 Benoît L. Salomon
Benoît L. Salomon Mathieu Leclerc
Mathieu Leclerc Jimena Tosello4
Jimena Tosello4 Emilie Ronin
Emilie Ronin Eliane Piaggio
Eliane Piaggio José L. Cohen
José L. Cohen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 12 March 2018
Sec. Immunological Tolerance and Regulation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00444
This article is part of the Research TopicThe Role of TNF-TNFR2 Signal in Immunosuppressive Cells and its Therapeutic ImplicationsView all 20 articles
Tumor necrosis factor α (TNF) is a potent pro-inflammatory cytokine that has deleterious effect in some autoimmune diseases, which led to the use of anti-TNF drugs in some of these diseases. However, some rare patients treated with these drugs paradoxically develop an aggravation of their disease or new onset autoimmunity, revealing an immunosuppressive facet of TNF. A possible mechanism of this observation is the direct and positive effect of TNF on regulatory T cells (Tregs) through its binding to the TNF receptor type 2 (TNFR2). Indeed, TNF is able to increase expansion, stability, and possibly function of Tregs via TNFR2. In this review, we discuss the role of TNF in graft-versus-host disease as an example of the ambivalence of this cytokine in the pathophysiology of an immunopathology, highlighting the therapeutic potential of triggering TNFR2 to boost Treg expansion. We also describe new targets in immunotherapy of cancer, emphasizing on the putative suppressive effect of TNF in antitumor immunity and of the interest of blocking TNFR2 to regulate the Treg compartment.
Tumor necrosis factor α is a pleiotropic cytokine produced by various cell types and involved in a wide range of pathological processes [for review, see Ref (1, 2)]. It has been initially considered as a pro-inflammatory molecule. However, preclinical and clinical data have shown that it also mediates a paradoxical anti-inflammatory and immunomodulatory effect. Indeed, in murine models of type 1 diabetes or lupus nephritis, TNF may have a protective effect (3–7). Moreover, new onset or exacerbation of chronic inflammatory and autoimmune diseases has been observed in patients treated with anti-TNF therapies (8–14). We will describe below in detail the case of graft-versus-host disease (GVHD) as an example of the ambivalent action of TNF in an immunopathology.
Tumor necrosis factor α binds to two receptors, namely, TNF receptor type 1 (TNFR1) and TNFR2 (Figure 1). Unlike TNFR1 that has a ubiquitous expression, TNFR2 is expressed by some immune cells, preferentially by a fraction of Tregs, some endothelial cells, and cells of the nervous tissue (2, 15). Several mechanisms have been proposed to explain the suppressive action of TNF. It was shown that chronic stimulation with TNF may inactivate TCR signaling (16) or induce T cell exhaustion (17). Alternatively, the cytokine may kill CD8+ T cells, a phenomenon emphasized for autoreactive cells (18). Besides these cell-intrinsic mechanisms, TNF may exert its suppressive activity by stimulating cells that have immunosuppressive action, such as myeloid-derived suppressor cells (MDSCs) (19, 20). Finally, the pioneer works of Chen and Oppenheim suggested that this immunosuppressive effect of TNF could be related to a direct activation of Tregs (15, 21). This latter mechanism, which is the most studied one and supported by data obtained by different groups, is detailed below. Generally, the suppressive action of TNF is considered to be mediated by its interaction with TNFR2 since TNFR2 signaling appears to be protective in various immunopathologies and several of the mechanisms described earlier are TNFR2 dependent (22). However, whereas TNF/TNFR1 interaction has been mostly described to be pro-inflammatory, TNFR1 triggering may also inhibit IL-12/IL-23 p40 expression by macrophages (23). This mechanism may explain the paradoxical expansion of Th1/Th17 cells following anti-TNF treatment in patients with autoimmune diseases who do not respond to this therapy (24, 25).
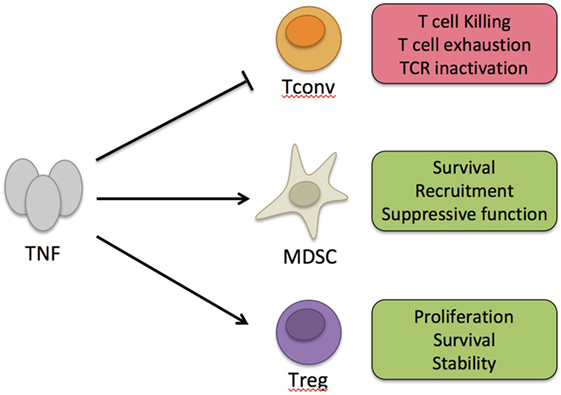
Figure 1. Immunosuppressive action of tumor necrosis factor α (TNF). TNF can exert its immunosuppressive activity by intrinsic negative effect on conventional T cells (Tconvs) activation or by boosting suppressive cells, such as myeloid-derived suppressor cells (MDSC) or regulatory T cells (Tregs). On Tconvs, long-term effect of TNF may promote killing, exhaustion, or TCR inactivation. On MDSC, TNF may boost their activity by promoting their survival, local recruitment, or suppressive function. On Tregs, TNF may promote their proliferation, survival, and stability.
TNFR2 expression is upregulated in activated Tregs and can be detected in activated conventional T cells (Tconvs), although at lower levels than in activated Tregs. Some other members of the TNFR family, such as GITR, OX40, or 4-1BB, are also preferentially expressed by Tregs, and their expression is also upregulated upon activation (26). Remarkably, in transcriptomic analyses, comparing Tregs and Tconvs of lymphoid tissues, TNFR2, OX40, and GITR belong to the Treg signature and their expression correlates with low DNA methylation in Tregs suggesting that their transcription is at least partly regulated at the epigenetic level (27, 28). These three molecules are expressed early in the Treg lineage, since the thymic Treg progenitor stage, and their expression is essential for Treg development (29). In mice lymphoid tissues or in human blood, TNFR2 is expressed by the fraction of activated Tregs expressing high levels of other activation markers such as CTLA-4 (30). TNFR2 expression remarkably identifies a subset of Tregs with the highest suppressive capacity (21, 30, 31).
The direct effect of TNF on TNFR2-expressing Tregs has been studied by Chen and Oppenheim in vitro and has been reviewed elsewhere (32). Briefly, TNF increases proliferation, survival, stability, expression of CD25, Foxp3, and activation markers, as well as suppressive function of mouse Tregs (15, 26, 30, 31). Many of these effects of TNF, notably on proliferation, could be reproduced with human Tregs (32–35). However, some studies claim that TNF inhibits the suppressive activity of human Tregs (36–39). The interpretation of some of these studies was complicated by the fact that TNF can render Tconvs more refractory to the Treg-mediated suppression. After extensive and careful exploration of this question, we could conclude that TNF does not inhibit the suppressive activity of human Tregs (35).
The in vivo role of TNFR2 on Treg biology has been more difficult to evaluate because of the absence of a conditional knockout of TNFR2 in Tregs. However, there is strong evidence that TNF can boost Treg expansion in different inflammatory contexts (40). We showed that TNF, probably produced by Tconvs, stimulated Treg proliferation during type 1 diabetes (41). Others observed a similar phenomenon during septic shock, infectious disease, or immune response (15, 42, 43). Also, TNFR2-deficient Tregs lost their capacity to control colitis, which was associated with reduced survival and stability compared with wild-type control Tregs (31, 44). The critical role of TNFR2 expressed by Tregs has been also studied in the context of GVHD and cancer and will be specifically discussed below. Overall, among all the effects of TNF on Treg biology, its capacity to increase proliferation is the most convincing since it has been reported in many in vitro and in vivo studies performed by different groups using mouse and human Tregs. The evidence that this cytokine also increases Treg survival and stability is quite convincing and its effect on Treg function requires further investigation.
Tumor necrosis factor α plays a key role in acute GVHD (aGVHD), a systemic and highly inflammatory complication that occurs after allogeneic hematopoietic stem cell transplantation (allo-SCT) (45). TNF indeed plays a major role at different steps of this pathological process in which donor T cells recognize as foreign host healthy tissues and eventually cause their destruction (Figure 2). In this line, clinical studies have clearly demonstrated a positive correlation between soluble TNFR1 levels measured 7 days after transplant and the time to onset and severity of aGVHD (46, 47). The increase in TNFR1 levels between baseline and day 7 was not only an independent predictor of aGVHD but also of transplant-related mortality and overall survival. Also, a rise in TNF, as measured by protein levels in peripheral blood, RNA transcription levels, or flow cytometry, precedes the onset of aGVHD, before peaking at the time of its development (48–50). Overall, the results of these clinical studies have led to the integration of TNFR1 as part of a biomarker panel that can discriminate patients with and without aGVHD, and predict survival (51).
