 Xiaoyong Chen
Xiaoyong Chen Shasha Liu2,3
Shasha Liu2,3 Mohsan Ullah Goraya
Mohsan Ullah Goraya Mohamed Maarouf
Mohamed Maarouf Shile Huang
Shile Huang Ji-Long Chen
Ji-Long Chen- 1Key Laboratory of Fujian-Taiwan Animal Pathogen Biology, College of Animal Sciences, Fujian Agriculture and Forestry University, Fuzhou, China
- 2CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences (CAS), Beijing, China
- 3University of Chinese Academy of Sciences, Beijing, China
- 4Department of Biochemistry and Molecular Biology, Louisiana State University Health Sciences Center, Shreveport, LA, United States
Influenza A viruses (IAVs) are contagious pathogens responsible for severe respiratory infection in humans and animals worldwide. Upon detection of IAV infection, host immune system aims to defend against and clear the viral infection. Innate immune system is comprised of physical barriers (mucus and collectins), various phagocytic cells, group of cytokines, interferons (IFNs), and IFN-stimulated genes, which provide first line of defense against IAV infection. The adaptive immunity is mediated by B cells and T cells, characterized with antigen-specific memory cells, capturing and neutralizing the pathogen. The humoral immune response functions through hemagglutinin-specific circulating antibodies to neutralize IAV. In addition, antibodies can bind to the surface of infected cells and induce antibody-dependent cell-mediated cytotoxicity or complement activation. Although there are neutralizing antibodies against the virus, cellular immunity also plays a crucial role in the fight against IAVs. On the other hand, IAVs have developed multiple strategies to escape from host immune surveillance for successful replication. In this review, we discuss how immune system, especially innate immune system and critical molecules are involved in the antiviral defense against IAVs. In addition, we highlight how IAVs antagonize different immune responses to achieve a successful infection.
Introduction
Influenza viruses belong to the Orthomyxoviridae family, which is characterized by a segmented, negative sense, and single-stranded RNA (ssRNA) genome (1). They are categorized into four genera (type A, B, C, and D), among which influenza A virus (IAV) can infect a wide spectrum of animal species (2, 3). The IAV genome is about 13,500 bases long and composed of eight ribonucleoprotein (RNP) units that encode at least 17 distinct proteins, including recently identified NS3, M42, PA-N182, and PA-N155 (4–6). IAVs can be further classified on the basis of the molecular structure and genetic characteristics of hemagglutinin (HA) and neuraminidase (NA) proteins. To date, 16 HA subtypes and 9 NA subtypes have been identified to be circulating in animals and humans (7). Besides, two HA- and NA-like subtypes designating IAV-like viruses (H17N10 and H18N11) have recently been discovered in bats (8). Co-infection with multiple virus strains in individuals can result in re-assortment (antigenic shift) of genes to produce novel subtypes that could give rise to a global influenza outbreak (9). There are approximately 5 million clinical infection cases caused by influenza viruses every year and 250,000–500,000 deaths resulted from the annual epidemics around the globe, particularly in people over 65 years old who account for 90% of all influenza-associated deaths in the USA (10). Moreover, in March 2013, residents in China exhibiting signs of respiratory infection were first reported to be infected by a novel re-assorted IAV of avian-origin, which was isolated from infected patients and identified as H7N9 (11).
There are several important IAV-encoded proteins that have been reported to be associated with the virus pathogenesis and host immune response to the viral infection. It has been revealed that changes at amino acid level in the viral proteins are related to increased disease severity and immune evasion in humans or avian caused by IAVs. For example, HA is the most abundant surface glycoprotein of the virus that has the ability to attach the host cell, causing cellular fusion and viral entry (12). In addition, HA contains epitopes which are key to trigger the production of neutralizing antibodies by B cells. Thus, the epitopes of HA are the dominant determinants that affect viral mutation and recombination mechanisms (13). The high variability of HA allows IAVs to escape from host immune surveillance and results in influenza seasonal epidemics. NA, the second most abundant glycoprotein, cleaves sialic acid (SA) moieties, promotes the release of nascent virions, and facilitates IAV dispersion (14). NA also plays crucial roles in the viral infection and HA-mediated membrane fusion by binding to SA receptors (15, 16). Respiratory epithelial cells constitutively expressed mucin glycoproteins at their surface that include MUC5AC, MUC5B, and MUC1, which play an important role in restricting IAV infection (17–19). For example, these mucin glycoproteins are rich in SA, which act as viral receptor decoys and prevent the viral binding to the target cells (19–21). However, NA can degrade these mucins and thus attenuate their action (21, 22). Moreover, it is shown that amino acid residues 147 and 151 in NA protein are critical for its interaction with SA. For example, D151G mutation in NA is responsible for its binding to avian α2-3 and human α2-6 SA, promoting the association of H3N2 virus with SA receptors. However, this mutation decreases the enzymatic activity of NA needed to detach the HA from its receptors (22, 23).
Nonstructural protein-1 (NS1), a multiple function protein, contains two functional domains (N-terminal RNA-binding domain and C-terminal effector domain). NS1 is a major inhibitor of host innate immune response. For example, it suppresses the production and signaling of type I interferons (IFNs) (24). In addition, NS1 can trigger the apoptosis of human airway epithelial cells via a caspase-dependent mechanism during the IAV infection (25). Matrix protein 2 (M2) forms tetrameric proton channels that are responsible to maintain pH across the viral envelope following the viral endocytosis, and help to release the uncoated viral RNP into the cytoplasm and nuclear import to start viral replication. M2 helps to hold the optimum high pH of the trans-Golgi network for HA-induced fusion and prevents premature conformational changes of HA (26). PB1-F2 is encoded by the alternate open reading frame of PB1 gene of IAV. PB1-F2 with N66S mutation binds with mitochondrial antiviral signaling protein (MAVS) and inhibits the initiation of IFNs. The 1918 deadly influenza strain H1N1 and H5N1 with N66S mutation increase the production of pro-inflammatory cytokines and enhance viral replication in the lung (27, 28).
During infection, host innate immunity provides the first line of defense and triggers pro-inflammatory responses (29). Adaptive immunity also plays a critical role in the clearance of viral pathogens during the later stages of infection. Additionally, respiratory mucosal immunity is induced in the related mucosal tissues during the IAV infection and involved in antiviral defense. In spite of several immune mechanisms to neutralize invasive pathogens or restrict viral replication, IAVs still have evolved diverse strategies to evade host immunity and can establish successful infection. Here, we review how host immune system responds to IAV infection and how IAVs evade the host immune surveillance.
Innate Immune Response to the IAV Infection
IAVs Target and Enter Host Cells
Influenza A viruses primarily target and infect airway and alveolar epithelial cells, which contain the SA glycans as receptors, thus causing alveolar epithelial injury and eventually failure of gas exchange (30, 31). Hence, human IAV infection may lead to acute respiratory distress syndrome (ARDS) and even death (32, 33). Various subtypes of IAVs have different abilities to attach human upper respiratory tract (URT). For example, H1N1 adsorbs abundant ciliated epithelial cells and goblet cells, whereas H5N1 hardly attaches to these cells in human URT (30). In contrast, H5N1 infects alveolar macrophages as well as alveolar epithelial cells (34, 35). Additionally, human and avian IAVs could target and infect various cells in the lower respiratory tract (LRT). It has been observed that H1N1 and H3N2 attach more abundantly to human trachea and bronchi and adsorb more cell types than H5N1 (36). Of note, low pathogenic (LP) avian IAVs generally do not cause a severe pneumonia because they bind human submucosal gland cells and their mucus which can restrain and remove these viruses before approaching LRT (37). However, high pathogenic (HP) H5N1 is able to infect type II pneumocytes as these cells possess an active metabolism, therefore providing a possibility to develop severe pneumonia (35). Moreover, it has been shown that H5N1 reduces proliferation of infected endothelial cells and causes excessive production of cytokines, leading to the lung damage (38, 39).
Hemagglutinin protein on the viral envelope can recognize SA receptors on the surface of the host cells, which is the most crucial step in the process of IAV invasion into an organism (40, 41). Influenza viruses have two common cellular receptors: SA α-2,3 galactose (SAα-2,3-Gal) and SA α-2,6 galactose (SAα-2,6-Gal) (42). It has been found that human influenza strains preferentially attach to the SAα-2,6-Gal receptor (43). The 2,6 SA receptors are present on respiratory epithelial cells of the human URT, while 2,3 SA receptors are found on epithelial cells of the birds, pigs, and in the LRT of humans (44). Thus, variation of cell surface receptors contributes as a major barrier to cross-species and zoonotic transmissions of influenza virus. Hydrolysis of HA0 precursor gives rise to a dimer HA1–HA2 linked by disulfide bonds. HA1 binds to cellular receptors and HA2 facilitates the fusion of the virus to cellular membrane (45). It is also found that HA of influenza virus binds to C-type lectin as an alternative of SA (46). Virus attachment to host cell induces endocytosis using the clatherin and clavoline-dependent mechanism. Following the endocytosis, release of viral particle is pH-dependent physiological event that occurs at late lysosome (47). Low pH of endosome opens the M2 proton channel for proton flux into the virus and triggers the uncoating and subsequent release of viral RNPs (48). The free viral RNA is imported to the nucleus by interacting with the cellular importin-α/β for replication (49, 50). Although it is recognized that both virus preference for SA receptor and host restriction factors are critical for IAV binding and entry into host cells, further studies are needed to unravel the complicated mechanisms underlying the interaction of IAVs with human airway cells.
Activation of Innate Immune Signaling upon Intracellular Detection of IAV Infection
The innate immune response is the first line of defense against viral infection which is rapid in response, but nonspecific. During the IAV infection, viral conserved components called pathogen associated molecular patterns (PAMPs) are recognized by host pathogen recognition receptors (PRRs), such as retinoic acid-inducible gene-I protein (RIG-I) and toll-like receptor (TLR), leading to activation of innate immune signaling that finally induces the production of various cytokines and antiviral molecules (51, 52). These PAMPs have certain characteristic of viral RNA that are not shared by cellular RNAs, such as regions of double-stranded RNA (dsRNA) or the presence of a 5’-triphosphate group (53, 54).
