 David Michalovich
David Michalovich Sergey Nejentsev
Sergey Nejentsev- 1Refractory Respiratory Inflammation Discovery Performance Unit, GlaxoSmithKline, Stevenage, United Kingdom
- 2Department of Medicine, University of Cambridge, Cambridge, United Kingdom
Activated PI3 kinase delta syndrome (APDS) is a primary immunodeficiency caused by dominant mutations that increase activity of phosphoinositide-3-kinase δ (PI3Kδ). APDS can be caused by mutations in the PIK3CD gene that encodes PI3Kδ catalytic subunit p110δ (APDS1) or mutations in the PIK3R1 gene that encodes regulatory subunit p85α (APDS2). APDS research advanced rapidly after the initial discovery in 2013. More than 200 APDS patients have been identified around the world. Multiple novel APDS mutations were reported and molecular mechanisms leading to PI3Kδ activation have been elucidated. The finding of APDS significantly increased our understanding of the role of PI3Kδ in the human immune system. Perhaps most importantly, discovery of the molecular basis of this primary immunodeficiency suggested that APDS patients, who previously received only non-specific therapy, could be treated by a novel class of drugs that inhibits PI3Kδ activity. This led to the ongoing clinical trials of selective PI3Kδ inhibitors in APDS patients. Overall, the APDS story provides an excellent example of translational research, beginning with patients who had an unknown disease cause and leading to a novel specific knowledge-based treatment.
Introduction
Primary immunodeficiencies (PIDs) are a group of disorders that cause immune dysfunction and manifest with increased susceptibility to infections. Many PIDs are monogenic diseases. To date, mutations in more than 300 genes have been shown to cause various PIDs (1). Activated PI3 kinase delta syndrome (APDS) is a PID that results from gain-of-function mutations in genes encoding the phosphoinositide-3-kinase δ (PI3Kδ). This review will focus on the APDS mutations, phenotypes of the disease, and current therapeutic approaches.
Phosphoinositide-3-kinase δ is a class IA lipid kinase that phosphorylates phosphatidylinositol-4,5-bisphosphate [PtdIns(4,5)P2 or PIP2] to produce phosphatidylinositol-3,4,5-trisphosphate [PtdIns(3,4,5)P3 or PIP3]. There are three class IA PI3Ks in mammalian cells: α, β, and δ. Each class IA PI3K is composed of a catalytic subunit: p110α, p110β, or p110δ (encoded by genes PIK3CA, PIK3CB, and PIK3CD, respectively), and one of the five regulatory subunits: p85α, p55α, p50α (all encoded by different transcripts of the PIK3R1 gene), p85β (encoded by the PIK3R2 gene), or p55γ (encoded by the PIK3R3 gene). The regulatory subunit stabilizes the catalytic subunit to prevent its proteasomal degradation, inhibits activity of the catalytic subunit, and recruits it to the plasma membrane (2). Catalytic subunits p110α and p110β are broadly expressed, while p110δ is mainly expressed in cells of the hematopoietic system, primarily lymphocytes and myeloid cells (3). In immune cells, PI3Kδ is activated downstream of cytokine receptors, toll-like receptors, B-cell and T-cell receptors, and Ras superfamily of small GTPases (4). PIP3 produced by PI3Ks activates kinases PDK1 and AKT, leading to the activation of mTOR complex 1 and inhibition of FOXO family of transcription factors. In lymphocytes, PIP3 activates kinases BTK and ITK that mediate activation of phospholipase Cγ and other proteins (3). PIP3 is dephosphorylated to PIP2 by a phosphatase PTEN.
APDS Mutations
In 2013, two groups, one in Cambridge (UK) and the other in Bethesda (USA), used whole-exome-sequencing analysis of PID patients with unknown etiology and reported a novel PID caused by rare heterozygous germline gain-of-function mutations in the PIK3CD gene (5, 6). The mutations led to the increased PI3Kδ activity and the disease was called APDS (5) or p110δ-activating mutation causing senescent T cell, lymphadenopathy, and immunodeficiency (PASLI) (6) (OMIM #615513). Subsequently, rare heterozygous germline mutations in the PIK3R1 gene were described that also resulted in an increased PI3Kδ activity and immune deficiency, phenocopying patients with the PIK3CD mutations. This disorder has been termed APDS2 or PASLI-R1 (7, 8) (OMIM #616005). Now, a PID caused by activating mutations in the PIK3CD gene is referred to as APDS1 and both diseases together are known as APDS.
