 Xu Wang
Xu Wang Deyu Chen
Deyu Chen- Institute of Oncology, Affiliated Hospital of Jiangsu University, Zhenjiang, China
Purinergic signaling, which utilizes nucleotides (particularly ATP) and adenosine as transmitter molecules, plays an essential role in immune system. In the extracellular compartment, ATP predominantly functions as a pro-inflammatory molecule through activation of P2 receptors, whereas adenosine mostly functions as an anti-inflammatory molecule through activation of P1 receptors. Neutrophils are the most abundant immune cells in circulation and have emerged as an important component in orchestrating a complex series of events during inflammation. However, because of the destructive nature of neutrophil-derived inflammatory agents, neutrophil activation is fine-tuned, and purinergic signaling is intimately involved in this process. Indeed, shifting the balance between P2 and P1 signaling is critical for neutrophils to appropriately exert their immunologic activity. Here, we review the role of purinergic signaling in regulating neutrophil function, and discuss the potential of targeting purinergic signaling for the treatment of neutrophil-associated infectious and inflammatory diseases.
Introduction
Purinergic signaling is among the most primitive signal transduction systems in evolutionary history (1). In humans, purinergic receptors (P2 and P1 receptors) are expressed in virtually all tissues and cell types, and they mediate a wide range of physiological and pathophysiological responses, such as neurotransmission, hypertension, inflammation, and cancer (2). Nucleotides (particularly ATP) and nucleosides (ADO), the basic elements of all living organisms, are well known for their function in energy metabolism. Notably, in the extracellular compartment, ATP and ADO are critical signal transduction molecules that participate in a wealth of different cellular responses through the activation of P2Rs and P1Rs, respectively (3).
The immune system is a tightly regulated and integrated cellular network that functions to preserve and restore homeostasis. The purinergic signaling system is an evolutionarily selected system that fine-tunes immune cell functions (4). Indeed, P2R- and P1R-mediated purinergic signaling frequently shows opposing effects in terms of modulating immune cell functions (5). Specifically, ATP-mediated P2 receptor signaling prevalently facilitates immune cell activation, whereas ADO-mediated P1R signaling mostly restricts immune cell activation (2). Shifting the balance from pro-inflammatory P2R signaling to anti-inflammatory P1R signaling or vice versa may have important consequences on the immune response outcome (3). Neutrophils are the most abundant immune cells in human blood and have emerged as an important component in orchestrating a complex series of events during inflammation (6). However, because of their short lifespan and how difficult they are to maintain in in vitro culture and to genetically manipulate, neutrophils are largely ignored in the purinergic signaling field. Recently, a wealth of pharmacologic and genetic evidence has shifted this paradigm by extending the role of purinergic signaling in neutrophils (7–9). Indeed, the coordinated interplay between P2 and P1 purinergic signaling is critical for neutrophils to effectively initiate their immunologic activity and restore tissue homeostasis. Here, we review the mounting evidence of neutrophil purinergic signaling and highlight their therapeutic potential in the treatment of neutrophil-associated infectious and inflammatory diseases.
Extracellular ATP and ADO
In mammalian cells, ATP is synthesized by glycolysis or oxidative phosphorylation and stored at a high intracellular concentration of ~5 mM. In the physiological state, extracellular ATP is present in very minute amounts of ~10 nM due to plasma membrane-anchored ectonucleotidases (10). However, in pathological conditions, extracellular ATP concentrations are dramatically increased. Using a plasma membrane luciferase (pmeLUC) probe to monitor ATP levels in situ, investigators have demonstrated that the extracellular ATP level is nearly undetectable in healthy tissues, whereas in pathological conditions, such as graft-versus-host disease, acute hepatitis, and even primary tumor or metastases, the extracellular ATP concentration can be as high as a few hundred micromolar (11).
Under extreme conditions, such as trauma, ischemia, and infection, cellular necrosis will release large amounts of ATP from intracellular storage pools (Figure 1) (3). However, in most cases, extracellular ATP release is finely controlled by diffusion through plasmalemmal channels or exocytotic release from ATP-rich vesicles (2). Connexins and pannexin hemichannels are widely expressed throughout various cell types, including inflammatory cells, endothelial cells, and epithelial cells (12). Among them, connexin 43 (Cx43) and pannexin 1 (Panx1) hemichannels are the most studied. They were originally recognized as gap junction proteins that form non-covalent linkages between two cells and mediate cell-to-cell communications (12). However, isolated hemichannels can function as plasmalemmal channels between the cytoplasm and the extracellular space, thereby controlling ATP release (12). During cell apoptosis, Panx1 is cleaved by executioner enzymes of apoptosis (caspase 3 and 7) to generate a truncated and activated subunit that regulates ATP release from apoptotic cells (13). Recent evidence has demonstrated that Cx43 and Panx1 hemichannels are expressed in neutrophils and associated with the autocrine purinergic signaling that regulates neutrophil chemotaxis (8, 14). In addition, the exocytotic release of ATP from secretory vesicles (SVs) that specifically accumulate and store ATP is an important source of extracellular ATP. For example, dense granules in platelets contain large amounts of ATP (15). Once activated, ATP is readily secreted from platelets through exocytosis to mediate its biological functions in both autocrine and paracrine manners (15). Additional evidence shows that lysosomes are ATP-rich vesicles that can act as an important source of extracellular ATP through lysosomal exocytosis (16). Notably, pathogenic microorganisms, including bacteria and fungi, can release extracellular ATP (17). However, their roles in mediating host purinergic signaling have not been clarified.
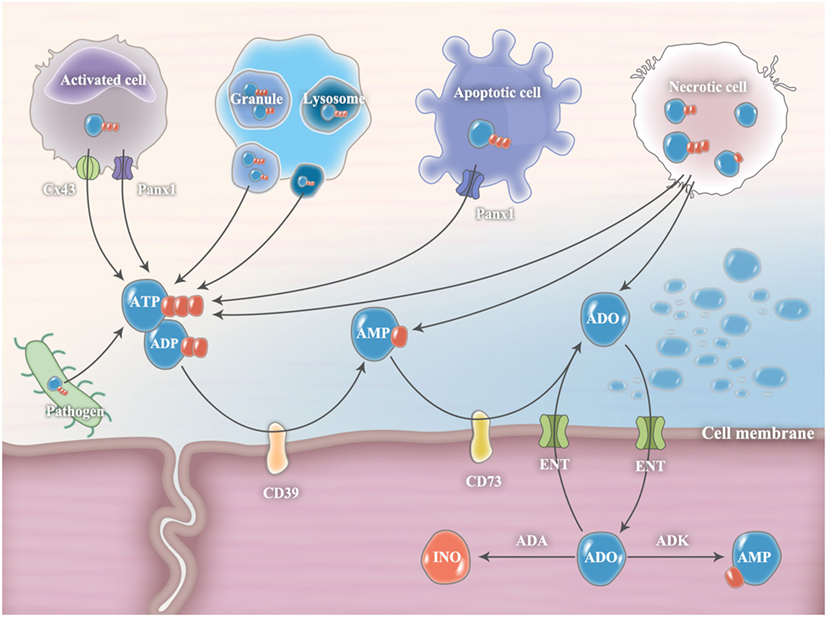
Figure 1. Release and metabolism of extracellular ATP and ADO. In pathological conditions, extracellular ATP release is finely controlled by diffusion through connexin 43 (Cx43) or pannexin 1 (Panx1) hemichannels and exocytotic release from ATP-rich vesicles (such as granule and lysosome). During cell apoptosis, Panx1 is cleaved by executioner enzymes of apoptosis (caspase 3 and 7) to generate a truncated and activated subunit that regulates ATP release from apoptotic cells. Under extreme conditions, cellular necrosis will release large amounts of ATP from intracellular storage pools. Pathogenic microorganisms, including bacteria and fungi, are also important sources of extracellular ATP. Extracellular ADP can be released from ADP-rich granules or necrotic cells. In the extracellular milieu, ATP and ADP levels are tightly controlled by plasma membrane ectonucleotidases, CD39 and CD73, which convert ATP/ADP to AMP and subsequently convert AMP to ADO, respectively. The accumulated ADO will be gradually transferred from the extracellular compartment into the intracellular compartment through equilibrative nucleoside transporters (ENTs) and subsequently metabolized to inosine (INO) by adenosine deaminase (ADA) or to AMP by adenosine kinase (ADK).
