
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 31 January 2018
Sec. Cancer Immunity and Immunotherapy
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00151
This article is part of the Research Topic Targeting Metabolism in Cancer Immunotherapy View all 8 articles
 Lilla Hornyák1†
Lilla Hornyák1† Nikoletta Dobos2†
Nikoletta Dobos2† Gábor Koncz3Zsolt Karányi1,4
Gábor Koncz3Zsolt Karányi1,4 Dénes Páll4Zoltán Szabó5
Dénes Páll4Zoltán Szabó5 Gábor Halmos2
Gábor Halmos2 Lóránt Székvölgyi1*
Lóránt Székvölgyi1*
Tumors are composed of abnormally transformed cell types and tissues that differ from normal tissues in their genetic and epigenetic makeup, metabolism, and immunology. Molecular compounds that modulate the immune response against neoplasms offer promising new strategies to combat cancer. Inhibitors targeting the indoleamine-2,3-dioxygenase 1 enzyme (IDO1) represent one of the most potent therapeutic opportunities to inhibit tumor growth. Herein, we assess the biochemical role of IDO1 in tumor metabolism and immune surveillance, and review current diagnostic and therapeutic approaches that are intended to increase the effectiveness of immunotherapies against highly aggressive and difficult-to-treat IDO-expressing cancers.
Indolamine-2,3-dioxygenase 1 is a cytosolic enzyme with a heme (Fe2+) prosthetic group that catalyzes the first and rate-limiting step of tryptophan (Trp) catabolism (Figure 1A). IDO1 converts the essential amino acid Trp to kynurenine (Kyn) by cleaving the 2,3-double bond of the indole ring while a molecular oxygen (O2) merges into the unsealed molecule. The product is N-formylkynurenine that becomes rapidly and spontaneously transformed into Kyn (1). In the next steps, Kyn is further converted to other active metabolites, such as hydroxykynurenine, anthranilic acid, kynurenic acid, 3-hydroxyanthranilic acid, quinolinic acid, and picolinic acid (Figure 1A). The two end-products of the pathway are NAD+ and ATP that both fuel cellular metabolism (2).
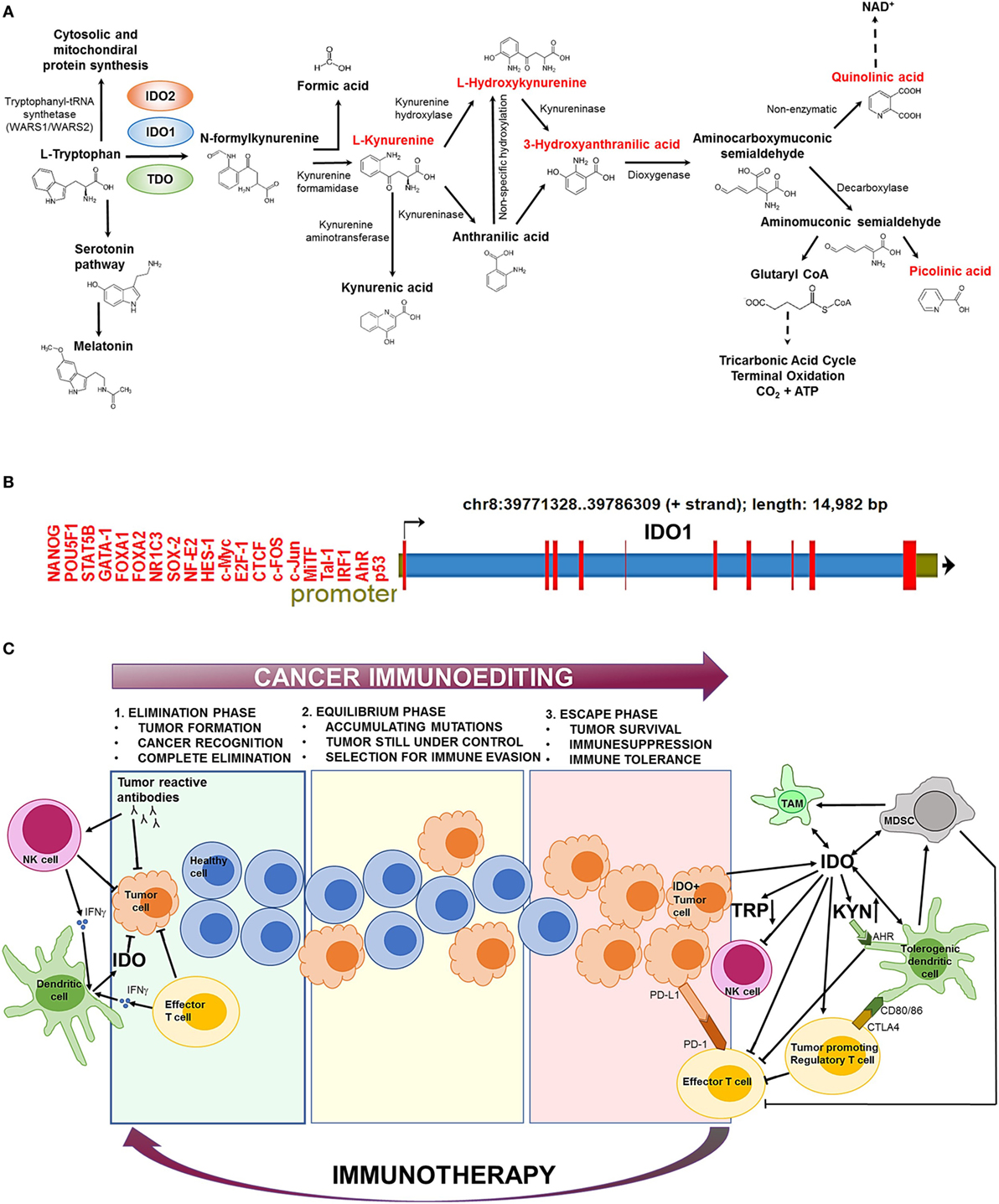
Figure 1. The biochemical function and regulation of indolamine-2,3-dioxygenase 1 (IDO1). (A) The kynurenine (Kyn) pathway of tryptophan (Trp) catabolism. l-Trp is metabolized in three separate biochemical pathways (indicated by arrows). In the Kyn pathway, IDO1/IDO2 and tryptophan-2,3-dioxygenase (TDO) catalyze the first and rate-limiting step of Trp degradation that gives rise to N-formylkynurenine. N-formylkynurenine is then transformed into l-Kyn and formic acid by kynurenine formamidase. l-Kyn is converted to anthranilic acid by kynureninase or l-hydroxykynurenine by kynurenine hydroxylase. Non-specific hydroxylation of anthranilic acid results in l-hydroxykynurenine. Kynureninase converts l-hydroxykynurenine to 3-hydroxyanthranilic acid that is further metabolized by hydroxyanthranilate dioxygenase to aminocarboxymuconic semialdehyde. The semialdehyde spontaneously forms quinolinic acid that is a precursor of NAD+ synthesis, or a decarboxylase enzyme converts it to aminomuconic semialdehyde. Aminomuconic semialdehyde is then converted to picolinic acid or glutaryl-CoA that is metabolized in the tricarbonic acid cycle and terminal oxidation. Metabolites that are highlighted in red have been directly implicated in immunosuppressive mechanisms and cancer development. (B) The structure of the IDO1 gene. IDO1 is located on chromosome 8 [39771328–39786309 forward (+) strand; 14,982 base pairs] comprising 10 exonic region (red bars). The promoter region (green section upstream the transcription start site) contains several transcription factor-binding sites that have been identified by ChIP sequencing. ChIP peaks were collected from the GTRD database of transcription binding sites (3). Only normal (non-transformed) cell types were considered. (C) The role of IDO1 in cancer immunoediting. In the first phase of immunoediting (“elimination”), sporadically arising transformed cells are destroyed by the innate and adaptive immune systems. Activated B cells produce tumor reactive antibodies to