
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 08 March 2018
Sec. Immunological Memory
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00461
This article is part of the Research Topic Orchestration of an Immune Response to Respiratory Pathogens View all 15 articles
 Ian M. Orme*
Ian M. Orme* Marcela I. Henao-Tamayo
Marcela I. Henao-Tamayo
The purpose of vaccination against tuberculosis and other diseases is to establish a heightened state of acquired specific resistance in which the memory immune response is capable of mediating an accelerated and magnified expression of protection to the pathogen when this is encountered at a later time. In the earliest studies in mice infected with Mycobacterium tuberculosis, memory immunity and the cells that express this were definable both in terms of kinetics of emergence, and soon thereafter by the levels of expression of markers including CD44, CD62L, and the chemokine receptor CCR7, allowing the identification of effector memory and central memory T cell subsets. Despite these initial advances in knowledge, more recent information has not revealed more clarity, but instead, has created a morass of complications—complications that, if not resolved, could harm correct vaccine design. Here, we discuss two central issues. The first is that we have always assumed that memory is induced in the same way, and consists of the same T cells, regardless of whether that immunity is generated by BCG vaccination, or by exposure to M. tuberculosis followed by effective chemotherapy. This assumption is almost certainly incorrect. Second, a myriad of additional memory subsets have now been described, such as resident, stem cell-like, tissue specific, among others, but as yet we know nothing about the relative importance of each, or whether if a new vaccine needs to induce all of these, or just some, to be fully effective.
The purpose of vaccination is to establish a long-lived state of immunological memory to a given pathogen which can mediate an accelerated response to that pathogen if it returns (1). That immunological memory exists in some sort of form is long known; Thucydides, in describing the Peloponnesian war in 430 BC wrote that a plague affecting the citizens of Athens never attacked the same man twice. Far more recently, Panum, a Danish doctor, observed that elderly residents of the Faroe Islands exposed to measles in 1781 were immune to a second outbreak 65 years later. However, it was only 50 years or so ago that the work of Gowans began to focus down on a particular white blood cell, the lymphocyte, as the actual mediator of immunity, and only 30 years since the first T cell transfer studies (2) suggested that one component of the host response in mice infected with Mycobacterium tuberculosis exhibited a longer lived phenotype, with subsequent studies (3) showing that it remained present even if the infection was cleared by chemotherapy, indicating a long-lived phenotype not dependent on continued exposure to specific antigen.
These early studies with M. tuberculosis made the reasonable assumption that generation of long-lived immunity was mediated by a discrete T cell, the memory T cell. This assumption, we now know, was wrong, and the field since then began to recognize that there are at least two major subsets of memory cells, distinguishable both in terms of phenotype and tissue distribution. Then, more recently, this has further evolved into evidence for further subsets, as will be discussed below.
A further issue regards the system/model in which one can study these cells in the context of tuberculosis. Chronic disease, which can be studied in mice, generates cells in the lungs that have phenotypic characteristics of memory immunity (4, 5). In turn, BCG vaccination induces memory T cells in relatively similar numbers to chronic disease in terms of memory T cell subsets. Infection with M. tuberculosis followed by clearance with drugs induces a strong memory T cell response, but if these animals are re-challenged, then the two major memory T cell subsets are both triggered to essentially equivalent levels (6). In the latter case, we would expect this immunity to be stable and result in further expansion of memory immunity, but in fact the reverse is true. This further illustrates our minimal understanding of these events.
As our knowledge of memory immunity developed, the concept quickly emerged that memory cells “marginate”—move from their initial sites of sensitization and spread out across the body to provide an early warning system should their specific pathogen reappear (7, 8). Memory B cells become distributed throughout the lymph node system, and T cells have an even wider distribution within lymph nodes and peripheral lymphatic tissues. This redistribution includes a particular emphasis on the two main mucosal tissues in the gut and the respiratory tract. The important findings of Sallusto and her colleagues (9–11) that there were two separate main subsets of memory T cells provide an additional element of overall design to this complex system, based upon a division of labor in which effector memory T cells (TEM) protected the periphery while central memory T cells (TCM) represented a “rapid response team” based in more central tissues such as the spleen and bone marrow.
As this concept of margination developed into the newer TEM/TCM model, it was still unclear to what degree each population remained essentially cessile, or whether despite a favored niche (gut, lung, for example), they continued to have some degree of recirculation properties. This is still very much under investigation today and has led to the potential identification of further subsets of T cells, discussed below.
As noted above, there is good consensus that there are at least two major subsets of memory T cells (9, 11, 12). TEM are found in peripheral sites such as the lungs, gut, and skin, where they represent a “first line of defense,” whereas TCM are found in lymphoid organs such as the spleen and the bone marrow, and are thought to represent the second line should pathogens reach that far. This paradigm has proved to be workable and useful and is further helped by a clear phenotypic difference between the two—TEM are CD44hi CD62Llo CCR7lo while TCM are CD44hi CD62Lhi CCR7hi. Memory T cells in general can express an array of co-stimulatory molecules, including CD27, CD28 [which appears critical (13)], ICOS, 4-1BB, OX40, and CD40L, and various regulatory markers such as PD-1, BTLA, and CTLA-4.TEM are CD44hi CD62Llo, T-betint, CD27+, and KLRG-1neg. They are more responsive to IL-2R signaling, express higher T-bet levels, but lowered Bcl-6 and CXCR5, whereas TCM are the reverse. TEM lack CCR7, and can rapidly produce key cytokines including IFNγ and IL-2.
Our general concept is that infection with a given pathogen generates the clonal expansion of antigen-specific lymphocytes, which differentiate into effector cells. If/when the pathogen is cleared, the response contracts as most of these cells die, but some cells remain viable and become long-lived memory cells (14). If the pathogen then reappears, there is a subsequent transition in which memory cells become secondary effectors, exhibiting kinetics far faster than the emergence of primary response effectors (15). As yet, however, there still is no clear consensus on whether memory cells arise from a small percentage of effector cells, or arise independently, nor is there much known about the fate of re-stimulated memory cells and the secondary effectors some of them then become.
Memory T cells arise after stimulation by common gamma-chain cytokines, which triggers homeostatic expansion of this population. Signals from MHC molecules are required, as is CD70 engagement, as well as production of autocrine IL-2 to prevent clonal contraction; this also depresses potential apoptosis while upregulating expression of the IL-7 receptor (16). In the case of CD8+ TCM, these cells require exposure to IL-7 and IL-15 to survive in a state of interphase and undergo occasional cell division without requiring signals from MHC molecules. This is different to other memory subsets, indicating that homeostatic control of the memory response is heterogeneous (17).
There are currently two main models of memory T cell generation, and which one applies to our models of tuberculosis infection is still currently unknown. In the first model, TM differentiation occurs concomitantly with effector T cell generation right from the offset, when antigen presentation determines early programming of cells that will become memory cells, and which then become dominant after primary effector immunity has contracted. In the second model, TM arise later, possibly during the contraction phase, and either arises independently of effector T cells, or from a longer lived subset of them. There is no doubt that, at this time, the signals that control memory immunity development, maintenance, longevity, and function, are as yet still poorly defined (18), plus it is also probably true to say that we know much more about CD8 memory as opposed to CD4 (19, 20). A current concept is that competition for limited amounts of antigen presented by MHC-II is thought to be a limiting factor in CD4 memory T cell generation, with the model predicting that the more activated the cell becomes, the less likely it will become a TM (suggesting control by receptor signaling strength). If the antigen availability is high enough, T cell priming occurs rapidly in the presence of activated dendritic cell (DC), and this thought to drive effector memory T cell emergence, whereas the generation of central memory cells may be driven by more mature DC. This seems consistent with our knowledge that BCG is only slowly cleared from vaccinated mice, so sufficient antigen potentially remains to generate TEM after initial immunity has contracted. This contrasts with various virus infection models (21), as well as malaria models (22), in which rapid clearance of the infection and rapid contraction of effector immunity favors TCM generation (15, 23–25). At the cellular level, this further correlates with evidence for increased sensitivity of TCR/MHC engagement driving high-affinity TM cells, more efficient TCR triggering, altered CD3 clustering, and increased Zap70 signaling.
