 Erich Piovan
Erich Piovan Valeria Tosello1
Valeria Tosello1 Alberto Amadori
Alberto Amadori Paola Zanovello
Paola Zanovello- 1UOC Immunologia e Diagnostica Molecolare Oncologica, Istituto Oncologico Veneto IOV—IRCCS, Padova, Italy
- 2Dipartimento di Scienze Chirurgiche, Oncologiche e Gastroenterologiche, Università di Padova, Padova, Italy
The NOTCH signaling pathway is a conserved signaling cascade that regulates many aspects of development and homeostasis in multiple organ systems. Aberrant activity of this signaling pathway is linked to the initiation and progression of several hematological malignancies, exemplified by T-cell acute lymphoblastic leukemia (T-ALL). Interestingly, frequent non-mutational activation of NOTCH1 signaling has recently been demonstrated in B-cell chronic lymphocytic leukemia (B-CLL), significantly extending the pathogenic significance of this pathway in B-CLL. Leukemia patients often present with high-blood cell counts, diffuse disease with infiltration of the bone marrow, secondary lymphoid organs, and diffusion to the central nervous system (CNS). Chemokines are chemotactic cytokines that regulate migration of cells between tissues and the positioning and interactions of cells within tissue. Homeostatic chemokines and their receptors have been implicated in regulating organ-specific infiltration, but may also directly and indirectly modulate tumor growth. Recently, oncogenic NOTCH1 has been shown to regulate infiltration of leukemic cells into the CNS hijacking the CC-chemokine ligand 19/CC-chemokine receptor 7 chemokine axis. In addition, a crucial role for the homing receptor axis CXC-chemokine ligand 12/CXC-chemokine receptor 4 has been demonstrated in leukemia maintenance and progression. Moreover, the CCL25/CCR9 axis has been implicated in the homing of leukemic cells into the gut, particularly in the presence of phosphatase and tensin homolog tumor suppressor loss. In this review, we summarize the latest developments regarding the role of NOTCH signaling in regulating the chemotactic microenvironmental cues involved in the generation and progression of T-ALL and compare these findings to B-CLL.
Introduction
The NOTCH signaling cascade is an evolutionarily conserved signaling pathway that in mammals consists of a family of four transmembrane receptors (NOTCH1, NOTCH2, NOTCH3, and NOTCH4) (1) and five ligands of the Delta-Serrate-Lag family [jagged 1 (JAG1), JAG2, delta-like 1 (DLL1), DLL3 and DLL4] (2). This signaling system plays a crucial role in regulating development and tissue homeostasis (3). Given the important role played by NOTCH signaling in regulating key cellular traits such as differentiation, proliferation, and apoptosis, it is perhaps not surprising that deregulation of NOTCH has been implicated in the pathogenesis of a variety of malignancies (4, 5). In this regard, the most firmly established evidence for altered NOTCH signaling in cancer is represented by activating NOTCH1 receptor mutations found in over 50–60% of T-cell acute lymphoblastic leukemia (T-ALL) cases (6). In addition, 8–30% of T-ALLs harbor mutations in F-box and WD repeat domain containing 7 (FBXW7), a protein that normally promotes NOTCH1 ubiquitination and degradation, which lead to increased NOTCH1 protein stability (7, 8). Moreover, paracrine mechanisms that result in NOTCH1 or NOTCH3 signaling upregulation or rare mutations in NOTCH3 (9) could contribute to T-ALL. Further, aberrant expression of the NOTCH ligand, DLL4, may contribute to NOTCH1-driven leukemias (10). Thus, the majority of T-ALL cases have hyper-activation of the NOTCH signaling pathway. Interestingly, activating mutations affecting NOTCH1 are also present in 4–13% of B-cell chronic lymphocytic leukemia (B-CLL) cases (11, 12), and very recently frequent non-mutational NOTCH1 activation in B-CLL has also been reported, irrespective of NOTCH1 mutational status (13). However, differently from T-ALL, the specific role of NOTCH1 signaling in the pathogenesis of B-CLL remains to be established. T-ALL is an aggressive hematological malignancy arising from the malignant transformation and subsequent clonal expansion of immature T-cell precursors. Clinically, T-ALL patients present with diffuse infiltration of the bone marrow (BM) by immature T-cell blasts, high-blood cell counts (hyperleukocytosis) with extramedullary infiltration of lymph nodes and other organs such as the central nervous system (CNS), and the presence of mediastinal masses (14). T-ALL may arise in the BM from thymus settling progenitors endowed with T-lineage potential or thymus resident T-cell precursor cells. These transformed T lymphoblasts under the influence of oncogenic NOTCH1 activation and collaborating oncogenes spread infiltrating BM cavities and/or thymus with extensive disease already at time of diagnosis. In addition, leukemic cells invade other tissues such as liver, spleen, lymph nodes, and CNS. B-CLL, on the other hand, is a common hematological malignancy characterized by the clonal expansion of non-functional CD5+ B cells in the BM and lymph nodes (15). The putative normal counterparts of this disease, although debated, are considered naïve and memory B cells (16, 17). Interestingly, B-CLL cells in the lymph node are known to harbor frequent NOTCH1 activation independent of mutations (18) and recent findings have shown that NOTCH1 is physiologically expressed and activated in the cells of origin of B-CLL (13). Additionally, approximately 50% of B-CLL cases without NOTCH1 mutations express the active form of NOTCH1 ICN1 (intracellular portion of NOTCH1), bringing NOTCH1 signaling to the forefront also in this disease.
Chemokines and their receptors, in particular so-called “homeostatic chemokines” which normally orchestrate leukocyte trafficking and homing during development, have been recently implicated in directing organ-specific metastasis (19, 20). Mechanistic insights on the trafficking of NOTCH-dependent leukemia cells to target organs are still ill-defined, however, recent reports have highlighted the importance of some homing receptors and their ligands (Figure 1) such as: (i) CC-chemokine ligand 19 (CCL19)/CC-chemokine receptor 7 (CCR7) (21); (ii) CXC-chemokine ligand 12 (CXCL12)/CXC-chemokine receptor 4 (CXCR4) (22–24); and (iii) CCL25/CCR9 (25). As leukemic relapse remains a major cause of treatment failure in childhood ALL, with leukemic relapses directly linked to the survival of blasts in the BM and/or distant sites such as CNS, the identification of targetable mechanisms behind this phenomenon are of clear impact.
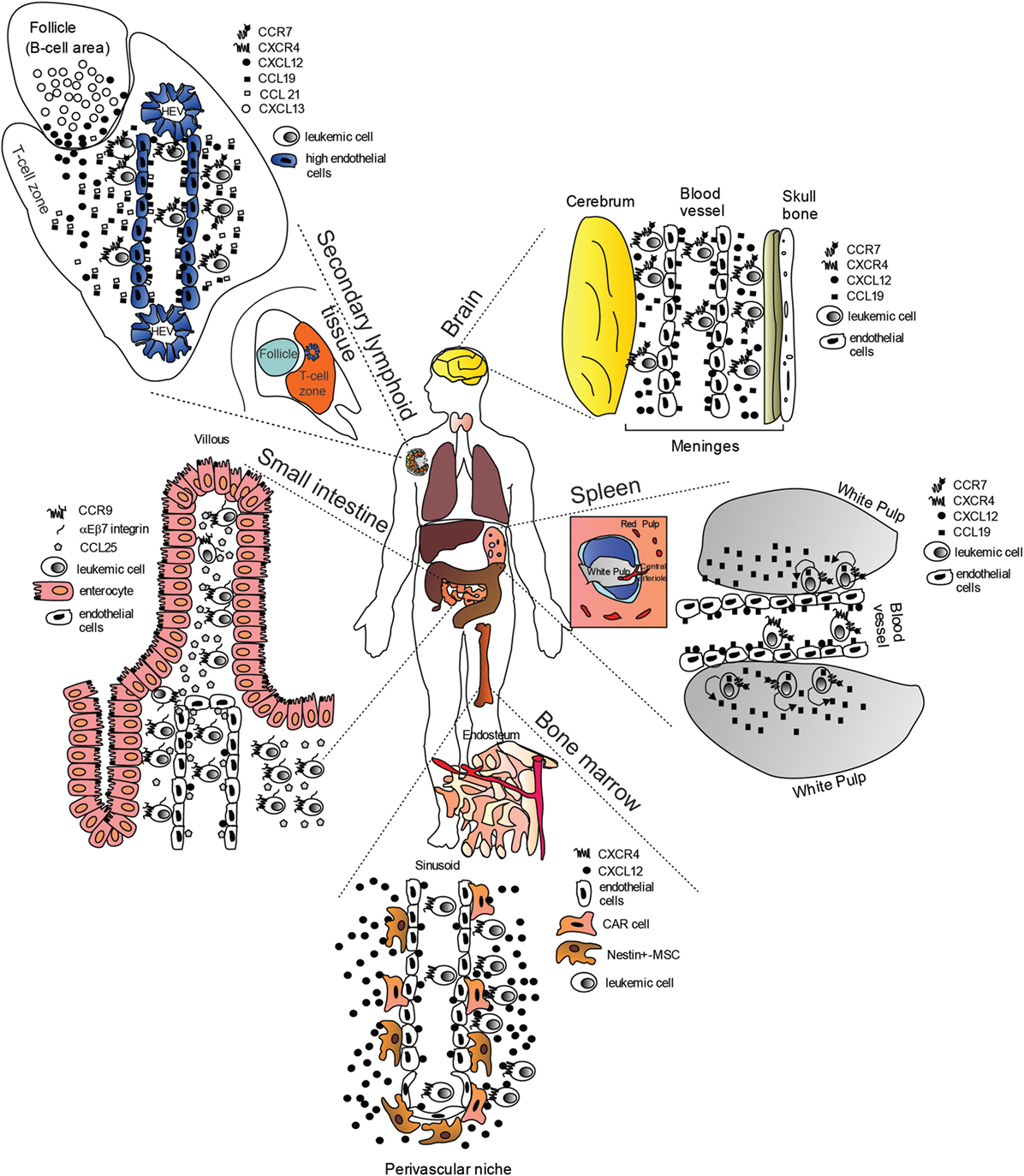
Figure 1. “Cellular highways” hijacked by leukemic cells implicated in T-cell acute lymphoblastic leukemia dissemination (many of the findings may also apply to B-cell chronic lymphocytic leukemia). Under physiological conditions, homeostatic chemokines control cellular migration by directing cells expressing specific chemokine receptors to appropriate locations expressing their cognate chemokine ligands. These cellular highways are also used by leukemic cells. In the brain, CC-chemokine ligand 19 (CCL19) and CXC-chemokine ligand 12 (CXCL12) recruit CC-chemokine receptor 7 (CCR7)- and CXC-chemokine receptor 4 (CXCR4)-expressing leukemic cells from blood vessels. In the spleen, CCL19 recruits CCR7-expressing leukemic cells from blood vessels possibly in combination with CXCL12. Migrated leukemic cells may then activate an autocrine/paracrine secretion of CCL19. CCR7-expressing leukemic cells together with CD62L (not shown) and CXCR4, gain access to secondary lymphoid organs such as lymph nodes (shown) via interactions with CCL19, CCL21, peripheral lymph node vascular addressin (not shown) and CXCL12 presented on high-endothelial venules (HEV). Here, leukemic cells are retained, proliferate, and completely substitute the normal tissue architecture. In the bone marrow (BM), CXCR4-expressing leukemic cells are probably initially recruited to the perivascular niche expressing high levels CXCL12, where a leukemic niche is established. Inhibitors of the CXCL12/CXCR4 interaction release leukemic cells from their BM niche, and allow these cells to enter the blood stream. In the small intestine, CCR9-expressing leukemia cells (together with αEβ7 integrin) are recruited by CCL25, where the presence of phosphoinositide-3 kinase-AKT pathway activation contributes to confer a proliferative advantage to leukemic cells in an otherwise non-supportive microenvironment.