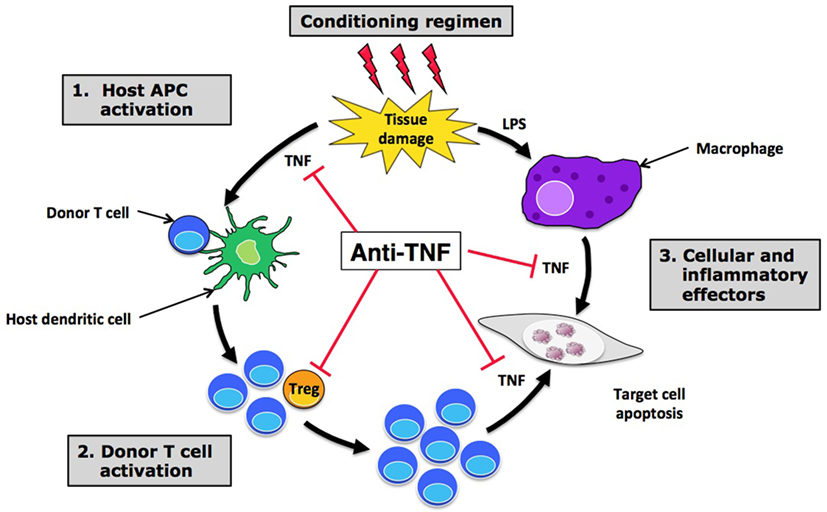
Figure 2. Hope and disappointment in targeting tumor necrosis factor α (TNF) in graft-versus-host disease (GVHD). Anti-TNF treatments are able to block the effect of TNF at different steps of acute GVHD pathophysiology, including initial host APC activation (1), effector T cell recruitment and activation in target tissues (2), and direct cell necrosis (3). By inhibiting TNF ligation to TNFR2 expressed by regulatory T cells (Tregs), anti-TNF treatments could also have a deleterious effect on these suppressive cells, leading to an increased expansion and activation of alloreactive donor T cells that may be responsible for the disappointing results observed with anti-TNF treatments in this setting. Abbreviations: APC, antigen-presenting cell; LPS, lipopolysaccharide.
The key role of TNF in aGVHD pathophysiology logically led researchers and physicians to try to block this cytokine to decrease inflammation and consequently to prevent or treat aGVHD. Along this line, most of the clinical trials focused on two molecules: infliximab—a monoclonal antibody (mAb) that binds TNF—and etanercept—a soluble TNFR that competes with cellular receptors for TNF binding. The great hope risen by TNF targeting in aGVHD during the first decade of this century has unfortunately faded rapidly due to somewhat disappointing results of clinical studies. Indeed, clinical trials failed to prove any benefit in adding infliximab to standard treatment, both for aGVHD prophylaxis and treatment (52, 53). Only small retrospective studies have shown promising response rates for the treatment of steroid-refractory aGVHD, mostly in case of intestinal tract involvement (54–57). However, the benefit of infliximab in steroid-refractory aGVHD does not seem to be superior to the one observed with other drugs available (58), even though prospective randomized trials are lacking. Moreover, other studies have shown that responses after infliximab therapy are poorly sustained and have raised concern over a heightened risk of severe infections (59, 60).
Regarding etanercept, a single center prospective study showed a promising response rate when combining etanercept with standard high-dose corticosteroids for first-line treatment of aGVHD compared with a cohort of contemporaneous case-matched patients treated with high-dose corticosteroids alone (61). However, the higher response rate observed with etanercept did not translate into a significantly superior survival at 6 months from aGVHD onset. Moreover, a multicenter prospective randomized “pick the winner” study comparing four promising molecules in combination with corticosteroids for first-line aGVHD treatment identified mycophenolate mofetil, and not etanercept, as the most promising agent (62). However, mycophenolate mofetil failed to prove any benefit in the subsequent multicenter, randomized, double-blinded, and placebo-controlled phase 3 trial evaluating its addition to standard corticosteroids (63). In the setting of steroid-refractory aGVHD, two small single center studies have shown only a modest effect of etanercept with few complete responses (64, 65). As for infliximab, efficacy seemed to be higher in case of gut involvement. Finally, a phase 2 study involving 100 patients also evaluated etanercept as part of aGVHD prophylaxis, in combination with tacrolimus and low-dose methotrexate (66). Once again, the benefit of etanercept was not obvious, as its addition to standard prophylaxis did not affect the overall risk of grade 2–4 aGVHD, as compared with a control cohort of 161 previously reported patients. Only a potential benefit among non-total-body-irradiated patients was suggested in this study. TNFR1 plasma level monitoring can also be used to evaluate and/or predict response to treatment with etanercept, as a significant reduction in these levels has been observed in responding patients (61, 66). To summarize, the current place of anti-TNF treatments in the arsenal of aGVHD is only limited to a therapeutic option for steroid-refractory disease, mostly in case of intestinal tract involvement. A possible explanation of this failure is that blocking TNF would also impact on the TNFR2-dependent Treg boost that is protective in GVHD as suggested by experimental data discussed below.
Regulatory T cells modulate alloreactivity during allo-SCT. Cell therapy using Tregs efficiently control GVHD (67, 68) whereas Treg deletion can be used to boost the graft-versus-leukemia (GVL) effect (69). Thus, some research teams envisioned TNFR2 as a potential target to act directly on Tregs in this setting and modulate alloreactivity with either TNFR2 agonists or antagonists. In this regard, three important studies were published almost simultaneously in 2016 (70–72).
In a murine model of aGVHD prevention relying on Treg infusion, we have clearly shown using three different experimental approaches that the protective effect mediated by therapeutic Tregs was dependent on TNF produced by pathogenic Tconv and TNFR2 expressed by Tregs (71). Indeed, when blocking the TNF/TNFR2 interaction with an anti-TNFR2 mAb, or when using either TNFR2-deficient therapeutic Tregs or TNF-deficient Tconvs, aGVHD prevention was abolished in all cases, highlighting a boost of alloreactivity after TNF/TNFR2 blockade. Moreover, Treg and Tconv phenotypes were also modified, with the former displaying decreased expression of activation and suppression markers while the latter showed increased production of pro-inflammatory cytokines.
The second study was published by Chopra and colleagues, who developed a TNFR2 agonist called STAR2 (70). In vitro, STAR2 was able to stimulate expansion and activation of Tregs, an effect not observed with Tconvs. This selective Treg expansion and activation was also triggered in vivo, when mice were treated with STAR2 intraperitoneal injections. Most of all, in a murine model of aGVHD, pretransplant administration of STAR2 to recipient mice protected from aGVHD and significantly increased survival. The protective effect of STAR2 was associated with a preserved GVL effect and had no deleterious effect on posttransplant anti-cytomegalovirus immune reconstitution.
Finally, in the study of Pierini and colleagues, therapeutic Tregs were preincubated in vitro with TNF (+IL-2) for a short period (72). This TNF priming resulted in a higher expression of Foxp3 and activation/suppression markers by Tregs and a higher proliferation rate. Most interestingly, when such “TNF-primed Tregs” were infused to recipient mice in an aGVHD murine model, this resulted in prolonged survival, increased weight gain, and improved GVHD clinical score, even at the very low 1:10 Treg:Tconv ratio. In this study also, the beneficial effect of TNF priming did not come with a detrimental loss of the GVL effect.
Altogether, the results of these three studies pave the way for TNFR2 targeting to modulate alloreactivity after allo-SCT (Figure 3A). Additional preclinical data are needed, especially regarding the effect of TNFR2 agonists and antagonists on various human cell types (Tconvs, Tregs, and cancer cells) in vitro, before the start-up of clinical trials evaluating their efficacy and safety for prevention and/or treatment of aGVHD and posttransplant relapse of hematologic disease, respectively. Notably, in the setting of aGVHD prevention, single center clinical trials have shown the high potential of Treg cell therapy (73, 74). However, adoptive transfer of such cells is limited by the small proportion of Tregs among peripheral blood mononuclear cells (PBMCs) that necessitates an ex vivo culture for expansion before infusion to the patient. In this regard, the direct administration of a TNFR2 agonist to the patient to selectively activate and expand in vivo Tregs with the highest suppressive capacity holds the promise of a more simple, costless, less time consuming, and possibly more efficient method.
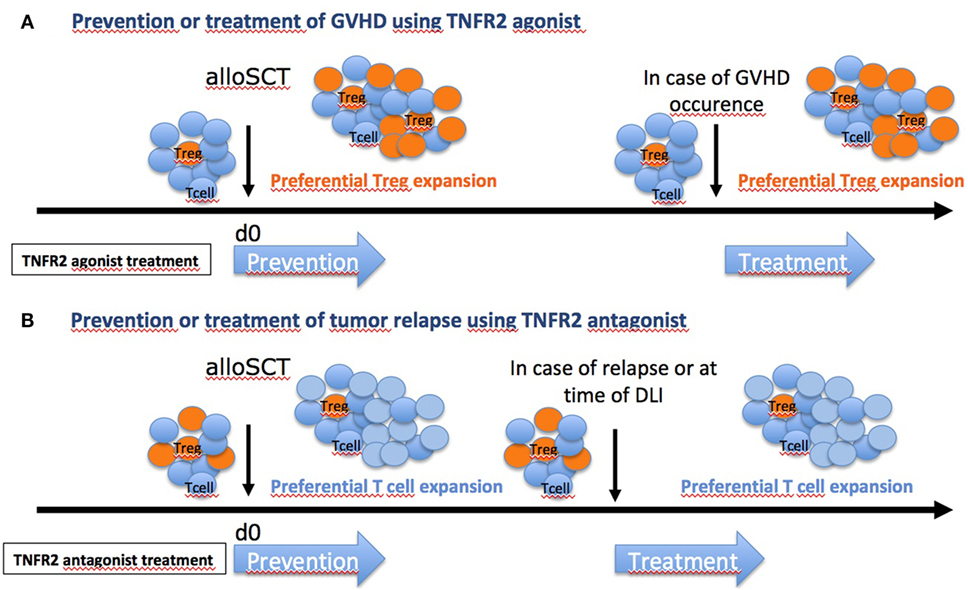
Figure 3. Hope in targeting TNFR2 [and regulatory T cells (Tregs)] in graft-versus-host disease (GVHD). Depending on the clinical situation and the risk for the patient to develop or not GVHD, different therapeutic strategies could be envisaged. (A) For patients with elevated risk of GVHD (unrelated donor or one or several HLA mismatch), TNFR2 agonist could be administered to recipients before allo-SCT, as shown previously (73), or at time of grafting to boost Tregs. Patients could also be treated at time of GVHD occurrence. (B) For patients with elevated risk of relapse (aggressive leukemia, geno-identical allo-SCT), anti-TNFR2 could be administered to recipients at time of grafting to inhibit Tregs. In case of tumor relapse, patients could also be treated at time of donor lymphocyte infusion (DLI) to block Treg effect.