eradicate most transformed cells. Natural killer (NK) cells and effector T cells release inflammatory cytokines, such as IFN-γ, which activate dendritic cells (DCs) that secrete low levels of IDO1. IDO1 depletes the essential amino acid Trp from the tumor microenvironment that inhibits tumor growth. In the “equilibrium” phase, surviving tumor cells are still controlled by the immune system; however, they rapidly accumulate mutations. When the immune system can no longer block the abnormal and autonomous growth of “edited” cells, the tumor becomes clinically manifested (“escape”). The escape phase is associated with high IDO1 level that is primarily produced by tumor cells and tolerogenic immune cells [e.g., tolerogenic DCs, myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs)]. Trp depletion and Kyn accumulation lead to immunosuppression and tolerogenicity by inhibiting effector T cell and NK cell functions and stimulating regulatory T cells. IDO1 also promotes the expansion and activation of MDSCs and induces polarization of macrophages to a tolerogenic phenotype. Increased Kyn levels activate the aryl hydrocarbon receptor (AhR) that switch the activity of DCs from immunogenic to tolerogenic. Elevated CTLA4 expression of regulatory T cells results in further increase of IDO1 secretion by DCs. IDO1-induced expansion and activation of regulatory T cells, tolerogenic DCs, and MDSCs suppress the activity of antitumor effector T cells. Other immunosuppressive agents (e.g., PD-L1/PD-1, CTLA4) also inhibit effector T cell functions. Oncological immunotherapy aims to reverse immunoediting (backward arrow) by inhibiting and activating local immunosuppressive and tumor eradication mechanisms, respectively.
In humans, IDO1 has an evolutionary paralog (indolamine-2,3-dioxygenase 2; IDO2) and a functional ortholog (tryptophan-2,3-dioxygenase; TDO) that catalyze the same biochemical reaction; however, IDO2 and TDO show high tissue specificity and much lower expression level than IDO1 that significantly restrict their activity (2, 4). TDO is transcribed only in the liver1 [protein-level expression is not established (5)] and its major function is to control the free Trp concentration of the blood (4). IDO2 mRNA is expressed at low levels in the placenta and liver (protein expression is not known2), while IDO1 shows a high protein expression in the peripheral lymph organs (lymph nodes, spleen and tonsils3).
The activity of IDO1 is mainly regulated at the transcriptional level (Figure 1B). Bona fide transcription factor binding sites have been detected by ChIP-seq for a large catalog of human transcription factors (3). The identified gene regulatory proteins involve (i) NF- κB, which allows IDO1 mRNA expression regulation through the interferon pathway (6), (ii) the aryl hydrocarbon receptor (AhR) that binds to putative dendritic-cell responsive elements and promotes the l-Kyn-dependent induction of IDO1 (7, 8), and (iii) CTCF that mediates IDO1 expression via long-range chromatin looping interactions between the promoter and distal enhancer regions (9).
Superimposed on the transcriptional control of IDO1, specific posttranslational mechanisms also operate that affect the activity and half-life of the enzyme. For instance, the diffusible messenger nitrogen monoxide (NO) reacts with the heme cofactor of IDO1 generating ferric (Fe3+) heme and nitrate (NO3), which leads to the dose-dependent and reversible inhibition of enzymatic activity (10, 11). It has been also shown that endogenous NO production accelerates the proteasomal degradation of IDO1 (12). Other antioxidants like the anti-inflammatory agent pyrrolidine dithiocarbamate (13) restrict the availability of heme and thereby block holoenzyme assembly (14). In inflammatory conditions, NO and superoxide anions are simultaneously produced in large amounts, which rapidly generate the highly reactive peroxynitrite anion. Peroxynitrite inhibits IDO1 via the nitration of critical tyrosine residues (Tyr15, Tyr345, and Tyr353), without affecting IDO1 protein level (15, 16).
