
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 11 April 2018
Sec. Primary Immunodeficiencies
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00735
 Sira Nanthapisal1,2*
Sira Nanthapisal1,2* Despina Eleftheriou1
Despina Eleftheriou1 Kimberly Gilmour3
Kimberly Gilmour3 Valentina Leone4Radhika Ramnath5Ebun Omoyinmi1Ying Hong1
Valentina Leone4Radhika Ramnath5Ebun Omoyinmi1Ying Hong1 Nigel Klein1
Nigel Klein1 Paul A. Brogan1
Paul A. Brogan1
Cutaneous leukocytoclastic vasculitis arises from immune complex deposition and dysregulated complement activation in small blood vessels. There are many causes, including dysregulated host response to infection, drug reactions, and various autoimmune conditions. It is increasingly recognised that some monogenic autoinflammatory diseases cause vasculitis, although genetic causes of vasculitis are extremely rare. We describe a child of consanguineous parents who presented with chronic cutaneous leukocytoclastic vasculitis, recurrent upper respiratory tract infection, and hypocomplementaemia. A homozygous p.His380Arg mutation in the complement factor I (CFI) gene CFI was identified as the cause, resulting in complete absence of alternative complement pathway activity, decreased classical complement activity, and low levels of serum factor I, C3, and factor H. C4 and C2 levels were normal. The same homozygous mutation and immunological defects were also identified in an asymptomatic sibling. CFI deficiency is thus now added to the growing list of monogenic causes of vasculitis and should always be considered in vasculitis patients found to have persistently low levels of C3 with normal C4.
Leukocytoclastic vasculitis (LV) is the term commonly used to describe histopathological findings of small vessel vasculitis characterized by leukocytoclasis and the presence of nuclear debris from infiltrating neutrophils. LV commonly affects the skin (cutaneous LV, CLV), and typically presents as palpable purpura of the lower extremities (1, 2). Circulating immune complexes, sometimes triggered by drugs or infections, are deposited within small blood vessels and incite an inflammatory response which includes activation of complement, resulting in small vessel vasculitis (3–5). CLV should not be regarded as a specific diagnosis in itself, since there are many underlying causes, including the primary systemic vasculitides [such as the antineutrophil cytoplasmic antibody (ANCA) associated vasculitides, IgA vasculitis, and many others]; drug reactions and other causes of hypersensitivity vasculitis; and various autoimmune connective tissue diseases (1, 6, 7). Increasingly, however, it is recognised that vasculitis (including LV) may be a presenting feature of an ever expanding list of monogenic autoinflammatory diseases, including: deficiency of adenosine deaminase type 2 (8, 9); STING-associated vasculitis of infancy (10); and other emerging genetic immunodysregulatory diseases (11); monogenic defects in complement (12, 13); and miscellaneous even rarer genetic syndromes (14). Most cases of CLV are, however, ultimately deemed idiopathic (1, 15).
The complement system is a complex and evolutionarily ancient component of the innate immune system. It is the first line of defense against invading organisms or foreign proteins, and enhances antibody function and phagocytosis (16). Activation of classical, alternative, or lectin complement pathways are documented in several vasculitides including urticarial vasculitis (17, 18), and ANCA-associated vasculitis (19). The complement system is composed of more than 30 proteins that function as complement cascade components, or system regulators (20). Genetic mutations of [including loss-of-function or gain-of-function (GOF) mutations], or autoantibodies directed to specific complement proteins can result in a broad spectrum of severe immunological sequelae that includes: recurrent bacterial infections, autoimmunity, paroxysmal nocturnal haemoglobinuria, glomerulonephritis, atypical haemolytic uremic syndrome (aHUS; typically associated with mutation in factor H, heterozygous mutations in: factor I, C3 or factor B; and mutations leading to membrane co-factor protein deficiency), and hereditary angioedema (associated with C1 esterase inhibitor deficiency) (20–22). Herein, we describe a child with recessive mutation in complement factor I (CFI) with deficiency of CFI causing chronic CLV and recurrent bacterial infections.
