 Anusree Mahanta1,2†
Anusree Mahanta1,2† Piyali Ganguli3,4†
Piyali Ganguli3,4† Pankaj Barah1
Pankaj Barah1 Ram Rup Sarkar3,4Neelanjana Sarmah1
Ram Rup Sarkar3,4Neelanjana Sarmah1 Saurav Phukan1Mayuri Bora1
Saurav Phukan1Mayuri Bora1 Shashi Baruah1*
Shashi Baruah1*
- 1Department of Molecular Biology and Biotechnology, Tezpur University, Tezpur, India
- 2Institute of Stem Cell Biology and Regenerative Medicine, Bengaluru, India
- 3Chemical Engineering and Process Development, CSIR- National Chemical Laboratory, Pune, India
- 4Academy of Scientific and Innovative Research (AcSIR), CSIR-NCL Campus, Pune, India
Diseases by protozoan pathogens pose a significant public health concern, particularly in tropical and subtropical countries, where these are responsible for significant morbidity and mortality. Protozoan pathogens tend to establish chronic infections underscoring their competence at subversion of host immune processes, an important component of disease pathogenesis and of their virulence. Modulation of cytokine and chemokine levels, their crosstalks and downstream signaling pathways, and thereby influencing recruitment and activation of immune cells is crucial to immune evasion and subversion. Many protozoans are now known to secrete effector molecules that actively modulate host immune transcriptome and bring about alterations in host epigenome to alter cytokine levels and signaling. The complexity of multi-dimensional events during interaction of hosts and protozoan parasites ranges from microscopic molecular levels to macroscopic ecological and epidemiological levels that includes disrupting metabolic pathways, cell cycle (Toxoplasma and Theileria sp.), respiratory burst, and antigen presentation (Leishmania spp.) to manipulation of signaling hubs. This requires an integrative systems biology approach to combine the knowledge from all these levels to identify the complex mechanisms of protozoan evolution via immune escape during host–parasite coevolution. Considering the diversity of protozoan parasites, in this review, we have focused on Leishmania and Plasmodium infections. Along with the biological understanding, we further elucidate the current efforts in generating, integrating, and modeling of multi-dimensional data to explain the modulation of cytokine networks by these two protozoan parasites to achieve their persistence in host via immune escape during host–parasite coevolution.
Introduction
Parasitic protozoa are responsible for some of the major diseases of humans affecting several million people each year resulting in significant morbidity and mortality and loss of economic activity. There have been some gains in reducing the incidence of these diseases owing to better intervention strategies, but in absence of effective vaccines, diseases like malaria, leishmaniasis, trypanosomiasis still pose a major public health problem. These protozoans typically establish chronic infections validating their success in evasion and manipulation of host defense and of metabolic processes for their survival, proliferation, and transmission. Many of these pathogenic protozoa have adapted to intracellular habitat as seen in infections by Plasmodium spp., Leishmania spp., and others. The intracellular niche makes them vulnerable to lysosomal enzymes, reactive oxygen intermediates, and detection by cytosolic sensors of infection, but also offers some protection from adaptive immunity (1). This dynamic host–pathogen interaction, leads to the activation of a series of intracellular and intercellular biochemical signaling processes leading to synthesis of diffusible effector molecules that includes cytokines and reactive oxygen species. “The earliest stages of infection are a parasite’s first opportunity to establish itself within its host and conversely, it is also the host’s chance to mount a rapid and effective response to clear, or at least control the infection” (2). Recent studies demonstrate that pathogens including protozoa modulate the host cell environment by manipulating the host transcriptome by epigenetic modifications besides targeting the major signaling hubs of metabolic, immune, and cell cycle processes to promote their growth, multiplication and survival (3–9). Many protozoans secrete effector molecules that actively modulate host immune transcriptome to alter cytokine levels and signaling either to escape immune processes as in liver stages of P. falciparum or to drive their growth as seen in the blood stages of this pathogen.
Considering the diversity of protozoan pathogenesis, this review will focus on manipulation and hijacking of cytokine networks by Leishmania and Plasmodium spp. for their survival in human host. We will highlight few recently published representative omics and systems biology based studies on Leishmania and Plasmodium parasites, toward understanding modulation of cytokine and chemokine networks in the host by the parasite to achieve their persistence in host via immune escape.
Cytokines and Cytokine Regulation
Cytokines are small molecules of the immune system, synthesized by various cell types that by virtue of binding to their receptors present on a multitude of cells mediate immune cell activation, differentiation, and cross talk to maintain immune homeostasis (10, 11). Synthesis and regulation of cytokine expression depends on the type of stimulus, cell type, and its state of activation (12–14). Expression of cytokine genes is also regulated by epigenetic modifications that include DNA methylation, histone modifications, and higher order chromatin interactions (15, 16) and posttranscriptional regulation by micro RNA-mediated mechanisms (16–19). Differentiation of immune cells as in T cell subpopulations and macrophage phenotypes is determined and regulated by cytokine environment (4, 16, 20, 21) and epigenetic modifications at cytokine gene loci (22, 23). Cytokine crosstalk between IFNα/β and TNF-α was noted to be at level of chromatin wherein IFNs in addition to regulating interferon signaling genes, also potentiated the TNF genes (4). Similarly, emerging data suggest extensive crosstalk between NLR family proteins of inflammation complex for IL-1β and IL-18 secretion and other cytokines integrated signalosome facilitating integration of diverse pathways for optimal immune response (24). H3K27, methyltransferase enhancer of zeste homolog 1 is reported to promote TLR-triggered inflammatory cytokine production by suppressing the TLR negative regulator toll-interacting protein, thereby contributing to the full activation of the innate immune response against invading pathogens (25).
Cytokine Signaling Manipulation by Protozoan Pathogens
Intracellular protozoa modulate cytokine gene expression and signaling by some common themes that include targeting of transcription factors (15, 23) phosphorylation status of signaling molecules like STATs, immune check point molecules like CTLA-4 and PD-1 to drive regulatory pathways (26) as well as kinases (5, 6, 27). The pathways usually targeted by pathogens include NF-κB, cell cycle, interferons, MAP Kinase JAK–STAT and pathways mediated by TLR and NLR receptors because of their wide range of functionality and core association with the host genome (28–30).
