 Priyadharshini Devarajan
Priyadharshini Devarajan Michael C. Jones
Michael C. Jones Olivia Kugler-Umana
Olivia Kugler-Umana Allen M. Vong
Allen M. Vong Susan L. Swain
Susan L. Swain
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 26 March 2018
Sec. Immunological Memory
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00596
This article is part of the Research Topic Orchestration of an Immune Response to Respiratory Pathogens View all 15 articles
Although much is known about the mechanisms by which pathogen recognition drives the initiation of T cell responses, including those to respiratory viruses, the role of pathogen recognition in fate decisions of T cells once they have become effectors remains poorly defined. Here, we review our recent studies that suggest that the generation of CD4 T cell memory is determined by recognition of virus at an effector “checkpoint.” We propose this is also true of more highly differentiated tissue-restricted effector cells, including cytotoxic “ThCTL” in the site of infection and TFH in secondary lymphoid organs. We point out that ThCTL are key contributors to direct viral clearance and TFH to effective Ab response, suggesting that the most protective immunity to influenza, and by analogy to other respiratory viruses, requires prolonged exposure to antigen and to infection-associated signals. We point out that many vaccines used today do not provide such prolonged signals and suggest this contributes to their limited effectiveness. We also discuss how aging impacts effective CD4 T cell responses and how new insights about the response of aged naive CD4 T cells and B cells might hold implications for effective vaccine design for both the young and aged against respiratory viruses.
An effective immune response to respiratory pathogens, such as Influenza A virus (IAV), requires coordination between the innate and adaptive immune system. Host defense to influenza infection begins when lung airway epithelial cells, dendritic cells (DC), and alveolar macrophages alert the host to the presence of virus through the activation of pattern recognition receptors (PRR) (1). This triggers the production of inflammatory cytokines, which activates antigen-presenting cells (APC). APC migrate to secondary lymphoid organs where they present antigen to T and B cells as soon as 2 days postinfection (dpi) (2). In a primary infection, CD4 and CD8 effectors are the most important for clearing virus, with Ab arising later. Effectors soon contract and a cohort become memory T cells that can persist in the host long-term and provide durable protection against the same virus.
Thereafter, re-encounter with the same virus goes largely unnoticed since neutralizing long-lived Ab, produced by long-lived plasma cells (LLPC), rapidly clears virus. However, when virus surface proteins hemaggluttinin (HA) and neuraminidase (NA) can mutate sufficiently and escape recognition by the Ab. IAV also escape when new strain with a distinct HA and NA subtype (heterosubtypic) develops or by a high dose of exposure that overcomes Ab. When virus is incompletely cleared, memory T cells and B cells are induced to mount a secondary response, making secondary effectors that can protect against a broader range of influenza strains (3). Most studies have focused on T cell activation during priming, but it is unclear what continued signals are needed into the effector phase to generate memory. Although live virus is cleared between 10 and 13 dpi, viral antigen presentation is detected for up to 21 dpi (4), indicating Ag recognition could continue to drive T cell responses.
Indeed antigen presentation through 8 dpi drives effector CD8 T cells to expand and promotes memory formation (5, 6). We found that antigen presentation throughout the effector response was required for almost all CD4 T cell memory generated by IAV infection (7, 8). Antigen recognition induced autocrine CD4 effector IL-2 production that acted at days 6–8 postinfection at this “checkpoint” to downregulate Bim and to upregulate the IL-7Ra. These signals along with antigen recognition, promoted survival of CD4 effectors and drove their transition to memory (7, 8). Infection was not required at this stage, since antigen-pulsed APC were sufficient to support optimal memory CD4 T cell generation and maintenance that was able to protect a naïve host from an otherwise lethal influenza challenge (7). Antigen engagement at the memory checkpoint specifically upregulated the expression of CD25, Bcl-6, and phosphorylated STAT3 in the effector CD4 T cells (8). CD25 expression is needed for efficient IL-2R signaling, so that IL-2 can prevent effector cell death (9). Costimulation through CD27 is also important at this juncture (7) for the most efficient formation of CD4 memory, just as it is during priming (10). Correspondingly, a subset of DCs that express CD70 post-priming has also been identified which correlate with CD27+CD8 effector expansion late into the IAV response (11).