Intracellularly, ADO is generated from hydrolysis of AMP by 5-nucleotidase or by hydrolysis of S-adenosyl-homocysteine (SAH) by SAH hydrolase (18). The intracellular adenosine concentration is quite variable. Minor changes in steady-state ATP levels (~5 mM) translate into major changes in intracellular adenosine concentrations (19). In the physiological state, extracellular ADO (~10 nM) is released constitutively from multiple cell types because of the ubiquitous presence of equilibrative nucleoside transporters (ENTs) (20). From the basal level, the extracellular ADO concentration can increase substantially (~100 times higher) in pathological conditions. The sources of extracellular adenosine are either from cell necrosis, equilibrative transporters, or ATP/ADP/AMP hydrolysis by nucleotidases (21). The accumulated ADO will be gradually transferred from the extracellular compartment into the intracellular compartment through ENTs and subsequently metabolized to INO by ADA or to AMP by adenosine kinase (ADK) (21).
In the extracellular milieu, ATP and ADP levels are tightly controlled by plasma membrane ectonucleotidases, such as nucleoside triphosphate diphosphohydrolase 1 (NTPDase1, also known as CD39) and ecto-5′-nucleotidase (also known as CD73), which convert ATP/ADP to AMP and subsequently convert AMP to ADO, respectively (5). The CD39/CD73 pathway is a critical checkpoint, driving a shift from an ATP-induced pro-inflammatory environment to an anti-inflammatory milieu induced by ADO (5). Both CD39 and CD73 are expressed in neutrophils and appear to be critical players in the regulation of neutrophil activation (7). In addition to CD39 and CD73, nucleotide pyrophosphatases and phosphodiesterases (NPPs), alkaline phosphatases (ALP), acid phosphatases (ACP), and ectokinases can also degrade extracellular nucleotides and nucleosides; however, their roles in regulating immune responses are less well understood (3).
Purinergic Receptors
Separate families of receptors for ATP (P2 receptors) and ADO (P1 receptors) were first identified in 1978. According to their transduction mechanism and molecular cloning, P2 receptors are subsequently divided into ionotropic P2XRs and metabotropic P2YRs. At present, seven P2XRs (P2X1–7R), eight P2YRs (P2Y1/2/4/6/11/12/13/14R), and four P1 receptors (A1/A2A/A2B/A3R) have been cloned and characterized (2).
P2XRs
Ionotropic P2XRs are ATP-gated trimeric ion channels (3). P2XRs share a similar tertiary topology with an intracellular NH2 and a longer COOH terminus, a large extracellular loop responsible for ligand binding, and two transmembrane-spanning regions (TM1 and TM2) (3). TM1 is involved in channel gating and a helix of TM2 forms channel pore (3). Following ATP binding, P2XRs become permeable to Na+, K+, and Ca2+. Moreover, activation of P2X7R forms a large pore that allows the passage of molecules as large as 900 Da, which is associated with the release of pro-inflammatory cytokines (22).
P2YRs
P2YRs belong to the δ-branch of class A G-protein-coupled receptor (GPCR) family and contain seven hydrophobic transmembrane regions connected by three extracellular loops and three intracellular loops (3). Based on the phylogenetic and structural divergence, two distinct P2YR subgroups have been identified (23). The first group contains P2Y1/2/4/6/11Rs, with a sequence homology of 35–52% in amino-acid composition and the presence of a Y–Q/K–X–X–R defining motif in the transmembrane α-helix 7. The second group contains P2Y12/13/14Rs, with a sequence homology of 47–48% and the presence of the K–E–X–X–L motif in transmembrane α-helix 7. Moreover, the two subgroups also differ in their primary coupling to G-proteins (23). P2Y1/2/4/6/11Rs primarily couple to Gq/G11 and initiate phospholipase C/inositol trisphosphate/diacylglycerol pathway to increase intracellular calcium, whereas P2Y12/13/14Rs principally couple to Gi/0 and inhibit adenylate cyclase (AC) to decrease intracellular cyclic AMP (cAMP). Additional evidence shows that P2Y11R uses Gs to stimulate AC and increase intracellular cAMP.
P1Rs
P1Rs belong to classical G-protein-coupled metabotropic receptors, and are single polypeptides comprising seven α-helices oriented perpendicular to the plasmalemma (24). There is a close similarity in sequence of A1R, A2AR, A2BR, and A3R. A2AR shares a sequence identity of 46, 37, or 31% with A2BR, A1R, and A3R, respectively (24). ADO is the endogenous ligand for all four ADO receptors. A1R and A3R inhibit adenylyl cyclase activity through Gi G-proteins, and stimulate phospholipase C/inositol trisphosphate/diacylglycerol pathway via Gβγ G-proteins (4). A2AR and A2BR primarily couple to Gs G-proteins to increase AC activity (4). Moreover, all four ADO receptors couple to mitogen-activated protein kinases (MAPKs), which renders them more sophisticated biological functions (4).
Expression of Purinergic Receptors in Neutrophils
In neutrophils, expression analyses have been performed for all purinergic receptors, except P2Y12R (Table 1). Convincing data obtained from mRNA, protein, and functional assays have demonstrated that P2X1R, P2X7R, P2Y2R, P2Y14R, and all four ADO receptors are expressed in neutrophils. Expression evidence for other purinergic receptors is relatively weak, and additional work is needed to further confirm whether P2X2-6Rs, P2Y1R, P2Y4R, P2Y6R, P2Y11R, P2Y12R, and P2Y13R are expressed in neutrophils.
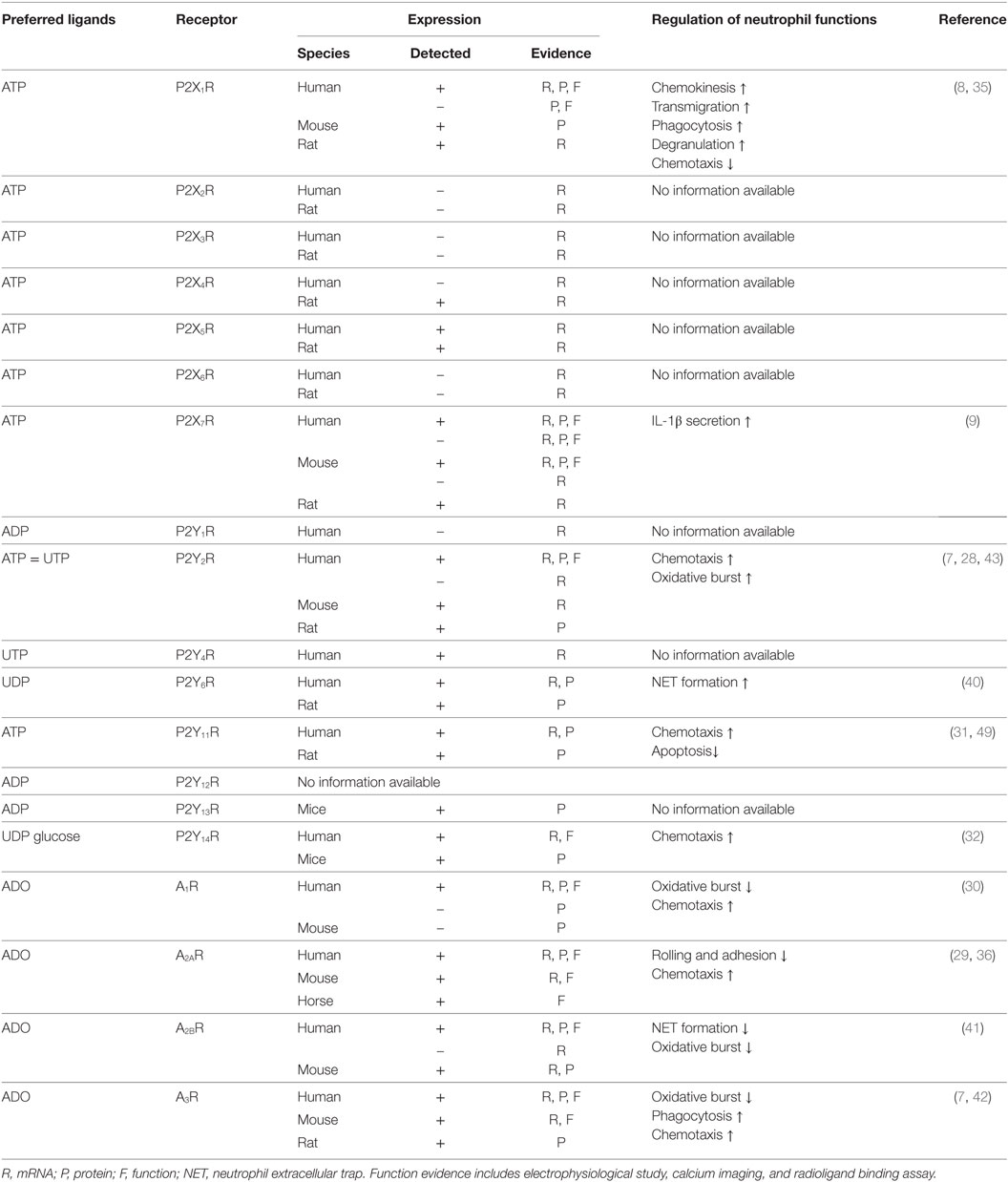
Table 1. Expression and/or function of purinergic receptors in neutrophils.