Toxoplasma spp. secrete dense granular protein (GRA) and Rhoptry proteins that activate host kinases and possess kinase activity, respectively, into host cell, which by phosphorylating STAT3 and STAT6, nuclear translocation of NF-κB or activation status of MAPK pathways modulate the levels of IL-4, IL-6, IL-12, and IFN-g (31–35). “T. gondii inhibitor of STAT1 transcriptional is another secretory protein that recruits the host nucleosome remodeling and deaceytlase complex to block STAT1-mediated gene transcription” (36). Trypanasoma cruzi modulates NF-κB pathway by TLR and NLR mediated signaling for favorable cytokine environment (37–39) However, the protozoa is also reported to manipulate TGF β pathway (40) and also induces the production of IL-10 (40, 41) and arginase for its survival and replication.
Plasmodium and Host Inflammatory Response
Malaria, caused by Plasmodium spp. of Apicomplexa phylum, has been the strongest evolutionary selective force in recent human history and has shaped human genome (42) and is one of the major causes of mortality of children below 5 years of age particularly in WHO African region, taking the life of a child every 2 min (43). The life cycle of the parasite is complex and completed in multiple stages in the human and in the mosquito (female Anopheles spp.) hosts with stage specific gene and protein signatures (44). Briefly, sporozoites inoculated into human host by bite of infected mosquito travel to liver to mature into merozoites that infect RBCs to continue asexual cycle and also develop into gametocytes which, after fertilization in mosquito gut, develop and mature into sporozoites.
During the liver stages of the parasite, the host immune response tends to be tolerogenic and circumsporozoite protein was seen to inhibit NADPH oxidase and IL-12 and suppressed IL-6 and TNF-α secretion with simultaneous increase of IL-10 levels, allowing parasite to escape detection by immune system (45, 46).
Inflammation is recognized as pivotal feature of immune response to blood stages of Plasmodium infection (47). Notably, clinical manifestations of the disease are related to erythrocytic stage of infection. An early and finely balanced inflammatory response with increase in levels of pro-inflammatory IL-12, IFN-γ, TNF-α, IL-1β, and IL-6 and of anti-inflammatory IL-10 and TGF-β is essential for resolution of parasitemia and of disease (48–52). However, pathological activation of exaggerated levels of the very same pro-inflammatory cytokines (cytokine storm) concomitant with lower levels of regulatory mechanisms has been attributed to severe and cerebral malaria syndromes (14, 53–57). A recent study examined the levels of different biomarkers of immune response and found high concentrations of sCDI63 and Fractalkine, which are involved in immune response downregulation and modulation of anti-inflammatory responses in asymptomatic malaria (58). These authors also reported high levels of Neopterin, which is related to increased cell-mediated immune responses and macrophage activation in severe and cerebral malaria patients, indicating an overall sustained state of inflammation supporting the hypothesis of intense and prolonged inflammatory response in severe and in cerebral malaria patients.
The question then arises is that why and how would the parasite drive intense inflammatory response that has the potential to be fatal which could limit parasite transmission and hence not be in interest of the pathogen? The answer appears to lie in (a) enhanced expression of adhesion molecules on endothelial cells by pro-inflammatory cytokines (IFNγ and TNFα) (59) and (b) by requirement for endothelial adhesion mediated by P. falciparum membrane protein 1 (PfEMP1) with CD36 and endothelial protein C receptor (EPCR) (60, 61). From the parasite view, endothelial sequestration is essential to escape clearance in spleen and to facilitate falciparum merozoite maturation. The highly diverse PfEMP1 proteins encoded by parasite var genes contain a Duffy-binding like and cysteine-rich interdomain region (CIDR) domains. Most CIDRα1 domains bind to EPCR and CIDRα2–6 bind CD36 (60, 61). Notably, interaction of EPCR with its ligand the activated protein C (APC) has a role in anti-inflammatory, coagulation homeostasis, and endothelial barrier protection functions (62) and its blockade of these functions by PfEMP1–EPCR interaction that is postulated to contribute to cerebral malaria pathology (59, 61). Interestingly, Smith et al. (61) found increased association of severe malaria with EPCR binding CIDRα1domain containing isolates supporting the contention. Interactions with CD36 are also reported to inhibit IL-12 synthesis and suppressing dendritic cell (DC) maturation and T cell activation.
It is, therefore, not unimaginable that parasite manipulates NF-κB and Type 1 interferon pathway to drive inflammation. Plasmodium-derived PAMPs that include GPI anchors, CpG motifs, AT-rich motifs, and haemazoin are sensed by PRRs of host that include TLRs, NLRs, and AIM2 on cells of monocyte/macrophage lineage and on DCs (61, 63–65). These ligand–receptor interactions initiate MyD88 and STING-IRF3 mediated downstream signaling leading to activation of NF-κB and IRF3 pathways and synthesis of pro-inflammatory cytokines and interferon α/β (55, 65–68). It is the exaggerated activation of these pathways “mediated by IFN-γ pro-inflammatory priming with extreme levels of pro-inflammatory mediators” with concomitant loss of regulatory cytokines that drives malaria pathogenesis (46, 57, 68). It has also been proposed that in addition to driving inflammation, P. falciparum by downregulating GATA3 expression suppresses IL-10 and SOCS3 that are necessary to control inflammation, possibly by exploiting the IFNα/β pathway as summarized in Figure 1.