In the context of TH1 responses, Harrington and others (26) have provided evidence using cytokine reporter mice, which indicates that memory T cells arise from IFNγ-positive primary effector cells—a more satisfactory explanation than the scenario in which memory cells arise despite minimal reaction to the pathogen.
As discussed above, there is general consensus that TEM populate the periphery as a “first line of defense,” whereas TCM occupy a more centralized distribution in the spleen, bone marrow, and lymph nodes (8, 9, 11, 12, 27–29). Despite this, the overall nomenclature is becoming more complicated, with cells in the lungs described (30–32) as resident T cells (TRM), which may also include memory precursor effector cells—as well as in addition, spleen-associated stem cell-like memory cells (TSCM), which we may have accidentally tripped over in 2005 (33), and since (5), although these tend primarily to primarily located in the spleen and lymph nodes.
The idea of memory T cell margination inevitably evolved into our current concept of resident memory T cells, cells expressing specific receptors/ligands directing them into specific tissues, in which they then become retained (34). The dynamics of these events are still unclear, given that we still do not know the distinction between truly migratory TEM and TCM—coming in and out of lymphoid tissues, or feeding peripheral sites from central reservoirs, versus cells that are truly resident—a confusion that exists in reviews even now (35). However, as parabiosis studies indicate (in virus infection models at least), a long-lived resident/cessile population exists and may be regarded as a separate subset to TEM and TCM, leading to the recent suggestion (30) that CD27 and CD43 staining could be used to better define these. In the context of CD8 cells, expression of CD103 and CD69 seem reasonable markers, although this is yet to be firmly established for CD4 T cells (7) In addition, CD69 seems key to maintaining TRM, in the context of preventing tissue egress (36). Exactly “where” in the lungs such cells reside is still unclear, but associated with the base of the bronchial epithelium seems one possibility (Figure 1).
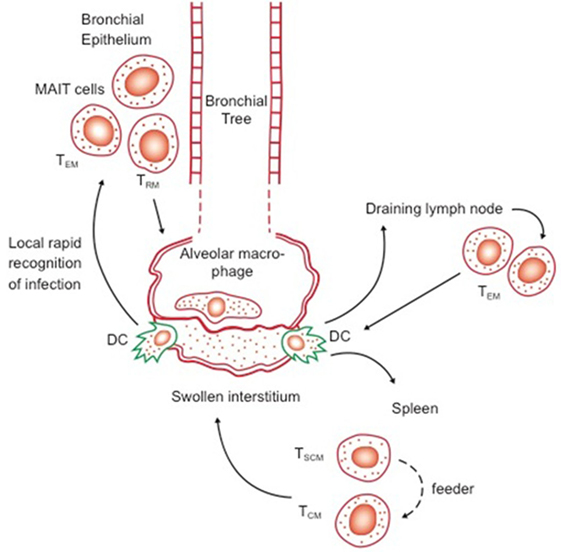
Figure 1. Possible distribution of key reactive lymphocytes in the bronchial epithelium (first line of defense) and lymph nodes and spleen, in memory immune animals. Transport of bacilli and/or antigen out of the alveolus or swollen interstitium by dendritic cells (DC) will trigger responses by local responses by effector memory T cells (TEM) and resident T cells (TRM), as well as potentially by local mucosal-associated invariant T cells, as well as rapid responses by TEM and central memory T cells (TCM) from the draining lymph nodes and spleen. Whether TSCM act directly or feed the expansion of TCM is as yet unknown.
In fact, the TRM subset may be far more widely distributed in the body than previously thought, given that recent estimates have calculated that a similar subset found in skin dermal tissues may be in excess of 1010! As such, therefore, this represents a truly huge peripheral defense system. Although the majority of data to date has focused on CD8-positive skin-resident memory T cells, as above, these subsets differ phenotypically from TEM and TCM (37, 38). Again, they express CD103, which is consistent with this molecule being able to bind to E-cadherin expressed by skin keratinocytes, as is CD49a, which binds to various types of collagens, and they express other “stay put” ligands as well such as CCR4 and E-selectin.
In our own studies in BCG vaccinated mice, we made the unusual observation that a CD4+ CD62Llo CCR7+ subset slowly increased in number over time (5). Interesting, a more recent study looking at human skin CD4 subsets, a population with the same phenotype was observed in the dermis (39), with about a third of these also expressing CD69. The authors of this study named these “migratory memory T cells” and argued that they may represent a subset that recirculates bidirectionally between the skin and the blood or the lymphatics (40). While obviously not proven the cells, we observed in the lungs may be a subset of TRM with increased migratory properties. While confusing, this illustrates that the whole memory T cell population is probably a very dynamic event, rather than all cells finding a tissue niche and just staying there.
Cells now identified as stem cell-like memory T cells [TSCM] followed the realization that they had similarities to conventional hematopoetic stem cells; they share similar core transcriptional signatures, can readily proliferate, and gave rise to heterogenous cellular progeny (41)—in fact, they can reconstitute the full diversity of memory T cells. TSCM have a naïve-like phenotype and express high levels of the SCA-1 marker, CD122 (IL-2Rβ), and CXCR3 [14].
The concept of TSCM first arose in studies in 2005 (42) looking at CD8 responses in a mouse model of human GVHD. These cells had a “naïve/unactivated” CD44lo CD62Lhi phenotype, but they were long lived, required IL-15 (suggesting they were memory cells), had the ability to efficiently self-renew, and were multi-potential, in that they could apparently give rise to both effector and central memory populations—even while maintaining their own pool by self-renewal. Whereas initially, it was thought that these were just a component of the central memory response, they were shown subsequently (43) to be a distinct subset with a much higher proliferative capacity. More recently, a similar CD8 subset has been observed in macaques (44).
Available information to that point was limited to CD8 responses, but in 2011, a CD4 T cell subset with stem cell-like properties was identified in a tumor rejection model in mice (45). This subset was further identified as a component of the TH17 response based on secretion of this cytokine. This was of interest, not the least because TH17 are highly “plastic” in that they possess substantial flexibility in their developmental programming, and as a result, can acquire properties similar to TH1 T cells. In the tumor models, TH17 can also directly mediate rejection by themselves (with the caveat that they first have to be polarized in vitro), and can express both IFNγ and IL-17. Finally, these studies found parallels between CD4 and CD8 memory cells with stem cell-like properties, in terms of shared signaling via the Wnt/β-catenin pathway (the expression of which can be used to help identify them). Similar observations have been made using human cells, in which, inhibition of mTOR signaling or the Wntβ-catenin system induces the expansion of TSCM (46).
