 Tamás Németh1,2*
Tamás Németh1,2* Krisztina Futosi1,2
Krisztina Futosi1,2 Kata Szilveszter1,2
Kata Szilveszter1,2 Olivér Vilinovszki1
Olivér Vilinovszki1 Levente Kiss-Pápai1,2
Levente Kiss-Pápai1,2 Attila Mócsai1,2*
Attila Mócsai1,2*
- 1Department of Physiology, Semmelweis University School of Medicine, Budapest, Hungary
- 2MTA-SE “Lendület” Inflammation Physiology Research Group of the Hungarian Academy of Sciences and Semmelweis University, Budapest, Hungary
Autoantibody production and autoantibody-mediated inflammation are hallmarks of a number of autoimmune diseases. The K/BxN serum-transfer arthritis is one of the most widely used models of the effector phase of autoantibody-induced pathology. Several hematopoietic lineages including neutrophils, platelets, and mast cells have been proposed to contribute to inflammation and tissue damage in this model. We have previously shown that the Syk tyrosine kinase is critically involved in the development in K/BxN serum-transfer arthritis and bone marrow chimeric experiments indicated that Syk is likely involved in one or more hematopoietic lineages during the disease course. The aim of the present study was to further define the lineage(s) in which Syk expression is required for autoantibody-induced arthritis. To this end, K/BxN serum-transfer arthritis was tested in conditional mutant mice in which Syk was deleted in a lineage-specific manner from neutrophils, platelets, or mast cells. Combination of the MRP8-Cre, PF4-Cre, or Mcpt5-Cre transgene with floxed Syk alleles allowed efficient and selective deletion of Syk from neutrophils, platelets, or mast cells, respectively. This has also been confirmed by defective Syk-dependent in vitro functional responses of the respective cell types. In vivo studies revealed nearly complete defect of the development of K/BxN serum-transfer arthritis upon neutrophil-specific deletion of Syk. By contrast, Syk deletion from platelets or mast cells did not affect the development of K/BxN serum-transfer arthritis. Our results indicate that autoantibody-induced arthritis requires Syk expression in neutrophils, whereas, contrary to prior assumptions, Syk expression in platelets or mast cells is dispensable for disease development in this model.
Introduction
A number of autoimmune diseases, including rheumatoid arthritis, systemic lupus erythematosus, small vessel vasculitis, or pemphigoid diseases, are characterized by production of autoantibodies against various autoantigens of the mammalian body (1). Those autoantibodies are thought to contribute to the autoimmune disease pathogenesis, either directly by engagement of their target autoantigens (activating or function-blocking autoantibodies), or by triggering an inflammatory reaction and concomitant tissue damage caused by the infiltrating inflammatory cells.
The K/BxN serum-transfer arthritis is one of the most widely used mouse model of autoantibody-induced tissue damage. This model is initiated by systemic injection of serum from so-called K/BxN mice in which the expression of a specific T-cell-receptor transgene on an autoimmunity-prone genetic background leads to the generation of high titers of autoantibodies against the ubiquitously expressed glucose 6-phosphate isomerase enzyme (2–5). Transferring those autoantibodies with the K/BxN serum to naive animals triggers robust inflammation of the distal joints and of other tissues. K/BxN serum-transfer arthritis is triggered by immune complex (IC) deposition and concomitant activation of Fcγ-receptors (5). A number of hematopoietic lineages are thought to be involved in the development of K/BxN serum-transfer arthritis. The role of neutrophils is indicated by the fact that antibody-mediated depletion (6) or genetic deletion (7, 8) of neutrophils prevents arthritis development in this model. Arthritis development was also reduced in mast cell-deficient KitW/W-v mice (9) suggesting an important role of mast cells. In addition, platelets were proposed to be required for the development of K/BxN serum-transfer arthritis by releasing platelet-derived microparticles upon collagen-induced activation in the synovial tissue (10).
Syk is a nonreceptor tyrosine kinase primarily expressed in cells of the hematopoietic lineage (11). It mediates signaling by a number of cell surface receptors including B-cell-receptors (12, 13), Fcγ- and Fcε-receptors (14–18), β2 and β3 integrins (19–21), C-type lectins (11, 22), and other receptors coupled to immunoreceptor tyrosine-based activation motifs (ITAMs) (11, 23). Given its role in various hematopoietic lineages and signaling downstream of diverse cell surface receptors, Syk is indispensable for a number of in vivo processes including B-cell development (12, 13), various inflammatory disease processes (17, 24, 25), antifungal immunity (26), or lymph vessel development (27). Based on its central role in the immune system, Syk has been proposed as a therapeutic target in various autoimmune and inflammatory diseases (11, 28).
We have previously shown that Syk is critically involved in arthritis development in the autoantibody-induced K/BxN serum transfer model (25). Our additional studies indicated that Syk is involved in a pathway downstream of Fc-receptors and Src-family kinases (29) and activates further downstream processes through PLCγ2 (30) and CARD9 (31). However, it is at present incompletely understood in which lineage(s) Syk needs to be expressed for arthritis development in this model. Bone marrow chimeric experiments suggested the role for Syk in one or more hematopoietic lineages (25). Several lines of evidence suggest an important role for Syk in neutrophils (19, 31, 32). An important role for GpVI, an ITAM-coupled collagen receptor on platelets, for the development of K/BxN serum-transfer arthritis (10) suggested a role for Syk in platelets for disease development in this model (33). Finally, the proposed role of mast cells (9, 34) and the critical role for Syk in mast cell activation (14, 18) raised the possibility that Syk expression in mast cells contributes to development of K/BxN serum-transfer arthritis.
The above studies prompted us to perform lineage-specific deletion of Syk from neutrophils, platelets, and mast cells, and to test the effect of those mutations on the development of autoantibody-induced arthritis in the K/BxN serum-transfer model. Our results indicate an important role for Syk expression in neutrophils whereas, contrary to our expectations, Syk expression in platelets or mast cells appears to be dispensable for arthritis development in this model.
Materials and Methods
Animals
Mice carrying a deleted Syk allele (Syktm1Tyb, referred to as Syk−) (12) were kept in heterozygous form and used to obtain Syk−/− and control fetuses for fetal liver transplantation (19, 25). Lineage-specific deletion of Syk was achieved by crossing MRP8-Cre (35), PF4-Cre (36), or Mcpt5-Cre transgenic mice (37) with animals carrying a floxed Syk allele (Syktm1.2Tara, referred to as Sykflox) (38) to obtain MRP8-Cre+Sykflox/flox, PF4-Cre+Sykflox/flox, and Mcpt5-Cre+Sykflox/flox mice, referred to as SykΔPMN, SykΔPLT, and SykΔMC animals, respectively. Mice carrying the KRN T-cell-receptor transgene (2) were maintained in heterozygous form by mating with C57BL/6 mice. All transgenic mice were backcrossed to the C57BL/6 genetic background for at least six generations. Genotyping was performed by allele-specific PCR.
