
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 12 February 2018
Sec. HIV and AIDS
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00214
 Alessandra Bandera1*†
Alessandra Bandera1*† Michela Masetti2,3†
Michela Masetti2,3† Massimiliano Fabbiani1
Massimiliano Fabbiani1 Mara Biasin2
Mara Biasin2 Antonio Muscatello1
Antonio Muscatello1 Nicola Squillace1
Nicola Squillace1 Mario Clerici3,4
Mario Clerici3,4 Andrea Gori1,5*
Andrea Gori1,5* Daria Trabattoni2
Daria Trabattoni2
Background: Inflammasome-mediated activation of caspase-1 regulates inflammatory responses and pyroptosis. We analyzed possible associations between inflammasome-related genes and immune reconstitution in HIV-infected antiretroviral therapy (ART)-treated patients.
Methods: Cross-sectional, case–control study. HIV-infected patients on ART for ≥24 months with HIV-RNA<50 cp/mL for ≥12 months were enrolled and defined as immunological responders (IR) or non-responders (INR) if CD4 count was ≥500 or ≤350 cells/μL, respectively. Expression of inflammasome genes, caspases 1, 3, 4, 5 and γ-interferon-inducible protein 16 (IFI16) was measured in unstimulated and LPS- or aldrithiol-2-treated HIV-1BaL virions-stimulated peripheral blood mononuclear cells. Microbial translocation markers were evaluated.
Results: Thirty-nine patients (22 IRs; 17 INRs) were enrolled. LPS-stimulated inflammasome genes were significantly upregulated in INRs. Whereas HIV-1BaL stimulation induced (NOD)-like receptor (NLR) family pyrin domain containing 3 (NLRP3) expression in both IRs and INRs, NLRP3 and IL-18 expression was significantly increased in INRs compared to IRs. Significant higher caspase-1 expression was seen as well, whereas caspase 3, 4, and 5 expression was similar in both groups. No differences in microbial translocation markers (LPS and soluble CD14) were detected in the two groups.
Conclusion: Upregulation of NLRP3 and caspase-1 is observed in INR patients. This could play a role in persistent immune activation that characterize INRs. Caspase-1 upregulation could induce CD4 T-cell loss via pyroptosis, contributing to unsatisfactory CD4 T-cells recovery.
Summary: Higher levels of the inflammasome genes, NLRP3 and caspase-1, are observed in ART-treated patients who lack immunological recovery. Inflammasome upregulation in immunological non-responders (INR) may be partly responsible for the persistence of immune activation and to CD4 T-cell loss via pyroptosis.
Elevated levels of inflammation and immune activation, which can persevere in HIV-infected individuals on virologically effective antiretroviral therapy (ART), have been associated to morbidity and mortality from non-AIDS events, such as cardiovascular disease, neurocognitive dysfunction, and kidney and bone disorders (1, 2). These events are more frequently observed in HIV-infected patients unable to reach a CD4+ T-cell count of 500/mm3, thus defined as INR.
Increases in immune activation (3) and inflammation (4) are currently believed to be the principal correlates of poor immunological response to ART, affecting T-cell pool homeostasis and modifying T lymphocytes counts at peripheral and thymic levels. Peculiar host genetic factors, as polymorphisms of genes of the pathway relative to inflammation/apoptosis (5) or genes implicated in T-cell homeostasis, such as IL7R (6), have also been correlated to suboptimal CD4+ T-cell reconstitution.
Inflammasome activation is essential for host response to pathogens, but recently clear roles for the inflammasome in the pathogenesis of inflammatory diseases has been evidenced in several studies [rev. in Ref. (7)]. The nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family, pyrin domain containing 3 (NLRP3) inflammasome is the prevalently studied among inflammasomes. Upon inflammasome activation, the NLRP3 protein recruits the inflammasome adaptor protein apoptosis-associated speck-like protein (ASC), which contains a C-terminal caspase recruitment domain, which interacts with pro-caspase-1 inducing its cleavage and activation. Inflammasome-dependent caspase-1 activity results in a specific inflammatory form of cell death known as pyroptosis, where dying cells release their cytoplasmic contents, including inflammatory cytokines, into the extracellular space (8, 9). Molecular mechanisms of chronic inflammation observed in HIV-infected patients successfully treated with ART are not clear. Recently, pyroptosis has been implicated in CD4 + T-cell loss even in absence of ongoing productive HIV replication (10–14).
As the role of the inflammasome system in the lack of immune reconstitution during ART has not been investigated till now, we analyzed possible associations between inflammasome activity, caspase-1 activation, pyroptosis, and immune reconstitution in HIV-infected ART-treated patients. Specifically, we assessed the expression of NLRP3 inflammasome, caspases 1, 3, 4, 5, and pro-inflammatory cytokines in peripheral blood mononuclear cells (PBMCs) of HIV-infected patients under suppressive ART and investigated their association with CD4+ T-cell count reconstitution.
Cross-sectional, case–control study in which 39 HIV-infected ART-treated patients have been enrolled at the Unit of Infectious Diseases, San Gerardo Hospital, Monza, Italy. Patients identified as cases were HIV-infected patients defined as INR; controls were HIV-infected patients defined as immunological responders (IRs).