Pathogen recognition receptors have the ability to distinguish self from non-self molecules within the infected cells. RIG-I is the main receptor to recognize the intracellular ssRNA and transcriptional intermediates of IAVs in the infected host cells (Figure 1). Non-self RNA and transcriptional products of IAVs in the cytoplasm are also sensed by melanoma differentiation-associated gene 5 (55). Following the recognition of PAMPs, RIG-I is activated and its caspase activation and recruitment domains (CARDs) are exposed. Then the CARD is modulated by dephosphorylation or ubiquitination by E3 ligases, such as TRIM-containing protein 25 (TRIM25) (56). Thus, CARD-dependent association of RIG-I and MAVS trigger the downstream transduction signaling at the outer mitochondrial membrane (57). Subsequently, the transcription factors, including interferon regulatory factor 3 (IRF3) and IRF7, and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) are activated, causing the expression of a variety of IFNs and cytokines (Figure 1) (58).
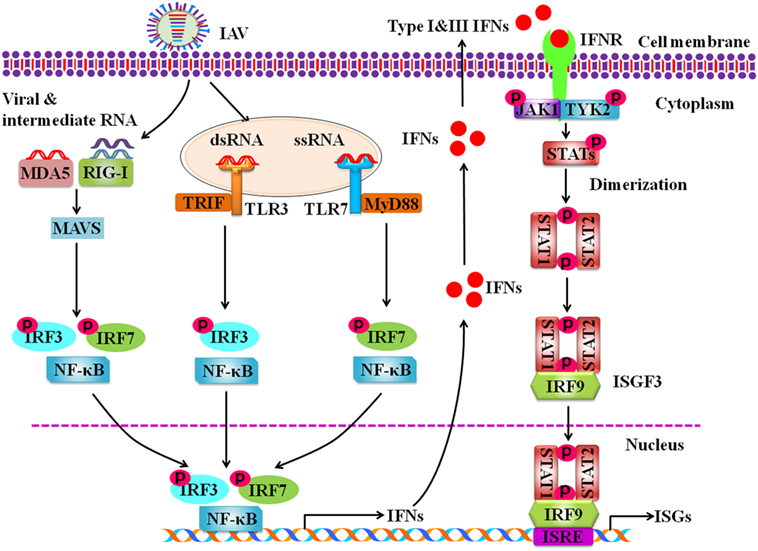
Figure 1. Schematic diagram for innate immune response against influenza A virus (IAV) infection. Intracellular detection of IAV infection by the host pathogen recognition receptors (PRRs) activates transcription factors nuclear factor kappa-light-chain-enhancer of activated B cells, interferon regulatory factor 3 (IRF3), and IRF7. The PRRs include retinoic acid-inducible gene-I protein, melanoma differentiation-associated gene 5, and toll-like receptors. Then these activated transcription factors trigger the expression of type I and type III interferons (IFNs). IFNs secreted by the infected cells interact with their receptors, which results in activation of Janus kinase-signal transducer and activator of transcription signaling pathway that governs the expression of various IFN-stimulated genes.
Toll-like receptors are critical PRRs that sense the pathogen outside of cell membrane and internally at endosomes and lysosomes (59). TLRs expressed on cell membrane are TLR1, 2, 4, 5, and 6 that recognize PAMPs derived from bacteria, fungi, and protozoa, while TLR3, 7, 8, and 9 are expressed on the surface of endosomes and lysosomes and exclusively recognize nucleic acid PAMPs derived from various viruses, including IAVs (60–62). TLR3, TLR7, and TLR8 are involved in sensing the IAV components in cytoplasmic endosomes during the virus replication. It is known that TLR3 recognizes dsRNA in endosomes (63). Interestingly, it was shown that TLR3 may recognize recently unidentified RNA structures that are present in phagocytosed cells infected with IAVs (64). In plasmacytoid dendritic cells (pDCs), TLR7 recognizes the ssRNA of the influenza virions that are taken up into the endosomes (65). Then downstream signaling of TLR7 is activated via the adaptor protein myeloid differentiation factor 88 in pDCs, which results in the activation of either NF-κB or IRF7 to induce the expression of pro-inflammatory cytokines and type I IFNs, respectively (65). In macrophages and DCs, TLR3 interacts with TIR-domain-containing adapter-inducing interferon-β. Such interaction results in activation of the serine–threonine kinases IκKε (IKKε) and TBK1 that phosphorylate IRF3 to regulate the expression of IFN-β (66). In human monocytes and macrophages, TLR8 is stimulated by its ligand ssRNA, leading to the production of IL-12. However, the relationship between TLR8 and IAV infection has not been defined (67).
Moreover, some NOD-like receptors, such as NOD-like receptor family pyrin domain containing 3 (NLRP3, also known as cryopyrin) and NLR apoptosis inhibitory protein 5, have been observed to be activated upon cellular infection with IAV (68). NLRP3 is expressed by number of cells, including DCs, macrophages, neutrophils, monocytes, and human pulmonary epithelial cells (69, 70). Three signals are required for activation of inflamosome to trigger cytokine production. First, NLRP3 is activated through pathogen detection, which induces the expression of the genes encoding pro-IL-1β, pro-IL-18, and pro-caspase-1 (71). Second, the IAV-encoded M2 ion channel is required to trigger NLRP3 inflammasome activation and cleavage of pro-IL-1β and pro-IL-18 (72). Recently, it was revealed that accumulation of IAV PB1-F2 in the lysosome of macrophages acted as the third signal leading to activation of the NLRP3 inflammasome (73).
Antiviral Molecules Involved in Innate Immunity against IAV Infection
Activation of specific transcription factors including, NF-κB, IRF3, and IRF7 during IAV infection results in translocation of these factors into the nucleus where they initiate the transcription of genes encoding IFNs and pro-inflammatory cytokines (TNF, IL6, IL1β, etc.). It is well known that type I IFNs, such as IFN-α and IFN-β, and type III IFNs also known as interferon lambdas (IFN-λ1, IFN-λ2, IFN-λ3, IFN-λ4) play important roles in antiviral response in both virus-infected and uninfected cells (74). Infection with IAV induces robust expression of type I and type III IFN genes (75). Following the expression, IFN-α and IFN-β interact with IFN-α/β receptors (IFNAR), while IFN-λs interact with IFNL receptors (IFNLR) in an autocrine or paracrine manner, which activate Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling pathway. Phosphorylated STAT1 and STAT2 bind with IRF9 to form a complex ISG factor 3 (ISGF3). ISGF3 translocates into nucleus and binds with IFN-stimulated response element, which triggers the transcription of numerous IFN-stimulated genes (ISGs) (Figure 1) (76). Previous studies suggest that type I and type III IFNs provide similar defense against IAV infection in wild-type mice (77). It was further shown that IAV infection induced expression of same type of ISGs in epithelial cells of wild type, IFNAR- or IFNLR-deficient mice (78). Only when both IFN-α/β and IFN-λ receptors in mice were knocked out, the animals failed to restrict non-pathogenic influenza virus (77). In spite of similar role of IFNα/β and IFN-λs to a certain extent, some notable differences exist. For example, it has been observed that mice infected with influenza virus showed higher pulmonary inflammation and mortality after treatment with IFNα, while IFN-λ remained protective (79, 80).
These ISGs target different steps of IAV life cycle. For example, viral entry into cells can be restricted by several ISGs, including Mx family, interferon-induced transmembrane protein family (IFITMs), cholesterol 25-hydroxylase (CH25H), and TRIM proteins. Mx family is comprised of MxA and MxB in human, Mx1 and Mx2 in mice. Mx proteins are produced by various cells, such as hepatocytes, DCs, endothelial cells, and immune cells (81). Mx proteins were the first ISGs identified to restrict IAV infection of mice (82). Recently, a study showed that MxA could retain incoming viral genome in the cytoplasm of human cell (83). In addition, nuclear Mx1 in mice impedes the process of early transcription of IAV activated by the polymerase in the nucleus (84). It is thought that the sensitivity of IAVs to MxA depends on their nucleocapsid proteins, and usually avian strains of IAVs are more sensitive to MxA than human strains (85, 86). However, role of MxB in humans or Mx2 in mice during IAV infection is poorly understood (87).
Interferon-induced transmembrane protein families are known as new ISGs that restrict early viral entry by altering the cellular membrane properties like cell adhesion, fluidity, and spontaneous curvatures (88, 89). It has been found that IFITM proteins restrict the replication of IAVs by interfering virus–host cell fusion following viral attachment and endocytosis (90). Another antiviral ISG, CH25H is an integral element of cellular membranes and upregulated by IFN signaling. CH25H enzymatic activity converts cholesterol to soluble 25-hydroxycholesterol (25HC), which is involved in antiviral defense against enveloped viruses, including influenza virus through blocking viral fusion. Recently, it was suggested that high concentration of 25HC causes physical changes of cellular membrane properties to prevent viral fusion (91). Moreover, previous studies showed that IFN-activated STAT1 bound to the promoter proximal region of the Ch25h gene to stimulate the production of 25HC that enhanced innate immune response against IAV (92, 93). In addition, TRIM proteins play multiple roles in antiviral immunity. TRIM25, an E3 ubiquitin ligase, is considered to regulate the re-localization of RIG-I to mitochondrion and signal transduction to MAVS for innate immune response against the viral infection (94). TRIM22 blocks IAV genome encapsidation and degrades nucleoprotein of IAV by polyubiquitination (95). TRIM32 binds with influenza PB1 RNA polymerase, reduces the polymerase activity, and thus restricts the viral replication (96).
There are increasing number of ISGs that regulate viral mRNA expression and protein translation. For example, zinc finger antiviral protein (ZAP), oligoadenylate synthase and ribonuclease L (OAS-RNase L), PKR, and ISG15 are involved in the regulation of IAV mRNA levels and protein synthesis. ZAPs inhibit the expression of IAV PB2 and PA proteins by reducing the viral mRNA expression and blocking its translation (97, 98). OAS-RNase L can destroy viral RNA in the cytosol of host cells and finally halt the protein synthesis process and viral replication. Mice with reduced expression of RNase L are more prone to influenza virus infection (99). PKR is expressed by all kind of cells and upregulated by type I and type III IFNs (100). PKR expressed in inactive form is activated by influenza virus infection. PKR is a known anti-IAV factor that binds to viral dsRNA and suppresses viral protein synthesis. Genetically deficient PKR mice are highly susceptible to influenza virus (101). ISG15 is an ubiquitin-like protein and restrict viral replication by interfering with virus release and translation of viral proteins (102).