Wild type (WT) control C57BL/6 mice were purchased from Charles River or the Hungarian National Institute of Oncology (Budapest, Hungary). NOD mice, as well as a congenic strain carrying the CD45.1 allele on the C57BL/6 genetic background (B6.SJL-Ptprca) were purchased from the Jackson Laboratory.
Mice were kept in individually sterile ventilated cages (Tecniplast) in a conventional facility. All animal experiments were approved by the Animal Experimentation Review Board of the Semmelweis University.
Bone marrow chimeras were generated by intravenous injection of unfractionated bone marrow or fetal liver cells into recipients carrying the CD45.1 allele on the C57BL/6 genetic background, which were lethally irradiated before by 11 Gy from a 137Cs source using a Gamma-Service Medical (Leipzig, Germany) D1 irradiator. 4 weeks after transplantation, peripheral blood samples were stained for Ly6G and CD45.2 (Clones 1A8 and 104, respectively; both from BD Biosciences) and analyzed by a BD Biosciences FACSCalibur flow cytometer as previously described (see Figure S1A in Supplementary Material) (29).
K/BxN Serum-Transfer Arthritis
Mice carrying the KRN T-cell receptor transgene on the C57BL/6 genetic background were mated with NOD mice to obtain transgene-positive (arthritic) K/BxN and transgene-negative (non-arthritic) BxN mice (2, 30). The presence of the transgene was determined by allele-specific PCR and confirmed by phenotypic assessment. Blood was taken by retroorbital bleeding and sera from arthritic and control mice were pooled separately.
Arthritis was induced by a single intraperitoneal injection of 300 μl K/BxN (arthritic) or BxN (control) serum into intact mice or bone marrow chimeras, followed by daily assessment of arthritis severity for 2 weeks as described (30, 31, 39). Visible clinical signs were scored on a 0–10 scale by two investigators blinded for the origin and treatment of the mice. Ankle thickness was measured by a spring-loaded caliper (Kroeplin).
Isolation and Activation of Neutrophils, Platelets, and Mast Cells
Mouse neutrophils were isolated from the bone marrow of the femurs and tibias of intact mice or chimeras by hypotonic lysis followed by Percoll (GE Healthcare) gradient centrifugation using sterile and endotoxin-free reagents as described (18, 31, 39). Cells were kept at room temperature in Ca2+- and Mg2+-free medium until use and prewarmed to 37°C prior to activation. Neutrophil assays were performed at 37°C in HBSS supplemented with 20 mM HEPES, pH 7.4. Adhesion-dependent superoxide release by neutrophils was followed by a cytochrome c reduction test from 100 μl aliquots of 4 × 106/ml cells plated on fibrinogen (Calbiochem) coated surfaces in the presence of 50 ng/ml murine TNF-α (PeproTech) as described (39).
Platelets were isolated from peripheral blood by mild centrifugation in the presence of heparin. For an in vitro aggregation assay (40), platelets were divided into two groups, one labeled with an anti-CD9-PE (Clone EM-04; Abcam) and the other one with an anti-CD9-APC (Clone eBioKMC8; eBioscience) antibody. The two differently labeled groups were mixed in equal volumes and were activated by 50 ng/ml Convulxin (Enzo Life Sciences) at 37°C while shaking at 700 rpm for 5 min. The reaction was stopped by BD FACS Lysing Solution (BD Biosciences). The samples were analyzed by flow cytometry, where platelets were identified according to their forward and side scatter characteristics. Aggregation was determined as the percentage of CD9-PE/CD9-APC double positive events (40).
Mast cells were cultured from the bone marrow in the presence of 5 ng/ml murine IL-3 and 20 ng/ml stem cell factor (both from PeproTech). The purity of the cultures was tested by an anti-FcεR antibody (Clone MAR-1; eBioscience) by flow cytometry. For in vitro activation, mast cells were first incubated with an anti-dinitrophenyl (DNP) IgE antibody (Clone SPE-7) at a final concentration of 0.5 μg/ml overnight at 37°C on fetal bovine serum (FBS)-coated plates, followed by the crosslinking of Fcε receptors by the addition of 100 ng/ml DNP-human serum albumin to the cell suspensions (both reagents from Sigma-Aldrich). After 30 min, the cells were washed and mast cells were kept in Dulbecco’s Modified Eagle’s Medium (DMEM; Sigma-Aldrich) overnight at 37°C on FBS-coated plates. The release of the inflammatory mediator MIP-1α was tested from the cell-free supernatants by a commercial ELISA kit (R&D Systems) according to the manufacturer’s instructions. The absence of Syk did not have a major effect on neutrophil, platelet, or mast cell development and numbers (data not shown).
Biochemical Studies
For analysis of protein contents, neutrophils, platelets, and mast cells were lysed in 100 mM NaCl, 30 mM Na-HEPES (pH 7.4), 20 mM NaF, 1 mM Na-EGTA, 1% Triton X-100, 1 mM benzamidine, freshly supplemented with 0.1 U/ml Aprotinin, 1:100 Mammalian Protease Inhibitor Cocktail, 1:100 Phosphatase Inhibitor Cocktail 2, 1 mM PMSF, and 1 mM Na3VO4 (all from Sigma-Aldrich). After removal of insoluble material, lysates were boiled in sample buffer. Whole cell lysates were run on SDS-PAGE and immunoblotted using antibodies against Syk (Clone N19; Santa Cruz) or β-actin (Clone AC-74; Sigma-Aldrich) followed by peroxidase-labeled secondary antibodies (GE Healthcare). The signal was then developed using the ECL system (GE Healthcare) and exposed to X-ray film.
Presentation of the Data and Statistical Analysis
Experiments were performed the indicated number of times. Quantitative graphs and kinetic curves show mean and SEM from all independent in vitro experiments or from all individual mice from the indicated number of experiments. Statistical analyses were carried out by the STATISTICA software using two-way (factorial) ANOVA, with treatment and genotype being the two independent variables. In case of kinetic assays, area under the curve was used for statistical analysis. P values below 0.05 were considered statistically significant.
Results
K/BxN Serum-Transfer Arthritis in Syk−/− Bone Marrow Chimeras
To test the role of Syk within hematopoietic lineage cells, we generated bone marrow chimeric mice by transplanting Syk−/− or WT control fetal liver cells into lethally irradiated recipients. As shown in Figure 1A, injection of arthritogenic K/BxN serum into WT control chimeras triggered robust inflammation of the ankle joints whereas no such response could be observed in Syk−/− bone marrow chimeras which carry Syk-deficient hematopoietic tissues. Quantitative kinetic analysis of the clinical scoring of arthritis (Figure 1B) or the ankle thickness (Figure 1C) revealed that Syk−/− bone marrow chimeras were completely protected from arthritis development in this model (p = 3 × 10−6 and p = 1.3 × 10−3, respectively). Those results confirmed our prior studies showing critical role for Syk in the hematopoietic compartment in K/BxN serum-transfer arthritis (25).