Regulation of Neutrophil Function by Purinergic Receptors
Purinergic Regulation of Neutrophil Chemotaxis
Chemotaxis denotes the ability of cells to sense gradients, polarize, and directionally migrate within a chemotactic gradient field. Recent studies have highlighted the critical role of autocrine purinergic signaling in directing neutrophil chemotaxis (Table 1) (25). During chemotaxis, neutrophils require polarization, with an anterior pseudopod at the leading edge to sense chemoattractant gradients and a posterior pseudopod at the trailing edge to maintain orientated migration (26). Once neutrophils sense the chemoattractants, Panx1 hemichannels rapidly translocate to the leading edge and release mitochondria-derived ATP from pseudopod protrusions (27). The extracellular release of ATP serves as an autocrine messenger to amplify chemotaxis signals through activation of P2Y2R-mediated mTOR signaling at the leading edge (28). Extracellular ATP and positive feedback via the P2Y2R receptor constitutes a mechanism that is required for neutrophil gradient sensing (7). Then, released ATP is hydrolyzed to ADO in situ by neutrophil membrane-associated CD73, which subsequently activates neutrophil A3R at the leading edge to drive a second round of signal amplification (7). The second amplification step is equally important to the initial amplification that facilitates neutrophil chemotaxis because it controls migration speed (7). At the trailing edge, diffused or locally generated ADO activates A2AR and triggers cAMP/PKA signaling to inhibit P2Y2R-mediated mTOR signaling (29). The activation of A2AR maintains cell polarization and promotes uropod retraction. In polarized neutrophils, the P2Y2R receptor remains uniformly distributed across the cell membrane, whereas CD73 and A3R accumulate at the leading edge, and A2AR translocates to the trailing edge.
The described autocrine purinergic signaling axis plays crucial roles in mediating neutrophil chemotaxis in a chemotactic gradient field. In addition to P2Y2R, A3R, and A2AR, other purinergic receptors also mediate neutrophil chemotaxis. Activation of P2X1R by extracellular ATP fails to induce neutrophil directional chemotaxis, but it can enhance neutrophil chemokinesis (random cell migration) through Rho kinase signaling (8). Moreover, the lipopolysaccharide (LPS)-induced autocrine release of ATP inhibits neutrophil chemotaxis in a chemotactic gradient field via P2X1R (8). LPS activates neutrophil Cx43 hemichannels to release extracellular ATP, which binds to P2X1R and initiates Ca2+ influx. The Ca2+ influx subsequently inhibits neutrophil chemotaxis by enhancing myosin light chain phosphorylation, which interferes with neutrophil polarization. Knockout of P2X1R reverses LPS-inhibited neutrophil chemotaxis. In contrast to P2X1R, A1R facilitates neutrophil chemotaxis in the presence of LPS (30). ADO and an agonist to A1R can restore neutrophil chemotaxis by inhibiting the LPS-induced p38 kinase. Additional studies indicate that neutrophil P2Y11R and P2Y14R receptors also enhance neutrophil chemotaxis, but the mechanisms are not well understood (31, 32).
Purinergic Regulation of Neutrophil Rolling, Adhesion, and Transmigration
Neutrophils are typically the first immune cells that are recruited to inflammatory sites (33). In most tissues, the neutrophil recruitment cascade involves the following commonly recognized steps: tethering, rolling, adhesion, crawling, and subsequent transmigration (34). A recent in vivo study demonstrated that LPS-induced neutrophil rolling and adhesion in cremaster muscle postcapillary venules does not differ between WT and P2rx1−/− mice (35). However, neutrophil transmigration is inhibited in P2rx1−/− mice, indicating that P2 × 1 signaling may participate in the neutrophil recruitment cascade by promoting neutrophil transmigration but not rolling and adhesion (Table 1). Using adoptive transfer of neutrophils from WT and P2rx1−/− mice into WT mice, the results demonstrated that the absence of the P2X1R in neutrophils, but not in vascular endothelial cells or other immune cells, is responsible for neutrophil emigration from venules. In contrast to P2X1R, neutrophil-expressed A2AR appears to inhibit the neutrophil recruitment cascade. Using an A2AR agonist to initiate A2AR signaling, β2 integrin-mediated neutrophil rolling and adhesion are markedly inhibited both in TNF-α-challenged murine cremaster muscle postcapillary venules and in ex vivo flow chamber models (36).
Purinergic Regulation of Neutrophil Phagocytosis and Neutrophil Extracellular Traps (NETs)
Neutrophils are professional phagocytes that are endowed with a unique capacity to engulf and thereby eliminate pathogens and cell debris. The discovery of NETs has extended the understanding of neutrophil antimicrobial strategies (37). NETs are large, extracellular, web-like structures that are composed of decondensed chromatin and neutrophil antimicrobial factors. NETs can not only trap and kill a variety of microbes but also activate and regulate innate and adaptive immunity (38). Recent studies identified a cell-autonomous, microbe size-sensing mechanism that allows neutrophils to sense pathogen size and respond by phagocytosing small pathogens or selectively releasing NETs in response to large pathogens (39). Phagocytosis and NETs are both crucial for neutrophils to efficiently eliminate invading pathogens.
As a potent activator of neutrophils, LPS enhances neutrophil phagocytosis of Escherichia coli (E. coli) in humans (8). However, the promotive effects of LPS are abolished when a P2X1R antagonist is introduced (8). Because LPS induces neutrophils to release extracellular ATP, these results suggest that autocrine activation of P2 × 1 signaling may be essential for enhancing neutrophil phagocytosis (Table 1) (8). UDP is the natural ligand to P2Y6R. While UDP itself fails to initiate NET formation in human neutrophils, UDP-mediated P2Y6 signaling is involved in the monosodium urate crystal-induced formation of NETs (40). When incubated with Klebsiella pneumoniae, neutrophil A2BR expression is increased by approximately 500-fold (41). In addition, neutrophils from Adora2b−/− mice show a greatly enhanced ability to kill bacteria compared with that of neutrophils from WT mice. The following studies demonstrate that A2BR impairs the neutrophils’ ability to kill Klebsiella pneumoniae by suppressing NET formation but not phagocytosis. Neutrophil A3R enhances bacterial clearance, and activating human neutrophils with an A3R agonist promotes the formation of neutrophil filipodia-like projections, which are named cytonemes (42). The formation of these structures enables neutrophils to sample, capture and “reel in” pathogens to induce phagocytosis.
Purinergic Regulation of Neutrophil Oxidative Burst
Superoxide serves as a potent antimicrobial agent to kill microbial pathogens and modulates multiple signaling pathways. Because of the destructive nature of superoxide, the oxidative burst is fine-tuned, and purinergic signaling is intimately involved in this process (Table 1). P2Y2R is an activator of neutrophil oxidative burst (43). Knockdown of P2Y2R in differentiated neutrophil-like HL-60 cells (dHL-60) significantly inhibits fMLP-induced oxidative burst. Monosodium urate crystals can induce neutrophil oxidative burst, but an antagonist to P2Y6R suppresses this monosodium urate crystal-induced neutrophil oxidative burst (40). Notably, exogenous ATP was not administered in these two studies, and autocrine activation of neutrophil P2Y2R and P2Y6R may amplify the oxidative burst. In contrast to P2Y2R and P2Y6R, A2BR, and A3R inhibit the neutrophil oxidative burst. Agonists to A2BR or A3R significantly inhibit stimulus-induced superoxide production in WT neutrophils but not in Adora2b−/− or Adora3−/− neutrophils (44, 45).
Purinergic Regulation of Neutrophil Degranulation
Neutrophil granules, including primary granules (PGs), secondary granules (SGs), tertiary granules (TGs), and SVs, are formed sequentially during granulopoiesis (46). Neutrophil granules contain a multitude of antimicrobial peptides and proteolytic enzymes. These proteins enable neutrophils to exert their bactericidal and immunologic functions but are potentially harmful to the host if released inappropriately. Purinergic signaling plays bidirectional roles in regulating neutrophil degranulation (Table 1). An in vitro study showed that fMLP-induced neutrophil degranulation can be further enhanced by a non-hydrolyzable ATP analog, ATPγS, but not by hydrolyzable ATP (47). Instead, hydrolyzable ATP suppresses fMLP-induced neutrophil degranulation. Given the potent hydrolytic activity of neutrophil membrane ectonucleotidases, which converts ATP to ADO, the following studies reveal that the inhibitory effect on neutrophil degranulation is induced by the hydrolysis products of ATP, ADO. Furthermore, with the application of selective agonists and antagonists, a recent study indicated that LPS-induced autocrine release of ATP promotes neutrophil exocytosis of SVs, TGs, and SGs via activation of P2X1R (8). The described bidirectional effects of purinergic signaling on neutrophil degranulation may be required for neutrophils to appropriately release their granule contents to regulate their antimicrobial activity during infection and avoid damaging healthy tissues.