Hypoxia also leads to reduced IDO1 expression and, therefore, reduced Kyn production (17). Low IDO1 activity during the hypoxic phase promotes the activation of immune cells (18); however, hypoxic conditions also augment the secretion of effector CD4(+) T-cell cytokines, especially IFN-gamma, which in turn upregulates IDO1 mRNA expression (19).
At the protein level, IDO1 is mainly regulated by proteasomal degradation in response to immunogenic stimuli. Suppressor of cytokine signaling 3 (SOCS3), for instance, binds to IDO1 under inflammatory conditions and targets the IDO1/SOCS3 complex for polyubiquitination and proteasomal digestion. IDO1 degradation converts tolerogenic dendritic cells (DCs) into immunogenic cells and, therefore, a prerequisite for normal dendritic-cell function (20). Activated AhR is another component of the ubiquitin/proteasome system that contributes to the regulatory proteolysis of IDO1 and other proteins (8) that affect IDO1’s half-life.
Indoleamine-2,3-dioxygenase 1 contains two phosphorylatable tyrosine residues (Y115 and Y253) that modulate its enzymatic activity (21). Phosphorylation of any of these residues results in conformational changes in IDO1 and blocks the catalytic activity of IDO1 (22). In addition to the regulation of catalytic activity, these motifs act as docking sites for various molecular partners that either prolong or shorten IDO1’s half-life and maintain its immunoregulatory effects or stimulate inflammatory responses, respectively (8). For example, IL-6 triggers the phosphorylation of the Y253 residue of IDO1 that recruits the ECS (Elongin-Cullin-SOCS) E3 ligase complex, targeting IDO/SOCS3 for proteasomal degradation (23). On the contrary, phosphorylation of the Y115 residue allows the binding of SH2 domain tyrosine phosphatases SHP1 and SHP2, while SOCS3 becomes excluded. Hence, the TGF-β/Fyn/SHP axis activates the non-canonical NF-κB pathway that upregulates IDO1 production. Recently, ligand-bound AhR and Arginase 1 have been also shown to promote IDO1 phosphorylation through Src kinases, activating the signaling function of IDO1 that leads to de novo synthesis of the enzyme via TGF-β production (8, 24, 25).
The IDO1 pathway was originally described as an innate immune mechanism that defended the host organism against infections (26). The immunoprotective role of IDO1 was directly supported by the anti-pathogen effect of Trp metabolites (l-Kyn, l-hydroxykynurenine, 3-hydroxyanthranilic acid, quinolinic acid, picolinic acid) that prevented the proliferation and spread of intracellular pathogens (4, 27). Subsequent studies, however, identified tissue macrophages producing high levels of IDO1 upon IFN-γ stimulation that strongly inhibited the proliferation of effector T cells (28). It was also shown that accumulating Trp metabolites induced the differentiation of regulatory T cells and apoptosis of effector T cells that gave rise to immunosuppression (4, 26). l-Kyn is particularly toxic to lymphocytes (2) and induces the differentiation of regulatory T cells via AhR binding (29). l-hydroxykynurenine aids the suppression of CD4+ T cells and promotes the action of regulatory T cells (30). 3-hydroxyanthranilic acid modulates the immune functions of monocytes and lymphocytes, induces the apoptosis of effector T cells, and promotes the proliferation of regulatory T cells. Quinolinic acid stimulates the cell cycle of cancer cells and contributes to the acquisition of multidrug resistance against chemotherapeutic agents (29). Picolinic acid inhibits effector T cell proliferation (31). Later, it has become clear that the balance between the immunoprotective and immunosuppressive roles of IDO1 and Trp metabolites is tightly controlled by the stoichiometry of available local factors (e.g., IL-6, IL-12, CD40, IFN-γ, CTLA4, Foxo3a, IL-10, and PD-1) (26, 32). The resultant effect of these local activities modulates IDO1 expression and helps maintain global immune homeostasis and peripheral immune tolerance.
There are many pathologic diseases that are associated with increased IDO1 activity, including atherosclerosis, obesity, autoimmunity, major infections (e.g., community-acquired pneumonia, tuberculosis, listeriosis, influenza, HBV, HCV, HIV, sepsis), rejection of organ transplants, and cancer (2, 27). Originally, IDO1 has been considered as an anti-cancer molecule, proposing that increased IDO1 activity of antigen-presenting cells depletes the essential amino acid Trp from tumor cells and inhibits their growth. However, with the discovery of IDO1-mediated immunosuppressive functions, the pro-cancer activity of the enzyme has been recognized (33). IDO1 is overexpressed in more than 50% of tumors (34) that utilize IDO1-associated immunosuppressive mechanisms to promote their spread and survival (35). In cervical cancer, for instance, IDO1 shows a significantly higher mRNA transcription and protein expression level than in normal cervix, and also in comparison to other cancers (36). The extent of IDO overexpression also depends on the type of tumor and risk factors that reach patients in their life. For instance, oral squamous cell carcinoma (OSCC) was compared in never-smokers and never-drinkers (NSND) with smoker and drinker (SD) patients. In NSND patients suffering from OSCC, expression of IDO1 was significantly higher than in SDs (37).
Indoleamine-2,3-dioxygenase1 production is elevated upon (i) IFN-γ production of effector T cells (2), (ii) inflammatory cytokine production of innate immune cells (6, 38), (iii) IL-10 and IL-27 stimulation (39), (iv) CTLA4 expression on Treg cells causing increased IDO1 secretion by dendritic cells (DCs) (40), and (v) TGF-β, IL-10 and adenosine production of Treg and other immunosuppressive cells (40–42), (vi) cyclooxygenase-2 (COX-2) and prostaglandin E2 (PGE2) stimuli that are mediated through the PKC, PI3K, and MAPK pathways (several types of tumors carry PI3K or MAPK oncogenic mutations leading to constitutive IDO1 expression.) (43).
The mechanism of IDO1-elicited immunosuppression is not fully understood; however, increased IDO1 and Kyn levels are known to inhibit natural killer (NK) cell function (44, 45), prevent the activation of effector T cells, stimulate the activation of Treg cells (35, 46) and the differentiation of tolerogenic DCs (47), and promote the expansion and activation of myeloid-derived suppressor cells (48). Furthermore, Trp depletion inhibits mechanistic target of rapamycin complex 1 that stimulates T cell apoptosis and antigen-presenting cell-mediated inflammation (1, 49).