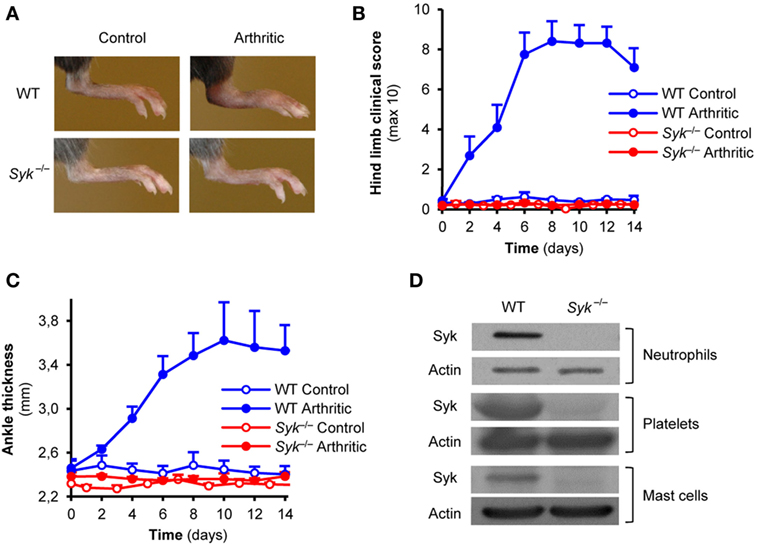
Figure 1. Autoantibody-induced arthritis in Syk-deficient bone marrow chimeras. Wild type (WT) and Syk−/− bone marrow chimeras were injected with BxN (Control) or K/BxN (Arthritic) serum intraperitonally on day 0. Arthritis development was followed by photographing on day 7 (A), clinical scoring of the hind limbs (B), and ankle thickness measurement (C). Panel (D) shows the absence of the Syk tyrosine kinase from whole cell lysates of Syk−/− neutrophils, platelets, and mast cells. Photos are representative of, and quantitative data show mean and SEM from, four control and four to five arthritic serum-treated individual mice per group from two independent experiments. Western blot images are representative of two to three independent experiments. See the text for actual p values.
Syk Is Expressed in Neutrophils, Platelets, and Mast Cells
Prior studies suggested a role for neutrophils, platelets, and mast cells in K/BxN serum-transfer arthritis (6–10, 34), as well as the functional role for Syk in those cells (14, 16, 18–20). To confirm the presence of Syk in those lineages and its deletion from Syk−/− cells, we tested the expression of Syk in primary neutrophils and platelets, or bone marrow-derived mast cells, from WT and Syk−/− bone marrow chimeras. As shown in Figure 1D, Syk was present in all three cell types derived from WT but not Syk−/− bone marrow chimeras (see the entire blots in Figures S1B–D in Supplementary Material).
Efficacy and Specificity of Syk Deletion From Neutrophils, Platelets, and Mast Cells
To test the role of Syk in a linage-specific manner, we turned to Cre-lox-mediated lineage-specific conditional deletion of Syk. To this end, we generated mice carrying the Sykflox/flox mutation along with a neutrophil-specific MRP8-Cre (SykΔPMN), the platelet-specific PF4-Cre (SykΔPLT), or the mast cell-specific Mcpt5-Cre (SykΔMC) transgenes. We then isolated neutrophils or platelets, and cultured bone marrow-derived mast cells, from those animals. As shown in Figure 2A, Syk expression was strongly reduced in SykΔPMN, but was not affected in SykΔPLT or SykΔMC neutrophils. Similarly, Syk was absent from SykΔPLT but not from SykΔPMN or SykΔMC platelets (Figure 2B). Finally, Syk expression was abrogated in SykΔMC but not in SykΔPMN or SykΔPLT mast cells (Figure 2C; see the entire blots in Figure S2 in Supplementary Material). Those results confirm both the efficacy and the specificity of the SykΔPMN, SykΔPLT, and SykΔMC mutations.
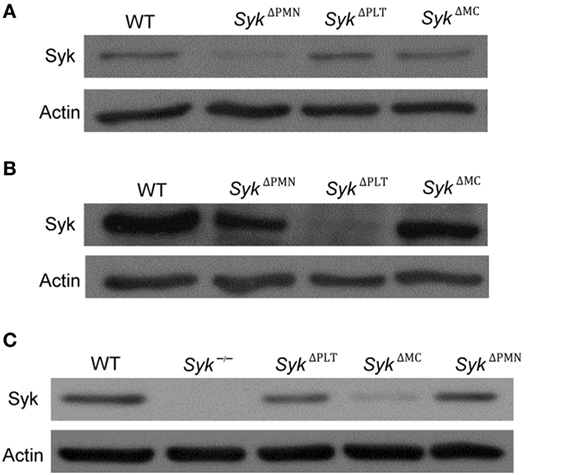
Figure 2. The efficacy and specificity of lineage-specific Syk deletion from different cell types. The efficacy and specificity of lineage-specific deletion was tested by immunoblotting of whole cell lysates of neutrophils (A), platelets (B), and mast cells (C) derived from wild type (WT), SykΔPMN, SykΔPLT, or SykΔMC mice or from Syk−/− bone marrow chimeras. Blots are representative of three independent experiments.
Lineage-Specific Deletion of Syk Abrogates Functional Responses of Neutrophils, Platelets, and Mast Cells
To test the functional efficacy of lineage-specific Syk deletion, we also tested supposedly Syk-dependent functional responses of neutrophils, platelets, and mast cells. Superoxide release of neutrophils plated on a fibrinogen surface in the presence of a soluble TNF stimulus is mediated by β2 integrins in a supposedly Syk-dependent manner (19). As shown in Figure 3A, this response was nearly completely blocked in neutrophils from SykΔPMN animals (p = 0.02). Convulxin is a snake venom toxin activating the Fc-receptor-related collagen receptor GpVI on platelets in a Syk-dependent manner (40, 41). As shown in Figure 3B, convulxin induced aggregation of WT but not SykΔPLT platelets (p = 0.03). Crosslinking of IgE bound to the surface of mast cells triggers release of various proinflammatory mediators through Fcε-receptors in a Syk-dependent manner (14). As shown in Figure 3C, MIP-1α release induced by IgE crosslinking of mast cells was abrogated by the SykΔMC mutation (p = 1.3 × 10−4). Those results indicate that lineage-specific deletion of Syk from neutrophils, platelets, or mast cells leads to the expected functional consequences in those cells.