Deregulation of NOTCH1 Signaling in Lymphoid Leukemias
NOTCH alterations can be found in a broad spectrum of hematological tumors [reviewed in Ref. (26, 27)]. In particular, NOTCH1 and to a lesser extent also NOTCH2, resulted the most frequently mutated. NOTCH1 is well known for its role as a master player in the pathogenesis of T-ALL as demonstrated by the high incidence of mutations in this disease (6). Most of these mutations cluster in the negative regulatory region (NRR), which prevents the extracellular receptor from being cleaved by the Disintegrin and metalloproteinase domain-containing protein 10 in the absence of ligand. These mutations mainly include missense substitutions or short insertions or deletions, which lead to receptor destabilization and ligand-independent activation (28). Other mutations in NOTCH1 truncate the PEST [proline (P), glutamic acid (E), serine (S), threonine (T)-rich protein sequence] domain through non-sense or frameshift events that lead to premature STOP codons in the C-terminal portion of NOTCH1 and increase half-life of ICN1. In addition, in a significant fraction of T-ALL cases, loss of function mutations or deletions in FBXW7 gene, an ubiquitin ligase implicated in ICN1 turnover, contribute to activation of NOTCH1 signaling in this malignancy (7, 8). Importantly, in about 20% of T-ALL cases, NOTCH1 signaling results strongly activated by the cooperativity of both mechanisms because of dual mutations affecting the NRR and PEST regions of NOTCH1 or the NRR domain together with the FBXW7 mutations (6–8). The importance of NOTCH1 mutations has also been extensively validated in murine mouse models of T-ALL. Forced expression of activated forms of Notch1 in murine hematopoietic progenitors determine T-ALL with a penetrance that depends on the strength of oncogenic Notch1 alleles (29, 30). In addition, numerous T-ALL mouse models showed Notch1 alterations as significant events in T-ALL progression (31, 32). In the context of NOTCH signaling, a role of Notch3 was also established with transgenic mice expressing ICN3 developing T-ALL with high penetrance, demonstrating a potential role for Notch3 in T-ALL (33). In addition, the human T-ALL cell line TALL1, which has wild-type Notch1 but is sensitive to γ-secretase inhibitors (GSI), carries an NRR mutation in NOTCH3 gene and shows ICN3 overexpression (9, 34). In T-ALL, the oncogenic function of NOTCH1 has been extensively studied and is linked to its capacity to regulate crucial signaling pathways and genes such as nuclear factor-κB (NF-κB), MYC, IGF-1R, and IL-7R all of which contribute to tumor growth and progression (35–39). NOTCH1 also regulates two families of transcriptional repressors Hes and Hey/Hers which in turn exert several downstream effects of NOTCH1 signaling. In particular, Hes1 sustains the phosphoinositide-3 kinase (PI3K)-AKT pathway and NF-κB activation through the direct suppression of phosphatase and tensin homolog (PTEN) and CYLD, respectively (40, 41). Moreover, Hes1 negatively regulates apoptosis of T-ALL cells through the repression of the BBC3/Puma pro-apoptotic factor (42). In addition to the consolidated function of NOTCH1 signaling in promoting anabolic processes and growth, NOTCH1 has been found to regulate some chemokine receptors (CCR5, CCR7, and CCR9; see below) thus orchestrating cell migration in specific microenvironments (21, 43).
As described above, NOTCH1 mutations have also been described in B-CLL (11, 12). Mutational activation of NOTCH1 has been found in about 8% of B-CLL at diagnosis and at significantly higher frequency during disease progression toward Richter transformation (about 30%), as well as in chemo-refractory B-CLL (about 20%). Differently from T-ALL, NOTCH1 mutations clustered uniquely in the PEST domain and the 2-bp frameshift deletion (ΔCT7544–7545, P2515fs) is present in about 80% of cases, making it a potential target for screening and specific targeted therapies. Consistent with the association of NOTCH1 mutations with clinically aggressive forms of the disease, B-CLL with NOTCH1 mutations at diagnosis have a poor prognosis similar to B-CLL carrying TP53 disruption and NOTCH1 mutations and TP53 disruption tended to distribute in a mutually exclusive pattern (44). The functional role of NOTCH1 mutations in B-CLL is not completely understood. A recent study showed that ICN1 is expressed in about 50% of peripheral blood B-CLL cases that present wild-type NOTCH1, suggesting that alternative mechanisms are involved in NOTCH1 activation in B-CLL (13). Moreover, independent from the mutational status, ICN1+ cases expressed a NOTCH1 gene signature and were sensitive to GSI. Notably, NOTCH1 regulated genes included those with a crucial role in the pathogenesis of B-CLL, including CCND3, BCL2, MCL1, BCR signaling pathway genes, and NF-κB pathway members.
Chemokines and Chemokine Receptors
Chemokines are small, secreted cytokines with chemotactic properties that are best known for their capacity to mediate immune cell trafficking and lymphoid tissue homeostasis (45, 46). This subfamily of cytokines which comprise over 48 ligands regulate cell trafficking and positioning by activating 20 seven-transmembrane spanning G-protein-coupled chemokine receptors (GPCR). In addition, chemokines can also bind to non-G-protein-coupled seven-transmembrane spanning receptors called atypical chemokine receptors (ACKR), which due to their incapacity to interact with Gi proteins are supposed to act mainly as decoy receptors, scavenging chemokines to help maintain chemokine gradients in tissues. Chemokines are subdivided into four classes based on the position of the first two cysteine (C) residues at their N-terminal protein sequence: CC-chemokines, CXC-chemokines, XC-chemokines, and CX3C-chemokines. The chemokine receptor nomenclature is based on the chemokine subclass specificity of the receptor, where L (ligand) is replaced by R (receptor) (47). There is an important degree of promiscuity in the chemokine superfamily, with numerous ligands binding different receptors and vice versa (46). Functionally, chemokines can be divided into “inflammatory” (induced upon inflammation) and “homeostatic” (constitutively expressed in specific tissues or cells) (48). Metaphorically, we can imagine our body as containing “cellular highways” regulated mainly by “homeostatic” chemokines and their receptors through which cells travel to reach specific locations within the body. In this system, chemokines can be envisioned as “traffic directors” responsible for sending cells expressing appropriate chemokine receptors to specific sites. Leukemia cells “hijack” this system to disseminate throughout the body and ensure their survival beyond the primary tumor site (19).