The index case was a female born to consanguineous Pakistani parents. She was well during the first year of life and tolerated her primary immunisations including BCG. From the second year of life she developed recurrent episodes of purulent otitis media (4 to 6 episodes per year) requiring multiple courses of antibiotics; although bacterial otitis was inconsistently documented, Staphylococcus aureus was detected on at least one occasion. She developed hearing loss secondary to these episodes, and required hearing aids. At the age of 3 years she developed a recurrent vasculitic rash affecting her arms, legs, and lower abdomen with palpable purpura (Figure 1A) this was associated with abdominal pain and arthralgia of the knees suggestive of a diagnosis of chronic IgA vasculitis (previously referred to as Henoch Schönlein purpura). Renal function and blood pressure were normal, and there was no evidence of proteinuria. Skin biopsy at that stage revealed a dermal perivascular (mainly surrounding capillaries) and interstitial infiltrate composed predominantly of neutrophils and nuclear dust (Figure 1B). Direct immunofluorescence for IgG, IgA, IgM, and C3 was negative. A second skin biopsy revealed the same picture. At the age of 4 years she developed pneumonia complicated by empyema that required drainage and intravenous antibiotics, although no organism was cultured during that episode. Between the age of 5 and 6 years she developed two further episodes of lower respiratory tract infection requiring antibiotics, and had persistent cutaneous vasculitis despite intermittent courses of oral prednisolone (1–2 mg/kg/day), and daily azathioprine (1–3 mg/kg). Therefore, at the age of 7 years she was referred to Great Ormond Street Hospital NHS Foundation Trust (GOSH) for further investigation of persistent cutaneous vasculitis and recurrent infection.
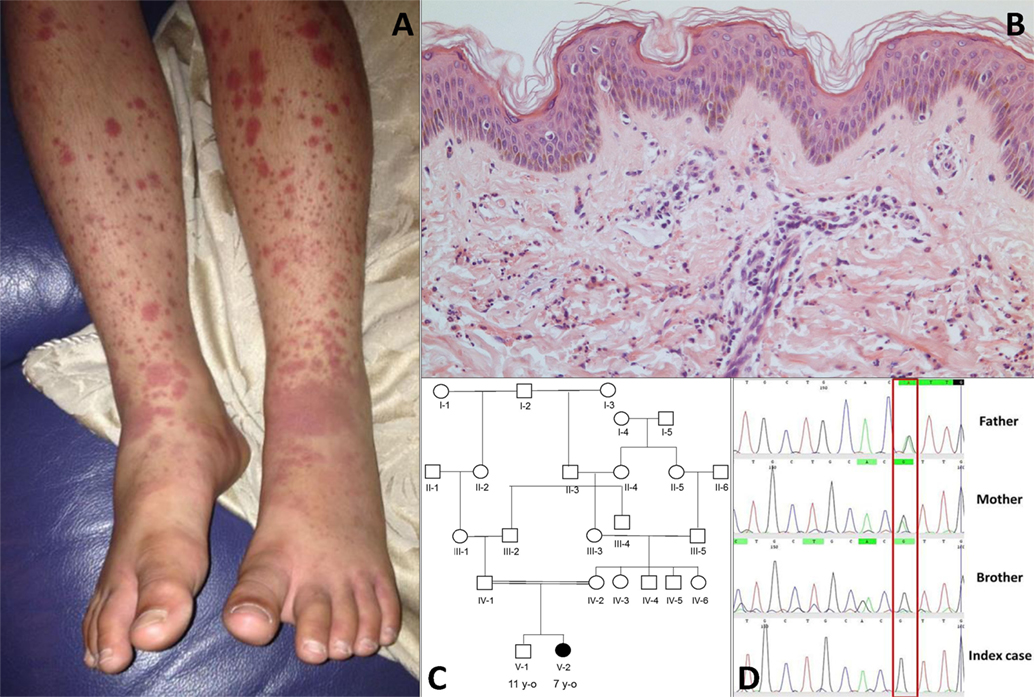
Figure 1. (A) Cutaneous vasculitis over both lower extremities of the index case [V-2; (C)]. (B) Skin biopsy of the lesion showing a dermal perivascular and interstitial infiltrate of predominantly neutrophils and nuclear dust which is compatible with leukocytoclastic vasculitis (haematoxylin & eosin stain, high magnification). (C) Family pedigree. (D) Sanger sequencing confirmed a homozygous G/G mutation (single black line) at position 1,139 of complement factor I gene in the index case and the asymptomatic brother; a heterozygous A/G state (Green/black line) was confirmed in both parents.
The family history (Figure 1C) revealed that parents were first cousins and were clinically well; her brother (aged 11 years) was also fit and well. Moreover, after specific enquiry, there was no history of recurrent or severe infections in the brother (V-1), or in either parent. There was also no confirmed family history of autoimmunity or immunodeficiency. On review of systems of the index case, there was no current history of prolonged or periodic fevers, arthralgia, or other systemic features. Physical examination demonstrated an injected left middle ear with effusion; and vasculitic rash affecting the lower limbs (Figure 1A). Blood pressure was normal, and there was no significant proteinuria or other organ specific involvement with vasculitis.