It is unclear if a “niche” exists for TSCM. The bone marrow and spleen may serve this purpose, and it has been suggested that they can be found associated with fibroblastic reticular cells within lymph nodes (which provide them with signals) (41). Their patterns of systemic recirculation and tissue distributions seem similar to naïve T cells, and recent studies (47) in humans seem to indicate a state of continuous flux and renewal of TSCM, even in elderly individuals.
TSCM are minimally differentiated phenotypically and functionally, and appear to fall midway between naïve and conventional memory cells. Right now, the debate continues as to where TSCM fit in the overall cell family tree. A working model is that TSCM arise from naïve T cells and act as feeder cells for other more differentiated cells. Because TSCM and TCM share similar abilities for rapid cell expansion, then, it is proposed that TSCM turn into TCM as needed. TCM, then, in turn, seed peripheral sites with TEM—now expressing ligands needed to identify target sites (lungs, gut, skin, etc.) where many probably become cessile TRM. If the pathogen reappears, these subsets mediate a rapid response, with many becoming secondary effectors. However, this is far from clear, since it is possible that TSCM can give rise to TEM as well. For instance, in our chronic tuberculosis murine model (5), one would predict that TSCM would increase the numbers of TCM present in lungs as the infection progressed, but this does not happen.
A further question yet to be determined is whether there is any relationship between memory CD44lo CD62Lhi TSCM cells and CD4 cells secreting IL-17 (TH17). Recent studies have shown (48) that TH17 can be directly protective in a mouse model of tuberculosis. In that study, Rag−/− mice were infused with TH17 or TH1 CD4 T cells and both showed cell activation and subsequent protection against a challenge infection. Interestingly, while immunity transferred by TH1 cells was transient and contracted, TH17 cell transfer was stable and gave much longer survival (very consistent with the idea that TSCM are an “early” memory response). The only drawback was an increasingly florid inflammatory infiltrate characterized by an excess of neutrophils [as our laboratory has also noted (6)]. These data, the authors concluded, directly indicated that TH17 cells had the capacity to transfer protective immunity. The question yet unanswered is whether this was due to TH17 cells directly (perhaps acquiring an IL-17/IFNγ double phenotype?), or mediated by a TSCM subset arising from the overall TH17 population (45)? More recently, an important study from Denmark showed (49) that two vaccine candidates could induce TH17 responses in mice, that these were long lived (18 months), stable, and upon challenge started to show traits associated with TH1 responses. Thus, if TH17 cells have a memory component, is this part of or independent of the TSCM/TCM/TEM interrelationship discussed above?
Finally, follicular helper T cells (Tfh cells) control germinal center host responses at both the cellular and humoral levels. There is increasing information that this includes a memory T cell component (memory Tfh). These cells are driven by Bcl-6, and by exposure to IL-6 and IL-21 (50). Recent evidence supports the view that antigen-specific memory Tfh cells express CXCR5, but lack Bcl-6, ICOS, and expression of several other markers seen in primary responses.
As recently extensively reviewed (1, 51–54), any successful vaccine against tuberculosis will need to generate memory T cells. We now know that the T cell response is phenotypically heterogeneous, and in the context of memory in tuberculosis, it is almost certain that more than one subset is both involved and important. The primary focus has been on the CD4-based immune response—since CD8 responses seem to play a much more minor role—and in particular on CD4 cells that secrete IFNγ.
It goes without saying that understanding memory immunity in the context of M. tuberculosis infection is imperative if we are going to design better vaccines, compounded by the fact that results from some of the current candidates are rather underwhelming. However, the blunt fact is that our knowledge of this parameter is woeful, and mainstream Immunology is gradually discovering new subsets of memory T cells on a steady basis. The TB research field is responding to this partially, certain vaccines induce TCM, BCG given by a different route induces TRM, and so forth, but what is utterly lacking is an understanding of which ones are the most important. Added to this complexity is the vaccine “type” itself (54)—live vaccine A might work best if it induces TM subset X, while protein in adjuvant candidate B works best if it induces TM subset Y. If there are, say, five TM subsets, do we need to trigger all of them, just 3 or 4, or is just one? Does there need to be a balance between them? Are we deluding ourselves by continually comparing experimental outcomes directly to BCG (as our positive control in animal models)? For instance, candidate X might take much longer to induce TM compared to BCG, but in long-term studies, the longevity of the immunity it generates may be far superior. However, because of the standard assays we employ (mostly short, usually for economic reasons), we would miss the latter and discard this candidate (55–57).
If there was (as we believed for a while) a single “memory T cell” subset, then matters would be uncomplicated—one could perform parallel studies of vaccination with candidates followed by challenge, then determine which vaccine gave the strongest and most long-lasting memory immunity. This is unfortunately now not the case, however, because there appear to be multiple subsets involved, and we do not know which ones are critical for protection in vaccinated individuals, nor is there much consensus. This has the potential for being a further serious impediment for TB vaccine development.
In the first attempt to define host memory immunity involved in resistance to tuberculosis after vaccination, Lefford et al. gave rats the BCG vaccine, then used treatment with isoniazid to remove any surviving vaccine bacilli (58). He then challenged these animals and showed that they had increased resistance, thus showing that a form of immunity persisted even when the antigenic stimulus had been removed. A similar approach was then tried a decade later, but here, mice were directly infected with M. tuberculosis before the application of isoniazid chemotherapy. As before, these animals showed evidence of substantially increased resistance to a homologous challenge, and parallel passive cell transfer experiments showed that this resistance was mediated by CD4 T cells (3). This was followed by studies showing that the length of time needed to establish a state of memory immunity after BCG vaccination was inversely proportional to the vaccine dose, but that once established the level of immunity was equivalent (59). Soon thereafter, it was shown (60) that memory immunity involved T cells that were antigen-specific (in this case, to ESAT) and secreted IFNγ. In addition, in one of the first applications of flow cytometry to these questions, it was found that T cells changed their phenotype over the course of infection, with the gradual emergence of cells expressing the CD44hi CD45RBlo phenotype (61).
Mice are usually vaccinated with BCG by the subcutaneous route, at a dose of ~106. Some initial protection can be detected 10–15 days later, but an initial peak is not reached until ~25–30 days, with the latter being a conventional time point used in most vaccine testing studies (62–64) despite the fact that assays at this time are measuring effector T cell activity, not memory. BCG given via this route can occasionally reach the spleen and sometimes the lungs in very small numbers, and so, protective T cells present in this organ 2–3 months later can reasonably be regarded as memory T cells.
If after this time, the animal is infected with M. tuberculosis, there is an accelerated expansion of activated T cells, many of which are secreting IFNγ, and more rapid control of the infection as illustrated lower bacterial loads in the lungs, and by smaller and more lymphocytic granulomas, indicating accelerated expression of memory immunity. The source of this immunity was investigated more recently in mice that were vaccinated with BCG but not subsequently challenged (5). This showed that in such mice there was a slow increase over the first 100 days in the numbers of T cells recoverable from the lungs that had a CD44hi CD62Llo phenotype (and, therefore, TEM under the Sallusto definition); moreover, 90% of these cells were CD4-positive. Only 10% or so expressed a CD44hi CD62Lhi TCM phenotype. These observations led to the hypothesis that BCG predominantly established a TEM population in the lungs, and this subsequently led to the proposal of a hypothesis that the apparently lower induction of any TCM may reflect an inherent weakness of the vaccine (65).
