 Sundary Sormendi
Sundary Sormendi Ben Wielockx
Ben Wielockx- Heisenberg Research Group, Institute of Clinical Chemistry and Laboratory Medicine, Technische Universität Dresden, Dresden, Germany
Low oxygen tension or hypoxia is a determining factor in the course of many different processes in animals, including when tissue expansion and cellular metabolism result in high oxygen demands that exceed its supply. This is mainly happening when cells actively proliferate and the proliferating mass becomes distant from the blood vessels, such as in growing tumors. Metabolic alterations in response to hypoxia can be triggered in a direct manner, such as the switch from oxidative phosphorylation to glycolysis or inhibition of fatty acid desaturation. However, as the modulated action of hypoxia-inducible factors or the oxygen sensors (prolyl hydroxylase domain-containing enzymes) can also lead to changes in enzyme expression, these metabolic changes can also be indirect. With this review, we want to summarize our current knowledge of the hypoxia-induced changes in metabolism during cancer development, how they are affected in the tumor cells and in the cells of the microenvironment, most prominently in immune cells.
Introduction
Metabolism is the set of chemical processes by which energy homeostasis is maintained, allowing cells to adjust to the needs that the surrounding environment demands. By adjusting their metabolic pathway network, cells are able to adapt to nutrients and deprived oxygen availability, as well as to adequately respond to different cell signals. During the past few years, the importance of the metabolic state of a cell and how this exerts differentiation and functionality during physiological and pathological processes has become evident. Indeed, metabolic reprogramming is considered a hall-mark of cancer progression (1, 2). Recently, new progresses in molecular biology and high-throughput molecular analyses revealed that many of the signaling pathways, which are altered by gene mutations can regulate cell metabolism. However, the oncogenic transformation process not only involves cancer cells, but it also alters their tumor microenvironment (TME), which includes stromal and infiltrating immune cells (3). Although in this context their metabolism has received less attention, they signify a rich cell population in many solid tumors. Moreover, the metabolic changes that these cells endure have been shown to have a great impact on their contribution during tumor development. These metabolic changes not only translate in different cell functionality, but they are also important in establishing a pro-tumoral “metabolite crosstalk.” According to this idea, it has been shown that specific excreted metabolites, including lactate (4) are exploited or signal to particular cells. Also the connection between metabolism and signal transduction within the neoplastic area is known. We will, therefore, summarize the most essential and relevant studies in the field of cancer-related metabolism, highlighting the regulating properties of hypoxia pathway proteins.
Hypoxia Pathway Proteins in Cancer
Solid tumors are characterized by rapid cell growth that is not equally compensated with a functionally effective and efficient vasculature. This poor vessel irrigation leads to a highly heterogeneous tumor mass with variable oxygen pressure and nutrient levels that cancer cells as well as TME cells need to overcome. Vaupel and colleagues recognized that the partial pressure of oxygen (pO2) within human cancers is significantly lower than in surrounding tissue. This so-called intra-tumoral hypoxia is associated with increased risk of local spread, metastasis, and patient mortality (5). Indeed, a complex pathway exists that regulates the adaptive response to hypoxia. The master regulators of the cellular response to hypoxia constitute a heterodimeric complex formed by a constitutively expressed nuclear HIFβ, and a cytoplasmic oxygen-dependent HIFα (HIF-1α, HIF-2α, and HIF-3α) subunit. Stabilization of HIFα is regulated by a group of oxygen and iron dependent enzymes, known as hypoxia-inducible factor (HIF)-prolyl hydroxylase domain enzymes (PHD1–3). Therefore, under physiological oxygen concentrations PHDs hydroxylate two prolyl residues of HIFα, which allows binding of the Von Hippel–Lindau tumor-suppressor protein, leading to subsequent ubiquitination and proteasomal degradation of this alpha subunit. However, in hypoxia PHDs are much less active, allowing gene transcription regulation by the HIF isoforms, with overlapping, distinct or even opposite roles (6) (Figure 1). Since its discovery, regulation of the hypoxia pathway has been strongly related to cancer development. It cannot only modulate survival and proliferation of cancer cells, activation of this pathway can induce angiogenesis, escape from immune-surveillance, epithelial-to-mesenchymal transition, and even distant metastasis (7, 8). Due to the central regulatory role of PHD2 (9) in the hypoxia pathway, several studies have focused on this isoform. In this regard, we were able to demonstrate that loss of PHD2 in tumor cells leads to decreased tumor growth, depending on an anti-proliferative effect of TGFβ activation through matrix metalloproteinases, but not HIF-1α (10, 11). More recently, using a spontaneous breast cancer mouse model, Kuchnio and colleagues showed that PHD2 haplo-deficiency in cancer cells reduce metastasis via two mechanisms: (1) by decreasing cancer-associated fibroblasts (CAF) activation due to a reduced secretion of TGFβ by cancer cells, matrix production and contraction by CAFs and (2) by improving vessel normalization (12). As mentioned earlier, newly formed vessels are often disorganized, immature, and leaky. Mice heterozygous for PHD2 are protected from distant metastasis due to endothelial normalization in a HIF2-α-dependent manner (13). Branco-Price and colleagues described that deficiency of HIF-1α in the endothelium diminishes NO synthesis, resulting in retarded tumor cell migration and consequent tumor cell metastasis. However, loss of HIF-2α had a reversed effect (14).
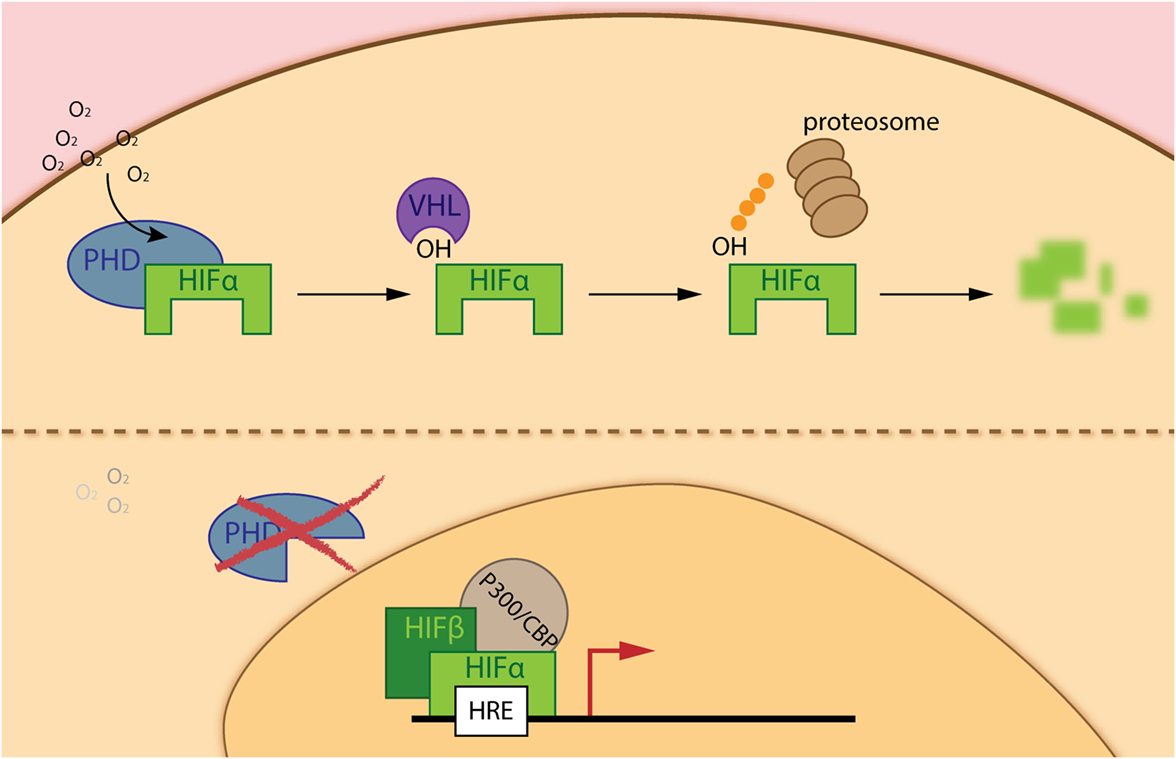
Figure 1. The hypoxia inducible factor (HIF) pathway. Under adequate oxygen pressure, prolyl-hydroxylase domain enzymes (PHDs) hydroxylate two prolyl residues on the α-subunit of the hypoxia-inducible transcription factors (HIF-α). The hydroxylated residues are recognized by the ubiquitin ligase Von Hippel–Lindau, leading to subsequent degradation of HIF-α via the proteasome (upper part of the figure). However, during hypoxia (deprived oxygen pressure), PHDs are inactive and HIF-α is able to translocate into the nucleus, interacts with the HIF-β subunit and P300/CBP enabling binding to hypoxia-responsive elements (HRE) in the promotor region of genes implicated in the hypoxia response (lower part of the figure).