Several clinical trials have clearly demonstrated that modulation of the immune response can improve the overall survival of advanced stage cancer patients (Figure 4) (75, 76). Indeed, since the approval of a-CTLA-4 antibody treatment for metastatic melanoma in 2011 (77–79), the field has witnessed the advent of numerous therapeutic approaches modulating the immune response (80, 81). Blockade of programmed death 1 and its major ligand PD-L1 has given impressive and durable clinical results (82–84) and fueled clinical evaluation (85) of (i) new inhibitory checkpoint targets, such as LAG-3 (86), TIM-3 (87), VISTA (88), and TIGIT (89), (ii) agonistic antibodies targeted to co-stimulatory receptors, such as 4-1BB (90), GITR (91), CD40 (92), and OX40 (93), (iii) cell-based therapies using dendritic cells, tumor-infiltrating lymphocytes (TILs), and genetically engineered T cells (CAR-T cells) (94), (iv) immune modulators such as innate ligands (95), and (v) vaccines, notably directed to neo-epitopes (94, 96). Along these lines, dozens of antitumor immunotherapeutic approaches have been already approved by regulatory agencies and thousands of such clinical trials are currently ongoing. Nevertheless, only 20–40% of patients benefit from these therapies, and some cases of resistance have been described (97–99).
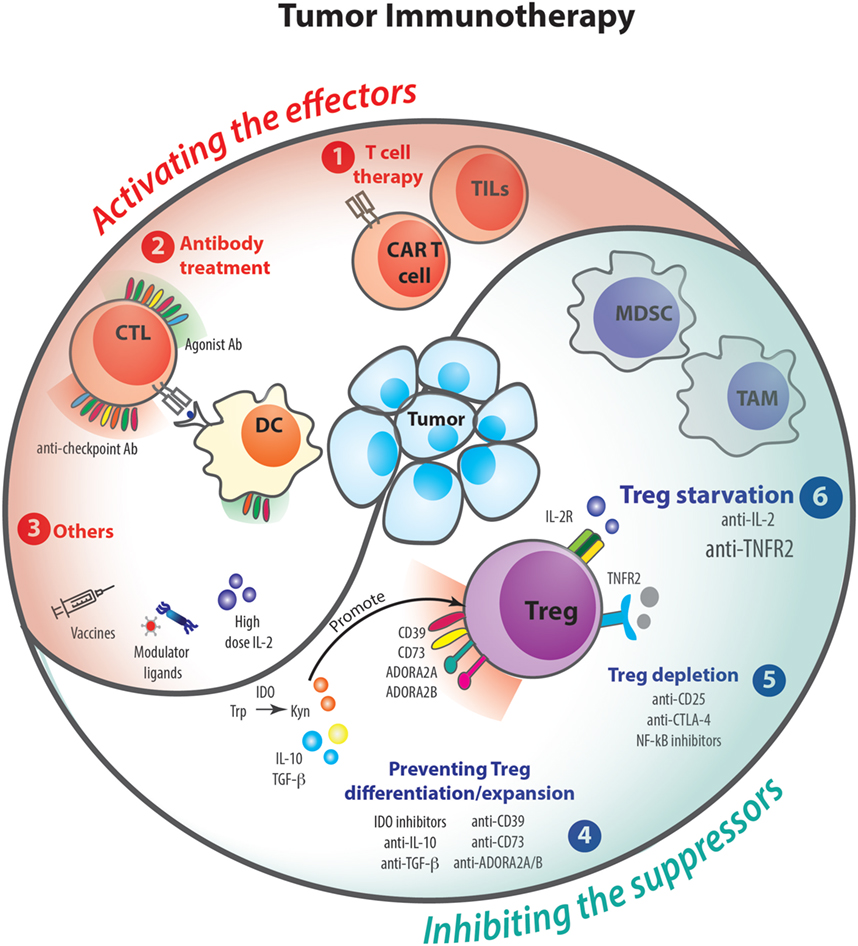
Figure 4. Tumor immunotherapies and regulatory T cells (Tregs). Over the past decades, several clinical trials and animal models have demonstrated that therapies acting on the immune response can help to fight against cancer. To control tumoral process, immunotherapies can either activate the effector arm of the immune response (1–3) or inhibit the suppressor mechanisms (4–6). The following therapies that potentiate T cell responses have been proposed: tumor-infiltrating lymphocytes (TILs) and CAR T cell thrapies (1); agonist and anti-checkpoint antibody treatments (2); other therapies such as vaccines, modulator ligands, and high doses of IL-2 (3). On the other hand, cancers promote suppressor mechanisms involving Tregs or myeloid-derived suppressor cell (MDSC), which are able to inhibit effector cells. Some treatments are being tested to modulate Treg suppression by preventing their differentiation/expansion (4), Treg depletion (5), or starvation (6).
Most of the abovementioned treatments are thought to work mainly by (re)-activating the cytotoxic arm of the immune response (100–102), namely, CD8+ T cells and NK cells; and by rescuing them from exhaustion (103, 104). Nevertheless, as the antitumoral immune response is also highly curtailed by Tregs, overcoming Treg-mediated immunosuppression in the tumor microenvironment (105, 106) represents a sound alternative for successful cancer immunotherapy. Of note, this can be obtained either by depleting Tregs or by inhibiting their function in vivo (107).
The first proof of the beneficial effect of Treg depletion on the antitumoral response was brought forward by Onizuka et al. (108). They showed that administration of an anti-CD25 antibody (mAb; PC61) had prophylactic, although not therapeutic efficacy, probably due to the concurrent elimination of CD25-expressing activated effector lymphocytes. More recently, the group of Quezada (109) has shown that anti-CD25 antibody-mediated Treg depletion can be ineffective due to the high expression of the inhibitory Fc receptor FcgRIIb by cells present in the tumor microenvironment. Consequently, anti-CD25 antibodies designed to avoid FcgRIIb binding induced massive Treg depletion in the tumor and led to impressive tumor regression. Also, specific depletion of Tregs in transgenic DEREG mice (110), which express a diphtheria toxin receptor under the control of the Foxp3 regulatory sequences, resulted in a partial regression of established melanoma that correlated with CD8+ T cell accumulation in the tumor. Furthermore, mouse studies point out that anti-CTLA-4 antibodies mainly act by eliminating or inhibiting the tumor-associated Tregs (which highly express this molecule) rather than by reinvigorating exhausted T cells (111, 112). Indeed, controversial results have been observed with Daclizumab (an anti-CD25 antibody) and with a fusion protein between the IL-2 and the diphtheria toxin (Ontak) (113–118). Thus, direct proofs of the beneficial effect of Treg depletion in human are still missing for solid cancers, and there are to date no clinical tools that specifically target this population. This point is more advanced in the field of onco-hematology. With the intent of preventing or treating post allo-SCT relapse of hematologic disease, GVL effect can be activated by donor lymphocyte infusion (DLI). In this setting, the harmful effect of Tregs after DLI was suggested by a study in which the authors quantified Tregs in DLI products and demonstrated that patients with a durable complete remission of their malignancy after DLI had received a lower number of Tregs (119). This observation led to the idea of depleting Tregs to improve responses to DLIs, an approach that was successfully tested in a clinical trial in which a magnetic depletion of CD25+ cells was performed on donor PBMCs before their infusion to recipients that were considered “alloreactivity resistant” (69).
Besides Treg depletion, tumor-associated Tregs can be therapeutically targeted by the modulation of the tumor microenvironment. Indeed, cancer cells produce metabolites, cytokines, and growth factors that can (i) promote Treg accumulation and expansion, (ii) enhance Treg function, and even (iii) induce Treg conversion from conventional CD4+ T cells (96). Among them, adenosine—generated upon catabolism of ATP by the ectoenzymes CD39 and CD73—and kynurenines—generated upon catabolism of tryptophan by the indoleamine 2,3-dioxygenase (IDO) enzyme—favor the accumulation, conversion, and expansion of Tregs and suppression of effector T cells (120). Accordingly, IDO inhibitors and either antagonists of A2A/A2B adenosine receptor or anti-CD39 and anti-CD73 antibodies significantly decrease the rate of Treg peripheral conversion and impair tumor growth (108, 121–124). Furthermore, therapeutic agents targeting these molecules in combination with immune checkpoint inhibitors show additive or synergistic effects in experimental tumor models, and their combination is currently under clinical investigation (96, 125). In addition, therapies aiming at inhibition of CD4+ T cell differentiation into Tregs have been tested. Among them, the effects of neutralizing antibodies or pharmacologic inhibitors of IL-10 and TGF-β have been evaluated in preclinical and clinical settings (126–128). These studies have demonstrated both pro- and antitumoral effects, probably due to their complex involvement in immune and non-immune processes. Moreover, there are not consistent data on the effect of these therapies on Tregs. Overall, manipulation of Treg induction and function through inhibition of metabolic and biochemical pathways active in the tumor microenvironment represent an alternative immunotherapeutic approach. Nevertheless, the significant side effects associated with the involvement of these pathways in different physiological processes must be taken into consideration.