Recent studies have drawn attention to the route of BCG administration. While an earlier study (66) saw no difference between the efficacy of BCG in mice given the vaccine by aerosol or subcutaneously, more recent studies have suggested otherwise. In a potentially important breakthrough, Perdomo and her colleagues (67) demonstrated the induction of TRM in the lungs following instillation of BCG via the trachea, with these cells expressing CXCR3, CD103, and CD69; in addition, these cells were IFNγ-positive. However, these data should be viewed in comparison with results from the same laboratory in which a new rBCG vaccine candidate preferentially elicited CCR7+ TCM T cells (68). In contrast, in a rhesus macaque model, Sharpe and her colleagues (69) found that intravenous BCG vaccination was the most effective, with strong induction of IFNγ- and TNFα-positive TEM. If anything, this further emphasizes our point above that different candidates can make different memory responses, and that even the same type of vaccine can give different responses when given by different routes.
After 40–50 days following low dose aerosol infection with M. tuberculosis the bacterial load in the lungs stabilizes at around one million bacteria. This establishes a chronic disease state in which there is a progressive but very slow increase in bacterial numbers over the next 100–200 days, concomitant with a slow degeneration of the lung granulomatous structures (70). Over this time, there are dynamic changes, both in terms of T cell subsets and macrophage/dendritic cell populations (71).
As with BCG vaccination, most cells recoverable from the lungs over this time are CD44hi CD62Llo CD4 T cells. Most of these are certainly TEM/TRM subsets, but there is probably a further subset of secondary effector cells as well due to the continued presence of antigen. This is mediated by IFNγ, as illustrated by the continued presence of macrophages staining positive for NOS2 (72).
The distinction between TEM and effector cells is further suggested by the observation (73) that activated T cells in the chronically infected lung are PD-1+, and transition into KLRG-1+ “terminally differentiated” cells (but not the reverse). It is increasing clear that these PD-1+ cells, once thought to be exhausted cells, in fact represent a major lung T cell subset, with further analysis (74) showing that these cells depend on ICOS and Bcl-6 expression. However, one note that these results are different to a further study in which KLRG-1-negative CD4 cells predominated; these expressed CD62Lhi and presumably, therefore, are part of the TCM response (75).
Given these discrepant findings, it is clear that the role of KLRG-1 and PD-1 subsets needs to be further clarified—an obvious starting point being their comparison in chronic infection models versus those of vaccination. A further complication is the newly proposed distinction between “parenchymal” and “intravascular” T cells in the lungs of M. tuberculosis-infected mice (76). In that model, cells expressing KLRG-1 and IFNγ are retained in the vasculature, whereas cells that mediate protection—most of them IFNγ-negative—are found in the lung parenchyma (76, 77). However, not only is this a misuse of the term “parenchymal,”1 but the actual flow cytometry staining technique, which is based on the injection a few minutes earlier with anti-CD45, does not take into account lung blood capillary transit time (78). This takes a finite time (neutrophils, which are similar size take several hours) because the lymphocyte has to deform so it can pass into the capillary. Lymphocytes are 6–8 µm in diameter and can be twice as large if activated blast cells, and have to pass through capillaries that are only 5-µm in diameter (much narrower than capillaries in other body organs). Our own interpretation of these observations (see Figure 2) is that the cells that stain positive for IFNγ are actually at the proximal end of the capillary bed—and have not reached the lesions yet—whereas the “parenchymal” cells in the lesions are IFNγ low or negative because they have already released this cytokine. In addition, we would propose that KLRG-1 expression is to enable cells to bind cadherins in the extracellular matrix to contribute to the developing granuloma.
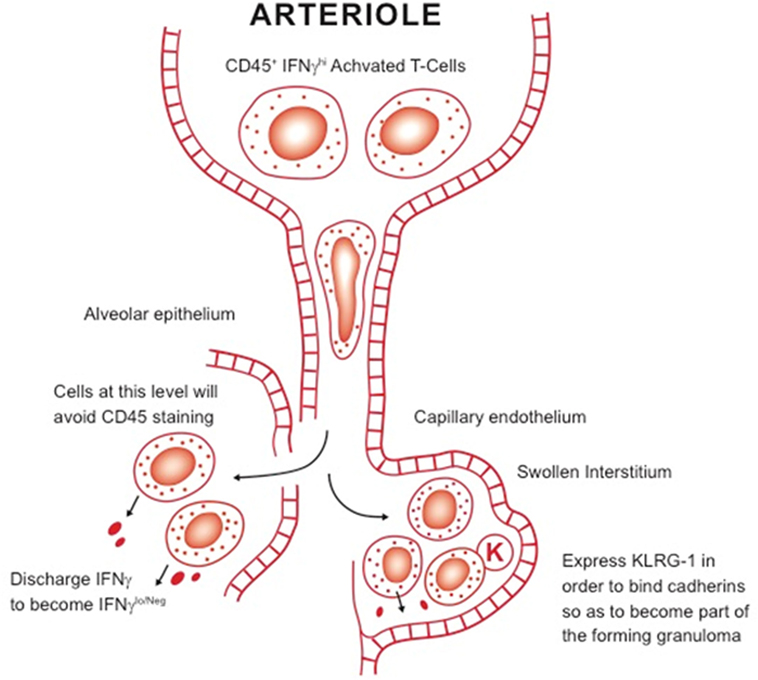
Figure 2. Our working hypothesis for the distinction between intravascular T cells and other subsets in the lungs. Large activated blast lymphocytes plug the proximal end of the lung blood capillary system, where they are readily stained by anti-CD45 antibody. These cells have to considerably deform before they can be pushed by hydrostatic pressure through the system. It is not clear, however, how long this process takes but it could be several hours. Once they encounter sites of infection, they pass out of the blood capillary and either cross the damaged alveolar epithelium or pass into the swollen interstitium. After encountering infected local macrophages, they release IFNγ, thus becoming negative upon staining for this cytokine. In addition, some express KLRG-1, needed for the cell to bind cadherins in the extracellular matrix, facilitating granuloma formation.
Given the belief that memory immunity becomes established once the pathogen has been cleared and antigen is no longer available, early studies on memory immunity after M. tuberculosis infection consisted of initial infection then clearance of this infection by treating the animal with isoniazid chemotherapy. These showed (3) that the accelerated resistance conferred upon these animals was mediated by a long lived T cell population that was CD4-positive and not actively proliferating (prior to stimulation). In addition to acquired specific resistance, a component of non-specific resistance was also demonstrable in adoptive cell transfer studies. In keeping with the growing concept at the time (79) that secreted proteins rather than constitutive proteins of the bacillus drove initial protective immunity, triggering of the memory T cell response required the live organism [as indeed was shown earlier in a classical study on this topic (80)].
For many years, “endogenous reinfection” was considered to be the primary cause of secondary disease or relapse in tuberculosis patients, and this formed the cornerstone of experimental models for some considerable time (81). Over the past decade, concomitant with the worsening epidemic in areas such as Southern Africa, it has become apparent that exogenous reinfection is probably the main cause of secondary disease (82), and in fact, it has been demonstrated that patients treated successfully for tuberculosis are at much higher risk of catching it again (83).
This latter result troubled us, because our animal model studies suggested that such people would be expected to be more resistant, not less. To revisit this, we infected mice with the virulent HN878 Beijing strain, and then rescued these mice by chemotherapy. When these animals were re-challenged, they were highly resistant and we observed the potent emergence of both TEM and TCM memory T cell subsets, both IFNγ-positive (6). However, 30–40 days later, the numbers of these cells progressively declined, and the mice developed a diffuse tuberculous pneumonitis, which was fatal. Examination of the declining T cell numbers showed that many were PD-1hi [an expression we know can be reversed by chemotherapy (84)]. It is unclear if this represents exhausted cells—given the new information about PD-1 discussed above—but what it does illustrate is that memory immunity, thought to be long-lived and stable, may not be at all.