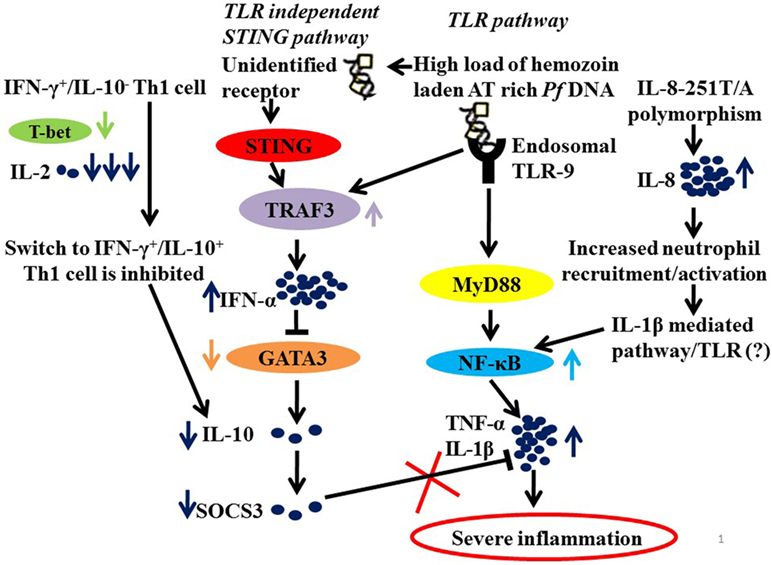
Figure 1. A hypothetical model summarizing the probable mechanisms of severe inflammation in malaria. Parasite molecules like Haemazoin, Pf AT-rich DNA recruited by TLR and TLR independent (STING) pathways (63, 69). High load of Pf AT rich DNA would lead to increased levels of TRAF3 and of IFN-α. And IFN-α, in turn, suppresses GATA3 expression in Th2 cells resulting in low levels of IL-10 and hence down regulated SOCS3 (68). In addition, low levels of IL-2 and T-bet fail to mediate switch from IFN-γ+/IL-10− to IFN-γ+/IL-10+ Th1 cells that requires T-bet and IL-2 levels, also explain low levels of IL-10. Finally, downregulated SOCS3, which is known to mediate the anti-inflammatory functions of IL-10, fails to regulate an exaggerated proinflammatory response. Another contributory role to severe inflammation in malaria is the high prevalence of IL-8-251T/A, which increases IL-8 expression for enhanced recruitment and activation of inflammatory cells neutrophils resulting in increased activation of NF-κB via IL-1β-mediated pathway.
Leishmania: T Cell Differentiation and Cross Regulation of Cytokine Signaling
Leishmaniasis caused by Leishmania spp. is a public health problem with 1.3 million reported Leishmaniasis cases worldwide which is intensified by availability of few effective drugs (70) and vaccine (71, 72). Being an intracellular parasite, it needs to overcome host-resistance mechanisms and exploit host environment for survival. From the parasite context, metabolism of Leishmania possesses a unique metabolic organization that can re-route metabolites, the uptake of which is constrained in different host environments toward synthesis of specific biomass metabolites; thereby providing novel mechanisms for metabolic adaptations (73, 74). From the host context, the contribution of specific virulence factors in immune suppression or the inability of the host to generate a sufficient immune response against the parasite, which promotes infection. Survival strategy of Leishmania is to modulate the signaling pathways of the macrophages after entering the phagolysosome. Depending on the type of infection and the parasite burden, either Th-1 healing or the Th-2 non-healing immune responses are generated, but detailed mechanism is poorly explored. This can be largely understood with respect to the interaction of parasite molecules with the host signaling pathways to suppress host immunity against infection (71).
During invasion, the surface molecules of Leishmania interact with the toll-like-receptor proteins present on the macrophages membrane (75). The activation of the TLRs triggers the downstream signaling pathways such as the RAS–RAF-mediated MAPK pathway, canonical and non-canonical NF-κB pathway, JAK–STAT pathway, PI3K–PLC Gamma pathway, and the JNK pathway (76). Subsequently several transcription factors, such as ERK1/2, NF-κB, NFAT, AP1, STAT3, are activated that initiate the synthesis and secretion of several cytokines, growth factors, chemokines and antimicrobicidal molecules which are responsible for the host immune responses during the infection (77).
However, during chronic infection (Figure 2), the antigenic molecules of the Leishmania parasite activate the phosphatase proteins in the macrophage, e.g., SHP-1 and PTP1B, which leads to the dephosphorylation and deactivation of selected signaling pathways (78). This leads to downregulation of expression of iNOS and nitric oxide in the infected macrophages, thereby compromising microbicidal functions of the cell and creating an immune-suppressed condition, which is favorable for the continued survival of the pathogen inside APC. Simultaneously, the production of the cytokines, such as IL-12 and TNF-α, gets severely reduced. Such changes in the cytokine expression pattern of the antigen-presenting cells leads to the alteration of the phenotypic responses of the T-cells that now start showing a bias toward the non-healing Th-2 immune response that is characterized by an increased production of IL-4, IL-10, IL-13, and TGF-β cytokines (79), and the suppression of IFN-γ that regulates the healing Th-1 response (71). The transcription factors T-bet and GATA3 play a pivotal role in the regulation of the Th-1/Th-2 ratio during the infection (80). Leishmania also inhibits the ability of the host cell for antigen presentation to other immune cells, by repressing the MHC class II gene expression (81) and by modulating the interaction of the co-stimulatory molecules B7-1/CD28 (82) and CD40/CD40L (83).
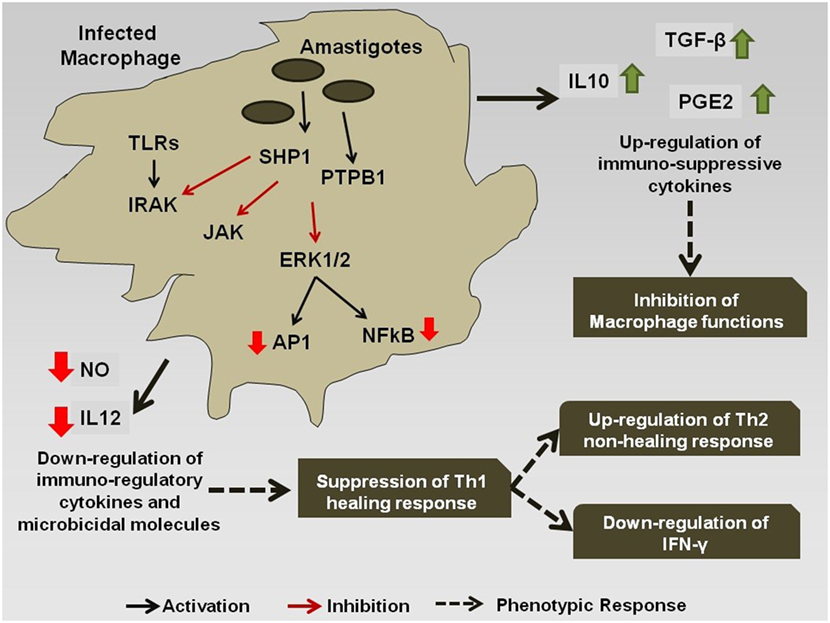
Figure 2. Immuno-modulation by Leishmania parasite: Leishmania antigens interfere with the signaling cascade of the macrophage and promote the Th-2 non-healing response that helps in the survival of the parasite inside the host.