Inclusion criteria were: men and women >18 years of age, HIV positivity tested with ELISA and confirmed by Western Blot, treatment with combined ART (i.e., >3 antiretroviral drugs), duration of ART > 24 months, plasma HIV-RNA < 50 cp/mL for at least 12 months, CD4 + T lymphocyte count<350/mmc if INR, CD4 + T lymphocyte count >500/mmc if IRs, informed consent signed. Immunological response to ART was considered optimal when CD4+ T-cell counts were above 500 cells/mmc as these levels of immune reconstitution are associated with morbidity and mortality rates close to uninfected individuals (15).
Exclusion criteria were as follows: presence of actual opportunistic AIDS-related diseases, HCV chronic infection, chronic inflammatory disorders, and ongoing immunosuppressive therapy.
This study was conducted in accordance with the recommendations of the Ethical Committee of our Institution (Comitato Etico Brianza, Italy) including written informed consent from all patients. All subjects provided written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Ethical Committee of Brianza on 29th May 2014.
Whole blood was collected by venipuncture in Vacutainer tubes containing EDTA (Becton Dickinson, Rutherford, NJ, USA). Plasma was stored at −80°C for further analysis and PBMCs were separated on lymphocyte separation medium (Cedarlane laboratories Limited, Hornby, ON, Canada) and washed twice in phosphate-buffered saline (PBI, Milan, Italy). Number of viable leukocytes was determined by automated cell counter ADAM-MC (Digital Bio, NanoEnTek Inc., Korea).
Peripheral blood mononuclear cells were cultured in RPMI medium supplemented with 1% l-glutamine (Sigma-Aldrich, Milan, Italy), 1% penicillin/streptomycin (Sigma-Aldrich, Milan, Italy), and 10% Human AB Serum (Euroclone, Milan, Italy). PBMCs were incubated for 3 h either in absence or in presence of LPS (1 µg/ml) (Sigma-Aldrich, Milan, Italy) and for 24 h either in absence or in presence of aldrithiol-2 (AT2 treated)- HIV-1BaL virions (300 ng/ml).
Analyses were performed both in basal and stimulated conditions. mRNA was extracted from 1 × 106 PBMCs by using the acid guanidinium thiocyanate–phenol–chloroform method, dissolved in RNase-free water, and purified from genomic DNA with RNase-free DNase (New England BioLabs, Ipswich, MA, USA). One microgram of RNA was reverse-transcribed into first-strand cDNA in a 20-µl final volume containing 1 µM random hexanucleotide primers, 1 µM oligo (dT), and 200 U of Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI, USA). Inflammasome signaling pathways were analyzed by a PCR array including a set of 84 optimized real-time PCR primer assays on 96-well plates (SABiosciences Corporation, Frederick, MD, USA) according to manufacturer’s instructions. The experiments have been run on samples pooled into two distinct groups (INRs and IRs) on the basis of condition (unstimulated, LPS-stimulated and AT2-treated HIV-1BaL stimulated) and represent the mean value of the different targets analyzed in each group. The results were calculated relative to the housekeeping genes GAPDH and β-actin. Data were analyzed by SABioscences online software and only variables with at least a twofold increase in their value are presented, as previously described (16, 17).
For Real-Time PCR (96 CFX Connect Bio-Rad) experiments, reactions were performed using a SYBR Green PCR mix (iTaq™ Universal SYBR Green Supermix, Bio-Rad). GAPDH and β-Actin were used as housekeeping genes and the mean Ct of these two genes was used as housekeeping gene. Results are expressed as fold change 2^ΔΔCt and were calculated according to Livak’s method (18). Primers sequences were the following: GAPDH primers: forward CGGATTTGGTCGTATTGGG, reverse GCTTCCCGTTCTCAGCCTTG; β-actin primers: forward ATGCCCAGGAAGGAAGGCTG, reverse GGGAAATCGTGCGTGACATT; IL-1β primers: forward TTCTGCTTGAGAGGTGCTGATG, reverse TGTCCTGCGTGTTGAAAGATGA; IL-18 primers: forward GCTGCTGAACCAGTAGAAGAC, reverse CCGATTTCCTTGGTCAATGAAGA. For detection of NLRP3, Caspase-1, Caspase-3, Caspase-4, Caspase-5 and γ-interferon-inducible protein 16 (IFI16) primers were purchased already optimized (PrimePCR, Bio-Rad, Segrate, Italy).
Caspase-1 was measured by Quantikine ELISA Kit (R&D Systems; Minneapolis, MN, USA) according to manufacturer’s instructions. Plasma samples were diluted 1:2 prior to assay.
LPS was measured by Limulus Amebocyte Lysate (LAL) Chromogenic Endpoint Assay (Hycult Biotechnology, Uden, The Netherlands). Samples, standards, and reagents were prepared according to manufacturer’s instructions. Plasma samples were heated at 75°C for 5 min in order to neutralize endotoxin inhibiting compounds and diluted 1:5 in endotoxin free water prior to assay. Soluble CD14 (sCD14) was measured by Quantikine ELISA Kit (R&D Systems, Minneapolis, MN, USA), following manufacturer’s instructions; plasma samples were diluted 1:400 prior to assay.
This is a pilot study without sample size computation. Potential differences of inflammasome activation between the two groups (INR vs IR) have been evaluated.