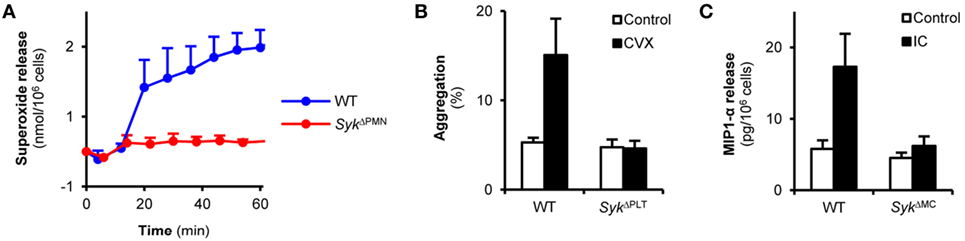
Figure 3. Syk is indispensable for immunoreceptor tyrosine-based activation motif-mediated in vitro cellular responses of neutrophils, platelets, and mast cells. (A) Wild type (WT) or SykΔPMN neutrophils were plated on fibrinogen-coated surfaces in the presence of TNF-α and their superoxide release was measured by a cytochrome c reduction test. Control data points were subtracted. (B) WT or SykΔPLT platelets were isolated from peripheral blood, labeled by two different fluorochrome-conjugated CD9 antibodies and were activated by convulxin (CVX) for 5 min. Aggregation was measured as the percentage of CD9-PE/CD9-APC double positive events. (C) WT or SykΔMC bone marrow-derived mast cells were incubated with anti-DNP IgE antibodies followed by an Fcε receptor crosslinking step with DNP-HSA. MIP-1α levels were determined from the cell-free supernatant by an ELISA assay. Kinetic curves and graphs represent mean and SEM from three (A,C) or six (B) samples from three (A) or two (B,C) independent experiments. See the text for actual p values. DNP, dinitrophenyl; HSA, human serum albumin, IC, immune complex.
Neutrophil-Specific Deletion of Syk Abrogates Autoantibody-Induced Arthritis
We next tested the consequence of neutrophil-specific deletion of Syk on the development of K/BxN serum-transfer arthritis. As shown in Figure 4A, arthritogenic K/BxN serum triggered visible arthritis development in WT animals. However, no signs of arthritis could be observed in SykΔPMN animals (Figure 4A). Quantitative kinetic analysis revealed that SykΔPMN mice were nearly completely protected from development of clinical signs of arthritis (Figure 4B; p = 1.5 × 10−5) and arthritis-induced ankle swelling (Figure 4C; p = 1.7 × 10−3). Similar results could be observed when testing a larger cohort of bone marrow chimeras generated by transplanting WT or SykΔPMN bone marrow cells into lethally irradiated WT recipients (Figures 4D,E; p = 6.2 × 10−7 and p = 3.2 × 10−6, respectively). Those results indicate a critical role for Syk expression within neutrophils for the development of autoantibody-induced arthritis in vivo.
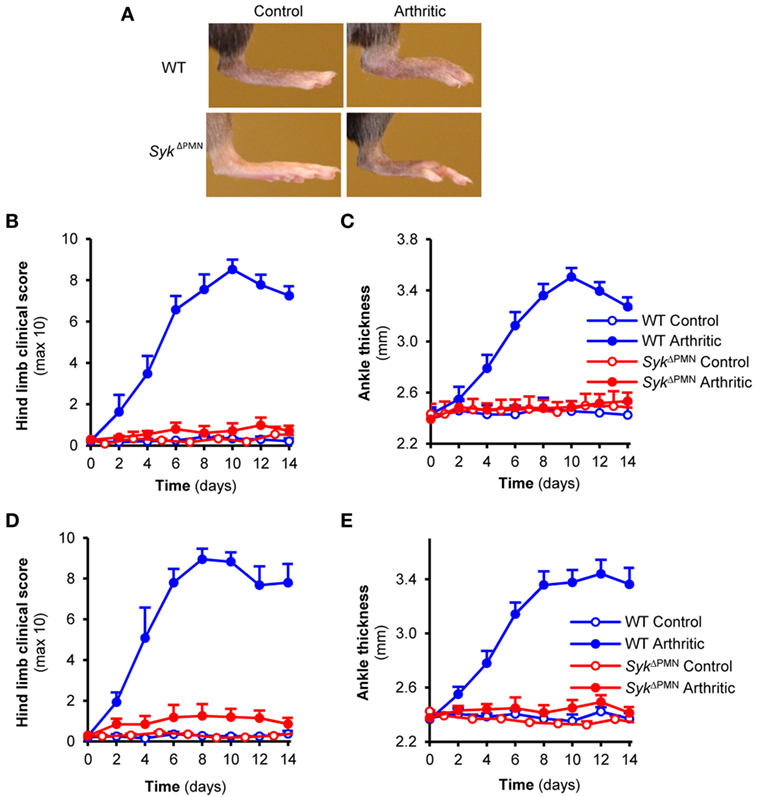
Figure 4. Neutrophil-specific Syk deletion attenuates experimental arthritis. Wild type (WT) and SykΔPMN intact animals (A–C) or bone marrow chimeras (D,E) were injected with BxN (Control) or K/BxN (Arthritic) serum intraperitonally on day 0. Arthritis development was followed by photographing (A), clinical scoring of the hind limbs (B,D), and ankle thickness measurement (C,E). Quantitative data show mean and SEM from three control and five to six arthritic serum-treated individual mice per group from three independent experiments (B,C) or from five control and five to seven arthritic serum-treated mice per group from three independent experiments (D,E). See the text for actual p values.
Normal Arthritis Development Upon Platelet-Specific Deletion of Syk
Boilard et al. previously showed that genetic deletion of the Syk-coupled GpVI collagen receptor of platelets strongly reduced arthritis development in the K/BxN serum-transfer model (10), suggesting an important role for Syk expression in platelets in this model (33). To test this hypothesis experimentally, we tested K/BxN serum-transfer arthritis in SykΔPLT mice in which Syk was deleted in a platelet-specific manner. Contrary to our expectations, platelet-specific Syk deletion did not affect the development of visual signs of arthritis in our model (Figure 5A). Quantitative kinetic analysis did not reveal any effect of the SykΔPLT mutation on the clinical appearance (Figure 5B; p = 0.51) or on the ankle thickness increase (Figure 5C; p = 0.76) either. Similar results were obtained when using bone marrow chimeras generated by transplanting WT or SykΔPLT bone marrow cells into lethally irradiated WT recipients (Figures 5D,E; p = 0.49 and p = 0.9, respectively). Those results, together with the lack of Syk (Figure 2B) and the defective Syk-dependent functional activation (Figure 3B) of SykΔPLT platelets indicate that Syk expression in platelets is not required for the development of K/BxN serum-transfer arthritis.