CXCL12/CXCR4–CXCR7 Signaling
The stromal cell-derived factor-1 (or CXCL12) initially thought to selectively interact with CXCR4, but now known to signal also through CXCR7 or ACKR3 (49), is widely expressed in numerous tissues and cells, including immature osteoblasts and endothelial cells (EC) within the BM, stromal cells in thymus, lungs, liver, brain, and lymph nodes. CXCR4 is also broadly expressed and is frequently overexpressed in cancer (50). Under homeostasis, the CXCL12/CXCR4 axis is crucial for the homing of hematopoietic progenitor cells (HPC) in the BM and their mobilization into the periphery (51). HPC reside in BM “niches” or specialized areas consisting of diverse cells regulating self-renewal, proliferation, and survival of HPC (52). At least two distinct BM niches have been identified, called “osteoblastic/endosteal” and “vascular” niches. In the hypoxic endosteal niche, osteoblasts lining the endosteum are responsible for HPC retention and quiescence maintenance through the intervention of numerous molecules including granulocyte colony-stimulating factor, bone morphogenetic protein, JAG-1/NOTCH1, Angiopoietin-1/Tie2, and osteopontin signaling (53). The vascular niche, localized at the sinusoidal walls, which includes EC, regulates proliferation, differentiation, and mobilization of HPC by secreting stimulatory and inhibitory soluble factors (54). A third niche, formed by CXCL12-abundant reticular cells (CAR), is located in central areas of the BM thus surrounding sinusoidal EC. These CAR cells, which comprise reticular Nestin+-mesenchymal stromal cells, as well as leptin receptor positive perivascular stromal cells (55, 56), are essential for the earliest stages of lymphoid development and express high levels of CXCL12, stem cell factor, interleukin-7, Angiopoietin-1, Fms-Related Tyrosine Kinase 3 Ligand, vascular cell adhesion molecule 1, and osteopontin (57–59). These reticular cells promote HPC retention and proliferation.
It is becoming increasingly evident that leukemic cells (and leukemic stem cells) actively interact with the BM microenvironment to promote their proliferation and survival at the expense of normal hematopoiesis (60). Indeed, using a Notch-1-dependent mouse model it has been found that TALL cells suppress normal hematopoiesis through the remodeling of the BM microenvironment by hijacking the proliferative vascular niche and repressing the endosteal/osteoblastic niche (61). The depletion of osteoblasts was due to the aberrant activation of Notch in these cells probably through Hes1-mediated repression of Runx2 transcriptional activity (61). This activation of Notch signaling in osteoblasts (possibly through increased expression of JAG1 or inflammatory cytokines) was associated with a reduced expression of CXCL12 within the stem/perivascular niche. Ultimately, one could envision a feedback loop where leukemic T-cell blasts disrupt homeostatic stem/lymphoid niches leading to compromised hematopoiesis while promoting their own Notch-dependent outgrowth.
T-cell lineage cell production relies on the thymic colonization by BM-exported early progenitors (thymus-seeding progenitors) expressing P-selectin glycoprotein ligand-1 and the chemokine receptors CCR7, CCR9, CXCR4, and possibly CCR5 (62, 63). These cells enter the thymus at the cortico-medullary junction where they undergo T-cell development. In the thymus, CXCL12 seems expressed throughout the cortex (64) by cortical thymic epithelial cells and together with the ligands for CCR7 (CCL21/CCL19) and CCR9 (CCL25) (65) contribute to the gradients required for the step-wise migration of immature thymocytes through the cortex toward the medulla. It has been found that chemokine receptor expression is very dynamic during T-cell development, in fact CCR7 is downregulated during double negative (DN) stages such that pre-positive selection double positive (DP) thymocytes are CCR7−, while CD4 and CD8 single positive (SP) thymocytes generated after positive selection re-express CCR7 prior to entering the medulla for tolerance induction (64, 66). On the other hand, DN and DP thymocytes express both CCR9 and CXCR4. The CXCL12/CXCR4 axis seems to have a role beyond acting as a retention signal that maintains DP thymocytes in the cortex, as it critically impacts on the proliferation and survival of DN thymocytes during β-selection acting as a co-stimulator of the pre-T-cell receptor (67). Moreover, CXCL12 may also act as a chemorepellent during the exit of mature SP cells from the thymus into the bloodstream, a process called chemofugetaxis (68). Recently, however, it has been suggested that following positive selection, CXCR4 high CCR9+CD69− DP cells downregulate CXCR4 to become CXCR4 low CCR9+ CD69+ DP cells and subsequently CD4+ and CD8+ SP cells with very low/undetectable CXCR4 surface expression. Thus, unlike for the DN thymic compartment, CXCR4 expression in DP cells may be dispensable for downstream αβ-T-cell development (64).
CXC-chemokine ligand 12 modulates cancer biology principally through two mechanisms: (i) direct/autocrine effects promoting cancer cell growth, metastasis, and angiogenesis; (ii) by indirect/paracrine effects, including recruitment of CXCR4+ cancer cells to CXCL12-expressing organs (BM, liver, thymus, lymph nodes, brain, among others) or CXCR4-expressing stromal cells to tumor sites (69). CXCR4 is overexpressed in many human cancers (70), with numerous studies demonstrating differential expression patterns (nuclear, cytoplasmic, and membrane) which translated in differences in biological behavior of cancers (71). Thus, membrane and/or cytoplasmic CXCR4 promotes tumor cell proliferation and metastasis, while nuclear CXCR4 is ineffective in explicating these functions. The role for CXCL12/CXCR4 axis in the infiltration of extramedullary sites, which commonly express significant levels of CXCL12 is supported by the correlation between high-surface CXCR4 expression by ALL cells (including T-ALL cells) and infiltration of extramedullary organs such as spleen and liver (72).
Recently, Pitt et al. (22) demonstrated that mouse Notch1-dependent T-ALL cells were directly interacting with CXCL12-producing vascular EC, and that this contact was necessary for leukemia maintenance and progression. In addition, murine and human T-ALL cells were shown to express increased cell-surface CXCR4 compared with mature peripheral T cells. Interestingly, this increased expression was not present at the transcript level, suggesting a non-transcriptional mechanism. Indeed, CXCR4 cell-surface expression, results from a balance between endocytosis, intracellular trafficking, and recycling, as well as gene expression (73, 74). CXCR4 internalization requires phosphorylation of its C-terminus, followed by ubiquitination and subsequent β-arrestin-dependent sorting into early endosomes, which are then processed into late endosomes or multivescicular bodies and further fused with lysosomes, ultimately leading to receptor and ligand degradation. The maturation of endosomes entails a cascade controlled by Rab small GTP-ases (75). CXCR4 internalization also depends on a dileucine motif within the C-terminal tail of CXCR4 (76) and numerous proteins including cortactin (77) and PIM1 (73) have been shown to regulate CXCR4 recycling and cell-surface expression. Remarkably, defects in endocytic trafficking of CXCR4 may contribute to increased surface expression and cancer progression (78). In acute myeloid leukemia (AML), a link has been found between PIM1 kinase activity and the surface expression and function of the CXCR4 receptor, with PIM1 expression levels correlating with CXCR4 surface expression (73). Indeed, PIM1 can phosphorylate serine 339 in the C-terminal domain of the CXCR4 receptor (a site critical for receptor recycling) contributing to high-CXCR4 surface expression and function at least in AML and B-CLL (79) cells. Along these lines, it has recently been shown that calcineurin (a serine/threonine protein phosphatase) previously associated with leukemia initiating cell activity (80), affects CXCR4 cell-surface expression at least partially through increased cortactin expression and thus CXCR4 recycling (23). CXCR4 expression was found to be essential for T-ALL maintenance and progression (22, 23) with loss of CXCL12/CXCR4 signaling leading to reduced Myc expression (a transcription factor directly regulated by NOTCH1) and previously linked to leukemia initiating cell activity in T-ALL. Surprisingly, although NOTCH1 has been reported to regulate numerous chemokine receptors in T-ALL (CCR5, CCR7, and CCR9; see below) this is not true for CXCR4 (21, 43), suggesting that NOTCH1 activation is not responsible for the increased CXCR4 expression. Differently in B-CLL cells, which also express high levels of surface CXCR4 and where the CXCL12/CXCR4 axis is regarded as a retention signal in tissue niches, CXCR4 has been shown to be a direct NOTCH1 target (13), suggesting a fundamental role of the NOTCH1-CXCR4 axis in the dissemination of B-CLL cells to lymphoid organs. Some of the main consequences on the biological behavior of T-ALL and B-CLL cells determined by NOTCH1 signaling are summarized in Table 1.
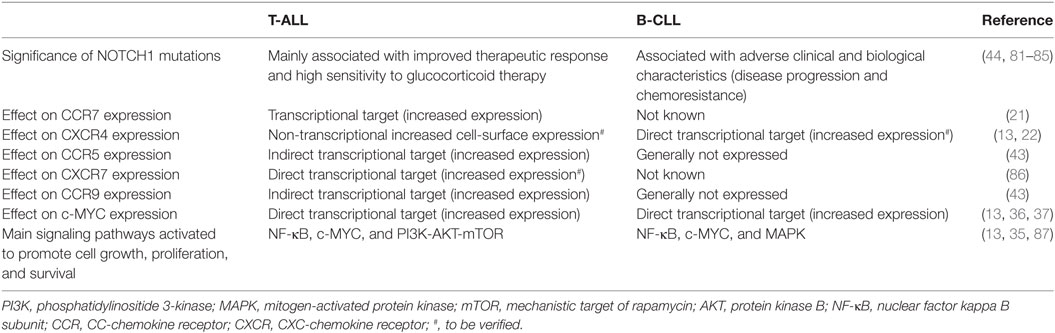
Table 1. Functional similarities and differences determined by NOTCH1 in influencing the biological behavior of T-cell acute lymphoblastic leukemia (T-ALL) and B-cell chronic lymphocytic leukemia (B-CLL) cells.