The HIF-pathway proteins not only regulate growth and dissemination of cancer cells, but they also control the tumor-associated immune cells (15, 16). Therefore, many different groups have been focusing on what role the hypoxia pathway proteins play in the two major contrasting forces throughout tumor development: (1) anti-tumor defense and (2) suppression by the immune system. Concerning the latter, tumor-associated macrophages (TAMs) or pro-tumoral macrophages help the tumor to grow. Actually, most of the studies performed relate to the role of the HIF-pathway proteins in TAMs during cancer development. The first studies on this were focused on the role of the HIF transcription factors, revealing that loss of HIF-1α in TAMs increases M2 polarization and pro-angiogenic responses. Moreover, these TAMs overexpress HIF-2α, which correlates with poor patient prognosis (17). In line with this, HIF-2α deficiency in macrophages reduces TAM infiltration into hepatocellular carcinoma in mice (18). In addition, in a transgenic mouse model of breast carcinoma development (MMTV-PyMT), Doedens and colleagues demonstrated that targeted deletion of HIF-1α in macrophages leads to reduced breast tumor growth. Indeed, their work strongly proposes a HIF1-α-dependent macrophage-mediated T cell suppression (19). Furthermore, our research group demonstrated that PHD2 deficiency in myeloid and T cells is a pre-requisite to diminish tumor volume due to increased death of cancer cells (20). Nevertheless, Clever et al. recently reported for the first time a clear role for PHDs in regulating T cell anti-tumoral response. In this study, wild-type and PHD1–3 T cell triple knock-out mice showed similar subcutaneous B16 tumor growth, while the triple PHD KO mice were significantly protected from tumor colonization in the lung (21).
Since hypoxia constitutes one of the hallmarks of solid tumors, and oxygen availability has a direct effect on cell metabolism, it is not surprising that numerous authors have described the reciprocal regulation that HIFs exert on metabolic reprogramming of cancer cells and immune response in the TME and vice versa (22–25). In this regard, oxygen not only regulates PHD activity directly (6), CO2 production during mitochondrial respiration through the TCA cycle can also suppress HIF activity in high concentrations. The mechanism behind this process still needs to be clarified, but it seems that acidification inhibits protein synthesis (mTOR inhibition) and HIF1 α is extremely sensitive to protein synthesis (26). In addition, ROS production during oxidative metabolism influences HIF activity (27, 28), as well as accumulation of specific immunometabolites such as α-ketoglutarate (α-KG), fumarate, and succinate (29–32).
Cancer Cell Metabolism
The Warburg effect is found to be one of the most striking metabolic shifts that healthy normal cells undergo during tumorigenesis (33). This effect of aerobic glycolysis, described by Warburg already in 1920s still forms a hot-topic of tumor metabolism nowadays. This process defines that cancer cells predominantly obtain their energy (in terms of ATP production) through the glycolytic pathway rather than the TCA cycle, even in the presence of adequate oxygen levels (33). But why would cancer cells use glycolysis when energy production is inefficient? Despite the low amount of ATP produced by glycolysis (2 ATP molecules per glucose molecule in glycolysis versus 36 molecules of ATP in TCA), the efficiency of this process relies on faster kinetics of glycolysis, producing a comparable amount of ATP by either form of glucose metabolism during the same period of time (34). This also implies that nutrients are conserved for biosynthesis of nucleic acids, lipids, and amino acids to support cell growth, rather than oxidized in mitochondria for maximal ATP production (35–41). Moreover, this high glycolytic rate entails a great lactate excretion, leading to increased TME acidosis, which alters the tumor stroma interface allowing enhanced invasiveness (42, 43). The presence of variable levels of lactate and hypoxia constitutes one of the main reasons for tumor heterogeneity. Indeed, “metabolic symbiosis” among hypoxic and aerobic cells within the tumor mass has been demonstrated. Lisanti and coworkers described “the Reversed Warburg effect” in which CAFs perform aerobic glycolysis and provide cancer cells with metabolites for oxidative phosphorylation (OxPhos) (44, 45) (Figure 2). In this pro-tumoral “metabolite crosstalk,” lactate produced by hypoxic cells is taken up by aerobic cells, which use it as their principal substrate for OxPhos. Lactate recycling is not new, and is well known from the Cori cycle in the liver (46). Sonveaux et al. showed that human cancer cells cultured under hypoxic conditions convert glucose to lactate and excrete it, while aerobic cancer cells take this lactate back up via monocarboxylate transporter 1 (MCT1) and utilize it for OxPhos (4). Another important glycolysis-related enzyme is pyruvate kinase (PK), which catalyzes the final glycolytic reaction. Therefore, reduction of PK activity causes a build-up of glycolytic intermediates that are redirected toward biosynthesis. Elevated expression of the isoform PKM2 has been demonstrated in several types of cancer, including colon, kidney, lung, and breast (47). Several studies have shown that PKM2 directly regulates the Warburg effect, since the knock-out of this enzyme reduces glucose uptake and lactate production, increasing oxygen consumption, and finally reducing tumorigenesis (48–50). In addition to this, Luo et al. reported that hydroxylation of PKM2 by PHD3 allows its binding to HIF-1α, enhancing expression of HIF-1α targeted genes (51). In addition, HIF-1α restricts OXPHOS and regulates the expression of pyruvate dehydrogenase kinase (PDK), an enzyme that phosphorylates and inactivates pyruvate dehydrogenase. The latter limits pyruvate utilization for OxPhos (52). Furthermore, active Akt2 accumulates in the mitochondria during hypoxia and phosphorylates pyruvate dehydrogenase kinase 1 (PDK1) on Thr346 to inactivate the pyruvate dehydrogenase complex (53). Regarding the HIF pathway, cancer cells present frequent activation of the PI3K–mTOR axis, which functions as a nutrient sensor pathway. mTOR activation favors HIFα activity and promotes tumor angiogenesis. Thus, it has been shown that loss of the mTOR inhibitor TSC2 (tuberous sclerosis complex 2 protein) results in the accumulation of HIF-1α and increased expression VEGF (54). Another study relates mTOR-mediated regulation of HIF-1α to the pathogenesis and increased angiogenesis in chronic myelogenous leukemia (55) (Figure 2).