Purinergic Regulation of Neutrophil Apoptosis
Circulating neutrophils have a very short lifespan of 8–20 h and do not proliferate (48). However, under inflammation and other pathologic states, neutrophil lifespan is markedly prolonged (48). Extracellular ATP is a critical regulator that inhibits neutrophil apoptosis. Even a 10-min exposure to ATP is sufficient to cause a sustained delay of neutrophil apoptosis in humans (49). Using various selective purinergic receptor antagonists, investigators have identified that ATP-mediated delay of neutrophil apoptosis is P2Y11R-dependent (49). P2Y11R mediates the anti-apoptotic effect of ATP through elevation of neutrophil intracellular cAMP and activation of the subsequent cAMP-dependent protein kinases.
Purinergic Signaling Shapes Neutrophil Immunity in Pathological Conditions
Bacterial Infection and Sterile Inflammation
Neutrophils are the first line of defense within the immune system, and these cells infiltrate diseased tissues to eliminate invading pathogens and necrotic debris, with the aim of restoring tissue homeostasis (50). During Streptococcus pneumoniae corneal infection in mice, neutrophils are the predominant P2X7R-expressing inflammatory cells to infiltrate the infection (9). Knockout of P2X7R has no influence on neutrophil infiltration into the infected cornea but significantly impairs bacterial clearance. The following adoptive transfer experiments using WT or P2rx7−/− neutrophils indicated that P2X7R-expressing neutrophils are required for bacterial clearance. Neutrophil P2X7R exacerbates the local immune responses by mediating ATP-induced NLRP3 inflammasome activation and IL-1β secretion. In contrast to P2X7R, neutrophil A2BR impairs antimicrobial activity during Klebsiella pneumoniae infection (41). Adora2b−/− mice demonstrate enhanced bacterial clearance but unaltered neutrophil infiltration in infected lungs, compared with WT mice. Furthermore, WT recipients of Adora2b−/− BM show a comparable degree of protection compared with global Adora2b−/− knockout mice. The enhanced bactericidal effects appear to be associated with neutrophils, because neutrophils from Adora2b−/− mice demonstrate enhanced extracellular bactericidal activity via generation of NETs.
In thermal injury-induced mice with sterile liver inflammation, selective inhibition or knockout of P2X7R results in reduced hepatic neutrophil recruitment (51). BM chimeric mice demonstrate that P2X7R targets cells that are of hematopoietic origin but are not neutrophils because isolated P2rx7−/− neutrophils are recruited equivalently to inflammatory foci after adoptive transfer. Significant ATP release and increased expression of P2Y2R in mice liver are observed during concanavalin A-mediated hepatitis (52). Selective inhibition or knockout of P2Y2R protects against hepatitis and neutrophil infiltration. Chimeric mice demonstrate that P2Y2R is required in BM-derived cells for hepatic neutrophil infiltration and subsequent liver damage. Inhibition of P2X3R and P2X2/3R with a selective antagonist reduces rat neutrophil infiltration into stimulus-induced inflamed knee joints (53) but not skin and subcutaneous tissues (54, 55). Similarly, P2X7R antagonism inhibits rat neutrophil infiltration into inflamed knee joints (56), but knockout of P2X7R has no effect on skin neutrophil infiltration (57). The variations between neutrophil infiltration in joints and skin tissues are probably caused by specific chemoattractants and the order of their action during neutrophil recruitment at different tissues (58).
Acute Lung Injury (ALI)
Acute lung injury is characterized by lung edema, endothelial and epithelial injury and an excessive infiltration of neutrophils into the interstitium and bronchoalveolar space (59). Neutrophils play a key role in the progression of ALI (60). Neutrophils infiltrate lung tissues, occlude pulmonary capillaries, release cytotoxic mediators such as proteolytic enzymes and superoxide, and thereby destroy lung tissues.
Intratracheal administration of LPS is classically used to induce ALI. With this experimental model, several genetic and pharmacological studies have confirmed that inappropriate activation of P2R signaling is associated with neutrophil-induced hyperinflammation and tissue damage. Knockout or antagonism of mouse P2X7R inhibits neutrophil recruitment into the lung and protects against ALI (61–63). LPS provides a dual signal for alveolar macrophages and induces cytokine production via TLR4/MyD88 signaling and necrosis via P2X7R/CD14 signaling, which promotes the release of pro-IL-1α and subsequently activates the IL-1 receptor on endothelial cells to induce tight junction opening and allow neutrophil infiltration into the lungs (62). In addition, the P2X7R-induced shedding of soluble VCAM-1 from alveolar epithelial type-I cells functions as a chemoattractant to recruit neutrophils during ALI (63). The pulmonary neutrophil recruitment induced by intratracheal LPS can also be inhibited in mice that receive either with P2Y1R or P2Y14R antagonists (64). Activation of P2Y1R and P2Y14R in platelets induces a platelet-dependent neutrophil motility toward macrophage-associated chemokines. In contrast to P2R signaling, activation of P1R signaling frequently interferes with neutrophil trafficking during ALI. During exposure to LPS, mouse lung A1R expression is upregulated (65). However, when neutrophils are depleted before LPS inhalation, the induction of A1R is attenuated, suggesting that neutrophils induce A1R in the inflamed lungs (65). Moreover, LPS-induced transmigration of neutrophils into the interstitium and bronchoalveolar lavage is further elevated in Adora1−/− mice but can be reversed by an A1R agonist in WT mice. These findings were further confirmed by in vitro experiments showing that chemokine-induced transmigration is reduced when neutrophils are pretreated with an A1R agonist and that an A1R agonist can prevent pulmonary endothelial cells from LPS-induced cellular remodeling and cell retraction. Lung A2AR expression is increased when mice are exposed to intratracheal LPS (66). LPS-induced neutrophil accumulation in the pulmonary circulation, but not interstitium and bronchoalveolar lavage, is significantly greater in Adoa2a−/− mice than in WT mice (66). In chimeric mice lacking A2AR on BM-derived cells, neutrophil migration into the bronchoalveolar lavage is increased, and an A2AR agonist reduces LPS-induced neutrophil recruitment only when A2AR is expressed on BM-derived cells (66, 67). Similar results are observed with mice that selectively lack A2AR on myeloid cells; these mice show increased migration of neutrophils into the bronchoalveolar lavage (66). Taken together, these data indicate that A2AR activation on BM-derived cells is critical for pulmonary neutrophil infiltration during ALI. Intratracheal administration of LPS induces A2BR expression in the lung, and Adoa2b−/− mice show an increase in neutrophil migration into the pulmonary interstitium (68). Using chimeric mice, investigators identified that A2BR on BM-derived cells is crucial for inhibiting neutrophil migration during ALI (68, 69). However, in a two-hit ALI model where intratracheal LPS treatment is followed by injurious mechanical ventilation, tissue-specific Adora2b−/− in alveolar epithelial cells, but not myeloid lineage or endothelial cells, closely resembles the observed detrimental effects of global Adora2b−/− (70). Differences in the pathological environments induced by mechanical ventilation may contribute to this discrepancy.
In severe pulmonary infection-induced ALI models, P2X7R is involved in the exacerbated inflammatory injuries and neutrophil infiltration. Severe tuberculosis and pulmonary injury are caused when mice are infected with hypervirulent Mycobacterium bovis (71). Chimeric mice that lack P2X7R in BM-derived cells show alleviated pulmonary injury, which demonstrates that P2X7R in BM-derived cells plays a critical role in the progression of severe tuberculosis. An exacerbated immune response is one of the main causes of influenza virus-induced lung damage during infection (72). Knockout of P2X7R results in a better outcome in response to influenza virus infection, as characterized by reduced lung pathology and neutrophil infiltration. Moreover, the absence of P2X7R or inhibition of P2X7R activation by selective antagonist or apyrase suppresses pulmonary inflammatory responses and neutrophil infiltration in the early phase of acute adenoviral infection (73).
Acute lung injury represents the primary complication in sepsis during the sequential development of multiple organ dysfunction (74). In intraperitoneal LPS injection-induced SIRS-associated ALI, P2X1R mediates pulmonary neutrophil infiltration. Lecut et al. reported that a deficiency of P2X1R augments neutrophil accumulation in the lungs and tissue damage (75), whereas Maître et al. observed that deficiency of P2X1R leads to strongly reduced neutrophil accumulation in the lungs and less tissue damage (76). The differences could be related to the experimental design (e.g., monitoring of age, sex, and weight matching) or to the dose and serotype of LPS. Further investigations will be necessary to confirm these findings (76). Expression of lung A2AR is upregulated in mice that are intravenously injected with LPS (77). LPS-induced accumulation of pulmonary neutrophils is further enhanced in Adora2a−/− mice but can be reversed in WT mice by an A2AR agonist, indicating the protective effects of A2AR against ALI. Cecal ligation and puncture (CLP) is an alternative approach to induce SIRS-associated ALI. Performing CLP in P2ry2−/− and Adora3−/− mice, investigators observed decreased recruitment of neutrophils into the lungs and attenuated development of ALI compared with WT mice (78). Similar results were observed in experiments using P2ry12−/−mice, and antagonists of P2Y12R succeeded in reversing the pathological changes in WT mice (79, 80). These studies demonstrate the stimulatory role of P2Y2R, A3R, and P2Y12R in neutrophil infiltration during ALI.