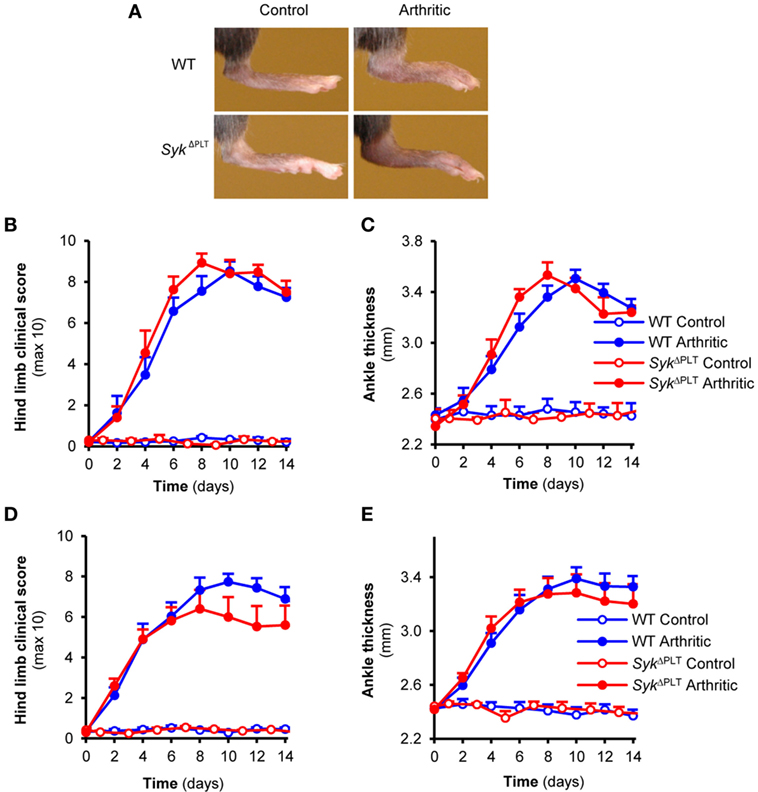
Figure 5. Platelet-specific Syk deletion has no effect on autoantibody-induced arthritis. Wild type (WT) and SykΔPLT intact animals (A–C) or bone marrow chimeras (D,E) were injected with BxN (Control) or K/BxN (Arthritic) serum intraperitonally on day 0. Arthritis development was followed by photographing (A), clinical scoring of the hind limbs (B,D), and ankle thickness measurement (C,E). Quantitative data show mean and SEM from three control and five arthritic serum-treated individual mice per group from three independent experiments (B,C) or from six control and eight to nine arthritic serum-treated mice per group from three independent experiments (D,E). See the text for actual p values.
Mast Cell-Specific Syk Deletion Does Not Affect Autoantibody-Induced Arthritis
Mast cells are one of the major targets of Syk function (14, 18) and they have also been proposed to participate in the development of K/BxN serum-transfer arthritis (9, 34). Therefore, we hypothesized that Syk expression in mast cells may be required for arthritis development in this model. To this end, we tested the development of K/BxN serum-transfer arthritis in SykΔMC mice. As shown in Figure 6A, the SykΔMC mutation did not affect the development of visible signs of arthritis in our model. Quantitative kinetic analysis did not reveal any inhibition of arthritis development either when scoring clinical signs of arthritis (Figure 6B; p = 0.38) or when measuring arthritis-induced increase of ankle thickness (Figure 6C; p = 0.37). By contrast, there was even a tendency of earlier arthritis development in the SykΔMC animals (Figures 6B,C), raising the possibility of a negative role of Syk expressed in mast cells. Because of the radioresistance of mast cells, no bone marrow chimeras have been generated using SykΔMC mice. The lack of inhibition of arthritis in SykΔMC animals, together with the dramatic reduction of Syk expression (Figure 2C) and Syk-mediated functional responses (Figure 3C) in SykΔMC mast cells indicates that Syk expression within mast cells is dispensable for arthritis development in the K/BxN serum-transfer model.
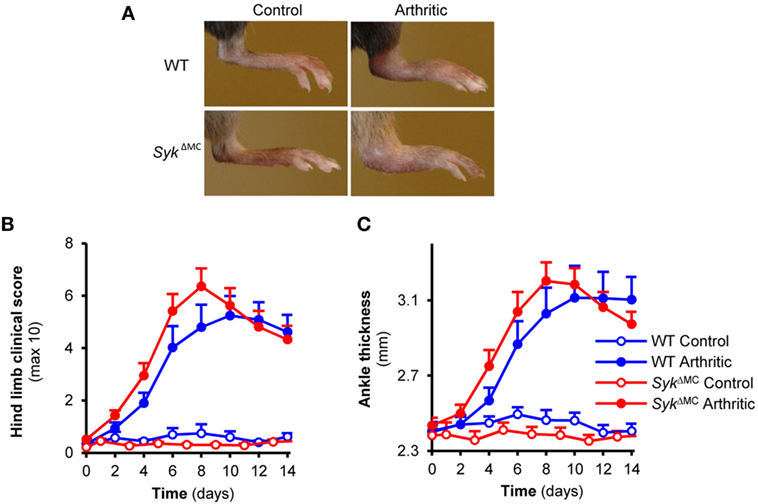
Figure 6. Mast cell-specific Syk deletion does not affect the effector phase of experimental arthritis. Wild type (WT) and SykΔMC animals were injected with BxN (Control) or K/BxN (Arthritic) serum intraperitonally on day 0. Arthritis development was followed by photographing (A), clinical scoring of the hind limbs (B), and ankle thickness measurement (C). Quantitative data show mean and SEM from seven to nine control and twelve to thirteen arthritic serum-treated individual mice per group from four independent experiments. See the text for actual p values.
Discussion
The Syk tyrosine kinase is critically involved in various inflammatory disease processes including the development of autoantibody-induced arthritis and dermatitis models (11, 17, 25). Given the wide expression of Syk in practically all hematopoietic lineages (11), understanding Syk function in a lineage-specific manner is of particular importance. Our results presented in this work indicate that of the three most prominent Syk-expressing lineages supposedly involved in the development of autoantibody-induced arthritis, Syk expression in neutrophils is critical, whereas that in platelets or mast cells is dispensable, for the development of K/BxN serum-transfer arthritis.
We and others have shown that Syk plays a critical role in various functional responses of neutrophils (16, 17, 19, 42, 43) without affecting neutrophil development (17, 19). Neutrophils have also been shown to be critical for the development of autoantibody-induced arthritis (6–8), likely at least in part through IgG IC-mediated activation of Fcγ-receptors expressed on the neutrophil cell surface (29), as well as by yet incompletely understood neutrophil-mediated initial vascular changes (8). Based on those studies, we hypothesized that Syk expression within neutrophils is critical for autoantibody-induced arthritis development. Our results confirmed that hypothesis, and they were also in line with prior studies from other groups (32) and our own analysis of neutrophil-specific deletion of the CARD9 adapter protein, a supposedly downstream effector of Syk (31). Though it is at present incompletely understood how Syk within neutrophils participates in autoantibody-induced arthritis development, our prior studies showing defective release of proinflammatory mediators by Syk-deficient neutrophils despite normal intrinsic migratory capacity of the cells (17, 19, 31) suggest that Syk, similar to Src-family kinases (29), participates in the amplification of neutrophil recruitment by neutrophil-derived proinflammatory mediators (44).