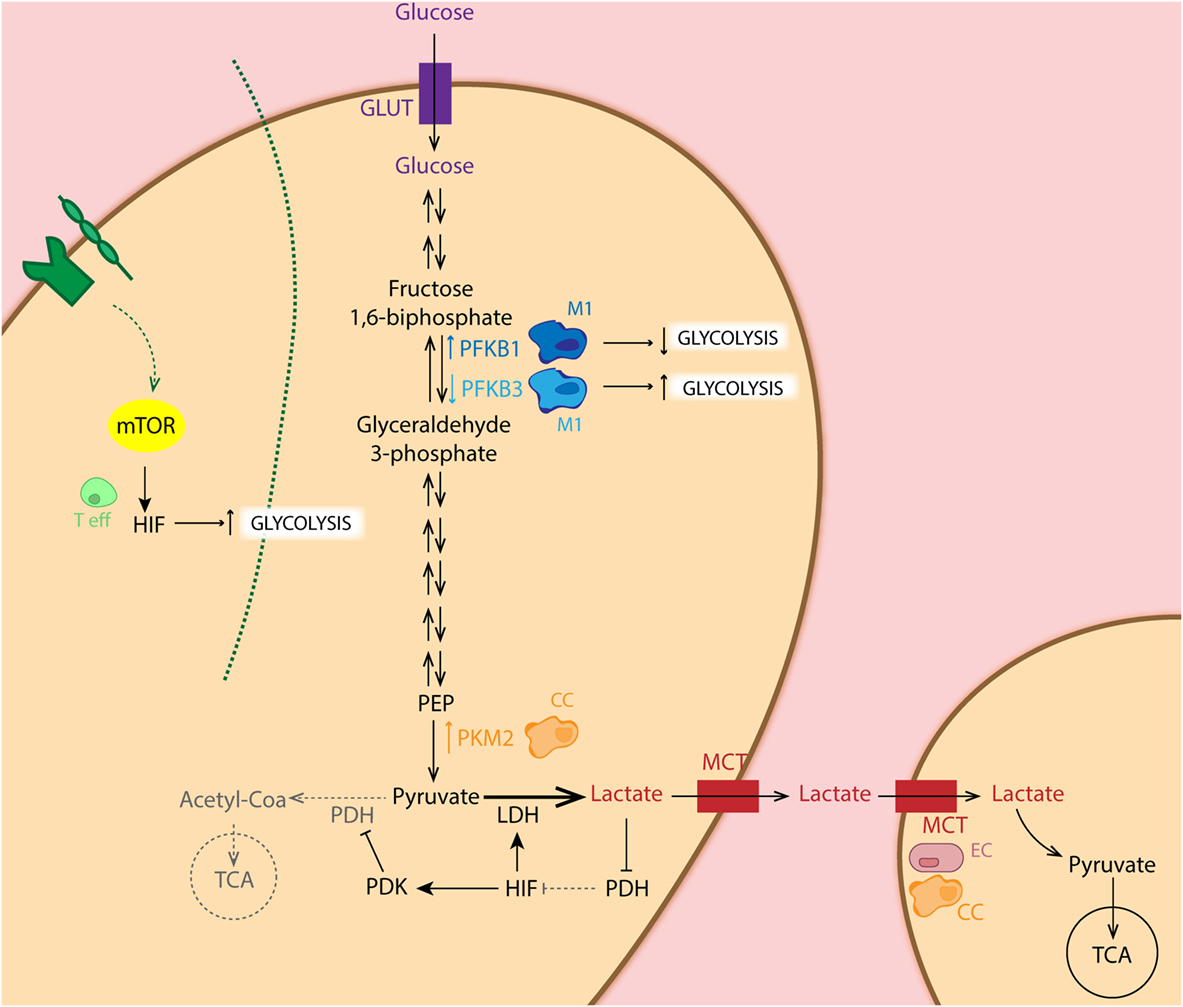
Figure 2. Schematic representation of the glycolytic pathway in different cell types. Metabolic intermediates such as lactate inhibit the PHD function, leading to accumulation of hypoxia-inducible factor (HIF) which regulates expression of several glycolytic enzymes. Importantly, HIF balances glucose metabolism: (1) by inducing expression of LDH, leading to conversion of pyruvate to lactate, (2) as well as dampening the entry of pyruvate in the TCA cycle through inhibition of PDH by pyruvate dehydrogenase kinase (PDK) overexpression. Particularly, cancer cells (CC: orange) show increased expression of PKM2, the limiting rate enzyme of glycolysis, favoring glucose metabolism. CCs as well as endothelial cells (ECs: pink) show increase expression of MCT, facilitating lactate uptake for oxidative phosphorylation. Pro-tumoral M2 macrophages (light blue) present increased expression of phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) isoform, enhancing glycolysis in these cells, whereas anti-tumoral M1 (dark blue) express the low activity isoform PFKFB1. Activation of T cells (green) via TCR-CD28 leads to enhanced glycolysis essential for their effector functions in an mTOR/HIF-dependent manner.
Previous studies have shown that specific metabolites are able to directly regulate the hypoxia pathway. Therefore, loss-of-function mutations of the tumor suppressor genes encoding the succinate dehydrogenase complex and fumarate hydratase lead to the accumulation of succinate or fumarate, resulting in HIF stabilization through inhibition of PHDs (56). Also other intracellular metabolites, such as pyruvate, lactate and oxaloacetate block PHD-mediated inhibition of HIF-1α underlying its prominent basal activity, commonly seen in many highly glycolytic cancer cells. This suggests that enhancement of HIF-1 by glucose metabolites may constitute a feed-forward signaling mechanism involved in malignant progression (57). In addition, isocitrate dehydrogenases 1 and 2 (IDH1 and IDH2) are frequently mutated in cancer. These enzymes function at the intersection of different processes, including oxygen-sensing signal transduction, cellular defense against oxidative stress, oxidative respiration, and cellular metabolism in lipid synthesis. The mutated forms of IDHs (58) produce 2-HG instead of α-KG essential for PHD function (59). Indeed, it has been shown that 2-HG can either inhibit or activate PHD-driven hydroxylation of HIF in an enantiomer-specific way (60).
Since the discovery of the Warburg effect, mitochondrial dysfunction was designated as a metabolic hallmark of cancer cells. However, earlier studies provided genetic evidence that mitochondrial metabolism is essential for tumorigenesis (61–63). Indeed, cancer cells generate an abundant amount of NADPH in the mitochondria and the cytosol to sustain high antioxidant activity and prevent the build-up of potentially detrimental ROS (64, 65).
Although anaerobic glycolysis is an acclaimed feature of cancer cells, this is not the only metabolic alteration in the transformed cells. In fact, for tumor cells to proliferate, fatty acid (FA) synthesis (for membrane biogenesis) as well as glutaminolysis (for amino acid precursors) has been reported to be affected during tumorigenesis (37, 66, 67). It has been shown that lipid production is critical for cancer cell survival, while the expression of the central lipogenic enzyme fatty acid synthase (FASN) is strongly correlated with cancer progression (68, 69). FAs used for cancer cells during lipogenesis can be endogenously derived from citrate in the TCA cycle, but they can also be seized from exogenous sources. To obtain free FA from circulation, lipoprotein lipase (LPL) hydrolyzes circulating triglycerides. Then, free FAs are imported into the cell via the FA translocase CD36. Both proteins LPL and CD36 are widely expressed in breast, liposarcoma, and prostate tumor samples (70) (Figure 3). In addition to this, lipid metabolism plays an important role in preventing ER stress, as it appears that balancing saturated and unsaturated lipid species are required, due to the lipotoxic effects of the former. Therefore, desaturation of de novo synthesized lipids by oxygen-dependent stearyl-Coenzyme A (CoA) desaturases (SCDs) plays a critical role for cancer cell survival. In this regard, SCD1-mediated lipid desaturation has been found to be a critical determinant of cancer cell survival downstream SREBP transcription factors (which are regulated by the mTORC1 pathway) (71). In fact, unsaturated lipid deficiency of hypoxic cells has been shown to cause cell death by ER stress and activation of the unfolded protein response in an mTORC1-dependent manner (72, 73). Another important lipid-metabolism-related enzyme is acetyl-CoA synthase 2 (ACSS2). ACSS2 converts acetate to acetyl-CoA, which is used as a nutritional source by cancer cells supporting biosynthesis of membrane phospholipids. Moreover, it is an epigenetic regulator in histone acetylation. It was also shown that hypoxia enhances the expression of ACSS2, which has been related to poor prognosis in breast cancer patients (74). The importance of lipid metabolism alterations in cancer cells relays not only on the role of lipids for biogenesis but also on their capacity to signal. In this respect, it has been shown that in human cancer cells as well as primary tumors, monoacylglycerol lipase (MAGL) is vastly overexpressed. This enzyme regulates a FA network that drives oncogenic signaling lipids, which promotes migration, invasion, survival, and in vivo tumor growth (75).