Ischemia-Reperfusion (IR) Injury
Ischemia-reperfusion injury occurs when blood and concomitant oxygen return to tissues after the initial insult of ischemia or lack of oxygen, which is a common complication following myocardial infarction, transplantation, stroke and trauma (81). Restoration of circulation and reoxygenation are frequently associated with inflammatory responses and subsequent tissue injuries, rather than recovery of normal tissue functions (81). IR injury is recognized as a complex cascade of events, involving numerous biochemical compounds that are released in response to ischemia and interactions between vascular endothelial cells and immune cells (81). Neutrophil infiltration is a hallmark of IR injury and represents an important component in the protracted inflammatory response and its severity (82). The profound effects of purinergic signaling on neutrophils during IR injury have been identified.
Convincing evidence obtained from Adora1−/− mice demonstrates that A1R signaling is essential for protecting against multiple IR-induced organ injuries (83–85). Adora1−/− mice exhibit significantly higher neutrophil infiltration and tissue inflammatory responses during IR-induced renal, pulmonary and hepatic injuries than WT mice (83–85). In addition, A1R agonists can maintain the neutrophil infiltration and tissue inflammatory responses in WT mice but not in Adora1−/− mice (84, 85). In parallel with the A1R agonist, an A1R allosteric enhancer that selectively increases the efficacy of endogenous ADO–A1R interaction protects against neutrophil infiltration and renal IR injury in WT mice but not in Adora1−/− mice (83). Suppressed neutrophil infiltration and the accompanying beneficial effects have been achieved during renal, pulmonary and myocardial IR injuries by administration of A2AR agonists in various experimental animals (86–88). Nevertheless, when the agonist is applied in Adora2a−/− mice or paired with a specific antagonist, suppression of neutrophil infiltration and all the protective effects are eliminated (88, 89). These findings reveal the roles of A2AR signaling in inhibiting neutrophil infiltration and protecting against tissue injuries during renal, pulmonary and myocardial IR. Agonism of A3R provides significant protection against neutrophil infiltration, lung inflammation and dysfunction after pulmonary IR injury in WT mice but not Adora3−/− mice (90). Further in vitro transwell assays have shown that an A3R agonist inhibits neutrophil transmigration, suggesting that the protective effects of A3R may be due to the direct prevention of neutrophil activation. In addition, A3R activation reduces neutrophil infiltration into IR-injured myocardium in WT mice but not in global A3R deficient mice or chimeric mice that lack of A3R in BM-derived cells (22). Subsequent experiments using in vitro transwell assays achieved results that are consistent with previous studies, indicating that neutrophil-expressed A3R is essential for inhibiting neutrophil infiltration.
In contrast to the effects of A1R, A2AR, and A3R for suppressing neutrophil activation during IR injury, A2BR exhibits a bidirectional role in myocardial and pulmonary IR injury. Evidence from chimeric mice that lack A2BR in BM cells shows that A2BR is protective when activated on BM-derived cells (most likely neutrophils) in myocardial IR injury (91). The following study that used mice with a specific deletion of A2BR in the myeloid lineage or either endothelial or myocardial cells demonstrated that myeloid lineage cells are necessary to provide cardioprotection during IR injury and inhibit neutrophil accumulation (92). Adoptive transfer of Adora2b−/− neutrophils into neutrophil-depleted mice further confirmed the role of neutrophil-expressed A2BR in cardioprotection against IR injury (92). In contrast to myocardial IR injury, A2BR appears to promote pulmonary IR injury and inflammatory responses by stimulating the production of cytokines and neutrophil infiltration (93, 94). However, the pro-inflammatory effects of A2BR are likely specific for resident pulmonary cells, but not BM-derived neutrophils, based on the data obtained from BM chimeric mice (94).
Compared with the considerable evidence showing that ADO-driven P1 signaling mediates neutrophil activation and infiltration during IR injury, few studies have focused on the effects of ATP-induced P2R signaling on neutrophil function during IR injury. A recent report demonstrated that antagonism of P2X7R ameliorates IR-induced renal neutrophil infiltration and tissue injury, and similar results were achieved by knockout of P2X7R (95). Chimeric mice with P2X7R deficiency in BM cells further confirm that activation of P2X7R in BM-derived cells is essential for renal neutrophil infiltration during IR injury. Additional studies are needed to further clarify the effects of other P2 receptors on neutrophils during IR injury.
Concluding Remarks and Future Perspectives
The past 5 years have brought major advances in the identification of numerous types of neutrophil purinergic receptors and in the understanding of their functions in coordinating the appropriate immune response against invading pathogens or diseased tissue. The fine-tuned balance between P2R and P1R signaling appears to be critical for shaping neutrophil plasticity and heterogeneity to orchestrate a complex series of events during inflammation (Figure 2). Several drugs that target purinergic signaling (such as adenosine and clopidogrel) have already used in patients. More selective and effective pharmacological tools are continually being developed, and increasing developments in the field of neutrophil purinergic signaling may be further exploited in the treatment of patients with inflammatory or infectious diseases in the near future. In keeping with this goal, key future challenges will be to understand the nature of the neutrophil purinergic signaling repertoire during in vivo infectious and inflammatory conditions, how purinergic receptors are integrated with other pattern recognition receptors to control neutrophil function and the dynamic biochemistry of ATP and ADO in the extracellular environment.
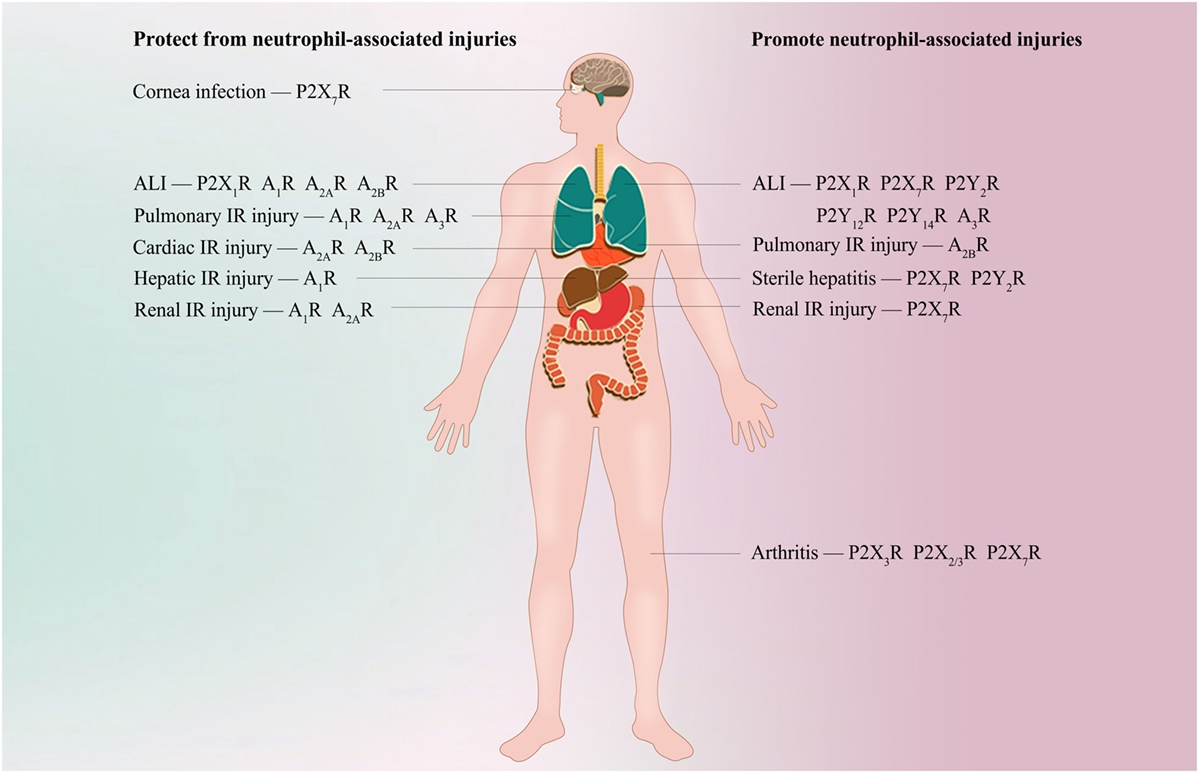
Figure 2. Potential purinergic targets for treating neutrophil-associated diseases. During bacterial clearance in Streptococcus pneumoniae corneal infection, agonism of neutrophil P2X7R is required for bacterial clearance. Inappropriate activation of P2X7R, P2Y2R, P2Y12R, P2Y14R, and A3R is associated with neutrophil-induced hyperinflammation and tissue damage during acute lung injury (ALI); however, A1R, A2AR, and A2BR are essential for protecting against ALI. Specific antagonism of P2Y2R, P2Y12R, P2Y14R, and A3R or specific agonism of A1R, A2AR, and A2BR alleviates neutrophil recruitment into the lung and protects against ALI. P2X1R exhibits a bidirectional role in neutrophil accumulation and tissue damage during ALI, but the detailed mechanisms are unclear. A1R, A2AR, and A3R signaling frequently inhibits neutrophil infiltration and protects against tissue injuries during pulmonary, cardiac, hepatic, or renal ischemia-reperfusion (IR) injury. However, evidence shows that A2BR is protective when activated on BM-derived cells (most likely neutrophils) in myocardial IR injury, but promote pulmonary IR injury when activated on resident pulmonary cells. During renal IR, antagonism of P2X7R ameliorates neutrophil infiltration and renal injury. P2X7R or P2Y2R is required for hepatic neutrophil infiltration and subsequent liver damage in thermal or chemical injury-induced sterile hepatitis, respectively. Specific inhibition of P2X7R or P2Y2R is shown to protect against neutrophil infiltration and liver inflammatory injuries. Antagonism of P2X3R, P2X2/3R, and P2X7R reduces neutrophil infiltration into inflamed knee joints in arthritis.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by the National Natural Science Foundation of China, No. 81701945 and 81572956.