On top of the abovementioned strategies designed to disarm Tregs for therapeutic aims, “cytokine starvation or cytokine deviation” represents an alternative promising approach. Namely, deprivation of Tregs from IL-2 and TNF—two key cytokines essential for their biology—should lead to Treg dysfunction or death. Clinical manipulation of IL-2 levels remains complex as IL-2 can act both as an immune stimulating or suppressive cytokine, depending on the dose. On one hand, low-doses of IL-2 favor Treg survival and suppressive function and lead to a better control of autoimmune and inflammatory diseases (129–131). On the other hand, high-dose IL-2 administration boosts effector immunity and, consequently, enhances antiviral or antitumoral responses (132, 133). Noteworthy, in the cancer setting, low efficacy of high-dose IL-2 administration (134) can be explained in part by the unwanted effect of IL-2 on Tregs, which constitutively express the high affinity IL-2 receptor [composed by three subunits: IL-2-Rα (CD25), IL-2Rβ and IL-2Rγ] (135). For efficient antitumoral effect, there is a need to activate CD8+ T and NK cells, which also respond to IL-2 through the intermediate affinity IL-2 receptor, composed of IL-2Rβ and IL-2Rγ (136, 137). Interestingly, to prevent the IL-2 critical signal on Tregs, IL-2/anti-IL-2 antibody complexes, formed by an anti-IL-2 antibody acting as a CD25 mimotope hampering IL-2 fixation to CD25, were used to redirect IL-2 action to CD8+ T and NK cells (138). Of note, mutant IL-2 proteins have been designed to bear reduced binding affinity to CD25 and preserved affinity for IL-2Rβ, endowing them with preferential action on NK and CD8+ T cells. As for IL-2, depriving Tregs from TNF may also impair their function and improve antitumoral responses as detailed below. Thus, starvation of cytokine, such as IL-2, may emerge as a new firearm among the arsenal of immunotherapeutic strategies, which either alone or in combination, enrich the picture of immune checkpoint inhibitors available to fight cancers.
As suggested by its name, TNF was described initially as a killer of cancer cells. We now know that this cytokine plays a complex role in cancer and tumor immunity because of its pleiotropic effect and the fact that it has two receptors. Actually, TNF is even considered mostly as a pro-tumor cytokine. Numerous mouse studies have shown that anti-TNF drugs reduced tumor growth in different types of cancers. This deleterious effect of TNF was further supported in TNF knockout mice that display reduced tumor growth (139–146). The individual role of TNFR1 and TNFR2 was assessed in knockout mice in some of these studies.
The pro-tumoral effect of TNF has been explained by different mechanisms (Figure 5). TNF may directly promote cell transformation by activating oncogenes and inducing DNA damage (147). It may stimulate cell proliferation favoring cell transformation and neovascularization that is critical in cancer development (144, 146, 148). TNF may also promote growth of tumors that benefit from inflammatory cytokines and chemokines by recruiting neutrophils and macrophages (139, 141, 145, 149). Also, TNF may favor tumor invasiveness and metastasis by stimulating matrix metalloproteinase production and vascular permeability (150). When analyzed, the role of TNFR1 rather than TNFR2 was involved in these different mechanisms.
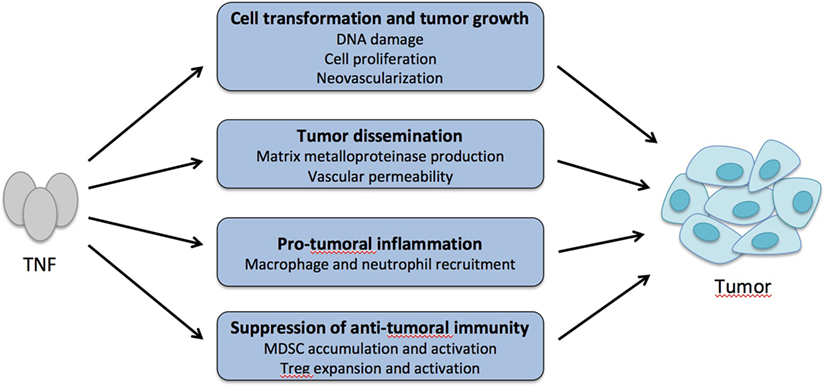
Figure 5. Tumor necrosis factor α (TNF) is a pro-tumoral cytokine. TNF may promote cell transformation and tumor growth by increasing DNA damage and mutations, abnormal cell proliferation, and neovascularization. TNF may also favor tumor cell dissemination by increasing matrix metalloproteinase production and vascular permeability and leakiness. By recruiting macrophages and neutrophils in the tumor environment that release inflammatory cytokines and chemokines, TNF may promote growth of tumors that respond to these inflammatory factors. Finally, by boosting the activity of myeloid-derived suppressor cells (MDSC) and regulatory T cells (Treg) in the tumor environment, TNF may indirectly suppress antitumor immunity.
In mouse models of cancers, reduced tumor growth in mice treated by anti-TNF drugs or in TNFR2 knockout mice was associated with decreased numbers of MDSCs suggesting that TNF increases survival, recruitment, or function of MDSCs that suppress antitumor immunity (Figure 5) (20, 140, 143, 151). In a mouse model of melanoma, TNF injection favored tumor metastasis by acting on TNFR2-expressing hematopoietic cells, which was associated with an increase of Tregs (142). A similar mechanism was observed in models of colorectal cancer and hepatocarcinoma, since the tumor-dependent Treg expansion was abolished with an anti-TNFR2 mAb. Also, pretreatment of Tregs with TNF increased their capacity to suppress antitumor immunity after adoptive transfer (152).
In the setting of hematologic tumor relapse after allo-SCT, a similar approach using an anti-TNFR2 blocking mAb or TNFR2 antagonist could be considered to inactivate the deleterious effect of Tregs (Figure 3B). These molecules may be administered directly to the recipient to prevent or treat hematologic relapse, with or without a combined DLI, or even be used to preincubate donor PBMCs before infusion, to inactivate Tregs contained in the product.
All the above studies were performed in mice. What do we know about the role of TNF in cancer in patients? It is well described that some cancers, such as colorectal cancer and hepatocarcinoma, benefit from chronic inflammation. Importantly, recent meta-analyses of patients receiving anti-TNF treatment because of their autoimmune diseases did not show an increased risk of cancer development (153, 154). Also, because of the beneficial effect of anti-TNF administration in preclinical mouse models, some patients with advanced cancers received TNF blockers. In this phase II trial, infliximab and etanercept were well tolerated (155, 156). The possible effects of these treatments have been studied in vitro or in vivo after xeno-transplantation in immunodeficient mice. Results indicated that blocking TNF may reduce tumor growth, which is associated with reduced tumor dissemination, angiogenesis, and infiltration with myeloid cells (157–159). Finally, TNF may suppress antitumor immunity by boosting Tregs via TNFR2 since high amounts of TNFR2+ Tregs were associated with more severe lung and ovarian cancer (160, 161).
It has to be emphasized that these studies that provide possible mechanisms to explain the supratumoral effect of TNF were only based on correlations or in vitro observations. None of them has provided definitive in vivo proofs because of the pleiotropic effect of TNF. This would have required, for instance, conditional deletion of TNFR in a cell subset. However, based on what is known on the effect of TNF on Tregs and of Tregs on antitumoral immunity (see above), the possibility that TNF inhibits antitumor immunity by boosting Tregs is a very attractive hypothesis that may play a major role in some cancer types.
Immunotherapy of cancers is a promising land but unfortunately only a minority of patients responds to these treatments. Among multiple targets that are being tested, TNFR2 is an attractive one. Indeed, TNF blockade may have different impacts by limiting cell transformation, neovascularization, or pro-tumoral inflammation and may boost antitumor immunity by acting on MDSC or Tregs. Recent works suggest that targeting TNFR2-expressing Tregs would be a safe and efficient way to stimulate antitumor immunity. Future experiments and clinical trials are required to validate this new therapy.
BS, EP, and JC organized the plan and structure of the manuscript, and all the authors contributed to the redaction.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by Fondation pour la recherche médicale (Equipe FRM 2015), Agence Nationale de la Recherche (ANR-RF-2015-01), Fédération Leucémie Espoir, INCA/DGOS PRT-K 2015, and Fondation Ramsay Générale de Santé.