In addition, many other ISGs are also involved in innate immunity against IAV infection. These include viperin, tetherin, and so on (103, 104). It has been shown that overexpression of viperin (also known as RSAD2) restricts the release of influenza virus by affecting the formation of lipid rafts specific microdomains that are particular budding sites of the virus (105). Tetherin is another potential host antiviral factor. In 2008, it was reported that tetherin inhibited retrovirus release (106). Tetherin appeared to limit cellular export of viral progenies by internalizing and degrading them exported to the surface of infected cells (107). It was also found that tetherin restrained the influenza virus by tethering and degrading newly budded viral particles (108). Recently, it has been known that tetherin was able to suppress the budding of several laboratory oriented and seasonal influenza strains, but unable to restrict pandemic influenza A/Hamburg/4/2009 and wild-type influenza virus particles (103, 104, 108).
Cells Involved in Innate Immunity against the IAV Infection
Airway epithelial cells are the first target of IAVs. These cells produce antiviral and chemotactic molecules that initiate immune responses by rapid recruitment of innate effector cells, such as NK cells, monocytes, and neutrophils. All cell types have their own unique mechanisms to interact with virus-infected cells to limit viral replication, and also prime adaptive immune cells for antigen-specific immunity and memory. Tumor necrosis factor alpha (TNF-α) and IL-1 induce endothelial adhesion molecules, which trigger the migration of innate immune cells, such as macrophages, blood borne DCs, and natural killer (NK) cells to the site of infection.
Alveolar macrophages are critical for limiting viral spread. Activated macrophages phagocytose IAV-infected cells and thus limit viral spread and regulate the following adaptive immune response (109). Monocytes derived from bone marrow precursors circulate in bloodstream. During IAV infection, MCP-1 (CCL-2) produced by infected epithelial cells attracts alveolar macrophages and monocytes via their CCR2 receptors (110). NK cells are important cytotoxic lymphocytes of innate immune system to eliminate IAV infection. It has been revealed that lysis of IAV-infected cells is mediated by the binding of IAV-HA with the cytotoxicity NKp44 and NKp46 receptors (111). Expression of IAV-HA on the surface of infected cells is recognition signal for NK cells, and thereby NK cells target and lyse the infected cells (112, 113).
Dendritic cells, the specialized antigen-presenting cells, bridge up the innate and adaptive immune responses during the IAV infection. Adaptive immune response begins when naïve and memory T lymphocytes recognize viral antigens presented by DCs. In the naïve steady state, DCs are orchestrated underneath the respiratory tract, including the airway epithelial tissue, lung parenchyma, and the alveolar spaces of the lungs (114, 115), where they constantly monitor for invading pathogens by their dendrites that are extended to airway lumen through the tight junctions of epithelial cells. Upon infection with IAV, the conventional DCs (cDCs) migrate from lungs to lymph nodes through interaction between CCR7 and its ligand CCL19 and CCL21 (116). In the lymph nodes, cDCs present antigens derived from IAV to T lymphocytes (117, 118). The self-infected DCs degrade the viral protein into immune peptides. Immune peptides (epitopes) in the cytosol are exported to the endoplasmatic reticulum, where they bind with major histocompatibility complex (MHC) class I molecule. Following the binding with epitopes, MHC class I is transported to the cell membrane via the Golgi complex for recognition by virus-specific CD8+ cytotoxic T cells (CTL). However, viral proteins degraded in endosomes/lysosomes are associated with MHC class II molecule. These complexes are presented on the cell membrane for recognition by CD4+ T helper (Th) cells. This process may lead to B cell proliferation and maturation to antibody producing plasma cells (119). In addition, DCs can exert cytolytic activity and contribute to the formation of bronchus-associated lymphoid tissue (BALT) during the IAV infection (120).
Adaptive Immunity Against the IAV Infection
T cells and B cells play key roles in adaptive immunity against the IAV infection. T cells are mainly known as CD4+ T and CD8+ T cells. CD8+ T cells differentiate into cytotoxic T lymphocytes (CTLs), which produce cytokines and effector molecules to restrict viral replication and kill virus-infected cells. Therefore, T cells are crucial for the restriction of viral infection. Upon infection with IAV, naïve CD8+ T cells are activated by DCs migrated from lungs to T-cell zone of the draining lymph nodes, leading to T-cell proliferation and differentiation into CTLs (121, 122). Moreover, type I IFNs, IFN-γ, IL-2, and IL-12 also help CD8+ T cells to differentiate into CTLs (123, 124). IFN-λs were also shown to enhance the T-cell proliferation during influenza virus vaccination (125). CTLs decrease the expression of CCR7 and upregulate the expression of CXCR3 and CCR4, which enables their migration from lymph nodes to the lungs where they kill IAV-infected cells.
Mechanism by which CTLs function is well understood. Upon targeting the virus-infected cells, CTLs produce cytotoxic granules that contain molecules like perforin and granzymes (e.g., GrA and GrB). Perforin binds target cells to form pores on the cell membrane that promote passive diffusion of granzymes to induce apoptosis. It has also been found that GrA can restrict virus replication via cleavage of viral and host cell proteins that are involved in protein synthesis (126, 127). In addition, CTLs have the ability to induce apoptosis by expressing cytokines, such as TNF, FASL, and TRAIL, which recruit death receptors in IAV-infected cells (128). Post-infection virus-specific CTLs and DCs circulate in blood, lymphoid organs, and the site of infection (117, 129). These memory CTL cells are quick in response to secondary IAV infection, and the activation and differentiation process received during first infection affects their proficiency and efficiency during a secondary infection (130). Although neutralizing antibodies protected from second infection with the same serotype of IAV, CTLs are specific for epitopes in conserved IAV proteins, such as NP, M1, and PA. Therefore, the CTL response is heterosubtypic in nature (131).
Studies have shown that IAV-specific CD8+ T cells can last for 2 years in murine models (132). The cytotoxicity of the memory CD8+ T cells decreases significantly, which is related to their declined target competence and reduced cytolytic molecule expression (131). Autophagy plays a critical role in the establishment of memory CD8+ T cells, as Atg7-deficient mice are unable to form CD8+ T cell memory against IAV infection (133). Notably, IAV-specific memory CD8+ T cells in the nasal epithelia prevent the spread of the virus from the URT to the lung, thus blocking the development of pulmonary disease (134). Besides, lung-resident memory CD8+ T cells can defend against heterologous IAV infection, via restraining viral replication and facilitating viral elimination (135). Additionally, lung-resident monocytes support to establish lung-resident CD8+ T cell during IAV infection (136).
CD4+ T cell is another important type of immune cells that is involved in adaptive immunity against the IAV infection. CD4+ T cells can also target IAV-infected epithelial cells through MHC class II and induce MHC class II expression in epithelial cells in murine models (137, 138). Multiple co-stimulatory ligands expressed by CD4+ T cells contribute to B cell activation and antibody production, among which CD40 ligand (CD40L) is noteworthy (139). CD40L has been shown to enhance immune response against the highly mutated HA protein of IAV (140). Similar to CD8+ T cells, CD4+ T cells are activated by DCs that migrate from the lung to the draining lymph nodes during the IAV infection (141, 142). CD4+ T cells differentiate into Th1 cells in response to IAV infection, according to their stimulators, including antigen, co-stimulatory molecules, and cytokines secreted by DCs, epithelial cells, and inflammatory cells (143, 144). Th1 effector CD4+ T cells express antiviral cytokine, such as IFN-γ, TNF, and IL-2 (145), and activate alveolar macrophages (146). The IL-2 and IFN-γ produced by Th1 cells regulate CD8+ T-cell differentiation to clear the viral infection (147, 148). CD4+ T cells are also able to differentiate into Th2, Th17, regulatory T cells (Treg cells), follicular helper T cells, and sometimes as killer cells (149). Th2 cells bind to virus-derived MHC class II-associated peptides by antigen-presenting cells and produce IL-4 and IL-13 to promote B cell responses predominantly (150). It has been observed that Th17 and Treg cells are involved in regulating cellular immunity against IAV infection (151). Although it is known that CD4+ T cells can direct CD8+ T cell responses by secreting various cytokines, the precise roles of CD4+ T cells to facilitate and regulate CD8+ T cell responses to IAV infection remain elusive, because primary CD8+ T cell response against IAV infection could be initiated independently of CD4+ T cells in mice (152).
B cells are indispensable for priming the defense against infection with heterosubtypic influenza virus strains. In cooperation with memory T cells, naïve B cells reduce morbidity and promote recovery upon heterosubtypic infection (153). At the same time, non-neutralizing antibodies generated by B cells facilitate viral elimination and accelerate memory CD8+ T cell expansion after heterosubtypic infection (153). In addition, IAV-specific antibody-dependent cell-mediated cytotoxicity (ADCC) also plays a role in the cross-reaction against diverse HA subtypes (154). Though IgA is important in the protection against IAV infection in the respiratory tract, IgG is the dominant antibody in this process. Additionally, some studies have implied that IgG could inhibit pathogenesis involving influenza, while IgA is more crucial for the inhibition of transmission of IAVs (155).
Till now, the lifespan and response speed of both memory B cells and plasma cells are foremost in the induction of protective antibody response by IAV vaccines. However, in the elderly, the memory B cells are maintained, but the antibody response is not maintained even upon multiple IAV immunizations. This suggests a potential defect with aging in the development of plasma cells (156). A study has shown that autophagy is involved in maintaining memory B cells to counteract IAV infection; Atg7-deficient mice exhibits loss of memory B cells, causing reduced secondary antibody response to IAV infection and displaying severe lung damage (157).