Since the initial publications, 10 activating missense mutations have been reported in the PIK3CD gene resulting in APDS1 (5, 6, 9–15) (Figure 1). The E1021K variant in the C-lobe of the p110δ kinase domain is by far the most frequently reported APDS mutation. In the p110δ protein, E1021K is positioned similar to the somatic mutation H1047R of another PI3K isoform, p110α. Both E1021K and H1047R increase PI3K activity by enhancing association of the catalytic subunits with membranes and facilitating more effective phosphorylation of PIP2 (5, 16–18). The R929C mutation in the C-lobe of the p110δ kinase domain may also act in a similar manner (14). Other p110δ mutations located in the C2 domain (N334K, C416R) and the helical domain (E525K) likely interfere with inhibitory contacts between p110δ and p85α (18). Interestingly, activating somatic mutations of the homologous amino-acid residues of p110α (N345, C420, and E545) have been also found in tumors. The recently identified E81K and G124D mutations in the adapter-binding domain and the linker between the adapter-binding and the Ras-binding domains may affect the orientation of the adapter-binding domain and hence interaction between p110δ and p85α (11).
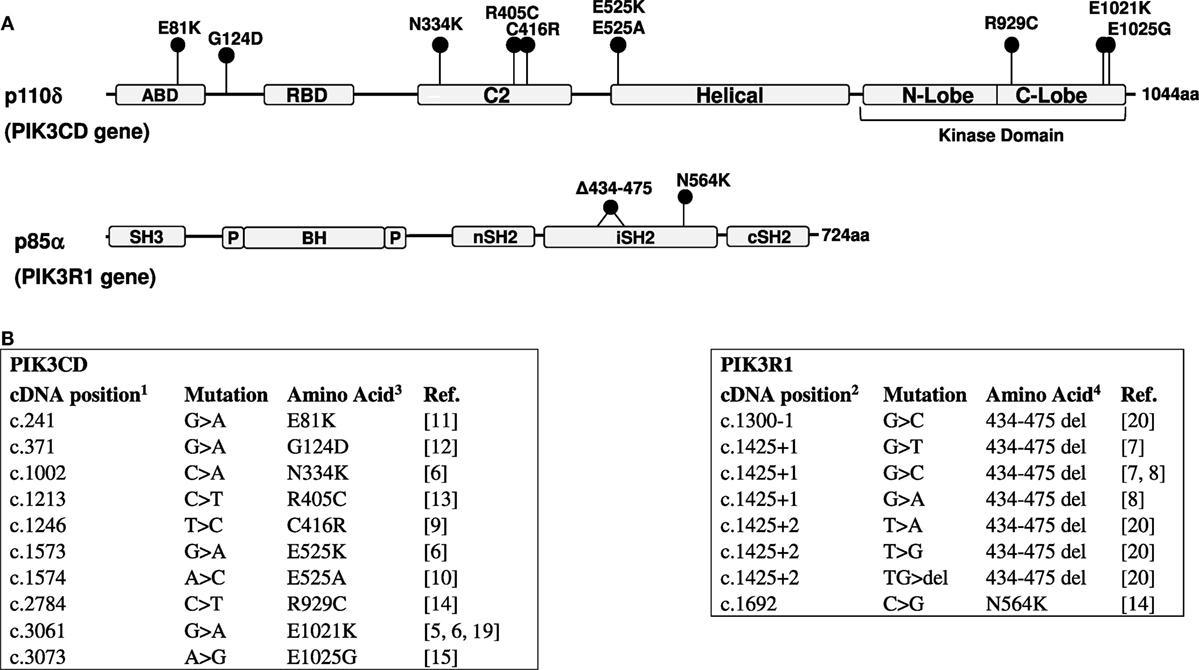
Figure 1. (A) Domain structure of the p110δ and p85α proteins and positions of mutations. ABD, adaptor-binding domain; RBD, Ras-binding domain; BH, breakpoint cluster region homology domain; P, proline-rich regions. (B) Activated PI3 kinase delta syndrome mutations in the PIK3CD (5, 6, 9–15, 19) and PIK3R1 (7, 8, 14, 20) genes. 1—NM_005026; 2—NM_181523 (RefSeq); 3—O00329; 4—P27986 (UniProt).