1. Aggarwal BB. Tumour necrosis factors receptor associated signalling molecules and their role in activation of apoptosis, JNK and NF-kappaB. Ann Rheum Dis (2000) 59(Suppl 1):i6–16. doi:10.1136/ard.59.suppl_1.i6
2. Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol (2003) 3:745–56. doi:10.1038/nri1184
3. Gordon C, Ranges GE, Greenspan JS, Wofsy D. Chronic therapy with recombinant tumor necrosis factor-alpha in autoimmune NZB/NZW F1 mice. Clin Immunol Immunopathol (1989) 52:421–34. doi:10.1016/0090-1229(89)90157-8
4. Jacob CO, Aiso S, Michie SA, McDevitt HO, Acha-Orbea H. Prevention of diabetes in nonobese diabetic mice by tumor necrosis factor (TNF): similarities between TNF-alpha and interleukin 1. Proc Natl Acad Sci U S A (1990) 87:968–72. doi:10.1073/pnas.87.3.968
5. Jacob CO, McDevitt HO. Tumour necrosis factor-alpha in murine autoimmune ‘lupus’ nephritis. Nature (1988) 331:356–8. doi:10.1038/331356a0
6. Wu AJ, Hua H, Munson SH, McDevitt HO. Tumor necrosis factor-alpha regulation of CD4+CD25+ T cell levels in NOD mice. Proc Natl Acad Sci U S A (2002) 99:12287–92. doi:10.1073/pnas.172382999
7. Yang XD, Tisch R, Singer SM, Cao ZA, Liblau RS, Schreiber RD, et al. Effect of tumor necrosis factor alpha on insulin-dependent diabetes mellitus in NOD mice. I. The early development of autoimmunity and the diabetogenic process. J Exp Med (1994) 180:995–1004. doi:10.1084/jem.180.3.995
8. De Bandt M, Sibilia J, Le Loet X, Prouzeau S, Fautrel B, Marcelli C, et al. Systemic lupus erythematosus induced by anti-tumour necrosis factor alpha therapy: a French national survey. Arthritis Res Ther (2005) 7:R545–51. doi:10.1186/ar1715
9. De Rycke L, Baeten D, Kruithof E, Van den Bosch F, Veys EM, De Keyser F. The effect of TNFalpha blockade on the antinuclear antibody profile in patients with chronic arthritis: biological and clinical implications. Lupus (2005) 14:931–7. doi:10.1191/0961203305lu2240rr
10. Ko JM, Gottlieb AB, Kerbleski JF. Induction and exacerbation of psoriasis with TNF-blockade therapy: a review and analysis of 127 cases. J Dermatolog Treat (2009) 20:100–8. doi:10.1080/09546630802441234
11. Mohan N, Edwards ET, Cupps TR, Oliverio PJ, Sandberg G, Crayton H, et al. Demyelination occurring during anti-tumor necrosis factor alpha therapy for inflammatory arthritides. Arthritis Rheum (2001) 44:2862–9. doi:10.1002/1529-0131(200112)44:12<2862::AID-ART474>3.0.CO;2-W
12. Ramos-Casals M, Brito-Zeron P, Cuadrado MJ, Khamashta MA. Vasculitis induced by tumor necrosis factor-targeted therapies. Curr Rheumatol Rep (2008) 10:442–8. doi:10.1007/s11926-008-0072-z
13. Ramos-Casals M, Brito-Zeron P, Soto MJ, Cuadrado MJ, Khamashta MA. Autoimmune diseases induced by TNF-targeted therapies. Best Pract Res Clin Rheumatol (2008) 22:847–61. doi:10.1016/j.berh.2008.09.008
14. van Oosten BW, Barkhof F, Truyen L, Boringa JB, Bertelsmann FW, von Blomberg BM, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology (1996) 47:1531–4. doi:10.1212/WNL.47.6.1531
15. Chen X, Baumel M, Mannel DN, Howard OM, Oppenheim JJ. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol (2007) 179:154–61. doi:10.4049/jimmunol.179.1.154
16. Bazzaz JT, Amoli MM, Taheri Z, Larijani B, Pravica V, Hutchinson IV. TNF-alpha and IFN-gamma gene variation and genetic susceptibility to type 1 diabetes and its microangiopathic complications. J Diabetes Metab Disord (2014) 13:46. doi:10.1186/2251-6581-13-46
17. Beyer M, Abdullah Z, Chemnitz JM, Maisel D, Sander J, Lehmann C, et al. Tumor-necrosis factor impairs CD4(+) T cell-mediated immunological control in chronic viral infection. Nat Immunol (2016) 17:593–603. doi:10.1038/ni.3399
18. Ban L, Zhang J, Wang L, Kuhtreiber W, Burger D, Faustman DL. Selective death of autoreactive T cells in human diabetes by TNF or TNF receptor 2 agonism. Proc Natl Acad Sci U S A (2008) 105:13644–9. doi:10.1073/pnas.0803429105
19. Hu X, Li B, Li X, Zhao X, Wan L, Lin G, et al. Transmembrane TNF-alpha promotes suppressive activities of myeloid-derived suppressor cells via TNFR2. J Immunol (2014) 192:1320–31. doi:10.4049/jimmunol.1203195
20. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest (2012) 122:4094–104. doi:10.1172/JCI64115
21. Chen X, Subleski JJ, Kopf H, Howard OM, Mannel DN, Oppenheim JJ. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol (2008) 180:6467–71. doi:10.4049/jimmunol.180.10.6467
22. Faustman DL, Davis M. TNF receptor 2 and disease: autoimmunity and regenerative medicine. Front Immunol (2013) 4:478. doi:10.3389/fimmu.2013.00478
23. Notley CA, Inglis JJ, Alzabin S, McCann FE, McNamee KE, Williams RO. Blockade of tumor necrosis factor in collagen-induced arthritis reveals a novel immunoregulatory pathway for Th1 and Th17 cells. J Exp Med (2008) 205:2491–7. doi:10.1084/jem.20072707
24. Bystrom J, Clanchy FI, Taher TE, Mangat P, Jawad AS, Williams RO, et al. TNFalpha in the regulation of Treg and Th17 cells in rheumatoid arthritis and other autoimmune inflammatory diseases. Cytokine (2018) 101:4–13. doi:10.1016/j.cyto.2016.09.001
25. Xueyi L, Lina C, Zhenbiao W, Qing H, Qiang L, Zhu P. Levels of circulating Th17 cells and regulatory T cells in ankylosing spondylitis patients with an inadequate response to anti-TNF-alpha therapy. J Clin Immunol (2013) 33:151–61. doi:10.1007/s10875-012-9774-0
26. Hamano R, Huang J, Yoshimura T, Oppenheim JJ, Chen X. TNF optimally activatives regulatory T cells by inducing TNF receptor superfamily members TNFR2, 4-1BB and OX40. Eur J Immunol (2011) 41:2010–20. doi:10.1002/eji.201041205
27. Arvey A, van der Veeken J, Plitas G, Rich SS, Concannon P, Rudensky AY. Genetic and epigenetic variation in the lineage specification of regulatory T cells. Elife (2015) 4:e07571. doi:10.7554/eLife.07571
28. Delacher M, Imbusch CD, Weichenhan D, Breiling A, Hotz-Wagenblatt A, Trager U, et al. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat Immunol (2017) 18:1160–72. doi:10.1038/ni.3799
29. Mahmud SA, Manlove LS, Schmitz HM, Xing Y, Wang Y, Owen DL, et al. Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nat Immunol (2014) 15:473–81. doi:10.1038/ni.2849
30. Chen X, Subleski JJ, Hamano R, Howard OM, Wiltrout RH, Oppenheim JJ. Co-expression of TNFR2 and CD25 identifies more of the functional CD4+FOXP3+ regulatory T cells in human peripheral blood. Eur J Immunol (2010) 40:1099–106. doi:10.1002/eji.200940022
31. Chen X, Wu X, Zhou Q, Howard OM, Netea MG, Oppenheim JJ. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. J Immunol (2013) 190:1076–84. doi:10.4049/jimmunol.1202659
32. Chen X, Oppenheim JJ. The phenotypic and functional consequences of tumour necrosis factor receptor type 2 expression on CD4(+) FoxP3(+) regulatory T cells. Immunology (2011) 133:426–33. doi:10.1111/j.1365-2567.2011.03460.x
33. Okubo Y, Mera T, Wang L, Faustman DL. Homogeneous expansion of human T-regulatory cells via tumor necrosis factor receptor 2. Sci Rep (2013) 3:3153. doi:10.1038/srep03153
34. Torrey H, Butterworth J, Mera T, Okubo Y, Wang L, Baum D, et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci Signal (2017) 10:eaaf8608. doi:10.1126/scisignal.aaf8608
35. Zaragoza B, Chen X, Oppenheim JJ, Baeyens A, Gregoire S, Chader D, et al. Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat Med (2016) 22:16–7. doi:10.1038/nm.4019
36. Nagar M, Jacob-Hirsch J, Vernitsky H, Berkun Y, Ben-Horin S, Amariglio N, et al. TNF activates a NF-kappaB-regulated cellular program in human CD45RA- regulatory T cells that modulates their suppressive function. J Immunol (2010) 184:3570–81. doi:10.4049/jimmunol.0902070
37. Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med (2013) 19:322–8. doi:10.1038/nm.3085
38. Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood (2006) 108:253–61. doi:10.1182/blood-2005-11-4567
39. Zanin-Zhorov A, Ding Y, Kumari S, Attur M, Hippen KL, Brown M, et al. Protein kinase C-theta mediates negative feedback on regulatory T cell function. Science (2010) 328:372–6. doi:10.1126/science.1186068
40. Baeyens A, Saadoun D, Billiard F, Rouers A, Gregoire S, Zaragoza B, et al. Effector T cells boost regulatory T cell expansion by IL-2, TNF, OX40, and plasmacytoid dendritic cells depending on the immune context. J Immunol (2015) 194:999–1010. doi:10.4049/jimmunol.1400504