The need for cognate interaction of effectors with Ag–APC during this phase potentially allows for the influence by Ag dose/avidity and by co-stimulatory ligands on the APC dependent on pathogen-associated molecular patterns (PAMPs). This ensures that strong memory develops only when there is no longer infection at the early effector phase, when necessary because of unresolved threats (Figure 1). We suggest that defining pathways that drive optimum CD4 and CD8 T cell memory will inform vaccine design to produce more protective T cell memory.
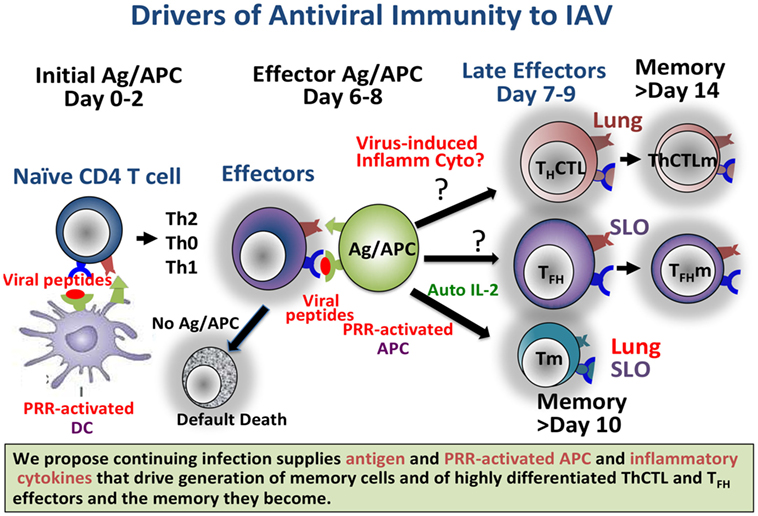
Figure 1. Continuing infection, producing abundant antigen and pattern recognition receptor (PRR)-activated APC, and we propose inflammatory cytokines, drives generation of memory cells and of highly differentiated ThCTL and TFH effectors and the memory they become. Providing such signals in a vaccine setting should result in longer-lasting more protective immunity. Known and likely roles for viral Ag and for PRR in the response to Influenza A virus are depicted.
CD4 T cells responding to viral infections differentiate into a heterogeneous population of effector cells, with subsets that mediate viral clearance through distinct mechanisms including those that are cytokine-mediated (Th1, Th2, Th17) and that kill infected cells (ThCTL), as well as indirect mechanisms of help for B cell differentiation (TFH) in germinal centers (GC) (3, 9, 12). Protection against lethal challenge with influenza can be mediated by synergy of CD4 T effectors acting via generation of Ab and by perforin-mediated lysis due to ThCTL (13).
It is well established that TFH are required for GC formation and that they support GC B cell responses leading to isotype switching, somatic hypermutation, and selection of high affinity with the production of LLPC and memory B cells (14). As they recognize antigen on GC B cells, the TFH in turn become GC-TFH and later can become memory TFH (15, 16). The LLPC are responsible for producing the long-lived Ab that provides most rapid protection against viral infection. Thus, the tissue-restricted recognition of Ag by TFH is critical to GC-TFH development, for subsequent TFH memory and for long-lived Ab-mediated protection. The critical TFH functions and their transition to memory have been well reviewed (14–16). Understanding what signals from cognate interaction of TFH and GCB are needed and how long they are needed is crucial to maximizing immunity.
Another tissue-restricted CD4 effector population is the cytotoxic CD4 T cells, ThCTL (17, 18). ThCTL lyse target cells by the same mechanisms utilized by cytotoxic CD8 T cells, including the perforin-mediated pathway. ThCTL are generated during influenza and many other viral infections (19). After IAV infection they are found in the lung and bronchoalveolar lavage (19, 20), suggesting they are restricted to the sites of infection. Two markers of ThCTL have been found: CRTAM and NKG2C/E. MHC Class I-restricted T cell associated molecule (CRTAM) can be expressed by IAV-specific CD4 T cells upon activation. CRTAM+ CD4 cells, upregulate expression of granzymes and display peptide specific cytotoxicity, indicating these cells are ThCTL (21). While CRTAM marks cytotoxic CD4 T cells, its expression requires ex vivo activation, making tracking ThCTL more difficult.