Because of the apparent dominance of TEM in peripheral non-lymphoid sites, it has been suggested (7) that a vaccine that elicits and maintains high-frequency TEM populations in the lungs would provide a more efficient degree of protection against M. tuberculosis infection. A further issue is the cytokine-secreting properties of the cells our vaccine induces, and to date, human studies have produced ambiguous results (85, 86). Similarly, it has been argued that IFNγ is of little importance in the expression of protective immunity in the mouse lung (77), although this stunning conclusion was drawn from an adoptive transfer study comparing very high numbers of bacteria in the lungs in which an “area under the curve” analysis was performed (the reader can draw their own conclusions as to whether this is an appropriate use of this test), and which of course is utterly contrary to classical studies using gene disrupted mice.
That aside, at the clinical trial level, there is no consensus as to whether a vaccine driving TH1 responses is essential, and indeed excessive T cell activation may be detrimental (86). In addition, humans are not mice and cannot be dissected, and a poor TH1 signal in the blood may simply reflect the fact that the cells expressing these are doing their job in lung lesions rather than recirculating. Moreover, some have also pointed out that IFNγ is not always essential for TM function (87), and we ourselves have recently suggested the idea (51) of an “rapid influx and weight of numbers” model that does not necessarily involve cytokines at all.
Evidence is starting to appear in favor of our own (65) argument that BCG is a poor vaccine because of lack of TCM generation, with reports that a new rBCG candidate that generates CXCR5 + TCM (68), plus the demonstration that boosting of BCG with the new fusion H56 generates expansion of Bcl-6 + CD4 cells (88), and data showing that the new live attenuated candidate SO2 also pushes TCM responses (89). Given our own published viewpoint, this is obviously encouraging. That said, the use of CXCR5 may be a confounding issue. CXCR5 may well be a marker of TCM, but it is also implicated as a marker of Tfh (90). CD4 cells expressing CXCR5 associate with “follicle-like” cell aggregates in the mouse granuloma (91), and express CD44hi, ICOS, and PD-1 [in addition, about half express the orphan receptor ROR-γt (88)]. In fact, the presence of these cells might explain the mysterious arrival of B cell follicles into lung granulomas (92). Although CXCR5-gene KO mice still control M. tuberculosis infection in the lungs, they do so less efficiently, but in this regard, a more recent study (74) has questioned their importance. Moreover, in a malaria model (93), CXCR5+ cells have been implicated as precursors of both TCM and Tfh cells. As with other models, if the parasite is rapidly cleared, this favors TCM generation.
Regarding CD4 cells, we have shown that these are the predominant subset in the lungs of BCG vaccinated mice, and >90% are of the TEM phenotype (5). However, it is unknown if these are truly resident TRM or are slowly recirculating through the lung lymphoid tissues. This idea has been addressed elsewhere by Gebhardt et al. (94) who have predicted that this subset would be slowly lost over time, an idea in keeping with our own hypothesis (65) regarding the gradual disappearance of BCG mediated protection in children/young adults.
Can BCG or M. tuberculosis directly induce TRM? In the case of BCG, it appears so, but also seems to depend on the route by which the vaccine is given. As noted above, very recently Perdomo et al. (67) demonstrated that T cells induced in the lungs after the vaccine was injected into the trachea expressed CXCR3 (promoting ingress into these tissues), produced IFNγ, and stained CD103+CD69+. While some concern remains as to whether CD103 is a consistent marker of TRM [30], these are interesting findings, consistent with the hypothesis that BCG induces TRM, and hopefully will be further confirmed in the near future if they can be shown to have the distinct transcriptional signature that appears to specifically identify this subset, which distinguishes these from other CD44hi CD62Llo memory subsets. Clearly, expression of the CD103 integrin indicates an intention to “stay put,” and these cells are also CCR7lo and S1PR1lo, molecules needed for tissue exit.
Generating TRM is a reasonable starting point, but would amplifying this subset rather than others result in local immunity but no immunity to bacteria that we know disseminate via the blood and lymphatics? We know that subcutaneous BCG generates a strong TEM response in the lungs, and in fact, it has been proposed (7) that because exposure to M. tuberculosis involves prolonged antigen stimulation and considerable inflammation, the generation of high-frequency TEM could help counteract the immune-evasion properties of the bacillus. Our own experience indicates that BCG does indeed do this in the mouse model, but since the infection is “controlled and contained” but not eradicated, then, this is only a pyrrhic victory.
The ultimate ambition here is to find a vaccine of some sort that generates a form of immunity that will recognize the presence of the bacillus before it gets into the interstitial space (from whence macrophages/dendritic cells will carry them off to the lymph nodes) (95). While from an immunological point of view one might speculate that this could be achieved, unfortunately, the anatomy of the lung precludes this. Alveolar macrophages prowl the lung epithelial surface through a sea of surfactant, and probably most of the time, kill ingested M. tuberculosis, but when they fail to do so, no sort of back-up protective mechanism seems to exist.
Recently, the existence of non-classical lymphocyte-like cell populations has been identified in gut mucosa, and there appear to be complimentary populations existing in the lungs. Among these, a subset that occurs in relatively high frequency are mucosal-associated invariant T cells. These possess a semi-invariant MHC alpha chain, indicating a restricted recognition pattern, and can secrete protective cytokines (96). In general, these cells are perceived as part of a “first line innate” system, and there is no evidence they can be manipulated or differentiated into a memory cell population by vaccination (53). One can make a similar argument for other tissue-resident innate cells, such as NK cells, NKT cells, and innate lymphoid cells.
There is no avoiding the fact that TB vaccine development has been glacial. Only one candidate has been fully evaluated in clinical trials (as a BCG boosting vaccine), and this candidate failed (97, 98). The reaction to this has ranged from the very unhelpful “never should have been tried in the first place” to a more sober explanation from our laboratory that argued that the vaccine was tested in a region where the M. tuberculosis strains were low fitness, making BCG boosting impossible (99). Several other candidates exist and are apparently moving slowly through the pipeline, but calls from some of us to test them jointly in head to head evaluations in different animal models in laboratories that have no vested interest themselves have been ignored for over a decade.
If there is room for optimism, it reflects the fact that we are starting to consider if different subsets of memory T cells could be specifically targeted; as noted above, BCG given via the trachea generates a much stronger TRM response. This translated into improved protection after aerosol challenge, although whether this actually reflects local “nonspecific resistance” due to the presence of BCG in the lungs (absent in mice vaccinated subcutaneously) remains unclear.
As noted above, to find ways to stop the TB bacillus dead in its tracks will require a paradigm shift in our thinking. The presence of the bacillus is not even recognized until it uses its ESX system to break through the alveolar epithelium and into the interstitium, and even then, there is a favorable lag period while some bacilli are transported to draining lymph nodes to sensitize T cells, then, a further delay before these enter the blood and find their way back to the sites of bacterial implantation. By this time, as necrosis-prone animal models such as the guinea pig illustrate, the damage has already been done.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Collen Rodrigues for the illustrations.
1. Kirman JR, Henao Tamayo MI, Agger EM. The memory immune response to tuberculosis. 2nd ed. In: Jacobs WR Jr, McShane H, Mizrahi V, Orme IM, editors. Tuberculosis and the Tubercle Bacillus. Washington, DC: ASM Press (2017). p. 95–116.