41. Grinberg-Bleyer Y, Saadoun D, Baeyens A, Billiard F, Goldstein JD, Gregoire S, et al. Pathogenic T cells have a paradoxical protective effect in murine autoimmune diabetes by boosting Tregs. J Clin Invest (2010) 120:4558–68. doi:10.1172/JCI42945
42. Joedicke JJ, Myers L, Carmody AB, Messer RJ, Wajant H, Lang KS, et al. Activated CD8+ T cells induce expansion of Vbeta5+ regulatory T cells via TNFR2 signaling. J Immunol (2014) 193:2952–60. doi:10.4049/jimmunol.1400649
43. Myers L, Joedicke JJ, Carmody AB, Messer RJ, Kassiotis G, Dudley JP, et al. IL-2-independent and TNF-alpha-dependent expansion of Vbeta5+ natural regulatory T cells during retrovirus infection. J Immunol (2013) 190:5485–95. doi:10.4049/jimmunol.1202951
44. Housley WJ, Adams CO, Nichols FC, Puddington L, Lingenheld EG, Zhu L, et al. Natural but not inducible regulatory T cells require TNF-alpha signaling for in vivo function. J Immunol (2011) 186:6779–87. doi:10.4049/jimmunol.1003868
45. Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet (2009) 373:1550–61. doi:10.1016/S0140-6736(09)60237-3
46. Choi SW, Kitko CL, Braun T, Paczesny S, Yanik G, Mineishi S, et al. Change in plasma tumor necrosis factor receptor 1 levels in the first week after myeloablative allogeneic transplantation correlates with severity and incidence of GVHD and survival. Blood (2008) 112:1539–42. doi:10.1182/blood-2008-02-138867
47. Kitko CL, Paczesny S, Yanik G, Braun T, Jones D, Whitfield J, et al. Plasma elevations of tumor necrosis factor-receptor-1 at day 7 postallogeneic transplant correlate with graft-versus-host disease severity and overall survival in pediatric patients. Biol Blood Marrow Transplant (2008) 14:759–65. doi:10.1016/j.bbmt.2008.04.002
48. Fowler DH, Foley J, Whit-Shan Hou J, Odom J, Castro K, Steinberg SM, et al. Clinical “cytokine storm” as revealed by monocyte intracellular flow cytometry: correlation of tumor necrosis factor alpha with severe gut graft-versus-host disease. Clin Gastroenterol Hepatol (2004) 2:237–45. doi:10.1016/S1542-3565(04)00011-4
49. Holler E, Kolb HJ, Moller A, Kempeni J, Liesenfeld S, Pechumer H, et al. Increased serum levels of tumor necrosis factor alpha precede major complications of bone marrow transplantation. Blood (1990) 75:1011–6.
50. Ritchie D, Seconi J, Wood C, Walton J, Watt V. Prospective monitoring of tumor necrosis factor alpha and interferon gamma to predict the onset of acute and chronic graft-versus-host disease after allogeneic stem cell transplantation. Biol Blood Marrow Transplant (2005) 11:706–12. doi:10.1016/j.bbmt.2005.05.015
51. Paczesny S, Krijanovski OI, Braun TM, Choi SW, Clouthier SG, Kuick R, et al. A biomarker panel for acute graft-versus-host disease. Blood (2009) 113:273–8. doi:10.1182/blood-2008-07-167098
52. Couriel DR, Saliba R, de Lima M, Giralt S, Andersson B, Khouri I, et al. A phase III study of infliximab and corticosteroids for the initial treatment of acute graft-versus-host disease. Biol Blood Marrow Transplant (2009) 15:1555–62. doi:10.1016/j.bbmt.2009.08.003
53. Hamadani M, Hofmeister CC, Jansak B, Phillips G, Elder P, Blum W, et al. Addition of infliximab to standard acute graft-versus-host disease prophylaxis following allogeneic peripheral blood cell transplantation. Biol Blood Marrow Transplant (2008) 14:783–9. doi:10.1016/j.bbmt.2008.04.006
54. Couriel D, Saliba R, Hicks K, Ippoliti C, de Lima M, Hosing C, et al. Tumor necrosis factor-alpha blockade for the treatment of acute GVHD. Blood (2004) 104:649–54. doi:10.1182/blood-2003-12-4241
55. Jacobsohn DA, Hallick J, Anders V, McMillan S, Morris L, Vogelsang GB. Infliximab for steroid-refractory acute GVHD: a case series. Am J Hematol (2003) 74:119–24. doi:10.1002/ajh.10392
56. Patriarca F, Sperotto A, Damiani D, Morreale G, Bonifazi F, Olivieri A, et al. Infliximab treatment for steroid-refractory acute graft-versus-host disease. Haematologica (2004) 89:1352–9.
57. Sleight BS, Chan KW, Braun TM, Serrano A, Gilman AL. Infliximab for GVHD therapy in children. Bone Marrow Transplant (2007) 40:473–80. doi:10.1038/sj.bmt.1705761
58. Martin PJ, Rizzo JD, Wingard JR, Ballen K, Curtin PT, Cutler C, et al. First- and second-line systemic treatment of acute graft-versus-host disease: recommendations of the American society of blood and marrow transplantation. Biol Blood Marrow Transplant (2012) 18:1150–63. doi:10.1016/j.bbmt.2012.04.005
59. Yalniz FF, Hefazi M, McCullough K, Litzow MR, Hogan WJ, Wolf R, et al. Safety and efficacy of infliximab therapy in the setting of steroid-refractory acute graft-versus-host disease. Biol Blood Marrow Transplant (2017) 23(9):1478–84. doi:10.1016/j.bbmt.2017.05.001
60. Yang J, Cheuk DK, Ha SY, Chiang AK, Lee TL, Ho MH, et al. Infliximab for steroid refractory or dependent gastrointestinal acute graft-versus-host disease in children after allogeneic hematopoietic stem cell transplantation. Pediatr Transplant (2012) 16:771–8. doi:10.1111/j.1399-3046.2012.01756.x
61. Levine JE, Paczesny S, Mineishi S, Braun T, Choi SW, Hutchinson RJ, et al. Etanercept plus methylprednisolone as initial therapy for acute graft-versus-host disease. Blood (2008) 111:2470–5. doi:10.1182/blood-2007-09-112987
62. Alousi AM, Weisdorf DJ, Logan BR, Bolanos-Meade J, Carter S, Difronzo N, et al. Etanercept, mycophenolate, denileukin, or pentostatin plus corticosteroids for acute graft-versus-host disease: a randomized phase 2 trial from the blood and marrow transplant clinical trials network. Blood (2009) 114:511–7. doi:10.1182/blood-2009-03-212290
63. Bolanos-Meade J, Logan BR, Alousi AM, Antin JH, Barowski K, Carter SL, et al. Phase 3 clinical trial of steroids/mycophenolate mofetil vs steroids/placebo as therapy for acute GVHD: BMT CTN 0802. Blood (2014) 124:3221–7. doi:10.1182/blood-2014-06-577023
64. Busca A, Locatelli F, Marmont F, Ceretto C, Falda M. Recombinant human soluble tumor necrosis factor receptor fusion protein as treatment for steroid refractory graft-versus-host disease following allogeneic hematopoietic stem cell transplantation. Am J Hematol (2007) 82:45–52. doi:10.1002/ajh.20752
65. Park JH, Lee HJ, Kim SR, Song GW, Lee SK, Park SY, et al. Etanercept for steroid-refractory acute graft versus host disease following allogeneic hematopoietic stem cell transplantation. Korean J Intern Med (2014) 29:630–6. doi:10.3904/kjim.2014.29.5.630
66. Choi SW, Stiff P, Cooke K, Ferrara JL, Braun T, Kitko C, et al. TNF-inhibition with etanercept for graft-versus-host disease prevention in high-risk HCT: lower TNFR1 levels correlate with better outcomes. Biol Blood Marrow Transplant (2012) 18:1525–32. doi:10.1016/j.bbmt.2012.03.013
67. Cohen JL, Trenado A, Vasey D, Klatzmann D, Salomon BL. CD4(+)CD25(+) immunoregulatory T Cells: new therapeutics for graft-versus-host disease. J Exp Med (2002) 196:401–6. doi:10.1084/jem.20020090
68. Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med (2002) 196:389–99. doi:10.1084/jem.20020399
69. Maury S, Lemoine FM, Hicheri Y, Rosenzwajg M, Badoual C, Cherai M, et al. CD4+CD25+ regulatory T cell depletion improves the graft-versus-tumor effect of donor lymphocytes after allogeneic hematopoietic stem cell transplantation. Sci Transl Med (2010) 2:41ra52. doi:10.1126/scitranslmed.3001302
70. Chopra M, Biehl M, Steinfatt T, Brandl A, Kums J, Amich J, et al. Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J Exp Med (2016) 213:1881–900. doi:10.1084/jem.20151563
71. Leclerc M, Naserian S, Pilon C, Thiolat A, Martin GH, Pouchy C, et al. Control of GVHD by regulatory T cells depends on TNF produced by T cells and TNFR2 expressed by regulatory T cells. Blood (2016) 128:1651–9. doi:10.1182/blood-2016-02-700849
72. Pierini A, Strober W, Moffett C, Baker J, Nishikii H, Alvarez M, et al. TNF-alpha priming enhances CD4+FoxP3+ regulatory T-cell suppressive function in murine GVHD prevention and treatment. Blood (2016) 128:866–71. doi:10.1182/blood-2016-04-711275
73. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117:1061–70. doi:10.1182/blood-2010-07-293795
74. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood (2011) 117:3921–8. doi:10.1182/blood-2010-10-311894
75. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi:10.1038/nrc3239
76. Alexander W. The checkpoint immunotherapy revolution: what started as a trickle has become a flood, despite some daunting adverse effects; new drugs, indications, and combinations continue to emerge. P T (2016) 41:185–91.
77. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364:2517–26. doi:10.1056/NEJMoa1104621
79. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi:10.1056/NEJMoa1003466
80. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348:56–61. doi:10.1126/science.aaa8172
81. Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science (2013) 342:1432–3. doi:10.1126/science.342.6165.1432
82. Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol (2012) 24:207–12. doi:10.1016/j.coi.2011.12.009
83. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol (2017) 18:153–67. doi:10.1038/nri.2017.108
84. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol (2010) 28:3167–75. doi:10.1200/JCO.2009.26.7609
85. Burugu S, Dancsok AR, Nielsen TO. Emerging targets in cancer immunotherapy. Semin Cancer Biol (2017). doi:10.1016/j.semcancer.2017.10.001
86. He Y, Rivard CJ, Rozeboom L, Yu H, Ellison K, Kowalewski A, et al. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci (2016) 107:1193–7. doi:10.1111/cas.12986
87. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev (2017) 276:97–111. doi:10.1111/imr.12520
88. Dempke WCM, Fenchel K, Uciechowski P, Dale SP. Second- and third-generation drugs for immuno-oncology treatment-the more the better? Eur J Cancer (2017) 74:55–72. doi:10.1016/j.ejca.2017.01.001
89. Manieri NA, Chiang EY, Grogan JL. TIGIT: a key inhibitor of the cancer immunity cycle. Trends Immunol (2017) 38:20–8. doi:10.1016/j.it.2016.10.002
90. Bartkowiak T, Curran MA. 4-1BB Agonists: multi-potent potentiators of tumor immunity. Front Oncol (2015) 5:117. doi:10.3389/fonc.2015.00117
91. Knee DA, Hewes B, Brogdon JL. Rationale for anti-GITR cancer immunotherapy. Eur J Cancer (2016) 67:1–10. doi:10.1016/j.ejca.2016.06.028
92. Moran AE, Kovacsovics-Bankowski M, Weinberg AD. The TNFRs OX40, 4-1BB, and CD40 as targets for cancer immunotherapy. Curr Opin Immunol (2013) 25:230–7. doi:10.1016/j.coi.2013.01.004
93. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol (2015) 5:34. doi:10.3389/fonc.2015.00034
94. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol (2016) 13:273–90. doi:10.1038/nrclinonc.2016.25
95. Liu Y, Zeng G. Cancer and innate immune system interactions: translational potentials for cancer immunotherapy. J Immunother (2012) 35:299–308. doi:10.1097/CJI.0b013e3182518e83
96. Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer (2017) 17:286–301. doi:10.1038/nrc.2017.17
97. Morrissey KM, Yuraszeck TM, Li CC, Zhang Y, Kasichayanula S. Immunotherapy and novel combinations in oncology: current landscape, challenges, and opportunities. Clin Transl Sci (2016) 9:89–104. doi:10.1111/cts.12391
98. Ramos RN, Piaggio E, Romano E. Mechanisms of resistance to immune checkpoint antibodies. Handb Exp Pharmacol (2017):1–20. doi:10.1007/164_2017_11
99. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168:707–23. doi:10.1016/j.cell.2017.01.017
100. Goldszmid RS, Dzutsev A, Trinchieri G. Host immune response to infection and cancer: unexpected commonalities. Cell Host Microbe (2014) 15:295–305. doi:10.1016/j.chom.2014.02.003
101. Medler TR, Cotechini T, Coussens LM. Immune response to cancer therapy: mounting an effective antitumor response and mechanisms of resistance. Trends Cancer (2015) 1:66–75. doi:10.1016/j.trecan.2015.07.008
102. Shalapour S, Karin M. Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest (2015) 125:3347–55. doi:10.1172/JCI80007
103. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol (2015) 36:265–76. doi:10.1016/j.it.2015.02.008
104. Zarour HM. Reversing T-cell dysfunction and exhaustion in cancer. Clin Cancer Res (2016) 22:1856–64. doi:10.1158/1078-0432.CCR-15-1849
105. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res (2017) 27:109–18. doi:10.1038/cr.2016.151
106. Wang H, Franco F, Ho PC. Metabolic regulation of Tregs in cancer: opportunities for immunotherapy. Trends Cancer (2017) 3:583–92. doi:10.1016/j.trecan.2017.06.005
107. Colombo MP, Piconese S. Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev Cancer (2007) 7:880–7. doi:10.1038/nrc2250
108. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res (1999) 59:3128–33.
109. Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, et al. Fc-Optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity (2017) 46:577–86. doi:10.1016/j.immuni.2017.03.013
110. Klages K, Mayer CT, Lahl K, Loddenkemper C, Teng MW, Ngiow SF, et al. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res (2010) 70:7788–99. doi:10.1158/0008-5472.CAN-10-1736
111. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med (2009) 206:1717–25. doi:10.1084/jem.20082492
112. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A (2015) 112:6140–5. doi:10.1073/pnas.1417320112
113. Attia P, Maker AV, Haworth LR, Rogers-Freezer L, Rosenberg SA. Inability of a fusion protein of IL-2 and diphtheria toxin (denileukin diftitox, DAB389IL-2, ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J Immunother (2005) 28:582–92. doi:10.1097/01.cji.0000175468.19742.10
114. Barnett B, Kryczek I, Cheng P, Zou W, Curiel TJ. Regulatory T cells in ovarian cancer: biology and therapeutic potential. Am J Reprod Immunol (2005) 54:369–77. doi:10.1111/j.1600-0897.2005.00330.x
115. Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest (2005) 115:3623–33. doi:10.1172/JCI25947
116. Mahnke K, Schonfeld K, Fondel S, Ring S, Karakhanova S, Wiedemeyer K, et al. Depletion of CD4+CD25+ human regulatory T cells in vivo: kinetics of Treg depletion and alterations in immune functions in vivo and in vitro. Int J Cancer (2007) 120:2723–33. doi:10.1002/ijc.22617
117. Vlad G, Ho EK, Vasilescu ER, Fan J, Liu Z, Cai JW, et al. Anti-CD25 treatment and FOXP3-positive regulatory T cells in heart transplantation. Transpl Immunol (2007) 18:13–21. doi:10.1016/j.trim.2007.03.001
118. Waldmann TA. Daclizumab (anti-Tac, Zenapax) in the treatment of leukemia/lymphoma. Oncogene (2007) 26:3699–703. doi:10.1038/sj.onc.1210368
119. Hicheri Y, Bouchekioua A, Hamel Y, Henry A, Rouard H, Pautas C, et al. Donor regulatory T cells identified by FoxP3 expression but also by the membranous CD4+CD127low/neg phenotype influence graft-versus-tumor effect after donor lymphocyte infusion. J Immunother (2008) 31:806–11. doi:10.1097/CJI.0b013e318184908d
120. Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol (2016) 37:193–207. doi:10.1016/j.it.2016.01.002
121. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev (2017) 276:121–44. doi:10.1111/imr.12528
122. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol (2012) 3:190. doi:10.3389/fimmu.2012.00190
123. Platten M, von Knebel Doeberitz N, Oezen I, Wick W, Ochs K. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front Immunol (2014) 5:673. doi:10.3389/fimmu.2014.00673
124. Sitkovsky M, Ohta A. Targeting the hypoxia-adenosinergic signaling pathway to improve the adoptive immunotherapy of cancer. J Mol Med (Berl) (2013) 91:147–55. doi:10.1007/s00109-013-1001-9
125. Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD. Combination immunotherapy: a road map. J Immunother Cancer (2017) 5:16. doi:10.1186/s40425-017-0218-5
126. Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov (2012) 11:790–811. doi:10.1038/nrd3810
127. Sato T, Terai M, Tamura Y, Alexeev V, Mastrangelo MJ, Selvan SR. Interleukin 10 in the tumor microenvironment: a target for anticancer immunotherapy. Immunol Res (2011) 51:170–82. doi:10.1007/s12026-011-8262-6
128. Van der Jeught K, Bialkowski L, Daszkiewicz L, Broos K, Goyvaerts C, Renmans D, et al. Targeting the tumor microenvironment to enhance antitumor immune responses. Oncotarget (2015) 6:1359–81. doi:10.18632/oncotarget.3204
129. Perol L, Lindner JM, Caudana P, Nunez NG, Baeyens A, Valle A, et al. Loss of immune tolerance to IL-2 in type 1 diabetes. Nat Commun (2016) 7:13027. doi:10.1038/ncomms13027