The difference in the antigenic challenge posed to the host gives rise to differences in expression of the macrophage proteins, as seen in visceral versus the cutaneous infections (84). The difference in macrophage protein expression profile, as exemplified by increased production of COX2 and PGE2 production in case of L. donovani infection (as opposed to L. major) (85) indicates different Leishmania species selectively activate or inhibits different host pathways due to differences in the antigenic challenge. Also, it has been observed in a study that L. donovani, which is known to cause visceral leishmaniasis, may in rare cases give rise to cutaneous leishmaniasis (86). This behavior of L. donovani infection may be attributed to host’s resistance to the disease which restricts the spread of the infection to the visceral organs and keeps it localized to cutaneous regions (86).
The CD4+ CD25+ regulatory T cells also play a major role in regulating the persistence of the parasite L. major inside the host. Inhibition of the T-reg promoting cytokines such as IL-10 leads to the clearance of the pathogen from the host (87). However, during Leishmaniasis the low production of the IFN-γ and IL-12 cytokines leads to the increased proliferation of the T-reg cells that leads to the re-activation of the Leishmania parasites inside the host (87).
Systems Biology Based Integrative Approaches for Understanding the Host–Parasite Interaction and Co-Evolutionary Patterns in Protozoan Diseases
During the interaction of hosts and protozoan parasites, both employ mutual selective pressures on each other, which may facilitate rapid reciprocal adaptation. Different stages of the parasite life cycle introduce another layer of complexity (88). Significant amount of molecular, omics, clinical, epidemiological as well as ecological data has been generated at in vitro and in vivo levels using various pathogens and respective diseases. Integrative analysis of such discretely generated and located data from the host and protozoan parasite variants, in laboratory as well as natural populations is the most essential necessity to identify the complex mechanisms of protozoan evolution via immune escape during host–parasite coevolution. Public resources such as EuPathDB (89), Pathogen–Host Interactions (90), ProtozoaDB (91), together with protozoan species-specific databases are tremendously useful to collect useful information for initiating systems based integrative analysis. The key steps in such integrative approach involves data generation/data collection, data organization, data integration, integrative network construction, network analyses, and finally computer-based mathematical simulation and predictive modeling (92). As an example, using a reconstructed genome scale metabolic model of Leishmania infantum adaptations, (73) have identified the robustness of the parasite metabolic network against accidental errors and demonstrated the wide array of choices for the parasite to achieve optimal survival (73).
Recent advancement in RNA-Seq based techniques has facilitated the simultaneous sequencing of both host and parasite (including non-model parasites) transcriptomes (93). In a first of its kind RNA-seq experiment in control human neutrophils during priming with pro-inflammatory cytokines (TNF-α and GM-CSF), Wright et al. have shown the rapid expression of a common set of transcripts for cytokines, chemokines, and cell surface receptors (CXCL1, CXCL2, IL1A, IL1B, IL1RA, ICAM1) (94). They have demonstrated the utility of this approach to define functional changes in neutrophils following cytokine exposure. During a mega scale analysis of 116 malaria patients and infecting P. falciparum parasite, Yamagishi et al. have identified variable behaviors of the field malaria parasites, which were far more complex than those observed under laboratory conditions (95). Pittman et al. have generated a large scale T. gondii–host interactome, using dual transcriptional profiling of mice and parasite during acute and chronic infection (96) to demonstrate the influence of parasite development on host gene transcription as well as the epigenetic influence of the host environment on parasite gene transcription. Various systems-wide studies on malaria parasites have reported posttranscriptional (97) and translational (98) control at various points of the parasite lifecycle. One of such controlling mechanism is translational delay, by which protein expression in parasite is actively suspended for expressed mRNA transcripts. It was shown in P. falciparum that by suppressing more than 30% of its genes, the parasite rapidly adapts to new environments within the host by remaining undetected to the host immune system and undergo developmental switching in order to survive (99).
Conclusion and Future Perspectives
There is large apparent heterogeneity in offense strategies employed by the protozoan pathogen in human infections. In contrast to this, there appears to be a broad consensus on the major signaling hubs manipulated by the pathogens. It would be worthwhile to dissect the host–pathogen interactions at cellular, molecular, and systems level to discriminate between infections that are virulent with potential for fatal outcomes from asymptomatic or uncomplicated infections with limited morbidity. It may be hypothesized that immuno regulatory mechanisms that confer disease tolerance are distinct from immune and metabolic responses to severe diseases and demand to be determined by large global studies employing different protozoan pathogen systems. However, despite the availability of huge amount of multi-dimensional data in host–protozoan interaction, functional characterization, and annotation of parasite genomes is severely limited by lack of both genetic tools and resources in protozoa. Given the size, heterogeneity and complexity of the host–parasite interaction data, development of new computational tools and user-friendly methods for integrating heterogeneous “Big Data” will facilitate to fill up the missing links. This will be beneficial for better understanding of the evolutionary arm race between the host and the parasite, and finally for the efficient management and control of the protozoan diseases in humans.
Author Contributions
SB: manuscript design and contributed the introduction and sections on cytokines, malaria and Toxoplasma and future perspectives, PB: contributed the section on systems biology-based integrative approaches to understand host–parasite interaction and future perspectives, RRS: contributed in the sections on Leishmaniasis and Trypanasoma, AM: contributed to the section on malaria, NS: contributed to the section on cytokines and cytokine regulation, SP: contributed to the section on Toxoplasma, MB: contributed to the section on Toxoplasma, and PG: contributed in the sections on Leishmaniasis and Trypanasoma. SP and MB: contributed to section on Cytokine Signalling Manipulation by Protozoan parasites.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer DN and handling editor declared their shared affiliation.
Acknowledgments
We would like to thank all the participants in the study and the Clinical Collaborators in the grant and infrastructure support of University Grants Commission. This study was funded by Department of Biotechnology (Grant Nos: BT/22/NE/2011, BT/CP/11/NE/TBP/2010 and BT/PR14958/BID/7/537/2015).