NKG2A/C/E is a family of C type lectin receptors found on NK cells and CD8 T cells (22, 23). NKG2C/E, however, can be found on CD4 T cells directly ex vivo from infected mouse lungs (20). Isolating CD4 T cells based on their expression of NKG2C/E, indicates that cytotoxic activity ex vivo, as well as increased expression of perforin and granzyme is found only in the NKG2C/E-expressing effectors. NKG2C/Epos ThCTL, readily degranulate and secrete high levels of IFNg when they recognize Ag, consistent with their potent antiviral activity (20). Although effector CD4 T cells infiltrate the tissues throughout the body (24), ThCTL are only found in the lung and we find that they upregulate a gene expression program consistent with tissue residency (20). Further, Ab to CD4 injected intravenously to assess relative accessibility at the effector stage, indicates most ThCTL are inaccessible, suggesting they are within the lung tissue (20). This location in sites of infection is important as local lung-resident memory CD4 T cells promote better protection than splenic memory CD4 cells against IAV infection (25).
Due to their protective capacity, in both mice and humans infected with influenza (13, 26, 27), it is important to identify the factors that support generation of ThCTL. ThCTL do not require standard polarizing cytokines during initial activation, but do depend on IL-2 (17). We find that CD4 T cells need Blimp-1 as a transcription factor to enable the cytotoxic phenotype. Loss of Blimp-1 in CD4 T cells leads to reduced ThCTL in the lungs (20), and, in others studies, reduced ability to prevent weight loss after influenza (26). Bcl-6, a transcriptional repressor of Blimp-1, is in contrast critical for TFH generation (28–30), underlining the diverse transcriptional regulation of ThCTL versus TFH. We note that the restriction of ThCTL to sites of infection and their late appearance (20) suggest that they, like TFH, may require late cognate interactions to direct their final differentiation (Figure 1).
The innate immune system plays a critical role in initiating the cascade of the adaptive responses to viruses. Toll-, RIG-I- and Nod-like receptors (TLR, RLR, or NLR) on the initial infected cells are triggered by PAMP to signal the first wave of inflammatory cytokine production. These in turn activate various APC to effectively initiate T cell priming. IAV infection activates innate pathways primarily via the PRR, such as TLR3, TLR7, RIG-I, and Nlrp3. Previous reviews have discussed the role of PRR-signaling in IAV infection in detail (31, 32). However, little is known beyond the role of the PRR pathways acting early in priming and initiation of T cell responses (31–33). Here, we discuss recent advances in the fields of CD4 memory, TFH and ThCTL that are making it clear that PRR pathways play a more global role in shaping CD4 effector and memory responses.
We find that the generation of CD4 memory does not require infection during the effector phase, as activated APC presenting peptides are sufficient to drive in vivo generated CD4 effectors to become memory in uninfected mice (8). However, the role of PRR pathways in generating specialized CD4 memory responses such as TFH memory, ThCTL memory and CD4 TRM is only now being studied. The gamma-chain cytokines, IL-2, IL-7, and IL-15, each play important roles in T cell memory (7, 34, 35). PRR pathways can induce high levels of IL-15 during infection (35). While we know that constitutive levels of IL-15 and IL-7 maintain homeostatic memory CD8 and CD4 T cell populations (35), the role of high levels of PRR-signaling such as that leading to type I IFN and other proinflammatory cytokines during active infection remains unclear. We find that IL-15 is required during the effector phase of the CD4 response for the generation of an IL-2 independent CD4 TRM population (36). Another study indicates local inflammatory cues from IL-12 and IFNβ, made by intestinal macrophages, are involved in differentiation and persistence of the CD8 TRM populations (37). While multiple PRR pathways promote T cell memory, the causal relationship between the memory subsets and the specific PRR has only been shown indirectly through the requirement for innate cytokines.
The TLR9 adjuvant CpG acts on TLR9/MyD88 signaling in both DC and B cells to promote optimum TFH generation (37). Type I IFN produced by PRR-signaling, also promotes the TFH genetic program. Type I IFNα/β induced Bcl-6, CXCR5, and PD-1 expression in CD4 T cells that were activated in vitro by Ab to CD3 and CD28, by a STAT-1-dependent pathway (38). During persistent LCMV infection, chronic Type I IFN supports TFH formation (39). On the other hand, another study showed that the absence of STAT3 in CD4 T cells during an acute LCMV infection resulted in reduced TFH differentiation caused by increased Type I IFN production, thus suggesting that Type I IFN indirectly inhibited TFH differentiation (40). Type I IFNs have widely variable effects on T cell activation depending on whether they are present before activation, during activation or after activation and depending on whether they are present acutely or chronically (41). Thus, it is likely that the disparate impacts of Type I IFN on TFH generation in the studies above are due to differences in model systems used.