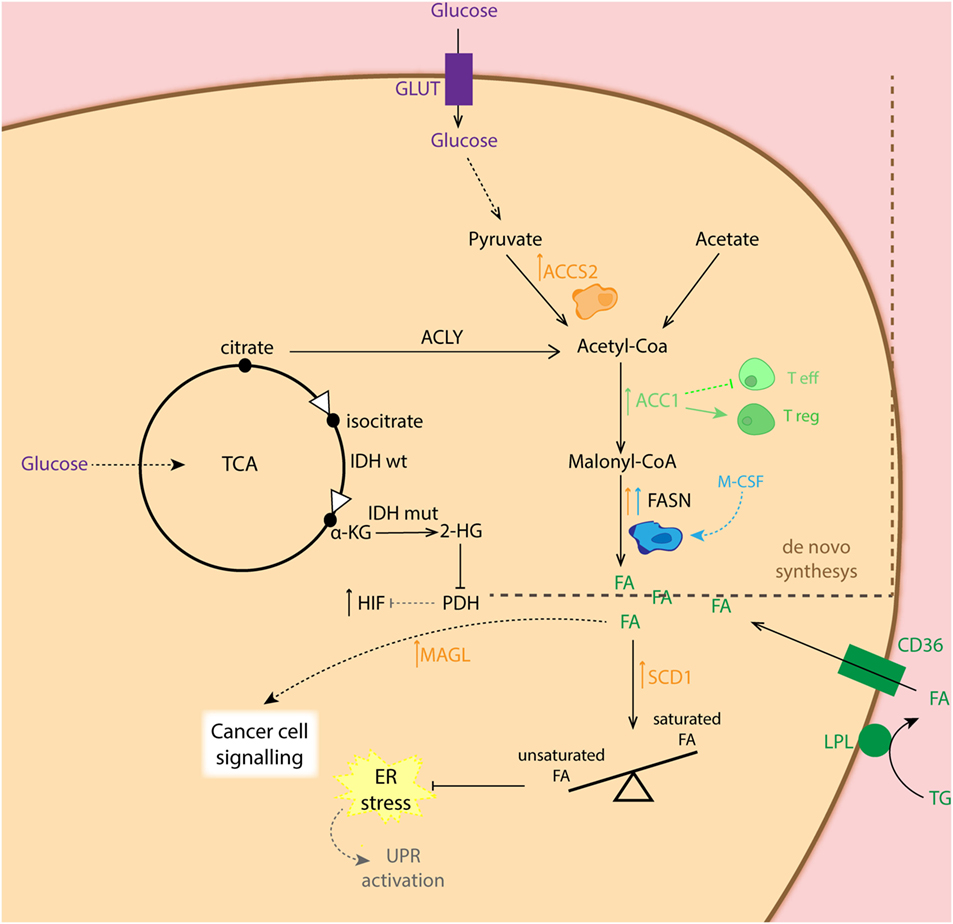
Figure 3. Schematic representation of the fatty acid (FA) synthesis in different cell types. Cancer cells (CC: orange) show increased expression of ACCS2 favoring conversion of pyruvate to acetyl-coA as well as fatty acid synthase (FASN) for conversion of malonyl-coA to FA. The latter, is also overexpressed in M2 macrophages (blue). CCs can also obtain FA from exogenous sources via converting triglycerides (TG) into FA, which are taken up by the CD36 transporter. Overexpression of monoacylglycerol lipase (MAGL) in CC is related to synthesis of lipids involved in cell signaling. Increased expression of SCD1 in CC balances higher presence of unsaturated FA essential for maintenance of ER homeostasis. Enhanced expression of ACC1 shifts T cell differentiation toward T regulatory cells (Treg: dark green).
As mention before, tumors are glutamine addicted. Cancer cells display high rates of glutaminolysis in order to obtain several precursors needed for supporting robust proliferation. In this regard, c-Myc has been shown to directly upregulate glutamine-metabolizing enzymes, such as glutaminase, which leads to fast integration of nitrogens and carbons in the anabolic network (76–78). Interestingly, it has been reported that the c-Myc function is directly regulated by both HIF-1α and HIF-2α (79, 80). The reductive metabolism of glutamine, mediated by IDH1, adds extensively to lipogenesis in cancer cells (81) (Figure 4), and which is partially facilitated by an increase in PDK1 (53, 82) and c-Myc (76, 83) in a HIF1-dependent manner, but it is primarily determined by the relative abundance of citrate and α-KG (83, 84). In addition, Kynurenine is another oncometabolite, which was defined as a tryptophan metabolite made from indoleamine-2,3-dioxygenase (IDO) (85), and known for its robust immunosuppressive effects (86) (Figure 4).
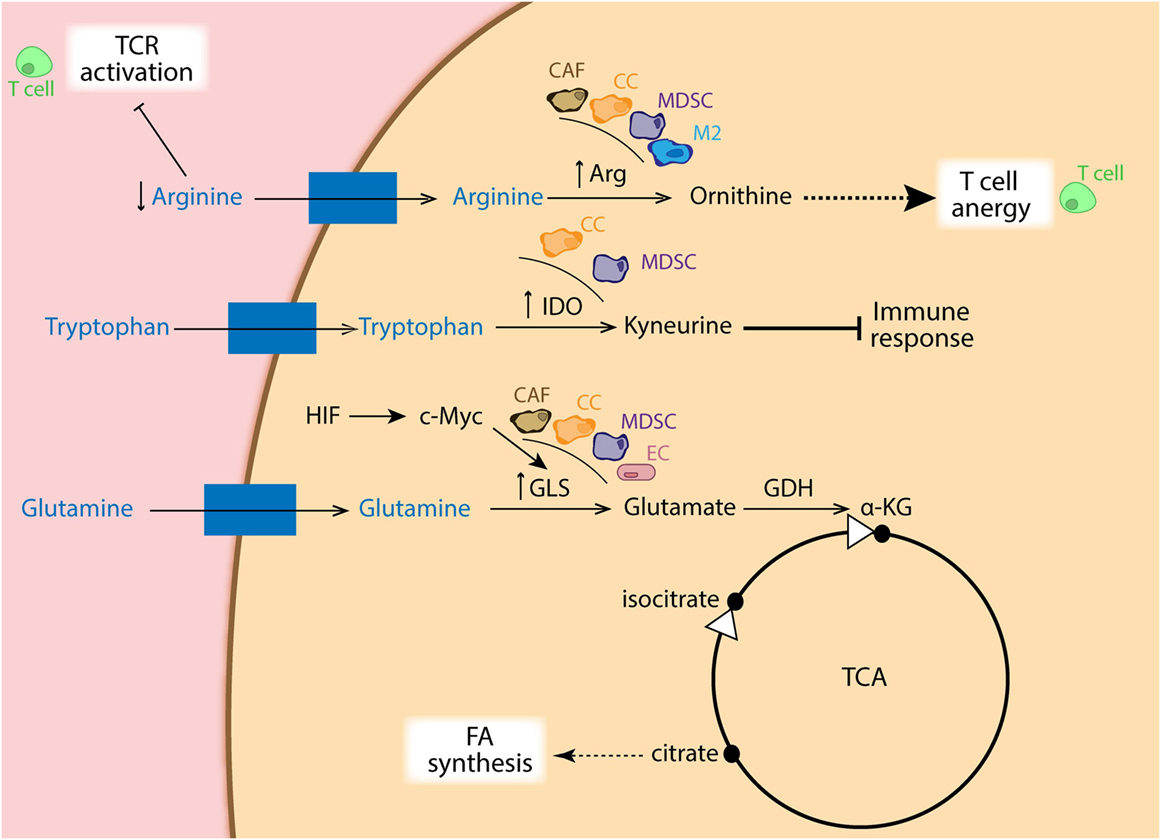
Figure 4. Schematic representation of amino acid metabolism in different cell types. Cancer cells (CCs: orange), cancer-associated fibroblasts (CAFs: brown), myeloid-derived suppressor cells (MDSCs: purple), as well as M2 macrophages (blue) show increased expression of arginase, depleting the surrounding environment of arginine, which is essential for TCR activation in T cells (green). Ornithine coming from arginase metabolism also dampens T cell activation. Moreover, increased expression of indoleamine-2,3-dioxygenase (IDO) in CC and MDSCs favors catabolism of tryptophan into kyneurine, with immune suppressive effects. In addition, overexpression of GLS via HIF/c-Myc regulation in CA, CAFs, MDSCs, and endothelial cells (ECs: pink) generates glutamate, which is used to replenish intermediates of the TCA cycle, favoring FA synthesis from citrate.