In contrast to our neutrophil-specific deletion studies, our platelet-specific deletion experiments did not support our hypothesis based on literature data. Though Syk is not required for platelet development (20), it plays a critical role in various platelet functions (11) including αIIbβ3 integrin-dependent platelet spreading (20), responses mediated by the hemITAM-coupled C-type lectin CLEC-2 (45), as well as signaling downstream of GpVI, an ITAM-coupled collagen-receptor of platelets (40, 41). GpVI is closely related to Fcα-receptors and it is directly associated with, and supposedly signals through, the ITAM-containing Fc-receptor γ-chain (FcRγ) (40, 41, 46–49). Platelets and, specifically, GpVI has been shown to play a critical role in the development of K/BxN serum-transfer arthritis (10), suggesting that Syk expression downstream of platelet GpVI is critically involved in arthritis development in this model (33). Our results of normal arthritis development upon platelet-specific deletion of Syk (Figure 5) despite practically complete lack of Syk from platelets (Figure 2) and completely defective GpVI-mediated in vitro platelet function (Figure 3) argues against that hypothesis. There are several possible explanations for those findings. Though GpVI is associated with the ITAM-containing FcRγ adapter, it may be able to bypass the ITAM-Syk pathway under certain conditions, using FcRγ as a chaperone required for cell surface expression but not as an ITAM-mediated signaling adaptor. Platelets have also been proposed to interact with fibroblast-like synoviocytes in a COX-1-dependent manner which is independent of platelet GpVI or FcRγ expression (50). This pathway may be able to compensate for the defective GpVI–FcRγ–Syk pathway upon platelet-specific Syk deletion. We also cannot exclude the possibility that GpVI needs to be expressed in a non-platelet lineage to support autoantibody-induced arthritis in mice. Finally, technical details such as a role for the small remaining Syk expression after Cre-mediated Syk deletion, or different experimental conditions may also account for the different conclusions drawn from our study and from those proposing a critical role for the platelet GpVI–FcRγ–Syk pathway in autoantibody-induced arthritis (10, 33). It should also be mentioned that our study focused on visible signs of arthritis and therefore we cannot exclude the possibility that Syk expression in platelets modulates the inflammation process by a mechanism not clearly visible by macroscopic inspection.
In the third part of our study, we tested the role of Syk in mast cells during autoantibody-induced arthritis. Syk has been shown to play a critical role in mast cell function without affecting mast cell survival (14, 18) and mast cells were proposed to be important players in autoantibody-induced arthritis development (9). Therefore, we hypothesized that Syk expression in mast cells may play a role in the development of K/BxN serum-transfer arthritis. Our results showing normal arthritis development in that model upon mast cell-specific Syk deletion (Figure 6) despite strongly reduced Syk expression (Figure 2) and defective Syk-mediated functional responses (Figure 3) in mast cells argue against that possibility. There are several possible explanations for those findings. Since the mechanism of how mast cells contribute to IgG autoantibody-induced disease pathogenesis is incompletely understood, it is possible that mast cells use a Syk-independent signal transduction pathway during K/BxN serum-transfer arthritis (e.g., when mast cells are not directly activated by the autoantibody-containing ICs, but rather indirectly through Syk-independent chemokine, cytokine, or PRR pathways). It should also be mentioned that follow-up studies have questioned the critical role of mast cells in autoantibody-induced arthritis development (51, 52), pointing to difficulties of the interpretation of data obtained with different mast cell-deficient mouse strains. Indeed, our limited preliminary studies also suggested that the role of mast cells is highly dependent on the experimental conditions used for triggering autoantibody-induced arthritis in mice (Z. Jakus and A. M., unpublished observations). Finally, given that our experiments focused on visible signs of inflammation, we cannot exclude the possibility that Syk expression in mast cells may modulate arthritis development or the overall inflammation process in a manner not clearly visible by macroscopic assessment.
Besides neutrophils, platelets, and mast cells, Syk is also expressed in other lineages possibly involved in arthritis development. B-cells are one of the most prominent lineages requiring Syk function (12, 13). However, it is unlikely that Syk expressed in B-cells contributes to K/BxN serum-transfer arthritis since that model mimics the post-immunization effector phase of autoimmune arthritis and it develops normally even in the absence of B-cells in μMT-deficient or Rag-deficient mice (3). Macrophages have been proposed to be important players in the development of K/BxN serum-transfer arthritis (53). Unfortunately, currently available techniques do not allow the proper analysis of the in vivo relevance of Syk expression within macrophages because of the limited spectrum/specificity of the available macrophage-specific Cre-expressing mouse strains (54). We have previously shown that Syk is critically involved in osteoclast development and function (23). Though understanding the role of Syk in arthritis-induced bone erosions would be of clear importance, this question is beyond the scope of the present study focusing on the inflammatory aspect of autoantibody-induced disease processes.
Taken together, our results provide understanding of the role of Syk in autoantibody-induced arthritis at the cellular lineage level. Our findings indicate a critical role for Syk expression in neutrophils, but refute prior assumptions for the role of Syk in platelets and argue against a role for Syk expression in mast cells. Those results will strongly contribute to the understanding of the pathomechanism of autoantibody-mediated disease processes at the cellular and molecular level.
Ethics Statement
All animal experiments were approved by the Animal Experimentation Review Board of the Semmelweis University.
Author Contributions
TN and AM conceived the study, designed the experiments, and wrote the manuscript. TN, KF, KS, OV, and LK-P performed the experiments. TN, KF, and AM analyzed and interpreted the data. AM supervised the project.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer KB and handling editor declared their shared affiliation.
Acknowledgments
We thank Edina Simon for expert technical assistance; Gábor Bánhegyi for access to equipment; Diane Mathis, Christophe Benoist, Victor Tybulewicz, Emmanuelle Passegue, Axel Roers, and Alexander Tarakhovsky for transgenic animals. This work was supported by the Lendület program of the Hungarian Academy of Sciences (LP2013-66/2013 to AM), the Hungarian National Research, Development and Innovation Office (NVKP_16-2016-1-0039 to AM), the Hungarian Ministry of National Economy (VEKOP-2.3.2-16-2016-00002 to AM), and the János Bolyai Research Scholarships of the Hungarian Academy of Sciences (to TN and KF). AM was a recipient of a Wellcome Trust International Senior Research Fellowship (Grant No. 087782).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fimmu.2018.00555/full#supplementary-material.
