 Marco A. Moro-García
Marco A. Moro-García Juan C. Mayo2
Juan C. Mayo2 Rosa M. Sainz
Rosa M. Sainz Rebeca Alonso-Arias
Rebeca Alonso-Arias
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 01 March 2018
Sec. Inflammation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00339
This article is part of the Research TopicThe Aging Immune System and HealthView all 23 articles
T lymphocytes, from their first encounter with their specific antigen as naïve cell until the last stages of their differentiation, in a replicative state of senescence, go through a series of phases. In several of these stages, T lymphocytes are subjected to exponential growth in successive encounters with the same antigen. This entire process occurs throughout the life of a human individual and, earlier, in patients with chronic infections/pathologies through inflammatory mediators, first acutely and later in a chronic form. This process plays a fundamental role in amplifying the activating signals on T lymphocytes and directing their clonal proliferation. The mechanisms that control cell growth are high levels of telomerase activity and maintenance of telomeric length that are far superior to other cell types, as well as metabolic adaptation and redox control. Large numbers of highly differentiated memory cells are accumulated in the immunological niches where they will contribute in a significant way to increase the levels of inflammatory mediators that will perpetuate the new state at the systemic level. These levels of inflammation greatly influence the process of T lymphocyte differentiation from naïve T lymphocyte, even before, until the arrival of exhaustion or cell death. The changes observed during lymphocyte differentiation are correlated with changes in cellular metabolism and these in turn are influenced by the inflammatory state of the environment where the cell is located. Reactive oxygen species (ROS) exert a dual action in the population of T lymphocytes. Exposure to high levels of ROS decreases the capacity of activation and T lymphocyte proliferation; however, intermediate levels of oxidation are necessary for the lymphocyte activation, differentiation, and effector functions. In conclusion, we can affirm that the inflammatory levels in the environment greatly influence the differentiation and activity of T lymphocyte populations. However, little is known about the mechanisms involved in these processes. The elucidation of these mechanisms would be of great help in the advance of improvements in pathologies with a large inflammatory base such as rheumatoid arthritis, intestinal inflammatory diseases, several infectious diseases and even, cancerous processes.
Inflammation is the process in which leukocytes and plasma proteins are recruited from blood into tissues, accumulated and then activated to elicit an adequate immune response. Inflammation is triggered by recognition of pathogen-associated molecular patterns and damage-associated molecular patterns from injured tissues during innate immune responses and it is refined and prolonged during adaptive immune responses. Many of these reactions involve cytokines which are produced by dendritic cells, macrophages, and other types of cells during innate immune reactions. The leukocytes that are mainly recruited in inflammation are neutrophils, and monocytes (Figure 1A).
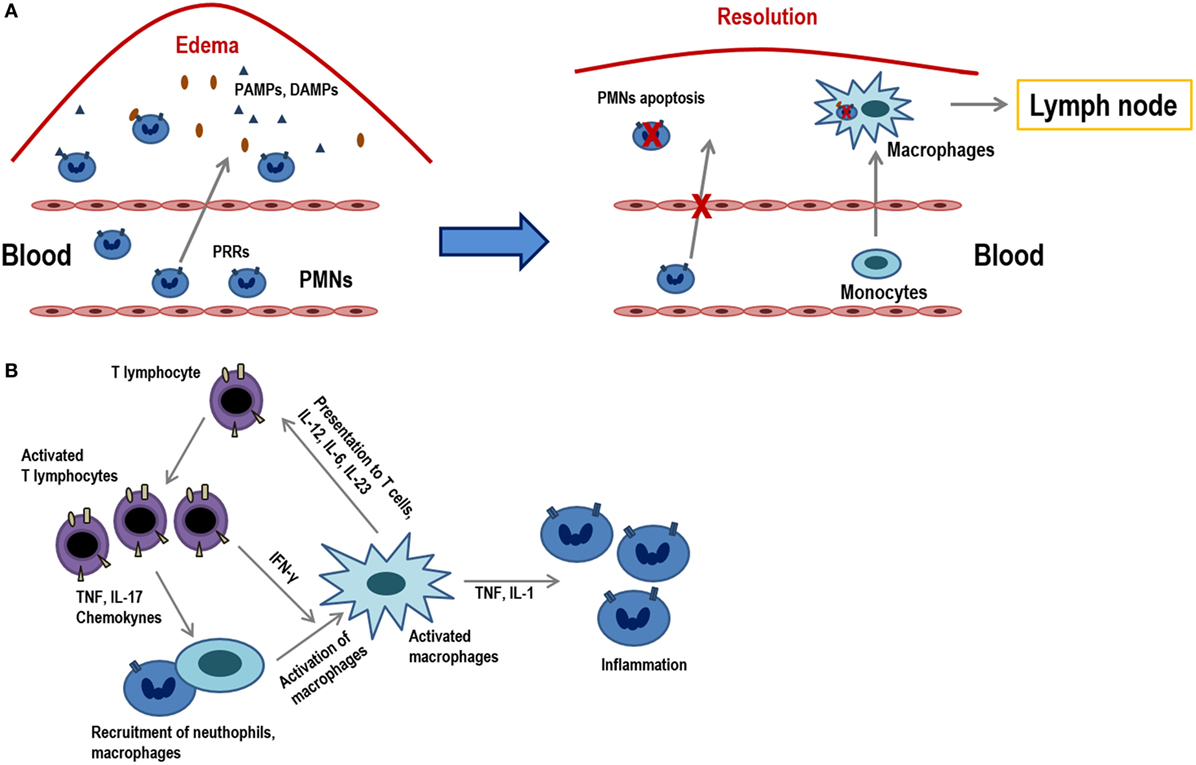
Figure 1. Acute inflammation vs. chronic inflammation. (A) The onset of acute inflammation is characterized by the accumulation of polymorphonuclear neutrophils (PMNs) and monocytes that rapidly convert into tissue macrophages, as well as the appearance of edema due to damage in the inflamed tissues. Damage-associated molecular patterns and pathogen-associated molecular patterns are recognized by pattern recognition receptors (PRRs) and attract PMNs to the place of infection. PMNs represent the first line of defense in infected tissues as they eliminate much of the pathogens or harmful materials by phagocytosis. When inflammation occurs, neutrophils suffer apoptosis and they are ingested by macrophages that migrate to the lymph nodes where they will present the antigens. (B) Activated T lymphocytes produce cytokines (TNF, IL-17, chemokines) that recruit macrophages and others (IFN-γ) which activate them. T lymphocyte subpopulations (Th1, Th2, Th17, etc.) produce diverse types of cytokines and, in turn, activated macrophages and stimulate T lymphocytes via the presentation of antigens and through different cytokines (IL-12, IL-6, IL-23). These macrophages also act on neutrophils by releasing molecules, such as TNF and IL-1.
Inflammation can be sensed in the nearby lymph nodes and thus influence recruitment and activation of lymphocytes in the nodes. Peripheral tissue inflammation, which usually accompanies infections, causes a significant increase of blood flow into lymph nodes and consequently an increase in T lymphocyte influx into lymph nodes draining at the site of inflammation. T lymphocytes are fully activated only when a foreign peptide is recognized in the context of the innate immune system activation by a pathogen or by some other causes of inflammation. In this pro-inflammatory environment, co-stimulatory ligands and increase in the expression MHC class I and II molecules are induced in antigen-presenting cells (APCs), which are necessary for an optimal T lymphocyte activation to occur. There are also many inflammatory mediators and cytokines that attract T lymphocytes, activating them through their antigenic receptors (1).
Although innate immune stimuli may contribute to chronic inflammation, the adaptive immune system may also be involved because T lymphocyte-producing cytokines are powerful inducers of inflammation. In this scenario, macrophages are activated by type 1 helper T lymphocytes (Th1 cells), both through cell contact and through IFN-γ secretion (2).
When cells that responded to the inflammatory environment cannot eliminate pathogens, the acute inflammatory condition can become a chronic condition (Figure 1B). In addition to a local or systemic inflammatory status, this chronic phase is characterized by a maintained leukocyte infiltrate within the injured tissues. This low-grade inflammation is prevalent during aging and it is, therefore, denominated as “inflammaging.” Furthermore, this phenomenon can also be observed in chronic infections, autoimmunity diseases, other chronic inflammatory pathologies or cancer. Consequently, all of them are characterized by persistent antigens that induce a sustained inflammation concomitant with a marked differentiation of the adaptive immunity, mainly in T lymphocytes. These highly differentiated cells in turn contribute to perpetuate the process by producing increased levels of proinflamatory cytokines. During the last stages of differentiation, the pro-inflammatory environment may be responsible for an inefficient response of T lymphocytes, as it is shown in older individuals (3). Indeed, it has been demonstrated that a reduced cytokine-related JAK–STAT signaling is correlated with chronic inflammation and age-associated morbidities (4).
T lymphocytes are produced in the bone marrow from where they migrate to the thymus for completing the maturation process. Then, naïve T lymphocytes recirculate between blood and secondary lymphoid organs until they contact their specific antigen adequately and properly. Upon contact, they proliferate and acquire properties to assemble an appropriate immune response. After antigen elimination, part of these cells remains as memory cells, with its own homeostasis and proliferation, though most of the effective cells die. Memory cells display a series of migratory and functional features that allow them to mount a quick response after the reencounter with the antigen. Therefore, the adaptive immune response presents two main advantages for the individual. On the one hand, it allows to create a specific immune response against the invading pathogen, with which it will finish it in a very effective way. On the other hand, a set of memory cells is formed to endure for many years thus affording protection from new reinfection by the same pathogen.
T lymphocytes can be categorized using a combination of different surface markers (CD45RA, CCR7) in distinct groups depending on their functionality. These categories are the naïve (CD45RA+CCR7+), effector memory (EM, CD45RA−CCR7−), central memory (CM, CD45RA−CCR7+), and effector memory RA (EMRA, CD45RA+CCR7−) populations (5). The effector T lymphocytes are a quite heterogeneous population and the use of two markers (CD27 and CD28) allows categorizing this population into other subpopulations; (CD28−CD27−) is the population more differentiated of all (6).
The step from naïve T lymphocytes to effector and memory T lymphocytes is one of the most fundamental processes in the T lymphocyte-mediated immunity and requires proliferative, metabolic, and oxidative adaptations.
The number of naïve T lymphocytes remains stable in number and diversity along the time, when there are no involved powerful immune responses. However, T lymphocytes are neither a lethargic nor an immovable cellular population and indeed this naïve T lymphocyte pool is continuously interacting with other cells through homeostatic signs. The number of T lymphocytes in the periphery is almost constant although many new naïve cells appear every day from the thymus, especially during the early ages of the individual. Therefore, this homeostasis requires a strict control as it is very important to maintain this constant number along the time. Furthermore, the half-life of naïve T lymphocytes, roughly over 50 days, is quite longer than that of other cellular populations (7–9). Survival of naïve T lymphocytes requires signals mediated by the interaction of T lymphocyte receptor (TCR)–peptide–MHC and some cytokines, principally IL-7. This cytokine is particularly important for the correct T lymphocyte homeostasis but its concentration is very low. Thus, all the naïve T lymphocytes including the recent thymic emigrants (RTEs) compete for IL-7. If these cells do not receive enough IL-7 signal, they would die (Figure 2A). The necessity of soluble mediators to intervene in the homeostasis of naïve T lymphocytes has been evidenced by the capacity of various cytokines to avoid apoptosis of naïve T lymphocytes. Among these cytokines are IL-4, IL-6, lymphopoietin, and IL-7 (10–12), the latter being the one which plays a main role (13–15). Supporting this, when an IL-7 blocking antibody is injected or naïve cells are transfer into IL-7-deficient mice, survival of this subset is greatly diminished (13, 16–18). It is also believed that IL-7 is a limiting factor for determining the final size of the total lymphocyte pool, as it has been verified in several experiments in which the number of lymphocytes significantly increases in IL-7-overexpressing mice (19, 20). RTEs express low levels of IL-7 receptors, but they are more responsive to the cytokine than mature naïve T lymphocytes. Mechanistically, it has been demonstrated that IL-7 signaling in RTEs preferentially upregulates the antiapoptotic protein Bcl-2 expression, resulting in a decrease in cell apoptosis, but without an increase in proliferative effects. In contrast, mature naïve cells show a decrease in Bcl-2 expression. However, mature naïve cells have a greater proliferative response in the presence of IL-7 (21).