CXC-chemokine ligand 12 binding to CXCR4 triggers receptor homo- and heterodimerization, often with CXCR7 (a second chemokine receptor for CXCL12; discussed below), depending on the levels of co-expression (88). The binding of CXCL12 to CXCR4 initiates divergent signaling events that result in numerous responses (possibly cell-type specific) such as chemotaxis, cell survival, and/or proliferation, increase in intracellular calcium and gene transcription (Figure 2). CXCR4 is a GPCR that uses trimeric G-proteins constituted mainly of a Gαi subunit which inhibits adenyl cyclase activity and to a lesser extent a Gαq subunit which activates phospholipase C-β, which leads to inositol 1,4,5 trisphosphate and diacylglycerol production. Ultimately, these events lead to activation of the transcription factor NF-κB, the tyrosine kinase PYK2, Janus kinase-signal transducer and activator of transcription and PI3K-AKT pathways. The βγ dimer instead is mainly involved in Ras activation of ERK1/2 MAPK and activation of PI3K through direct interaction of the βγ dimer with ion channels. Moreover, following ligand-induced CXCR4 phosphorylation by G-protein receptor kinases the interaction with β-arrestin not only mediates clathrin-dependent endocytosis (see above) but also promotes the activation of MAPKs (p38, ERK1/2) and CXCL12-dependent chemotaxis (89). Recently, CXCR7 has been identified as a second receptor for CXCL12, showing a 10-fold higher affinity for this ligand than CXCR4 (49). This receptor is a member of the ACKR subgroup as it does not activate G-proteins after ligand binding (48). This receptor also binds CXCL11 (known ligand of CXCR3) with low affinity and macrophage migration inhibitory factor (90, 91). CXCR7 has been implicated in cell survival and adhesion (92). CXCR7 can act as a scavenger receptor or decoy receptor that removes CXCL12 from the extracellular milieu. Binding of ligands (CXCL12 or CXCL11) to CXCR7 promotes their internalization (49), ligand trafficking to lysosomes (where ligands are degraded), and CXCR7 recycling back to the cell membrane (93). Such CXCR7-dependent regulation of local CXCL12 availability ultimately leads to reduced CXCL12/CXCR4 signaling. On the other hand, the CXCL12 scavenging function of CXCR7 may positively regulate CXCR4-mediated migration by preventing down-regulation of CXCR4 surface expression and function following the exposure to excessive CXCL12 concentrations. In contrast, in cells with primarily intracellular CXCR7 expression and high-CXCR4 surface expression, CXCR7 blockade was not able to alter CXCR4-mediated phosphorylation of ERK and AKT, suggesting that CXCR7 was not necessary for CXCR4 signaling (94). Emerging evidence suggests that CXCR7 is a fully signaling receptor independent of G proteins and can activate intracellular signaling pathways such as AKT, MAPK, Janus kinase-signal transducer, and activator of transcription 3 either by direct modulation, through a β-arrestin-dependent pathway or after heterodimerization with CXCR4 (95). Thus, the relative expression levels of CXCR4 and CXCR7 could critically influence the cellular response to CXCL12. Recently, CXCR7 expression has been found to be very low in normal BM CD34+ cells compared with high levels of expression of this receptor in malignant ALL cells and cell lines (96, 97). In addition, particularly high levels of CXCR7 transcript were found in the T-ALL subtype. Analysis of the cellular distribution of CXCR7 in T-ALL cell lines disclosed a rather heterogeneous pattern with a sizable fraction being intracellular in Jurkat cells differently from MOLT4 cells. Interestingly, this different cellular distribution did not modify the functional consequences of CXCR7 silencing, as both cell lines exhibited reduced cell migration in the presence of a CXCL12 gradient (97). Notably, through the use of Notch pathway inhibitors, Asters group has identified a subset of Notch-binding sites in leukemia cell genomes that are dynamic, rapidly changing in occupancy when Notch signaling is modulated (86). These dynamic NOTCH1 sites are highly associated with genes that are directly regulated by Notch and mainly lie in large regulatory switches (termed superenhancers), characterized by exceptionally broad and high levels of H3K27 acetylation (98). The CXCR7 gene was found to be among these genes with high-dynamic regulatory potential and that are up-regulated following GSI washout in CUTLL1 cells (86). As the list of genes with highly dynamic regulatory potential are enriched for previously identified putative direct NOTCH1 target genes, it will be interesting to validate CXCR7 as a NOTCH1 direct target as this could add a further layer of complexity to the role played by NOTCH1 in promoting T-ALL retention/dissemination.
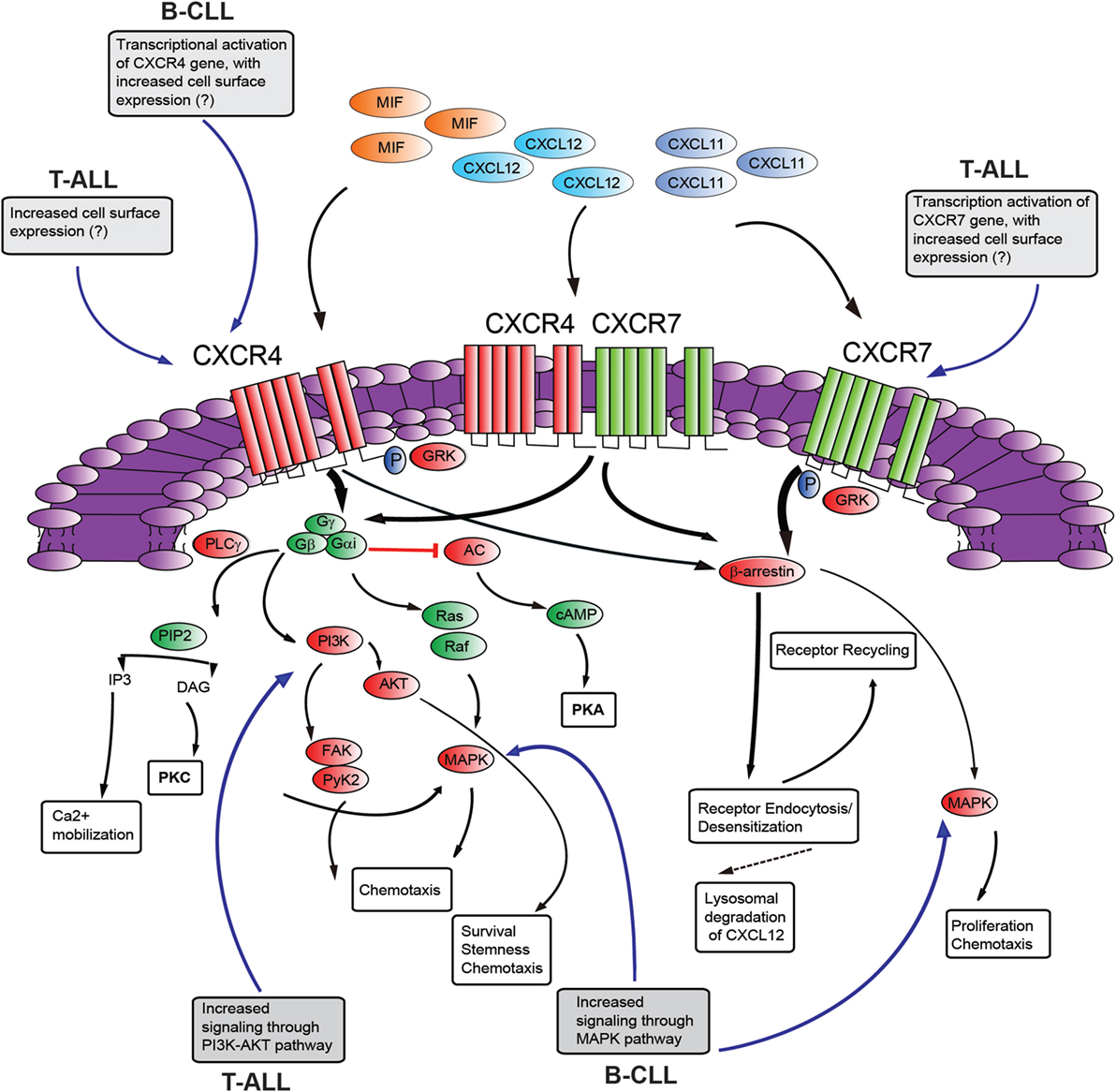
Figure 2. Schematic diagram of putative CXCR4–CXCR7 crosstalk affecting signaling pathways. The influence of NOTCH signaling on the main aspects of this signaling axis is shown in gray boxes [differences between T-cell acute lymphoblastic leukemia (T-ALL) and B-cell chronic lymphocytic leukemia (B-CLL) is presented]. CXCL12 employs two distinct receptors, CXCR4 and CXCR7 which can form homodimers or heterodimers. Additionally, CXCR4 and CXCR7 can act as receptors for macrophage migration inhibitory factor (MIF), while CXCR7 can also bind to CXCL11. Commonly, stimulation of CXCR4 leads to G-protein-coupled chemokine receptors (GPCR) signaling through phosphoinositide-3 kinase (PI3K)/AKT, PLC/IP3, MAPK pathways, and mobilization of Ca2+ from intracellular sources. CXCR4/CXCR7 heterodimerization attenuates GPCR signaling, promoting β-arrestin mediated signaling. Activation of CXCR7 triggers β-arrestin mediated signaling. Internalization of the receptors CXCR4 and CXCR7, and subsequent recycling to the cell surface, is also mediated by β-arrestin. Binding of CXCL12 to CXCR7 promotes internalization and scavenging (lysosomal degradation) of CXCL12. AC, adenylyl cyclase; cAMP, cyclic adenosyl monophosphate; PKA, protein kinase A; PLC, phospholipase C; GRK, GPCR kinase; PI3K, phosphatidylinositide 3-kinase; Gα/Gβ/Gγ, heterotrimeric G-protein consisting of subunits α, β, and γ; PIP2, phosphatidylinositol 4,5-bisphosphate; IP3, inositol 1,4,5-bisphosphate; AKT, protein kinase B; MAPK, mitogen-activated protein kinase; FAK, focal adhesion kinase; Pyk-2, proline rich kinase-2; DAG, diacylglycerol; PKC, protein kinase C. “?”, not known; black, pathway activation; red, pathway repression.