References
1. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol (2016) 16:177–92. doi:10.1038/nri.2016.4
2. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med (2012) 367:2322–33. doi:10.1056/NEJMra1205750
3. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature (2014) 509:310–7. doi:10.1038/nature13085
4. Antonioli L, Blandizzi C, Pacher P, Hasko G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer (2013) 13:842–57. doi:10.1038/nrc3613
5. Antonioli L, Pacher P, Vizi ES, Hasko G. CD39 and CD73 in immunity and inflammation. Trends Mol Med (2013) 19:355–67. doi:10.1016/j.molmed.2013.03.005
6. Scapini P, Cassatella MA. Social networking of human neutrophils within the immune system. Blood (2014) 124:710–9. doi:10.1182/blood-2014-03-453217
7. Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science (2006) 314:1792–5. doi:10.1126/science.1132559
8. Wang X, Qin W, Xu X, Xiong Y, Zhang Y, Zhang H, et al. Endotoxin-induced autocrine ATP signaling inhibits neutrophil chemotaxis through enhancing myosin light chain phosphorylation. Proc Natl Acad Sci U S A (2017) 114:4483–8. doi:10.1073/pnas.1616752114
9. Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E. Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1beta secretion in response to ATP. Nat Commun (2016) 7:10555. doi:10.1038/ncomms10555
10. Baron L, Gombault A, Fanny M, Villeret B, Savigny F, Guillou N, et al. The NLRP3 inflammasome is activated by nanoparticles through ATP, ADP and adenosine. Cell Death Dis (2015) 6:e1629. doi:10.1038/cddis.2014.576
11. Morciano G, Sarti AC, Marchi S, Missiroli S, Falzoni S, Raffaghello L, et al. Use of luciferase probes to measure ATP in living cells and animals. Nat Protoc (2017) 12:1542–62. doi:10.1038/nprot.2017.052
12. Esseltine JL, Laird DW. Next-generation connexin and pannexin cell biology. Trends Cell Biol (2016) 26:944–55. doi:10.1016/j.tcb.2016.06.003
13. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate ’find-me’ signal release and membrane permeability during apoptosis. Nature (2010) 467:863–7. doi:10.1038/nature09413
14. Bao Y, Chen Y, Ledderose C, Li L, Junger WG. Pannexin 1 channels link chemoattractant receptor signaling to local excitation and global inhibition responses at the front and back of polarized neutrophils. J Biol Chem (2013) 288:22650–7. doi:10.1074/jbc.M113.476283
15. Schumacher D, Strilic B, Sivaraj KK, Wettschureck N, Offermanns S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell (2013) 24:130–7. doi:10.1016/j.ccr.2013.05.008
16. Wang Y, Martins I, Ma Y, Kepp O, Galluzzi L, Kroemer G. Autophagy-dependent ATP release from dying cells via lysosomal exocytosis. Autophagy (2013) 9:1624–5. doi:10.4161/auto.25873
17. Mempin R, Tran H, Chen C, Gong H, Kim Ho K, Lu S. Release of extracellular ATP by bacteria during growth. BMC Microbiol (2013) 13:301. doi:10.1186/1471-2180-13-301
18. Borea PA, Gessi S, Merighi S, Varani K. Adenosine as a multi-signalling guardian angel in human diseases: when, where and how does it exert its protective effects? Trends Pharmacol Sci (2016) 37:419–34. doi:10.1016/j.tips.2016.02.006
19. Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets – what are the challenges? Nat Rev Drug Discov (2013) 12:265–86. doi:10.1038/nrd3955
20. Valdes R, Elferich J, Shinde U, Landfear SM. Identification of the intracellular gate for a member of the equilibrative nucleoside transporter (ENT) family. J Biol Chem (2014) 289:8799–809. doi:10.1074/jbc.M113.546960
21. Wirsdorfer F, de Leve S, Cappuccini F, Eldh T, Meyer AV, Gau E, et al. Extracellular adenosine production by ecto-5’-nucleotidase (CD73) enhances radiation-induced lung fibrosis. Cancer Res (2016) 76:3045–56. doi:10.1158/0008-5472.CAN-15-2310
22. Ge ZD, van der Hoeven D, Maas JE, Wan TC, Auchampach JA. A(3) adenosine receptor activation during reperfusion reduces infarct size through actions on bone marrow-derived cells. J Mol Cell Cardiol (2010) 49:280–6. doi:10.1016/j.yjmcc.2010.01.018
23. Alencar AKN, Montes GC, Barreiro EJ, Sudo RT, Zapata-Sudo G. Adenosine receptors as drug targets for treatment of pulmonary arterial hypertension. Front Pharmacol (2017) 8:858. doi:10.3389/fphar.2017.00858
24. Jespers W, Schiedel AC, Heitman LH, Cooke RM, Kleene L, van Westen GJP, et al. Structural mapping of adenosine receptor mutations: ligand binding and signaling mechanisms. Trends Pharmacol Sci (2018) 39:75–89. doi:10.1016/j.tips.2017.11.001
25. Tweedy L, Knecht DA, Mackay GM, Insall RH. Self-generated chemoattractant gradients: attractant depletion extends the range and robustness of chemotaxis. PLoS Biol (2016) 14:e1002404. doi:10.1371/journal.pbio.1002404
26. Weninger W, Biro M, Jain R. Leukocyte migration in the interstitial space of non-lymphoid organs. Nat Rev Immunol (2014) 14:232–46. doi:10.1038/nri3641
27. Bao Y, Ledderose C, Seier T, Graf AF, Brix B, Chong E, et al. Mitochondria regulate neutrophil activation by generating ATP for autocrine purinergic signaling. J Biol Chem (2014) 289:26794–803. doi:10.1074/jbc.M114.572495
28. Bao Y, Ledderose C, Graf AF, Brix B, Birsak T, Lee A, et al. mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J Cell Biol (2015) 210:1153–64. doi:10.1083/jcb.201503066
29. Liu L, Das S, Losert W, Parent CA. mTORC2 regulates neutrophil chemotaxis in a cAMP- and RhoA-dependent fashion. Dev Cell (2010) 19:845–57. doi:10.1016/j.devcel.2010.11.004
30. Xu X, Zheng S, Xiong Y, Wang X, Qin W, Zhang H, et al. Adenosine effectively restores endotoxin-induced inhibition of human neutrophil chemotaxis via A1 receptor-p38 pathway. Inflamm Res (2017) 66:353–64. doi:10.1007/s00011-016-1021-3
31. Alkayed F, Kashimata M, Koyama N, Hayashi T, Tamura Y, Azuma Y. P2Y11 purinoceptor mediates the ATP-enhanced chemotactic response of rat neutrophils. J Pharmacol Sci (2012) 120:288–95. doi:10.1254/jphs.12173FP
32. Barrett MO, Sesma JI, Ball CB, Jayasekara PS, Jacobson KA, Lazarowski ER, et al. A selective high-affinity antagonist of the P2Y14 receptor inhibits UDP-glucose-stimulated chemotaxis of human neutrophils. Mol Pharmacol (2013) 84:41–9. doi:10.1124/mol.113.085654
33. de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol (2016) 16:378–91. doi:10.1038/nri.2016.49
34. Pruenster M, Kurz AR, Chung KJ, Cao-Ehlker X, Bieber S, Nussbaum CF, et al. Extracellular MRP8/14 is a regulator of beta2 integrin-dependent neutrophil slow rolling and adhesion. Nat Commun (2015) 6:6915. doi:10.1038/ncomms7915
35. Darbousset R, Delierneux C, Mezouar S, Hego A, Lecut C, Guillaumat I, et al. P2X1 expressed on polymorphonuclear neutrophils and platelets is required for thrombosis in mice. Blood (2014) 124:2575–85. doi:10.1182/blood-2014-04-571679
36. Yago T, Tsukamoto H, Liu Z, Wang Y, Thompson LF, McEver RP. Multi-Inhibitory Effects of A2A adenosine receptor signaling on neutrophil adhesion under flow. J Immunol (2015) 195:3880–9. doi:10.4049/jimmunol.1500775