Funding
This work was supported by the Department of Biotechnology Institutional Biotech Hub (Grant no: BT/22/NE/2011) and the Department of Biotechnology (Grant no: BT/CP/11/NE/TBP/2010). This work supported with a grant provided to RRS by the Department of Biotechnology, Government of India project [BT/PR14958/BID/7/537/2015]. The authors also acknowledge the infrastructure support form University Grants Commission. PG acknowledges the Junior Research Fellowship from CSIR, India.” and SB acknowledges clinical support from Prof. Kakati, AMC&H, Dibrugarh.
References
1. Sacks D, Sher A. Evasion of innate immunity by parasitic protozoa. Nat Immunol (2002) 3:1041–7. doi:10.1038/ni1102-1041
2. McGovern KE, Wilson EH. Dark side illuminated: imaging of Toxoplasma gondii through the decades. Parasit Vectors (2013) 6:334. doi:10.1186/1756-3305-6-334
3. de Monerri NCS, Kim K. Pathogens hijack the epigenome A new twist on host-pathogen interactions. Am J Pathol (2014) 184:897–911. doi:10.1016/j.ajpath.2013.12.022
4. Park SH, Kang K, Giannopoulou E, Qiao Y, Kang K, Kim G, et al. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat Immunol (2017) 18:1104. doi:10.1038/ni.3818
5. Bougdour A, Tardieux I, Hakimi MA. Toxoplasma exports dense granule proteins beyond the vacuole to the host cell nucleus and rewires the host genome expression. Cell Microbiol (2014) 16:334–43. doi:10.1111/cmi.12255
6. Schneider AG, Abdallah DSA, Butcher BA, Denkers EY. toxoplasma gondii triggers phosphorylation and nuclear translocation of dendritic cell STAT1 while simultaneously blocking IFN gamma-induced STAT1 transcriptional activity. PLoS One (2013) 8. doi:10.1371/journal.pone.0060215
7. Escoll P, Mondino S, Rolando M, Buchrieser C. Targeting of host organelles by pathogenic bacteria: a sophisticated subversion strategy. Nat Rev Microbiol (2016) 14:5–19. doi:10.1038/nrmicro.2015.1
8. Hanein D. Toxoplasma gondii targets the host actin cytoskeleton during invasion, GO figure. Biophys J (2013) 104:140a. doi:10.1016/j.bpj.2012.11.796
9. Lee SH, Lee BY, Min DY, Kim JM, Ahn MH. Role of cytoskeleton in host cell invasion by intracellular protozoa Toxoplasma gondii. J Microbiol Biotechnol (2002) 12:628–34.
10. Jones MJ, Fejes AP, Kobor MS. DNA methylation, genotype and gene expression: who is driving and who is along for the ride? Genome Biol (2013) 14:126. doi:10.1186/gb-2013-14-7-126
11. Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science (2010) 327:291–5. doi:10.1126/science.1183021
12. Mayer-Barber KD, Yan B. Clash of the cytokine titans: counter-regulation of interleukin-1 and type I interferon-mediated inflammatory responses. Cell Mol Immunol (2017) 14:22–35. doi:10.1038/cmi.2016.25
13. Kubo M, Motomura Y. Transcriptional regulation of the anti-inflammatory cytokine IL-10 in acquired immune cells. Front Immunol (2012) 3:275. doi:10.3389/fimmu.2012.00275
14. Freitas do Rosario AP, Lamb T, Spence P, Stephens R, Lang A, Roers A, et al. IL-27 promotes IL-10 production by effector Th1 CD4+ T cells: a critical mechanism for protection from severe immunopathology during malaria infection. J Immunol (2012) 188:1178–90. doi:10.4049/jimmunol.1102755
15. Falvo JV, Jasenosky LD, Kruidenier L, Goldfeld AE. Epigenetic control of cytokine gene expression: regulation of the TNF/LT locus and T helper cell differentiation. Adv Immunol (2013) 118:37–128. doi:10.1016/B978-0-12-407708-9.00002-9
16. Mia S, Warnecke A, Zhang XM, Malmstrom V, Harris RA. An optimized protocol for human M2 macrophages using M-CSF and IL-4/IL-10/TGF-beta yields a dominant immunosuppressive phenotype. Scand J Immunol (2014) 79:305–14. doi:10.1111/sji.12162
17. Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell (2009) 136:26–36. doi:10.1016/j.cell.2008.12.027
18. Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol (2008) 9:839–45. doi:10.1038/ni.f.209
19. Asirvatham AJ, Magner WJ, Tomasi TB. miRNA regulation of cytokine genes. Cytokine (2009) 45:58–69. doi:10.1016/j.cyto.2008.11.010
20. McGovern KE, Wilson EH. Role of chemokines and trafficking of immune cells in parasitic infections. Curr Immunol Rev (2013) 9:157–68. doi:10.2174/1573395509666131217000000
21. Diaz-Gandarilla JA, Osorio-Trujillo C, Hernandez-Ramirez VI, Talamas-Rohana P. PPAR activation induces M1 macrophage polarization via cPLA(2)-COX-2 inhibition, activating ROS production against Leishmania mexicana. Biomed Res Int (2013) 2013:215283. doi:10.1155/2013/215283
22. Lee CG, Sahoo A, Im SH. Epigenetic regulation of cytokine gene expression in T lymphocytes. Yonsei Med J (2009) 50:322–30. doi:10.3349/ymj.2009.50.3.322
23. Zhu JF, Jankovic D, Oler AJ, Wei G, Sharma S, Hu GQ, et al. The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity (2012) 37:660–73. doi:10.1016/j.immuni.2012.09.007