Paradoxically, the adaptive and innate immune systems that primarily protect against cancer development drive the formation of the highly aggressive and difficult-to-treat IDO1+ tumors. The genetic and biochemical characteristics of the emerging tumor is established by the process of “immunoediting” that prevents and promotes tumor formation at the same time, involving three consecutive stages called “elimination,”, “equilibrium,” and “escape” (50) (Figure 1C). In the first phase (“elimination”), most transformed cells are efficiently recognized and destroyed by the action of effector cells as NK and T cells (50). At this stage, IDO1 is produced at low levels within the tumor microenvironment and inhibits tumor proliferation (33). In the “equilibrium” phase, surviving tumor cells become “edited” by the continuous attack of the immune system and accumulate mutations (51). In the “escape” phase, IDO1 is produced in large quantities by tumor cells and tolerogenic immune cells that are recruited to the tumor microenvironment (52, 53). Increased IDO1 activity leads to elevated Kyn production that prevents effector T cell (35, 46) and NK cell functions (44, 45). In parallel, IDO1 induces the activation and expansion of Treg cells (26), DCs (47), and MDCSs (48) that further suppress the function of antitumor T cells. These mechanisms collectively establish an immunosuppressive tumor microenvironment that supports tumor growth. IDO1 positivity is strongly associated with multidrug resistance of tumors and inversely correlates with patient survival (54). Therefore, timely diagnosis and therapeutic correction of IDO+ tumors are of crucial importance to prevent clinical manifestation.
Indoleamine-2,3-dioxygenase 1 overexpression increases the relative concentration of Kyn compared to Trp, hence Kyn/Trp ratio can be used as a prognostic clinico-pathological marker to monitor cancer invasiveness and progression. Accordingly, increased systemic Kyn/Trp ratio and elevated IDO1 activity have been associated with poor prognosis and low survival of patients diagnosed with cervical cancer and glioblastoma multiforme (55, 56). For the sensitive detection of Trp metabolites in IDO1+ tumor tissues, a wealth of Trp-based radiotracers has been developed for positron emission tomography imaging. Radioactive Trp analogs as α-[11C]methyl-l-tryptophan (AMT) and l- and d-1-[18F]fluoroethyl-tryptophan provide useful information about response to immunotherapy, but they are also crucial for the preclinical and clinical validation of novel IDO1 inhibitors (57, 58).
Protein expression of IDO1 was found to be high in a number of tumor samples (36, 56); therefore, IDO1 may be a relevant therapeutic target to abrogate immune suppression (59). Currently, several IDO inhibitors undergo clinical evaluation (60) and many of them are now in Phase II clinical trials (Table 1). Most inhibitors were designed to block the Kyn pathway (e.g., epacadostat, indoximod, GDC-0919, and an IDO1 peptide-based vaccine; Figure 1A) that suspends immunosuppression (1). Some of the tested compounds are used alone, or in combination with immunotherapy (CTLA4, PD-1 blockade), chemotherapy, adoptive transfer, COX-2 inhibitors (e.g., celecoxib), membrane-associated PGE2 synthase inhibitors (e.g., MF63), PGE2 receptor (EP4) competitive antagonists (e.g., GW627368X), and PI3K inhibitors (43, 59). The latter combinative therapies are intended to improve the inhibition of local immunosuppression around tumor tissues and to enhance tumor eradication (61).
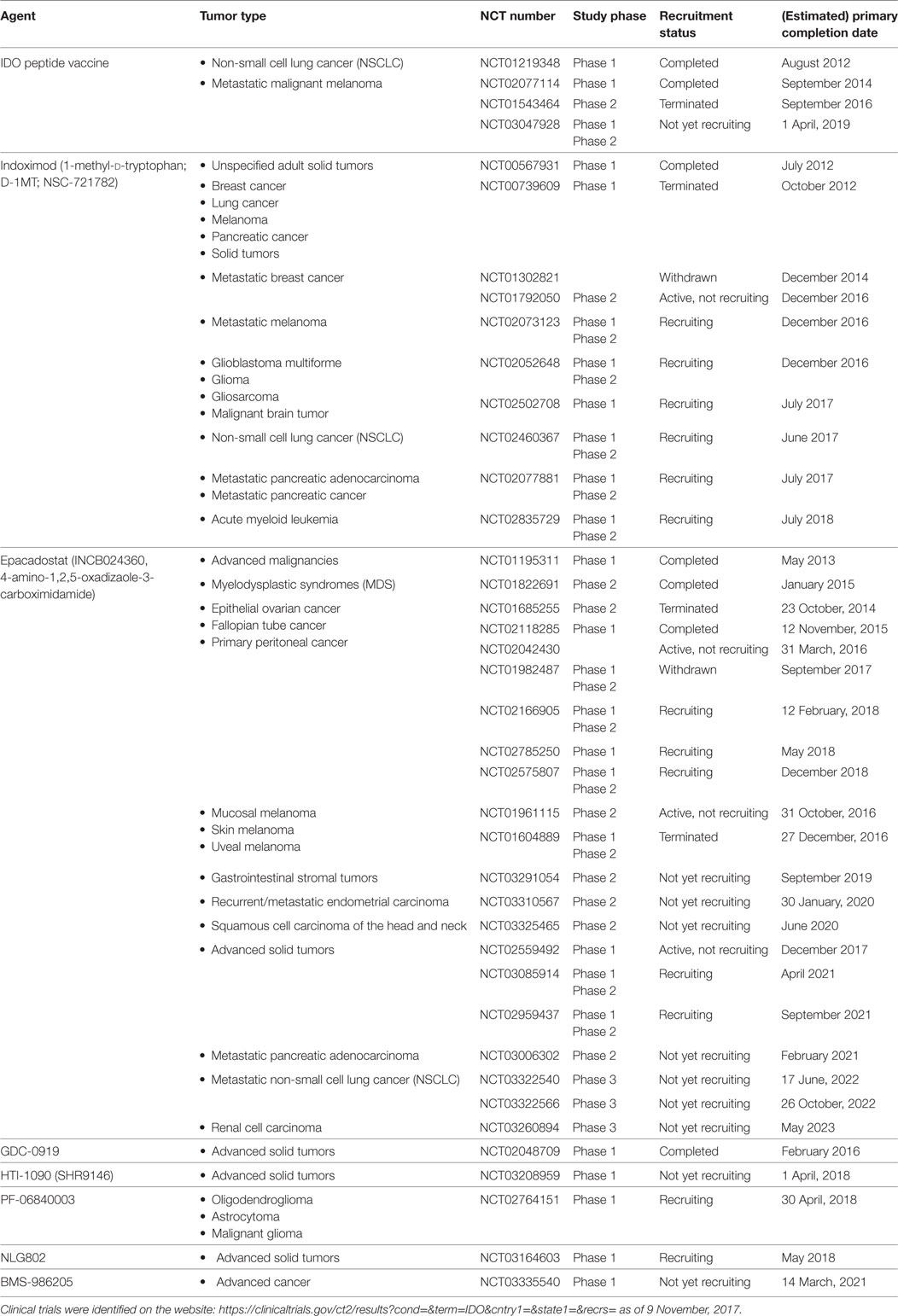
Table 1. Clinical trials of indoleamine-2,3-dioxygenase (IDO) inhibitors in cancer therapy.