Several mutations causing APDS2 were identified in the PIK3R1 gene (Figure 1). These include one missense mutation and seven mutations affecting the splice sites of exon 11 (coding exon 10), one affecting the splice acceptor site, and six affecting the splice donor site. All splice site mutations lead to the skipping of exon 11 and an in-frame deletion of 42 amino-acid residues in positions 434–475 within the inter-SH2 coiled-coil domain of p85α. The additional N564K variant in p85α is also found in the inter-SH2 domain (14). The inter-SH2 domain of p85α is known to inhibit the catalytic p110 subunit by interacting with its C2 domain (2). Interestingly, the APDS2 mutations in p85α lead to the disease that phenocopy APDS1, despite that p85α is ubiquitously expressed and interacts not only with p110δ but also with p110α and p110β. However, it has been demonstrated that the 42 amino-acid deletion in p85α effectively disrupts inhibitory interactions between p85α and p110δ, leading to a strong basal activation of PI3Kδ, while it only weakly increases PI3Kα activity (18). This differential effect explains why the impact of this mutation is largely restricted to the immune system.
Mutations that cause APDS have been found in patients from different countries around the world. So far, more than 200 APDS patients carrying activating mutations in the PIK3CD and PIK3R1 genes have been identified. None of these variants were found in large cohorts of healthy subjects, e.g., they are absent from the largest human exome and whole-genome database gnomAD that includes more than 138,000 subjects (21). In several families, APDS mutations were shown to appear de novo among children, while being absent in their parents (5, 6, 10, 20), and long-range haplotype analysis in families with the E1021K mutation showed no founder effect (5). These findings indicate that APDS mutations appear recurrently in human populations. It is possible that activation of PI3Kδ provides selective advantages to cells during gametogenesis. Current data show that APDS mutations have high penetrance, e.g., out of the 53 subjects from 30 APDS1 families only one adult carrier of the E1021K mutation had no reported health issues (22). The true prevalence of APDS is not known. In the original study, heterogeneous cohorts comprising 184 PID patients were screened for the E1021K mutation and 17 APDS patients from seven unrelated families were identified (5). However, these cohorts included multiple patients with hyper-IgM syndrome and, therefore, were enriched for APDS mutations. Another study screened 669 patients with undefined PIDs for the N334K, C416R, E525K, and E1021K mutations in PIK3CD and the PIK3R1 splice site mutations and found only PIK3CD mutations in three siblings diagnosed with common variable immune deficiency (CVID) and two sporadic cases with combined immunodeficiency (23). Thus, prevalence of APDS may vary considerably between different PID cohorts.
APDS Phenotypes
Two comprehensive studies of APDS cohorts have been carried out recently and characterized its clinical and immunological manifestations (Table 1). One study examined the phenotypes of 53 patients with APDS1 (50 subjects with E1021K and 3 subjects E525K mutations) (22). The other studied 36 patients with APDS2 (20). Almost all APDS1 and APDS2 patients suffered from recurrent respiratory infections caused by bacterial pathogens, mainly Streptococcus pneumoniae and Haemophilus influenzae. Bronchiectasis was a common complication of lung infections affecting up to 60% of APDS1 patients. Interestingly, the majority of bronchiectasis patients had normal IgG levels and diagnosing PID in such subjects may not have been straightforward. Therefore, screening bronchiectasis patients without a clear PID for APDS mutations can reveal unrecognized APDS cases. Severe, persistent, or recurrent herpes virus infections, including EBV, CMV, HSV, and VZV infections, were found in 49% APDS1 and 31% APDS2 patients and were associated with lymphadenopathy. Immunologically, increased frequency of transitional B cells was often observed in APDS patients (Table 1). Many patients also had increased serum IgM levels and, therefore, some of the patients previous were diagnosed with hyper-IgM syndrome (5). Approximately one third of APDS1 patients and 17% of APDS2 patients had autoimmune or autoinflammatory manifestations. High incidence of lymphomas was also recorded in APDS patients (20, 22, 24). Unexpectedly, neurodevelopmental delay was found to be a relatively frequent manifestation in both APDS cohorts (Table 1), which may suggest an important role of PI3Kδ in the development of central nervous system that was not recognized previously.