In addition to this, metabolic fitness relates to an increased HIF signaling in tumor cells, which permits the cancer cells to strive better for crucial metabolites, such as glucose and glutamine, than the stromal cells (87). This competition for nutrients has been demonstrated in the exhaustion of tumor-associated lymphocytes, suggesting a metabolic associated immune suppression (88, 89).
Tumor Microenvironment
Although tumor cells have been the main subject of study in cancer research, stromal cells and infiltrating immune cells have gained great interest, over the past years. Furthermore, the intriguing crosstalk of tumor cells with the TME or even different components of the TME among each other (as introduced in the previous section), regulate a vast amount of processes during tumor development (3). In this section, we will discuss several metabolic adaptations of different cell types of the TME (Figure 5).
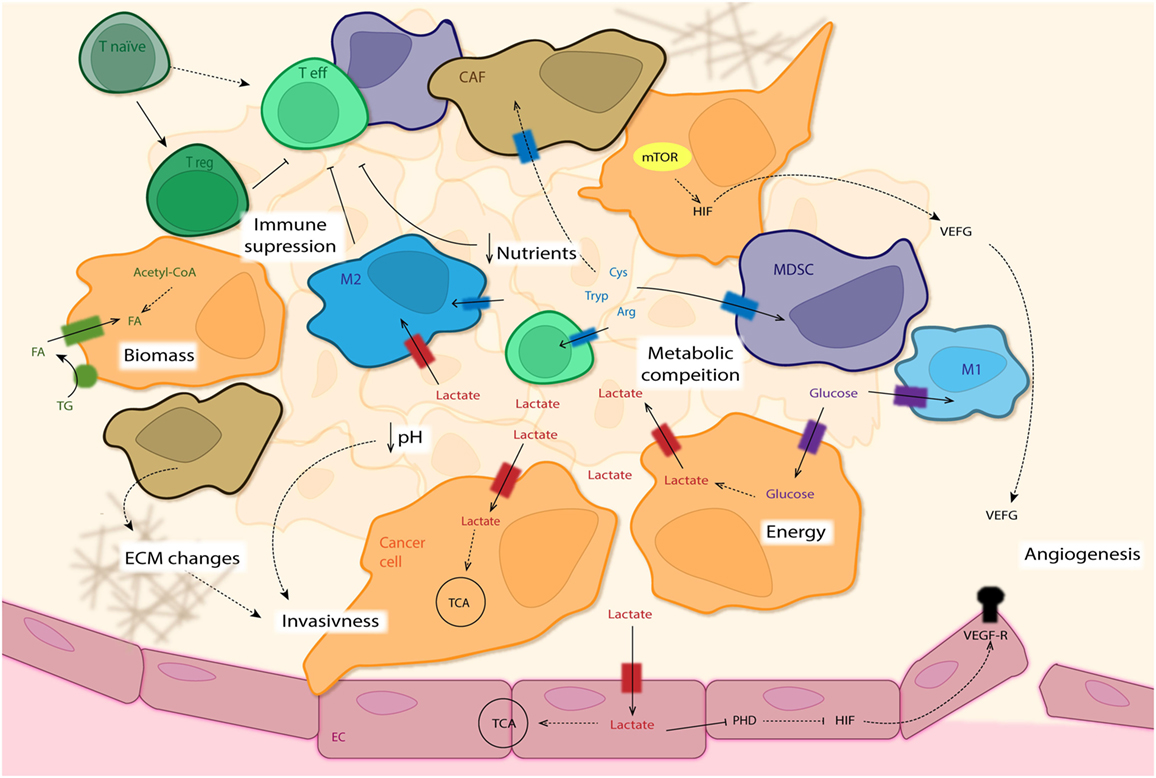
Figure 5. The tumor microenvironment (TME). This overview summarizes the intricate interactions of the different cells types that constitute the tumor mass. Some cancer cells (CC: orange) use glycolysis as main source for energy production (independently of oxygen availability—Warburg effect), excreting lactate and leading to acidification of the surrounding area, which finally promotes invasiveness of CC. Other CCs as well as M2 macrophages (blue) and endothelial cells (ECs: pink) with access to adequate levels of oxygen use this lactate for oxidative phosphorylation (OxPhos). Lactate in ECs leads to inhibition of PHD and subsequent activation of HIF, enhancing expression of VEGF-Rs (among others) and promoting angiogenesis. Uptake of fatty acids (FA) as well as de novo synthesis is used for new biomass required for fast CC proliferation. Next to amino acid depletion in the tumor mass by M2 cells, cancer-associated fibroblasts (CAF: brown) and myeloid-derived suppressor cells (MDSCs: purple) establish a metabolic competition between pro- and anti-tumor forces present in the TME. The absence of nutrients needed for T effector cell activation (Teff: light green) leads to their anergy, with a differentiation shift toward T regulatory cells (Treg: dark green). The lack of nutrients leads to activation of the mTOR pathway in CCs and production of VEGF (among others) via HIF regulation, promoting angiogenesis.