CXCL19/CCR7 Signaling
This signaling axis is physiologically important for its role in the development of immune responses, as it normally recruits activated dendritic cells and naïve T cells (expressing CCR7) to draining lymph nodes (expressing high levels of the ligands CCL19/CCL21), thus initiating an adaptive immune response (99). In tumors, CCR7 is often overexpressed and its expression mostly correlates with lymph node metastasis (100). Many leukemia and lymphomas also express CCR7, and this may account for their tropism for lymph nodes (especially T-cell zones) (101). Additionally in B-CLL, the interaction between CXCR5 (expressed at high levels in B-CLL, but not T-ALL cells) and its ligand CXCL13 (produced by resident stromal cells) is responsible for recruiting leukemic cells to lymphoid organs and possibly orchestrates the establishment and maintenance of proliferation centers (pseudofollicles) within these tissues (102). T-ALL patients have increased risk of CNS involvement at diagnosis or relapse, with the mechanisms behind this tropism still ill-defined. Possible entry routes for leukemic cells in the CNS include dissemination to the subarachnoid space from the BM of the skull via the bridging veins or from the cerebrospinal fluid via the choroid plexus; through brain capillaries to the cerebral parenchyma; infiltration of meninges via bony lesions of the skull and possibly traumatic lumbar puncture (24, 103). Buonamici et al. (21) showed that CCR7 signaling regulates CNS infiltration of leukemic T cells, using oncogenic Notch1 mouse models. Indeed, gene expression profiling of uncommitted hematopoietic progenitors expressing oncogenic Notch1 (Notch1-IC) showed significant upregulation of Ccr7. NOTCH1-dependent regulation of CCR7 was confirmed in T-ALL cell lines and primary T-ALL samples. Furthermore, overexpression of mouse ccr7 in a T-ALL cell line not expressing CCR7 (DND41) licenses these cells to specifically infiltrate the brain, possibly through interaction with CCL19 expressed on brain EC. Interestingly, using ccr6−/−, ccr7−/−, and cxcr4−/− fetal liver progenitors transduced with oncogenic Notch1-IC, cxcr4 rather than ccr7 was implicated in CNS infiltration by T-ALL cells, in addition to BM engraftment (24). Significantly, in primary T-ALL samples, high CCR7/CXCR4 mRNA levels correlated with increased risk of CNS involvement (104), although only CCR7 expression had an independent predictive impact on CNS status. Taken together, these data suggest that both CXCR4 and CCR7 play a role in the recruitment of leukemic T cells to the CNS.
The spleen is an important organ involved in extramedullary hematopoiesis and is frequently infiltrated in numerous lymphoid malignancies. There is a high incidence of splenomegaly in ALL, especially T-ALL, with the presence of splenomegaly associated with poorer prognosis of leukemia patients (105). Recent findings from Notch1-dependent leukemia models (106), suggest that the higher levels of CCL19 found in the splenic microenvironment compared with BM could be responsible for the initial homing of these leukemic cells to the spleen (given their expression of CCR7), and at the same time the splenic microenvironment could stimulate the expression of CCL19 by T-ALL cells establishing a positive feed-back loop, leading to further recruitment of leukemic cells to the spleen (106).
CCL25/CCR9 Signaling
The CCL25/CCR9 chemokine axis normally influences the homing, development, and homeostasis of T cells (107). CCR9 is expressed on the majority of immature DP (CD4+CD8+) thymocytes, and then is downregulated during their transition to mature SP CD4+ or CD8+ stage (108). Also, approximately half of all γδ TCR+ thymocytes and peripheral γδ-T cells express functional CCR9 (109). The ligand of CCR9, CCL25, is highly expressed not only by cortical and medullary thymic epithelial cells but also by epithelial cells of the small intestine (108). Intriguingly, a case report of a pediatric T-ALL expressing CCR9 (and CD103 or αEβ7 integrin) at diagnosis, that switched to acute myeloid leukemia at relapse with disease localization to the gut has been reported (110), suggesting a role for CCR9 in the gut tropism of these leukemic cells. Recently, an elegant study found that conditional postnatal knockdown of Pten (shPten) in the hematopoietic compartment produced a highly disseminated T-ALL with the majority of leukemias harboring activating mutations in the Notch1 PEST domain (25). These shPten leukemias expressed high levels of CCR9 and showed marked dissemination to the intestine (and liver). Surprisingly, PTEN reactivation had no effect on tumor growth in the lymph nodes or spleen, while it markedly decreased tumor infiltration into intestine and liver, suggesting that the impact of Pten expression on disease progression is dictated by the anatomical site of leukemic disease. Subsequent experiments to determine how PTEN influences T-ALL homing and survival in the intestine disclosed that reduced PTEN expression (through Pten knockdown) sensitized leukemia cells to CCL25-induced Akt phosphorylation leading to their increased migration in transwell assays, and this effect was largely abrogated following PTEN re-expression (25). These findings suggest that leukemic cells with PTEN suppression or loss are facilitated in dissemination to distant sites such as the intestine (if they express CCR9) and amplify weak environmental cues (such as CCL25 signaling) that enable their survival. Consistent with this notion, stimulation of T-ALL cells with CCL25 has been reported to enhance their resistance to TNF-α mediated apoptosis (through the induction of the inhibitor of apoptosis protein Livin) partly through the activation of c-jun-NH2-kinase 1 (111). Interestingly, the Notch pathway has been shown to indirectly control the expression levels of CCR9 (and CCR5) in T-ALL cell lines and patient-derived primary leukemia cells, and subsequent biological effects such as cell proliferation and migration (43). It could thus be speculated that PTEN suppression together with NOTCH1 activation (frequently observed in human T-ALL) could cooperate to enhance migration to specific anatomical sites such as the intestine (through the increased expression of selected chemokine receptors such as CCR9) and confer a proliferative advantage in an otherwise non-supportive microenvironment (CCL25-expressing sites).
Conclusion and Perspectives
ALL is the most common malignancy in children, with 15% showing markers for the T-lineage (T-ALL). Of these, approximately 20% still die due to disease relapse. Instead in adults, T-ALL represents around 25% of ALL cases, with approximately 50% dying due to disease relapse notwithstanding current combination chemotherapy (112, 113). B-CLL is the most common human leukemia in adults, with patients often presenting an indolent course, surviving for a number of years with relatively mild symptoms (15). In ALL, leukemia relapses have been directly linked to the survival of blasts in organs such as CNS or testes in addition to BM (103). Infiltration of distant organs such as CNS is frequently observed in T-ALL and is an important obstacle for long-term remission. Many genes are implicated in the pathogenesis of T-ALL, including NOTCH, with NOTCH1 mutations being identified in over half of T-ALL patients (6). Although the mechanisms of normal T-cell homing to lymphoid organs and trans-endothelial migration are relatively well known, the mechanisms exploited by leukemic T cells to gain access to target organs remain elusive. Homeostatic chemokines are considered pivotal molecules in promoting metastasis in solid tumors (19), and may help to account for the non-random metastatic destinations encountered in different neoplasia. In B-CLL, NOTCH1 activation probably reflects the constitutive, dysregulated expression of a physiological signal (13). NOTCH1 mutations in T-ALL hijack the physiological role of NOTCH signaling during thymocyte development (114) with oncogenic NOTCH1 alterations expressed in HPC often used as models of human T-ALL to gain mechanistic insights. Mainly through the use of these NOTCH1-dependent leukemias it is emerging that homeostatic chemokines and their receptors are critically involved not only in dictating medullary and extramedullary dissemination but also directly affecting the viability and growth of nascent leukemic niches. Recent studies showing that surface chemokine receptor expression and function may not correlate with mRNA transcript levels and that defects in recycling or endocytic trafficking of chemokine receptors may contribute to cancer progression add a new layer of complexity to the mechanisms acting to fine-tune the functional consequences of chemokine signaling. Thus, future studies evaluating the significance of chemokine receptor expression/signaling will need to go beyond mRNA expression levels, but will also have to take into account receptor phosphorylation, ubiquitination, recycling, and internalization rates. In particular, it may also be worth revisiting the role of CXCL12 biology in T-ALL (and possibly B-CLL) from the CXCR7 perspective. Intriguing are also recent observations that anti-tumor therapies (radiation and chemotherapy, among others) promote a hypoxic environment (19), which through the stabilization of hypoxia-inducible factors can increase the expression of chemokine receptors such as CXCR4 (115); conversely, other chemotherapies can downregulate chemokine receptor expression (19). Thus, some current therapies aimed at killing tumor cells may actually promote a more aggressive phenotype in the surviving cells (116). In B-CLL, the Bruton’s tyrosine kinase inhibitor, Ibrutinib, has been shown to determine early lymphocytosis and organomegaly reduction followed by normal cell count restoration, possibly in part due to its effects on CXCR4 expression (117). The effects of contemporary chemotherapy regimens used in T-ALL on chemokine receptor expression remain to be elucidated. Comprehensively, although numerous studies have focused on the role of chemokine receptors and their regulation by NOTCH, much less is known on downstream signals such as integrin activation or actin remodeling dynamics.