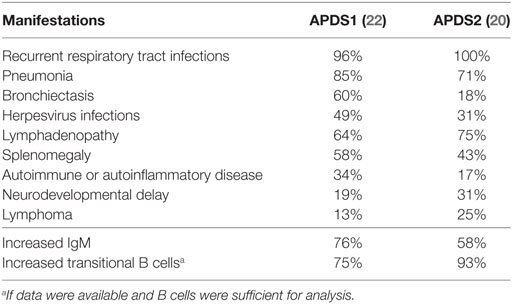
Table 1. Characteristic clinical and immunological features of activated PI3 kinase delta syndrome (APDS).
Thus, APDS manifests as a PID with a high rate of recurrent respiratory tract infections, often leading to bronchiectasis, herpes virus infections, lymphadenopathy, splenomegaly, increased risk of lymphomas, frequent autoimmune manifestations, and, occasionally, developmental delay. In addition, APDS2 patients had a high frequency of growth retardation (45%), a feature that was not found in APDS1 patients. This difference may reflect impaired interactions of p85α with p110α and p110β catalytic subunits. Of note, a number of other dominant germline mutations, which reside within the nSH2 and iSH2 domains of p85α and reduce PI3K signaling, are known to cause the SHORT syndrome that includes short stature, hyperextensibility of joints, hernia, ocular depression, Rieger anomaly, and teething delay (25–28) (OMIM #269880). A single patient with a homozygous loss-of-function mutation in the PIK3R1 gene was described that resulted in the absent p85α and reduced expression of p110δ. The patient had B lymphopenia and hypogammaglobulinemia and suffered from recurrent Campylobacter bacteremia and inflammatory bowel disease (29) (OMIM #615214). Also, a patient with biallelic loss-of-function mutations in the PIK3CD gene and reduced p110δ expression was reported to have B lymphopenia and hypogammaglobulinemia, sinopulmonary infections, septic arthritis, inflammatory bowel disease, and autoimmune hepatitis (30). Therefore, although p110δ deficiency also leads to a PID, its phenotype is different from APDS.
Therapies for APDS Patients—Precision Medicine for a Rare Disease
Treatment regimes for APDS patients include antibiotic prophylaxis and immunoglobulin replacement therapy. Hematopoietic stem cell transplantation (HSCT) has been successful in several APDS patients and can be a treatment option, especially in young patients (20, 22). Immunosuppressive therapies aimed at reducing lymphoproliferation have included treatment with rituximab (anti-CD20 monoclonal antibody) and rapamycin to target the activation of the mTOR pathway. Treatment with rapamycin led to the improvement of immunological markers and a reduction in splenomegaly and lymphadenopathy (6). Nevertheless, the discovery of the APDS etiology and the causative role of mutations that activate PI3Kδ opened an opportunity for a novel specific treatment using selective PI3Kδ inhibitors. This class of drugs has been developed for cancer treatment (31), as well as inflammatory disorders, such as rheumatoid arthritis, asthma, and chronic obstructive pulmonary disease (COPD) (32–34). One of the PI3Kδ inhibitors, idelalisib, has been approved for treatment of chronic lymphocytic leukemia and non-Hodgkin lymphoma (35, 36). Idelalisib (previously known as GS-1101) reduced the catalytic activity of mutant PI3Kδ as efficiently as the activity of the wild type PI3Kδ (5, 18). PI3Kδ inhibitors also normalized PI3Kδ hyperactivation in cells of APDS patients in vitro (5–8, 37). These results opened way for clinical trials of PI3Kδ inhibitors in APDS patients.