Descriptive results have been presented as mean ± SE upon description of data distribution (median, percentiles, minimum, maximum), medians with interquartile range (IQR). Inferential statistics using either parametric (unpaired Student’s t-test) or non-parametric tests (Mann–Whitney t-test) have been used, as appropriate. Sample distributions were assessed by KS normality test. Variables associated with inflammasome gene expression were also evaluated by univariate and multivariate linear regression analysis. Data were analyzed by GraphPad Prism analysis software.
Thirty-nine HIV-infected patients, 17 INRs and 22 IRs, were enrolled in the study. Demographic characteristics of patients are shown in Table 1. Median age was 47 years, time of HIV infection diagnosis 10 years, time with HIV-RNA<50 cp/mL 57 months. INR patients were older (median 60 vs 43 years, p < 0.001) and, as expected, had a higher prevalence of past AIDS-defining illnesses (76 vs 18%, p < 0.001). Nadir CD4 T-cell count was significantly lower in INRs as compared to IRs [median 44 (IQR 25–83) cells/μL vs 196 (IQR 85–298) cells/μL, p < 0.01] Median CD4 count was 295 (IQR 256–343) cells/μL in INRs vs 840 (IQR 718–1131) cells/μL in IRs, median CD4 percentage was 19% (IQR 16–21) in INRs, and 34% (IQR 28–40) in IRs. No significant differences were observed between the two groups when years from HIV diagnosis, duration of HIV-RNA suppression, years from ART initiation and current regimen of AR were analyzed.
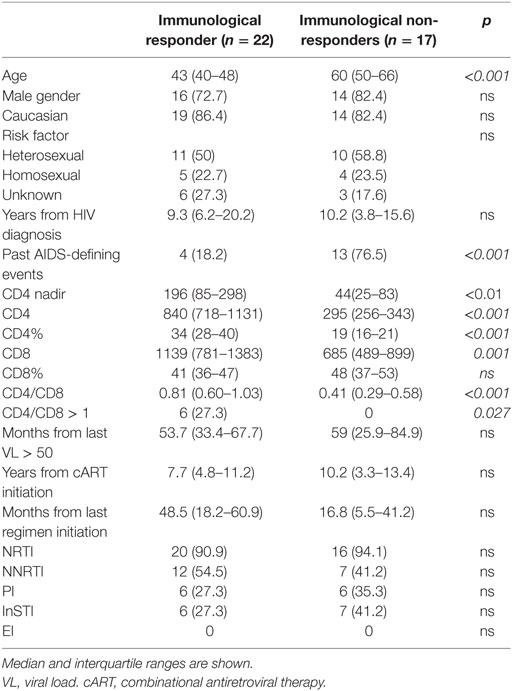
Table 1. Clinical and demographic characteristics of HIV-infected patients enrolled in the study.
To evaluate the expression of the inflammasome pathway, we performed a real-time PCR Array on PBMCs that were unstimulated or stimulated with either LPS or AT2-treated HIV-1BaL; samples were pooled into two distinct groups IRs and INRs (Figure 1). LPS is an activator of inflammasome response, whereas stimulation with AT2-treated HIV was performed in order to evaluate if the virus can potentially trigger an inflammasome response even in the context of chronic and unproductive HIV infection. Upon LPS stimulation, INRs showed an upregulation (nfold LPS-stimulated vs unstimulated >2) of many targets, including caspase-1, IL-1β, IL-18, NLRP3, and other pro-inflammatory cytokines, such as TNF and IL-6 and kinases involved in cellular death pathways such as RIPK1 and RIPK2 compared to IRs. AT2-HIV1-BaL stimulation was less potent in triggering inflammasome responses, but we could still detect an upregulation (nfold AT2-HIV-1 stimulation vs unstimulated >2) for caspase-1 both in IRs and INRs, which is interesting considering its pivotal role in pyroptotic cell death and inflammation.