Figure S1. Flow cytometric analysis of bone marrow chimeras and Western blot images with more details. (A) Flow cytometric analysis of CD45.2-positive donor-derived neutrophils in the peripheral blood of wild type (WT) and SykΔPMN bone marrow chimeras 4 weeks after bone marrow transplantation. (B–D) Detailed Western blot images showing the expression of Syk in neutrophil (B), platelet (C), or mast cell (D) lysates from Figure 1D.
Figure S2. Detailed Western blot images showing the efficacy and specificity of lineage-specific Syk deletion in whole cell lysates of neutrophils (A), platelets (B), and mast cells (C) from Figure 2.
References
1. Suurmond J, Diamond B. Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest (2015) 125:2194–202. doi:10.1172/JCI78084
2. Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell (1996) 87:811–22. doi:10.1016/S0092-8674(00)81989-3
3. Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity (1999) 10:451–61. doi:10.1016/S1074-7613(00)80045-X
4. Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science (1999) 286:1732–5. doi:10.1126/science.286.5445.1732
5. Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, et al. Arthritis critically dependent on innate immune system players. Immunity (2002) 16:157–68. doi:10.1016/S1074-7613(02)00275-3
6. Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol (2001) 167:1601–8. doi:10.4049/jimmunol.167.3.1601
7. Jonsson H, Allen P, Peng SL. Inflammatory arthritis requires Foxo3a to prevent Fas ligand-induced neutrophil apoptosis. Nat Med (2005) 11:666–71. doi:10.1038/nm1248
8. Binstadt BA, Patel PR, Alencar H, Nigrovic PA, Lee DM, Mahmood U, et al. Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat Immunol (2006) 7:284–92. doi:10.1038/ni1306
9. Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science (2002) 297:1689–92. doi:10.1126/science.1073176
10. Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science (2010) 327:580–3. doi:10.1126/science.1181928
11. Mócsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol (2010) 10:387–402. doi:10.1038/nri2765
12. Turner M, Mee PJ, Costello PS, Williams O, Price AA, Duddy LP, et al. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature (1995) 378:298–302. doi:10.1038/378298a0
13. Cheng AM, Rowley B, Pao W, Hayday A, Bolen JB, Pawson T. Syk tyrosine kinase required for mouse viability and B-cell development. Nature (1995) 378:303–6. doi:10.1038/378303a0
14. Costello PS, Turner M, Walters AE, Cunningham CN, Bauer PH, Downward J, et al. Critical role for the tyrosine kinase Syk in signalling through the high affinity IgE receptor of mast cells. Oncogene (1996) 13:2595–605.
15. Crowley MT, Costello PS, Fitzer-Attas CJ, Turner M, Meng F, Lowell C, et al. A critical role for Syk in signal transduction and phagocytosis mediated by Fcγ receptors on macrophages. J Exp Med (1997) 186:1027–39. doi:10.1084/jem.186.7.1027
16. Kiefer F, Brumell J, Al-Alawi N, Latour S, Cheng A, Veillette A, et al. The Syk protein tyrosine kinase is essential for Fcγ receptor signaling in macrophages and neutrophils. Mol Cell Biol (1998) 18:4209–20. doi:10.1128/MCB.18.7.4209
17. Németh T, Virtic O, Sitaru C, Mócsai A. The Syk tyrosine kinase is required for skin inflammation in an in vivo mouse model of epidermolysis bullosa acquisita. J Invest Dermatol (2017) 137:2131–9. doi:10.1016/j.jid.2017.05.017
18. Mócsai A, Zhang H, Jakus Z, Kitaura J, Kawakami T, Lowell CA. G-protein-coupled receptor signaling in Syk-deficient neutrophils and mast cells. Blood (2003) 101:4155–63. doi:10.1182/blood-2002-07-2346
19. Mócsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity (2002) 16:547–58. doi:10.1016/S1074-7613(02)00303-5
20. Obergfell A, Eto K, Mócsai A, Buensuceso C, Moores SL, Brugge JS, et al. Coordinate interactions of Csk, Src, and Syk kinases with αIIbβ3 initiate integrin signaling to the cytoskeleton. J Cell Biol (2002) 157:265–75. doi:10.1083/jcb.200112113
21. Jakus Z, Fodor S, Abram CL, Lowell CA, Mócsai A. Immunoreceptor-like signaling by β2 and β3 integrins. Trends Cell Biol (2007) 17:493–501. doi:10.1016/j.tcb.2007.09.001
22. Werninghaus K, Babiak A, Gross O, Holscher C, Dietrich H, Agger EM, et al. Adjuvanticity of a synthetic cord factor analogue for subunit Mycobacterium tuberculosis vaccination requires FcRγ-Syk-Card9-dependent innate immune activation. J Exp Med (2009) 206:89–97. doi:10.1084/jem.20081445
23. Mócsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, Spusta SC, et al. The immunomodulatory adapter proteins DAP12 and Fc receptor γ-chain (FcRγ) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci U S A (2004) 101:6158–63. doi:10.1073/pnas.0401602101
24. Hirahashi J, Mekala D, Van Ziffle J, Xiao L, Saffaripour S, Wagner DD, et al. Mac-1 signaling via Src-family and Syk kinases results in elastase-dependent thrombohemorrhagic vasculopathy. Immunity (2006) 25:271–83. doi:10.1016/j.immuni.2006.05.014
25. Jakus Z, Simon E, Balázs B, Mócsai A. Genetic deficiency of Syk protects mice from autoantibody-induced arthritis. Arthritis Rheum (2010) 62:1899–910. doi:10.1002/art.27438
26. Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature (2009) 459:433–6. doi:10.1038/nature07965
27. Abtahian F, Guerriero A, Sebzda E, Lu MM, Zhou R, Mócsai A, et al. Regulation of blood and lymphatic vascular separation by signaling proteins SLP-76 and Syk. Science (2003) 299:247–51. doi:10.1126/science.1079477
28. Geahlen RL. Getting Syk: spleen tyrosine kinase as a therapeutic target. Trends Pharmacol Sci (2014) 35:414–22. doi:10.1016/j.tips.2014.05.007
29. Kovács M, Németh T, Jakus Z, Sitaru C, Simon E, Futosi K, et al. The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. J Exp Med (2014) 211:1993–2011. doi:10.1084/jem.20132496