Respiratory Mucosal Immunity Against the IAV Infection
Lymphoid Tissues and Immunoglobins in the Respiratory Tract Involved in Immunity against the IAV Infection
The nasal openings and URT are the main entry sites for IAVs and mucosal immune system also acts as the first line to limit the IAV infection apart from innate immunity. Secretory IgA (s-IgA) and IgM are the major neutralizing antibodies present on mucosa to prevent viral entry. Nasal secretions contain IgA which can neutralize HA and NA of IAVs (129, 158). During primary infection with IAVs, all three major immunoglobulin classes (IgG, IgA, and IgM) are present in mucosal secretion to limit the infection, though IgA and IgM are higher in concentration than IgG (159). It is thought that IgM response is dominant during primary infection, whereas during secondary infection IgG response is dominant for immunoglobin secretion (2, 119, 121). In the URT, mucosal response is induced in the nasopharyngeal-associated lymphoid tissues (NALT) (160–162). When antigens are pinocytosed or phagocytosed by macrophages present on the NALT, they interact with local T and B cells, resulting in development of a large number of IgA Ab-forming cell (IgA-AFC) precursors (163, 164). The primed T and B cells migrate from NALT to the lungs via general circulation, where they differentiate into specific IgA-AFC to secrete antiviral antibodies. Thus, NALT appears to be initial inductive site for secretion of s-IgA against IAV infection. In the LRT, mucosal immune responses occur in the BALT (165). BALT is the site for AFC development and production of mucosal s-IgA against IAV infection (166).
Role of s-IgA Antibody in Defense against the IAV Infection
Secretory IgA is the primary isotype detected at the mucosal surface (167), which contributes to mucosal protection through its distinct ability to remove an agent before it traverses the mucosal barrier and infects the cell (168). By covering the viral surface, s-IgA prevents the influenza virions from adhering to the susceptible cells, and thus inhibits their invading host cells and neutralizes the viruses without complement participation. Investigations have been demonstrated that s-IgA plays vital roles both in protection against homologous IAV infection and in cross-protection against URT infection by the viral variants (169). Generally, parenteral administration of IAV vaccine leads to the generation of serum IgG, but not s-IgA, while s-IgA and IgG are both induced by intranasal administration (168, 170). Further, polymeric s-IgA is involved in defending against influenza in humans. Moreover, the quaternary structure of the polymeric s-IgA seems to play a key role in protecting human URT from influenza, and have more neutralizing capacity against IAVs than dimeric s-IgA (171).
Escape of IAVs from Host Immune Surveillance
To establish a successful infection, IAVs have evolved multiple strategies to circumvent the host immunity. For example, it is well known that IAV infection triggers robust production of IFNs that induce the expression of numerous antiviral molecules or ISGs. Although IFNs have a strong antiviral activity, they cannot fully control IAV infection due to the virus-mediated suppression of IFNs signaling. The mechanisms by which IAVs escape from host antiviral immune responses are discussed here.
The Antagonism of Major IAV Proteins
Hemagglutinin of IAVs has been shown to facilitate IFNAR ubiquitination and degradation, reducing the levels of IFNAR, and thus suppressing the expression of IFN-stimulated antiviral proteins (172). It has been described that two discrete antigenic sites, H9-A and H9-B, may provide a novel mechanism for H9N2 virus to counteract humoral immunity (173). In addition, a study has shown that the escape of H5N1 from vaccine-mediated immunity is caused by the addition of N-glycosylation sites on the globular head of HA (174). In contrast, antibody response against NA of IAV cannot inhibit viral infection, but restrain its diffusion, thus lowering the severity of influenza. IAVs employ NA protein to block the recognition of HA by natural cytotoxicity receptors, NKp46, and NKp44 receptors and evade the NKp46-mediated elimination, leading to minimized clearance of infected cells by NK cells (175).
Nonstructural protein-1 of IAVs is the most important IFNs antagonist protein, acting on multiple targets and suppressing the host IFN response. Viral RNA invading the host cell causes RIG-I ubiquitination by a RING-finger E3 ubiquitin ligase named as TRIM25, which is essential for RIG-I signaling pathway to trigger host antiviral innate immunity (94, 176). However, NS1 protein can inhibit the TRIM25-mediated RIG-I ubiquitination, thereby blocking RIG-I activation (177). Moreover, NS1 has an inhibitory effect on protein kinase RNA-activated (also known as protein kinase R, PKR), but the effect relies on the induced expression of vault RNAs (a kind of small non-coding RNA with approximately 100 bases). They are initially described as fornix RNP complex components (178). Through NS1 protein, influenza virus induces the expression of vault RNA that inhibits the activation of PKR and the production of IFNs and ultimately promotes the replication of the virus. In a recent reverse genetic investigation, it was found that after interfering with NS1, the phosphorylation level of PKR dramatically increased, which was attenuated by forced expression of vault RNAs (179). These data indicate that IAV has evolved a critical mechanism by which NS1-mediated PKR inhibition is mediated by upregulation of the host factor vault RNAs that inactivates PKR and blocks the production of downstream effector molecules of IFNs.
In addition, studies have shown that through the interaction with IκB kinases (IKK) α and β, two important kinases in NF-κB pathway, NS1 protein can block the phosphorylation of these kinases and eventually destroy the NF-κB complex predominating in nucleus as well as the expression of downstream genes (180, 181). Also, through the JAK-STAT pathway, NS1 protein can block IFN-mediated downstream signaling pathway and weaken the antiviral effect mediated by the downstream effector molecules induced by IFNs. Specifically, NS1 acts mainly by lowering the phosphorylation levels of STAT1, STAT2, and STAT3, preventing STAT2 from entering into the nucleus to bind to the DNA sequence of ISGs promoter region, leading to reduced expression of ISGs (182). Importantly, NS1 is not only involved in host innate immunity, but also affects adaptive immunity via modulating the maturation and the capacity of DCs to induce T cell responses (183). Evidence also indicates that influenza virus NS1 can bind to cellular double-stranded DNA (dsDNA), counteract the recruitment of RNA polymerase II (Pol II) to DNA, and finally block the transcription of IFNs and ISGs (184).
The Antagonism of Other IAV Proteins
Studies have found that PB1-F2 protein has a mitochondrial positioning signal, via interacting with MAVS, to counteract RLR-mediated activation of IFN signaling pathway (185). Investigation on the interaction between the virus and host by systematic biology analysis has revealed that PB2 protein, a member of the viral polymerase complex, also plays roles in IFN antagonism (186). Furthermore, PB2 interacts with the MAVS to evade from the host IFN antiviral response, which is similar to the action mode of PB1-F2 protein (187). Recently, viral M2 protein has been found to interfere with the host autophagy (188, 189). These studies have suggested that viral M2 may inhibit the activation of TLR pathway and the generation of IFNs via blocking the host autophagy.
Conclusion
It is well known that host immune response to IAV infection comprises multiple intricate processes that coordinate together to play significant roles in the protection of host. Given the high mutation rate of IAVs, it is necessary to have effective vaccination strategies that can induce robust production of specific antibodies and long-lived T cell response to defend against the viral infection. Since host innate immunity is also critical for anti-IAV infection, further efforts are needed to utilize the current knowledge and technology to enhance the host innate immunity for control of the disease. While our understanding of the IAV-host interaction has increased profoundly, extensive studies are required to better understand the dynamics of host immune system upon detection of the evolved IAVs. Bridging these gaps will pave the way not only for designing better vaccines and effective vaccination strategies but also for developing novel antiviral agents.
Author Contributions
XC, SL, and MG performed systematic literature review and wrote the manuscript. MM and SH revised the manuscript. J-LC organized and provided the frame for the manuscript and critically revised the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer HZ and handling editor declared their shared affiliation.
Funding
This work was supported by National Key Research and Development Program of China (2016YFD0500206, 2016YFD0500205, and 2016YFC1200304), Natural Science Foundation of China (91640101), and National Key National Basic Research Program (973) of China (2015CB910502).