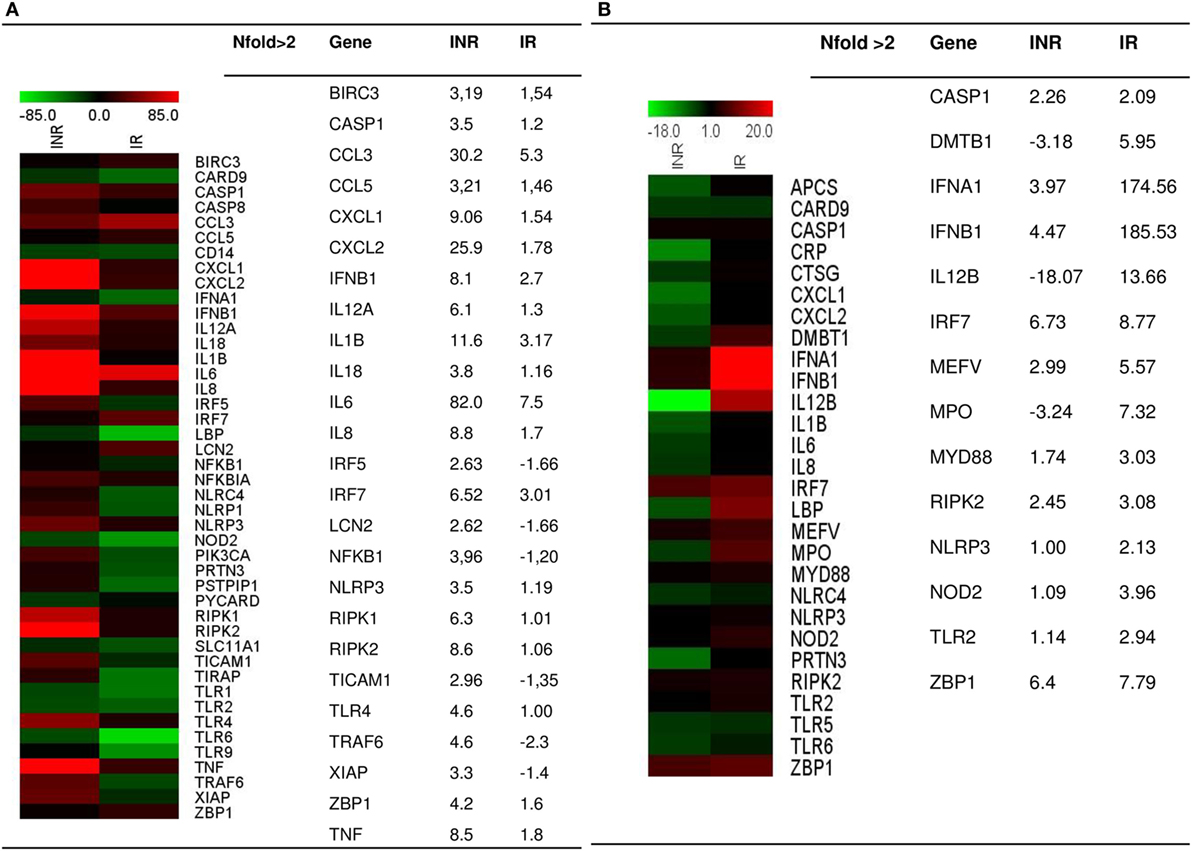
Figure 1. Inflammasome pathway. Real-time PCR Array on LPS- or AT2-treated-HIV-1BaL-stimulated peripheral blood mononuclear cells (PBMCs) in INR and IR HIV-infected patients. Samples were pooled into two distinct groups (IRs and INRs) and experiment was done in triplicate. Gene expression (nfold) is shown as a color scale from green to red (MEV multiple experiment viewer software). (A) Nfold LPS-stimulated PBMCs vs unstimulated PBMCs; (B) Nfold AT2-treated-HIV-1BaL PBMCs vs unstimulated PBMCs. Only targets showing at least >2-fold modulation are shown in table.
Given the results of the qRT-PCR array analysis, we further characterized the expression of inflammasome genes in each subject included in the study either in unstimulated or in LPS- or AT2-HIV-1BaL-stimulated PBMCs. In particular NLRP3, IL1β, IL-18, CASPASE-1, and IFI16 gene expression was evaluated (Figure 2). Upon LPS stimulation, we observed a significant increase of IL-1β (p < 0.05), IL-18, and caspase-1 (p < 0.02) in INRs compared to IRs. Upon AT2-HIV-1BaL stimulation, we observed significant increases of NLRP3 (p < 0.05), IL-18 (p < 0.02) and caspase-1 (p < 0.02) in INRs compared to IRs. Of note, NLRP3 expression was significantly higher (p = 0.02) in INRs compared to IRs in unstimulated cells and this association was confirmed also after adjustment for either age (mean change +2.052, 95% CI 0.208–3.896, p = 0.03) or CD4 + T-cell nadir (mean change 2.222, 95% CI 0.277–4.167, p = 0.027). Altogether these data point out that a higher degree of inflammasome activation is seen in INRs compared to IRs. Of note, the gene expression of caspase-1, which is the key effector molecule of inflammasome pathway and pyroptotic cell death, was significantly higher in INRs in each condition analyzed. This association was confirmed after adjustment for CD4 + T-cells nadir value (p = 0.039, mean change = 15.850, 95% CI 0.860–30.839 for LPS stimulation; p = 0.032, mean change = 6.785, 95% CI 0.612–12.958 for AT2-stimulation) but not after adjustment for age. Expression levels of IFI16 were similar in both groups analyzed.