Laboratory investigations (Table S1 in Supplementary Material) revealed a high erythrocyte sedimentation rate of 60 mm/hr, and normal C-reactive protein of 6 mg/L [reference range (RR) <20]. All full blood count parameters were within normal limits. Rheumatoid factor was positive (59 international units/ml; RR 0–20). Other autoantibodies (antinuclear antibody; ANCA, including anti-proteinase 3 and anti-myeloperoxidase; anti-double stranded DNA; anticardiolipin; and against extractable nuclear antigens) were all negative. Notably, she had a persistently low serum C3 of 0.22 g/L (RR 0.75–1.65 g/L) with normal C4 of 0.36 g/L (RR 0.1–0.54 g/L) and decreased function of alternative (0% of healthy control) and classical (5% of healthy control) complement pathways. Low C3, normal C4, absent alternative complement pathway activity, and markedly decreased classical complement pathway activity persisted dur-ing follow up (measured on four separate occasions over 2 years), and prompted more detailed scrutiny of the complement pathway.
Table 1 summarizes the genotype and results of complement studies. All experimental work was performed with ethical approval (ethics number 08H071382) with written informed consent from all adult participants, and parental consent, and assent for children. Whole-exome sequencing (WES) was performed in the index case. C3 and C4 measurements were performed by the immunology laboratory at GOSH using a standard nephelometry assay (BN II Siemens Healthcare, UK). Classical and alternative complement assays (EuroDiagnostics, Sweden) and MBL assays were performed by enzyme-linked immunosorbent assay (ELISA). CFI and H were measured at Pathology Imperial College Healthcare, NHS, UK using ELISA (Binding Site, UK). C1q was measured by radial immunodiffusion and C1q antibodies by ELISA at the Protein Reference Unit in Sheffield, UK.
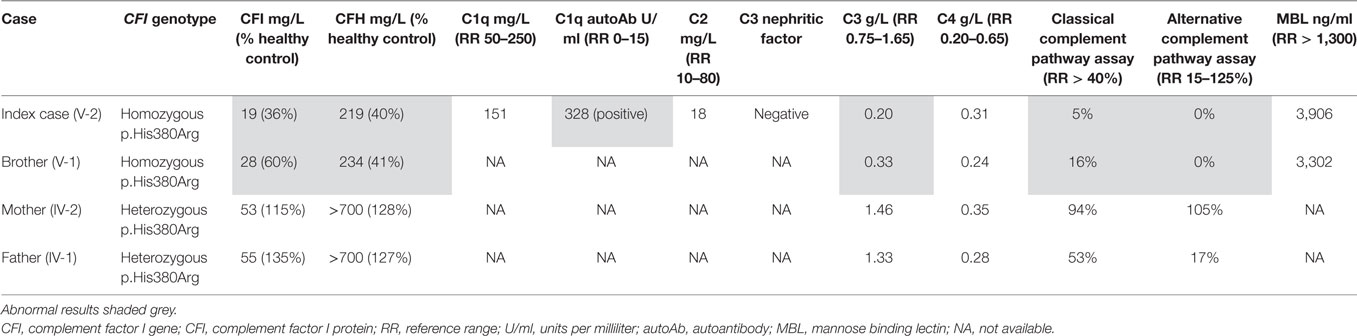
Table 1. Complement assays in the index case and other family members.
Whole-exome sequencing of the proband revealed a homozygous missense mutation (c.1139A > G; p.His380Arg) in the CFI gene that encodes CFI, an important complement regulatory factor that downregulates complement activation by cleaving C3b and C4b (23). This p.His380Arg mutation was predicted to be deleterious by in silico prediction algorithms, including Polyphen-2, SIFT, or GERP++, and is highly conserved between organisms according to the PhyloP algorithm. This homozygous mutation was confirmed to be present using Sanger sequencing in the index case (Figure 1C), and has very recently been described in homozygotic state in a single case with CFI deficiency (24). There were no other class 4 or 5 genetic variants (25) detected by WES in any of the complement pathway genes (including CFH), or in any other gene in the index case. Sanger sequencing in other family members confirmed that both parents were heterozygous for this mutation; and that her asymptomatic brother (V-1) was also homozygous for this mutation (Figure 1D). Subsequently, detailed complement studies were undertaken in the index case and the other first-degree family members. These results are summarized in Table 1. Both the index case (V-2) and the asymptomatic brother (V-1) had markedly low CFI levels, whilst the level in the parents was not reduced compared to controls. The C3 level in the index case and the brother were also reduced, but was normal in both parents. The classical and alternative complement assays were reduced in the index case, and brother; both parents had normal complement assays for all functional assays and individual components tested.