24. Barker BR, Taxman DJ, Ting JP. Cross-regulation between the IL-1beta/IL-18 processing inflammasome and other inflammatory cytokines. Curr Opin Immunol (2011) 23:591–7. doi:10.1016/j.coi.2011.07.005
25. Liu Y, Zhang Q, Ding Y, Li X, Zhao D, Zhao K, et al. Histone lysine methyltransferase Ezh1 promotes TLR-triggered inflammatory cytokine production by suppressing Tollip. J Immunol (2015) 194:2838–46. doi:10.4049/jimmunol.1402087
26. Wykes MN, Lewin SR. Immune checkpoint blockade in infectious diseases. Nat Rev Immunol (2018) 18:91–104. doi:10.1038/nri.2017.112
27. Cheeseman K, Weitzman JB. Host-parasite interactions: an intimate epigenetic relationship. Cell Microbiol (2015) 17:1121–32. doi:10.1111/cmi.12471
28. Ghosh D, Stumhofer JS. Do you see what I see: recognition of protozoan parasites by toll-like receptors. Curr Immunol Rev (2013) 9:129–40. doi:10.2174/1573395509666131203225929
29. Sibley LD. Invasion and intracellular survival by protozoan parasites. Immunol Rev (2011) 240:72–91. doi:10.1111/j.1600-065X.2010.00990.x
30. Kim BH, Shenoy AR, Kumar P, Bradfield CJ, MacMicking JD. IFN-inducible GTPases in host cell defense. Cell Host Microbe (2012) 12:432–44. doi:10.1016/j.chom.2012.09.007
31. Du J, An R, Chen L, Shen Y, Chen Y, Cheng L, et al. Toxoplasma gondii virulence factor ROP18 inhibits the host NF-kappaB pathway by promoting p65 degradation. J Biol Chem (2014) 289:12578–92. doi:10.1074/jbc.M113.544718
32. Saeij JPJ, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature (2007) 445:324–7. doi:10.1038/nature05395
33. Rosowski EE, Lu D, Julien L, Rodda L, Gaiser RA, Jensen KD, et al. Strain-specific activation of the NF-kappaB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J Exp Med (2011) 208:195–212. doi:10.1084/jem.20100717
34. Franco M, Shastri AJ, Boothroyd JC. Infection by Toxoplasma gondii specifically induces host c-Myc and the genes this pivotal transcription factor regulates. Eukaryot Cell (2014) 13:483–93. doi:10.1128/EC.00316-13
35. Peixoto L, Chen F, Harb OS, Davis PH, Beiting DP, Brownback CS, et al. Integrative genomic approaches highlight a family of parasite-specific kinases that regulate host responses. Cell Host Microbe (2010) 8:208–18. doi:10.1016/j.chom.2010.07.004
36. Bougdour A, Durandau E, Brenier-Pinchart MP, Ortet P, Barakat M, Kieffer S, et al. Host cell subversion by Toxoplasma GRA16, an exported dense granule protein that targets the host cell nucleus and alters gene expression. Cell Host Microbe (2013) 13:489–500. doi:10.1016/j.chom.2013.03.002
37. Cardoso MS, Reis-Cunha JL, Bartholomeu DC. Evasion of the immune response by Trypanosoma cruzi during acute infection. Front Immunol (2015) 6:659. doi:10.3389/fimmu.2015.00659
38. Rodrigues MM, Oliveira AC, Bellio M. The immune response to Trypanosoma cruzi: role of toll-like receptors and perspectives for vaccine development. J Parasitol Res (2012) 2012:507874. doi:10.1155/2012/507874
39. Silva GK, Gutierrez FR, Guedes PM, Horta CV, Cunha LD, Mineo TW, et al. Cutting edge: nucleotide-binding oligomerization domain 1-dependent responses account for murine resistance against Trypanosoma cruzi infection. J Immunol (2010) 184:1148–52. doi:10.4049/jimmunol.0902254
40. DosReis GA. Evasion of immune responses by Trypanosoma cruzi, the etiological agent of Chagas disease. Braz J Med Biol Res (2011) 44:84–90. doi:10.1590/S0100-879X2011007500005
41. Cribb P, Perdomo V, Alonso VL, Manarin R, Barrios-Payan J, Marquina-Castillo B, et al. Trypanosoma cruzi high mobility group B (TcHMGB) can act as an inflammatory mediator on mammalian cells. Plos Negl Trop Dis (2017) 11:e0005350. doi:10.1371/journal.pntd.0005350
42. Hedrick PW. Population genetics of malaria resistance in humans. Heredity (2011) 107:283–304. doi:10.1038/hdy.2011.16
43. World Health Organization. Malaria Fact Sheet No 94. Available from www.who.int/mediacentre/factsheets/fs375/en/
44. Joice R, Narasimhan V, Montgomery J, Sidhu AB, Oh K, Meyer E, et al. Inferring developmental stage composition from gene expression in human malaria. PLoS Comput Biol (2013) 9:e1003392. doi:10.1371/journal.pcbi.1003392
45. Zheng H, Tan Z, Xu W. Immune evasion strategies of pre-erythrocytic malaria parasites. Mediators Inflamm (2014) 2014:362605. doi:10.1155/2014/362605
46. Bertolino P, Bowen DG. Malaria and the liver: immunological hide-and-seek or subversion of immunity from within? Front Microbiol (2015) 6:41. doi:10.3389/fmicb.2015.00041
47. Clark IA, Budd AC, Alleva LM, Cowden WB. Human malarial disease: a consequence of inflammatory cytokine release. Malar J (2006) 5:85. doi:10.1186/1475-2875-5-85