References
1. Nturibi E, Bhagwat AR, Coburn S, Myerburg MM, Lakdawala SS. Intracellular colocalization of influenza viral RNA and Rab11A is dependent upon microtubule filaments. J Virol (2017) 91(19):e1179–1117. doi:10.1128/jvi.01179-17
2. Cox RJ, Brokstad KA, Ogra P. Influenza virus: immunity and vaccination strategies. Comparison of the immune response to inactivated and live, attenuated influenza vaccines. Scand J Immunol (2004) 59(1):1–15. doi:10.1111/j.0300-9475.2004.01382.x
3. Zhai SL, Zhang H, Chen SN, Zhou X, Lin T, Liu R, et al. Influenza D virus in animal species in Guangdong Province, Southern China. Emerg Infect Dis (2017) 23(8):1392–6. doi:10.3201/eid2308.170059
4. Ghedin E, Sengamalay NA, Shumway M, Zaborsky J, Feldblyum T, Subbu V, et al. Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature (2005) 437(7062):1162–6. doi:10.1038/nature04239
5. Goraya MU, Wang S, Munir M, Chen JL. Induction of innate immunity and its perturbation by influenza viruses. Protein Cell (2015) 6(10):712–21. doi:10.1007/s13238-015-0191-z
6. Vasin AV, Temkina OA, Egorov VV, Klotchenko SA, Plotnikova MA, Kiselev OI. Molecular mechanisms enhancing the proteome of influenza A viruses: an overview of recently discovered proteins. Virus Res (2014) 185(7):53–63. doi:10.1016/j.virusres.2014.03.015
7. Medina RA, García-Sastre A. Influenza A viruses: new research developments. Nat Rev Microbiol (2011) 9(8):590–603. doi:10.1038/nrmicro2613
8. Ma W, Garcíasastre A, Schwemmle M. Expected and unexpected features of the newly discovered bat influenza A-like viruses. PLoS Pathog (2015) 11(6):e1004819. doi:10.1371/journal.ppat.1004819
9. Zeng LY, Yang J, Liu S. Investigational hemagglutinin-targeted influenza virus inhibitors. Expert Opin Investig Drugs (2017) 26(1):63–73. doi:10.1080/13543784.2017.1269170
10. Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, et al. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA (2003) 289(2):179. doi:10.1001/jama.289.2.179
11. Li J, Yu XF, Pu XY, Xie L, Sun YX, Xiao HX, et al. Environmental connections of novel avian-origin H7N9 influenza virus infection and virus adaptation to the human. Sci China Life Sci (2013) 56(6):485–92. doi:10.1007/s11427-013-4491-3
12. Zhang H, Hale BG, Xu K, Sun B. Viral and host factors required for avian H5N1 influenza A virus replication in mammalian cells. Viruses (2013) 5(6):1431–46. doi:10.3390/v5061431
13. Wang X, Qi S, Ye Z, Ying H, Na S, Du Y, et al. Computational approach for predicting the conserved B-cell epitopes of hemagglutinin H7 subtype influenza virus. Exp Ther Med (2016) 12(4):2439. doi:10.3892/etm.2016.3636
14. Wang S, Li H, Chen Y, Wei H, Gao GF, Liu H, et al. Transport of influenza virus neuraminidase (NA) to host cell surface is regulated by ARHGAP21 and Cdc42 proteins. J Biol Chem (2012) 287(13):9804–16. doi:10.1074/jbc.M111.312959
15. Lin YP, Gregory V, Collins P, Kloess J, Wharton S, Cattle N, et al. Neuraminidase receptor binding variants of human influenza A(H3N2) viruses resulting from substitution of aspartic acid 151 in the catalytic site: a role in virus attachment? J Virol (2010) 84(13):6769. doi:10.1128/JVI.00458-10
16. Zhu X, Mcbride R, Nycholat CM, Yu W, Paulson JC, Wilson IA. Influenza virus neuraminidases with reduced enzymatic activity that avidly bind sialic acid receptors. J Virol (2012) 86(24):13371. doi:10.1128/JVI.01426-12
17. Roy MG, Livraghibutrico A, Fletcher AA, Mcelwee MM, Evans SE, Boerner RM, et al. Muc5b is required for airway defence. Nature (2014) 505(7483):412–6. doi:10.1038/nature12807
18. Ehre C, Worthington EN, Liesman RM, Grubb BR, Barbier D, O’Neal WK, et al. Overexpressing mouse model demonstrates the protective role of Muc5ac in the lungs. Proc Natl Acad Sci U S A (2012) 109(41):16528–33. doi:10.1073/pnas.1206552109
19. Mcauley JL, Corcilius L, Tan HX, Payne RJ, Mcguckin MA, Brown LE. The cell surface mucin MUC1 limits the severity of influenza A virus infection. Mucosal Immunol (2017) 10(6):1581–93. doi:10.1038/mi.2017.16
20. Duez JM, Sixt N, Péchinot A. Influenza virus infection: don’t forget the role of the mucociliary system! J Antimicrob Chemother (2009) 63(2):421–2. doi:10.1093/jac/dkn468
21. Cohen M, Zhang XQ, Senaati HP, Chen HW, Varki NM, Schooley RT, et al. Influenza A penetrates host mucus by cleaving sialic acids with neuraminidase. Virol J (2013) 10(1):321. doi:10.1186/1743-422X-10-321
22. Yang J, Liu S, Du L, Jiang S. A new role of neuraminidase (NA) in the influenza virus life cycle: implication for developing NA inhibitors with novel mechanism of action. Rev Med Virol (2016) 26(4):242–50. doi:10.1002/rmv.1879
23. Gulati S, Smith DF, Cummings RD, Couch RB, Griesemer SB, St GK, et al. Human H3N2 influenza viruses isolated from 1968 to 2012 show varying preference for receptor substructures with no apparent consequences for disease or spread. PLoS One (2013) 8(6):e66325. doi:10.1371/journal.pone.0066325
24. Qian W, Wei X, Guo K, Li Y, Lin X, Zou Z, et al. The C-terminal effector domain of non-structural protein 1 of influenza A virus blocks IFN-β production by targeting TNF receptor-associated factor 3. Front Immunol (2017) 8:779. doi:10.3389/fimmu.2017.00779
25. Lam WY, Tang JW, Yeung AC, Chiu LC, Sung JJ, Chan PK. Avian influenza virus A/HK/483/97(H5N1) NS1 protein induces apoptosis in human airway epithelial cells. J Virol (2008) 82(6):2741. doi:10.1128/JVI.01712-07
26. Sugrue RJ, Hay AJ. Structural characteristics of the M2 protein of influenza A viruses: evidence that it forms a tetrameric channel. Virology (1991) 180(2):617. doi:10.1016/0042-6822(91)90075-M
27. Fukuyama S, Kawaoka Y. The pathogenesis of influenza virus infections: the contributions of virus and host factors. Curr Opin Immunol (2011) 23(4):481–6. doi:10.1016/j.coi.2011.07.016
28. Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog (2007) 3(10):e141. doi:10.1371/journal.ppat.0030141
29. Wei H, Wang S, Chen Q, Chen Y, Chi X, Zhang L, et al. Suppression of interferon lambda signaling by SOCS-1 results in their excessive production during influenza virus infection. PLoS Pathog (2014) 10(1):e1003845. doi:10.1371/journal.ppat.1003845
30. Van RD, den Bakker MA, Leijten LM, Chutinimitkul S, Munster VJ, de Wit E, et al. Seasonal and pandemic human influenza viruses attach better to human upper respiratory tract epithelium than avian influenza viruses. Am J Pathol (2010) 176(4):1614–8. doi:10.2353/ajpath.2010.090949
31. Shinya K, Ebina M, Yamada S, Ono M, Kasai N, Kawaoka Y. Avian flu: influenza virus receptors in the human airway. Nature (2006) 4(4):321–6. doi:10.1038/440435a
32. Herold S, Becker C, Ridge KM, Budinger GR. Influenza virus-induced lung injury: pathogenesis and implications for treatment. Eur Respir J (2015) 45(5):1463. doi:10.1183/09031936.00186214
33. Hogan BL, Barkauskas CE, Chapman HA, Epstein JA, Jain R, Hsia CC, et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell (2014) 15(2):123. doi:10.1016/j.stem.2014.07.012
34. Nicholls JM, Chan MCW, Chan WY, Wong HK, Cheung CY, Kwong DLW, et al. Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nat Med (2007) 13(2):147–9. doi:10.1038/nm1529
35. Van RD, Munster VJ, De WE, Rimmelzwaan GF, Fouchier RA, Osterhaus AD, et al. H5N1 virus attachment to lower respiratory tract. Science (2006) 312(5772):399. doi:10.1126/science.1125548
36. Van RD, Munster VJ, De WE, Rimmelzwaan GF, Fouchier RA, Osterhaus AD, et al. Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am J Pathol (2007) 171(4):1215–23. doi:10.2353/ajpath.2007.070248
37. Olofsson S, Kumlin U, Dimock K, Arnberg N. Avian influenza and sialic acid receptors: more than meets the eye? Lancet Infect Dis (2005) 5(3):184–8. doi:10.1016/S1473-3099(05)70026-8
38. Zeng H, Pappas C, Belser JA, Houser KV, Zhong W, Wadford DA, et al. Human pulmonary microvascular endothelial cells support productive replication of highly pathogenic avian influenza viruses: possible involvement in the pathogenesis of human H5N1 virus infection. J Virol (2012) 86(2):667–78. doi:10.1128/JVI.06348-11
39. Kuiken T, Riteau B, Fouchier RA, Rimmelzwaan GF. Pathogenesis of influenza virus infections: the good, the bad and the ugly. Curr Opin Virol (2012) 2(3):276–86. doi:10.1016/j.coviro.2012.02.013
40. Xu R, Mcbride R, Paulson JC, Basler CF, Wilson IA. Structure, receptor binding, and antigenicity of influenza virus hemagglutinins from the 1957 H2N2 pandemic. J Virol (2010) 84(4):1715. doi:10.1128/JVI.02162-09
41. Gao Y, Zhang Y, Shinya K, Deng G, Jiang Y, Li Z, et al. Identification of amino acids in HA and PB2 critical for the transmission of H5N1 avian influenza viruses in a mammalian host. PLoS Pathog (2009) 5(12):e1000709. doi:10.1371/journal.ppat.1000709
42. Xiong X, Martin SR, Haire LF, Wharton SA, Daniels RS, Bennett MS, et al. Receptor binding by an H7N9 influenza virus from humans. Nature (2013) 499(7459):496. doi:10.1038/nature12372
43. Byrdleotis L, Cummings RD, Steinhauer DA. The interplay between the host receptor and influenza virus hemagglutinin and neuraminidase. Int J Mol Sci (2017) 18(7):1541. doi:10.3390/ijms18071541
44. de Graaf M, Fouchier RA. Role of receptor binding specificity in influenza A virus transmission and pathogenesis. EMBO J (2014) 33(8):823–41. doi:10.1002/embj.201387442
45. Chen J, Lee KH, Steinhauer DA, Stevens DJ, Skehel JJ, Wiley DC. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell (1998) 95(3):409. doi:10.1016/S0092-8674(00)81771-7
46. Upham JP, Pickett D, Irimura T, Anders EM, Reading PC. Macrophage receptors for influenza A virus: role of the macrophage galactose-type lectin and mannose receptor in viral entry. J Virol (2010) 84(8):3730–7. doi:10.1128/JVI.02148-09
47. Fontana J, Cardone G, Heymann JB, Winkler DC, Steven AC. Structural changes in influenza virus at low pH characterized by cryo-electron tomography. J Virol (2012) 86(6):2919–29. doi:10.1128/JVI.06698-11
48. Lamb RA, Krug RM. Orthomyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, Fields BN, editors. Fields Virology. Philadelphia: Lippincott-Raven Press (1996).
49. Flint SJ. Principles of Virology: Molecular Biology, Pathogenesis, and Control. Washington DC: ASM Press (2000). p. 942–3.