Currently, clinically approved targeted therapies to impede organ infiltration in acute leukemia are lacking. Of the chemokine axes that can be targeted, the CXCL12/CXCR4–CXCR7 axis seems most promising in T-ALL, as monotherapy with the selective CXCR4 antagonist, AMD3465, was highly effective in suppressing human disease in a xenograft model (22). However, monotherapy with CXCR4 inhibitors in other malignancies has more modest anti-leukemic effects (including B-CLL), as it mainly sensitized leukemic cells to conventional or targeted therapies through the mobilization of the leukemic cells into the periphery (102, 118, 119). Thus, it is likely that combination therapies comprising chemokine receptors antagonists together with conventional chemotherapeutic agents or specific targeted therapies such as NOTCH1 inhibitors will be required to eradicate the disease and prevent relapse.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Funding support provided in part by Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR) Ex 60%, Progetto di Ricerca di Ateneo (PRAT; Università di Padova; CDA #152403), and Istituto Oncologico Veneto 5x1000 fund to EP.
References
1. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science (1999) 284(5415):770–6. doi:10.1126/science.284.5415.770
2. D’Souza B, Miyamoto A, Weinmaster G. The many facets of Notch ligands. Oncogene (2008) 27(38):5148–67. doi:10.1038/onc.2008.229
3. Andersson ER, Sandberg R, Lendahl U. Notch signaling: simplicity in design, versatility in function. Development (2011) 138(17):3593–612. doi:10.1242/dev.063610
4. Koch U, Radtke F. Notch and cancer: a double-edged sword. Cell Mol Life Sci (2007) 64(21):2746–62. doi:10.1007/s00018-007-7164-1
5. Radtke F, Raj K. The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer (2003) 3(10):756–67. doi:10.1038/nrc1186
6. Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science (2004) 306(5694):269–71. doi:10.1126/science.1102160
7. O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med (2007) 204(8):1813–24. doi:10.1084/jem.20070876
8. Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med (2007) 204(8):1825–35. doi:10.1084/jem.20070872
9. Bernasconi-Elias P, Hu T, Jenkins D, Firestone B, Gans S, Kurth E, et al. Characterization of activating mutations of NOTCH3 in T-cell acute lymphoblastic leukemia and anti-leukemic activity of NOTCH3 inhibitory antibodies. Oncogene (2016) 35(47):6077–86. doi:10.1038/onc.2016.133
10. Xiong H, Maraver A, Latkowski JA, Henderson T, Schlessinger K, Ding Y, et al. Characterization of two distinct lymphoproliferative diseases caused by ectopic expression of the Notch ligand DLL4 on T cells. PLoS One (2013) 8(12):e84841. doi:10.1371/journal.pone.0084841
11. Fabbri G, Rasi S, Rossi D, Trifonov V, Khiabanian H, Ma J, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med (2011) 208(7):1389–401. doi:10.1084/jem.20110921
12. Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature (2011) 475(7354):101–5. doi:10.1038/nature10113
13. Fabbri G, Holmes AB, Viganotti M, Scuoppo C, Belver L, Herranz D, et al. Common nonmutational NOTCH1 activation in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A (2017) 114(14):E2911–9. doi:10.1073/pnas.1702564114
14. Attarbaschi A, Mann G, Dworzak M, Wiesbauer P, Schrappe M, Gadner H. Mediastinal mass in childhood T-cell acute lymphoblastic leukemia: significance and therapy response. Med Pediatr Oncol (2002) 39(6):558–65. doi:10.1002/mpo.10164
15. Pekarsky Y, Zanesi N, Croce CM. Molecular basis of CLL. Semin Cancer Biol (2010) 20(6):370–6. doi:10.1016/j.semcancer.2010.09.003
16. Klein U, Tu Y, Stolovitzky GA, Mattioli M, Cattoretti G, Husson H, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med (2001) 194(11):1625–38. doi:10.1084/jem.194.11.1625
17. Seifert M, Sellmann L, Bloehdorn J, Wein F, Stilgenbauer S, Durig J, et al. Cellular origin and pathophysiology of chronic lymphocytic leukemia. J Exp Med (2012) 209(12):2183–98. doi:10.1084/jem.20120833
18. Onaindia A, Gomez S, Piris-Villaespesa M, Martinez-Laperche C, Cereceda L, Montes-Moreno S, et al. Chronic lymphocytic leukemia cells in lymph nodes show frequent NOTCH1 activation. Haematologica (2015) 100(5):e200–3. doi:10.3324/haematol.2014.117705
19. Zlotnik A, Burkhardt AM, Homey B. Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol (2011) 11(9):597–606. doi:10.1038/nri3049
20. Chow MT, Luster AD. Chemokines in cancer. Cancer Immunol Res (2014) 2(12):1125–31. doi:10.1158/2326-6066.CIR-14-0160
21. Buonamici S, Trimarchi T, Ruocco MG, Reavie L, Cathelin S, Mar BG, et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature (2009) 459(7249):1000–4. doi:10.1038/nature08020
22. Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, et al. CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell (2015) 27(6):755–68. doi:10.1016/j.ccell.2015.05.002
23. Passaro D, Irigoyen M, Catherinet C, Gachet S, Da Costa De Jesus C, Lasgi C, et al. CXCR4 is required for leukemia-initiating cell activity in T Cell acute lymphoblastic leukemia. Cancer Cell (2015) 27(6):769–79. doi:10.1016/j.ccell.2015.05.003
24. Jost TR, Borga C, Radaelli E, Romagnani A, Perruzza L, Omodho L, et al. Role of CXCR4-mediated bone marrow colonization in CNS infiltration by T cell acute lymphoblastic leukemia. J Leukoc Biol (2016) 99(6):1077–87. doi:10.1189/jlb.5MA0915-394R
25. Miething C, Scuoppo C, Bosbach B, Appelmann I, Nakitandwe J, Ma J, et al. PTEN action in leukaemia dictated by the tissue microenvironment. Nature (2014) 510(7505):402–6. doi:10.1038/nature13239
26. Gu Y, Masiero M, Banham AH. Notch signaling: its roles and therapeutic potential in hematological malignancies. Oncotarget (2016) 7(20):29804–23. doi:10.18632/oncotarget.7772
27. Chiang MY, Radojcic V, Maillard I. Oncogenic Notch signaling in T-cell and B-cell lymphoproliferative disorders. Curr Opin Hematol (2016) 23(4):362–70. doi:10.1097/MOH.0000000000000254
28. Gordon WR, Roy M, Vardar-Ulu D, Garfinkel M, Mansour MR, Aster JC, et al. Structure of the Notch1-negative regulatory region: implications for normal activation and pathogenic signaling in T-ALL. Blood (2009) 113(18):4381–90. doi:10.1182/blood-2008-08-174748
29. Pear WS, Aster JC, Scott ML, Hasserjian RP, Soffer B, Sklar J, et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med (1996) 183(5):2283–91. doi:10.1084/jem.183.5.2283
30. Chiang MY, Xu L, Shestova O, Histen G, L’Heureux S, Romany C, et al. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J Clin Invest (2008) 118(9):3181–94. doi:10.1172/JCI35090
31. Ashworth TD, Pear WS, Chiang MY, Blacklow SC, Mastio J, Xu L, et al. Deletion-based mechanisms of Notch1 activation in T-ALL: key roles for RAG recombinase and a conserved internal translational start site in Notch1. Blood (2010) 116(25):5455–64. doi:10.1182/blood-2010-05-286328
32. Chiang MY, Wang Q, Gormley AC, Stein SJ, Xu L, Shestova O, et al. High selective pressure for Notch1 mutations that induce Myc in T-cell acute lymphoblastic leukemia. Blood (2016) 128(18):2229–40. doi:10.1182/blood-2016-01-692855
33. Bellavia D, Campese AF, Alesse E, Vacca A, Felli MP, Balestri A, et al. Constitutive activation of NF-kappaB and T-cell leukemia/lymphoma in Notch3 transgenic mice. EMBO J (2000) 19(13):3337–48. doi:10.1093/emboj/19.13.3337
34. Xu X, Choi SH, Hu T, Tiyanont K, Habets R, Groot AJ, et al. Insights into autoregulation of Notch3 from structural and functional studies of its negative regulatory region. Structure (2015) 23(7):1227–35. doi:10.1016/j.str.2015.05.001
35. Vilimas T, Mascarenhas J, Palomero T, Mandal M, Buonamici S, Meng F, et al. Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat Med (2007) 13(1):70–7. doi:10.1038/nm1524
36. Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A (2006) 103(48):18261–6. doi:10.1073/pnas.0606108103
37. Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev (2006) 20(15):2096–109. doi:10.1101/gad.1450406
38. Medyouf H, Gusscott S, Wang H, Tseng JC, Wai C, Nemirovsky O, et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. J Exp Med (2011) 208(9):1809–22. doi:10.1084/jem.20110121
39. Trimarchi T, Bilal E, Ntziachristos P, Fabbri G, Dalla-Favera R, Tsirigos A, et al. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell (2014) 158(3):593–606. doi:10.1016/j.cell.2014.05.049
40. Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med (2007) 13(10):1203–10. doi:10.1038/nm1636
41. Espinosa L, Cathelin S, D’Altri T, Trimarchi T, Statnikov A, Guiu J, et al. The Notch/Hes1 pathway sustains NF-kappaB activation through CYLD repression in T cell leukemia. Cancer Cell (2010) 18(3):268–81. doi:10.1016/j.ccr.2010.08.006
42. Schnell SA, Ambesi-Impiombato A, Sanchez-Martin M, Belver L, Xu L, Qin Y, et al. Therapeutic targeting of HES1 transcriptional programs in T-ALL. Blood (2015) 125(18):2806–14. doi:10.1182/blood-2014-10-608448
43. Mirandola L, Chiriva-Internati M, Montagna D, Locatelli F, Zecca M, Ranzani M, et al. Notch1 regulates chemotaxis and proliferation by controlling the CC-chemokine receptors 5 and 9 in T cell acute lymphoblastic leukaemia. J Pathol (2012) 226(5):713–22. doi:10.1002/path.3015
44. Rossi D, Rasi S, Fabbri G, Spina V, Fangazio M, Forconi F, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood (2012) 119(2):521–9. doi:10.1182/blood-2011-09-379966
45. Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol (2004) 22:891–928. doi:10.1146/annurev.immunol.22.012703.104543
46. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol (2014) 32:659–702. doi:10.1146/annurev-immunol-032713-120145
47. Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, et al. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev (2000) 52(1):145–76.
48. Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, et al. International union of basic and clinical pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev (2014) 66(1):1–79. doi:10.1124/pr.113.007724
49. Balabanian K, Lagane B, Infantino S, Chow KY, Harriague J, Moepps B, et al. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem (2005) 280(42):35760–6. doi:10.1074/jbc.M508234200
50. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer (2004) 4(7):540–50. doi:10.1038/nrc1388
51. Sahin AO, Buitenhuis M. Molecular mechanisms underlying adhesion and migration of hematopoietic stem cells. Cell Adh Migr (2012) 6(1):39–48. doi:10.4161/cam.18975
52. Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med (2014) 20(8):833–46. doi:10.1038/nm.3647
53. Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell (2010) 7(3):391–402. doi:10.1016/j.stem.2010.06.020
54. Kopp HG, Hooper AT, Avecilla ST, Rafii S. Functional heterogeneity of the bone marrow vascular niche. Ann N Y Acad Sci (2009) 1176:47–54. doi:10.1111/j.1749-6632.2009.04964.x
55. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity (2006) 25(6):977–88. doi:10.1016/j.immuni.2006.10.016
56. Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature (2010) 466(7308):829–34. doi:10.1038/nature09262
57. Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature (2013) 495(7440):231–5. doi:10.1038/nature11885
59. Cordeiro Gomes A, Hara T, Lim VY, Herndler-Brandstetter D, Nevius E, Sugiyama T, et al. Hematopoietic stem cell niches produce lineage-instructive signals to control multipotent progenitor differentiation. Immunity (2016) 45(6):1219–31. doi:10.1016/j.immuni.2016.11.004
60. Ayala F, Dewar R, Kieran M, Kalluri R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia (2009) 23(12):2233–41. doi:10.1038/leu.2009.175
61. Wang W, Zimmerman G, Huang X, Yu S, Myers J, Wang Y, et al. Aberrant Notch signaling in the bone marrow microenvironment of acute lymphoid leukemia suppresses osteoblast-mediated support of hematopoietic niche function. Cancer Res (2016) 76(6):1641–52. doi:10.1158/0008-5472.CAN-15-2092
62. Robertson P, Means TK, Luster AD, Scadden DT. CXCR4 and CCR5 mediate homing of primitive bone marrow-derived hematopoietic cells to the postnatal thymus. Exp Hematol (2006) 34(3):308–19. doi:10.1016/j.exphem.2005.11.017
63. Zlotoff DA, Sambandam A, Logan TD, Bell JJ, Schwarz BA, Bhandoola A. CCR7 and CCR9 together recruit hematopoietic progenitors to the adult thymus. Blood (2010) 115(10):1897–905. doi:10.1182/blood-2009-08-237784
64. Lucas B, White AJ, Parnell SM, Henley PM, Jenkinson WE, Anderson G. Progressive changes in CXCR4 expression that define thymocyte positive selection are dispensable for both innate and conventional alphabetaT-cell development. Sci Rep (2017) 7(1):5068. doi:10.1038/s41598-017-05182-7
65. Zhang SL, Wang X, Manna S, Zlotoff DA, Bryson JL, Blazar BR, et al. Chemokine treatment rescues profound T-lineage progenitor homing defect after bone marrow transplant conditioning in mice. Blood (2014) 124(2):296–304. doi:10.1182/blood-2014-01-552794
66. Halkias J, Melichar HJ, Taylor KT, Ross JO, Yen B, Cooper SB, et al. Opposing chemokine gradients control human thymocyte migration in situ. J Clin Invest (2013) 123(5):2131–42. doi:10.1172/JCI67175
67. Trampont PC, Tosello-Trampont AC, Shen Y, Duley AK, Sutherland AE, Bender TP, et al. CXCR4 acts as a costimulator during thymic beta-selection. Nat Immunol (2010) 11(2):162–70. doi:10.1038/ni.1830
68. Poznansky MC, Olszak IT, Evans RH, Wang Z, Foxall RB, Olson DP, et al. Thymocyte emigration is mediated by active movement away from stroma-derived factors. J Clin Invest (2002) 109(8):1101–10. doi:10.1172/JCI0213853
69. Duda DG, Kozin SV, Kirkpatrick ND, Xu L, Fukumura D, Jain RK. CXCL12 (SDF1alpha)-CXCR4/CXCR7 pathway inhibition: an emerging sensitizer for anticancer therapies? Clin Cancer Res (2011) 17(8):2074–80. doi:10.1158/1078-0432.CCR-10-2636
70. Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res (2014) 124:31–82. doi:10.1016/B978-0-12-411638-2.00002-1
71. Guo F, Wang Y, Liu J, Mok SC, Xue F, Zhang W. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene (2016) 35(7):816–26. doi:10.1038/onc.2015.139
72. Crazzolara R, Kreczy A, Mann G, Heitger A, Eibl G, Fink FM, et al. High expression of the chemokine receptor CXCR4 predicts extramedullary organ infiltration in childhood acute lymphoblastic leukaemia. Br J Haematol (2001) 115(3):545–53. doi:10.1046/j.1365-2141.2001.03164.x
73. Grundler R, Brault L, Gasser C, Bullock AN, Dechow T, Woetzel S, et al. Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated homing and migration. J Exp Med (2009) 206(9):1957–70. doi:10.1084/jem.20082074
74. Kumar A, Kremer KN, Dominguez D, Tadi M, Hedin KE. Galpha13 and Rho mediate endosomal trafficking of CXCR4 into Rab11+ vesicles upon stromal cell-derived factor-1 stimulation. J Immunol (2011) 186(2):951–8. doi:10.4049/jimmunol.1002019
75. Mizuno-Yamasaki E, Rivera-Molina F, Novick P. GTPase networks in membrane traffic. Annu Rev Biochem (2012) 81:637–59. doi:10.1146/annurev-biochem-052810-093700
76. Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol (2007) 69:451–82. doi:10.1146/annurev.physiol.69.022405.154712
77. Luo C, Pan H, Mines M, Watson K, Zhang J, Fan GH. CXCL12 induces tyrosine phosphorylation of cortactin, which plays a role in CXC chemokine receptor 4-mediated extracellular signal-regulated kinase activation and chemotaxis. J Biol Chem (2006) 281(40):30081–93. doi:10.1074/jbc.M605837200
78. Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M, et al. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell (2004) 6(5):459–69. doi:10.1016/j.ccr.2004.09.027
79. Decker S, Finter J, Forde AJ, Kissel S, Schwaller J, Mack TS, et al. PIM kinases are essential for chronic lymphocytic leukemia cell survival (PIM2/3) and CXCR4-mediated microenvironmental interactions (PIM1). Mol Cancer Ther (2014) 13(5):1231–45. doi:10.1158/1535-7163.MCT-13-0575-T
80. Gachet S, Genescà E, Passaro D, Irigoyen M, Alcalde H, Clémenson C, et al. Leukemia-initiating cell activity requires calcineurin in T-cell acute lymphoblastic leukemia. Leukemia (2013) 27(12):2289–300. doi:10.1038/leu.2013.156
81. Breit S, Stanulla M, Flohr T, Schrappe M, Ludwig WD, Tolle G, et al. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood (2006) 108(4):1151–7. doi:10.1182/blood-2005-12-4956
82. Park MJ, Taki T, Oda M, Watanabe T, Yumura-Yagi K, Kobayashi R, et al. FBXW7 and NOTCH1 mutations in childhood T cell acute lymphoblastic leukaemia and T cell non-Hodgkin lymphoma. Br J Haematol (2009) 145(2):198–206. doi:10.1111/j.1365-2141.2009.07607.x
83. Asnafi V, Buzyn A, Le Noir S, Baleydier F, Simon A, Beldjord K, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood (2009) 113(17):3918–24. doi:10.1182/blood-2008-10-184069
84. Kox C, Zimmermann M, Stanulla M, Leible S, Schrappe M, Ludwig WD, et al. The favorable effect of activating NOTCH1 receptor mutations on long-term outcome in T-ALL patients treated on the ALL-BFM 2000 protocol can be separated from FBXW7 loss of function. Leukemia (2010) 24(12):2005–13. doi:10.1038/leu.2010.203
85. Balatti V, Bottoni A, Palamarchuk A, Alder H, Rassenti LZ, Kipps TJ, et al. NOTCH1 mutations in CLL associated with trisomy 12. Blood (2012) 119(2):329–31. doi:10.1182/blood-2011-10-386144