130. Perol L, Martin GH, Maury S, Cohen JL, Piaggio E. Potential limitations of IL-2 administration for the treatment of experimental acute graft-versus-host disease. Immunol Lett (2014) 162:173–84. doi:10.1016/j.imlet.2014.10.027
131. Perol L, Piaggio E. New molecular and cellular mechanisms of tolerance: tolerogenic actions of IL-2. Methods Mol Biol (2016) 1371:11–28. doi:10.1007/978-1-4939-3139-2_2
132. Blattman JN, Grayson JM, Wherry EJ, Kaech SM, Smith KA, Ahmed R. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat Med (2003) 9:540–7. doi:10.1038/nm866
133. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol (2014) 192:5451–8. doi:10.4049/jimmunol.1490019
134. Zloza A, Dharmadhikari ND, Huelsmann EJ, Broucek JR, Hughes T, Kohlhapp FJ, et al. Low-dose interleukin-2 impairs host anti-tumor immunity and inhibits therapeutic responses in a mouse model of melanoma. Cancer Immunol Immunother (2017) 66:9–16. doi:10.1007/s00262-016-1916-4
135. Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity (2010) 33:153–65. doi:10.1016/j.immuni.2010.08.004
136. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science (2006) 311:1924–7. doi:10.1126/science.1122927
137. Letourneau S, Krieg C, Pantaleo G, Boyman O. IL-2- and CD25-dependent immunoregulatory mechanisms in the homeostasis of T-cell subsets. J Allergy Clin Immunol (2009) 123:758–62. doi:10.1016/j.jaci.2009.02.011
138. Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, et al. Antibodies to interleukin-2 elicit selective T cell subset potentiation through distinct conformational mechanisms. Immunity (2015) 42:815–25. doi:10.1016/j.immuni.2015.04.015
139. Arnott CH, Scott KA, Moore RJ, Robinson SC, Thompson RG, Balkwill FR. Expression of both TNF-alpha receptor subtypes is essential for optimal skin tumour development. Oncogene (2004) 23:1902–10. doi:10.1038/sj.onc.1207317
140. Atretkhany KS, Nosenko MA, Gogoleva VS, Zvartsev RV, Qin Z, Nedospasov SA, et al. TNF neutralization results in the delay of transplantable tumor growth and reduced MDSC accumulation. Front Immunol (2016) 7:147. doi:10.3389/fimmu.2016.00147
141. Charles KA, Kulbe H, Soper R, Escorcio-Correia M, Lawrence T, Schultheis A, et al. The tumor-promoting actions of TNF-alpha involve TNFR1 and IL-17 in ovarian cancer in mice and humans. J Clin Invest (2009) 119:3011–23. doi:10.1172/JCI39065
142. Chopra M, Riedel SS, Biehl M, Krieger S, von Krosigk V, Bauerlein CA, et al. Tumor necrosis factor receptor 2-dependent homeostasis of regulatory T cells as a player in TNF-induced experimental metastasis. Carcinogenesis (2013) 34:1296–303. doi:10.1093/carcin/bgt038
143. Ham B, Wang N, D’Costa Z, Fernandez MC, Bourdeau F, Auguste P, et al. Receptor-2 facilitates an immunosuppressive microenvironment in the liver to promote the colonization and growth of hepatic metastases. Cancer Res (2015) 75:5235–47. doi:10.1158/0008-5472.CAN-14-3173
144. Knight B, Yeoh GC, Husk KL, Ly T, Abraham LJ, Yu C, et al. Impaired preneoplastic changes and liver tumor formation in tumor necrosis factor receptor type 1 knockout mice. J Exp Med (2000) 192:1809–18. doi:10.1084/jem.192.12.1809
145. Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest (2008) 118:560–70. doi:10.1172/JCI32453
146. Sangaletti S, Tripodo C, Ratti C, Piconese S, Porcasi R, Salcedo R, et al. Oncogene-driven intrinsic inflammation induces leukocyte production of tumor necrosis factor that critically contributes to mammary carcinogenesis. Cancer Res (2010) 70:7764–75. doi:10.1158/0008-5472.CAN-10-0471
147. Yan B, Wang H, Rabbani ZN, Zhao Y, Li W, Yuan Y, et al. Tumor necrosis factor-alpha is a potent endogenous mutagen that promotes cellular transformation. Cancer Res (2006) 66:11565–70. doi:10.1158/0008-5472.CAN-06-2540
148. Li B, Vincent A, Cates J, Brantley-Sieders DM, Polk DB, Young PP. Low levels of tumor necrosis factor alpha increase tumor growth by inducing an endothelial phenotype of monocytes recruited to the tumor site. Cancer Res (2009) 69:338–48. doi:10.1158/0008-5472.CAN-08-1565
149. Erdman SE, Rao VP, Poutahidis T, Rogers AB, Taylor CL, Jackson EA, et al. Nitric oxide and TNF-alpha trigger colonic inflammation and carcinogenesis in Helicobacter hepaticus-infected, Rag2-deficient mice. Proc Natl Acad Sci U S A (2009) 106:1027–32. doi:10.1073/pnas.0812347106
150. Stathopoulos GT, Kollintza A, Moschos C, Psallidas I, Sherrill TP, Pitsinos EN, et al. Tumor necrosis factor-alpha promotes malignant pleural effusion. Cancer Res (2007) 67:9825–34. doi:10.1158/0008-5472.CAN-07-1064
151. Ba H, Li B, Li X, Li C, Feng A, Zhu Y, et al. Transmembrane tumor necrosis factor-alpha promotes the recruitment of MDSCs to tumor tissue by upregulating CXCR4 expression via TNFR2. Int Immunopharmacol (2017) 44:143–52. doi:10.1016/j.intimp.2016.12.028
152. Chang LY, Lin YC, Chiang JM, Mahalingam J, Su SH, Huang CT, et al. Blockade of TNF-alpha signaling benefits cancer therapy by suppressing effector regulatory T cell expansion. Oncoimmunology (2015) 4:e1040215. doi:10.1080/2162402X.2015.1040215
153. Chen Y, Friedman M, Liu G, Deodhar A, Chu CQ. Do tumor necrosis factor inhibitors increase cancer risk in patients with chronic immune-mediated inflammatory disorders? Cytokine (2016) 101:78–88. doi:10.1016/j.cyto.2016.09.013
154. Micic D, Komaki Y, Alavanja A, Rubin DT, Sakuraba A. Risk of cancer recurrence among individuals exposed to antitumor necrosis factor therapy: a systematic review and meta-analysis of observational studies. J Clin Gastroenterol (2017). doi:10.1097/MCG.0000000000000865
155. Harrison ML, Obermueller E, Maisey NR, Hoare S, Edmonds K, Li NF, et al. Tumor necrosis factor alpha as a new target for renal cell carcinoma: two sequential phase II trials of infliximab at standard and high dose. J Clin Oncol (2007) 25:4542–9. doi:10.1200/JCO.2007.11.2136
156. Madhusudan S, Foster M, Muthuramalingam SR, Braybrooke JP, Wilner S, Kaur K, et al. A phase II study of etanercept (Enbrel), a tumor necrosis factor alpha inhibitor in patients with metastatic breast cancer. Clin Cancer Res (2004) 10:6528–34. doi:10.1158/1078-0432.CCR-04-0730
157. Kulbe H, Chakravarty P, Leinster DA, Charles KA, Kwong J, Thompson RG, et al. A dynamic inflammatory cytokine network in the human ovarian cancer microenvironment. Cancer Res (2012) 72:66–75. doi:10.1158/0008-5472.CAN-11-2178
158. Kulbe H, Thompson R, Wilson JL, Robinson S, Hagemann T, Fatah R, et al. The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res (2007) 67:585–92. doi:10.1158/0008-5472.CAN-06-2941
159. Liu F, Ai F, Tian L, Liu S, Zhao L, Wang X. Infliximab enhances the therapeutic effects of 5-fluorouracil resulting in tumor regression in colon cancer. Onco Targets Ther (2016) 9:5999–6008. doi:10.2147/OTT.S109342
160. Govindaraj C, Scalzo-Inguanti K, Madondo M, Hallo J, Flanagan K, Quinn M, et al. Impaired Th1 immunity in ovarian cancer patients is mediated by TNFR2+ Tregs within the tumor microenvironment. Clin Immunol (2013) 149:97–110. doi:10.1016/j.clim.2013.07.003
Keywords: tumor necrosis factor α, TNFR2, regulatory T cells, cancer, graft-versus-host disease, immunotherapy
Citation: Salomon BL, Leclerc M, Tosello J, Ronin E, Piaggio E and Cohen JL (2018) Tumor Necrosis Factor α and Regulatory T Cells in Oncoimmunology. Front. Immunol. 9:444. doi: 10.3389/fimmu.2018.00444
Received: 26 December 2017; Accepted: 19 February 2018;
Published: 12 March 2018
Edited by:
Xin Chen, University of Macau, ChinaReviewed by:
Richard Williams, University of Oxford, United KingdomCopyright: © 2018 Salomon, Leclerc, Tosello, Ronin, Piaggio and Cohen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benoît L. Salomon, YmVub2l0LnNhbG9tb25AdXBtYy5mcg==
†Co-seniors.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.