Complement factor I is located on chromosome 4q25 (26) and encodes CFI, a serum glycoprotein produced mainly in the liver by hepatocytes. CFI is an important complement activation regulator (18); the major transcript (NM_000204) consists of 13 exons corresponding to a single heavy chain and a single light chain on the protein domain, linked by two disulphide bonds (23). The light chain harbours the serine protease (SP) domain, responsible for the catalytic activity of CFI (27). CFI regulates the activation of both the classical and alternative complement systems, but is particularly important for regulating the alternative pathway. CFI deactivates C3b and C4b by cleaving C3b into iC3b and C3d and C4b into C4c and C4d. To perform its function, CFI requires several cofactors, such as C4b-binding protein (C4BP), complement factor H, complement receptor 1 (CR1/CD35), and membrane cofactor protein (MCP/CD46); Figure 2 (21). Mono-allelic or bi-allelic mutations in CFI result in two different phenotypes of immunological disease. Heterozygous mutations in CFI are associated with aHUS, a severe disease characterized by systemic thrombotic microangiopathy (28–30). Bi-allelic loss-of-function mutations in CFI cause CFI deficiency, with less than 20 mutations reported in less than 50 cases worldwide (23, 31–35). Persistent alternative complement pathway activation (low C3 with normal C4) provided the clue to CFI deficiency, which was confirmed using WES in the index case. Low C3 can also be caused by bi-allelic loss-of-function C3 mutations, or mono-allelic GOF C3 mutations (36). Our kindred did not have any mutations in C3 (data not shown), thus excluding these important differential diagnoses.
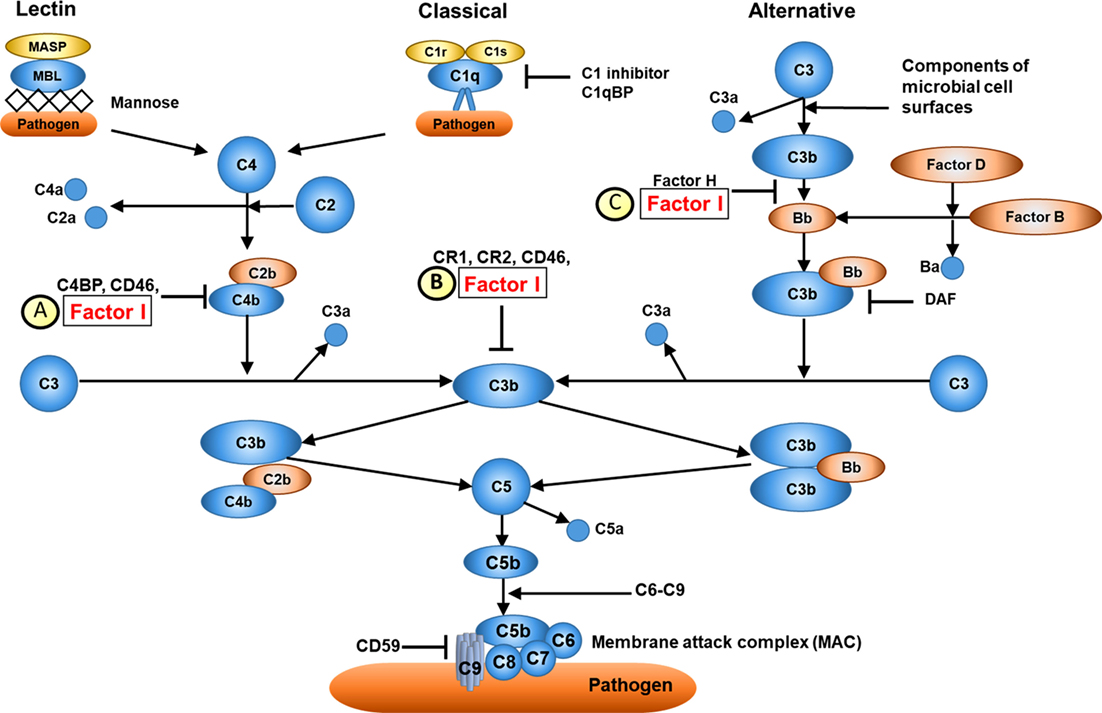
Figure 2. Overview of complement factor I (CFI) function. (A) CFI inhibits both the classical and lectin complement pathways by deactivation of C4bC2a complex. Surface-bound C4bC2a activating complex is bound by C4b-binding protein (C4BP) leading to the dissociation of C2a. CFI subsequently binds to the C4b-C4BP complex leading to the cleavage of C4b into C4c and C4d. (B) CFI deactivates complement receptor 1 (CR1)/CD35-bound C3b. Membrane cofactor protein (MCP/C46) binds to C3b-CR1 complex. The association of CFI results in the cleavage of C3b. (C) CFI inhibits the alternative complement pathway by deactivation of C3b: surface-bound C3b is cleaved into iC3B and C3d by factor H and CFI. Thus, whilst CFI is a major regulator of both the classical and alternative complement pathways, the alternative pathway is most affected, hence C3 is typically much lower than C4 in cases of CFI deficiency (see main text). Furthermore, since factor H is an important regulator of the alternative pathway, secondary factor H deficiency is observed in CFI deficiency (see main text).