Epacadostat (INCB024360) and indoximod (NLG8189 or 1-methyl-d-tryptophan) are the most common IDO inhibitors that are well tolerated and show obvious beneficial effects in cancer therapies; however, both have some major side effects. Epacadostat showed grade 3 and 4 adverse effects in patients with advanced malignancies, most frequently abdominal pain, hypokalemia, fatigue, and some minor effects involving nausea, decreased appetite, vomiting, constipation, diarrhea, dyspnea, back pain, and cough (62). Indoximod treatment also showed some major toxicities in a dose-escalation study, involving grade 1 fatigue and grade 2 hypophysitis (63). In combination with the microtubular poison docetaxel, the most common adverse effects of indoximod were fatigue, anemia, hyperglycemia, infection, and nausea (62, 63).
Combination of IFN-γ treatment with IDO1 inhibitors is a promising new cancer immunotherapeutic strategy that effectively enhances antitumor immunity and eliminates TRCs (i.e., stem cell-like cancer cells that are self-renewing, highly tumorigenic, and can repopulate tumors). In clinical practice, administration of IFN-γ with IDO1 inhibitors is the only oncolytic therapy that significantly disrupts TRCs. IFN-γ induces the entry of TRCs into dormancy, while IDO1 inhibitor-elicited immunosuppression allows effector T cells and NK cells to attack dormant TRCs (64). Monotherapies with IDO inhibitors or other combinative treatments usually terminate with failure because of immune evasion of TRCs, which leads to metastasis formation, tumor recurrence and multidrug resistance (64).
Mutational load/neoantigen-burden, basal level of tumor infiltrating T cells (TILs), differential expression of immune-checkpoint genes within the tumor tissue are important biomarkers that help predict the tumor’s predisposition toward immune-checkpoint inhibitors (ICIs) targeting IDO1, CTLA4, or PD-1 and increase the clinical success of immunotherapies. Most ICI-responsive cancers (e.g., lung and bladder cancers, melanoma) were associated with intrinsically high TIL numbers and high mutational load/neoantigen-burden, while other cancers (e.g., glioblastoma) were predicted to be ICI-resistant based on their biomarker profiles (37, 65).
Beyond the application of biomarkers and chemical inhibitors, IDO1 can be genetically targeted by genome editing tools that offer new therapeutic opportunities for cancer patients. In animal studies, genetic inhibition of IDO1 expression reactivated the antitumor immune response against IDO+ cancer cells and inhibited tumor growth (63). The shIDO-ST treatment, for instance, is based on a Salmonella typhimurium (ST) vector that codes for a small hairpin RNA targeting IDO1 (shIDO) (66). Intravenously injected shIDO-ST accumulated in the tumor tissues of mice, causing IDO1 silencing and concomitant infiltration and activation of polymorphonuclear neutrophil granulocytes (PMNs). PMNs produced reactive oxygen species that established a highly toxic microenvironment for tumor cell growth (67). A recent genomic sequencing study that combined large-scale tumor exome analysis with MHC I class prediction revealed a strong positive correlation between IDO1 expression, mutational burden, and neoantigen load in cervical cancers (36). The above studies collectively identify IDO1 as an attractive target to increase the effectiveness of cancer immunotherapies.
The mechanism of “cancer immunoediting” is the direct consequence of a T cell-dependent immunoselection process that drives the formation of IDO1+ tumors. The action of a competent immune system is, therefore, determinative for the acquisition of cancer immunogenicity. Important posttranslational control mechanisms affect the activity and half-life of IDO1 (e.g., NO, hypoxia, proteasomal degradation, phosphorylation) that should be considered in terms of cancer therapy. For instance, IDO1 inhibitors could be administered as co-therapeutic agents in the presence of redox regulators, IFN-γ, or anti-IL-6. Combining IDO1 drugs with the inhibition of specific transcription factors regulating IDO1 activity (e.g., AhR) may also improve the effectiveness and specificity of chemotherapies. Current genome editing and exome sequencing technologies offer promising new strategies to identify novel tumor-specific mutational antigens and thus expand the repertoire of tumor-specific immunotherapies.
All authors participated in the writing of this manuscript and agreed to be accountable for the content of the work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
LS received grants from the Hungarian Academy of Sciences (Lendület programme, LP2015-9/2015), NKFIH-ERC-HU-117670, and GINOP-2.3.2-15-2016-00024. GH was supported by the GINOP-2.3.2-15-2016-00043 project. GK was supported by NKFIH-K-125224. DP and ZS were supported by GINOP-2.3.2-15-2016-00062.