50. Sun X, Whittaker GR. Entry of influenza virus. Adv Exp Med Biol (2013) 790:72. doi:10.1007/978-1-4614-7651-1_4
51. Cao X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol (2016) 16(1):35. doi:10.1038/nri.2015.8
52. Ouyang J, Zhu X, Chen Y, Wei H, Chen Q, Chi X, et al. NRAV, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe (2014) 16(5):616–26. doi:10.1016/j.chom.2014.10.001
53. Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell (2010) 140(3):397–408. doi:10.1016/j.cell.2010.01.020
54. Baum A, Sachidanandam R, Garcíasastre A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U S A (2010) 107(37):16303. doi:10.1073/pnas.1005077107
55. Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science (2006) 314(5801):997–1001. doi:10.1126/science.1132998
56. Munir M. TRIM proteins: another class of viral victims. Sci Signal (2010) 3(118):jc2. doi:10.1126/scisignal.3118jc2
57. Yoneyama M, Onomoto K, Jogi M, Akaboshi T, Fujita T. Viral RNA detection by RIG-I-like receptors. Curr Opin Immunol (2015) 32:48. doi:10.1016/j.coi.2014.12.012
58. Hiscott J, Lin R, Nakhaei P, Paz S. MasterCARD: a priceless link to innate immunity. Trends Mol Med (2006) 12(2):53–6. doi:10.1016/j.molmed.2005.12.003
59. Takeshita F, Tanaka T, Matsuda T, Tozuka M, Kobiyama K, Saha S, et al. Toll-like receptor adaptor molecules enhance DNA-raised adaptive immune responses against influenza and tumors through activation of innate immunity. J Virol (2006) 80(13):6218. doi:10.1128/JVI.00121-06
60. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol (2010) 11(5):373. doi:10.1038/ni.1863
61. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805. doi:10.1016/j.cell.2010.01.022
62. Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol (2011) 30(1):16–34. doi:10.3109/08830185.2010.529976
63. Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5’-diphosphates. Nature (2014) 514(7522):372–5. doi:10.1038/nature13590
64. Schulz O, Diebold SS, Chen M, Näslund TI, Nolte MA, Alexopoulou L, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature (2005) 433(7028):887. doi:10.1038/nature03326
65. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, et al. Recognition of single-stranded RNA viruses by toll-like receptor 7. Proc Natl Acad Sci U S A (2004) 101(15):5598–603. doi:10.1073/pnas.0400937101
66. Le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, Chignard M, et al. Cutting edge: influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol (2007) 178(6):3368–72. doi:10.4049/jimmunol.178.6.3368
67. Ablasser A, Poeck H, Anz D, Berger M, Schlee M, Kim S, et al. Selection of molecular structure and delivery of RNA oligonucleotides to activate TLR7 versus TLR8 and to induce high amounts of IL-12p70 in primary human monocytes. J Immunol (2009) 182(11):6824. doi:10.4049/jimmunol.0803001
68. Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol (2014) 14(1):9. doi:10.1038/nri3565
69. Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, et al. Differential expression of NLRP3 among hematopoietic cells. J Immunol (2011) 186(4):2529–34. doi:10.4049/jimmunol.1002720
70. Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene E, Messling VV, et al. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog (2013) 9(4):e1003256. doi:10.1371/journal.ppat.1003256
71. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol (2009) 27(27):229. doi:10.1146/annurev.immunol.021908.132715
72. Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol (2010) 11(5):404–10. doi:10.1038/ni.1861
73. Mcauley JL, Tate MD, Mackenziekludas CJ, Pinar A, Zeng W, Stutz A, et al. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog (2013) 9(5):e1003392. doi:10.1371/journal.ppat.1003392
74. Shan NC, Xiao WZ, Li L, Bai YR, Bei H, Wen SH, et al. Evolution of IFN-λ in tetrapod vertebrates and its functional characterization in green anole lizard (Anolis carolinensis). Dev Comp Immunol (2016) 61:208. doi:10.1016/j.dci.2016.04.004
75. Wang J, Oberley-Deegan R, Wang S, Nikrad M, Funk C, Hartshorn K, et al. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-lambda 1) in response to influenza A infection. J Immunol (2009) 182(3):1296–304. doi:10.4049/jimmunol.182.3.1296
76. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32(1):513. doi:10.1146/annurev-immunol-032713-120231
77. Mordstein M, Kochs G, Dumoutier L, Renauld JC, Paludan SR, Klucher K, et al. Interferon-λ contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog (2008) 43(3):e1000151. doi:10.1371/journal.ppat.1000151
78. Crotta S, Davidson S, Mahlakoiv T, Desmet CJ, Buckwalter MR, Albert ML, et al. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog (2013) 9(11):e1003773. doi:10.1371/journal.ppat.1003773
79. Davidson S, Mccabe TM, Crotta S, Gad HH, Hessel EM, Beinke S, et al. IFNλ is a potent anti-influenza therapeutic without the inflammatory side effects of IFNα treatment. EMBO Mol Med (2016) 8(9):1099–112. doi:10.15252/emmm.201606413
80. Kim S, Kim MJ, Kim CH, Kang JW, Shin HK, Kim DY, et al. The superiority of IFN-lambda as a therapeutic candidate to control acute influenza viral lung infection. Am J Respir Cell Mol Biol (2016) 56(2):202–12. doi:10.1165/rcmb.2016-0174OC
81. Fernández M, Quiroga JA, Martin J, Herrero M, Pardo M, Horisberger MA, et al. In vivo and in vitro induction of MxA protein in peripheral blood mononuclear cells from patients chronically infected with hepatitis C virus. J Infect Dis (1999) 180(2):262. doi:10.1086/314859
82. Staeheli P, Haller O, Boll W, Lindenmann J, Weissmann C. Mx protein: constitutive expression in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell (1986) 44(1):147. doi:10.1016/0092-8674(86)90493-9
83. Xiao H, Killip MJ, Staeheli P, Randall RE, Jackson D. The human interferon-induced MxA protein inhibits early stages of influenza A virus infection by retaining the incoming viral genome in the cytoplasm. J Virol (2013) 87(23):13053. doi:10.1128/JVI.02220-13
84. Pavlovic J, Haller O, Staeheli P. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J Virol (1992) 66(4):2564.
85. Mänz B, Dornfeld D, Götz V, Zell R, Zimmermann P, Haller O, et al. Pandemic influenza A viruses escape from restriction by human MxA through adaptive mutations in the nucleoprotein. PLoS Pathog (2013) 9(3):e1003279. doi:10.1371/journal.ppat.1003279
86. Zimmermann P, Mänz B, Haller O, Schwemmle M, Kochs G. The viral nucleoprotein determines Mx sensitivity of influenza A viruses. J Virol (2011) 85(16):8133–40. doi:10.1128/JVI.00712-11
87. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol (2014) 14(5):315–28. doi:10.1038/nri3665
88. Evans SS, Lee DB, Han T, Tomasi TB, Evans RL. Monoclonal antibody to the interferon-inducible protein Leu-13 triggers aggregation and inhibits proliferation of leukemic B cells. Blood (1990) 76(12):2583–93.
89. Kelly JM, Gilbert CS, Stark GR, Kerr IM. Differential regulation of interferon-induced mRNAs and c-myc mRNA by alpha- and gamma-interferons. Eur J Biochem (1985) 153(2):367–71. doi:10.1111/j.1432-1033.1985.tb09312.x
90. Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell (2009) 139(7):1243–54. doi:10.1016/j.cell.2009.12.017
91. Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity (2013) 38(1):92–105. doi:10.1016/j.immuni.2012.11.005
92. Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, et al. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity (2013) 38(1):106–18. doi:10.1016/j.immuni.2012.11.004
93. Gold ES, Diercks AH, Podolsky I, Podyminogin RL, Askovich PS, Treuting PM, et al. 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proc Natl Acad Sci U S A (2014) 111(29):10666–71. doi:10.1073/pnas.1404271111
94. Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature (2007) 446(7138):916–20. doi:10.1038/nature05732
95. Di Pietro A, Kajaste-Rudnitski A, Oteiza A, Nicora L, Towers GJ, Mechti N, et al. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J Virol (2013) 87(8):4523–33. doi:10.1128/JVI.02548-12
96. Fu B, Wang L, Ding H, Schwamborn JC, Li S, Dorf ME. TRIM32 senses and restricts influenza A virus by ubiquitination of PB1 polymerase. PLoS Pathog (2015) 11(6):e1004960. doi:10.1371/journal.ppat.1004960
97. Tang Q, Wang X, Gao G. The short form of the zinc finger antiviral protein inhibits influenza A virus protein expression and is antagonized by the virus-encoded NS1. J Virol (2017) 91(2):e1909–16. doi:10.1128/jvi.01909-16
98. Liu C-H, Zhou L, Chen G, Krug RM. Battle between influenza A virus and a newly identified antiviral activity of the PARP-containing ZAPL protein. Proc Natl Acad Sci U S A (2015) 112(45):14048–53. doi:10.1073/pnas.1509745112
99. Silverman RH. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol (2007) 81(23):12720–9. doi:10.1128/jvi.01471-07
100. Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol (2006) 80(9):4501–9. doi:10.1128/JVI.80.9.4501-4509.2006
101. Dauber B, Martínez-Sobrido L, Schneider J, Hai R, Waibler Z, Kalinke U, et al. Influenza B virus ribonucleoprotein is a potent activator of the antiviral kinase PKR. PLoS Pathog (2009) 5(6):e1000473. doi:10.1371/journal.ppat.1000473
102. Yuan W, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J (2001) 20(3):362–71. doi:10.1093/emboj/20.3.362
103. Hu S, Yin L, Mei S, Li J, Xu F, Sun H, et al. BST-2 restricts IAV release and is countered by the viral M2 protein. Biochem J (2017) 474(5):715–30. doi:10.1042/bcj20160861