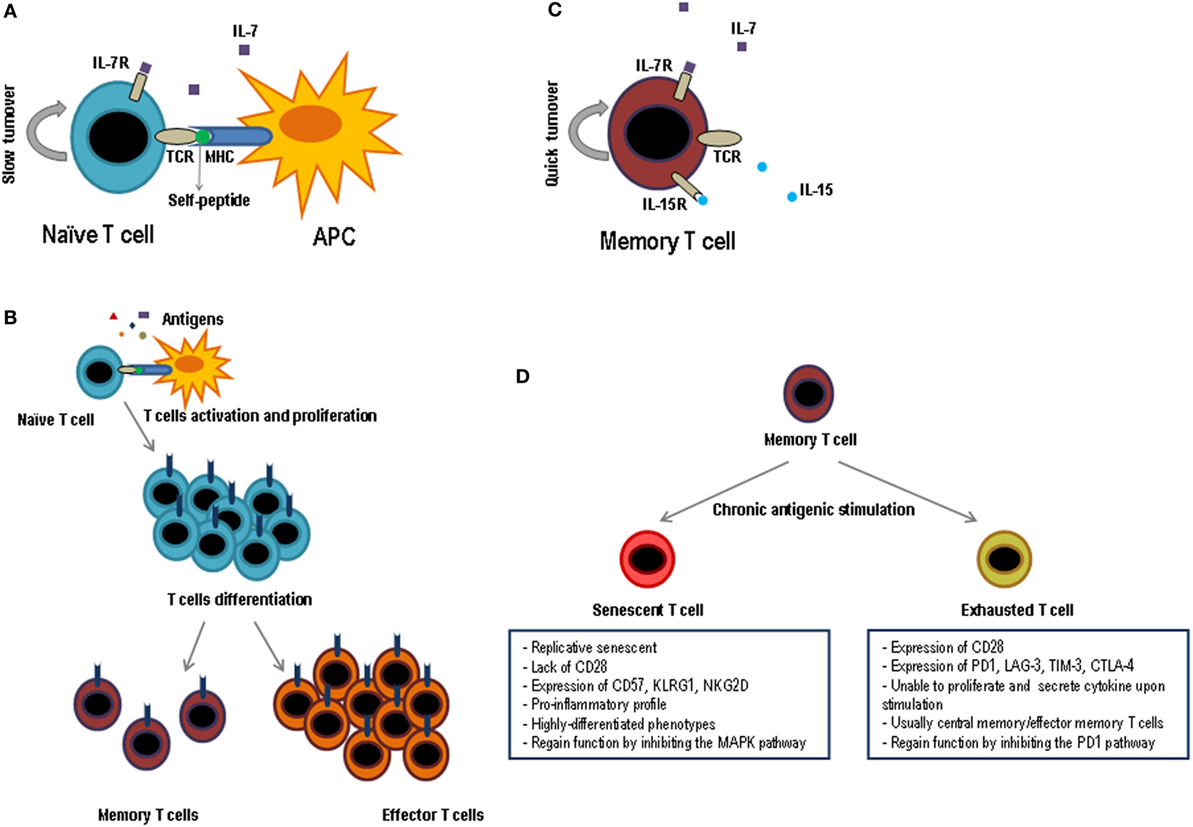
Figure 2. Functional phases of T lymphocytes. Schematic representation of the key factors for survival and maintenance of naïve cells (A) and memory (C); while naïve cells are dependent on IL-7 and the contact with self-peptides by MHC, memory cells are dependent on IL-7 and IL-15 and do not need the contact with autoantigens. (B) After contact with its specific antigen, naïve T lymphocytes proliferate exponentially and give rise to two differentiated populations, effector T lymphocytes and memory T lymphocytes. Effector cells will be eliminated as soon as the pathogen is destroyed, while memory cells will last a long time and they will be responsible for protecting the individual against new infections caused by the same pathogen. (D) When the memory cells are subjected to multiple cycles of activation by means of chronic antigens they can generate two types of T lymphocytes, exhausted T lymphocytes and senescent T lymphocytes. The principal characteristics of both cell types are shown in the figure.
During lymphocyte development in the thymus, thymocytes reacting to self-molecules are eliminated or believed to be induced to produce regulatory T lymphocytes. But, on the other hand, in order to overcome positive selection, thymocytes must be able to recognize low affinity self-peptide–MHC so they can in turn leave the thymus as naïve mature cells (22). This reactivity to self-peptide–MHC is decreased after positive selection (23, 24), but does not disappear in naïve T lymphocytes, since the TCR ζ-chain continues to be phosphorylated and this can only occur if the TCR meets self-peptide–MHC complexes (25, 26). Therefore, consistent data from multiple studies confirm that self-peptide–MHC complex interactions are important for long-term naive T lymphocyte survival; however, these interactions raise much discussion and controversy. Even though there is a wide knowledge about the interaction between IL-7R and TCR in the homeostasis of naïve T lymphocytes, interestingly many of the routes that define the interactions between them are not well defined, but it is clear that it must include a mutual regulation between these two routes. This is a particularly interesting topic and should receive attention in further studies.
There are several works focused on the effect of acute inflammation, for example after bacterial sepsis, in the population of naive T lymphocytes. Most of these studies state that the function of these cells remains unchanged, but concomitant with a decrease in their count after being exposed to high concentrations of pro-inflammatory cytokines (27–29). A few recent studies have shown that the sensitivity of CD8+ T lymphocytes to antigens is greatly increased if cytokines, such as IL-12, IL-18, and IFN-γ, are present prior to antigenic recognition (30, 31). However, only a very few studies have approached the effect of persistent inflammation on naive T lymphocytes, as it occurs in the context of aging or in the pro-inflammatory tumor microenvironment. Most of them suggest that there is a loss in the number of naïve T lymphocytes and a decrease in their functionality, although it is not clear the molecular mechanisms involved and more studies must be carried out (3, 32).
Naïve T lymphocytes present an amazing capacity to react against specific antigens through massive proliferation and differentiation to effector T lymphocytes. They are able to migrate to the infection sites and eliminate the triggering pathogen. Encounter with the antigen takes place in the secondary lymphoid organs, where APCs show them to the T lymphocytes. Then, a differentiation process begins; it is destined to produce large amounts of effective cells to fight against the pathogen with a clonal proliferative process but without coming to exhaustion. Interaction between T lymphocytes and APCs continues in tissues and so does the expansion and cellular differentiation, in an effort to contain the infection without damaging the tissues of the affected individual. Therefore, antigenic recognition by T lymphocytes provides a series of changes that leads to a clonal expansion of differentiated and effective T lymphocytes. The activation of T lymphocytes through its specific TCR can be detected seconds after antigenic contact, and it continues during hours and days in the context of pathology (33, 34) (Figure 2B).
The activation of lymphocytes after recognizing a peptide in the context of a MHC molecule (priming of naïve T lymphocytes) carries a series of processes of various types (genetics, proliferative, differentiation, biochemical) that lead to the formation of specific clones of effective lymphocytes, some of which will remain for long time in the form of memory cells. Before antigen exposure, the frequency of naive T lymphocytes specific for any antigen is 1 in 105 to 106 lymphocytes. After antigen exposure, the frequency of CD8+ T lymphocytes specific may increase to as many as 1 in 3 CD8+ T lymphocytes, representing a >50,000-fold expansion of antigen-specific CD8+ T lymphocytes, and the number of specific CD4+ cells increases up to 1 in 100 CD4+ lymphocytes may increase up to 5,000-fold.
It is well known that the single contact of T lymphocyte with its antigen is not enough to generate a cellular response but it rather causes the cell to enter into a refractory state and does not respond to any stimulus (35). This discovery led to the hypothesis that there should be other additional stimulatory signals that would enable T lymphocytes to become activated and exert their functions. Thus, when the CD28 molecule was identified as a co-stimulator of T lymphocyte function, the theory of the “two signals” was then reported, these two signals being necessary for the T lymphocytes activation. However, numerous evidences have suggested that other membrane-bound and soluble inflammatory signals are necessary to complete the activation of T lymphocytes, thus enforcing the “three signals” and “four signals” alternative theories. All experimental data seem to highlight the significant role of inflammatory mediators in the process of T lymphocytes differentiation into the effector population, resulting in a more adequate tool to respond to the aggression suffered (36, 37). On the other hand, there is also a great deal of data suggesting that TNF superfamily receptors (CD30, 41BB, OX-40, CD27) interact with their ligands in APCs (CD30L, 41BBL, OX-40L, CD70), which favors the survival of the activated cells and their passage into memory cells (38, 39).
T lymphocytes also have a large number of inhibitory molecules that help to regulate the cellular response, in a way that keeps this response not to be exaggerated and that would end up being harmful to the body. These inhibitory molecules act both, by limiting the co-stimulatory signals and by binding to the appropriate co-stimulatory receptors. The main inhibitory receptors belong to the CD28 family, including cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed death 1 (PD-1), both being involved in the phenomenon of tolerance. CTLA-4 is an inhibitory molecule expressed in activated T lymphocytes which causes an increase in the intracellular phosphatase activity, thus producing a decrease in the signals generated by the TCR and CD28 molecule. On the other hand, CTLA-4 acts also as a competitive receptor for the CD80/CD86 receptor, but with a higher affinity for these receptors than CD28 itself (40, 41). As a result, depending on their level of expression on the cell surface, CTLA-4 may interfere with CD80/CD86 binding. Several more inhibitors, such as lymphocyte activation gene 3, and V-domain Ig suppressor of T lymphocyte activation have been described, blocking these inhibitors by specific antibodies are being studied to increase the immune response in numerous cancers (42, 43).
The scenario in which the T lymphocyte response occurs may change in situations such as aging or in tumor microenvironment, where systemic or local inflammation is present (Figure 3). The levels of pro-inflammatory cytokines, such as IL-6, IL-1β, TNF-α, or GM-CSF, increase in these cases and this influences the lymphocyte response (3, 44). Dendritic cells are essential for T lymphocytes to be activated and differentiated as they present the antigen, producing co-stimulatory signals and essential cytokines. All these processes are highly dependent on the inflammatory environment mainly in chronic situation (45, 46). In the immune response during aging, myeloid dendritic cells in inflammatory environments have a decreased ability to present the antigens to CD4+ and CD8+ T lymphocytes (47). Moreover, most studies describe a reduced ability to produce cytokines by dendritic cells stimulated through toll-like receptors in vitro, a defect related to low response to vaccination in elderly (48, 49). TNF-α directly affects the immune response, in part by reducing the expression of the co-stimulatory molecule CD28 (50, 51). The pro-inflammatory environment may also be responsible for the attenuated response of T lymphocytes to cytokines, possibly due to the activation of negative regulatory pathways (4). According to the inflammatory environment in which lymphocytes are, the polarization of T lymphocytes will be different. In inflammatory environments, there is no evidence that Th1, Th2, or Th17 cells are diminished, but follicular T lymphocytes have been found to be less common in the elderly with systemic inflammation in response to vaccination (52, 53). It has been shown that CD4+ T lymphocytes activated in inflammatory environments are less responsive to type I interferons due to recruitment by the IFNR signaling complex of SHP1 (54). In conclusion, the inflammatory environment must be taken into account when evaluating immune responses. Individuals with high systemic and/or local inflammatory levels, as it occurs in the elderly and in cancer patients, may have an impaired response to cytokines and a poor response to antigens. In these cases, an anti-inflammatory treatment could be beneficial in trying to restore a correct immune response.
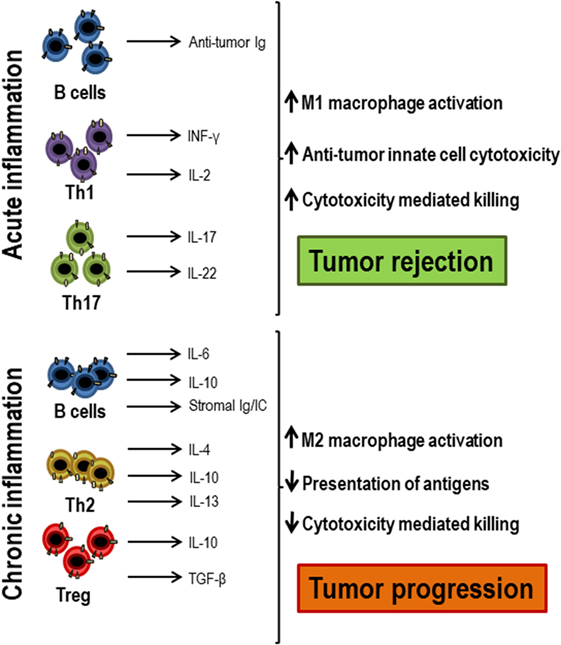
Figure 3. Acute and chronic inflammation participation in antitumor responses. When acute inflammation is activated by tumor cells, Th1 cells secrete antitumor cytokines, such as IFN-γ and IL-2, which, together with antitumor antibodies produced by B lymphocytes, exert an antitumor response and lead to tumor rejection, attracting innate immune cells and cytotoxic T lymphocytes. However, when chronic inflammation occurs in response to tumors, there is often an increase in regulatory T lymphocytes, Th2 cells, and activated B lymphocytes that secrete growth factors, such as IL-4, IL-6, IL-10, IL-13, TGF-β, and immunoglobulins that decrease both antigenic presentation and the activation of cytotoxic cells, favoring tumor progression.
The T lymphocyte-specific antigen response is characterized by clonal expansion, followed by a contraction in the number of specific cells and the formation of memory T lymphocytes. In this process, T lymphocytes acquire many key features to afford protection against re-infections, which are potentially deadly. Memory T lymphocytes are found in greater numbers than naïve T lymphocytes and in the presence of IL-7 and IL-15 these cells can be maintained for long periods of time without antigenic stimulation (55, 56) (Figure 2C). Central and effector memory T lymphocytes are less dependent on the contact with the MHC–peptide complex for survival than naïve T lymphocytes do (57). In addition, their self-renewal is three to four times faster than in naïve T lymphocytes and has a high proliferation rate in lymphopenia (58). The large number of memory cells, their high ability to reactivate, produce cytokines, and kill after antigenic stimulation and their distribution by almost all tissues makes memory T lymphocytes capable of protecting the individual much better than naïve T lymphocytes.
As previously mentioned, memory T lymphocytes can be divided into two populations, effector memory cells (EM, CD45RA−, CCR7−) and central memory cells (CM, CD45RA+, CCR7+), but other subpopulations can be established from the expression of CD27 and CD28 (5). EM are preferentially located in non-lymphoid and mucosal tissues and have a lower response threshold than CM, which are found mainly in secondary lymphoid organs and have a greater expansion capacity than EM.
There is no unanimity to describe the way by which T lymphocytes become effector and central memory cells (59). The most convincing hypothesis suggest a developmental trend from naïve T lymphocytes to memory cells, in which most memory cells have gone through a phase of effector cells. Some effector cells might then evolve into memory cells since some microarray studies have shown that passage of naïve cells to effectors and memory is a gradual step (59–61). Besides, several laboratories have used murine models of labeled effector T lymphocytes to track them, showing that most memory cells have indeed derived from these effector cells (62, 63). Thus, at least a set of memory cells that appear after an infection are generated from the effector cells.
Numerous studies have shown that exposure to a restricted inflammation increases the appearance of memory-like T lymphocytes when the intensity of inflammatory signals is controlled (59, 64). Several inflammatory molecules, such as interferon type I and IL-12 can be considered to act as the third signal (in addition to TCR and co-stimulation) and thus, to promote differentiation of T lymphocytes to effector cells (65).