48. Rovira-Vallbona E, Moncunill G, Bassat Q, Aguilar R, Machevo S, Puyol L, et al. Low antibodies against Plasmodium falciparum and imbalanced pro-inflammatory cytokines are associated with severe malaria in Mozambican children: a case-control study. Malar J (2012) 11:181. doi:10.1186/1475-2875-11-181
49. Anyona SB, Kempaiah P, Raballah E, Ouma C, Were T, Davenport GC, et al. Functional promoter haplotypes of interleukin-18 condition susceptibility to severe malarial anemia and childhood mortality. Infect Immun (2011) 79:4923–32. doi:10.1128/IAI.05601-11
50. Ayimba E, Hegewald J, Segbena AY, Gantin RG, Lechner CJ, Agosssou A, et al. Proinflammatory and regulatory cytokines and chemokines in infants with uncomplicated and severe Plasmodium falciparum malaria. Clin Exp Immunol (2011) 166:218–26. doi:10.1111/j.1365-2249.2011.04474.x
51. Boeuf PS, Loizon S, Awandare GA, Tetteh JKA, Addae MM, Adjei GO, et al. Insights into deregulated TNF and IL-10 production in malaria: implications for understanding severe malarial anaemia. Malar J (2012) 11:253. doi:10.1186/1475-2875-11-253
52. Mendonca VRR, Queiroz ATL, Lopes FM, Andrade BB, Barral-Netto M. Networking the host immune response in Plasmodium vivax malaria. Malar J (2013) 12:69. doi:10.1186/1475-2875-12-69
53. Ockenhouse CF, Hu WC, Kester KE, Cummings JF, Stewart A, Heppner DG, et al. Common and divergent immune response signaling pathways discovered in peripheral blood mononuclear cell gene expression patterns in presymptomatic and clinically apparent malaria. Infect Immun (2006) 74:5561–73. doi:10.1128/IAI.00408-06
54. Sharma S, DeOliveira RB, Kalantari P, Parroche P, Goutagny N, Jiang Z, et al. Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity (2011) 35:194–207. doi:10.1016/j.immuni.2011.05.016
55. Mahanta A, Kar SK, Kakati S, Baruah S. Heightened inflammation in severe malaria is associated with decreased IL-10 expression levels and neutrophils. Innate Immun (2015) 21:546–52. doi:10.1177/1753425914561277
56. Storm J, Craig AG. Pathogenesis of cerebral malaria-inflammation and cytoadherence. Front Cell Infect Microbiol (2014) 4:100. doi:10.3389/fcimb.2014.00100
57. Lourembam SD, Sawian CE, Baruah S. Dysregulation of cytokines expression in complicated falciparum malaria with increased TGF-beta and IFN-gamma and decreased IL-2 and IL-12. Cytokine (2013) 64:503–8. doi:10.1016/j.cyto.2013.08.007
58. Tahar R, Albergaria C, Zeghidour N, Ngane VF, Basco LK, Roussilhon C. Plasma levels of eight different mediators and their potential as biomarkers of various clinical malaria conditions in African children. Malar J (2016) 15:337. doi:10.1186/s12936-016-1378-3
59. Engwerda CR, Mynott TL, Sawhney S, De Souza JB, Bickle QD, Kaye PM. Locally up-regulated lymphotoxin alpha, not systemic tumor necrosis factor alpha, is the principle mediator of murine cerebral malaria. J Exp Med (2002) 195:1371–7. doi:10.1084/jem.20020128
60. Hsieh FL, Turner L, Bolla JR, Robinson CV, Lavstsen T, Higgins MK. The structural basis for CD36 binding by the malaria parasite. Nat Commun (2016) 7:12837. doi:10.1038/ncomms12837
61. Bernabeu M, Danziger SA, Avril M, Vaz M, Babar PH, Brazier AJ, et al. Severe adult malaria is associated with specific PfEMP1 adhesion types and high parasite biomass. Proc Natl Acad Sci U S A (2016) 113:E3270–9. doi:10.1073/pnas.1524294113
62. Sarangi PP, Lee HW, Kim M. Activated protein C action in inflammation. Br J Haematol (2010) 148:817–33. doi:10.1111/j.1365-2141.2009.08020.x
63. Gazzinelli RT, Kalantari P, Fitzgerald KA, Golenbock DT. Innate sensing of malaria parasites. Nat Rev Immunol (2014) 14:744–57. doi:10.1038/nri3742
64. Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol (2012) 13:325–32. doi:10.1038/ni.2231
65. Kalantari P, DeOliveira RB, Chan J, Corbett Y, Rathinam V, Stutz A, et al. Dual engagement of the NLRP3 and AIM2 inflammasomes by Plasmodium-derived hemozoin and DNA during malaria. Cell Rep (2014) 6:196–210. doi:10.1016/j.celrep.2013.12.014
66. Durai P, Govindaraj RG, Choi S. Structure and dynamic behavior of toll-like receptor 2 subfamily triggered by malarial glycosylphosphatidylinositols of Plasmodium falciparum. FEBS J (2013) 280:6196–212. doi:10.1111/febs.12541
67. Pichyangkul S, Yongvanitchit K, Kum-Arb U, Hemmi H, Akira S, Krieg AM, et al. Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a toll-like receptor 9-dependent pathway. J Immunol (2004) 172:4926–33. doi:10.4049/jimmunol.172.8.4926
68. Mahanta A, Baruah S. Lower expression of GATA3 and T-bet correlates with downregulated IL-10 in severe falciparum malaria. Clin Transl Immunology (2015) 4:e49. doi:10.1038/cti.2015.30
69. Shaham SH. IL-10 Regulation in Complicated Malaria. [dissertation/master’s thesis], Tezpur University.
70. Croft SL, Seifert K, Yardley V. Current scenario of drug development for leishmaniasis. Indian J Med Res (2006) 123:399–410.