CD4 T cells stimulated in vitro in the presence of IFNα and IL-2 have increased cytotoxic potential (26). CD4 T cells in the lungs of IFNAR-deficient mice also expressed lower levels of Granzyme B and perforin, suggesting ThCTL generation also may depend on inflammatory cytokines. In support of roles for inflammation acting on T effectors, gene profiling following IAV infection indicate innate cytokines including Type I and Type III IFNs, are produced well into the effector phase of the response (42). Since innate inflammatory cytokines, such as Type I IFN, can be pathological (43) it makes sense that their production requires PRR activation, present only during continuing infection and that they could, then help further drive the differentiation of specialized effector CD4 T cells to ensure pathogen clearance.
Innate pathways also play an indirect role in promoting continued antigen presentation by inducing inflammation and enhancing APC activation. A recent study showed that type 1 IFN has differential effects on APC subsets by inducing different levels of co-stimulatory ligands, such as various TNFSF ligands, CD80, and CD86 on inflammatory APCs versus classical DCs, which control priming of CD4 T cells (44). Expression of many of these TNFR ligands such as OX-40, CD27, and 4-1BBL have been correlated with protective antiviral T cell responses (45, 46).
In addition to the roles of PRR-stimulation in driving the final differentiation of TFH, discussed above, we recently showed that PRR-activated DC, acting as APC for CD4 T cells, could greatly augment both TFH generation, generation of IL-21 secreting CD4 effectors. This indirectly enhances generation of GCB cells and most importantly of long-lived Ab in response to inactivated IAV vaccine (47). Vaccines, especially traditional inactivated or subunit flu vaccines often result in weak, barely protective Ab levels and low frequency T cell memory, especially when given to the elderly (48). In contrast, live infection is able to generate very long-lived and effective immunity of all types, suggesting that vaccines will need to be modified to provide the key signals inherent in live infection in order to improve efficacy. We found that when peptide-pulsed DC were activated by TLR-signaling and used to prime CD4 T cells specific for IAV, high levels of inflammatory cytokine were produced during the CD4:Ag/APC interaction. This enhanced otherwise weak helper T and B cell responses by an IL-6 dependent mechanism. This effect was apparent even in an unmanipulated mouse, where TLR-activated DC, presenting inactivated IAV, enhanced B cell IgG Ab response over several months (47). PRR activation in conjunction with BcR-triggering also can directly activate B cells and promote their differentiation to AbSC. This synergistic signaling drives T-independent B cell responses, but can also be involved in conventional B cell (B2) responses (48, 49). Thus, the presence of PRR signals acts at multiple levels in DC and in B cells to promote the initial B cell response, and we propose also later to promote TFH, LLPC, and memory B cells.
Animals undergo a dramatic shift in their immune responses as they age. The number of naive T cells (50) and naive follicular B cells (FOB) decreases and remaining naive cells are less responsive (51). So while previously established T and B memory cells often remain largely functional, responses to new viruses or strains of viruses not previously encountered is compromised. Our studies show that aged CD4 T cells have reduced responsiveness to IL-6 (47, 52) leading to weak generation of helper subsets. Providing high Ag doses on PRR-activated APC, markedly enhanced aged naive CD4 helper responses and indirectly improved B cell responses by a mechanism dependent on APC produced IL-6 (47). This study provided clear evidence that PRR signals acting on APC were responsible for improved CD4 helper responses and supported the concept that greater PAMP signaling is required to prime naive T cells as they age.