Complement factor I deficiency results in uncontrolled amplification of C3 cleavage, which leads to consumptive C3 deficiency. As a result, patients with CFI deficiency have a defect in bacterial opsonisation leading to an increase in the susceptibility to recurrent pyogenic infections (Haemophilus influenzae, Neisseria meningitides, and Streptococcus pneumoniae), or non-bacterial infection. C3 consumption and dysregulation of the complement system may also lead to defects in clearance of immune complexes, and autoimmune organ injury, particularly glomerulonephritis or (more rarely) vasculitis (23, 30, 31, 34, 37, 38). The homozygous CFI p.His380Arg mutation we identified is a mutation in the light chain domain, which is responsible for the catalytic activity and disulfide bond subsection of CFI. Therefore, mutation of this position could affect both the catalytic cleavage function and post-translational structural modification of CFI. This amino acid is highly conserved across species, and the p.His380Arg mutation is predicted to be deleterious by all pathogenic prediction algorithms (Table S2 in Supplementary Material). Indeed, this mutation has recently been described as the cause of CFI deficiency in a 4-year-old patient (24) which taken alongside our report now irrefutably confirms its patho-genicity.
Functional complement assays of the index case (V-2) and the brother (V-1) revealed markedly decreased serum CFI, C3, CFH, and decreased activity of both the alternative and classical complement pathways. These results reflect the increased activation of both alternative and classical complement pathways. Known disease-causing mutations in CFI have been identified in both the light chain and heavy chains, and affect different functions and properties of CFI, including catalytic and secretory function (30). Persistently low C3 with normal C4 levels in our homozygous siblings suggests that the classical pathway was less compromised than the alternative pathway. This observation is explained by the fact that the classical pathway only requires very low concentrations (<0.1%) of its components to operate, whereas the alternative pathway requires higher concentrations (>5%) (39); hence, whilst CFI deficiency results in severe dysregulation of both alternative and classical pathways, the classical pathway is less affected and retains some function.
Dysregulation of the alternative complement pathway also explains the low factor H we observed (in the absence of any CFH mutations detected in this kindred) in our two patients with CFI deficiency (Table 1), since the principal function of factor H is to regulate the alternative pathway. Low levels of factor H are also described in other reports of CFI deficiency (24, 40, 41) and is secondary to consumption of the regulatory factor H presumably as a compensatory response to excessive and dysregulated alternate pathway activation.
The C1q antibodies we detected in our index case on a single occasion, in the face of normal C1q levels, are of dubious clinical significance and probably represent a non-specific epiphenomenon. In hypocomplementemic urticarial vasculitis there is a marked and selective reduction of serum C1q, in association with autoantibodies to the collagen-like region of C1q, which bind to C1q, activating the complement pathway (42, 43) Therefore, whilst we cannot completely exclude the possibility that C1q antibodies might play some secondary role to explain the vasculitic phenotype, the fact that our index case had normal C1q levels, low CFI, low C3, and low alternative pathway function which segregated perfectly with the genotype in this pedigree, and the finding of a second case of CFI recently published due to this same homozygous CFI mutation now irrefutably prove that it is indeed the CFI deficiency that is the main driver of the phenotype, and not primarily C1q autoantibodies.
Interestingly, the brother who also was homozygous for the p.His380Arg CFI mutation and had abnormal alternative and classical complement function, and low CFI, CFH, and C3 levels, is currently well, and without any history of recurrent infections. Completely asymptomatic, and mildly symptomatic individuals with confirmed CFI deficiency have also been previously described in other kindreds, but without any clear explanation for this variability in predisposition to infection (31, 44). Furthermore, reports of recurrent infections (including of the respiratory and urinary tract) have also been reported to occur with increased frequency in patients with heterozygotic CFI mutation and partial CFI deficiency (45). That said, there was no history of increased frequency or severity of infection in the heterozygotic parents of our kindred.
Therapeutic options for autoimmunity/autoinflammation associated with CFI deficiency are limited. Antibiotic prophylaxis and vaccination against encapsulated bacteria are pragmatic approaches to prevent infection (31, 44), which currently we have recommended for both the children described herein. It is also possible that institution of a tailored vaccination programme may improve outcomes for individuals with partial CFI deficiency as well (24, 45).