According to several studies, prolonged and/or intense exposure to the pro-inflammatory cytokine IL-12 promotes a preferential differentiation toward effector cells, rather than memory cells (66, 67). The effect of other inflammatory molecules on T lymphocytes may be indirect. For instance, an interesting study found a blockage in the contraction of T lymphocytes when there was a deficiency of IFN-γ (68). In this scenario, we believe that IFN-γ offers a competitive advantage for the appearance of memory T lymphocytes, in some types of immune responses (69). Some studies suggest that at least some level of inflammatory signal is necessary for the differentiation to memory T lymphocytes. T lymphocytes deprived of this inflammatory signal (third signal) or deficient in T-bet transcription factor showed a decreased ability to produce long-lived memory cells. T-bet is needed for the expression of the CD122 molecule, that is the beta chain of the cytokine receptors IL-15 and IL-2, and to be able to react to homeostasis mediated by IL-15 (66, 70, 71). The cytokine IL-15 not only can promote the division and proliferation of differentiated and memory T lymphocytes but it is also capable of increasing its functional capacities (72, 73).
The differentiation of memory T lymphocytes in the course of chronic infections or in the presence of persistent antigens (autoimmune diseases, tumors, or atherosclerotic diseases) is different from differentiation in acute infections and leads to a defect in the functionality of T lymphocytes. These cells phenotypically acquire expression of several typical cytotoxic cell markers, such as NKG2D, PD-1, CD56, CD16, KLRG1, etc. (74, 75). Unlike memory cells generated after the elimination of an acute infection, memory cells in situations where the antigen is present in a chronic form, and in an inflammatory environment, proliferate and increase in number because the continuous stimulus (76, 77). These subpopulations, in turn, have propensity to secrete pro-inflammatory cytokines, such as IFN-γ, IL-1, TNF-α, and IL-6, contributing to increase the systemic or local inflammation (78).
One of the most remarkable characteristics of the immunological memory system is the capacity of the antigen-primed T lymphocytes to carry out a rapid response after they encounter the same antigen again (79). Co-stimulation signals have been recognized as critical for optimal T lymphocyte responses and result from important interaction between receptors on the surface of T lymphocytes and their ligands on APCs. However, memory T lymphocytes exhibit more reduced dependency to co-stimulation, and a significant lower threshold to respond.
T lymphocytes must be able to grow exponentially upon the first, and especially the second and the following, encounter with an antigen, and when they are no longer needed some of them enter apoptosis and disappear. Like most normal cells, lymphocytes are able to pass through a limited number of division cycles. Limited number of cell cycle divisions is associated to cell senescence or cell stress, which ends up in cell death triggering. Similar to what occurs to normal cells that evolve toward a neoplastic state acquiring a high rate of proliferation, lymphoid cells are also able to constitutively activate telomerase (80). Upregulation of telomerase and consequently elongation of telomeres allows extending the cells lifespan (81, 82). When the mechanisms that compensate telomeric shortening disappear and when shortening reaches a critical point known, cells enter a state of growth arrest termed senescence (83, 84). Telomere size can be determined by analyzing the terminal restriction fragments (TRF) that contains the TTAGGG region. The length of the TRF varies depending on cell population and each chromosome within the same cell. The critical point for cells to enter senescence appears when the size of TRFs reach less than 6 kb (85). The overall finding from different studies is that T lymphocytes in humans can carry out a certain number of divisions, after which they can no longer be divided (86, 87). Importantly, the reach of the replicative senescence stage by T lymphocytes does not imply a loss of cell viability. Moreover, senescent T lymphocytes under suitable conditions can remain alive and metabolically active for a prolonged period of time (88, 89). This transitional state of lymphocytes is not, however, shared with stem cells or malignant tumor cells, which do not reach replicative senescence and have stable chromosomes despite intense division. Stem and malignant T lymphocytes maintain telomerase activity and, therefore, replicative capacity throughout their lives (90). Nevertheless, by using mice lacking telomerase, it has been demonstrated that telomere shortening shunts premalignant T lymphocytes into the senescent state, therefore, reducing tumorigenesis (91).
It is very important to distinguish undifferentiated and highly differentiated T lymphocytes in order to study telomerase activity in these two populations. The undifferentiated T lymphocytes (CD27 +CD28+) display longer telomeres than highly differentiated T lymphocytes (CD27−CD28−) while intermediate populations (CD27−CD28+ or CD27+CD28−) have telomere lengths between both, undifferentiated and highly differentiated cells (92, 93). In addition, the proliferation ratio in T lymphocytes is higher in highly differentiated CD45RA−CCR7− cells that have a lower telomeric length (92, 94). Telomerase activity correlates with the length of telomeres, thus this activity is greater in the more undifferentiated cells and much lower in the cells with high differentiation. In addition, as the cell ages, the ability to induce telomerase expression and activation is lost (92, 93, 95). Moreover, differences in the behavior of telomerase have been found between CD4+ and CD8+ T lymphocytes. Cultures of CD4+ and CD8+ T lymphocytes from the same subject that have encountered an antigen for the fourth time were unable to increase telomerase production. However, the CD4+ T lymphocytes had much higher telomerase activity than that of the CD8+ T lymphocytes from the same donor (96). Several studies have shown that homeostasis of CD4+ T lymphocytes is much more rigorous than that presented by CD8+ T lymphocytes. In addition, aging in lymphocytes was previously described in CD8+ T lymphocytes, since the changes observed in immunosenescence occur much earlier in these CD8+ T lymphocytes (97–99).
The signaling via the TCR and the co-stimulation with other molecules such as CD28 is essential for the induction of telomerase activity. This activity peaks 4–5 days after the TCR has been stimulated, but presents a decrease in its activity after 10 days (92, 100, 101). T lymphocytes can proliferate under stimulation of various cytokines without the mediation of TCR. This is homeostatic proliferation, the mechanism by which naïve and memory T lymphocytes are maintained in the periphery (8). The cytokines IL-7 and IL-15 have been related to telomerase activity in the CD4+ and CD8+ T lymphocytes, respectively (102, 103). Our laboratory has demonstrated that IL-15 has a preferential effect on the CD4+CD28− T lymphocyte population, which causes an increase in proliferation and specific responses of these cells (73). It has also been found inhibitory effects on telomerase activity by some cytokines such as IFN-α and TGF-β (104, 105).
Factors that promote repeated T lymphocyte stimulation, such as persistent antigen and chronic inflammation, appear to drive telomere loss and replicative senescence. Elderly individuals and cancer patients, often exhibit chronic inflammation characterized by immune system dysregulation with increased inflammatory cytokine production (73, 74, 106, 107) and redox imbalance due to reduced antioxidant defenses and overproduction of reactive oxygen species (ROS) (108). It is now recognized that chronic inflammation is a major risk factor for several age-associated diseases, including chronic obstructive pulmonary disease, neurodegeneration, obesity, and vascular disease (109, 110). Premature telomere erosion in peripheral blood lymphocytes is a common characteristic of these diseases as well as autoimmune syndromes (103). These findings suggest that telomere loss increases susceptibility to autoimmune disease and may be a predisposing factor for age-related inflammatory disease.
Restoring telomerase activity in T lymphocytes would have a great impact on human lives by restoring telomere shortening and, in turn, avoiding the deleterious effects of aged T lymphocytes. Several studies have shown that if the telomerase activity is preserved, the telomeric length is stabilized and replicative senescence can be delayed (111, 112). One possible solution to stimulate telomerase activity would be to eliminate senescent T lymphocytes from the bloodstream or bring them into apoptosis. Telomere loss could be stopped by inhibiting cytokines, such as TNF-α, mediating telomere shortening.
In a viral infection or cancerous environment where there is a permanent exposure to certain antigens and high inflammation, memory cells are highly affected (113). This alteration, known as T cell exhaustion, presents a series of characteristics: progressive loss of effector functions, upregulation and co-expression of many inhibitory receptors, alteration of transcription factors, dysfunctional metabolism, failure to enter a quiescent state, and response to normal homeostasis (114) (Figure 2D). Although the appearance of exhausted T lymphocytes was first described in an environment of viral infection, it has also been observed in the presence of cancer and in various inflammatory diseases. Initially, exhausted T lymphocytes have a defect in proliferative progression and a defect in the expression of telomerase, but the rest of their functions are completely conserved (115). Next, exhausted T lymphocytes, mainly due to continuous antigenic stimulation and a pro-inflammatory environment, enter into a state of differentiation where they gradually lose their effector functions, such as cytokine production and cytotoxic capacity, which prevents these cells from being effective against cancer or microbial infections (113). Exhausted T lymphocytes normally arise during high-grade chronic infections in highly pro-inflammatory environments, where the level and duration of antigenic stimulation are critical for this process to occur (116, 117).
Viral infections and cancerous tumors can cause the appearance of exhausted T lymphocytes, but not all cases lead to an exhausted T lymphocyte (113, 118, 119). The difference in the ability to produce exhausted T lymphocytes may be due to different T lymphocyte specificities. The most effective specificities in stopping pathogens inactivate them faster and destroy them quickly. Exhausted cells regain functionality and become typical memory T lymphocytes when viral infection is under control and in turn, antigen concentration decreases (120).
Exhaustion of T lymphocytes is accompanied by an increase in expression of inhibitory molecules, including PD-1, CTLA-4, LAG3, 2B4, CD160, and T lymphocyte immunoreceptor with immunoglobulin and ITIM domains (TIGIT). PD-1 is an inhibitory protein that intervenes in self-tolerance by inhibiting the activation of T lymphocytes using a similar mechanism as describe for CTLA-4 (121). After PD-1 interaction with its ligands, namely PD-L1 or PD-L2, TCR signaling is blocked by recruiting SHP-2 phosphatase and subsequent dephosphorylating of the antigen receptor (122). Interestingly, both ligands are often overexpressed in many tumor cells, but PD-1 is highly expressed in T lymphocytes from patients with different types of cancer. PD-1 ligand levels from tumor cells and PD-1 levels from T lymphocytes are usually correlated with the tumor aggressiveness and poor prognosis (121).
The proliferation of exhausted cells is partially compensated with PD-1 suppression, but telomerase activity is not restored (123). Many causes leading to T lymphocyte exhaustion are shared with those leading to T lymphocyte aging. Several epigenetic studies in naïve and central memory T lymphocytes reveal an age-associated loss of access of T lymphocytes promoters to their chromatin site of action, particularly to the NRF1-binding sites (124). This loss seems to intervene in the reduced expression of the genes of the mitochondrial respiratory chain and, therefore, in the deficient oxidative phosphorylation that can lead to death of the T lymphocytes (125). Repression of the molecule PGC1α, a cofactor of NRF1 activity, is an event that appears early in exhausted T lymphocytes, indicating a mechanistic overlap between T lymphocyte aging and exhaustion (126). Effector memory T lymphocytes of elderly individuals compared to effector memory cells in young people show few differences with respect to the accessibility of chromatin promoters. Thus, the memory T lymphocytes in elderly individuals do not exhibit the epigenetic marks of the exhausted T lymphocytes (124, 127). This observation may indicate that T lymphocyte aging affects mainly to naïve cells and central memory, possibly through pathways similar to those involved in T lymphocyte exhaustion. On the other hand, effector T lymphocytes in the elderly do not show any signs of exhaustion.
The epigenetic phenotype of the exhausted T lymphocytes remains stable when PD-1 is blocked and the restoration of function is, therefore, only transient in most T lymphocytes (128). This evidence leads us to consider whether treatment with PD-1 blockers decreases along with age due to aging of the exhausted T lymphocytes or rather because the responsive fraction of exhausted T lymphocytes to treatment decrease with age.
The appearance of exhausted T lymphocytes prevents the possibility of an adequate control of infections and malignant tumors. Therefore, if overexpressing pathways in the exhausted T lymphocytes could be modulated, for example, by inhibiting PD-1 and CTLA-4, their dysfunctional state could be reversed and immune responses invigorated.
T lymphocytes must carry out metabolic strategies throughout their differentiation process and because of the changing microenvironment in which they are located. The purpose of these adaptations is to meet energy and structural needs in the different stages of proliferation and to achieve functional responses according to the availability of nutrients.
Naïve T lymphocytes, since they exit the thymus as mature cells and during their travel throughout the secondary lymphoid tissues until encountering their specific antigen, show a reduced rate of cell division. Even more, it is considered that they are in a functional quiescence, which does not require a high-energy consumption. They use the available nutrients trying to obtain the highest energy yield. Glucose, fatty acids, and amino acids are metabolized until they enter into the tricarboxylic acid (TCA) cycle where ATP and reducing equivalents are generated, which subsequently increase the production of ATP upon entering the pathway of oxidative phosphorylation (129, 130).
When naïve T lymphocytes encounter APCs carrying their specific antigenic peptide, activation takes place in a lymph node. Clonal expansion of the antigen-specific T lymphocytes is mainly mediated by IL-2 and the subsequent functional differentiation adapts the response to the specific triggering pathogen. Proliferation leads to generate sufficient amount of specific T lymphocytes capable of eradicating the pathogen. Thus, T lymphocyte activation not only requires energy but also the production of precursors that support the explosive proliferation through the biosynthesis of the required cellular components, proteins, lipids, nucleic acids, etc. These processes imply a high increase in energy demands, resulting in a switch in the metabolic pathways of nutrient utilization. Initially, there is an increase in glucose uptake, by increasing the expression of glucose transporter (GLUT) such as GLUT-1 (131). Glucose is, therefore, essentially metabolized to lactate by aerobic glycolysis, a phenomenon known as “Wargurg Effect,” which was initially described in tumor cells and, more recently, also in T lymphocytes (132–134). It is currently considered that “T lymphocyte activation-induced metabolic reprogramming is reminiscent of the metabolic changes associated with oncogenic transformation” (135, 136). More than 90 years ago, Otto Warburg described that tumor cells, even in the presence of oxygen, showed a high rate of glycolysis. This so-called aerobic glycolysis is now recognized as one of the new hallmark of cancer capabilities (5). In differentiated cells, glucose renders through the TCA cycle an energetic yield of 36 molecules of ATP. And yet, cancer cells rather prefer to compromise a high ATP production in order to obtain other benefits, in terms of building blocks for their new daughter cells (137–139). Recently, a similar program has also been observed in all proliferative cells including T lymphocytes upon antigen activation. This metabolic adaptation ensures a high glycolytic rate, which is not inhibited by the production of mitochondrial ATP. Moreover, glycolysis inhibits apoptosis and contributes to the maintaining of mitochondrial membrane potential (137, 140). Regarding the immune system, metabolic reprogramming has also been linked to the acquisition of certain functional properties by T lymphocytes such as the secretion of IFN-γ (134, 141).