Stromal Cells
Endothelial Cells (ECs)
Endothelial cells are the best characterized cells of the TME. Despite the fact that ECs dispose of immediate access to oxygen in the blood, ECs are highly glycolytic, generating up to 85% of their ATP via glycolysis. Indeed, their glycolytic rate is comparable to that of cancer cells, and increases even during proliferation (90). Rather than acting as a bioenergetic power source, it has been reported that through the production of pro-angiogenic reactive oxygen species (ROS) mitochondria in ECs have a signaling function (91). In this regard, also the important role of the pentose phosphate pathway (PPP) as a weight against oxidative stress is noteworthy, as it controls redox homeostasis through NADPH production, together with ribose-5-phosphate for synthesis of lipids nucleotides and amino acids (92). Although glutamine and FA metabolism for ATP production in ECs is still under debate, glutaminolysis has been described to be essential for EC proliferation, as inhibition of glutaminase induces their senescence (93, 94). Also cholesterol synthesis has been shown to be crucial for vessel sprouting, as it enables the development of lipid rafts required for proper membrane localization and signaling of the VEGF receptors (95). At this point, it is important to note that VEGF creates a tip cell signal, required for vessel sprouting, a process during which ECs display exclusive patterns of cellular metabolism with high rates of glycolysis and reliant on FAO for nucleic acid synthesis and proliferation (94, 96). Since tip cells are located far from functional vessels, their activity is also regulated via the hypoxia pathway (97). In this regard, it has been shown that deletion of endothelial HIF-2α enhances angiogenesis, although the vessels were more disorganized and hypoxic (98, 99). In addition, PHD2+/− mice showed increased HIF-α stabilization, and normalization of the endothelium, increased oxygenation and reduced secondary metastasis (13). On top of this, it has been reported that the HIF subunits have opposing roles when it comes to the permeability of the endothelium. HIF-1α deficiency in ECs hampers tumor cell migration through endothelial layers, while loss of HIF-2α enhances tumor cell migration and metastasis (14). These contrasting effects may be directly related to the dramatic differences in inducible nitric oxide synthase (iNOS) expression in both deficient lines. In addition to this, a pro-tumoral “metabolite crosstalk” between EC and cancer cells has been reported. In this regard, Boidot et al. identified a direct link between the function of p53 and MCT1 expression, regulating the influx of lactate produced by cancer cells into EC (100). Later studies from the same group showed that internalized lactate through MCT1 by EC promote tumor angiogenesis through PHD2 inhibition, and activating HIF1 (101, 102). The same mechanism has been reported to trigger IκBα degradation, stimulating an autocrine pro-angiogenic NFκB/IL-8 pathway, and finally driving cell migration and tube formation (103). Furthermore, the receptor tyrosine kinases AXL, TIE2, and VEGFR-2 is activated by lactate in order to promote angiogenesis (104).
Fibroblasts
Another important stromal cell of the TME is the CAF. The importance of CAFs relies not only on their ability to produce growth factors and chemokines regulating other stromal cells and cancer cells but also in their capacity to modify the extracellular matrix (ECM), facilitating tumor angiogenesis and invasiveness (105). CAFs is actually a mix of myofibroblast-like cells that ascend different types of cells, including fibroblasts, bone-marrow-derived stromal cells, ECs, and adipocytes (106–108). Despite their relevance in regulating tumor development, studies on the metabolism of CAFs have been limited. Interestingly, proliferating fibroblasts produce biomass for a next proliferation, while quiescent fibroblasts use biomass to replace oxidized lipids and degraded proteins, as well as synthesis of ECM proteins. Hypoxia pathway proteins are an important stimulus of this process, since they increase the expression of remodeling enzymes leading to increased tumor rigorousness and enhanced metastasis (109). In this respect, reduced PHD2 activity led to a diminished CAF-induced ECM remodeling and diminished metastasis (12, 110). Independent of their activation state, healthy fibroblasts incorporate glucose carbons in the TCA cycle at comparable rates (111). Fibroblasts have also been described to replenish intermediates of the TCA by a process known as anaplerosis. In particular, anaplerotic flux from pyruvate to oxaloacetate via pyruvate carboxylase in quiescent fibroblasts ensures continuity of the TCA cycle, whereas proliferating fibroblasts primarily use glutamine for anaplerosis. Like cancer cells, proliferating fibroblasts rely on PPP for biosynthesis. By contrast, quiescent fibroblasts generate NADPH via the PPP, which is essential for their survival (111). Moreover, CAFs also perform FA synthesis, essential for their proliferation (112). Production of ROS by cancer cells inhibits PHD2 (with subsequent gain of function of HIF-1α) and enhances NO production by CAFs. This finally leads to dysfunctional mitochondria that are mitophaged, forcing CAFs to rely on glycolysis for ATP production, with accompanied increase in lactate production (113). High lactate production together with amino acids and keton bodies supply cancer cells with high-energy nutrients for oxidative metabolism in tumor oxygenated areas (114, 115). In addition, loss of HIF-1α in fibroblasts leads to vascular normalization, decreases hypoxia but increases breast cancer development (116). HIF1α-driven aerobic glycolysis in stromal cells supports cancer cell growth via the paracrine production of nutrients (such as lactate), which cancer cells can recycle (4, 102, 117, 118). As we will discuss in the following section, arginases (ArgI and ArgII) are important for immune suppression in the tumor, converting l-arginine to ornithine, resulting in T cell anergy and reduced anti-tumor response (119). CAFs that are localized to hypoxic regions in pancreatic tumors express high levels of ArgII, suggesting for CAF-mediated immunosuppression (120).
Immune Cells
Although numerous studies have indicated the involvement of almost every immune cells type, macrophages have been by far the most studied immune cell type during cancer development. As mentioned before, the importance of metabolism in regulating immune cell phenotype and function and its impact during tumor development is well known (121). Indeed, during the past years, a new field of study has emerged, focusing on the metabolism of the immune system also known as immunometabolism. This new area of research studies how changes in cell metabolism regulate the immune system during homeostasis as well as during inflammatory processes, including tumor-associated inflammation. Indeed, how metabolic changes in immune cells during tumor development regulate the contribution of these cells to disease progression has been the center of a great number of studies. As mentioned before, high levels of glycolysis but hampered angiogenesis inside hypoxic tumor areas can result in near glucose depletion and accumulation of waste products such as lactate. Hence, anti-tumoral immune cells infiltrating TME face significant metabolic challenges to mount and sustain against the tumor. In this regard, T cells have been extensively studied as the main force fighting tumor growth, characterized by particular metabolic shifts according to their activation state (Figure 5).
T Cells
Resting naïve T cells require low amounts of glucose, amino acids, and FAs to sustain basic energetic and minimal replacement demands. More than 90% of their ATP production comes from FAO and OxPhos, whereas glutaminolysis and PPP contribute to biosynthesis purposes. Upon activation, T cells increase glucose and glutamine catabolism for nucleotide and lipid synthesis that are essential for cell growth, while OxPhos for ATP production is maintained (122–125) (Figure 5). Indeed, glycolysis has been described to be essential for T cell effector functions, since its impairment suppresses proliferation. TCR–CD28 co-stimulation triggers the shift from naïve to effector T (Teff) cells through PI3K/Akt/mTOR pathway, and activation of cMyc and HIF-1α transcription factors. This promotes glycolytic gene expression and post-translational modification essential to drive aerobic glycolysis and amino acid metabolism in Teff cells, while suppressing catabolic FAO for ATP (122, 126–128). On the other hand, T regulatory (Treg) cells orchestrate a pro-tumoral environment by inhibiting effector T cell responses in the tumor area. Contrary to Teff, Treg cells rely on both FAO and OxPhos for ATP upon activation (127, 129, 130). This metabolic state allows Treg cells to survive tumor conditions and exert their immunosuppressive effect, whereas anti-tumor effector T cells would face impair TCR signaling due to lack of glucose (122, 123, 129–131). Indeed, Treg expansion has been linked to activation of the nutrient stress sensor AMPK. Thus, when the AMP:ATP ratio increases due to the lack of nutrients, AMPK favors oxidative catabolic pathways (132). This metabolic shift implies that AMPK can immediately impact the balance of Teff and Treg cells via mTORC1 inhibition (133). Upon CD3/CD28 activation, T cells accumulate metabolites involved in anabolic pathways increasing FAS (122). In addition, it has been shown that mTORC impairment compromise de novo lipid synthesis in T cells through induction of the transcription factor SREBP (134). The importance of lipid metabolism in T cell biology has been also reported at the level of the FAS limiting rate enzyme ACC1, which deletion interferes with differentiation of naïve to effector T cells (135). However, ACC1 deletion did not affect the ability of naïve T cells to proliferate and differentiate into Treg (135, 136), suggesting FAS as an important metabolic checkpoint during activation-induced differentiation into Teff cells. Similar to cancer cells, PI3K–mTOR axis stimulates HIFα activity, downstream of the TCR activation (137, 138). Also, IL6 stimulation of T cells leads to JAK–STAT pathway increased transcription of HIF mRNA (126, 139). As mentioned before, metabolites themselves can also act as signaling molecules. In this regard, decreased flux through the TCA cycle may lower succinate levels. It has indeed been shown that succinate can stabilize HIF-1α, inducing transcription of several inflammatory cytokines (32). Although there are not many studies on the role of the HIF-pathway in T cells during tumorigenesis, previous studies have reported a role for this pathway during T cell-mediated inflammation and differentiation (Th17 and Treg balance). Dang et al. reported that HIF-1α enhances Th17 differentiation by direct transcriptional activation of RORγt, while inducing FoxP3 proteasomal degradation and dampening Treg differentiation (126). During Th17 cell development, glycolysis rate is increased through mTOR–HIF-1α signaling induction (127). The tumor protecting or promoting role of Th17 is still controversial due to its differently described phenotypes [for review, see Ref. (140)].