1. Zhai L, Spranger S, Binder DC, Gritsina G, Lauing KL, Giles FJ, et al. Molecular pathways: targeting IDO1 and Other tryptophan dioxygenases for cancer immunotherapy. Clin Cancer Res (2015) 21:5427–33. doi:10.1158/1078-0432.CCR-15-0420
2. Bilir C, Sarisozen C. Indoleamine 2,3-dioxygenase (IDO): only an enzyme or a checkpoint controller? J Oncol Sci (2017) 3:52–6. doi:10.1016/j.jons.2017.04.001
3. Yevshin I, Sharipov R, Valeev T, Kel A, Kolpakov FGTRD. A database of transcription factor binding sites identified by ChIP-seq experiments. Nucleic Acids Res (2017) 45:D61–7. doi:10.1093/nar/gkw951
4. van Baren N, Van den Eynde BJ. Tryptophan-degrading enzymes in tumoral immune resistance. Front Immunol (2015) 6:34. doi:10.3389/fimmu.2015.00034
5. Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, et al. A subcellular map of the human proteome. Science (2017) 356:eaal3321. doi:10.1126/science.aal3321
6. Zulfiqar B, Mahroo A, Nasir K, Farooq RK, Jalal N, Rashid MU, et al. Nanomedicine and cancer immunotherapy: focus on indoleamine 2,3-dioxygenase inhibitors. Onco Targets Ther (2017) 10:463–76. doi:10.2147/OTT.S119362
7. Vogel CFA, Wu D, Goth SR, Baek J, Lollies A, Domhardt R, et al. Aryl hydrocarbon receptor signaling regulates NF-κB RelB activation during dendritic-cell differentiation. Immunol Cell Biol (2013) 91:568–75. doi:10.1038/icb.2013.43
8. Pallotta MT, Fallarino F, Matino D, Macchiarulo A, Orabona C. AhR-mediated, non-genomic modulation of IDO1 function. Front Immunol (2014) 5:497. doi:10.3389/fimmu.2014.00497
9. Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, et al. Chromatin architecture reorganization during stem cell differentiation. Nature (2015) 518:331–6. doi:10.1038/nature14222
10. Thomas SR, Mohr D, Stocker R. Nitric oxide inhibits indoleamine 2, 3-dioxygenase activity in interferon-gamma primed mononuclear phagocytes. J Biol Chem (1994) 269:14457–64.
11. Thomas SR, Terentis AC, Cai H, Takikawa O, Levina A, Lay PA, et al. Post-translational regulation of human indoleamine 2,3-dioxygenase activity by nitric oxide. J Biol Chem (2007) 282:23778–87. doi:10.1074/jbc.M700669200
12. Hucke C, MacKenzie CR, Adjogble KDZ, Takikawa O, Däubener W. Nitric oxide-mediated regulation of gamma interferon-induced bacteriostasis: inhibition and degradation of human indoleamine 2,3-dioxygenase. Infect Immun (2004) 72:2723–30. doi:10.1128/IAI.72.5.2723-2730.2004
13. Genes P, Liu SF, Ye X, Malik AB. Inhibition of NF-kB activation by pyrrolidine dithiocarbamate prevents in vivo expression of. Circulation (1999) 100:1330–7. doi:10.1161/01.CIR.100.12.1330
14. Thomas SR, Salahifar H, Mashima R, Hunt NH, Richardson DR, Stocker R. Antioxidants inhibit indoleamine 2,3-dioxygenase in IFN-gamma-activated human macrophages: posttranslational regulation by pyrrolidine dithiocarbamate. J Immunol (2001) 166:6332–40. doi:10.4049/jimmunol.166.10.6332
15. Fujigaki H, Saito K, Lin F, Fujigaki S, Takahashi K, Martin BM, et al. Nitration and inactivation of IDO by peroxynitrite. J Immunol (2006) 176:372–9. doi:10.4049/jimmunol.176.1.372
16. Grohmann U, Fallarino F, Bianchi R, Orabona C, Vacca C, Fioretti MC, et al. A defect in tryptophan catabolism impairs tolerance in nonobese diabetic mice. J Exp Med (2003) 198:153–60. doi:10.1084/jem.20030633
17. Schmidt SK, Ebel S, Keil E, Woite C, Ernst JF, Benzin AE, et al. Regulation of IDO activity by oxygen supply: inhibitory effects on antimicrobial and immunoregulatory functions. PLoS One (2013) 8:e63301. doi:10.1371/journal.pone.0063301
18. Herbert A, Ng H, Jessup W, Kockx M, Cartland S, Thomas SR, et al. Hypoxia regulates the production and activity of glucose transporter-1 and indoleamine 2,3-dioxygenase in monocyte-derived endothelial-like cells: possible relevance to infantile haemangioma pathogenesis. Br J Dermatol (2011) 164:308–15. doi:10.1111/j.1365-2133.2010.10086.x
19. Roman J, Rangasamy T, Guo J, Sugunan S, Meednu N, Packirisamy G, et al. T-cell activation under hypoxic conditions enhances IFN-γ secretion. Am J Respir Cell Mol Biol (2010) 42:123–8. doi:10.1165/rcmb.2008-0139OC
20. Orabona C, Pallotta MT, Volpi C, Fallarino F, Vacca C, Bianchi R, et al. SOCS3 drives proteasomal degradation of indoleamine 2, 3-dioxygenase (IDO) and antagonizes IDO-dependent tolerogenesis. Proc Natl Acad Sci U S A (2008) 105:20828–33. doi:10.1073/pnas.0810278105
21. Maria P, CIriana O, Claudia V, Ursula G, Paolo P, Francesca F. Proteasomal degradation of indoleamine 2,3-dioxygenase in CD8+ dendritic cells is mediated by suppressor of cytokine signaling 3 (SOCS3). Int J Tryptophan Res (2010) 3:91–7. doi:10.4137/IJTR.S3971
22. Albini E, Rosini V, Gargaro M, Mondanelli G, Belladonna ML, Pallotta MT, et al. Distinct roles of immunoreceptor tyrosine-based motifs in immunosuppressive indoleamine 2,3-dioxygenase 1. J Cell Mol Med (2017) 21:165–76. doi:10.1111/jcmm.12954