Other functional parallelism found is glutaminolysis, which also increases in response to T lymphocyte activation as well as after transformation of cancer cells. Glutamine is rapidly consumed by tumor cells, playing a key structural role as a nutrient in the biosynthesis of nucleotides. The concentration of glutamine has been shown to be limiting in the progression of the cell cycle. Glutamine deprivation leads to cell cycle arrest in some cell types. Glutamine is not only an important source of carbon and nitrogen for the synthesis of other amino acids but the α-ketoglutarate generated is an anaplerotic substrate of the TCA cycle. Thus, it may contribute to the generation of ATP when glucose is primarily derived toward this so-called aerobic glycolysis (142).
On the other hand, pentose phosphate pathway (PPP) is the main catabolic route that generates ribose, necessary for the synthesis of nucleotides, and NADPH, essential for proliferation as it provides the reduction equivalents for fatty acid and cholesterol biosynthesis. Furthermore, PPP plays an essential role in balancing the redox status by regulating the production of glutathione (143). The utilization of PPP is usually elevated in cancer cells and in proliferative T lymphocytes. The key enzymes in the pathway are overexpressed in cancer and oncogenes and tumor suppressors have been shown to regulate PPP activity.
Metabolic decisions are dependent on the co-stimulatory signals received by T lymphocytes, in particular on CD28, which mediates many of these processes via the PI3K–Akt–mTOR pathway. However, in cancer cells intracellular programs combine with extracellular signals, to achieve a significant independence from external requirements. Genetic alterations as common as PI3K and its negative regulator PTEN as well as gen amplifications of upstream receptor tyrosine kinases result in an increase in glucose uptake and in the metabolic reprogramming in several cancer cells (144). It is noteworthy to mention that the metabolic state of cancer cells has a potential influence on the surrounding cells. Thus, within the tumor microenvironment, cancer cells alter the metabolic composition of the extracellular milieu, affecting the signaling pathways that influence the infiltration of immune cells, including T infiltrating lymphocytes. mTOR is a central part of various signals that come from the immune microenvironment because mTOR works as a sensor of extracellular medium conditions. mTOR is an evolutionarily conserved serine/threonine kinase, which forms two complexes, mTORC1 and mTORC2, determined by the association with different adapters and scaffolding proteins. mTOR is responsible for integrating different responses upon receiving environmental signals and for the control of various cellular functions, such as growth, apoptosis, actin reorganization, metabolism, and ribosome genesis (145, 146). mTOR activation targets T lymphocyte metabolism, switching it toward glycolytic metabolism by induction of two major transcription factors, namely HIF1α and c-MYC (147, 148). HIF1α is stabilized under hypoxia but it can also be activated by mTOR even under aerobic conditions. It increases the expression of GLUTs and glycolytic enzymes, such as pyruvate dehydrogenase kinase 1, limiting the entry of pyruvate into TCA cycle and favoring its reduction to lactate. The deregulation of multiple elements of the mTOR pathway has been reported in many types of cancers, implying significant effects on tumor progression. Likewise, mTORC1 signaling controls transcription of many genes, some of which are involved in metabolic and biosynthetic pathways (149).
Signals derived from anaerobic conditions and nutrients availability may modulate the cytokine profile of T lymphocytes (150). CD4+ T lymphocyte differentiation into a specific effector phenotype (mainly Th1, Th2, Th17, Treg) will depend fundamentally on the cytokines present in the immunological microenvironment at the time of antigenic presentation. mTOR plays a key role in the differentiation into the effector phenotypes, but not into Treg, so mTOR1 is needed for differentiation toward Th1 and Th17, whereas mTOR2 regulates Th2 differentiation (151). Thus, mTOR coordinates the metabolic pathways and the differentiation of each subpopulation of CD4+ T lymphocytes (146). High levels of HIF1α direct the CD4+ T lymphocyte metabolism to the glycolytic pathway, favoring the activation of RORγt transcription factor and its differentiation toward a Th17 inflammatory phenotype. Inhibition of this pathway, even under conditions that promote Th17 differentiation, results in Treg lymphocytes (152, 153). Th1 also possess a high glycolytic rate, concomitant with a higher surface location of GLUT1 (154). In fact, there is a coordinated regulation between T lymphocyte metabolism and the IFN-γ production by Th1. GAPDH is attached to UA-rich regions located at the 3′ in the untranslated region of the IFN-γ mRNA in non-activated cells. When glycolytic metabolism is activated, this enzyme is involved and does not bind to the mRNA, allowing its translation and IFN-γ production (134).
It has also been shown that the limited availability of glutamine in the extracellular medium favors Treg differentiation, even in the presence of cytokines involved in Th1 differentiation (155). The reason seems to be the decrease in intracellular levels of α-ketoglutarate, a mTORC1 activator, the expression of T-bet transcription factor and, as a result, differentiation into Th1. Similarly, the production of GLUT1 receptor, essential for the development of Th1 responses, is increased, whereas Tregs are unaffected by deficiency in this receptor and maintain their inhibitory capacity on T lymphocytes independently of GLUT1 (156).
In CD8+ T lymphocytes, mTORC1–HIF1α is activated by a PI3K–Akt-independent pathway, through phosphoinositide-dependent kinase 1. Metabolically, it promotes the activity of glycolytic enzymes and at functional level, it activates cytolytic capacity, controls the migration, and inhibits the generation of memory cells. Meanwhile, mTORC2 activates oxidative metabolism (157, 158).
In other way, long-lived specific memory cells that circulate between the secondary lymphoid organs, blood, and tissues do not proliferate significantly and have a quiescent functional state with scarce or no cytokine production. The main difference with naïve cells is that memory cells need to be prepared to respond quickly and efficiently to a new contact with the antigen. Their metabolism is based on the oxidation of glucose and fatty acids and they are characterized by a high content of large mitochondria, which are generated by fusion of the individual organelle that support the energetic requirements in reactivations (159, 160). In addition, ATP obtained from glucose oxidation is used to synthesize fatty acids, which in turn will be oxidized. The motive of this futile cycle could be the maintenance of the mitochondrial activity in order to be ready to respond quickly to specific-antigen re-stimulation (161–163).
Following multiple antigenic challenge, mainly in the case of chronic viral and tumor antigens and in situations of chronic inflammation, the specific T lymphocytes go through successive phases of clonal division, which as mentioned above, changes its degree of differentiation and, in parallel, its phenotype and functional capacity. In this stage, metabolic switching occurs in favor of an oxidative phenotype and is unambiguously associated with increased mitochondrial ROS production. On the other hand, inhibition of fatty acid oxidation decreases NADPH and glutation (GSH) levels. However, ROS levels increase, suggesting that control of fatty acid oxidation, in addition to PPP, regulates NADPH levels, which is essential to regenerate GSH pools from the glutathione disulfide (164, 165).
Finally, T lymphocytes reach replicative senescence, characterized by constitutive p38 mitogen-activated protein kinase activation, telomeric shortening, the loss of telomerase activity, and reduced proliferative capacity in response to stimulation (123). This new situation is also associated with metabolic adaptations. Again, a link between T lymphocytes aging and bioenergetic status has been proposed, since glucose deprivation in non-senescent T lymphocytes induces the activation of p38, and its constitutive activation inducer, the metabolic sensor of intracellular levels of ATP AMPK (5′-monophosphate activated protein kinase). These processes lead to a reduction in telomerase activity and proliferation similar to those observed in senescent T lymphocytes (166, 167). The main metabolic pathway used by these cells at this stage is glycolysis. Senescent T lymphocytes show mitochondrial dysfunction and consequently produce higher levels of ROS and defective mitochondrial biogenesis, which may justify their metabolic switch. P38 inhibition leads to mitophagy and thereby nonfunctional mitochondria are eliminated and ROS production is reduced. However, the increase in energy needed to sustain proliferation continues to be obtained from glycolysis and not from oxidative phosphorylation (168). As mentioned previously, high levels of inflammation in certain chronic viral infections and in cancer, induces an exhausted stage, similar to happen in senescence (113, 169, 170). There are numerous links between the inhibition of T lymphocytes by CTLA-4 and PD-1 and metabolic signaling pathways. CTLA-4 interacts with PP2A (protein phosphatase 2), a negative regulator of AKT, mTOR, and MAPK signaling, whereas PD-1 inhibits AKT phosphorylation by preventing CD28-mediated activation of PI3K (171). It has been shown that PD-1 inhibits glycolysis and amino acid metabolism and promotes lipid metabolism, whereas CTLA-4 inhibits both processes and mitochondrial biogenesis in memory cells (172, 173). Since the main metabolic pathway of T lymphocytes during activation is the aerobic glycolysis, these molecules could be blocking differentiation into effector T lymphocytes, at least partially, by metabolic regulation. The increase in β-oxidation of fatty acids could be an explanation for the maintenance of these cells, despite being exhausted, and in addition to its ability to recover functionality when interaction between PD-1 and its ligands hangs (173). Figure 4 shows a diagram of the main lymphocyte metabolic pathways in their different differentiation stages.
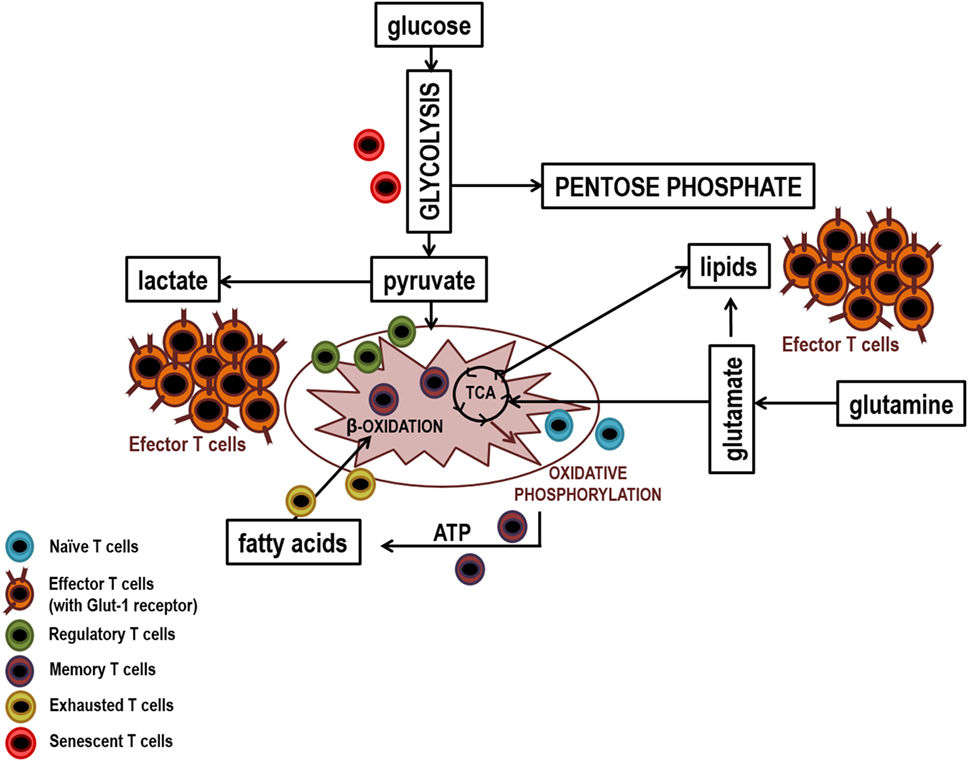
Figure 4. Main metabolic pathways of lymphocyte subpopulations. Naïve, memory, and regulatory T lymphocytes uptake low levels of glucose which it is metabolized by glycolysis and then the pyruvate generated is fully oxidized to CO2 in the mitochondria by the Krebs cycle. In contrast, effector lymphocytes have a large demand for glucose to maintain the biosynthetic processes that facilitate cellular growth, proliferation, and the synthesis of substantial quantities of effector molecules. Thus, effector lymphocytes show higher production of glucose transporters (GLUTs) and higher activity of glycolytic enzymes that facilitates elevated rates of glucose uptake and glycolytic flux. To sustain high rates of proliferation, pyruvate is converted to lactate and released out of cells. This prevents the accumulation of pyruvate which could result in the inhibition of the glycolysis pthway. Due to the high levels of glycolytic flux, glycolytic intermediates can be diverted into biosynthetic pathways to generate amino acids, lipids, and nucleotides in order to generate biomass. Senescent T lymphocytes use glycolysis extensively, partly because they have dysfunctional mitochondria and exhausted T lymphocytes mainly use lipid metabolism to carry out their poor cellular functions.
Relationship between oxidative stress and inflammation has been widely documented (174). Oxidative stress plays a pathogenic role in many chronic inflammatory diseases. Lower levels of GSH, an intracellular thiol antioxidant, causes ROS production, which results in imbalanced immune response and inflammation. Moreover, protein oxidations turn into release of inflammatory signal molecules and inflammatory stimuli induce the release of peroxiredoxin 2, a redox-active intracellular enzyme (175).
