18. doi:10.1016/j.jconrel.2014.07.040
152. Schinnerling K, García-González P, Aguillón JC. Gene expression profiling of human monocyte-derived dendritic cells—searching for molecular regulators of tolerogenicity. Front Immunol (2015) 6:528. doi:10.3389/fimmu.2015.00528
153. García-González PA, Schinnerling K, Sepúlveda-Gutiérrez A, Maggi J, Mehdi AM, Nel HJ, et al. Dexamethasone and monophosphoryl lipid A induce a distinctive profile on monocyte-derived dendritic cells through transcriptional modulation of genes associated with essential processes of the immune response. Front Immunol (2017) 8:1350. doi:10.3389/fimmu.2017.01350
154. Dáňová K, Grohová A, Strnadová P, Funda DP, Sumník Z, Lebl J, et al. Tolerogenic dendritic cells from poorly compensated type 1 diabetes patients have decreased ability to induce stable antigen-specific T cell hyporesponsiveness and generation of suppressive regulatory T cells. J Immunol (2017) 198(2):729–40. doi:10.4049/jimmunol.1600676
155. Lan YY, Wang Z, Raimondi G, Wu W, Colvin BL, de Creus A, et al. "Alternatively activated" dendritic cells preferentially secrete IL-10, expand Foxp3+CD4+ T cells, and induce long-term organ allograft survival in combination with CTLA4-Ig. J Immunol (2006) 177(9):5868–77. doi:10.4049/jimmunol.177.9.5868
156. Lee JH, Park CS, Jang S, Kim JW, Kim SH, Song S, et al. Tolerogenic dendritic cells are efficiently generated using minocycline and dexamethasone. Sci Rep (2017) 7(1):15087. doi:10.1038/s41598-017-15569-1
Keywords: acute phase proteins, inflammation, monocyte-derived dendritic cells, tolerance, immunotherapy
Citation: Serrano I, Luque A and Aran JM (2018) Exploring the Immunomodulatory Moonlighting Activities of Acute Phase Proteins for Tolerogenic Dendritic Cell Generation. Front. Immunol. 9:892. doi: 10.3389/fimmu.2018.00892
Received: 20 December 2017; Accepted: 10 April 2018;
Published: 30 April 2018
Edited by:
Catharien Hilkens, Newcastle University, United KingdomReviewed by:
Philippe Blancou, University of Nice Sophia Antipolis, FranceCopyright: © 2018 Serrano, Luque and Aran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Josep M. Aran, amFyYW5AaWRpYmVsbC5jYXQ=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.