We wondered if the reduced responses by B cells in the aged could also be enhanced by high Ag dose and PAMP stimulation. We also noted that T-independent B cell responses, do not require the age-compromised T helper cells, might provide immune protection in the aged. A subset of B cells termed “age associated B cells” (ABC) that lack both CD23 and CD21 increase with age in mice (53). Studies showed that some of the ABC can develop from antigen-experienced FOB suggesting they are memory-like B cells (54). However, we find that most ABC in unimmunized mice express only surface IgD and IgM and lack the expression of key B cell co-stimulatory and activation markers, suggesting they are naive (51). These sIgD+ ABC transferred to RAG-deficient mice, were driven by IAV infection into AbSC specific for IAV (51), indicating a T-independent pathway to Ab production. ABC identified in models of autoimmunity are dependent on BcR, TLR-7, and TLR-9 stimulation (53, 55), and memory ABC populations are implicated in mediating autoimmunity (56, 57). Thus like CD4 T cells, the ABC may require strong PRR-signaling to respond effectively. We propose that pathogens such as respiratory viruses can induce sIgD+ ABC to generate protective Ab responses even in the aged (51). We are investigating whether the sIgD + ABC response to IAV infection can indeed contribute to a protective Ab response and if this is dependent on high levels of Ag and PAMP signals.
It has become increasingly clear that vaccines, especially inactivated, subunit, or recombinant protein vaccines, often result in weak, barely protective Ab levels and low frequency T cell memory, and they are even less effective in the elderly (48, 58). On the other hand, LAIV are able to elicit superior lung specific responses and TRM (59). Various studies have indeed shown that that local antigen presentation and local inflammation are key factors that drive tissue-resident memory (60, 61). Live infection is able to generate effective immunity of all types that often lasts several decades or more, suggesting that modifications to vaccines to provide key signals inherent in live infection should be able to improve vaccine efficacy (62). Here, we suggest that strong signals like those from replicating pathogens, including high doses of antigen persisting to the peak of the immune response, along with high levels of PAMPs acting on innate and B cells, are necessary to optimally trigger T cell effector and memory generation and B cell response. Replicating viruses that facilitate high and continued levels of antigen presentation and inflammation, in addition to tissue localization of the immune response induced by intranasal administration, may be key factors that determine superior vaccine induced protection. Though further definition of these pathways is needed to best inform vaccine strategies, we propose that they can ultimately be used to drive generation of increased immunity against respiratory viruses.
SS, PD, MJ, AV, OK-U, and JX wrote various sections in the manuscript. SS and PD conceptualized and organized the article and coordinated author contributions. MJ finalized the manuscript for submission. All other authors did experimental work that influenced the thinking behind the article. All authors contributed to manuscript revision and have read and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors and their work are supported by grants from NIH (T32AI007349-27 awarded to AV and OK-U; Project 2 of U19AI109858; R01AI118820; Project 1 of P01AG0216 and Project 1 of P01AI46530; to SS).
1. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol (2014) 14(5):315–28. doi:10.1038/nri3665
2. Braciale TJ, Sun J, Kim TS. Regulating the adaptive immune response to respiratory virus infection. Nat Rev Immunol (2012) 12(4):295–305. doi:10.1038/nri3166
3. Strutt TM, McKinstry KK, Marshall NB, Vong AM, Dutton RW, Swain SL. Multipronged CD4(+) T-cell effector and memory responses cooperate to provide potent immunity against respiratory virus. Immunol Rev (2013) 255(1):149–64. doi:10.1111/imr.12088