Moreover, it has been reported that T cell activation is blocked due to disruption of the electron transport chain leading to impaired mitochondrial ROS production (141). Apart from this, the hypoxic environment within the tumor area may protect tumor cells from anti-tumor immunity by HIF-1α-dependent upregulation of PD-L1 on cancer cells, which inhibits PD-1 expressing T effector cells (142). Moreover, high lactate levels in the tumor area have been shown to suppress the PI3K/Akt/mTOR pathway inhibiting glycolysis, finally leading to impaired T cells (128, 143, 144). Glycolysis inhibition can lead to increase expression of PD-1, which is associated with T cell exhaustion and non-responsiveness through inhibition of TCR and CD28-mediated co-stimulation, helping the tumor to escape immune surveillance (145). Also tryptophan has gotten attention as a limiting amino acid in T cell activation. Tryptophan metabolism is mainly regulated by IDO, highly expressed by cancers, and in fact correlated with poor prognosis (146) (Figure 4). Using a mouse sarcoma model, Chang et al. showed that glucose restricts T cells, leading to hampered mTOR activity, glycolytic capacity, and INFγ production. The result is enhanced tumor progression. Checkpoint blockade using antibodies against CTLA4 PD1 and PDL1 restore all previous changes (88). Recent work supports the hypothesis that lactic acid blunts the immune response mediated by T and NK cells (147).
Macrophages
Historically, macrophages have been classified as M1 (classically activated) and M2 (alternatively activated) according to their pro- or anti-inflammatory state, respectively. However, more recently, the idea of a multidimensional spectrum rather than dual macrophage activated states has emerged (148, 149). Since specific stimuli induce specific functional outcomes, it is expected that different states of polarization present a particular metabolism. M1-phenotyped macrophages are highly glycolytic and characterized by an increased induction of the strong glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 isoform (PFKFB3) (150), conferring them an energetic advantage in hypoxic regions (151). This glycolytic state is mediated by the Akt/mTOR/HIF-1α pathway (32) and has been shown to induce TNF expression (152). The latter suggests a direct regulation of the inflammatory phenotype of macrophages depending on the glycolytic pathway. In relation to their anti-inflammatory role, M1 macrophages also use PPP and malic enzyme in order to produce high amounts of NO and ROS for killing pathogens, as well as NADPH to protect themselves from this high oxidative burst (153). On the other hand, M2 macrophages present high rates of FAO and OxPhos, with low glycolytic activity due to the expression of the weak glycolytic activator PFKFB1 isoform (154, 155) (Figure 2). O’Neill et al. showed that M2 polarization upon IL-4 signaling stimulates mitobiogenesis by upregulating PGCβ, enhancing the metabolic switch to FAO (155). Also, M2 macrophages reduce PPP flux and GSH via induction of carbohydrate kinase-like protein (CARKL) (156). Traditionally, TAMs have been related to a more M2 anti-inflammatory and pro-tumor phenotype. However, the fluctuating levels of lactate and oxygen in the heterogeneous tumor mass induce differential macrophage responses depending on their functional plasticity in tumor. In general, a lactic acid-induced polarization to M2 has been reported, inducing an immunosuppressive and tissue remodeling phenotype. This is characterized by the production of VEGF and arginase in a HIF-1α dependent manner (43, 121). Also, TAMs respond and adapt to different oxygen levels through activation of the HIF pathway. In this regard, it has been shown that expression of HIF-1α has a protective role in hypoxic areas, since loss of this transcription factor leads to decrease expression of IL-6, TNF, and iNOS, as well as increased CD206, all of them characteristic markers of the M2 phenotype (15, 17). TAMs are also characterized by high expression of arginase enzyme (induced by the high lactate levels in the TME). This has been shown to impair anti-tumor T cell function due to depletion of the arginine pool required for NO and protein synthesis leading to TCR function impairment (19, 43, 157, 158). Apart from this, recent studies have described the importance of iron metabolism in macrophage polarization. M1 macrophages express reduced levels of ferroportin, the iron transporter, but high levels of H-ferritin involved in iron storage, whereas M2 macrophages present the opposite profile. Therefore, iron sequestration in M1 macrophages is believed to restrict both bacterial and tumor growth, while M2 macrophages release iron, which promotes tissue repair and tumor cell proliferation (159, 160). In addition, iron constitutes a cofactor of the PHD enzymes in the hypoxia pathway. Therefore, intracellular iron levels directly regulate HIF-1α stability crucial for the survival and pro-tumor function of TAMs (161). In a tumor setting, TAMs also undergo changes in their lipid profile. It has been shown that M-CSF secreted from tumor cells leads to enhanced expression of FASN in macrophages, which polarize to an IL-10 expressing pro-tumoral phenotype (162).
Myeloid-Derived Suppressor Cells (MDSCs)
Another important immune cell type that has recently gained great attention is the MDSCs that, as its name indicates, is functionally defined by its potent immunosuppressive activity in both innate and adaptive immunity. This cell population comprises two major subsets: monocytic MDSCs (M-MDSCs) and polymorphonuclear MDSCs (PMN-MDSCs) (163). G-CSF has been described to play a critical role in differentiating and mobilizing bone marrow granulocytic precursors within tumors (164); whereas depending on the magnitude and context of the stimulus, GM-CSF can induce accumulation of these suppressor subsets thereby inhibiting proliferation as well as anti-tumor ability of neu-specific T cells (165, 166). In addition, it has been shown that IL-4Rα activation through IL-4 and IL-13 exposure evokes MDSCs suppressive mechanism in a STAT6-dependent manner (167, 168). MDSCs promote immune dysfunction by using different mechanisms, either directly via impairment of T cell amino acid metabolism or through regulation of oxidative stress, which finally interferes with T cell viability, migration, and activation. Also, MDSC are able to indirectly induce other immune regulatory cells, such as Treg cells and TAMs (169–171). The same as macrophages, MDSCs show high expression of arginase, depleting arginine from the TME essential for TCR activation and T cell proliferation (172, 173). MDSCs also sequester the amino acid cysteine, which is indispensable for T-cell activation (174) and expresses IDO enzyme for tryptophan catabolism (175, 176). Deprivation of the later has been shown to induce expansion of the Treg cell population (177). Combined expression of nitric oxide synthase, arginase, and NADPH oxidase confers MDSC important regulators of oxidative stress in TME (178–181). Therefore, presence of RNI (mainly derived from M-MDSC), and ROI (mainly from PMN-MDSC) downregulates TCR and IL2 receptor signaling, inhibiting T cell activation and proliferation (170). In addition to this, MDSCs show enhanced FA uptake and high expression of FAO enzymes, accompanied by an increased mitochondrial mass and oxygen consumption rate (182). Corzo et al. described the role of the hypoxia pathway in MDSCs, with HIF-1α as main responsible for MDSC differentiation and function in TME (183). In addition, HIF-1α-mediated expression of PD-L1 is essential for mediating MDSC immune suppression (as discussed in the previous sections) (184). Also hypoxia can enhance MDSC migration to the tumor site via HIF-1α-mediated production of chemokines (185, 186). Hypoxia also influences seeding of MDSCs in the pre-metastatic niche by stimulating increased secretion of lysyl oxidase (187–189). This process drives ECM remodeling in the metastatic niche and suppresses NK anti-tumor response (188).