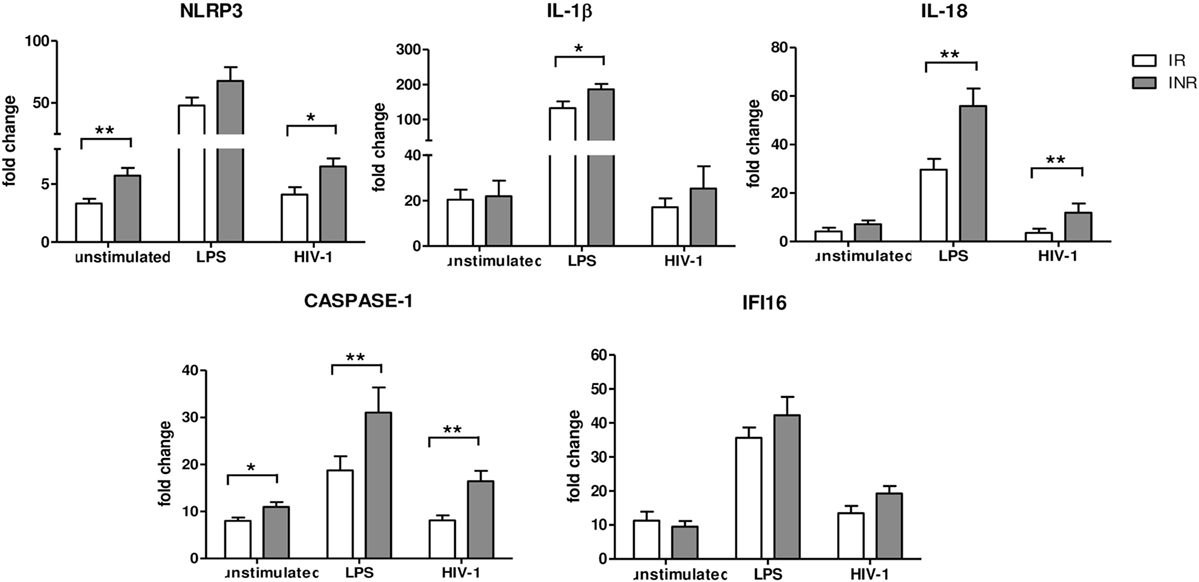
Figure 2. Inflammasome and pyroptosis gene expression. NLRP3, IL1β, IL-18, Caspase-1, and γ-interferon-inducible protein 16 (IFI16) expressions were quantified in peripheral blood mononuclear cells (PBMCs) from each patient enrolled in the study (n = 22 for IRs and n = 17 for INRs) either unstimulated or LPS or AT2-treated-HIV-1BaL stimulated. White bars represent IRs, gray bars represent INRs. Mean values and SEM are shown. *p < 0.05; **p < 0.02. Fold change expressed as 2ΔΔCt.
NLRP3 expression can be modulated also via a “non-canonical pathway,” which is mediated by caspase-4 and caspase-5. Therefore, we decided to evaluate caspase-4 and caspase-5 gene expression in INRs and IRs as well. Results showed that the expression of these caspases was comparable in both groups of patients, indicating that the “non-canonical” pathway is not involved in NLRP3 upregulation in these patients and in the context of suppressed viremia (Figure 3).
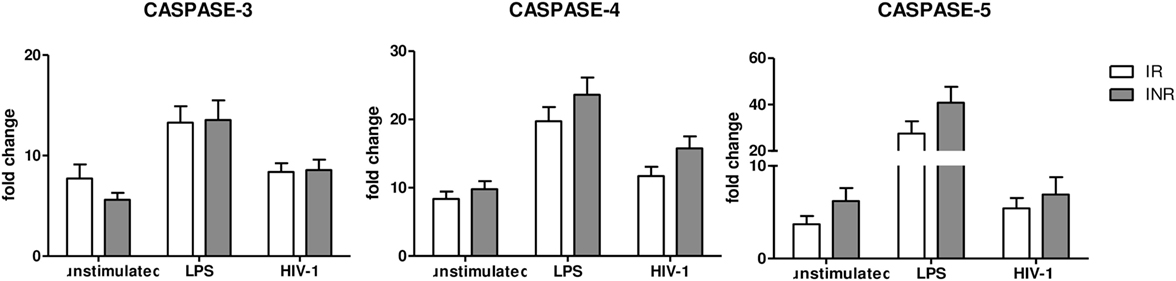
Figure 3. Caspase 3, 4, and 5 gene expression. Caspase 3, 4 and 5 expressions were quantified in peripheral blood mononuclear cells (PBMCs) from each patient enrolled in the study (n = 22 for immunological responders (IRs) and n = 17 for INRs) either unstimulated or LPS or AT2-treated-HIV-1BaL stimulated. Mean values and SEM are shown. White bars represent IRs, gray bars represent INRs. Fold change expressed as 2ΔΔCt.
Caspase-3 is associated with apoptosis, a form of programmed cell death that does not lead to inflammation. We observed no significant differences between the two groups in any of the conditions analyzed (Figure 3), suggesting that caspase-3-mediated apoptosis may be similar in INRs and IRs.
We further investigated caspase-1 expression in plasma of INRs and IRs. Results indicated that higher median levels of caspase-1 expression were observed in INRs compares to IRs (INRs: 526.7 vs IRs: 375.3 pg/ml); these differences approached but did not reach statistical significance (data not shown).
Microbial translocation has been suggested to be the main driver of immune activation in HIV-infected patients. We hypothesized that increased expression of some inflammasomes-related genes observed in INRs could be due to increased levels of microbial translocation. To this end, we analyzed plasma levels of sCD14 and LPS, two of the main hallmarks of microbial translocation. Unexpectedly, we could not detect any differences between the two groups of patients (data not shown, sCD14 median levels: IRs 1,626,000 vs INRs 1,723,000 pg/ml; LPS median levels: IRs 46.24 vs 47.87 EU/ml), indicating that microbial translocation is not the main driver of inflammasome upregulation in INRs and that, probably, does also not play a pivotal role in immune activation in ART-treated patients.
Our study provides insight into one of the major mechanisms involved in the lack of immune recovery in HIV-infected subjects experiencing virological suppression under ART regimens. Differently from other studies analyzing poor immune recovery, we applied strict patients’ selection criteria, and most of the characteristics influencing immune recovery that emerged from previous studies were considered in our population (age, sex, durations of infection and treatment, co-infections, duration of HIV-RNA suppression, and nadir CD4+ counts).