104. Gnirß K, Zmora P, Blazejewska P, Winkler M, Lins A, Nehlmeier I, et al. Tetherin sensitivity of influenza A viruses is strain specific: role of hemagglutinin and neuraminidase. J Virol (2015) 89(18):9178–88. doi:10.1128/jvi.00615-15
105. Wang X, Hinson ER, Cresswell P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe (2007) 2(2):96. doi:10.1016/j.chom.2007.06.009
106. Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature (2008) 451(7177):425. doi:10.1038/nature06553
107. Evans DT, Serra-Moreno R, Singh RK, Guatelli JC. BST-2/tetherin: a new component of the innate immune response to enveloped viruses. Trends Microbiol (2010) 18(9):388–96. doi:10.1016/j.tim.2010.06.010
108. Mangeat B, Cavagliotti L, Lehmann M, Gers-Huber G, Kaur I, Thomas Y, et al. Influenza virus partially counteracts restriction imposed by tetherin/BST-2. J Biol Chem (2012) 287(26):22015–29. doi:10.1074/jbc.M111.319996
109. Tumpey TM, Garcia-Sastre A, Taubenberger JK, Palese P, Swayne DE, Pantin-Jackwood MJ, et al. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol (2005) 79(23):14933–44. doi:10.1128/jvi.79.23.14933-14944.2005
110. Herold S, Von WW, Steinmueller M, Pleschka S, Kuziel WA, Mack M, et al. Alveolar epithelial cells direct monocyte transepithelial migration upon influenza virus infection: impact of chemokines and adhesion molecules. J Immunol (2006) 177(3):1817. doi:10.4049/jimmunol.177.3.1817
111. Mendelson M, Tekoah Y, Zilka A, Gershoni-Yahalom O, Gazit R, Achdout H, et al. NKp46 O-glycan sequences that are involved in the interaction with hemagglutinin type 1 of influenza virus. J Virol (2010) 84(8):3789. doi:10.1128/JVI.01815-09
112. Guo H, Kumar P, Malarkannan S. Evasion of natural killer cells by influenza virus. J Leukoc Biol (2011) 89(2):189–94. doi:10.1189/jlb.0610319
113. van Helden MJ, de Graaf N, Boog CJ, Topham DJ, Zaiss DM, Sijts AJ. The bone marrow functions as the central site of proliferation for long-lived NK cells. J Immunol (2012) 189(5):2333–7. doi:10.4049/jimmunol.1200008
114. Holt PG, Strickland DH, Wikström ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol (2008) 8(2):142–52. doi:10.1038/nri2236
115. Bahadoran A, Lee SH, Wang SM, Manikam R, Rajarajeswaran J, Raju CS, et al. Immune responses to influenza virus and its correlation to age and inherited factors. Front Microbiol (2016) 7:1841. doi:10.3389/fmicb.2016.01841
116. Heer AK, Harris NL, Kopf M, Marsland BJ. CD4+ and CD8+ T cells exhibit differential requirements for CCR7-mediated antigen transport during influenza infection. J Immunol (2008) 181(10):6984–94. doi:10.4049/jimmunol.181.10.6984
117. GeurtsvanKessel CH, Willart MA, van Rijt LS, Muskens F, Kool M, Baas C, et al. Clearance of influenza virus from the lung depends on migratory langerin+CD11b− but not plasmacytoid dendritic cells. J Exp Med (2008) 205(7):1621–34. doi:10.1084/jem.20071365
118. Hintzen G, Ohl L, del Rio ML, Rodriguez-Barbosa JI, Pabst O, Kocks JR, et al. Induction of tolerance to innocuous inhaled antigen relies on a CCR7-dependent dendritic cell-mediated antigen transport to the bronchial lymph node. J Immunol (2006) 177(10):7346–54. doi:10.4049/jimmunol.177.10.7346
119. van de Sandt CE, Kreijtz JH, Rimmelzwaan GF. Evasion of influenza A viruses from innate and adaptive immune responses. Viruses (2012) 4(9):1438–76. doi:10.3390/v4091438
120. GeurtsvanKessel CH, Bergen IM, Muskens F, Boon L, Hoogsteden HC, Osterhaus AD, et al. Both conventional and interferon killer dendritic cells have antigen-presenting capacity during influenza virus infection. PLoS One (2009) 4(9):e7187. doi:10.1371/journal.pone.0007187
121. Kreijtz JH, Fouchier RA, Rimmelzwaan GF. Immune responses to influenza virus infection. Virus Res (2011) 162(1–2):19–30. doi:10.1016/j.virusres.2011.09.022
122. Ho AW, Prabhu N, Betts RJ, Ge MQ, Dai X, Hutchinson PE, et al. Lung CD103+ dendritic cells efficiently transport influenza virus to the lymph node and load viral antigen onto MHC class I for presentation to CD8 T cells. J Immunol (2011) 187(11):6011–21. doi:10.4049/jimmunol.1100987
123. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity (2010) 32(1):79–90. doi:10.1016/j.immuni.2009.11.012
124. Whitmire JK, Tan JT, Whitton JL. Interferon-gamma acts directly on CD8+ T cells to increase their abundance during virus infection. J Exp Med (2005) 201(7):1053–9. doi:10.1084/jem.20041463
125. Egli A, Santer DM, O’Shea D, Barakat K, Syedbasha M, Vollmer M, et al. IL-28B is a key regulator of B- and T-cell vaccine responses against influenza. PLoS Pathog (2014) 10(12):e1004556. doi:10.1371/journal.ppat.1004556
126. van Domselaar R, Bovenschen N. Cell death-independent functions of granzymes: hit viruses where it hurts. Rev Med Virol (2011) 21(5):301–14. doi:10.1002/rmv.697
127. Andrade F. Non-cytotoxic antiviral activities of granzymes in the context of the immune antiviral state. Immunol Rev (2010) 235(1):128–46. doi:10.1111/j.0105-2896.2010.00909.x
128. Allie SR, Randall TD. Pulmonary immunity to viruses. Clin Sci (2017) 131(14):1737–62. doi:10.1042/cs20160259
129. Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol (2011) 12(7):639–46. doi:10.1038/ni.2053
130. van Gisbergen KP, Klarenbeek PL, Kragten NA, Unger PP, Nieuwenhuis MB, Wensveen FM, et al. The costimulatory molecule CD27 maintains clonally diverse CD8(+) T cell responses of low antigen affinity to protect against viral variants. Immunity (2011) 35(1):97–108. doi:10.1016/j.immuni.2011.04.020
131. Grant EJ, Quinones-Parra SM, Clemens EB, Kedzierska K. Human influenza viruses and CD8(+) T cell responses. Curr Opin Virol (2016) 16:132–42. doi:10.1016/j.coviro.2016.01.016
132. Valkenburg SA, Venturi V, Dang T, Bird NL, Doherty PC, Turner SJ, et al. Correction: early priming minimizes the age-related immune compromise of CD8+ T cell diversity and function. PLOS Pathog (2012) 8(2):e1002544. doi:10.1371/journal.ppat.1002544
133. Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, et al. Autophagy is a critical regulator of memory CD8+ T cell formation. Elife (2014) 3(3):e03706. doi:10.7554/eLife.03706
134. Pizzolla A, Tho N, Smith JM, Brooks AG, Kedzieska K, Heath WR, et al. Resident memory CD8(+) T cells in the upper respiratory tract prevent pulmonary influenza virus infection. Sci Immunol (2017) 2(12):eaam6970. doi:10.1126/sciimmunol.aam6970
135. Van BN, Harty JT. Influenza induced lung Trm: not all memories last forever. Immunol Cell Biol (2017) 95(8):651–5. doi:10.1038/icb.2017.32
136. Dunbar P, Wein AN, McMaster SR, Hayward SL, Kohlmeier JE. Tissue-resident monocytes promote the establishment of lung-resident CD8 T cell memory following influenza infection. J Immunol (2016) 196(1 Suppl):68.7.
137. Brown DM, Lee S, Garciahernandez ML, Swain SL. Multifunctional CD4 cells expressing gamma interferon and perforin mediate protection against lethal influenza virus infection. J Virol (2012) 86(12):6792–803. doi:10.1128/JVI.07172-11
138. McKinstry KK, Strutt TM, Kuang Y, Brown DM, Sell S, Dutton RW, et al. Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J Clin Invest (2012) 122(8):2847–56. doi:10.1172/jci63689
139. Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4? T cells in immunity to viruses. Nat Rev Immunol (2012) 12(2):136–48. doi:10.1038/nri3152
140. Yao Q, Fischer KP, Li L, Agrawal B, Berhane Y, Tyrrell DL, et al. Immunogenicity and protective efficacy of a DNA vaccine encoding a chimeric protein of avian influenza hemagglutinin subtype H5 fused to CD154 (CD40L) in Pekin ducks. Vaccine (2010) 28(51):8147–56. doi:10.1016/j.vaccine.2010.09.081
141. Ingulli E, Mondino A, Khoruts A, Jenkins MK. In vivo detection of dendritic cell antigen presentation to CD4(+) T cells. J Exp Med (1997) 185(12):2133–41. doi:10.1084/jem.185.12.2133
142. Lukens MV, Kruijsen D, Coenjaerts FE, Kimpen JL, van Bleek GM. Respiratory syncytial virus-induced activation and migration of respiratory dendritic cells and subsequent antigen presentation in the lung-draining lymph node. J Virol (2009) 83(14):7235–43. doi:10.1128/jvi.00452-09
143. Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY, Ferrante J, et al. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity (1996) 4(5):471–81. doi:10.1016/S1074-7613(00)80413-6
144. Pape KA, Khoruts A, Mondino A, Jenkins MK. Inflammatory cytokines enhance the in vivo clonal expansion and differentiation of antigen-activated CD4+ T cells. J Immunol (1997) 159(2):591–8.
145. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell (2000) 100(6):655–69. doi:10.1016/S0092-8674(00)80702-3
146. Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci U S A (2012) 109(11):4239–44. doi:10.1073/pnas.1114981109
147. Shu U, Kiniwa M, Wu CY, Maliszewski C, Vezzio N, Hakimi J, et al. Activated T cells induce interleukin-12 production by monocytes via CD40-CD40 ligand interaction. Eur J Immunol (1995) 25(4):1125–8. doi:10.1002/eji.1830250442
148. Stuber E, Strober W, Neurath M. Blocking the CD40L-CD40 interaction in vivo specifically prevents the priming of T helper 1 cells through the inhibition of interleukin 12 secretion. J Exp Med (1996) 183(2):693–8. doi:10.1084/jem.183.2.693
149. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol (2010) 28:445–89. doi:10.1146/annurev-immunol-030409-101212
150. Lamb JR, Eckels DD, Lake P, Johnson AH, Hartzman RJ, Woody JN. Antigen-specific human T lymphocyte clones: induction, antigen specificity, and MHC restriction of influenza virus-immune clones. J Immunol (1982) 128(1):233–8.