Regarding vaccination, although the production of specific antibodies against polysaccharides should not be affected by CFI deficiency, these individuals may have faster waning of antibody than healthy individuals (35), which could influence frequency of booster vaccinations. It is notable that treatment of the vasculitis with immunosuppression (azathioprine) in the index case was unsuccessful, and arguably could have contributed to the burden of infection. CFI replacement (e.g., with regular infusions of plasma) (41), would seem to have a logical approach, since it may control C3 consumption and ameliorate the vasculitic manifestations, although at the time of writing we have not yet fully explored this option. Last, we are unaware of any attempts to use liver transplantation to correct CFI deficiency, although at least in theory this might be a logical and definitive approach, albeit risky, and possibly not justifiable-based relative risks versus potential benefits in this context.
Thus, the spectrum of vasculitis caused by single gene mutations continues to expand, and CFI deficiency (or other genetic cause of alternative complement pathway dysregulation) should always be considered in patients with ANCA negative vasculitis, particularly when associated with low serum C3, but normal C4 levels.
Written informed consent was obtained from the participants for the publication of this case report.
SN and PB: design of the work, acquisition and analysis of data, drafting and revising manuscript, providing approval for publication. DE: design of the work, revising manuscript, providing approval for publication. KG: acquisition and analysis of data, revising manuscript, providing approval for publication. VL, RR, NK: acquisition of data, revising manuscript, providing approval for publication. EO and YH: design of the work, analysis of data, revising manuscript, providing approval for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Rosetrees Trust PhD studentship grant.
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.00735/full#supplementary-material.
1. Micheletti RG, Werth VP. Small vessel vasculitis of the skin. Rheum Dis Clin North Am (2015) 41:21–32, vii. doi:10.1016/j.rdc.2014.09.006
2. Sunderkotter C, Bonsmann G, Sindrilaru A, Luger T. Management of leukocytoclastic vasculitis. J Dermatolog Treat (2005) 16:193–206. doi:10.1080/09546630500277971
3. Carlson JA. The histological assessment of cutaneous vasculitis. Histopathology (2010) 56:3–23. doi:10.1111/j.1365-2559.2009.03443.x
4. Mackel SE, Jordon RE. Leukocytoclastic vasculitis. A cutaneous expression of immune complex disease. Arch Dermatol (1982) 118:296–301. doi:10.1001/archderm.1982.01650170010012
5. Palit A, Inamadar AC. Childhood cutaneous vasculitis: a comprehensive appraisal. Indian J Dermatol (2009) 54:110–7. doi:10.4103/0019-5154.53179
6. Krause K, Grattan CE, Bindslev-Jensen C, Gattorno M, Kallinich T, de Koning HD, et al. How not to miss autoinflammatory diseases masquerading as urticaria. Allergy (2012) 67:1465–74. doi:10.1111/all.12030
7. Ratzinger G, Zelger BG, Carlson JA, Burgdorf W, Zelger B. Vasculitic wheel – an algorithmic approach to cutaneous vasculitides. J Dtsch Dermatol Ges (2015) 13:1092–117. doi:10.1111/ddg.12859
8. Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med (2014) 6(370):921–31. doi:10.1056/NEJMoa1307362
9. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med (2014) 370:911–20. doi:10.1056/NEJMoa1307361
10. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med (2014) 371:507–18. doi:10.1056/NEJMoa1312625
11. Mauro A, Omoyinmi E, Sebire NJ, Barnicoat A, Brogan P. De Novo PTEN mutation in a young boy with cutaneous vasculitis. Case Rep Pediatr (2017) 2017:9682803. doi:10.1155/2017/9682803
12. Demirkaya E, Zhou Q, Smith CK, Ombrello MJ, Deuitch N, Tsai WL, et al. Deficiency of complement 1r subcomponent in early-onset systemic lupus erythematosus: the role of disease-modifying alleles in a monogenic disease. Arthritis Rheumatol (2017) 69(9):1832–9. doi:10.1002/art.40158
13. Kosaka S, Osada S, Kaneko T, Nishimura S, Kawana S. Cutaneous vasculitis and glomerulonephritis associated with C4 deficiency. Clin Exp Dermatol (2013) 38:492–5. doi:10.1111/j.1365-2230.2012.04423.x
14. Standing AS, Malinova D, Hong Y, Record J, Moulding D, Blundell MP, et al. Autoinflammatory periodic fever, immunodeficiency, and thrombocytopenia (PFIT) caused by mutation in actin-regulatory gene WDR1. J Exp Med (2017) 214:59–71. doi:10.1084/jem.20161228
15. Gyselbrecht L, De Keyser F, Ongenae K, Naeyaert JM, Praet M, Veys EM. Etiological factors and underlying conditions in patients with leucocytoclastic vasculitis. Clin Exp Rheumatol (1996) 14:665–8.
16. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I – molecular mechanisms of activation and regulation. Front Immunol (2015) 6:262. doi:10.3389/fimmu.2015.00262
17. Ballanti E, Perricone C, Greco E, Ballanti M, Di Muzio G, Chimenti MS, et al. Complement and autoimmunity. Immunol Res (2013) 56:477–91. doi:10.1007/s12026-013-8422-y
18. Chimenti MS, Ballanti E, Triggianese P, Perricone R. Vasculitides and the complement system: a comprehensive review. Clin Rev Allergy Immunol (2015) 49:333–46. doi:10.1007/s12016-014-8453-8
19. Jayne DR, Bruchfeld AN, Harper L, Schaier M, Venning MC, Hamilton P, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol (2017) 28(9):2756–67. doi:10.1681/ASN.2016111179
20. Botto M, Kirschfink M, Macor P, Pickering MC, Wurzner R, Tedesco F. Complement in human diseases: lessons from complement deficiencies. Mol Immunol (2009) 46:2774–83. doi:10.1016/j.molimm.2009.04.029
21. Degn SE, Jensenius JC, Thiel S. Disease-causing mutations in genes of the complement system. Am J Hum Genet (2011) 10(88):689–705. doi:10.1016/j.ajhg.2011.05.011
22. Mayilyan KR. Complement genetics, deficiencies, and disease associations. Protein Cell (2012) 3:487–96. doi:10.1007/s13238-012-2924-6
23. Nilsson SC, Sim RB, Lea SM, Fremeaux-Bacchi V, Blom AM. Complement factor I in health and disease. Mol Immunol (2011) 48:1611–20. doi:10.1016/j.molimm.2011.04.004
24. Franco-Jarava C, Alvarez de la Campa E, Solanich X, Morandeira-Rego F, Mas-Bosch V, Garcia-Prat M, et al. Early versus late diagnosis of complement factor I deficiency: clinical consequences illustrated in two families with novel homozygous CFI mutations. J Clin Immunol (2017) 37:781–9. doi:10.1007/s10875-017-0447-x
25. Wallis Y, Payne S, McAnulty C, Bodmer D, Sistermans E, Robertson K, et al. Practice Guidelines for the Evaluation of Pathogenicity and the Reporting of Sequence Variants in Clinical Molecular Genetics. Association for Clinical Genetic Science (ACGS), British Society for Genetic Medicine (2013). Available from: http://www.acgs.uk.com/media/774853/evaluation_and_reporting_of_sequence_variants_bpgs_june_2013_-_finalpdf.pdf
26. Morris KM, Aden DP, Knowles BB, Colten HR. Complement biosynthesis by the human hepatoma-derived cell line HepG2. J Clin Invest (1982) 70:906–13. doi:10.1172/JCI110687
27. Roversi P, Johnson S, Caesar JJ, McLean F, Leath KJ, Tsiftsoglou SA, et al. Structural basis for complement factor I control and its disease-associated sequence polymorphisms. Proc Natl Acad Sci U S A (2011) 2(108):12839–44. doi:10.1073/pnas.1102167108
28. Geelen J, van den Dries K, Roos A, van de Kar N, de Kat Angelino C, Klasen I, et al. A missense mutation in factor I (IF) predisposes to atypical haemolytic uraemic syndrome. Pediatr Nephrol (2007) 22:371–5. doi:10.1007/s00467-006-0320-2
29. Kavanagh D, Richards A, Noris M, Hauhart R, Liszewski MK, Karpman D, et al. Characterization of mutations in complement factor I (CFI) associated with hemolytic uremic syndrome. Mol Immunol (2008) 45:95–105. doi:10.1016/j.molimm.2007.05.004
30. Nilsson SC, Kalchishkova N, Trouw LA, Fremeaux-Bacchi V, Villoutreix BO, Blom AM. Mutations in complement factor I as found in atypical hemolytic uremic syndrome lead to either altered secretion or altered function of factor I. Eur J Immunol (2010) 40:172–85. doi:10.1002/eji.200939280
31. Alba-Dominguez M, Lopez-Lera A, Garrido S, Nozal P, Gonzalez-Granado I, Melero J, et al. Complement factor I deficiency: a not so rare immune defect: characterization of new mutations and the first large gene deletion. Orphanet J Rare Dis (2012) 18(7):42. doi:10.1186/1750-1172-7-42