Basal levels of ROS generated in response to endogenous and exogenous stimuli are crucial mediators of multiple cell processes such as growth, differentiation, or migration, but excessive production might induce cell death, apoptosis, and/or senescence. Oxidative stress triggered by the excessive ROS production, cause oxidative damage to cellular components such as DNA, proteins, or lipids, which is closely related to the pathogenesis of various diseases including cancer. In addition to exogenous ROS, major intracellular sources of ROS are NADPH oxidases and particularly mitochondria. Usually, physiologically generated ROS are balanced by non-enzymatic and enzymatic systems, such as, GSH, superoxide dismutases (SOD1/2), thioredoxins (Trx1/2), catalase, or peroxidases. In addition, NADPH, is one of the main thiol-dependent electron donors system in the cell and plays a critical role in the regulation of cellular redox environment and in a wide range of cellular pathways, including activation of transcription factors such as NF-κB, activator protein-1, p53, HIF-1, or the redox factor 1 (176). Elevated levels of ROS production have been considered an adverse event, playing an important role in tumor initiation and progression, but also in promoting inflammatory environments. However, ROS are now more widely recognized as important signaling molecules (177, 178). Redox signaling in cells by ROS such as hydrogen peroxide (H2O2) occurs through the reversible oxidation of cysteine thiol groups. A major cellular target of ROS is the thiol side chain (RSH) of cysteine, Cys sulfenic (Cys-SOH) and sulfinic (Cys-SO2H) acids have emerged as important mechanisms for regulation of protein function. These residues modifications result in reversible structural alterations that can modify protein function, which may imply either inactivation or gain of function (179).
The importance of ROS in immunity is exemplified by their generation and release in the form of an “oxidative burst” by phagocytic cells as part of the innate immune cell network to effectively destroy pathogens and clear debris. However, ROS exert, as it does in other cells, a dual role on T lymphocyte biology. Mild ROS are essential for T lymphocyte activation, expansion, and effector function (180–183). Still, elevated rate of ROS production or exposure and defective neutralization by antioxidant cellular systems, causes oxidative stress that compromises T lymphocyte proliferation and activity (182, 184). Balance between both situations may be fragile and the studies yield results that seem to be contradictory, probably due to different experimental conditions.
Generation of ROS and Ca2+ release from intracellular stores are direct consequences of TCR/CD28 stimulation. Both are essential for TCR signaling, particularly in activation-induced CD95L expression (185). 5-lipoxygenase, NOX-2 and mitochondrial complexes are the most important sources of ROS in T lymphocytes (186–188). Oxidative signals originated from Complex I of the ETC regulate T lymphocyte activation-induced expression of IL-2 and IL-4, whereas Complex III is required for CD4+ activation and antigen-specific T lymphocyte expansion (189, 190). It is well known that ROS can activate the transcription factor NF-κB, whereas chronic exposure to oxidative stress inhibits its phosphorylation and the activation of T lymphocytes (191, 192). Related to this, translocation of NF-κB to the nucleus occurs in a cytoplasmic oxidative environment; however, binding to DNA requires reducing environment. Very high levels of ROS might affect both compartments and in such circumstances NF-κB pathway will be inhibited (193, 194). On the contrary, reduced ROS production is associated with decreased phosphorylation of JNK and NF-κB and, therefore, low IFN-γ and CD39 expression in CD8+ T lymphocytes (195). Equivalent results have been found in other regulatory pathways, since the exposure to low levels of ROS stimulates mTORC1 while high concentrations or long-term ROS treatment decrease mTORC1 activity (196).
Reactive oxygen species are also implicated in T lymphocyte differentiation, and murine models with specific knockouts of NOX-2, such as gp91phox and p47phox, have been employed to test this association. p47phox deficiency leads to Th17 differentiation, because mice p47phox−/− have diminished expression of T-bet, STAT-1, and STAT-4 transcription factors, but increased phosphorylation of STAT-3. Additionally, it has been shown a reduced production of IL-2, IL-4, IFN-γ, TNF-α, and GM-CSF, but increased IL-10, IL-17, and TGF-β (188). On the contrary, lack of gp91phox leads to a Th1 phenotype with reduced GATA-3 expression and STAT-5 and STAT-6 phosphorylation but increased T-bet expression. These T lymphocytes produce less IL-4 and IL-5 but more IL-17 and IFN-γ (189, 197). Then, NOX-2-deficienT lymphocytes showed a decreased in IL-4 but an increased IL-17 production. Interestingly, NOX-2 is not required for the proper activation of primary murine T lymphocytes, as gp91phox−/− T lymphocytes have no defect in CD25 and CD69 expression, IL-2 production, or proliferation (187, 197).
CD4 T lymphocyte plasticity, switching from one lineage to another, may be affected by the oxidative microenvironment (37). As mentioned before, an oxidative microenvironment exerts opposite effects on cytokine secretion by Th1 compared to Th2 cells. When in vitro derived Th1 and Th2 clones or employed T lymphocytes derived from autoimmune thyroiditis to examine their ability to expand and produce cytokines in response to oxidative stress, low levels of H2O2 are able to reduce IFN-γ production by activated Th1 clones but to increase IL-4 secretion by activated Th2 clones (198). Besides, mitochondrial ROS can control T lymphocyte activation by upregulating IL-2 and IL-4 expression, and using T lymphocytes isolated from patients with atopic dermatitis, the inhibition of Complex I-mediated ROS blocks disease-associated spontaneous hyperexpression and TCR-induced expression of IL-4 (189).
In opposite to regulatory role of mild oxidation, oxidative stress shows important effects during T lymphocyte development and differentiation. Thymus-specific elevation of mitochondrial superoxide disrupts normal T lymphocyte development and impairs the function of the mammalian adaptive immune system (199).
The stage of differentiation largely determines sensitivity of individual T lymphocyte subsets to oxidative stress. The susceptibility of T lymphocytes to oxidative stress varies greatly depending on which stage of differentiation they are in (Figure 5). Effector cells are exposed to low oxidative environment, while memory cells are T lymphocytes found in the most oxidative environments. Some secreted cytokines can cause oxidative stress in T lymphocytes and in cancer cells. For example, tumors associated macrophages have been shown to induce sub-lethal oxidative stress in murine mammary cancer cells, maybe through the secretion of TNF-α. On the other hand, extracellular superoxide dismutases might finely tuning the levels of H2O2 in the extracellular milieu altering the proliferation and differentiation of immune cells (200). In fact, ROS may induce decreased viability in CD4+ T lymphocytes and the inhibition of DNA synthesis (181, 201). The latter is associated with alterations in the TCR signaling, including conformational changes of TCRζ and LCK, reduction of PLCγ-1 phosphorylation and calcium flux, and increased ERK phosphorylation. Moreover, it has been known that prolonged exposure to H2O2 suppress tyrosine phosphorylation, calcium flux, NFAT, and NF-κB activation, and IL-2 production (191).
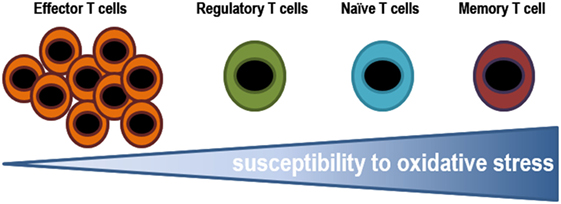
Figure 5. Susceptibility of T lymphocytes to oxidative stress. Effector cells are exposed to low oxidative environment, while memory cells are T lymphocytes found in the most oxidative environments. The ability of T lymphocytes to stand such oxidative conditions, in either an infectious disease state or a tumor microenvironment, might determine the pool of T lymphocytes.
In vitro assays testing the resistance of different subsets T lymphocytes to H2O2, it has been shown that it decreases from effector, to regulatory, naïve, and finally memory T lymphocytes (201). Effector T lymphocytes are able to withstand higher concentrations of ROS, which is probably essential to play their role in helping to phagocytes to eliminate pathogens (184). Whereas, human Tregs have higher thiol content and as a result, they are more resistant to cell death induced by H2O2 secreted by granulocytes than conventional T lymphocytes (201). Tregs suppress GSH synthesis and cysteine release by DCs in a CTLA-4-dependent manner. The resulting decrease in intracellular GSH leads to reduction in DNA synthesis in conventional T lymphocytes (191, 202), reduced levels leads to membrane displacement of LAT (central adapter protein in the TCR), and responsiveness T lymphocytes. A recent study showed that Tregs modulate GSH metabolism in T lymphocytes via cell contact and antigen-dependent, but not by an antigen-specific mechanism during suppression (202). The mechanism has been not identified yet but it could involve NADPH oxidase. Macrophages have also been shown to suppress T lymphocyte activation in vitro and in vivo through ROS (203) and, recent data demonstrates that macrophages induce Tregs via a ROS-dependent pathway that can be blocked by the NADPH oxidase inhibitor apocynin (204). In addition, scavenging enzymes that reduced oxidative intracellular milieu influence the regulation of T lymphocyte activity. Mitochondrial superoxide dismutase (MnSOD/SOD2) reduces T lymphocyte differentiation and functional ability by decreasing ROS levels (199, 205). Glutathione peroxidase-4 inhibits lipid peroxidation and plays a central role in the survival and the expansion of T lymphocytes.
In aging, the increase in oxidative stress and accumulated damage in the leukocytes appears to be related to the age-related deterioration of immune functions. A study by De la Fuente et al. reveals that the greater the cellular oxidative state and oxidative damage observed in immune cells of aged mice as well as in peripheral blood of elderly humans, were related with impaired immune responses (phagocytosis, chemotaxis, lymphoproliferation, etc.). However, in healthy centenarian individuals and in very long-lived mice, with preserved immune functions, they all presented a decreased expression of different inflammatory genes and highly controlled oxidative stress in their immune cells, which can partly explain their longevity (206–209). It is very important to emphasize in this context, that phagocytes, are postulated as the main responsible for oxidation-chronic inflammation stress that is associated with age and with immunosenescence (210). In the end, as a result of the oxidative damage that is related to aging, these cells could lose the ability to regulate their redox and inflammatory state, with the result of producing more and more oxidizing and inflammatory compounds, and thus contribute to the increase of oxidative and inflammatory stress. On the other hand, there are several studies performed on macrophages and peripheral blood neutrophils, in mice and humans, which have shown that these cells produce higher levels of oxidized compounds than those produced by lymphocytes and these levels increase with age. Furthermore, all these oxidative and inflammatory disbalances have been related to functional dysfunction of T lymphocytes.
On the other hand, different sources of ROS are involved in the activation-induced cell death (AICD) expression of Fas ligand (FasL) of T lymphocytes, and re-exposure to the specific antigen increases T lymphocyte sensitivity. First, H2O2 produced by DUOX-1 upon TCR serves to amplify proximal signaling events downstream of the TCR. Second, released from mitochondrial Complex I, potentially in response to ERK signaling, triggers the expression of FasL. Finally, Fas ligation activates NOX-2, which probably contributes to the execution of the apoptotic program via H2O2-mediated activation of AKT and the inhibition of MEK. Moreover, cell-intrinsic antioxidants, such as glutathione, vitamin E, MnSOD, and CuZnSOD, interfere with FasL expression, thus counteracting AICD (184).
The selective cell death of effector cells with the memory phenotype may influence the size of the eventual memory T lymphocyte pool and consequently the functional ability of the responding cells. More specifically, CTLs exhibiting an EM phenotype were preferentially sensitive to AICD, compared CTLs with a CM. This increased sensitivity of EM T lymphocytes to TCR-induced AICD might correlate to the reduced levels of thiols in CD45RO+ T lymphocytes as compared to CD45RA+ memory T lymphocytes (211, 212). The loss of thiols after proliferation on repeated TCR stimulation may relate to apoptosis susceptibility. In this way, naïve T lymphocytes have higher levels of surface thiols and higher production of intracellular GSH compared to the antigen-experienced T lymphocytes (213–215). In addition, scavengers could reduce ROS-induced apoptosis of naïve and memory T lymphocytes. In fact, the increase levels of reduced thiol groups and intracellular GSH in a T lymphocyte subset could be responsible for its increased ability to persist in an oxidative stress microenvironment.
Inflammation, both acute and chronic, both localized and systemic, plays a key role in the differentiation and ontogeny of T lymphocytes. From the moment of formation in the bone marrow until arrival to exhausted or senescent status, inflammation influences the development of T lymphocytes, and in turn the adaptive immune responses in the human body. The homeostasis and the way in which T lymphocytes respond to a specific antigen are influenced by the level of inflammation of the environment where the T lymphocytes are located. All the changes that occur along the ontogeny and differentiation of T lymphocytes require metabolic and oxidative adaptations. Despite the profound influence of inflammation in all these processes, little is known about the mechanisms through which it influences T lymphocytes; therefore, this is a research field with great practical applications to explore.
All authors have contributed equally to the elaboration of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Marisa Lopez-Cruzan (UT Health Science Center, Texas, EEUU) for her helpful assistance and review of the English grammar.
This work was supported by grant PI14/01566 from Plan Estatal de I + D + i 2013–2016, co-founded by “Instituto de Salud Carlos III” and by “Fondo Europeo de Desarrollo Regional (FEDER), from CONICYT (FONDECYT REGULAR 1151048),” and from “Ministerio de Economia y Competitividad, Gobierno de España” co-funded by FEDER (MINECO-17-BFI2016-79139-R).
1. Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol (2012) 30:1–22. doi:10.1146/annurev-immunol-100311-102839
2. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415(6871):536–41. doi:10.1038/415536a
3. Goronzy JJ, Weyand CM. Successful and maladaptive T cell aging. Immunity (2017) 46(3):364–78. doi:10.1016/j.immuni.2017.03.010
4. Shen-Orr SS, Furman D, Kidd BA, Hadad F, Lovelace P, Huang YW, et al. Defective signaling in the JAK-STAT pathway tracks with chronic inflammation and cardiovascular risk in aging humans. Cell Syst (2016) 3(4):374–84.e4. doi:10.1016/j.cels.2016.09.009
5. Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401(6754):708–12. doi:10.1038/44385
6. Koch S, Larbi A, Derhovanessian E, Ozcelik D, Naumova E, Pawelec G. Multiparameter flow cytometric analysis of CD4 and CD8 T cell subsets in young and old people. Immun Ageing (2008) 5:6. doi:10.1186/1742-4933-5-6
7. Hataye J, Moon JJ, Khoruts A, Reilly C, Jenkins MK. Naive and memory CD4+ T cell survival controlled by clonal abundance. Science (2006) 312(5770):114–6. doi:10.1126/science.1124228
8. Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity (2008) 29(6):848–62. doi:10.1016/j.immuni.2008.11.002
9. den Braber I, Mugwagwa T, Vrisekoop N, Westera L, Mögling R, de Boer AB, et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity (2012) 36(2):288–97. doi:10.1016/j.immuni.2012.02.006
10. Marrack P, Kappler J. Control of T cell viability. Annu Rev Immunol (2004) 22:765–87. doi:10.1146/annurev.immunol.22.012703.104554
11. Rathmell JC, Farkash EA, Gao W, Thompson CB. IL-7 enhances the survival and maintains the size of naive T cells. J Immunol (2001) 167(12):6869–76. doi:10.4049/jimmunol.167.12.6869
12. Rochman Y, Leonard WJ. The role of thymic stromal lymphopoietin in CD8+ T cell homeostasis. J Immunol (2008) 181(11):7699–705. doi:10.4049/jimmunol.181.11.7699
13. Vivien L, Benoist C, Mathis D. T lymphocytes need IL-7 but not IL-4 or IL-6 to survive in vivo. Int Immunol (2001) 13(6):763–8. doi:10.1093/intimm/13.6.763
14. Schluns KS, Kieper WC, Jameson SC, Lefrançois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol (2000) 1(5):426–32. doi:10.1038/80868
15. Hassan J, Reen DJ. IL-7 promotes the survival and maturation but not differentiation of human post-thymic CD4+ T cells. Eur J Immunol (1998) 28(10):3057–65. doi:10.1002/(SICI)1521-4141(199810)28:10<3057::AID-IMMU3057>3.0.CO;2-Z
16. Seddon B, Zamoyska R. TCR signals mediated by Src family kinases are essential for the survival of naive T cells. J Immunol (2002) 169(6):2997–3005. doi:10.4049/jimmunol.169.6.2997
17. Kondrack RM, Harbertson J, Tan JT, McBreen ME, Surh CD, Bradley LM. Interleukin 7 regulates the survival and generation of memory CD4 cells. J Exp Med (2003) 198(12):1797–806. doi:10.1084/jem.20030735
18. Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, et al. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci U S A (2001) 98(15):8732–7. doi:10.1073/pnas.161126098
19. Mertsching E, Burdet C, Ceredig R. IL-7 transgenic mice: analysis of the role of IL-7 in the differentiation of thymocytes in vivo and in vitro. Int Immunol (1995) 7(3):401–14. doi:10.1093/intimm/7.3.401
20. Kieper WC, Tan JT, Bondi-Boyd B, Gapin L, Sprent J, Ceredig R, et al. Overexpression of interleukin (IL)-7 leads to IL-15-independent generation of memory phenotype CD8+ T cells. J Exp Med (2002) 195(12):1533–9. doi:10.1084/jem.20020067
21. Kim HK, Waickman AT, Castro E, Flomerfelt FA, Hawk NV, Kapoor V, et al. Distinct IL-7 signaling in recent thymic emigrants versus mature naive T cells controls T-cell homeostasis. Eur J Immunol (2016) 46(7):1669–80. doi:10.1002/eji.201546214
22. Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol (2003) 21:139–76. doi:10.1146/annurev.immunol.21.120601.141107
23. Davey GM, Schober SL, Endrizzi BT, Dutcher AK, Jameson SC, Hogquist KA. Preselection thymocytes are more sensitive to T cell receptor stimulation than mature T cells. J Exp Med (1998) 188(10):1867–74. doi:10.1084/jem.188.10.1867
24. Lucas B, S˘tefanová I, Yasutomo K, Dautigny N, Germain RN. Divergent changes in the sensitivity of maturing T cells to structurally related ligands underlies formation of a useful T cell repertoire. Immunity (1999) 10(3):367–76. doi:10.1016/S1074-7613(00)80036-9
25. van Oers NS, Killeen N, Weiss A. ZAP-70 is constitutively associated with tyrosine-phosphorylated TCR zeta in murine thymocytes and lymph node T cells. Immunity (1994) 1(8):675–85. doi:10.1016/1074-7613(94)90038-8
26. Witherden D, van Oers N, Waltzinger C, Weiss A, Benoist C, Mathis D. Tetracycline-controllable selection of CD4(+) T cells: half-life and survival signals in the absence of major histocompatibility complex class II molecules. J Exp Med (2000) 191(2):355–64. doi:10.1084/jem.191.2.355
27. Condotta SA, Rai D, James BR, Griffith TS, Badovinac VP. Sustained and incomplete recovery of naive CD8+ T cell precursors after sepsis contributes to impaired CD8+ T cell responses to infection. J Immunol (2013) 190(5):1991–2000. doi:10.4049/jimmunol.1202379
28. Markwart R, Condotta SA, Requardt RP, Borken F, Schubert K, Weigel C, et al. Immunosuppression after sepsis: systemic inflammation and sepsis induce a loss of naive T-cells but no enduring cell-autonomous defects in T-cell function. PLoS One (2014) 9(12):e115094. doi:10.1371/journal.pone.0115094
29. Martin MD, Wirth TC, Lauer P, Harty JT, Badovinac VP. The impact of pre-existing memory on differentiation of newly recruited naive CD8 T cells. J Immunol (2011) 187(6):2923–31. doi:10.4049/jimmunol.1100698
30. Richer MJ, Nolz JC, Harty JT. Pathogen-specific inflammatory milieux tune the antigen sensitivity of CD8(+) T cells by enhancing T cell receptor signaling. Immunity (2013) 38(1):140–52. doi:10.1016/j.immuni.2012.09.017
31. Raué HP, Beadling C, Haun J, Slifka MK. Cytokine-mediated programmed proliferation of virus-specific CD8(+) memory T cells. Immunity (2013) 38(1):131–9. doi:10.1016/j.immuni.2012.09.019
32. Kerstein A, Schüler S, Cabral-Marques O, Fazio J, Häsler R, Müller A, et al. Environmental factor and inflammation-driven alteration of the total peripheral T-cell compartment in granulomatosis with polyangiitis. J Autoimmun (2017) 78:79–91. doi:10.1016/j.jaut.2016.12.004
33. Yoon H, Kim TS, Braciale TJ. The cell cycle time of CD8+ T cells responding in vivo is controlled by the type of antigenic stimulus. PLoS One (2010) 5(11):e15423. doi:10.1371/journal.pone.0015423
34. Au-Yeung BB, Zikherman J, Mueller JL, Ashouri JF, Matloubian M, Cheng DA, et al. A sharp T-cell antigen receptor signaling threshold for T-cell proliferation. Proc Natl Acad Sci U S A (2014) 111(35):E3679–88. doi:10.1073/pnas.1413726111
35. Jenkins MK, Chen CA, Jung G, Mueller DL, Schwartz RH. Inhibition of antigen-specific proliferation of type 1 murine T cell clones after stimulation with immobilized anti-CD3 monoclonal antibody. J Immunol (1990) 144(1):16–22.
36. Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med (2003) 197(9):1141–51. doi:10.1084/jem.20021910
37. O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science (2010) 327(5969):1098–102. doi:10.1126/science.1178334
38. Sanchez PJ, McWilliams JA, Haluszczak C, Yagita H, Kedl RM. Combined TLR/CD40 stimulation mediates potent cellular immunity by regulating dendritic cell expression of CD70 in vivo. J Immunol (2007) 178(3):1564–72. doi:10.4049/jimmunol.178.3.1564
39. Rogers PR, Croft M. CD28, Ox-40, LFA-1, and CD4 modulation of Th1/Th2 differentiation is directly dependent on the dose of antigen. J Immunol (2000) 164(6):2955–63. doi:10.4049/jimmunol.164.6.2955
40. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med (1991) 174(3):561–9. doi:10.1084/jem.174.3.561
41. Chikuma S. CTLA-4, an essential immune-checkpoint for T-cell activation. Curr Top Microbiol Immunol (2017) 410:99–126. doi:10.1007/82_2017_61
42. Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature (2004) 427(6970):154–9. doi:10.1038/nature02238
43. Memarnejadian A, Meilleur CE, Shaler CR, Khazaie K, Bennink JR, Schell TD, et al. PD-1 blockade promotes epitope spreading in anticancer CD8+ T cell responses by preventing fratricidal death of subdominant clones to relieve immunodomination. J Immunol (2017) 199(9):3348–59. doi:10.4049/jimmunol.1700643
44. Marelli G, Sica A, Vannucci L, Allavena P. Inflammation as target in cancer therapy. Curr Opin Pharmacol (2017) 35:57–65. doi:10.1016/j.coph.2017.05.007
45. Palucka K, Coussens LM, O’Shaughnessy J. Dendritic cells, inflammation, and breast cancer. Cancer J (2013) 19(6):511–6. doi:10.1097/PPO.0000000000000007
46. Veglia F, Gabrilovich DI. Dendritic cells in cancer: the role revisited. Curr Opin Immunol (2017) 45:43–51. doi:10.1016/j.coi.2017.01.002
47. Gupta S. Role of dendritic cells in innate and adaptive immune response in human aging. Exp Gerontol (2014) 54:47–52. doi:10.1016/j.exger.2013.12.009
48. Panda A, Qian F, Mohanty S, van Duin D, Newman FK, Zhang L, et al. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J Immunol (2010) 184(5):2518–27. doi:10.4049/jimmunol.0901022
49. Scheenstra MR, De Cuyper IM, Branco-Madeira F, de Bleser P, Kool M, Meinders M, et al. GATA1-deficient dendritic cells display impaired CCL21-dependent migration toward lymph nodes due to reduced levels of polysialic acid. J Immunol (2016) 197(11):4312–24. doi:10.4049/jimmunol.1600103
50. Bryl E, Vallejo AN, Weyand CM, Goronzy JJ. Down-regulation of CD28 expression by TNF-alpha. J Immunol (2001) 167(6):3231–8. doi:10.4049/jimmunol.167.6.3231
51. Bryl E, Vallejo AN, Matteson EL, Witkowski JM, Weyand CM, Goronzy JJ. Modulation of CD28 expression with anti-tumor necrosis factor alpha therapy in rheumatoid arthritis. Arthritis Rheum (2005) 52(10):2996–3003. doi:10.1002/art.21353
52. Herati RS, Reuter MA, Dolfi DV, Mansfield KD, Aung H, Badwan OZ, et al. Circulating CXCR5+PD-1+ response predicts influenza vaccine antibody responses in young adults but not elderly adults. J Immunol (2014) 193(7):3528–37. doi:10.4049/jimmunol.1302503
53. Linterman MA. How T follicular helper cells and the germinal centre response change with age. Immunol Cell Biol (2014) 92(1):72–9. doi:10.1038/icb.2013.77
54. Li G, Ju J, Weyand CM, Goronzy JJ. Age-associated failure to adjust type I IFN receptor signaling thresholds after T cell activation. J Immunol (2015) 195(3):865–74. doi:10.4049/jimmunol.1402389
55. Brincks EL, Woodland DL. Novel roles for IL-15 in T cell survival. F1000 Biol Rep (2010) 2:67. doi:10.3410/B2-67
56. Boyman O, Létourneau S, Krieg C, Sprent J. Homeostatic proliferation and survival of naive and memory T cells. Eur J Immunol (2009) 39(8):2088–94. doi:10.1002/eji.200939444
57. Caserta S, Zamoyska R. Memories are made of this: synergy of T cell receptor and cytokine signals in CD4(+) central memory cell survival. Trends Immunol (2007) 28(6):245–8. doi:10.1016/j.it.2007.04.006
58. Surh CD, Sprent J. Regulation of naive and memory T-cell homeostasis. Microbes Infect (2002) 4(1):51–6. doi:10.1016/S1286-4579(01)01509-X
59. Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity (2007) 27(3):393–405. doi:10.1016/j.immuni.2007.08.007
60. Opferman JT, Ober BT, Ashton-Rickardt PG. Linear differentiation of cytotoxic effectors into memory T lymphocytes. Science (1999) 283(5408):1745–8. doi:10.1126/science.283.5408.1745
61. Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med (2008) 205(3):625–40. doi:10.1084/jem.20071641
62. Harrington LE, Janowski KM, Oliver JR, Zajac AJ, Weaver CT. Memory CD4 T cells emerge from effector T-cell progenitors. Nature (2008) 452(7185):356–60. doi:10.1038/nature06672
63. Bannard O, Kraman M, Fearon DT. Secondary replicative function of CD8+ T cells that had developed an effector phenotype. Science (2009) 323(5913):505–9. doi:10.1126/science.1166831
64. Haring JS, Harty JT. Aberrant contraction of antigen-specific CD4 T cells after infection in the absence of gamma interferon or its receptor. Infect Immun (2006) 74(11):6252–63. doi:10.1128/IAI.00847-06
65. Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, et al. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev (2006) 211:81–92. doi:10.1111/j.0105-2896.2006.00382.x
66. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity (2007) 27(2):281–95. doi:10.1016/j.immuni.2007.07.010
67. Pearce EL, Shen H. Generation of CD8 T cell memory is regulated by IL-12. J Immunol (2007) 179(4):2074–81. doi:10.4049/jimmunol.179.4.2074
68. Badovinac VP, Harty JT. Adaptive immunity and enhanced CD8+ T cell response to Listeria monocytogenes in the absence of perforin and IFN-gamma. J Immunol (2000) 164(12):6444–52. doi:10.4049/jimmunol.164.12.6444
69. Whitmire JK, Eam B, Benning N, Whitton JL. Direct interferon-gamma signaling dramatically enhances CD4+ and CD8+ T cell memory. J Immunol (2007) 179(2):1190–7. doi:10.4049/jimmunol.179.2.1190
70. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol (2005) 6(12):1236–44. doi:10.1038/ni1268