151. Mukherjee S, Lindell DM, Berlin AA, Morris SB, Shanley TP, Hershenson MB, et al. IL-17-induced pulmonary pathogenesis during respiratory viral infection and exacerbation of allergic disease. Am J Pathol (2011) 179(1):248–58. doi:10.1016/j.ajpath.2011.03.003
152. La Gruta NL, Turner SJ. T cell mediated immunity to influenza: mechanisms of viral control. Trends Immunol (2014) 35(8):396–402. doi:10.1016/j.it.2014.06.004
153. Rangelmoreno J, Carragher DM, Misra RS, Kusser K, Hartson L, Moquin A, et al. B cells promote resistance to heterosubtypic strains of influenza via multiple mechanisms. J Immunol (2008) 180(1):454–63. doi:10.4049/jimmunol.180.1.454
154. Jegaskanda S, Vandenberg K, Laurie KL, Loh L, Kramski M, Winnall WR, et al. Cross-reactive influenza-specific antibody-dependent cellular cytotoxicity in intravenous immunoglobulin as a potential therapeutic against emerging influenza viruses. J Infect Dis (2014) 210(11):1811. doi:10.1093/infdis/jiu334
155. Seibert CW, Rahmat S, Krause JC, Eggink D, Albrecht RA, Goff PH, et al. Recombinant IgA is sufficient to prevent influenza virus transmission in guinea pigs. J Virol (2013) 87(14):7793–804. doi:10.1128/JVI.00979-13
156. Frasca D, Diaz A, Romero M, Blomberg BB. The generation of memory B cells is maintained, but the antibody response is not, in the elderly after repeated influenza immunizations. Vaccine (2016) 34(25):2834–40. doi:10.1016/j.vaccine.2016.04.023
157. Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med (2014) 20(5):503. doi:10.1038/nm.3521
158. Mazanec MB, Coudret CL, Fletcher DR. Intracellular neutralization of influenza virus by immunoglobulin A anti-hemagglutinin monoclonal antibodies. J Virol (1995) 69(2):1339–43.
159. Murphy BR, Clements ML. The systemic and mucosal immune response of humans to influenza A virus. Curr Top Microbiol Immunol (1989) 146:107–16.
160. Zuercher AW, Coffin SE, Thurnheer MC, Fundova P, Cebra JJ. Nasal-associated lymphoid tissue is a mucosal inductive site for virus-specific humoral and cellular immune responses. J Immunol (2002) 168(4):1796–803. doi:10.4049/jimmunol.168.4.1796
161. Fujimura Y, Takeda M, Ikai H, Haruma K, Akisada T, Harada T, et al. The role of M cells of human nasopharyngeal lymphoid tissue in influenza virus sampling. Virchows Arch (2004) 444(1):36–42. doi:10.1007/s00428-003-0898-8
162. Asanuma H, Thompson AH, Iwasaki T, Sato Y, Inaba Y, Aizawa C, et al. Isolation and characterization of mouse nasal-associated lymphoid tissue. J Immunol Methods (1997) 202(2):123–31. doi:10.1016/S0022-1759(96)00243-8
163. Harty JT, Badovinac VP. Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol (2008) 8(2):107–19. doi:10.1038/nri2251
164. Wu T, Hu Y, Lee YT, Bouchard KR, Benechet A, Khanna K, et al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol (2014) 95(2):215–24. doi:10.1189/jlb.0313180
165. Bienenstock J, Clancy R, Perey D. Bronchus-associated lymphoid tissue (BALT): its relation to mucosal immunity. Immunologic and Infectious Reactions in the Lung, (1976) 1:29–58.
166. Mikhak Z, Strassner JP, Luster AD. Lung dendritic cells imprint T cell lung homing and promote lung immunity through the chemokine receptor CCR4. J Exp Med (2013) 210(9):1855–69. doi:10.1084/jem.20130091
167. Chiu C, Ellebedy AH, Wrammert J, Ahmed R. B Cell Responses to Influenza Infection and Vaccination. Cham, ZG: Springer International Publishing (2014). 381 p.
168. Van RE, Ainai A, Suzuki T, Hasegawa H. Mucosal IgA responses in influenza virus infections; thoughts for vaccine design. Vaccine (2012) 30(40):5893. doi:10.1016/j.vaccine.2012.04.109
169. Asahi Y, Yoshikawa T, Watanabe I, Iwasaki T, Hasegawa H, Sato Y, et al. Protection against influenza virus infection in polymeric Ig receptor knockout mice immunized intranasally with adjuvant-combined vaccines. J Immunol (2002) 168(6):2930. doi:10.4049/jimmunol.168.6.2930
170. Akira A, Shin-Ichi T, Tadaki S, Elly VR, Ryo I, Takato O, et al. Intranasal vaccination with an inactivated whole influenza virus vaccine induces strong antibody responses in serum and nasal mucus of healthy adults. Hum Vaccin Immunother (2013) 9(9):1962–70. doi:10.4161/hv.25458
171. Suzuki T, Kawaguchi A, Ainai A, Tamura S, Ito R, Multihartina P, et al. Relationship of the quaternary structure of human secretory IgA to neutralization of influenza virus. Proc Natl Acad Sci U S A (2015) 112(25):7809. doi:10.1073/pnas.1503885112
172. Xia C, Vijayan M, Pritzl CJ, Fuchs SY, Mcdermott AB, Hahm B. Hemagglutinin of influenza A virus antagonizes type I interferon (IFN) responses by inducing degradation of type I IFN receptor 1. J Virol (2015) 90(5):2403–17. doi:10.1128/JVI.02749-15
173. Peacock T, Reddy K, James J, Adamiak B, Barclay W, Shelton H, et al. Antigenic mapping of an H9N2 avian influenza virus reveals two discrete antigenic sites and a novel mechanism of immune escape. Sci Rep (2016) 6:18745. doi:10.1038/srep18745
174. Hervé PL, Lorin V, Jouvion G, Da CB, Escriou N. Addition of N-glycosylation sites on the globular head of the H5 hemagglutinin induces the escape of highly pathogenic avian influenza A H5N1 viruses from vaccine-induced immunity. Virology (2015) 486(8):134–45. doi:10.1016/j.virol.2015.08.033
175. Baron Y, Seidel E, Tsukerman P, Mandelboim M, Mandelboim O. Influenza virus uses its neuraminidase protein to evade the recognition of two activating NK cell receptors. J Infect Dis (2014) 210(3):410. doi:10.1093/infdis/jiu094
176. Gack MU, Kirchhofer A, Shin YC, Inn KS, Liang C, Cui S, et al. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc Natl Acad Sci U S A (2008) 105(43):16743. doi:10.1073/pnas.0804947105
177. Gack MU, Albrecht RA, Urano T, Inn KS, Huang I, Carnero E, et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by RIG-I. Cell Host Microbe (2009) 5(5):439–49. doi:10.1016/j.chom.2009.04.006
178. Kedersha NL, Rome LH. Isolation and characterization of a novel ribonucleoprotein particle: large structures contain a single species of small RNA. J Cell Biol (1986) 103(3):699–709. doi:10.1083/jcb.103.3.699
179. Li F, Chen Y, Zhang Z, Ouyang J, Wang Y, Yan R, et al. Robust expression of vault RNAs induced by influenza A virus plays a critical role in suppression of PKR-mediated innate immunity. Nucleic Acids Res (2015) 43(21):10321–37. doi:10.1093/nar/gkv1078
180. Rückle A, Haasbach E, Julkunen I, Planz O, Ehrhardt C, Ludwig S. The NS1 protein of influenza A virus blocks RIG-I-mediated activation of the noncanonical NF-κB pathway and p52/RelB-dependent gene expression in lung epithelial cells. J Virol (2012) 86(18):10211–7. doi:10.1128/JVI.00323-12
181. Gao S, Song L, Li J, Zhang Z, Peng H, Jiang W, et al. Influenza A virus-encoded NS1 virulence factor protein inhibits innate immune response by targeting IKK. Cell Microbiol (2012) 14(12):1849. doi:10.1111/cmi.12005
182. Jia D, Rahbar R, Chan RW, Lee SM, Chan MC, Wang BX, et al. Influenza virus non-structural protein 1 (NS1) disrupts interferon signaling. PLoS One (2010) 5(11):e13927. doi:10.1371/journal.pone.0013927
183. Fernandez-Sesma A, Marukian S, Ebersole BJ, Kaminski D, Park M-S, Yuen T, et al. Influenza virus evades innate and adaptive immunity via the NS1 protein. J Virol (2006) 80(13):6295–304. doi:10.1128/JVI.02381-05
184. Anastasina M, May NL, Bugai A, Yu F, Söderholm S, Gaelings L, et al. Influenza virus NS1 protein binds cellular DNA to block transcription of antiviral genes. Biochim Biophys Acta (2016) 1859(11):1440–8. doi:10.1016/j.bbagrm.2016.09.005
185. Varga ZT, Ramos I, Hai R, Schmolke M, Garcíasastre A, Fernandezsesma A, et al. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog (2011) 7(6):e1002067. doi:10.1371/journal.ppat.1002067
186. Iwai A, Shiozaki T, Kawai T, Akira S, Kawaoka Y, Takada A, et al. Influenza A virus polymerase inhibits type I interferon induction by binding to interferon β promoter stimulator 1. J Biol Chem (2010) 285(42):32064–74. doi:10.1074/jbc.M110.112458
187. Grimm D, Staeheli P, Hufbauer M, Koerner I, Martínez-Sobrido L, Solórzano A, et al. Replication fitness determines high virulence of influenza A virus in mice carrying functional Mx1 resistance gene. Proc Natl Acad Sci U S A (2007) 104(16):6806–11. doi:10.1073/pnas.0701849104
188. Münz C. Influenza A virus lures autophagic protein LC3 to budding sites. Cell Host Microbe (2014) 15(2):130–1. doi:10.1016/j.chom.2014.01.014
Keywords: influenza A virus, immune response, innate immunity, adaptive immunity, immune evasion
Citation: Chen X, Liu S, Goraya MU, Maarouf M, Huang S and Chen J-L (2018) Host Immune Response to Influenza A Virus Infection. Front. Immunol. 9:320. doi: 10.3389/fimmu.2018.00320
Received: 28 July 2017; Accepted: 05 February 2018;
Published: 05 March 2018
Edited by:
Gayane Manukyan, Institute of Molecular Biology (NAS RA), ArmeniaReviewed by:
Jason Kindrachuk, University of Manitoba, CanadaHovakim Zakaryan, Institute of Molecular Biology (NAS RA), Armenia
Copyright: © 2018 Chen, Liu, Goraya, Maarouf, Huang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-Long Chen, Y2hlbmpsQGltLmFjLmNu