4. Jelley-Gibbs DM, Brown DM, Dibble JP, Haynes L, Eaton SM, Swain SL. Unexpected prolonged presentation of influenza antigens promotes CD4 T cell memory generation. J Exp Med (2005) 202(5):697–706. doi:10.1084/jem.20050227
5. Ballesteros-Tato A, Leon B, Lee BO, Lund FE, Randall TD. Epitope-specific regulation of memory programming by differential duration of antigen presentation to influenza-specific CD8(+) T cells. Immunity (2014) 41(1):127–40. doi:10.1016/j.immuni.2014.06.007
6. Leon B, Ballesteros-Tato A, Randall TD, Lund FE. Prolonged antigen presentation by immune complex-binding dendritic cells programs the proliferative capacity of memory CD8 T cells. J Exp Med (2014) 211(8):1637–55. doi:10.1084/jem.20131692
7. McKinstry KK, Strutt TM, Bautista B, Zhang W, Kuang Y, Cooper AM, et al. Effector CD4 T-cell transition to memory requires late cognate interactions that induce autocrine IL-2. Nat Commun (2014) 5:5377. doi:10.1038/ncomms6377
8. Bautista BL, Devarajan P, McKinstry KK, Strutt TM, Vong AM, Jones MC, et al. Short-lived antigen recognition but not viral infection at a defined checkpoint programs effector CD4 T cells to become protective memory. J Immunol (2016) 197(10):3936–49. doi:10.4049/jimmunol.1600838
9. Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4(+) T cells in immunity to viruses. Nat Rev Immunol (2012) 12(2):136–48. doi:10.1038/nri3152
10. Xiao Y, Peperzak V, Keller AM, Borst J. CD27 instructs CD4+ T cells to provide help for the memory CD8+ T cell response after protein immunization. J Immunol (2008) 181(2):1071–82. doi:10.4049/jimmunol.181.2.1071
11. Ballesteros-Tato A, Leon B, Lund FE, Randall TD. Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8(+) T cell responses to influenza. Nat Immunol (2010) 11(3):216–24. doi:10.1038/ni.1838
12. McKinstry KK, Strutt TM, Kuang Y, Brown DM, Sell S, Dutton RW, et al. Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J Clin Invest (2012) 122(8):2847–56. doi:10.1172/JCI63689
13. Brown DM, Dilzer AM, Meents DL, Swain SL. CD4 T cell-mediated protection from lethal influenza: perforin and antibody-mediated mechanisms give a one-two punch. J Immunol (2006) 177(5):2888–98. doi:10.4049/jimmunol.177.5.2888
14. Crotty S. A brief history of T cell help to B cells. Nat Rev Immunol (2015) 15(3):185–9. doi:10.1038/nri3803
15. Hale JS, Ahmed R. Memory T follicular helper CD4 T cells. Front Immunol (2015) 6:16. doi:10.3389/fimmu.2015.00016
16. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi:10.1146/annurev-immunol-031210-101400
17. Brown DM, Kamperschroer C, Dilzer AM, Roberts DM, Swain SL. IL-2 and antigen dose differentially regulate perforin- and FasL-mediated cytolytic activity in antigen specific CD4+ T cells. Cell Immunol (2009) 257(1–2):69–79. doi:10.1016/j.cellimm.2009.03.002
18. Brown DM, Lee S, Garcia-Hernandez Mde L, Swain SL. Multifunctional CD4 cells expressing gamma interferon and perforin mediate protection against lethal influenza virus infection. J Virol (2012) 86(12):6792–803. doi:10.1128/JVI.07172-11
19. Marshall NB, Swain SL. Cytotoxic CD4 T cells in antiviral immunity. J Biomed Biotechnol (2011) 2011:954602. doi:10.1155/2011/954602
20. Marshall NB, Vong AM, Devarajan P, Brauner MD, Kuang Y, Nayar R, et al. NKG2C/E marks the unique cytotoxic CD4 T cell subset, ThCTL, generated by influenza infection. J Immunol (2017) 198(3):1142–55. doi:10.4049/jimmunol.1601297
21. Takeuchi A, Badr Mel S, Miyauchi K, Ishihara C, Onishi R, Guo Z, et al. CRTAM determines the CD4+ cytotoxic T lymphocyte lineage. J Exp Med (2016) 213(1):123–38. doi:10.1084/jem.20150519
22. Vance RE, Kraft JR, Altman JD, Jensen PE, Raulet DH. Mouse CD94/NKG2A is a natural killer cell receptor for the nonclassical major histocompatibility complex (MHC) class I molecule Qa-1(b). J Exp Med (1998) 188(10):1841–8. doi:10.1084/jem.188.10.1841
23. Moser JM, Gibbs J, Jensen PE, Lukacher AE. CD94-NKG2A receptors regulate antiviral CD8(+) T cell responses. Nat Immunol (2002) 3(2):189–95. doi:10.1038/ni757