23. Orabona C, Pallotta M, Grohmann U. Different partners, opposite outcomes: a new perspective of the immunobiology of indoleamine 2,3-dioxygenase. Mol Med (2012) 18:834–42. doi:10.2119/molmed.2012.00029
24. Mondanelli G, Bianchi R, Pallotta MT, Orabona C, Albini E, Iacono A, et al. A relay pathway between arginine and tryptophan metabolism confers immunosuppressive properties on dendritic cells. Immunity (2017) 46:233–44. doi:10.1016/j.immuni.2017.01.005
25. Bessede A, Gargaro M, Pallotta MT, Matino D, Brunacci C, Bicciato S, et al. Aryl hydrocarbon receptor control of a disease tolerance defense pathway. Nature (2015) 511:184–90. doi:10.1038/nature13323.Aryl
26. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol (2013) 34:137–43. doi:10.1016/j.it.2012.10.001
27. Schmidt SV, Schultze JL. New insights into IDO biology in bacterial and viral infections. Front Immunol (2014) 5:384. doi:10.3389/fimmu.2014.00384
28. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science (1998) 281:1191–3. doi:10.1126/science.281.5380.1191
29. Heng B, Lim CK, Lovejoy DB, Bessede A, Gluch L, Guillemin GJ. Understanding the role of the kynurenine pathway in human breast cancer immunobiology. Oncotarget (2016) 7:6506–20. doi:10.18632/oncotarget.6467
30. Zaher SS, Germain C, Fu H, Larkin DFP, George AJT. 3-hydroxykynurenine suppresses CD4+ T-cell proliferation, induces T-regulatory-cell development, and prolongs corneal allograft survival. Invest Ophthalmol Vis Sci (2011) 52:2640–8. doi:10.1167/iovs.10-5793
31. Prodinger J, Loacker LJ, Schmidt RLJ, Ratzinger F, Greiner G, Witzeneder N, et al. The tryptophan metabolite picolinic acid suppresses proliferation and metabolic activity of CD4+ T cells and inhibits c-Myc activation. J Leukoc Biol (2016) 99:583–94. doi:10.1189/jlb.3A0315-135R
32. Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol (2002) 3:1097–101. doi:10.1038/ni846
33. Katz JB, Muller AJ, Prendergast GC. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol Rev (2008) 222:206–21. doi:10.1111/j.1600-065X.2008.00610.x
34. Löb S, Königsrainer A, Zieker D, Brücher BLDM, Rammensee HG, Opelz G, et al. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol Immunother (2009) 58:153–7. doi:10.1007/s00262-008-0513-6
35. Godin-Ethier J, Hanafi L-A, Piccirillo CA, Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Cancer Res (2011) 17:6985–91. doi:10.1158/1078-0432.CCR-11-1331
36. Qin Y, Ekmekcioglu S, Forget M-A, Szekvolgyi L, Hwu P, Grimm EA, et al. Cervical cancer neoantigen landscape and immune activity is associated with human papillomavirus master regulators. Front Immunol (2017) 8:689. doi:10.3389/fimmu.2017.00689
37. Foy JP, Bertolus C, Michallet MC, Deneuve S, Incitti R, Bendriss-Vermare N, et al. The immune microenvironment of HPV-negative oral squamous cell carcinoma from never-smokers and never-drinkers patients suggests higher clinical benefit of IDO1 and PD1/PD-L1 blockade. Ann Oncol (2017) 28:1934–41. doi:10.1093/annonc/mdx210
38. Rovira Gonzalez YI, Lynch PJ, Thompson EE, Stultz BG, Hursh DA. In vitro cytokine licensing induces persistent permissive chromatin at the Indoleamine 2,3-dioxygenase promoter. Cytotherapy (2016) 18:1114–28. doi:10.1016/j.jcyt.2016.05.017
39. Carbotti G, Barisione G, Airoldi I, Mezzanzanica D, Bagnoli M, Ferrero S, et al. IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget (2015) 6:43267–80. doi:10.18632/oncotarget.6530
40. Belladonna ML, Orabona C, Grohmann U, Puccetti P. TGF-β and kynurenines as the key to infectious tolerance. Trends Mol Med (2009) 15:41–9. doi:10.1016/j.molmed.2008.11.006
41. Yanagawa Y, Iwabuchi K, Onoé K. Co-operative action of interleukin-10 and interferon-γ to regulate dendritic cell functions. Immunology (2009) 127:345–53. doi:10.1111/j.1365-2567.2008.02986.x
42. Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood (2008) 112:1822–31. doi:10.1182/blood-2008-02-136325
43. Hennequart M, Pilotte L, Cane S, Hoffmann D, Stroobant V, De Plaen E, et al. Constitutive IDO1 expression in human tumors is driven by cyclooxygenase-2 and mediates intrinsic immune resistance. Cancer Immunol Res (2017) 5:695–709. doi:10.1158/2326-6066.CIR-16-0400
44. Pietra G, Vitale M, Moretta L, Mingari MC. How melanoma cells inactivate NK cells. Oncoimmunology (2012) 1:974–5. doi:10.4161/onci.20405
45. Wang D, Saga Y, Mizukami H, Sato N, Nonaka H, Fujiwara H, et al. Indoleamine-2,3-dioxygenase, an immunosuppressive enzyme that inhibits natural killer cell function, as a useful target for ovarian cancer therapy. Int J Oncol (2012) 40:929–34. doi:10.3892/ijo.2011.1295
46. Mellor AL, Keskin DB, Johnson T, Chandler P, Munn DH. Cells expressing indoleamine 2,3-dioxygenase inhibit T cell responses. J Immunol (2002) 168:3771–6. doi:10.4049/jimmunol.168.8.3771
47. Li Q, Harden JL, Anderson CD, Egilmez NK. Tolerogenic phenotype of IFN-γ-induced IDO+ dendritic cells is maintained via an autocrine IDO–kynurenine/AhR–IDO loop. J Immunol (2016) 197:962–70. doi:10.4049/jimmunol.1502615
48. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-expressed IDO recruits and activates MDSCs in a treg-dependent manner. Cell Rep (2015) 13:412–24. doi:10.1016/j.celrep.2015.08.077
49. Powell JJD, Pollizzi KNK, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev immunol (2012) 30:39–68. doi:10.1146/annurev-immunol-020711-075024.Regulation
50. Kim R, Emi M, Tanabe K. Cancer immunoediting: from immune surveillance to immune escape. Immunology (2007) 121:1–17. doi:10.1111/j.1365-2567.2007.02587.x
51. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol (2004) 22:329–60. doi:10.1146/annurev.immunol.22.012703.104803
52. Shou D, Liang W, Song Z, Yin J, Sun Q, Gong W. Suppressive role of myeloid-derived suppressor cells (MDSCs) in the microenvironment of breast cancer and targeted immunotherapies. Oncotarget (2016) 7:64505–11. doi:10.18632/oncotarget.11352
53. Zhao Q, Kuang D-M, Wu Y, Xiao X, Li X-F, Li T-J, et al. Activated CD69+ T cells foster immune privilege by regulating IDO expression in tumor-associated macrophages. J Immunol (2012) 188:1117–24. doi:10.4049/jimmunol.1100164
54. Okamoto A, Nikaido T, Ochiai K, Takakura S, Saito M, Aoki Y, et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin Cancer Res (2005) 11:6030–9. doi:10.1158/1078-0432.CCR-04-2671
55. Hascitha J, Priya R, Jayavelu S, Dhandapani H, Selvaluxmy G, Sunder Singh S, et al. Analysis of kynurenine/tryptophan ratio and expression of IDO1 and 2 mRNA in tumour tissue of cervical cancer patients. Clin Biochem (2016) 49:919–24. doi:10.1016/j.clinbiochem.2016.04.008
56. Zhai L, Ladomersky E, Lauing KL, Wu M, Genet M, Gritsina G, et al. Infiltrating T cells increase IDO1 expression in glioblastoma and contribute to decreased patient survival. Clin Cancer Res (2017) 23:6650–60. doi:10.1158/1078-0432.CCR-17-0120
57. Xin Y, Cai H. Improved radiosynthesis and biological evaluations of L- and D-1-[18F]fluoroethyl-tryptophan for PET imaging of IDO-mediated kynurenine pathway of tryptophan metabolism. Mol Imaging Biol (2017) 19:589–98. doi:10.1007/s11307-016-1024-z
58. Henrottin J, Lemaire C, Egrise D, Zervosen A, van Den Eynde B, Plenevaux A, et al. Fully automated radiosynthesis of N1-[18F]fluoroethyl-tryptophan and study of its biological activity as a new potential substrate for indoleamine 2,3-dioxygenase PET imaging. Nucl Med Biol (2016) 43:379–89. doi:10.1016/j.nucmedbio.2016.03.001
59. Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol (2016) 37:193–207. doi:10.1016/j.it.2016.01.002
60. Routy JP, Routy B, Graziani GM, Mehraj V. The kynurenine pathway is a double-edged sword in immune-privileged sites and in cancer: implications for immunotherapy. Int J Tryptophan Res (2016) 9:67–77. doi:10.4137/IJTR.S38355
61. Kozłowska A, Mackiewicz J, Mackiewicz A. Therapeutic gene modified cell based cancer vaccines. Gene (2013) 525:200–7. doi:10.1016/j.gene.2013.03.056
62. Brochez L, Chevolet I, Kruse V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur J Cancer (2017) 76:167–82. doi:10.1016/j.ejca.2017.01.011
63. Vacchelli E, Aranda F, Eggermont A, Sautès-Fridman C, Tartour E, Kennedy EP, et al. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology (2014) 3:e957994. doi:10.4161/21624011.2014.957994
64. Liu Y, Liang X, Yin X, Lv J, Tang K, Ma J, et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-γ-induced immunologic dormancy of tumor-repopulating cells. Nat Commun (2017) 8:15207. doi:10.1038/ncomms15207
65. Garg AD, Vandenberk L, Van Woensel M, Belmans J, Schaaf M, Boon L, et al. Preclinical efficacy of immune-checkpoint monotherapy does not recapitulate corresponding biomarkers-based clinical predictions in glioblastoma. Oncoimmunology (2017) 6:e1295903. doi:10.1080/2162402X.2017.1295903
66. Blache CA, Manuel ER, Kaltcheva TI, Wong AN, Ellenhorn JDI, Blazar BR, et al. Systemic delivery of Salmonella typhimurium transformed with IDO shRNA enhances intratumoral vector colonization and suppresses tumor growth. Cancer Res (2012) 72:6447–56. doi:10.1158/0008-5472.CAN-12-0193
Keywords: indoleamine-2,3-dioxygenase, gene expression, metabolism, immunotherapy, immune surveillance, cancer diagnostics, clinical trial
Citation: Hornyák L, Dobos N, Koncz G, Karányi Z, Páll D, Szabó Z, Halmos G and Székvölgyi L (2018) The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 9:151. doi: 10.3389/fimmu.2018.00151
Received: 12 December 2017; Accepted: 17 January 2018;
Published: 31 January 2018
Edited by:
Suhendan Ekmekcioglu, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Paolo Puccetti, University of Perugia, ItalyCopyright: © 2018 Hornyák, Dobos, Koncz, Karányi, Páll, Szabó, Halmos and Székvölgyi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lóránt Székvölgyi, bG9yYW50c3pAbWVkLnVuaWRlYi5odQ==
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.