30. Jakus Z, Simon E, Frommhold D, Sperandio M, Mócsai A. Critical role of phospholipase Cγ2 in integrin and Fc receptor-mediated neutrophil functions and the effector phase of autoimmune arthritis. J Exp Med (2009) 206:577–93. doi:10.1084/jem.20081859
31. Németh T, Futosi K, Sitaru C, Ruland J, Mócsai A. Neutrophil-specific deletion of the CARD9 gene expression regulator suppresses autoantibody-induced inflammation in vivo. Nat Commun (2016) 7:11004. doi:10.1038/ncomms11004
32. Elliott ER, Van Ziffle JA, Scapini P, Sullivan BM, Locksley RM, Lowell CA. Deletion of Syk in neutrophils prevents immune complex arthritis. J Immunol (2011) 187:4319–30. doi:10.4049/jimmunol.1100341
33. Boilard E, Blanco P, Nigrovic PA. Platelets: active players in the pathogenesis of arthritis and SLE. Nat Rev Rheumatol (2012) 8:534–42. doi:10.1038/nrrheum.2012.118
34. Nigrovic PA, Binstadt BA, Monach PA, Johnsen A, Gurish M, Iwakura Y, et al. Mast cells contribute to initiation of autoantibody-mediated arthritis via IL-1. Proc Natl Acad Sci U S A (2007) 104:2325–30. doi:10.1073/pnas.0610852103
35. Passegue E, Wagner EF, Weissman IL. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell (2004) 119:431–43. doi:10.1016/j.cell.2004.10.010
36. Tiedt R, Schomber T, Hao-Shen H, Skoda RC. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood (2007) 109:1503–6. doi:10.1182/blood-2006-04-020362
37. Scholten J, Hartmann K, Gerbaulet A, Krieg T, Muller W, Testa G, et al. Mast cell-specific Cre/loxP-mediated recombination in vivo. Transgenic Res (2008) 17:307–15. doi:10.1007/s11248-007-9153-4
38. Saijo K, Schmedt C, Su IH, Karasuyama H, Lowell CA, Reth M, et al. Essential role of Src-family protein tyrosine kinases in NF-κB activation during B cell development. Nat Immunol (2003) 4:274–9. doi:10.1038/ni893
39. Németh T, Futosi K, Hably C, Brouns MR, Jakob SM, Kovács M, et al. Neutrophil functions and autoimmune arthritis in the absence of p190RhoGAP: generation and analysis of a novel null mutation in mice. J Immunol (2010) 185:3064–75. doi:10.4049/jimmunol.0904163
40. Meinders M, Hoogenboezem M, Scheenstra MR, De Cuyper IM, Papadopoulos P, Németh T, et al. Repercussion of megakaryocyte-specific Gata1 loss on megakaryopoiesis and the hematopoietic precursor compartment. PLoS One (2016) 11:e0154342. doi:10.1371/journal.pone.0154342
41. Watson SP, Herbert JM, Pollitt AY. GPVI and CLEC-2 in hemostasis and vascular integrity. J Thromb Haemost (2010) 8:1456–67. doi:10.1111/j.1538-7836.2010.03875.x
42. Schymeinsky J, Sindrilaru A, Frommhold D, Sperandio M, Gerstl R, Then C, et al. The Vav binding site of the non-receptor tyrosine kinase Syk at Tyr 348 is critical for β2 integrin (CD11/CD18)-mediated neutrophil migration. Blood (2006) 108:3919–27. doi:10.1182/blood-2005-12-030387
43. Frommhold D, Mannigel I, Schymeinsky J, Mócsai A, Poeschl J, Walzog B, et al. Spleen tyrosine kinase Syk is critical for sustained leukocyte adhesion during inflammation in vivo. BMC Immunol (2007) 8:31. doi:10.1186/1471-2172-8-31
44. Németh T, Mócsai A. Feedback amplification of neutrophil function. Trends Immunol (2016) 37:412–24. doi:10.1016/j.it.2016.04.002
45. Suzuki-Inoue K, Fuller GL, Garcia A, Eble JA, Pohlmann S, Inoue O, et al. A novel Syk-dependent mechanism of platelet activation by the C-type lectin receptor CLEC-2. Blood (2006) 107:542–9. doi:10.1182/blood-2005-05-1994
46. Gibbins JM, Okuma M, Farndale R, Barnes M, Watson SP. Glycoprotein VI is the collagen receptor in platelets which underlies tyrosine phosphorylation of the Fc receptor γ-chain. FEBS Lett (1997) 413:255–9. doi:10.1016/S0014-5793(97)00926-5
47. Tsuji M, Ezumi Y, Arai M, Takayama H. A novel association of Fc receptor γ-chain with glycoprotein VI and their co-expression as a collagen receptor in human platelets. J Biol Chem (1997) 272:23528–31. doi:10.1074/jbc.272.38.23528
48. Clemetson JM, Polgar J, Magnenat E, Wells TN, Clemetson KJ. The platelet collagen receptor glycoprotein VI is a member of the immunoglobulin superfamily closely related to FcαR and the natural killer receptors. J Biol Chem (1999) 274:29019–24. doi:10.1074/jbc.274.41.29019
49. Kasirer-Friede A, Kahn ML, Shattil SJ. Platelet integrins and immunoreceptors. Immunol Rev (2007) 218:247–64. doi:10.1111/j.1600-065X.2007.00532.x
50. Boilard E, Larabee K, Shnayder R, Jacobs K, Farndale RW, Ware J, et al. Platelets participate in synovitis via Cox-1-dependent synthesis of prostacyclin independently of microparticle generation. J Immunol (2011) 186:4361–6. doi:10.4049/jimmunol.1002857
51. Zhou JS, Xing W, Friend DS, Austen KF, Katz HR. Mast cell deficiency in KitW-sh mice does not impair antibody-mediated arthritis. J Exp Med (2007) 204:2797–802. doi:10.1084/jem.20071391
52. Feyerabend TB, Weiser A, Tietz A, Stassen M, Harris N, Kopf M, et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity (2011) 35:832–44. doi:10.1016/j.immuni.2011.09.015
53. Solomon S, Rajasekaran N, Jeisy-Walder E, Snapper SB, Illges H. A crucial role for macrophages in the pathology of K/BxN serum-induced arthritis. Eur J Immunol (2005) 35:3064–73. doi:10.1002/eji.200526167
Keywords: Syk, arthritis, neutrophils, platelets, mast cells
Citation: Németh T, Futosi K, Szilveszter K, Vilinovszki O, Kiss-Pápai L and Mócsai A (2018) Lineage-Specific Analysis of Syk Function in Autoantibody-Induced Arthritis. Front. Immunol. 9:555. doi: 10.3389/fimmu.2018.00555
Received: 15 January 2018; Accepted: 05 March 2018;
Published: 19 March 2018
Edited by:
Ralf J. Ludwig, University of Lübeck, GermanyReviewed by:
Katja Bieber, University of Lübeck, GermanyMarko Radic, University of Tennessee College of Medicine, United States
Aaron James Marshall, University of Manitoba, Canada
Copyright: © 2018 Németh, Futosi, Szilveszter, Vilinovszki, Kiss-Pápai and Mócsai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tamás Németh, nemeth.tamas@med.semmelweis-univ.hu;
Attila Mócsai, mocsai.attila@med.semmelweis-univ.hu