Persistance of immune activation and high levels of inflammation have been identified as the underlying mechanisms inducing suboptimal immunological response to ART. We showed that NLRP3 expression is significantly increased in unstimulated or HIV-stimulated PBMCs of INRs as compared to patients who experienced complete immune recovery under ART. This result is of particular interest because, while there is ample evidence of inflammasome role during acute infection (19–22), there are no reports, to this date, on inflammasome expression during chronic infection. Importantly, we found that the condition of unsatisfactory CD4+ T-cell recovery under ART was associated with a significantly increased expression of NLRP3 even after adjustment for age and CD4+ T-cell nadir, which are known conditions able to influence immune recovery and immune activation in virologically suppressed patients. To characterize inflammasome expression in IRs and INRs, we stimulated PBMCs with LPS, which is known to be a potent activator of inflammasomes pathway. In LPS-stimulated PBMCs we observed that INRs were more responsive to LPS stimulation compared to IRs, suggesting a higher immune activation status, with an upregulation of NLRP3 and caspase-1 gene expression. In addition, we also reported a significant increase of inflammasome-related cytokines, such as IL-1β and IL-18 that are directly linked to inflammasome activation.
Next, we verified if HIV could be able to activate inflammasomes even in the settings of chronic infection as this issue has not yet been clearly addressed by other studies. Pontillo et al. reported that HIV is able to induce NLRP3 expression upon infection, but that fails to do so in chronically activated cells from infected individuals (19). Interestingly, we observed that HIV was indeed less efficient in stimulating the inflammasome pathway compared to LPS; however, we could still detect an upregulation of NLRP3, IL-1β, and IL-18 in INRs compared to IRs suggesting that inflammasome pathway is upregulated in these patients compared to IRs. Our results are apparently in contrast with pieces of evidence from a previous work by Nasi et al. who found that the expression of genes involved in inflammasome pathways did not significantly differ between HIV-infected patients under ART and healthy subjects (23). However, HIV-infected patients evaluated in that study had higher CD4+ T-cell count (about the range of normality) and inflammasome activation was analyzed only in basal conditions, whereas we found major differences after LPS- and HIV-stimulation between INR and IR patients.
Moreover, we found that INRs express significantly higher levels of caspase-1 either in unstimulated or LPS- or AT2-treated-HIV-1BaL treated cells, even after adjustment for CD4+ T-cell nadir, thus suggesting a role of caspase-1-dependent pyroptosis in CD4+ T-cell death and probably in the lack of immune recovery. In the age-adjusted analysis, the association between INRs and higher levels of caspase-1 was lost; however, our analysis was partly limited by the small sample size. This finding is consistent with recent studies in which pyroptosis has been shown as a major player in CD4+ T lymphocyte loss contributing to immune activation in HIV infection. It has also been reported that this process would take place primarily in the lymph node, rather than in the blood. Indeed, even though quiescent blood CD4+ T cell support HIV entry and fusion, they are also highly resistant to pyroptotic cell death and this is probably due to their deeper state of cellular rest (13). Nevertheless, we were able to observe an upregulation of genes involved in inflammasome pathway, especially caspase-1, and this could be due to the fact that we analyzed PBMCs, which are a heterogeneous cell population compromising not only CD4+ T cells but other lymphocytes and monocytes too. Monocytes are known for constitutive caspase-1 expression (24) and release of IL-1β upon TLR4 stimulation. We can hypothesize that t the higher expression levels of inflammasome components observed in INRs could depend on increased monocytes activation in INRs compared to IRs. Of note, the array data of LPS-stimulated PBMCs, showed a significant upregulation of TLR4 in INRs compared to IRs. In agreement with reports from Muñoz-Arias et al., (13), we could not detect any differences in IFI16 expression in PBMCs from INRs and IRs, which is probably due to the resting condition of blood CD4+ T cells.
Recently microbial translocation has been considered one of the main drivers of immune activation during HIV infection and so we decided to evaluate this phenomenon in our patients. We wondered if the up-regulation of the inflammasome pathway observed in INRs could be dependent on increased levels of microbial translocation. Surprisingly, we could not detect any differences in plasma levels of LPS and sCD14 between INRs and IRs, indicating that microbial translocation is not the main driver of inflammasome and immune activation in our cohort. Consistent with our results are also recent reports by Tincati et al. and Stiksrud et al. in which no differences in LPS and sCD14 plasma levels could be detected between INRs and IRs (25, 26). It has been suggested that probably while microbial translocation markers have been shown to predict clinical outcome in untreated HIV infection, their use in ART-treated patients may not be as accurate (27).