24. Roman E, Miller E, Harmsen A, Wiley J, Von Andrian UH, Huston G, et al. CD4 effector T cell subsets in the response to influenza: heterogeneity, migration, and function. J Exp Med (2002) 196(7):957–68. doi:10.1084/jem.20021052
25. Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL. Cutting edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol (2011) 187(11):5510–4. doi:10.4049/jimmunol.1102243
26. Hua L, Yao S, Pham D, Jiang L, Wright J, Sawant D, et al. Cytokine-dependent induction of CD4+ T cells with cytotoxic potential during influenza virus infection. J Virol (2013) 87(21):11884–93. doi:10.1128/JVI.01461-13
27. Wilkinson TM, Li CK, Chui CS, Huang AK, Perkins M, Liebner JC, et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med (2012) 18(2):274–80. doi:10.1038/nm.2612
28. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325(5943):1006–10. doi:10.1126/science.1175870
29. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science (2009) 325(5943):1001–5. doi:10.1126/science.1176676
30. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity (2009) 31(3):457–68. doi:10.1016/j.immuni.2009.07.002
31. Pulendran B, Maddur MS. Innate immune sensing and response to influenza. Curr Top Microbiol Immunol (2015) 386:23–71. doi:10.1007/82_2014_405
32. Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol (2015) 16(4):343–53. doi:10.1038/ni.3123
33. Jain A, Pasare C. Innate control of adaptive immunity: beyond the three-signal paradigm. J Immunol (2017) 198(10):3791–800. doi:10.4049/jimmunol.1602000
34. Read KA, Powell MD, McDonald PW, Oestreich KJ. IL-2, IL-7, and IL-15: multistage regulators of CD4(+) T helper cell differentiation. Exp Hematol (2016) 44(9):799–808. doi:10.1016/j.exphem.2016.06.003
35. Colpitts SL, Stoklasek TA, Plumlee CR, Obar JJ, Guo C, Lefrancois L. Cutting edge: the role of IFN-alpha receptor and MyD88 signaling in induction of IL-15 expression in vivo. J Immunol (2012) 188(6):2483–7. doi:10.4049/jimmunol.1103609
36. Strutt TM, Dhume K, Finn CM, Hwang JH, Castonguay C, Swain SL, et al. IL-15 supports the generation of protective lung-resident memory CD4 T cells. Mucosal Immunol (2017). doi:10.1038/mi.2017.101
37. Rookhuizen DC, DeFranco AL. Toll-like receptor 9 signaling acts on multiple elements of the germinal center to enhance antibody responses. Proc Natl Acad Sci U S A (2014) 111(31):E3224–33. doi:10.1073/pnas.1323985111
38. Nakayamada S, Poholek AC, Lu KT, Takahashi H, Kato M, Iwata S, et al. Type I IFN induces binding of STAT1 to Bcl6: divergent roles of STAT family transcription factors in the T follicular helper cell genetic program. J Immunol (2014) 192(5):2156–66. doi:10.4049/jimmunol.1300675
39. Osokine I, Snell LM, Cunningham CR, Yamada DH, Wilson EB, Elsaesser HJ, et al. Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc Natl Acad Sci U S A (2014) 111(20):7409–14. doi:10.1073/pnas.1401662111
40. Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity (2014) 40(3):367–77. doi:10.1016/j.immuni.2014.02.005
41. Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol (2015) 15(4):231–42. doi:10.1038/nri3806
42. Pommerenke C, Wilk E, Srivastava B, Schulze A, Novoselova N, Geffers R, et al. Global transcriptome analysis in influenza-infected mouse lungs reveals the kinetics of innate and adaptive host immune responses. PLoS One (2012) 7(7):e41169. doi:10.1371/journal.pone.0041169
43. Di Domizio J, Cao W. Fueling autoimmunity: type I interferon in autoimmune diseases. Expert Rev Clin Immunol (2013) 9(3):201–10. doi:10.1586/eci.12.106
44. Chang YH, Wang KC, Chu KL, Clouthier DL, Tran AT, Torres Perez MS, et al. dichotomous expression of TNF superfamily ligands on antigen-presenting cells controls post-priming anti-viral CD4(+) T cell immunity. Immunity (2017) 47(5):943–58e9. doi:10.1016/j.immuni.2017.10.014
45. Lin GH, Sedgmen BJ, Moraes TJ, Snell LM, Topham DJ, Watts TH. Endogenous 4-1BB ligand plays a critical role in protection from influenza-induced disease. J Immunol (2009) 182(2):934–47. doi:10.4049/jimmunol.182.2.934