71. Ganguli P, Chowdhury S, Sarkar RR. Identification of Th1/Th2 regulatory switch to promote healing response during leishmaniasis: a computational approach. EURASIP J Bioinform Syst Biol (2015) 2015(1):13. doi:10.1186/s13637-015-0032-7
72. Kumar R, Engwerda C. Vaccines to prevent leishmaniasis. Clin Transl Immunology (2014) 3:e13. doi:10.1038/cti.2014.4
73. Subramanian A, Sarkar RR. Revealing the mystery of metabolic adaptations using a genome scale model of Leishmania infantum. Sci Rep (2017) 7:10262. doi:10.1038/s41598-017-10743-x
74. Saunders EC, Ng WW, Kloehn J, Chambers JM, Ng M, McConville MJ. Induction of a stringent metabolic response in intracellular stages of Leishmania mexicana leads to increased dependence on mitochondrial metabolism. PLoS Pathog (2014) 10:e1003888. doi:10.1371/journal.ppat.1003888
75. Tuon FF, Amato VS, Bacha HA, Almusawi T, Duarte MI, Amato Neto V. Toll-like receptors and leishmaniasis. Infect Immun (2008) 76:866–72. doi:10.1128/IAI.01090-07
76. Faria MS, Reis FC, Lima AP. Toll-like receptors in Leishmania infections: guardians or promoters? J Parasitol Res (2012) 2012:930257. doi:10.1155/2012/930257
77. Bhardwaj S, Srivastava N, Sudan R, Saha B. Leishmania interferes with host cell signaling to devise a survival strategy. J Biomed Biotechnol (2010) 2010:109189. doi:10.1155/2010/109189
78. Shio MT, Hassani K, Isnard A, Ralph B, Contreras I, Gomez MA, et al. Host cell signalling and leishmania mechanisms of evasion. J Trop Med (2012) 2012:819512. doi:10.1155/2012/819512
79. Cummings HE, Tuladhar R, Satoskar AR. Cytokines and their STATs in cutaneous and visceral leishmaniasis. J Biomed Biotechnol (2010) 2010:294389. doi:10.1155/2010/294389
80. Diaz YR, Rojas R, Valderrama L, Saravia NG. T-bet, GATA-3, and Foxp3 expression and Th1/Th2 cytokine production in the clinical outcome of human infection with Leishmania (Viannia) species. J Infect Dis (2010) 202:406–15. doi:10.1086/653829
81. Olivier M, Gregory DJ, Forget G. Subversion mechanisms by which Leishmania parasites can escape the host immune response: a signaling point of view. Clin Microbiol Rev (2005) 18:293. doi:10.1128/CMR.18.2.293-305.2005
82. Kaye PM, Rogers NJ, Curry AJ, Scott JC. Deficient expression of co-stimulatory molecules on Leishmania-infected macrophages. Eur J Immunol (1994) 24:2850–4. doi:10.1002/eji.1830241140
83. Soong L, Xu JC, Grewal IS, Kima P, Sun J, Longley BJ Jr., et al. Disruption of CD40-CD40 ligand interactions results in an enhanced susceptibility to Leishmania amazonensis infection. Immunity (1996) 4:263–73. doi:10.1016/S1074-7613(00)80434-3
84. McMahon-Pratt D, Alexander J. Does the Leishmania major paradigm of pathogenesis and protection hold for New World cutaneous leishmaniases or the visceral disease? Immunol Rev (2004) 201:206–24. doi:10.1111/j.0105-2896.2004.00190.x
85. Gregory DJ, Sladek R, Olivier M, Matlashewski G. Comparison of the effects of Leishmania major or Leishmania donovani infection on macrophage gene expression. Infect Immun (2008) 76:1186–92. doi:10.1128/IAI.01320-07
86. McCall LI, Zhang WW, Matlashewski G. Determinants for the development of visceral leishmaniasis disease. PLoS Pathog (2013) 9:e1003053. doi:10.1371/journal.ppat.1003053
87. Mendez S, Reckling SK, Piccirillo CA, Sacks D, Belkaid Y. Role for CD4(+) CD25(+) regulatory T cells in reactivation of persistent leishmaniasis and control of concomitant immunity. J Exp Med (2004) 200:201–10. doi:10.1084/jem.20040298
88. Hill AV, Jepson A, Plebanski M, Gilbert SC. Genetic analysis of host-parasite coevolution in human malaria. Philos Trans R Soc Lond B Biol Sci (1997) 352:1317–25. doi:10.1098/rstb.1997.0116
89. Aurrecoechea C, Barreto A, Basenko EY, Brestelli J, Brunk BP, Cade S, et al. EuPathDB: the eukaryotic pathogen genomics database resource. Nucleic Acids Res (2017) 45:D581–91. doi:10.1093/nar/gkw1105
90. Urban M, Pant R, Raghunath A, Irvine AG, Pedro H, Hammond-Kosack KE. The pathogen-host interactions database (PHI-base): additions and future developments. Nucleic Acids Res (2015) 43:D645–55. doi:10.1093/nar/gku1165
91. Davila AM, Mendes PN, Wagner G, Tschoeke DA, Cuadrat RR, Liberman F, et al. ProtozoaDB: dynamic visualization and exploration of protozoan genomes. Nucleic Acids Res (2008) 36:D547–52. doi:10.1093/nar/gkm820
92. Swann J, Jamshidi N, Lewis NE, Winzeler EA. Systems analysis of host-parasite interactions. Wiley interdiscip Rev Syst Biol Med (2015) 7:381–400. doi:10.1002/wsbm.1311
93. Westermann AJ, Gorski SA, Vogel J. Dual RNA-seq of pathogen and host. Nat Rev Microbiol (2012) 10:618–30. doi:10.1038/nrmicro2852
94. Wright HL, Thomas HB, Moots RJ, Edwards SW. RNA-Seq reveals activation of both common and cytokine-specific pathways following neutrophil priming. PLoS One (2013) 8:e58598. doi:10.1371/journal.pone.0058598
95. Yamagishi J, Natori A, Tolba MEM, Mongan AE, Sugimoto C, Katayama T, et al. Interactive transcriptome analysis of malaria patients and infecting Plasmodium falciparum. Genome Res (2014) 24:1433–44. doi:10.1101/gr.158980.113
96. Pittman KJ, Aliota MT, Knoll LJ. Dual transcriptional profiling of mice and Toxoplasma gondii during acute and chronic infection. BMC Genomics (2014) 15:806. doi:10.1186/1471-2164-15-806
97. Doerig C, Rayner JC, Scherf A, Tobin AB. Post-translational protein modifications in malaria parasites. Nat Rev Microbiol (2015) 13:160–72. doi:10.1038/nrmicro3402
98. Cui LW, Lindner S, Miao J. Translational regulation during stage transitions in malaria parasites. Ann N Y Acad Sci (2015) 1342:1–9. doi:10.1111/nyas.12573
Keywords: cytokine networks, manipulation, Plasmodium, Leishmania, inflammation, signalling hubs, cross regulation, system biology
Citation: Mahanta A, Ganguli P, Barah P, Sarkar RR, Sarmah N, Phukan S, Bora M and Baruah S (2018) Integrative Approaches to Understand the Mastery in Manipulation of Host Cytokine Networks by Protozoan Parasites with Emphasis on Plasmodium and Leishmania Species. Front. Immunol. 9:296. doi: 10.3389/fimmu.2018.00296
Received: 14 November 2017; Accepted: 01 February 2018;
Published: 23 February 2018
Edited by:
Alexandre Morrot, Universidade Federal do Rio de Janeiro, BrazilReviewed by:
Danielle Oliveira Nascimento, Universidade Federal do Rio de Janeiro, BrazilElisangela Costa Da Silva, State University of Norte Fluminense, Brazil
Copyright: © 2018 Mahanta, Ganguli, Barah, Sarkar, Sarmah, Phukan, Bora and Baruah. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shashi Baruah, c2JhcnVhaEB0ZXp1LmVybmV0Lmlu, c2FzaGliYXJ1YWhAZ21haWwuY29t
†Joint first author