Two phase-II clinical trials are currently ongoing to study the safety, pharmacokinetics, pharmacodynamics, and efficacy of PI3Kδ inhibitors in APDS patients. Clinical trial NCT02435173 sponsored by Novartis uses an oral PI3Kδ inhibitor leniolisib (CDZ173) (38), while clinical trial NCT02593539 sponsored by GSK uses an inhaled PI3Kδ inhibitor nemiralisib (GSK2269557) (39) that had been originally developed for treatment of COPD (34). Recently, the clinical trial NCT02435173 has reported efficacy data from six APDS patients (37). The patients were part of a 12-week within subject dose-escalation study of oral leniolisib, administered twice daily. Leniolisib was well tolerated and the study reported normalization of circulating transitional and naïve B cells, reduction in senescent T cells, decrease in the elevated serum IgM levels, and inflammatory markers. After 12 weeks of treatment, lymph node and spleen sizes reduced by 39% and 40%, respectively (37). Normalization of immunophenotypes was most notable in the final 4-week dosing period. The study has now proceeded to a long-term treatment arm with patients receiving treatment for over 9 months (70-mg leniolisib, twice daily) and no significant adverse events have been detected (37). These exciting initial findings validate the focused approach to target the activated PI3Kδ in APDS patients. It will be of interest to see if the oral or inhaled inhibitors under development provide specific advantages for the APDS patients. Inhaled PI3Kδ inhibitors will have a different safety profile and may be appropriate for patients who are primarily affected by airway infections, potentially limiting progression of bronchiectasis.
Future Directions
Whole-exome and whole-genome sequencing of PID patients will likely identify novel variants in the PIK3CD and PIK3R1 genes and it remains essential to distinguish pathogenic mutations from neutral variants. Given that APDS is a rare monogenic disorder with high penetrance, variants that cause it are unlikely to be found in healthy subjects outside of patients’ families. Therefore, excluding variants detected in healthy cohorts, e.g., reported in the gnomAD database (21), will help initial screening of potential APDS-causing mutations. However, rare variants can still be neutral, so it will remain important to demonstrate that a novel candidate mutation leads to increased PI3K activity, e.g., by showing increased levels of PIP3 or phosphorylated AKT. The growing list of known APDS mutations will facilitate genetic diagnosis in future patients. Early diagnosis of APDS will be essential, as it will allow early therapy, e.g., HSCT or treatment with PI3Kδ inhibitors, which should prevent many APDS complications.
As our understanding of APDS improves, new questions emerge. With more APDS patients carrying various mutations being identified, it will be interesting to understand if specific mutations are associated with disease severity and clinical or immunological subphenotypes. The varying degree of disease severity in APDS patients raises the question as to whether rare activating mutations in genes encoding PI3Kδ or other proteins that regulate PI3K activity may be responsible for causing similar disorders, perhaps resembling only some of the APDS manifestations. In support of this hypothesis, loss-of-function mutations have been described in PTEN resulting in an APDS-like phenotype (10). Larger exome- or genome-sequencing studies in patients with diseases that resemble aspects of the APDS phenotype will be interesting to explore in this regard. These studies may reveal monogenic etiology in some of the patients with disorders, such as bronchiectasis. Furthermore, it is plausible that combinations of common polymorphisms in genes regulating PI3Kδ signaling may lead to its increased activity. Such subjects may be predisposed to APDS-like manifestations, e.g., bacterial respiratory infections, herpes virus infections, or bronchiectasis. Future genetic, biochemical, and immunological studies should address these questions.
In conclusion, the story of APDS illustrates how modern biomedical approaches led to the discovery of disease etiology in a group of uncharacterized patients and then provided a novel knowledge-based therapeutic strategy. Promising data emerging from the ongoing clinical trials of PI3Kδ inhibitors (37) rises the hope that the success of this approach may translate into therapies for APDS and, possibly, for APDS-like diseases in future.
Author Contributions
DM wrote the first draft of the manuscript and prepared Figure 1. SN edited the manuscript and prepared Table 1.
Conflict of Interest Statement
DM is an employee of GSK. SN is a recipient of a grant from the UK Medical Research Council and GSK to study APDS.
Funding
SN received support from the Wellcome Trust (095198/Z/10/Z), UK Medical Research Council grant MR/M012328/1 co-sponsored by GSK, and National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre.