In conclusion, it is tempting to speculate that higher inflammasome up-regulation observed in INRs may be due to residual HIV defective replication taking place in immune sanctuaries, which would lead to inflammation, immune activation and cell death. Of note, there have been reports showing that cell-to-cell transmission of HIV is not efficiently inhibited by ART and that it effectively triggers pyroptotic cell death of lymphoid-tissue-derived CD4+ T cells (11, 14), suggesting that depletion of CD4+ T cells would still take place in spite of suppressed viremia. In addition, it has been demonstrated that defective proviruses are able to transcribe novel protein-coding RNA species in HIV-infected patients on ART and with suppressed viremia (RNA levels <50 copies/ml) (28) and recent data presented at CROI 2017 demonstrated the production of HIV-1 proteins from these defective proviruses (29). These viral proteins produced even in the settings of suppressed viremia could be sensed by inflammasomes, leading to higher rates of immune activation during chronic infection and possibly to lack of immune response in INRs. The higher inflammasome upregulation observed in INRs may depend on an increased number of monocytes in these patients. It is of particular interest considering that activation of innate immunity has been linked to morbidity and mortality in the setting of ART-treated HIV infection (30, 31). Our data indicate that inflammasome and caspase-1 may be involved and play an important role in the lack of immunological response and higher immune activation status of INRs, however further investigations are necessary in order to fully prove this hypothesis. We expect that new studies on the molecular pathways involved in inflammasome, and pyroptotic cell death will shed light on molecular markers and features of these processes that could then be investigated in INRs.
This study was carried out in accordance with the recommendations of the Ethical Committee of our Institution (Comitato Etico Brianza, Italy) with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Ethical Committee of Brianza on 29th May 2014.
MM, MB, and DT performed the experiments; AB, MF, AM, and NS collected patients data and analyzed results; AB, AM, MC, AG, and DT designed the research; AB, DT, and MM wrote the paper.
This study was supported by a grant from Gilead Fellowship Program 2013, by a grant from ANLAIDS Lombardia.
This study was supported by a grant from Gilead Fellowship Program 2013 and by a grant from ANLAIDS Lombardia. MM was supported by a Ph.D. fellowship from University of Milan, PhD Course in Molecular and Translational Medicine, Milan, Italy. We particularly thank all the patients participating in the study, and the staff of the Unit of Infectious Diseases, San Gerardo Hospital, who cared for the patients.
1. Deeks SG, Lewin SR, Havlir DV. The end of AIDS: HIV infection as a chronic disease. Lancet (2013) 382:1525–33. doi:10.1016/S0140-6736(13)61809-7
2. Deeks SG, Tracy R, Douek DC. Systemic effects of inflammation on health during chronic HIV infection. Immunity (2013) 39:633–45. doi:10.1016/j.immuni.2013.10.001
3. Hunt PW, Martin JN, Sinclair E, Bredt B, Hagos E, Lampiris H, et al. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J Infect Dis (2003) 187:1534–43. doi:10.1086/374786
4. Wilson EM, Singh A, Hullsiek KH, Gibson D, Henry WK, Lichtenstein K, et al. Monocyte-activation phenotypes are associated with biomarkers of inflammation and coagulation in chronic HIV infection. J Infect Dis (2014) 210:1396–406. doi:10.1093/infdis/jiu275
5. Haas DW, Geraghty DE, Andersen J, Mar J, Motsinger AA, D’Aquila RT, et al. Immunogenetics of CD4 lymphocyte count recovery during antiretroviral therapy: an AIDS clinical trials group study. J Infect Dis (2006) 194(8):1098–107. doi:10.1086/507313
6. Rajasuriar R, Booth D, Solomon A, Chua K, Spelman T, Gouillou M. Biological determinants of immune reconstitution in HIV-infected patients receiving antiretroviral therapy: the role of interleukin 7 and interleukin 7 receptor alpha and microbial translocation. J Infect Dis (2010) 202:1254–64. doi:10.1086/656369
7. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi:10.1038/nri.2016.58
8. Hitzler I, Sayi A, Kohler E, Engler DB, Koch KN, Hardt WD. Caspase-1 has both proinflammatory and regulatory properties in Helicobacter infections, which are differentially mediated by its substrates IL-1β and IL-18. J Immunol (2012) 188:3594–602. doi:10.4049/jimmunol.1103212
9. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol (2006) 8:1812–25. doi:10.1111/j.1462-5822.2006.00751.x
10. Doitsh G, Galloway NLK, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature (2017) 505:509–14. doi:10.1038/nature22066
11. Doitsh G, Greene WC. Dissecting how CD4 T cells are lost during HIV infection. Cell Host Microbe (2016) 19:280–91. doi:10.1016/j.chom.2016.02.012
12. Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, et al. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science (2014) 343:428–32. doi:10.1126/science.1243640