2. Orme IM. The kinetics of emergence and loss of mediator T lymphocytes acquired in response to infection with Mycobacterium tuberculosis. J Immunol (1987) 138(1):293–8.
3. Orme IM. Characteristics and specificity of acquired immunologic memory to Mycobacterium tuberculosis infection. J Immunol (1988) 140(10):3589–93.
4. Cooper AM, Callahan JE, Keen M, Belisle JT, Orme IM. Expression of memory immunity in the lung following re-exposure to Mycobacterium tuberculosis. Tuber Lung Dis (1997) 78(1):67–73. doi:10.1016/S0962-8479(97)90017-4
5. Henao-Tamayo MI, Ordway DJ, Irwin SM, Shang S, Shanley C, Orme IM. Phenotypic definition of effector and memory T-lymphocyte subsets in mice chronically infected with Mycobacterium tuberculosis. Clin Vaccine Immunol (2010) 17(4):618–25. doi:10.1128/CVI.00368-09
6. Henao-Tamayo M, Obregon-Henao A, Ordway DJ, Shang S, Duncan CG, Orme IM. A mouse model of tuberculosis reinfection. Tuberculosis (Edinb) (2012) 92(3):211–7. doi:10.1016/j.tube.2012.02.008
7. Masopust D, Picker LJ. Hidden memories: frontline memory T cells and early pathogen interception. J Immunol (2012) 188(12):5811–7. doi:10.4049/jimmunol.1102695
8. Sallusto F, Lanzavecchia A, Araki K, Ahmed R. From vaccines to memory and back. Immunity (2010) 33(4):451–63. doi:10.1016/j.immuni.2010.10.008
9. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol (2004) 22:745–63. doi:10.1146/annurev.immunol.22.012703.104702
10. Sallusto F, Lanzavecchia A. Heterogeneity of CD4+ memory T cells: functional modules for tailored immunity. Eur J Immunol (2009) 39(8):2076–82. doi:10.1002/eji.200939722
11. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401(6754):708–12. doi:10.1038/44385
12. Sallusto F, Langenkamp A, Geginat J, Lanzavecchia A. Functional subsets of memory T cells identified by CCR7 expression. Curr Top Microbiol Immunol (2000) 251:167–71.
13. Ndlovu H, Darby M, Froelich M, Horsnell W, Luhder F, Hunig T, et al. Inducible deletion of CD28 prior to secondary Nippostrongylus brasiliensis infection impairs worm expulsion and recall of protective memory CD4(+) T cell responses. PLoS Pathog (2014) 10(2):e1003906. doi:10.1371/journal.ppat.1003906
14. Sprent J, Surh CD. T cell memory. Annu Rev Immunol (2002) 20:551–79. doi:10.1146/annurev.immunol.20.100101.151926
15. McKinstry KK, Strutt TM, Swain SL. The effector to memory transition of CD4 T cells. Immunol Res (2008) 40(2):114–27. doi:10.1007/s12026-007-8004-y
16. McKinstry KK, Strutt TM, Bautista B, Zhang W, Kuang Y, Cooper AM, et al. Effector CD4 T-cell transition to memory requires late cognate interactions that induce autocrine IL-2. Nat Commun (2014) 5:5377. doi:10.1038/ncomms6377
17. Boyman O, Letourneau S, Krieg C, Sprent J. Homeostatic proliferation and survival of naive and memory T cells. Eur J Immunol (2009) 39(8):2088–94. doi:10.1002/eji.200939444
18. Ahmed R, Bevan MJ, Reiner SL, Fearon DT. The precursors of memory: models and controversies. Nat Rev Immunol (2009) 9(9):662–8. doi:10.1038/nri2619
19. Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature (2017) 552(7685):362–7. doi:10.1038/nature24633
20. Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature (2017) 552(7685):404–9. doi:10.1038/nature25144
21. Dooms H, Abbas AK. Control of CD4+ T-cell memory by cytokines and costimulators. Immunol Rev (2006) 211:23–38. doi:10.1111/j.0105-2896.2006.00387.x
22. Gwyer Findlay E, Villegas-Mendez A, O’Regan N, de Souza JB, Grady LM, Saris CJ, et al. IL-27 receptor signaling regulates memory CD4+ T cell populations and suppresses rapid inflammatory responses during secondary malaria infection. Infect Immun (2014) 82(1):10–20. doi:10.1128/IAI.01091-13
23. McKinstry KK, Golech S, Lee WH, Huston G, Weng NP, Swain SL. Rapid default transition of CD4 T cell effectors to functional memory cells. J Exp Med (2007) 204(9):2199–211. doi:10.1084/jem.20070041
24. McKinstry KK, Strutt TM, Swain SL. Regulation of CD4+ T-cell contraction during pathogen challenge. Immunol Rev (2010) 236:110–24. doi:10.1111/j.1600-065X.2010.00921.x
25. Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4(+) T cells in immunity to viruses. Nat Rev Immunol (2012) 12(2):136–48. doi:10.1038/nri3152
26. Harrington LE, Janowski KM, Oliver JR, Zajac AJ, Weaver CT. Memory CD4 T cells emerge from effector T-cell progenitors. Nature (2008) 452(7185):356–60. doi:10.1038/nature06672
27. Lanzavecchia A, Sallusto F. Understanding the generation and function of memory T cell subsets. Curr Opin Immunol (2005) 17(3):326–32. doi:10.1016/j.coi.2005.04.010
28. Reiner SL, Sallusto F, Lanzavecchia A. Division of labor with a workforce of one: challenges in specifying effector and memory T cell fate. Science (2007) 317(5838):622–5. doi:10.1126/science.1143775
29. Sallusto F, Mackay CR, Lanzavecchia A. The role of chemokine receptors in primary, effector, and memory immune responses. Annu Rev Immunol (2000) 18:593–620. doi:10.1146/annurev.immunol.18.1.593
30. Carbone FR. Tissue-resident memory T cells and fixed immune surveillance in nonlymphoid organs. J Immunol (2015) 195(1):17–22. doi:10.4049/jimmunol.1500515
31. Carbone FR, Mackay LK, Heath WR, Gebhardt T. Distinct resident and recirculating memory T cell subsets in non-lymphoid tissues. Curr Opin Immunol (2013) 25(3):329–33. doi:10.1016/j.coi.2013.05.007
32. Mackay LK, Carbone FR. CD4 helpers put tissue-resident memory cells in their place. Immunity (2014) 41(4):514–5. doi:10.1016/j.immuni.2014.09.018
33. Kipnis A, Irwin S, Izzo AA, Basaraba RJ, Orme IM. Memory T lymphocytes generated by Mycobacterium bovis BCG vaccination reside within a CD4 CD44lo CD62 ligand(hi) population. Infect Immun (2005) 73(11):7759–64. doi:10.1128/IAI.73.11.7759-7764.2005
34. Bevan MJ. Memory T cells as an occupying force. Eur J Immunol (2011) 41(5):1192–5. doi:10.1002/eji.201041377
35. Mueller SN. Grand challenges in immunological memory. Front Immunol (2017) 8:385. doi:10.3389/fimmu.2017.00385
36. Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol (2016) 16(2):79–89. doi:10.1038/nri.2015.3
37. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol (2013) 14(12):1294–301. doi:10.1038/ni.2744
38. Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A (2012) 109(18):7037–42. doi:10.1073/pnas.1202288109
39. Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med (2015) 7(279):279ra39. doi:10.1126/scitranslmed.3010302
40. Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, et al. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity (2016) 45(6):1270–84. doi:10.1016/j.immuni.2016.10.018
41. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med (2011) 17(10):1290–7. doi:10.1038/nm.2446
42. Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat Med (2005) 11(12):1299–305. doi:10.1038/nm1326
43. Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med (2009) 15(7):808–13. doi:10.1038/nm.1982
44. Lugli E, Dominguez MH, Gattinoni L, Chattopadhyay PK, Bolton DL, Song K, et al. Superior T memory stem cell persistence supports long-lived T cell memory. J Clin Invest (2013) 123(2):594–9. doi:10.1172/JCI66327
45. Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity (2011) 35(6):972–85. doi:10.1016/j.immuni.2011.09.019
46. Scholz G, Jandus C, Zhang L, Grandclement C, Lopez-Mejia IC, Soneson C, et al. Modulation of mTOR signalling triggers the formation of stem cell-like memory T cells. EBioMedicine (2016) 4:50–61. doi:10.1016/j.ebiom.2016.01.019
47. Ahmed R, Roger L, Costa Del Amo P, Miners KL, Jones RE, Boelen L, et al. Human stem cell-like memory T cells are maintained in a state of dynamic flux. Cell Rep (2016) 17(11):2811–8. doi:10.1016/j.celrep.2016.11.037
48. Wozniak TM, Saunders BM, Ryan AA, Britton WJ. Mycobacterium bovis BCG-specific Th17 cells confer partial protection against Mycobacterium tuberculosis infection in the absence of gamma interferon. Infect Immun (2010) 78(10):4187–94. doi:10.1128/IAI.01392-09
49. Lindenstrom T, Woodworth J, Dietrich J, Aagaard C, Andersen P, Agger EM. Vaccine-induced th17 cells are maintained long-term postvaccination as a distinct and phenotypically stable memory subset. Infect Immun (2012) 80(10):3533–44. doi:10.1128/IAI.00550-12
50. Hale JS, Ahmed R. Memory T follicular helper CD4 T cells. Front Immunol (2015) 6:16. doi:10.3389/fimmu.2015.00016
51. Henao-Tamayo M, Ordway DJ, Orme IM. Memory T cell subsets in tuberculosis: what should we be targeting? Tuberculosis (Edinb) (2014) 94(5):455–61. doi:10.1016/j.tube.2014.05.001
52. Orme IM. Vaccine development for tuberculosis: current progress. Drugs (2013) 73(10):1015–24. doi:10.1007/s40265-013-0081-8
53. Orme IM. Vaccines to prevent tuberculosis infection rather than disease: physiological and immunological aspects. Tuberculosis (Edinb) (2014) 101:210–6. doi:10.1016/j.tube.2014.10.008
54. Orme IM. Tuberculosis vaccine types and timings. Clin Vaccine Immunol (2015) 22(3):249–57. doi:10.1128/CVI.00718-14
55. McShane H, Jacobs WR, Fine PE, Reed SG, McMurray DN, Behr M, et al. BCG: myths, realities, and the need for alternative vaccine strategies. Tuberculosis (Edinb) (2012) 92(3):283–8. doi:10.1016/j.tube.2011.12.003
56. Williams A, Hall Y, Orme IM. Evaluation of new vaccines for tuberculosis in the guinea pig model. Tuberculosis (Edinb) (2009) 89(6):389–97. doi:10.1016/j.tube.2009.08.004
57. Williams A, Orme IM. Animal models of tuberculosis: an overview. Microbiol Spectr (2016) 4(4):1–12. doi:10.1128/microbiolspec.TBTB2-0004-2015
58. Lefford MJ, McGregor DD, Mackaness GB. Properties of lymphocytes which confer adoptive immunity to tuberculosis in rats. Immunology (1973) 25(4):703–15.
59. Gruppo V, Orme IM. Dose of BCG does not influence the efficient generation of protective immunity in mice challenged with Mycobacterium tuberculosis. Tuberculosis (Edinb) (2002) 82(6):267–73. doi:10.1054/tube.2002.0340
60. Andersen P, Smedegaard B. CD4(+) T-cell subsets that mediate immunological memory to Mycobacterium tuberculosis infection in mice. Infect Immun (2000) 68(2):621–9. doi:10.1128/IAI.68.2.621-629.2000
61. Griffin JP, Orme IM. Evolution of CD4 T-cell subsets following infection of naive and memory immune mice with Mycobacterium tuberculosis. Infect Immun (1994) 62(5):1683–90.
62. Orme IM. Preclinical testing of new vaccines for tuberculosis: a comprehensive review. Vaccine (2006) 24(1):2–19. doi:10.1016/j.vaccine.2005.07.078
63. Orme IM, Ordway DJ. Animal models of mycobacteria infection. Curr Protoc Immunol (2011) 19:19.5. doi:10.1002/0471142735.im1905s94
64. Orme IM, Ordway DJ. Mouse and guinea pig models of tuberculosis. Microbiol Spectr (2016) 4(4):1–20. doi:10.1128/microbiolspec.TBTB2-0002-2015
65. Orme IM. The Achilles heel of BCG. Tuberculosis (Edinb) (2010) 90(6):329–32. doi:10.1016/j.tube.2010.06.002
66. Orme IM, Collins FM. Aerogenic vaccination of mice with Mycobacterium bovis BCG. Tubercle (1986) 67(2):133–40. doi:10.1016/0041-3879(86)90007-3
67. Perdomo C, Zedler U, Kuhl AA, Lozza L, Saikali P, Sander LE, et al. Mucosal BCG vaccination induces protective lung-resident memory T cell populations against tuberculosis. MBio (2016) 7(6):e01686–16. doi:10.1128/mBio.01686-16
68. Vogelzang A, Perdomo C, Zedler U, Kuhlmann S, Hurwitz R, Gengenbacher M, et al. Central memory CD4+ T cells are responsible for the recombinant Bacillus Calmette-Guerin deltaureC:hly vaccine’s superior protection against tuberculosis. J Infect Dis (2014) 210(12):1928–37. doi:10.1093/infdis/jiu347
69. Sharpe S, White A, Sarfas C, Sibley L, Gleeson F, McIntyre A, et al. Alternative BCG delivery strategies improve protection against Mycobacterium tuberculosis in non-human primates: protection associated with mycobacterial antigen-specific CD4 effector memory T-cell populations. Tuberculosis (Edinb) (2016) 101:174–90. doi:10.1016/j.tube.2016.09.004
70. Rhoades ER, Frank AA, Orme IM. Progression of chronic pulmonary tuberculosis in mice aerogenically infected with virulent Mycobacterium tuberculosis. Tuber Lung Dis (1997) 78(1):57–66. doi:10.1016/S0962-8479(97)90016-2
71. Gonzalez-Juarrero M, Shim TS, Kipnis A, Junqueira-Kipnis AP, Orme IM. Dynamics of macrophage cell populations during murine pulmonary tuberculosis. J Immunol (2003) 171(6):3128–35. doi:10.4049/jimmunol.171.6.3128
72. Cooper AM, Pearl JE, Brooks JV, Ehlers S, Orme IM. Expression of the nitric oxide synthase 2 gene is not essential for early control of Mycobacterium tuberculosis in the murine lung. Infect Immun (2000) 68(12):6879–82. doi:10.1128/IAI.68.12.6879-6882.2000