32. Bay JT, Katzenstein TL, Kofoed K, Patel D, Skjoedt MO, Garred P, et al. Novel CFI mutation in a patient with leukocytoclastic vasculitis may redefine the clinical spectrum of complement factor I deficiency. Clin Immunol (2015) 160:315–8. doi:10.1016/j.clim.2015.05.004
33. Haerynck F, Stordeur P, Vandewalle J, Van Coster R, Bordon V, De Baets F, et al. Complete factor I deficiency due to dysfunctional factor I with recurrent aseptic meningo-encephalitis. J Clin Immunol (2013) 33:1293–301. doi:10.1007/s10875-013-9944-8
34. Nilsson SC, Trouw LA, Renault N, Miteva MA, Genel F, Zelazko M, et al. Genetic, molecular and functional analyses of complement factor I deficiency. Eur J Immunol (2009) 39:310–23. doi:10.1002/eji.200838702
35. Ponce-Castro IM, Gonzalez-Rubio C, Delgado-Cervino EM, Abarrategui-Garrido C, Fontan G, Sanchez-Corral P, et al. Molecular characterization of complement factor I deficiency in two Spanish families. Mol Immunol (2008) 45:2764–71. doi:10.1016/j.molimm.2008.02.008
36. Jimenez-Reinoso A, Marin AV, Subias M, Lopez-Lera A, Roman-Ortiz E, Payne K, et al. Human plasma C3 is essential for the development of memory B, but not T, lymphocytes. J Allergy Clin Immunol (2018) 141(3):1151–4.e14. doi:10.1016/j.jaci.2017.09.037
37. Broderick L, Gandhi C, Mueller JL, Putnam CD, Shayan K, Giclas PC, et al. Mutations of complement factor I and potential mechanisms of neuroinflammation in acute hemorrhagic leukoencephalitis. J Clin Immunol (2013) 33:162–71. doi:10.1007/s10875-012-9767-z
38. Genel F, Sjoholm AG, Skattum L, Truedsson L. Complement factor I deficiency associated with recurrent infections, vasculitis and immune complex glomerulonephritis. Scand J Infect Dis (2005) 37:615–8. doi:10.1080/00365540510034536
39. Nita IM, Genel F, Nilsson SC, Smart J, Truedsson L, Choo S, et al. Molecular characterization of two novel cases of complete complement inhibitor factor I deficiency. Mol Immunol (2011) 48:1068–72. doi:10.1016/j.molimm.2011.01.012
40. Gonzalez-Rubio C, Ferreira-Cerdan A, Ponce IM, Arpa J, Fontan G, Lopez-Trascasa M. Complement factor I deficiency associated with recurrent meningitis coinciding with menstruation. Arch Neurol (2001) 58:1923–8. doi:10.1001/archneur.58.11.1923
41. Moller Rasmussen J, Teisner B, Jepsen HH, Svehag SE, Knudsen F, Kirstein H, et al. Three cases of factor I deficiency: the effect of treatment with plasma. Clin Exp Immunol (1988) 74:131–6.
42. Black AK. Urticarial vasculitis. Clin Dermatol (1999) 17:565–9. doi:10.1016/S0738-081X(99)00062-0
43. Wisnieski JJ, Jones SM. Comparison of autoantibodies to the collagen-like region of C1q in hypocomplementemic urticarial vasculitis syndrome and systemic lupus erythematosus. J Immunol (1992) 148:1396–403.
44. Sadallah S, Gudat F, Laissue JA, Spath PJ, Schifferli JA. Glomerulonephritis in a patient with complement factor I deficiency. Am J Kidney Dis (1999) 33:1153–7. doi:10.1016/S0272-6386(99)70155-1
Keywords: complement factor I, vasculitis, infection, complement deficiency, autoinflammation
Citation: Nanthapisal S, Eleftheriou D, Gilmour K, Leone V, Ramnath R, Omoyinmi E, Hong Y, Klein N and Brogan PA (2018) Cutaneous Vasculitis and Recurrent Infection Caused by Deficiency in Complement Factor I. Front. Immunol. 9:735. doi: 10.3389/fimmu.2018.00735
Received: 14 February 2018; Accepted: 26 March 2018;
Published: 11 April 2018
Edited by:
Mikko Risto Juhana Seppänen, Helsinki University Central Hospital, FinlandReviewed by:
Anete S. Grumach, Faculty of Medicine ABC, BrazilCopyright: © 2018 Nanthapisal, Eleftheriou, Gilmour, Leone, Ramnath, Omoyinmi, Hong, Klein and Brogan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sira Nanthapisal, bnNpcmFAdHUuYWMudGg=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.