46. Salek-Ardakani S, Flynn R, Arens R, Yagita H, Smith GL, Borst J, et al. The TNFR family members post-infection and CD27 link viral virulence to protective T cell vaccines in mice. J Clin Invest (2011) 121(1):296–307. doi:10.1172/JCI42056
47. Brahmakshatriya V, Kuang Y, Devarajan P, Xia J, Zhang W, Vong AM, et al. IL-6 production by TLR-activated APC broadly enhances aged cognate CD4 helper and B cell antibody responses in vivo. J Immunol (2017) 198(7):2819–33. doi:10.4049/jimmunol.1601119
48. Chen WH, Kozlovsky BF, Effros RB, Grubeck-Loebenstein B, Edelman R, Sztein MB. Vaccination in the elderly: an immunological perspective. Trends Immunol (2009) 30(7):351–9. doi:10.1016/j.it.2009.05.002
49. Pasare C, Medzhitov R. Control of B-cell responses by toll-like receptors. Nature (2005) 438(7066):364–8. doi:10.1038/nature04267
50. Tsukamoto H, Clise-Dwyer K, Huston GE, Duso DK, Buck AL, Johnson LL, et al. Age-associated increase in lifespan of naive CD4 T cells contributes to T-cell homeostasis but facilitates development of functional defects. Proc Natl Acad Sci U S A (2009) 106(43):18333–8. doi:10.1073/pnas.0910139106
51. Swain SL, Kugler-Umana O, Kuang Y, Zhang W. The properties of the unique age-associated B cell subset reveal a shift in strategy of immune response with age. Cell Immunol (2017) 321:52–60. doi:10.1016/j.cellimm.2017.05.009
52. Jones SC, Brahmakshatriya V, Huston G, Dibble J, Swain SL. TLR-activated dendritic cells enhance the response of aged naive CD4 T cells via an IL-6-dependent mechanism. J Immunol (2010) 185(11):6783–94. doi:10.4049/jimmunol.0901296
53. Hao Y, O’Neill P, Naradikian MS, Scholz JL, Cancro MP. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood (2011) 118(5):1294–304. doi:10.1182/blood-2011-01-330530
54. Russell Knode LM, Naradikian MS, Myles A, Scholz JL, Hao Y, Liu D, et al. Age-associated B cells express a diverse repertoire of VH and Vkappa genes with somatic hypermutation. J Immunol (2017) 198(5):1921–7. doi:10.4049/jimmunol.1601106
55. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood (2011) 118(5):1305–15. doi:10.1182/blood-2011-01-331462
56. Rubtsova K, Rubtsov AV, Thurman JM, Mennona JM, Kappler JW, Marrack P. B cells expressing the transcription factor T-bet drive lupus-like autoimmunity. J Clin Invest (2017) 127(4):1392–404. doi:10.1172/JCI91250
57. Naradikian MS, Hao Y, Cancro MP. Age-associated B cells: key mediators of both protective and autoreactive humoral responses. Immunol Rev (2016) 269(1):118–29. doi:10.1111/imr.12380
58. McElhaney JE, Kuchel GA, Zhou X, Swain SL, Haynes L. T-cell immunity to influenza in older adults: a pathophysiological framework for development of more effective vaccines. Front Immunol (2016) 7:41. doi:10.3389/fimmu.2016.00041
59. Zens KD, Chen JK, Farber DL. Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight (2016) 1(10). doi:10.1172/jci.insight.85832
60. Khan TN, Mooster JL, Kilgore AM, Osborn JF, Nolz JC. Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J Exp Med (2016) 213(6):951–66. doi:10.1084/jem.20151855
61. Bergsbaken T, Bevan MJ, Fink PJ. Local inflammatory cues regulate differentiation and persistence of CD8(+) tissue-resident memory T cells. Cell Rep (2017) 19(1):114–24. doi:10.1016/j.celrep.2017.03.031
Keywords: CD4 T cells, ThCTL, TFH, immune memory, influenza, virus, pathogen recognition
Citation: Devarajan P, Jones MC, Kugler-Umana O, Vong AM, Xia J and Swain SL (2018) Pathogen Recognition by CD4 Effectors Drives Key Effector and Most Memory Cell Generation Against Respiratory Virus. Front. Immunol. 9:596. doi: 10.3389/fimmu.2018.00596
Received: 01 December 2017; Accepted: 09 March 2018;
Published: 26 March 2018
Edited by:
Andrea Sant, David H. Smith Center for Vaccine Biology and Immunology, United StatesReviewed by:
Tania H. Watts, University of Toronto, CanadaCopyright: © 2018 Devarajan, Jones, Kugler-Umana, Vong, Xia and Swain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan L. Swain, c3VzYW4uc3dhaW5AdW1hc3NtZWQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.