37. Amulic B, Knackstedt SL, Abu Abed U, Deigendesch N, Harbort CJ, Caffrey BE, et al. Cell-cycle proteins control production of neutrophil extracellular traps. Dev Cell (2017) 43:449–62.e5. doi:10.1016/j.devcel.2017.10.013
38. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol (2017) 18(2):134–47. doi:10.1038/nri.2017.105
39. Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol (2014) 15:1017–25. doi:10.1038/ni.2987
40. Sil P, Hayes CP, Reaves BJ, Breen P, Quinn S, Sokolove J, et al. P2Y6 receptor antagonist MRS2578 inhibits neutrophil activation and aggregated neutrophil extracellular trap formation induced by gout-associated monosodium urate crystals. J Immunol (2017) 198:428–42. doi:10.4049/jimmunol.1600766
41. Barletta KE, Cagnina RE, Burdick MD, Linden J, Mehrad B. Adenosine A(2B) receptor deficiency promotes host defenses against gram-negative bacterial pneumonia. Am J Respir Crit Care Med (2012) 186:1044–50. doi:10.1164/rccm.201204-0622OC
42. Corriden R, Self T, Akong-Moore K, Nizet V, Kellam B, Briddon SJ, et al. Adenosine-A3 receptors in neutrophil microdomains promote the formation of bacteria-tethering cytonemes. EMBO Rep (2013) 14:726–32. doi:10.1038/embor.2013.89
43. Chen Y, Yao Y, Sumi Y, Li A, To UK, Elkhal A, et al. Purinergic signaling: a fundamental mechanism in neutrophil activation. Sci Signal (2010) 3:ra45. doi:10.1126/scisignal.2000549
44. van der Hoeven D, Wan TC, Gizewski ET, Kreckler LM, Maas JE, Van Orman J, et al. A role for the low-affinity A2B adenosine receptor in regulating superoxide generation by murine neutrophils. J Pharmacol Exp Ther (2011) 338:1004–12. doi:10.1124/jpet.111.181792
45. van der Hoeven D, Wan TC, Auchampach JA. Activation of the A(3) adenosine receptor suppresses superoxide production and chemotaxis of mouse bone marrow neutrophils. Mol Pharmacol (2008) 74:685–96. doi:10.1124/mol.108.048066
46. Larsen MT, Hager M, Glenthoj A, Asmar F, Clemmensen SN, Mora-Jensen H, et al. miRNA-130a regulates C/EBP-epsilon expression during granulopoiesis. Blood (2014) 123:1079–89. doi:10.1182/blood-2013-08-523233
47. Chen Y, Hashiguchi N, Yip L, Junger WG. Hypertonic saline enhances neutrophil elastase release through activation of P2 and A3 receptors. Am J Physiol Cell Physiol (2006) 290:C1051–9. doi:10.1152/ajpcell.00216.2005
48. Lahoz-Beneytez J, Elemans M, Zhang Y, Ahmed R, Salam A, Block M, et al. Human neutrophil kinetics: modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood (2016) 127:3431–8. doi:10.1182/blood-2016-03-700336
49. Vaughan KR, Stokes L, Prince LR, Marriott HM, Meis S, Kassack MU, et al. Inhibition of neutrophil apoptosis by ATP is mediated by the P2Y11 receptor. J Immunol (2007) 179:8544–53. doi:10.4049/jimmunol.179.12.8544
50. Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H, et al. Neutrophils: between host defence, immune modulation, and tissue injury. PLoS Pathog (2015) 11:e1004651. doi:10.1371/journal.ppat.1004651
51. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science (2010) 330:362–6. doi:10.1126/science.1195491
52. Ayata CK, Ganal SC, Hockenjos B, Willim K, Vieira RP, Grimm M, et al. Purinergic P2Y(2) receptors promote neutrophil infiltration and hepatocyte death in mice with acute liver injury. Gastroenterology (2012) 143:1620–9.e4. doi:10.1053/j.gastro.2012.08.049
53. Teixeira JM, Bobinski F, Parada CA, Sluka KA, Tambeli CH. P2X3 and P2X2/3 receptors play a crucial role in articular hyperalgesia development through inflammatory mechanisms in the knee joint experimental synovitis. Mol Neurobiol (2017) 54:6174–86. doi:10.1007/s12035-016-0146-2
54. Oliveira MC, Pelegrini-da-Silva A, Tambeli CH, Parada CA. Peripheral mechanisms underlying the essential role of P2X3,2/3 receptors in the development of inflammatory hyperalgesia. Pain (2009) 141:127–34. doi:10.1016/j.pain.2008.10.024
55. de Oliveira Fusaro MC, Pelegrini-da-Silva A, Araldi D, Parada CA, Tambeli CH. P2X3 and P2X2/3 receptors mediate mechanical hyperalgesia induced by bradykinin, but not by pro-inflammatory cytokines, PGE(2) or dopamine. Eur J Pharmacol (2010) 649:177–82. doi:10.1016/j.ejphar.2010.09.037
56. Teixeira JM, Dias EV, Parada CA, Tambeli CH. Intra-articular blockade of P2X7 receptor reduces the articular hyperalgesia and inflammation in the knee joint synovitis especially in female rats. J Pain (2017) 18:132–43. doi:10.1016/j.jpain.2016.10.008
57. Geraghty NJ, Mansfield KJ, Fuller SJ, Watson D, Sluyter R. The P2X7 receptor is not essential for development of imiquimod-induced psoriasis-like inflammation in mice. Purinergic Signal (2017) 13:405–15. doi:10.1007/s11302-017-9569-0
58. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13:159–75. doi:10.1038/nri3399
59. Nasseri S, Gurusamy M, Jung B, Lee D, Khang G, Doods H, et al. Kinin B1 receptor antagonist BI113823 reduces acute lung injury. Crit Care Med (2015) 43:e499–507. doi:10.1097/CCM.0000000000001268
60. Martin TR. Neutrophils and lung injury: getting it right. J Clin Invest (2002) 110:1603–5. doi:10.1172/JCI0217302
61. Wang S, Zhao J, Wang H, Liang Y, Yang N, Huang Y. Blockage of P2X7 attenuates acute lung injury in mice by inhibiting NLRP3 inflammasome. Int Immunopharmacol (2015) 27:38–45. doi:10.1016/j.intimp.2015.04.035
62. Dagvadorj J, Shimada K, Chen S, Jones HD, Tumurkhuu G, Zhang W, et al. Lipopolysaccharide Induces alveolar macrophage necrosis via CD14 and the P2X7 receptor leading to interleukin-1alpha release. Immunity (2015) 42:640–53. doi:10.1016/j.immuni.2015.03.007
63. Mishra A, Guo Y, Zhang L, More S, Weng T, Chintagari NR, et al. A critical role for P2X7 receptor-induced VCAM-1 shedding and neutrophil infiltration during acute lung injury. J Immunol (2016) 197:2828–37. doi:10.4049/jimmunol.1501041
64. Amison RT, Arnold S, O’Shaughnessy BG, Cleary SJ, Ofoedu J, Idzko M, et al. Lipopolysaccharide (LPS) induced pulmonary neutrophil recruitment and platelet activation is mediated via the P2Y1 and P2Y14 receptors in mice. Pulm Pharmacol Ther (2017) 45:62–8. doi:10.1016/j.pupt.2017.05.005
65. Ngamsri KC, Wagner R, Vollmer I, Stark S, Reutershan J. Adenosine receptor A1 regulates polymorphonuclear cell trafficking and microvascular permeability in lipopolysaccharide-induced lung injury. J Immunol (2010) 185:4374–84. doi:10.4049/jimmunol.1000433
66. Reutershan J, Cagnina RE, Chang D, Linden J, Ley K. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2a stimulation in lipopolysaccharide-induced lung injury. J Immunol (2007) 179:1254–63. doi:10.4049/jimmunol.179.2.1254
67. Aggarwal NR, D’Alessio FR, Eto Y, Chau E, Avalos C, Waickman AT, et al. Macrophage A2A adenosinergic receptor modulates oxygen-induced augmentation of murine lung injury. Am J Respir Cell Mol Biol (2013) 48:635–46. doi:10.1165/rcmb.2012-0351OC