86. Wang H, Zang C, Taing L, Arnett KL, Wong YJ, Pear WS, et al. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc Natl Acad Sci U S A (2014) 111(2):705–10. doi:10.1073/pnas.1315023111
87. Hales EC, Taub JW, Matherly LH. New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of gamma-secretase inhibitor resistant T-cell acute lymphoblastic leukemia. Cell Signal (2014) 26(1):149–61. doi:10.1016/j.cellsig.2013.09.021
88. Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood (2009) 113(24):6085–93. doi:10.1182/blood-2008-12-196618
89. Sun Y, Cheng Z, Ma L, Pei G. Beta-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem (2002) 277(51):49212–9. doi:10.1074/jbc.M207294200
90. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med (2007) 13(5):587–96. doi:10.1038/nm1567
91. Alampour-Rajabi S, El Bounkari O, Rot A, Muller-Newen G, Bachelerie F, Gawaz M, et al. MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP-70 signaling, and lymphocyte chemotaxis. FASEB J (2015) 29(11):4497–511. doi:10.1096/fj.15-273904
92. Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med (2006) 203(9):2201–13. doi:10.1084/jem.20052144
93. Luker KE, Steele JM, Mihalko LA, Ray P, Luker GD. Constitutive and chemokine-dependent internalization and recycling of CXCR7 in breast cancer cells to degrade chemokine ligands. Oncogene (2010) 29(32):4599–610. doi:10.1038/onc.2010.212
94. Shimizu S, Brown M, Sengupta R, Penfold ME, Meucci O. CXCR7 protein expression in human adult brain and differentiated neurons. PLoS One (2011) 6(5):e20680. doi:10.1371/journal.pone.0020680
95. Wurth R, Bajetto A, Harrison JK, Barbieri F, Florio T. CXCL12 modulation of CXCR4 and CXCR7 activity in human glioblastoma stem-like cells and regulation of the tumor microenvironment. Front Cell Neurosci (2014) 8:144. doi:10.3389/fncel.2014.00144
96. Tarnowski M, Liu R, Wysoczynski M, Ratajczak J, Kucia M, Ratajczak MZ. CXCR7: a new SDF-1-binding receptor in contrast to normal CD34(+) progenitors is functional and is expressed at higher level in human malignant hematopoietic cells. Eur J Haematol (2010) 85(6):472–83. doi:10.1111/j.1600-0609.2010.01531.x
97. Melo RCC, Longhini AL, Bigarella CL, Baratti MO, Traina F, Favaro P, et al. CXCR7 is highly expressed in acute lymphoblastic leukemia and potentiates CXCR4 response to CXCL12. PLoS One (2014) 9(1):e85926. doi:10.1371/journal.pone.0085926
98. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A (2010) 107(50):21931–6. doi:10.1073/pnas.1016071107
99. Forster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol (2008) 8(5):362–71. doi:10.1038/nri2297
100. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature (2001) 410(6824):50–6. doi:10.1038/35065016
101. Lopez-Giral S, Quintana NE, Cabrerizo M, Alfonso-Perez M, Sala-Valdes M, De Soria VG, et al. Chemokine receptors that mediate B cell homing to secondary lymphoid tissues are highly expressed in B cell chronic lymphocytic leukemia and non-Hodgkin lymphomas with widespread nodular dissemination. J Leukoc Biol (2004) 76(2):462–71. doi:10.1189/jlb.1203652
102. Burger JA. Chemokines and chemokine receptors in chronic lymphocytic leukemia (CLL): from understanding the basics towards therapeutic targeting. Semin Cancer Biol (2010) 20(6):424–30. doi:10.1016/j.semcancer.2010.09.005
103. Pui CH, Thiel E. Central nervous system disease in hematologic malignancies: historical perspective and practical applications. Semin Oncol (2009) 36(4 Suppl 2):S2–16. doi:10.1053/j.seminoncol.2009.05.002
104. Alsadeq A, Fedders H, Vokuhl C, Belau NM, Zimmermann M, Wirbelauer T, et al. The role of ZAP70 kinase in acute lymphoblastic leukemia infiltration into the central nervous system. Haematologica (2017) 102(2):346–55. doi:10.3324/haematol.2016.147744
105. Shuster JJ, Falletta JM, Pullen DJ, Crist WM, Humphrey GB, Dowell BL, et al. Prognostic factors in childhood T-cell acute lymphoblastic leukemia: a Pediatric Oncology Group study. Blood (1990) 75(1):166–73.
106. Ma S, Shi Y, Pang Y, Dong F, Cheng H, Hao S, et al. Notch1-induced T cell leukemia can be potentiated by microenvironmental cues in the spleen. J Hematol Oncol (2014) 7:71. doi:10.1186/s13045-014-0071-7
107. Uehara S, Grinberg A, Farber JM, Love PE. A role for CCR9 in T lymphocyte development and migration. J Immunol (2002) 168(6):2811–9. doi:10.4049/jimmunol.168.6.2811
108. Wurbel MA, Philippe JM, Nguyen C, Victorero G, Freeman T, Wooding P, et al. The chemokine TECK is expressed by thymic and intestinal epithelial cells and attracts double- and single-positive thymocytes expressing the TECK receptor CCR9. Eur J Immunol (2000) 30(1):262–71. doi:10.1002/1521-4141(200001)30:1<262::AID-IMMU262>3.0.CO;2-0
109. Uehara S, Song K, Farber JM, Love PE. Characterization of CCR9 expression and CCL25/thymus-expressed chemokine responsiveness during T cell development: CD3(high)CD69+ thymocytes and gammadeltaTCR+ thymocytes preferentially respond to CCL25. J Immunol (2002) 168(1):134–42. doi:10.4049/jimmunol.168.1.134
110. Annels NE, Willemze AJ, van der Velden VH, Faaij CM, van Wering E, Sie-Go DM, et al. Possible link between unique chemokine and homing receptor expression at diagnosis and relapse location in a patient with childhood T-ALL. Blood (2004) 103(7):2806–8. doi:10.1182/blood-2003-06-1812
111. Qiuping Z, Jei X, Youxin J, Wei J, Chun L, Jin W, et al. CC chemokine ligand 25 enhances resistance to apoptosis in CD4+ T cells from patients with T-cell lineage acute and chronic lymphocytic leukemia by means of livin activation. Cancer Res (2004) 64(20):7579–87. doi:10.1158/0008-5472.CAN-04-0641
112. Marks DI, Rowntree C. Management of adults with T-cell lymphoblastic leukemia. Blood (2017) 129(9):1134–42. doi:10.1182/blood-2016-07-692608
113. Karrman K, Johansson B. Pediatric T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer (2017) 56(2):89–116. doi:10.1002/gcc.22416
114. Sanchez-Martin M, Ferrando A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood (2017) 129(9):1124–33. doi:10.1182/blood-2016-09-692582
115. Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med (2003) 198(9):1391–402. doi:10.1084/jem.20030267
116. Muller A, Sonkoly E, Eulert C, Gerber PA, Kubitza R, Schirlau K, et al. Chemokine receptors in head and neck cancer: association with metastatic spread and regulation during chemotherapy. Int J Cancer (2006) 118(9):2147–57. doi:10.1002/ijc.21514
117. Chen SS, Chang BY, Chang S, Tong T, Ham S, Sherry B, et al. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia (2016) 30(4):833–43. doi:10.1038/leu.2015.316
118. Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, Frolova O, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood (2009) 113(24):6215–24. doi:10.1182/blood-2008-05-158311
Keywords: T-cell acute lymphoblastic leukemia, chemokines, CXC-chemokine receptor 4, stromal-derived factor-1, NOTCH, CXCR7, infiltration
Citation: Piovan E, Tosello V, Amadori A and Zanovello P (2018) Chemotactic Cues for NOTCH1-Dependent Leukemia. Front. Immunol. 9:633. doi: 10.3389/fimmu.2018.00633
Received: 24 January 2018; Accepted: 14 March 2018;
Published: 03 April 2018
Edited by:
Antonio Francesco Campese, Sapienza Università di Roma, ItalyReviewed by:
Alessandro Poggi, Ospedale Policlinico San Martino, ItalyAlex Yee-Chen Huang, Case Western Reserve University, United States
Copyright: © 2018 Piovan, Tosello, Amadori and Zanovello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erich Piovan, ZXJpY2gucGlvdmFuQHVuaXBkLml0;
Paola Zanovello, cGFvbGEuemFub3ZlbGxvQHVuaXBkLml0