71. Shaulov A, Murali-Krishna K. CD8 T cell expansion and memory differentiation are facilitated by simultaneous and sustained exposure to antigenic and inflammatory milieu. J Immunol (2008) 180(2):1131–8. doi:10.4049/jimmunol.180.2.1131
72. Ku CC, Murakami M, Sakamoto A, Kappler J, Marrack P. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science (2000) 288(5466):675–8. doi:10.1126/science.288.5466.675
73. Alonso-Arias R, Moro-García MA, Vidal-Castiñeira JR, Solano-Jaurrieta JJ, Suárez-García FM, Coto E, et al. IL-15 preferentially enhances functional properties and antigen-specific responses of CD4+CD28(null) compared to CD4+CD28+ T cells. Aging Cell (2011) 10(5):844–52. doi:10.1111/j.1474-9726.2011.00725.x
74. Alonso-Arias R, Moro-García MA, López-Vázquez A, Rodrigo L, Baltar J, García FM, et al. NKG2D expression in CD4+ T lymphocytes as a marker of senescence in the aged immune system. Age (Dordr) (2011) 33(4):591–605. doi:10.1007/s11357-010-9200-6
75. Moro-Garcia MA, Alonso-Arias R, Lopez-Larrea C. When aging reaches CD4+ T-cells: phenotypic and functional changes. Front Immunol (2013) 4:107. doi:10.3389/fimmu.2013.00107
76. Shin H, Blackburn SD, Blattman JN, Wherry EJ. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J Exp Med (2007) 204(4):941–9. doi:10.1084/jem.20061937
77. Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med (2006) 12(10):1198–202. doi:10.1038/nm1106-1329b
78. Davalos AR, Coppe JP, Campisi J, Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev (2010) 29(2):273–83. doi:10.1007/s10555-010-9220-9
79. Mehlhop-Williams ER, Bevan MJ. Memory CD8+ T cells exhibit increased antigen threshold requirements for recall proliferation. J Exp Med (2014) 211(2):345–56. doi:10.1084/jem.20131271
80. Martinez P, Blasco MA. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer (2011) 11(3):161–76. doi:10.1038/nrc3025
81. Klapper W, Qian W, Schulte C, Parwaresch R. DNA damage transiently increases TRF2 mRNA expression and telomerase activity. Leukemia (2003) 17(10):2007–15. doi:10.1038/sj.leu.2403086
82. Andrews NP, Fujii H, Goronzy JJ, Weyand CM. Telomeres and immunological diseases of aging. Gerontology (2010) 56(4):390–403. doi:10.1159/000268620
83. Blackburn EH. Telomeres and telomerase: their mechanisms of action and the effects of altering their functions. FEBS Lett (2005) 579(4):859–62. doi:10.1016/j.febslet.2004.11.036
84. Hodes RJ, Hathcock KS, Weng NP. Telomeres in T and B cells. Nat Rev Immunol (2002) 2(9):699–706. doi:10.1038/nri890
85. Campisi J. Cancer and ageing: rival demons? Nat Rev Cancer (2003) 3(5):339–49. doi:10.1038/nrc1073
86. Adibzadeh M, Mariani E, Bartoloni C, Beckman I, Ligthart G, Remarque E, et al. Lifespans of T lymphocytes. Mech Ageing Dev (1996) 91(2):145–54. doi:10.1016/0047-6374(96)01783-6
87. Perillo NL, Walford RL, Newman MA, Effros RB. Human T lymphocytes possess a limited in vitro life span. Exp Gerontol (1989) 24(3):177–87. doi:10.1016/0531-5565(89)90009-0
88. Wang E, Lee MJ, Pandey S. Control of fibroblast senescence and activation of programmed cell death. J Cell Biochem (1994) 54(4):432–9. doi:10.1002/jcb.240540410
89. Spaulding C, Guo W, Effros RB. Resistance to apoptosis in human CD8+ T cells that reach replicative senescence after multiple rounds of antigen-specific proliferation. Exp Gerontol (1999) 34(5):633–44. doi:10.1016/S0531-5565(99)00033-9
90. Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet (2005) 6(8):611–22. doi:10.1038/nrg1656
91. Feldser DM, Greider CW. Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell (2007) 11(5):461–9. doi:10.1016/j.ccr.2007.02.026
92. Plunkett FJ, Franzese O, Belaramani LL, Fletcher JM, Gilmour KC, Sharifi R, et al. The impact of telomere erosion on memory CD8+ T cells in patients with X-linked lymphoproliferative syndrome. Mech Ageing Dev (2005) 126(8):855–65. doi:10.1016/j.mad.2005.03.006
93. Fletcher JM, Vukmanovic-Stejic M, Dunne PJ, Birch KE, Cook JE, Jackson SE, et al. Cytomegalovirus-specific CD4+ T cells in healthy carriers are continuously driven to replicative exhaustion. J Immunol (2005) 175(12):8218–25. doi:10.4049/jimmunol.175.12.8218
94. Macallan DC, Wallace D, Zhang Y, De Lara C, Worth AT, Ghattas H, et al. Rapid turnover of effector-memory CD4(+) T cells in healthy humans. J Exp Med (2004) 200(2):255–60. doi:10.1084/jem.20040341
95. Fritsch RD, Shen X, Sims GP, Hathcock KS, Hodes RJ, Lipsky PE. Stepwise differentiation of CD4 memory T cells defined by expression of CCR7 and CD27. J Immunol (2005) 175(10):6489–97. doi:10.4049/jimmunol.175.10.6489
96. Valenzuela HF, Effros RB. Divergent telomerase and CD28 expression patterns in human CD4 and CD8 T cells following repeated encounters with the same antigenic stimulus. Clin Immunol (2002) 105(2):117–25. doi:10.1006/clim.2002.5271
97. Moro-García MA, Alonso-Arias R, López-Vázquez A, Suárez-García FM, Solano-Jaurrieta JJ, Baltar J, et al. Relationship between functional ability in older people, immune system status, and intensity of response to CMV. Age (Dordr) (2012) 34(2):479–95. doi:10.1007/s11357-011-9240-6
98. van Lier RA, ten Berge IJ, Gamadia LE. Human CD8(+) T-cell differentiation in response to viruses. Nat Rev Immunol (2003) 3(12):931–9. doi:10.1038/nri1254
99. Czesnikiewicz-Guzik M, Lee WW, Cui D, Hiruma Y, Lamar DL, Yang ZZ, et al. T cell subset-specific susceptibility to aging. Clin Immunol (2008) 127(1):107–18. doi:10.1016/j.clim.2007.12.002
100. Bellon M, Baydoun HH, Yao Y, Nicot C. HTLV-I Tax-dependent and -independent events associated with immortalization of human primary T lymphocytes. Blood (2010) 115(12):2441–8. doi:10.1182/blood-2009-08-241117
101. Scheuring UJ, Sabzevari H, Theofilopoulos AN. Proliferative arrest and cell cycle regulation in CD8(+)CD28(-) versus CD8(+)CD28(+) T cells. Hum Immunol (2002) 63(11):1000–9. doi:10.1016/S0198-8859(02)00683-3
102. Wallace DL, Bérard M, Soares MV, Oldham J, Cook JE, Akbar AN, et al. Prolonged exposure of naive CD8+ T cells to interleukin-7 or interleukin-15 stimulates proliferation without differentiation or loss of telomere length. Immunology (2006) 119(2):243–53. doi:10.1111/j.1365-2567.2006.02429.x
103. Li Y, Zhi W, Wareski P, Weng NP. IL-15 activates telomerase and minimizes telomere loss and may preserve the replicative life span of memory CD8+ T cells in vitro. J Immunol (2005) 174(7):4019–24. doi:10.4049/jimmunol.174.7.4019
104. Li H, Xu D, Li J, Berndt MC, Liu JP. Transforming growth factor beta suppresses human telomerase reverse transcriptase (hTERT) by Smad3 interactions with c-Myc and the hTERT gene. J Biol Chem (2006) 281(35):25588–600. doi:10.1074/jbc.M602381200
105. Reed JR, Vukmanovic-Stejic M, Fletcher JM, Soares MV, Cook JE, Orteu CH, et al. Telomere erosion in memory T cells induced by telomerase inhibition at the site of antigenic challenge in vivo. J Exp Med (2004) 199(10):1433–43. doi:10.1084/jem.20040178
106. Abedin S, Michel JJ, Lemster B, Vallejo AN. Diversity of NKR expression in aging T cells and in T cells of the aged: the new frontier into the exploration of protective immunity in the elderly. Exp Gerontol (2005) 40(7):537–48. doi:10.1016/j.exger.2005.04.012
107. Shinko D, Diakos CI, Clarke SJ, Charles KA. Cancer-related systemic inflammation: the challenges and therapeutic opportunities for personalized medicine. Clin Pharmacol Ther (2017) 102(4):599–610. doi:10.1002/cpt.789
108. Tarazona R, DelaRosa O, Alonso C, Ostos B, Espejo J, Peña J, et al. Increased expression of NK cell markers on T lymphocytes in aging and chronic activation of the immune system reflects the accumulation of effector/senescent T cells. Mech Ageing Dev (2000) 121(1–3):77–88. doi:10.1016/S0047-6374(00)00199-8
109. McElhaney JE, Effros RB. Immunosenescence: what does it mean to health outcomes in older adults? Curr Opin Immunol (2009) 21(4):418–24. doi:10.1016/j.coi.2009.05.023
110. Brown DM, Lee S, Garcia-Hernandez ML, Swain SL. Multifunctional CD4 cells expressing gamma interferon and perforin mediate protection against lethal influenza virus infection. J Virol (2012) 86(12):6792–803. doi:10.1128/JVI.07172-11
111. Choi J, Fauce SR, Effros RB. Reduced telomerase activity in human T lymphocytes exposed to cortisol. Brain Behav Immun (2008) 22(4):600–5. doi:10.1016/j.bbi.2007.12.004
112. Dagarag M, Evazyan T, Rao N, Effros RB. Genetic manipulation of telomerase in HIV-specific CD8+ T cells: enhanced antiviral functions accompany the increased proliferative potential and telomere length stabilization. J Immunol (2004) 173(10):6303–11. doi:10.4049/jimmunol.173.10.6303
114. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi:10.1038/nri3862
115. Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol (2011) 11(4):289–95. doi:10.1038/nri2959
116. Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A (2009) 106(21):8623–8. doi:10.1073/pnas.0809818106
117. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol (2009) 10(1):29–37. doi:10.1038/ni.1679
118. Zhou S, Ou R, Huang L, Price GE, Moskophidis D. Differential tissue-specific regulation of antiviral CD8+ T-cell immune responses during chronic viral infection. J Virol (2004) 78(7):3578–600. doi:10.1128/JVI.78.7.3578-3600.2004
119. Fuller MJ, Khanolkar A, Tebo AE, Zajac AJ. Maintenance, loss, and resurgence of T cell responses during acute, protracted, and chronic viral infections. J Immunol (2004) 172(7):4204–14. doi:10.4049/jimmunol.172.7.4204
120. Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature (2009) 458(7235):206–10. doi:10.1038/nature07662
121. Fessas P, Lee H, Ikemizu S, Janowitz T. A molecular and preclinical comparison of the PD-1-targeted T-cell checkpoint inhibitors nivolumab and pembrolizumab. Semin Oncol (2017) 44(2):136–40. doi:10.1053/j.seminoncol.2017.06.002
122. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi:10.1146/annurev.immunol.26.021607.090331
123. Henson SM, Macaulay R, Riddell NE, Nunn CJ, Akbar AN. Blockade of PD-1 or p38 MAP kinase signaling enhances senescent human CD8(+) T-cell proliferation by distinct pathways. Eur J Immunol (2015) 45(5):1441–51. doi:10.1002/eji.201445312
124. Moskowitz DM, Zhang DW, Hu B, Le Saux S, Yanes RE, Ye Z, et al. Epigenomics of human CD8 T cell differentiation and aging. Sci Immunol (2017) 2(8):eaag0192. doi:10.1126/sciimmunol.aag0192
125. Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell (2016) 166(1):63–76. doi:10.1016/j.cell.2016.05.035
126. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity (2016) 45(2):358–73. doi:10.1016/j.immuni.2016.07.008
127. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science (2017) 354(6316):1165–9. doi:10.1126/science.aae0491
128. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science (2016) 354(6316):1160–5. doi:10.1126/science.aaf2807
129. van der Windt GJ, Pearce EL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev (2012) 249(1):27–42. doi:10.1111/j.1600-065X.2012.01150.x
130. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science (2013) 342(6155):1242454. doi:10.1126/science.1242454
131. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16(6):769–77. doi:10.1016/S1074-7613(02)00323-0
132. Dimeloe S, et al. T-cell metabolism governing activation, proliferation and differentiation; a modular view. Immunology (2016) 150(1):35–44. doi:10.1111/imm.12655
133. Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci (2016) 41(3):211–8. doi:10.1016/j.tibs.2015.12.001
134. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi:10.1016/j.cell.2013.05.016
135. Dang CV. Links between metabolism and cancer. Genes Dev (2012) 26(9):877–90. doi:10.1101/gad.189365.112
136. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi:10.1126/science.1160809
137. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol (2011) 27:441–64. doi:10.1146/annurev-cellbio-092910-154237
138. Dimeloe S, Mehling M, Frick C, Loeliger J, Bantug GR, Sauder U, et al. The immune-metabolic basis of effector memory CD4+ T cell function under hypoxic conditions. J Immunol (2016) 196(1):106–14. doi:10.4049/jimmunol.1501766
139. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11(5):325–37. doi:10.1038/nrc3038
140. Maekawa Y, Ishifune C, Tsukumo S, Hozumi K, Yagita H, Yasutomo K. Notch controls the survival of memory CD4+ T cells by regulating glucose uptake. Nat Med (2015) 21(1):55–61. doi:10.1038/nm.3758
141. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol (2013) 14(10):1064–72. doi:10.1038/ni.2687
142. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol (2010) 185(2):1037–44. doi:10.4049/jimmunol.0903586
143. Lane AN, Fan TW. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res (2015) 43(4):2466–85. doi:10.1093/nar/gkv047
144. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab (2015) 23(1):27–47. doi:10.1016/j.cmet.2015.12.006
145. Linke M, Fritsch SD, Sukhbaatar N, Hengstschläger M, Weichhart T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett (2017) 591(19):3089–103. doi:10.1002/1873-3468.12711
146. Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev (2012) 249(1):43–58. doi:10.1111/j.1600-065X.2012.01152.x
147. Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell (2010) 39(2):171–83. doi:10.1016/j.molcel.2010.06.022
148. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11(2):85–95. doi:10.1038/nrc2981
149. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell (2007) 12(1):9–22. doi:10.1016/j.ccr.2007.05.008
150. Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol (2015) 6:1. doi:10.3389/fimmu.2015.00001
151. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol (2011) 12(4):295–303. doi:10.1038/ni.2005
152. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208(7):1367–76. doi:10.1084/jem.20110278
153. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi:10.1016/j.cell.2011.07.033
154. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi:10.4049/jimmunol.1003613
155. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Sig (2015) 8(396):ra97. doi:10.1126/scisignal.aab2610
156. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. doi:10.1016/j.cmet.2014.05.004
157. Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med (2012) 209(13):2441–53. doi:10.1084/jem.20112607
158. Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, et al. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J Clin Invest (2015) 125(5):2090–108. doi:10.1172/JCI77746
159. Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol (2014) 15(12):1104–15. doi:10.1038/ni.3031
160. Pollizzi KN, Sun IH, Patel CH, Lo YC, Oh MH, Waickman AT, et al. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8(+) T cell differentiation. Nat Immunol (2016) 17(6):704–11. doi:10.1038/ni.3438
161. Delgoffe GM, Powell JD. Feeding an army: the metabolism of T cells in activation, anergy, and exhaustion. Mol Immunol (2015) 68(2 Pt C):492–6. doi:10.1016/j.molimm.2015.07.026
162. O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity (2014) 41(1):75–88. doi:10.1016/j.immuni.2014.06.005
163. Almeida L, Lochner M, Berod L, Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol (2016) 28(5):514–24. doi:10.1016/j.smim.2016.10.009
164. Wahl DR, Petersen B, Warner R, Richardson BC, Glick GD, Opipari AW. Characterization of the metabolic phenotype of chronically activated lymphocytes. Lupus (2010) 19(13):1492–501. doi:10.1177/0961203310373109
165. Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim Biophys Acta (2011) 1807(6):726–34. doi:10.1016/j.bbabio.2010.10.022
166. Lanna A, Henson SM, Escors D, Akbar AN. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB 1 drives the senescence of human T cells. Nat Immunol (2014) 15(10):965–72. doi:10.1038/ni.2981
167. Akbar AN. The convergence of senescence and nutrient sensing during lymphocyte ageing. Clin Exp Immunol (2017) 187(1):4–5. doi:10.1111/cei.12876
168. Henson SM, et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8(+) T cells. J Clin Invest (2014) 124(9):4004–16. doi:10.1172/JCI75051
169. Doherty PC. Immune exhaustion: driving virus-specific CD8+ T cells to death. Trends Microbiol (1993) 1(6):207–9. doi:10.1016/0966-842X(93)90133-C
170. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol (2015) 36(4):265–76. doi:10.1016/j.it.2015.02.008
171. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25(21):9543–53. doi:10.1128/MCB.25.21.9543-9553.2005
172. Pedicord VA, Cross JR, Montalvo-Ortiz W, Miller ML, Allison JP. Friends not foes: CTLA-4 blockade and mTOR inhibition cooperate during CD8+ T cell priming to promote memory formation and metabolic readiness. J Immunol (2015) 194(5):2089–98. doi:10.4049/jimmunol.1402390
173. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi:10.1038/ncomms7692
174. Hussain T, Tan B, Yin Y, Blachier F, Tossou MC, Rahu N. Oxidative stress and inflammation: what polyphenols can do for us? Oxid Med Cell Longev (2016) 2016:7432797. doi:10.1155/2016/7432797
175. Salzano S, Checconi P, Hanschmann EM, Lillig CH, Bowler LD, Chan P, et al. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc Natl Acad Sci U S A (2014) 111(33):12157–62. doi:10.1073/pnas.1401712111
176. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov (2009) 8(7):579–91. doi:10.1038/nrd2803
177. Sainz RM, Lombo F, Mayo JC. Radical decisions in cancer: redox control of cell growth and death. Cancers (Basel) (2012) 4(2):442–74. doi:10.3390/cancers4020442
178. Miar A, Hevia D, Muñoz-Cimadevilla H, Astudillo A, Velasco J, Sainz RM, et al. Manganese superoxide dismutase (SOD2/MnSOD)/catalase and SOD2/GPx1 ratios as biomarkers for tumor progression and metastasis in prostate, colon, and lung cancer. Free Radic Biol Med (2015) 85:45–55. doi:10.1016/j.freeradbiomed.2015.04.001
179. Finkel T. Signal transduction by reactive oxygen species. J Cell Biol (2011) 194(1):7–15. doi:10.1083/jcb.201102095
180. Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol (2004) 5(8):818–27. doi:10.1038/ni1096
181. Cemerski S, Cantagrel A, Van Meerwijk JP, Romagnoli P. Reactive oxygen species differentially affect T cell receptor-signaling pathways. J Biol Chem (2002) 277(22):19585–93. doi:10.1074/jbc.M111451200
182. Gelderman KA, Hultqvist M, Holmberg J, Olofsson P, Holmdahl R. T cell surface redox levels determine T cell reactivity and arthritis susceptibility. Proc Natl Acad Sci U S A (2006) 103(34):12831–6. doi:10.1073/pnas.0604571103
183. Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med (2002) 195(1):59–70. doi:10.1084/jem.20010659
184. Belikov AV, Schraven B, Simeoni L. T cells and reactive oxygen species. J Biomed Sci (2015) 22:85. doi:10.1186/s12929-015-0194-3
185. Gülow K, Kaminski M, Darvas K, Süss D, Li-Weber M, Krammer PH. HIV-1 trans-activator of transcription substitutes for oxidative signaling in activation-induced T cell death. J Immunol (2005) 174(9):5249–60. doi:10.4049/jimmunol.174.9.5249
186. Los M, Schenk H, Hexel K, Baeuerle PA, Dröge W, Schulze-Osthoff K. IL-2 gene expression and NF-kappa B activation through CD28 requires reactive oxygen production by 5-lipoxygenase. EMBO J (1995) 14(15):3731–40.
187. Belikov AV, Schraven B, Simeoni L. TCR-triggered extracellular superoxide production is not required for T-cell activation. Cell Commun Signal (2014) 12:50. doi:10.1186/s12964-014-0050-1
188. Tse HM, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, et al. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J Immunol (2010) 185(9):5247–58. doi:10.4049/jimmunol.1001472
189. Kaminski MM, Sauer SW, Klemke CD, Süss D, Okun JG, Krammer PH, et al. Mitochondrial reactive oxygen species control T cell activation by regulating IL-2 and IL-4 expression: mechanism of ciprofloxacin-mediated immunosuppression. J Immunol (2010) 184(9):4827–41. doi:10.4049/jimmunol.0901662
190. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38(2):225–36. doi:10.1016/j.immuni.2012.10.020
191. Flescher E, Ledbetter JA, Schieven GL, Vela-Roch N, Fossum D, Dang H, et al. Longitudinal exposure of human T lymphocytes to weak oxidative stress suppresses transmembrane and nuclear signal transduction. J Immunol (1994) 153(11):4880–9.
192. Lahdenpohja N, Savinainen K, Hurme M. Pre-exposure to oxidative stress decreases the nuclear factor-kappa B-dependent transcription in T lymphocytes. J Immunol (1998) 160(3):1354–8.
193. Schreck R, Meier B, Männel DN, Dröge W, Baeuerle PA. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J Exp Med (1992) 175(5):1181–94. doi:10.1084/jem.175.5.1181
194. Suzuki YJ, Aggarwal BB, Packer L. Alpha-lipoic acid is a potent inhibitor of NF-kappa B activation in human T cells. Biochem Biophys Res Commun (1992) 189(3):1709–15. doi:10.1016/0006-291X(92)90275-P
195. Bai A, Moss A, Rothweiler S, Longhi MS, Wu Y, Junger WG, et al. NADH oxidase-dependent CD39 expression by CD8(+) T cells modulates interferon gamma responses via generation of adenosine. Nat Commun (2015) 6:8819. doi:10.1038/ncomms9819
196. Li M, Zhao L, Liu J, Liu A, Jia C, Ma D, et al. Multi-mechanisms are involved in reactive oxygen species regulation of mTORC1 signaling. Cell Signal (2010) 22(10):1469–76. doi:10.1016/j.cellsig.2010.05.015
197. Shatynski KE, Chen H, Kwon J, Williams MS. Decreased STAT5 phosphorylation and GATA-3 expression in NOX2-deficient T cells: role in T helper development. Eur J Immunol (2012) 42(12):3202–11. doi:10.1002/eji.201242659
198. Frossi B, De Carli M, Piemonte M, Pucillo C. Oxidative microenvironment exerts an opposite regulatory effect on cytokine production by Th1 and Th2 cells. Mol Immunol (2008) 45(1):58–64. doi:10.1016/j.molimm.2007.05.008
199. Case AJ, McGill JL, Tygrett LT, Shirasawa T, Spitz DR, Waldschmidt TJ, et al. Elevated mitochondrial superoxide disrupts normal T cell development, impairing adaptive immune responses to an influenza challenge. Free Radic Biol Med (2011) 50(3):448–58. doi:10.1016/j.freeradbiomed.2010.11.025
200. Griess B, Tom E, Domann F, Teoh-Fitzgerald M. Extracellular superoxide dismutase and its role in cancer. Free Radic Biol Med (2017) 112:464–79. doi:10.1016/j.freeradbiomed.2017.08.013
201. Mougiakakos D, Johansson CC, Kiessling R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood (2009) 113(15):3542–5. doi:10.1182/blood-2008-09-181040
202. Yan Z, Garg SK, Banerjee R. Regulatory T cells interfere with glutathione metabolism in dendritic cells and T cells. J Biol Chem (2010) 285(53):41525–32. doi:10.1074/jbc.M110.189944
203. Gelderman KA, Hultqvist M, Pizzolla A, Zhao M, Nandakumar KS, Mattsson R, et al. Macrophages suppress T cell responses and arthritis development in mice by producing reactive oxygen species. J Clin Invest (2007) 117(10):3020–8. doi:10.1172/JCI31935
204. Kraaij MD, Savage ND, van der Kooij SW, Koekkoek K, Wang J, van den Berg JM, et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc Natl Acad Sci U S A (2010) 107(41):17686–91. doi:10.1073/pnas.1012016107
205. Kamiński MM, Röth D, Sass S, Sauer SW, Krammer PH, Gülow K. Manganese superoxide dismutase: a regulator of T cell activation-induced oxidative signaling and cell death. Biochim Biophys Acta (2012) 1823(5):1041–52. doi:10.1016/j.bbamcr.2012.03.003
206. De la Fuente M, Miquel J. An update of the oxidation-inflammation theory of aging: the involvement of the immune system in oxi-inflamm-aging. Curr Pharm Des (2009) 15(26):3003–26. doi:10.2174/138161209789058110
207. Arranz L, Caamaño JH, Lord JM, De la Fuente M. Preserved immune functions and controlled leukocyte oxidative stress in naturally long-lived mice: possible role of nuclear factor kappa B. J Gerontol A Biol Sci Med Sci (2010) 65(9):941–50. doi:10.1093/gerona/glq101
208. de la Fuente M, Hernanz A, Guayerbas N, Alvarez P, Alvarado C. Changes with age in peritoneal macrophage functions. Implication of leukocytes in the oxidative stress of senescence. Cell Mol Biol (2004) 50:OL683–90.
209. Alonso-Fernández P, Puerto M, Maté I, Ribera JM, de la Fuente M. Neutrophils of centenarians show function levels similar to those of young adults. J Am Geriatr Soc (2008) 56(12):2244–51. doi:10.1111/j.1532-5415.2008.02018.x
210. De la Fuente M. Role of neuroimmunomodulation in aging. Neuroimmuno-modulation (2008) 15(4–6):213–23. doi:10.1159/000156465
211. Mehrotra S, Chhabra A, Chattopadhyay S, Dorsky DI, Chakraborty NG, Mukherji B. Rescuing melanoma epitope-specific cytolytic T lymphocytes from activation-induced cell death, by SP600125, an inhibitor of JNK: implications in cancer immunotherapy. J Immunol (2004) 173(10):6017–24. doi:10.4049/jimmunol.173.10.6017
212. Takahashi A, Hanson MG, Norell HR, Havelka AM, Kono K, Malmberg KJ, et al. Preferential cell death of CD8+ effector memory (CCR7-CD45RA-) T cells by hydrogen peroxide-induced oxidative stress. J Immunol (2005) 174(10):6080–7. doi:10.4049/jimmunol.174.10.6080
213. Franco R, Panayiotidis MI, Cidlowski JA. Glutathione depletion is necessary for apoptosis in lymphoid cells independent of reactive oxygen species formation. J Biol Chem (2007) 282(42):30452–65. doi:10.1074/jbc.M703091200
214. Mehrotra S, Mougiakakos D, Johansson CC, Voelkel-Johnson C, Kiessling R. Oxidative stress and lymphocyte persistence: implications in immunotherapy. Adv Cancer Res (2009) 102:197–227. doi:10.1016/S0065-230X(09)02006-5
Keywords: inflammation, T lymphocytes, differentiation, metabolic reprogramming, exhaustion, redox balance
Citation: Moro-García MA, Mayo JC, Sainz RM and Alonso-Arias R (2018) Influence of Inflammation in the Process of T Lymphocyte Differentiation: Proliferative, Metabolic, and Oxidative Changes. Front. Immunol. 9:339. doi: 10.3389/fimmu.2018.00339
Received: 06 November 2017; Accepted: 06 February 2018;
Published: 01 March 2018
Edited by:
Rafael Solana, Universidad de Córdoba, SpainReviewed by:
Sara Ferrando-Martinez, MedImmune, United StatesCopyright: © 2018 Moro-García, Mayo, Sainz and Alonso-Arias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rebeca Alonso-Arias, cmFsb25zb2FyaWFzQGhvdG1haWwuZXM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.