68. Konrad FM, Witte E, Vollmer I, Stark S, Reutershan J. Adenosine receptor A2b on hematopoietic cells mediates LPS-induced migration of PMNs into the lung interstitium. Am J Physiol Lung Cell Mol Physiol (2012) 303:L425–38. doi:10.1152/ajplung.00387.2011
69. Konrad FM, Meichssner N, Bury A, Ngamsri KC, Reutershan J. Inhibition of SDF-1 receptors CXCR4 and CXCR7 attenuates acute pulmonary inflammation via the adenosine A2B-receptor on blood cells. Cell Death Dis (2017) 8:e2832. doi:10.1038/cddis.2016.482
70. Hoegl S, Brodsky KS, Blackburn MR, Karmouty-Quintana H, Zwissler B, Eltzschig HK. Alveolar epithelial A2B adenosine receptors in pulmonary protection during acute lung injury. J Immunol (2015) 195:1815–24. doi:10.4049/jimmunol.1401957
71. Bomfim CCB, Amaral EP, Cassado ADA, Salles EM, do Nascimento RS, Lasunskaia E, et al. P2X7 receptor in bone marrow-derived cells aggravates tuberculosis caused by hypervirulent Mycobacterium bovis. Front Immunol (2017) 8:435. doi:10.3389/fimmu.2017.00435
72. Leyva-Grado VH, Ermler ME, Schotsaert M, Gonzalez MG, Gillespie V, Lim JK, et al. Contribution of the purinergic receptor P2X7 to development of lung immunopathology during influenza virus infection. MBio (2017) 8:e00229–17. doi:10.1128/mBio.00229-17
73. Lee BH, Hwang DM, Palaniyar N, Grinstein S, Philpott DJ, Hu J. Activation of P2X(7) receptor by ATP plays an important role in regulating inflammatory responses during acute viral infection. PLoS One (2012) 7:e35812. doi:10.1371/journal.pone.0035812
74. Hirano Y, Aziz M, Yang WL, Wang Z, Zhou M, Ochani M, et al. Neutralization of osteopontin attenuates neutrophil migration in sepsis-induced acute lung injury. Crit Care (2015) 19:53. doi:10.1186/s13054-015-0782-3
75. Lecut C, Faccinetto C, Delierneux C, van Oerle R, Spronk HM, Evans RJ, et al. ATP-gated P2X1 ion channels protect against endotoxemia by dampening neutrophil activation. J Thromb Haemost (2012) 10:453–65. doi:10.1111/j.1538-7836.2011.04606.x
76. Maître B, Magnenat S, Heim V, Ravanat C, Evans RJ, de la Salle H, et al. The P2X1 receptor is required for neutrophil extravasation during lipopolysaccharide-induced lethal endotoxemia in mice. J Immunol (2015) 194:739–49. doi:10.4049/jimmunol.1401786
77. He X, Hu JL, Li J, Zhao L, Zhang Y, Zeng YJ, et al. A feedback loop in PPARgamma-adenosine A2A receptor signaling inhibits inflammation and attenuates lung damages in a mouse model of LPS-induced acute lung injury. Cell Signal (2013) 25:1913–23. doi:10.1016/j.cellsig.2013.05.024
78. Inoue Y, Chen Y, Hirsh MI, Yip L, Junger WG. A3 and P2Y2 receptors control the recruitment of neutrophils to the lungs in a mouse model of sepsis. Shock (2008) 30:173–7. doi:10.1097/shk.0b013e318160dad4
79. Liverani E, Rico MC, Tsygankov AY, Kilpatrick LE, Kunapuli SP. P2Y12 receptor modulates sepsis-induced inflammation. Arterioscler Thromb Vasc Biol (2016) 36:961–71. doi:10.1161/ATVBAHA.116.307401
80. Rahman M, Gustafsson D, Wang Y, Thorlacius H, Braun OO. Ticagrelor reduces neutrophil recruitment and lung damage in abdominal sepsis. Platelets (2014) 25:257–63. doi:10.3109/09537104.2013.809520
81. Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest (2013) 123:92–100. doi:10.1172/JCI62874
82. Liao YH, Xia N, Zhou SF, Tang TT, Yan XX, Lv BJ, et al. Interleukin-17A contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and neutrophil infiltration. J Am Coll Cardiol (2012) 59:420–9. doi:10.1016/j.jacc.2011.10.863
83. Park SW, Kim JY, Ham A, Brown KM, Kim M, D’Agati VD, et al. A1 adenosine receptor allosteric enhancer PD-81723 protects against renal ischemia-reperfusion injury. Am J Physiol Renal Physiol (2012) 303:F721–32. doi:10.1152/ajprenal.00157.2012
84. Fernandez LG, Sharma AK, LaPar DJ, Kron IL, Laubach VE. Adenosine A1 receptor activation attenuates lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg (2013) 145:1654–9. doi:10.1016/j.jtcvs.2013.01.006
85. Kim J, Kim M, Song JH, Lee HT. Endogenous A1 adenosine receptors protect against hepatic ischemia reperfusion injury in mice. Liver Transpl (2008) 14:845–54. doi:10.1002/lt.21432
86. Tian Y, French BA, Kron IL, Yang Z. Splenic leukocytes mediate the hyperglycemic exacerbation of myocardial infarct size in mice. Basic Res Cardiol (2015) 110:39. doi:10.1007/s00395-015-0496-3
87. Sharma AK, Laubach VE, Ramos SI, Zhao Y, Stukenborg G, Linden J, et al. Adenosine A2A receptor activation on CD4+ T lymphocytes and neutrophils attenuates lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg (2010) 139:474–82. doi:10.1016/j.jtcvs.2009.08.033
88. Gazoni LM, Walters DM, Unger EB, Linden J, Kron IL, Laubach VE. Activation of A1, A2A, or A3 adenosine receptors attenuates lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg (2010) 140:440–6. doi:10.1016/j.jtcvs.2010.03.002
89. Sharma AK, Linden J, Kron IL, Laubach VE. Protection from pulmonary ischemia-reperfusion injury by adenosine A2A receptor activation. Respir Res (2009) 10:58. doi:10.1186/1465-9921-10-58
90. Mulloy DP, Sharma AK, Fernandez LG, Zhao Y, Lau CL, Kron IL, et al. Adenosine A3 receptor activation attenuates lung ischemia-reperfusion injury. Ann Thorac Surg (2013) 95:1762–7. doi:10.1016/j.athoracsur.2013.01.059
91. Koeppen M, Harter PN, Bonney S, Bonney M, Reithel S, Zachskorn C, et al. Adora2b signaling on bone marrow derived cells dampens myocardial ischemia-reperfusion injury. Anesthesiology (2012) 116:1245–57. doi:10.1097/ALN.0b013e318255793c
92. Seo SW, Koeppen M, Bonney S, Gobel M, Thayer M, Harter PN, et al. Differential tissue-specific function of adora2b in cardioprotection. J Immunol (2015) 195:1732–43. doi:10.4049/jimmunol.1402288
93. Huerter ME, Sharma AK, Zhao Y, Charles EJ, Kron IL, Laubach VE. Attenuation of pulmonary ischemia-reperfusion injury by adenosine A2B receptor antagonism. Ann Thorac Surg (2016) 102:385–93. doi:10.1016/j.athoracsur.2016.02.060
94. Anvari F, Sharma AK, Fernandez LG, Hranjec T, Ravid K, Kron IL, et al. Tissue-derived proinflammatory effect of adenosine A2B receptor in lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg (2010) 140:871–7. doi:10.1016/j.jtcvs.2010.06.051
Keywords: purinergic signaling, neutrophil, innate immune, inflammation, purinergic receptor
Citation: Wang X and Chen D (2018) Purinergic Regulation of Neutrophil Function. Front. Immunol. 9:399. doi: 10.3389/fimmu.2018.00399
Received: 15 January 2018; Accepted: 13 February 2018;
Published: 01 March 2018
Edited by:
Heiko Mühl, Goethe University Frankfurt, GermanyReviewed by:
Ben A. Croker, Harvard University, United StatesRonald Sluyter, University of Wollongong, Australia
Mausita Karmakar, Case Western Reserve University, United States
Copyright: © 2018 Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xu Wang, anNkeHd4QDEyNi5jb20=;
Deyu Chen, Y2RleXVAaG90bWFpbC5jb20=
†These authors have contributed equally to this work.