73. Reiley WW, Shafiani S, Wittmer ST, Tucker-Heard G, Moon JJ, Jenkins MK, et al. Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proc Natl Acad Sci U S A (2010) 107(45):19408–13. doi:10.1073/pnas.1006298107
74. Moguche AO, Shafiani S, Clemons C, Larson RP, Dinh C, Higdon LE, et al. ICOS and Bcl6-dependent pathways maintain a CD4 T cell population with memory-like properties during tuberculosis. J Exp Med (2015) 212(5):715–28. doi:10.1084/jem.20141518
75. Lindenstrom T, Knudsen NP, Agger EM, Andersen P. Control of chronic Mycobacterium tuberculosis infection by CD4 KLRG1-IL-2-secreting central memory cells. J Immunol (2013) 190(12):6311–9. doi:10.4049/jimmunol.1300248
76. Sakai S, Kauffman KD, Schenkel JM, McBerry CC, Mayer-Barber KD, Masopust D, et al. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J Immunol (2014) 192(7):2965–9. doi:10.4049/jimmunol.1400019
77. Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, et al. CD4 T Cell-derived IFN-gamma plays a minimal role in control of pulmonary Mycobacterium tuberculosis infection and must be actively repressed by PD-1 to prevent lethal disease. PLoS Pathog (2016) 12(5):e1005667. doi:10.1371/journal.ppat.1005667
78. Westermann J, Puskas Z, Pabst R. Blood transit and recirculation kinetics of lymphocyte subsets in normal rats. Scand J Immunol (1988) 28(2):203–10. doi:10.1111/j.1365-3083.1988.tb02432.x
79. Orme IM, Andersen P, Boom WH. T cell response to Mycobacterium tuberculosis. J Infect Dis (1993) 167(6):1481–97. doi:10.1093/infdis/167.6.1481
80. Lefford MJ, McGregor DD. The effect of chemotherapy for tuberculosis on immunity to tuberculosis. J Infect Dis (1974) 129(4):473–6. doi:10.1093/infdis/129.4.473
81. Smith DW, Wiegeshaus EH. What animal models can teach us about the pathogenesis of tuberculosis in humans. Rev Infect Dis (1989) 11(Suppl 2):S385–93. doi:10.1093/clinids/11.Supplement_2.S385
82. Chiang CY, Riley LW. Exogenous reinfection in tuberculosis. Lancet Infect Dis (2005) 5(10):629–36. doi:10.1016/S1473-3099(05)70240-1
83. Verver S, Warren RM, Beyers N, Richardson M, van der Spuy GD, Borgdorff MW, et al. Rate of reinfection tuberculosis after successful treatment is higher than rate of new tuberculosis. Am J Respir Crit Care Med (2005) 171(12):1430–5. doi:10.1164/rccm.200409-1200OC
84. Henao-Tamayo M, Irwin SM, Shang S, Ordway D, Orme IM. T lymphocyte surface expression of exhaustion markers as biomarkers of the efficacy of chemotherapy for tuberculosis. Tuberculosis (Edinb) (2011) 91(4):308–13. doi:10.1016/j.tube.2011.04.001
85. Ellis RD, Hatherill M, Tait D, Snowden M, Churchyard G, Hanekom W, et al. Innovative clinical trial designs to rationalize TB vaccine development. Tuberculosis (Edinb) (2015) 95(3):352–7. doi:10.1016/j.tube.2015.02.036
86. Fletcher HA, Snowden MA, Landry B, Rida W, Satti I, Harris SA, et al. T-cell activation is an immune correlate of risk in BCG vaccinated infants. Nat Commun (2016) 7:11290. doi:10.1038/ncomms11290
87. Wu CY, Kirman JR, Rotte MJ, Davey DF, Perfetto SP, Rhee EG, et al. Distinct lineages of T(H)1 cells have differential capacities for memory cell generation in vivo. Nat Immunol (2002) 3(9):852–8. doi:10.1038/ni832
88. Prota G, Christensen D, Andersen P, Medaglini D, Ciabattini A. Peptide-specific T helper cells identified by MHC class II tetramers differentiate into several subtypes upon immunization with CAF01 adjuvanted H56 tuberculosis vaccine formulation. Vaccine (2015) 33(48):6823–30. doi:10.1016/j.vaccine.2015.09.024
89. Nambiar JK, Pinto R, Aguilo JI, Takatsu K, Martin C, Britton WJ, et al. Protective immunity afforded by attenuated, PhoP-deficient Mycobacterium tuberculosis is associated with sustained generation of CD4+ T-cell memory. Eur J Immunol (2012) 42(2):385–92. doi:10.1002/eji.201141903
90. Marriott CL, Carlesso G, Herbst R, Withers DR. ICOS is required for the generation of both central and effector CD4(+) memory T-cell populations following acute bacterial infection. Eur J Immunol (2015) 45(6):1706–15. doi:10.1002/eji.201445421
91. Slight SR, Rangel-Moreno J, Gopal R, Lin Y, Fallert Junecko BA, Mehra S, et al. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J Clin Invest (2013) 123(2):712–26. doi:10.1172/JCI65728
92. Jaigirdar SA, MacLeod MK. Development and function of protective and pathologic memory CD4 T cells. Front Immunol (2015) 6:456. doi:10.3389/fimmu.2015.00456
93. Opata MM, Carpio VH, Ibitokou SA, Dillon BE, Obiero JM, Stephens R. Early effector cells survive the contraction phase in malaria infection and generate both central and effector memory T cells. J Immunol (2015) 194(11):5346–54. doi:10.4049/jimmunol.1403216
94. Gebhardt T, Mueller SN, Heath WR, Carbone FR. Peripheral tissue surveillance and residency by memory T cells. Trends Immunol (2013) 34(1):27–32. doi:10.1016/j.it.2012.08.008
95. Orme IM. A new unifying theory of the pathogenesis of tuberculosis. Tuberculosis (Edinb) (2014) 94(1):8–14. doi:10.1016/j.tube.2013.07.004
96. Napier RJ, Adams EJ, Gold MC, Lewinsohn DM. The role of mucosal associated invariant T cells in antimicrobial immunity. Front Immunol (2015) 6:344. doi:10.3389/fimmu.2015.00344
97. Tameris M, McShane H, McClain JB, Landry B, Lockhart S, Luabeya AK, et al. Lessons learnt from the first efficacy trial of a new infant tuberculosis vaccine since BCG. Tuberculosis (Edinb) (2013) 93(2):143–9. doi:10.1016/j.tube.2013.01.003
98. Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet (2013) 381(9871):1021–8. doi:10.1016/S0140-6736(13)60177-4
Keywords: vaccination, memory immunity, memory T cell subsets, BCG, Mycobacterium tuberculosis
Citation: Orme IM and Henao-Tamayo MI (2018) Trying to See the Forest through the Trees: Deciphering the Nature of Memory Immunity to Mycobacterium tuberculosis. Front. Immunol. 9:461. doi: 10.3389/fimmu.2018.00461
Received: 11 December 2017; Accepted: 21 February 2018;
Published: 08 March 2018
Edited by:
Andrea Sant, David H. Smith Center for Vaccine Biology and Immunology, United StatesReviewed by:
Tara Marlene Strutt, University of Central Florida, United StatesCopyright: © 2018 Orme and Henao-Tamayo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ian M. Orme, aWFuLm9ybWVAY29sb3N0YXRlLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.