References
1. Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol (2018) 38(1):129. doi:10.1007/s10875-017-0465-8
2. Burke JE, Williams RL. Synergy in activating class I PI3Ks. Trends Biochem Sci (2015) 40(2):88. doi:10.1016/j.tibs.2014.12.003
3. Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. PI3Kdelta and primary immunodeficiencies. Nat Rev Immunol (2016) 16(11):702. doi:10.1038/nri.2016.93
4. Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol (2013) 31:675. doi:10.1146/annurev-immunol-032712-095946
5. Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science (2013) 342(6160):866. doi:10.1126/science.1243292
6. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol (2014) 15(1):88. doi:10.1038/ni.2771
7. Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest (2014) 124(9):3923. doi:10.1172/JCI75746
8. Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med (2014) 211(13):2537. doi:10.1084/jem.20141759
9. Crank MC, Grossman JK, Moir S, Pittaluga S, Buckner CM, Kardava L, et al. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol (2014) 34(3):272. doi:10.1007/s10875-014-0012-9
10. Tsujita Y, Mitsui-Sekinaka K, Imai K, Yeh TW, Mitsuiki N, Asano T, et al. Phosphatase and tensin homolog (PTEN) mutation can cause activated phosphatidylinositol 3-kinase delta syndrome-like immunodeficiency. J Allergy Clin Immunol (2016) 138(6):1672. doi:10.1016/j.jaci.2016.03.055
11. Takeda AJ, Zhang Y, Dornan GL, Siempelkamp BD, Jenkins ML, Matthews HF, et al. Novel PIK3CD mutations affecting N-terminal residues of p110delta cause activated PI3Kdelta syndrome (APDS) in humans. J Allergy Clin Immunol (2017) 140(4):1152. doi:10.1016/j.jaci.2017.03.026
12. Heurtier L, Lamrini H, Chentout L, Deau MC, Bouafia A, Rosain J, et al. Mutations in the adaptor-binding domain and associated linker region of p110delta cause activated PI3K-delta syndrome 1 (APDS1). Haematologica (2017) 102(7):e278. doi:10.3324/haematol.2017.167601
13. Rae W, Gao Y, Ward D, Mattocks CJ, Eren E, Williams AP. A novel germline gain-of-function variant in PIK3CD. Clin Immunol (2017) 181:29. doi:10.1016/j.clim.2017.05.020
14. Wentink M, Dalm V, Lankester AC, van Schouwenburg PA, Scholvinck L, Kalina T, et al. Genetic defects in PI3Kdelta affect B-cell differentiation and maturation leading to hypogammaglobulineamia and recurrent infections. Clin Immunol (2017) 176:77. doi:10.1016/j.clim.2017.01.004
15. Dulau Florea AE, Braylan RC, Schafernak KT, Williams KW, Daub J, Goyal RK, et al. Abnormal B-cell maturation in the bone marrow of patients with germline mutations in PIK3CD. J Allergy Clin Immunol (2017) 139(3):1032. doi:10.1016/j.jaci.2016.08.028
16. Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang CH, et al. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A (2009) 106(40):16996. doi:10.1073/pnas.0908444106
17. Burke JE, Perisic O, Masson GR, Vadas O, Williams RL. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA). Proc Natl Acad Sci U S A (2012) 109(38):15259. doi:10.1073/pnas.1205508109
18. Dornan GL, Siempelkamp BD, Jenkins ML, Vadas O, Lucas CL, Burke JE. Conformational disruption of PI3Kdelta regulation by immunodeficiency mutations in PIK3CD and PIK3R1. Proc Natl Acad Sci U S A (2017) 114(8):1982. doi:10.1073/pnas.1617244114
19. Jou ST, Chien YH, Yang YH, Wang TC, Shyur SD, Chou CC, et al. Identification of variations in the human phosphoinositide 3-kinase p110delta gene in children with primary B-cell immunodeficiency of unknown aetiology. Int J Immunogenet (2006) 33(5):361. doi:10.1111/j.1744-313X.2006.00627.x
20. Elkaim E, Neven B, Bruneau J, Mitsui-Sekinaka K, Stanislas A, Heurtier L, et al. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase delta syndrome 2: a cohort study. J Allergy Clin Immunol (2016) 138(1):210. doi:10.1016/j.jaci.2016.03.022
21. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature (2016) 536(7616):285. doi:10.1038/nature19057
22. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase delta syndrome: a large patient cohort study. J Allergy Clin Immunol (2016) 139(2):597–606.e4. doi:10.1016/j.jaci.2016.06.021
23. Elgizouli M, Lowe DM, Speckmann C, Schubert D, Hulsdunker J, Eskandarian Z, et al. Activating PI3Kdelta mutations in a cohort of 669 patients with primary immunodeficiency. Clin Exp Immunol (2015) 183(2):221–9. doi:10.1111/cei.12706
24. Kracker S, Curtis J, Ibrahim MA, Sediva A, Salisbury J, Campr V, et al. Occurrence of B-cell lymphomas in patients with activated phosphoinositide 3-kinase delta syndrome. J Allergy Clin Immunol (2014) 134(1):233. doi:10.1016/j.jaci.2014.02.020
25. Thauvin-Robinet C, Auclair M, Duplomb L, Caron-Debarle M, Avila M, St-Onge J, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet (2013) 93(1):141. doi:10.1016/j.ajhg.2013.05.019
26. Dyment DA, Smith AC, Alcantara D, Schwartzentruber JA, Basel-Vanagaite L, Curry CJ, et al. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet (2013) 93(1):158. doi:10.1016/j.ajhg.2013.06.005
27. Koenig R, Brendel L, Fuchs S. SHORT syndrome. Clin Dysmorphol (2003) 12(1):45. doi:10.1097/00019605-200301000-00008
28. Chudasama KK, Winnay J, Johansson S, Claudi T, Konig R, Haldorsen I, et al. SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Genet (2013) 93(1):150. doi:10.1016/j.ajhg.2013.05.023
29. Conley ME, Dobbs AK, Quintana AM, Bosompem A, Wang YD, Coustan-Smith E, et al. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85alpha subunit of PI3K. J Exp Med (2012) 209(3):463. doi:10.1084/jem.20112533
30. Zhang K, Husami A, Marsh RA, Jordan MB. Identification of phosphoinositide-3-kinase (PI-3K) p110delta (PIK3CD) deficient individual. J Clin Immunol (2013) 33:673–4.
31. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov (2014) 13(2):140. doi:10.1038/nrd4204
32. Bartok B, Boyle DL, Liu Y, Ren P, Ball ST, Bugbee WD, et al. PI3 kinase delta is a key regulator of synoviocyte function in rheumatoid arthritis. Am J Pathol (2012) 180(5):1906. doi:10.1016/j.ajpath.2012.01.030
33. Sriskantharajah S, Hamblin N, Worsley S, Calver AR, Hessel EM, Amour A. Targeting phosphoinositide 3-kinase delta for the treatment of respiratory diseases. Ann N Y Acad Sci (2013) 1280:35–9. doi:10.1111/nyas.12039
34. Cahn A, Hamblin JN, Begg M, Wilson R, Dunsire L, Sriskantharajah S, et al. Safety, pharmacokinetics and dose-response characteristics of GSK2269557, an inhaled PI3Kdelta inhibitor under development for the treatment of COPD. Pulm Pharmacol Ther (2017) 46:69. doi:10.1016/j.pupt.2017.08.008
35. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med (2014) 370(11):997. doi:10.1056/NEJMoa1315226
36. Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med (2014) 370(11):1008. doi:10.1056/NEJMoa1314583
37. Rao VK, Webster S, Dalm V, Sediva A, van Hagen PM, Holland S, et al. Effective “activated PI3Kdelta syndrome”-targeted therapy with the PI3Kdelta inhibitor leniolisib. Blood (2017) 130(21):2307. doi:10.1182/blood-2017-08-801191
38. Hoegenauer K, Soldermann N, Zecri F, Strang RS, Graveleau N, Wolf RM, et al. Discovery of CDZ173 (leniolisib), Representing a structurally novel class of PI3K delta-selective inhibitors. ACS Med Chem Lett (2017) 8(9):975. doi:10.1021/acsmedchemlett.7b00293
Keywords: activated PI3 kinase delta syndrome, primary immunodeficiency, phosphoinositide-3-kinase δ, mutation, inhibitor
Citation: Michalovich D and Nejentsev S (2018) Activated PI3 Kinase Delta Syndrome: From Genetics to Therapy. Front. Immunol. 9:369. doi: 10.3389/fimmu.2018.00369
Received: 14 December 2017; Accepted: 09 February 2018;
Published: 27 February 2018
Edited by:
Stuart G. Tangye, Garvan Institute of Medical Research, AustraliaReviewed by:
Yuval Itan, Icahn School of Medicine at Mount Sinai, United StatesJames E. Thaventhiran, MRC Toxicology Unit (MRC), United Kingdom
Copyright: © 2018 Michalovich and Nejentsev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergey Nejentsev, c24yNjJAY2FtLmFjLnVr