13. Muñoz-Arias I, Doitsh G, Yang Z, Sowinski S, Ruelas D, Greene WC. Blood-derived CD4 T cells naturally resist pyroptosis during abortive HIV-1 infection. Cell Host Microbe (2015) 18:463–70. doi:10.1016/j.chom.2015.09.010
14. Galloway NLK, Doitsh G, Monroe KM, Yang Z, Muñoz-Arias I, Levy DN, et al. Cell-to-cell transmission of HIV-1 is required to trigger pyroptotic death of lymphoid-tissue-derived CD4 T Cells. Cell Rep (2015) 12:1555–63. doi:10.1016/j.celrep.2015.08.011
15. Lewden C, Chene G, Morlat P, Raffi F, Dupon M, Dellamonica P, et al. HIV-infected adults with a CD4 cell count greater than 500 cells/mm3 on long-term combination antiretroviral therapy reach same mortality rates as the general population. J Acquir Immune Defic Syndr (2007) 46(1):72–7. doi:10.1097/QAI.0b013e318134257a
16. Trabattoni D, Gnudi F, Ibba SV, Saulle I, Agostini S, Masetti M, et al. Thiazolides elicit anti-viral innate immunity and reduce HIV replication. Sci Rep (2016) 6:27148. doi:10.1038/srep27148
17. Rainone V, Schneider L, Saulle I, Ricci C, Biasin M, Al-Daghri NM, et al. Upregulation of inflammasome activity and increased gut permeability are associated with obesity in children and adolescents. Int J Obes (Lond) (2016) 40(6):1026–33. doi:10.1038/ijo.2016.26
18. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods (2001) 25:402–8. doi:10.1006/meth.2001.1262
19. Pontillo A, Silva LT, Oshiro TM, Finazzo C, Crovella S, Duarte JS. HIV-1 induces NALP3-inflammasome expression and interleukin-1β secretion in dendritic cells from healthy individuals but not from HIV-positive patients. AIDS (2012) 26:11–8. doi:10.1097/QAD.0b013e32834d697f
20. Pontillo A, Santillo BT, Duarte AJ, Oshiro TM. Differential inflammasome expression and IL-1β secretion in monocyte-derived dendritic cells differentiated with IL-4 or IFN-α. AIDS Res Ther (2013) 10:35. doi:10.1186/1742-6405-10-35
21. Hernandez JC, Latz E, Urcuqui-Inchima S. HIV-1 induces the first signal to activate the NLRP3 inflammasome in monocyte-derived macrophages. Intervirology (2014) 57:36–42. doi:10.1159/000353902
22. Guo H, Gao J, Taxman DJ, Ting JP, Su L. HIV-1 infection induces interleukin-1ß production via TLR8 protein-dependent and NLRP3 inflammasome mechanisms in human monocytes. J Biol Chem (2014) 289:21716–26. doi:10.1074/jbc.M114.566620
23. Nasi M, De Biasi S, Bianchini E, Digaetano M, Pinti M, Gibellini L, et al. Analysis of inflammasomes and antiviral sensing components reveals decreased expression of NLRX1 in HIV-positive patients assuming efficient antiretroviral therapy. AIDS (2015) 29:1937–41. doi:10.1097/QAD.0000000000000830
24. Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH. Differential requirement for the activation of the inflammasome for processing and release of IL-1β in monocytes and macrophages. Blood (2009) 113:2324–35. doi:10.1182/blood-2008-03-146720
25. Tincati C, Merlini E, Braidotti P, Ancona G, Savi F, Tosi D, et al. Impaired gut junctional complexes feature late-treated individuals with suboptimal CD4+ T-cell recovery upon virologically suppressive combination antiretroviral therapy. AIDS (2016) 30:991–1003. doi:10.1097/QAD.0000000000001015
26. Stiksrud B, Lorvik KB, Kvale D, Mollnes TE, Ueland PM, Trøseid M, et al. Plasma IP-10 is increased in immunological non responders and associated with activated regulatory T cells and persisting low CD4 counts. J Acquir Immune Defic Syndr (2016) 73:138–48. doi:10.1097/QAI.0000000000001080
27. Tincati C, Douek DC, Marchetti G. Gut barrier structure, mucosal immunity and intestinal microbiota in the pathogenesis and treatment of HIV infection. AIDS Res Ther (2016) 13:19. doi:10.1186/s12981-016-0103-1
28. Imamichi H, Dewar RL, Adelsberger JW, Rehm CA, O’Doherty U, Paxinos EE, et al. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc Natl Acad Sci U S A (2016) 113:8783–8. doi:10.1073/pnas.1609057113
29. Imamichi H, Adelsberger J, Lane CH. Production of HIV-1 proteins from “defective” HIV-1 provirus” [abstract 159]. Program and abstracts of CROI (Conference on Retroviruses and Opportunistic Infections). Seattle (2017). 73 p.
30. Longenecker C, Sullivan C, Baker JV. Immune activation and cardiovascular disease in chronic HIV infection. Curr Opin HIV AIDS (2016) 11(2):216–25. doi:10.1097/COH.0000000000000227
Keywords: inflammasome, caspase, pyroptosis, HIV, immune reconstitution
Citation: Bandera A, Masetti M, Fabbiani M, Biasin M, Muscatello A, Squillace N, Clerici M, Gori A and Trabattoni D (2018) The NLRP3 Inflammasome Is Upregulated in HIV-Infected Antiretroviral Therapy-Treated Individuals with Defective Immune Recovery. Front. Immunol. 9:214. doi: 10.3389/fimmu.2018.00214
Received: 09 November 2017; Accepted: 25 January 2018;
Published: 12 February 2018
Edited by:
Francesca Chiodi, Karolinska Institute (KI), SwedenReviewed by:
Vicente Estrada, Hospital Clínico San Carlos, SpainCopyright: © 2018 Bandera, Masetti, Fabbiani, Biasin, Muscatello, Squillace, Clerici, Gori and Trabattoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandra Bandera, YS5iYW5kZXJhQGFzc3QtbW9uemEuaXQ=;
Andrea Gori, YW5kcmVhLmdvcmlAdW5pbWliLml0
†These authors have contributed equally to the work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.