Neutrophils
During the past few years, the presence of tumor-associated neutrophils (TANs) has gained attention due to their pivotal role in tumor development. Indeed, a dual effect has been proposed for TANs during onset (190). In this regard, Fridlender et al. showed that in the absence of TGFβ, TANs encourage Teff response and anti-tumor activity, whereas in the presence of TGFβ they exhibit tumor promoting activity (191). Neutrophils comprise a significant proportion of the inflammatory infiltrate in cancerous lesions and high levels of blood neutrophils were observed in patients suffering from advanced stage tumors (192). In many cancer types, such as bronchoalveolar carcinoma (193), metastatic melanoma (192), and andrenal carcinoma (194), neutrophil accumulation was associated with increased aggressiveness and poor prognosis (195). By contrast, high neutrophil counts in gastric tumors correlate with favorable prognosis (196). Since neutrophils constitute an already mature population that does not proliferate, possible changes in their metabolism have not been studied in depth. Regarding their metabolism, neutrophils are strongly committed to glycolysis and PPP, whereas their few mitochondria are used for maintenance of the redox balance. It has been shown that their high rates of glycolysis are necessary for the generation of ATP, in which HIF-1α is critically involved by regulating the expression of key glycolytic enzymes (197). Apart from its main role in energy metabolism, glycolysis has been shown to be essential for some neutrophil functions such as oxidative burst and chemotaxis (198). Indeed, Thompson et al. reported that murine HIF-2α deficient inflammatory neutrophils displayed no impairment of chemotaxis, phagocytosis, or respiratory burst but elevated sensitivity to apoptosis leading to reduced neutrophilic inflammation (199). It has also been shown that hypoxia can promote neutrophil recruitment by modifying the adherence properties of ECs to neutrophils (200). In addition, FAS has been reported to have some relevance in neutrophil biology. In this regard, Lodhi et al. showed that peroxisomal lipid synthesis drives inflammation by supporting neutrophil membrane phospholipid composition as well as viability (201). Another important feature of neutrophils is the formation of neutrophil extracellular traps (NETs). Brinkmann et al. described for the first time that activated neutrophils are able to release their chromatin (DNA and histones) loaded with granule enzymes forming an extracellular mesh-like structure that can both trap and kill extracellular organisms (202). Glucose uptake, glycolysis, and a shift toward PPP have been shown to be essential for NETs formation (203, 204). Beyond their bactericidal role, NETs has been described to sequester tumor cells and promote metastasis (205). Also, association of adhesion molecule and cytokines to NETs has been related to cancer-induced organ failure (206).
Therapy Perspectives
Regarding tumor therapy, there are different approaches that can be used, including targeting of the cancer cells, or switching the nature of the immune cell to a more anti-tumoral state. During the past decade, a lot of attention has been given to try and selectively kill the tumor based on their metabolic alterations (207, 208). Indeed, increasing evidence supports the idea that dysregulated cellular metabolism is connected to drug resistance during cancer therapy. Therefore, combining cellular metabolism inhibitors with chemotherapeutic drugs constitutes a promising strategy to overcome this.
It has also become clear that there is much more the only Warburg effect when it comes to the metabolic rearrangements associated with malignant transformations. Indeed, there is also an increased flux through the PPP, higher rates of glutamine consumption and lipid biosynthesis, maintenance of redox homeostasis and limited levels of autophagy, at least in the first steps of oncogenesis (209–211).
Glycose Metabolism
Due to the high glycolic rate in developing tumors, regulation of this pathway in cancer cells has been considerately studied. Starting by targeting glucose intake, the GLUT1 inhibitor WZb117 has been shown to reduce ATP production and ER stress induction in cancer cells, with a synergistic anticancer effect in combination with cisplatin or paclitaxel (212). Also, under hypoxia conditions, Cao et al. reported that the GLUT1 inhibitor phloretin significantly enhances anticancer effects of the antibiotic daunorubicin, overcoming hypoxia-conferred drug resistance (213). In the same line, inhibition of glycolysis with 2-DG (glucose analog for hexokinase) in combination with radiation or chemotherapy treatments, enhance clinical efficacy of the latter (214). A great number of studies have focused on the importance that the highly expressed PKM2 enzyme in cancer cells has in conferring resistance to therapy (48, 215–217) (Figure 2). Inhibition of this last rate-limiting enzyme in the glycolytic pathway, increases apoptosis and inhibits proliferation during cisplatin (218), and docetaxel (219) treatment. In the last step of the glycolytic pathway, LDHA expression and activity has been reported to be higher in Taxol-resistant breast cancer cells. Inhibition of LDHA by oxamate (a pyruvate analog) in combination with paclitaxel treatment has shown synergistic effect on taxol-resistant cells by promoting apoptosis (220). Regulating the shift between glycolysis and TCA, PDK inhibits PDH conversion of pyruvate to acetyl-CoA. Inhibition of PDK3 (functional of pyruvate isoform even in high concentration) has shown to diminish hypoxia-induced resistance in cervical and colon cancer (221, 222).
Recent evidence also indicates that modulation of immunometabolism plays an important role in controlling immune responses against cancer progression. Indeed, several studies have focused on targeting metabolic pathways to enhance T cell function and persistence. One of the most promising is the use of PD-1 blocking antibodies to rescue T cell glycolysis and enhance Teff functions (223). By contrast, inhibition of mTOR (224, 225) or AMPK (226, 227) has been shown to lead to controversial results.
Lipid Metabolism
Since proliferation of cells requires the generation of novel phospholipid membranes, targeting de novo lipogenesis or steroidogenesis would also be a potential anticancer therapy approach (68). Several enzymes involved in these synthesis pathways, including FASN (228, 229), ACLY (230), ACCs (231), choline kinase (232, 233), monoglyceride lipase (75), and HMGCR (234) have been ascribed critical roles in oncogenesis or tumor progression in vivo, yet have not been tested in clinical settings. Indeed, FAS is significantly upregulated and correlates with poor prognosis in many types of cancer. Therefore, it is not surprising that several FAS inhibitors, such as cerulenin, C75, orlistat, C93, or GSL 837149a, have shown anti-tumor activity (Figure 3). In addition, the combination of FAS inhibition with docetaxel (235), trastuzumab (236), or adriamycin (237) treatment increases therapy sensitivity in breast cancer. Cancer cell metabolism is also highly dependent on glutaminolysis. It has been shown that glutamine in combination with leucine activates mTORC1 by enhancing glutaminolysis and α-ketoglutarate production (238). Targeting glutaminolysis by using the mTORC1 inhibitor rapamycin has been reported to enhance cisplatin treatment in gastric cancer (239). Pharmacological inhibition of FAO functions in MDSC, averts immune inhibitory pathways and decreases the production of inhibitory cytokines. Consequently, blocking FAO postponed tumor growth in a T-cell-dependent way, and increased the anti-tumor effect after adoptive T cell treatment (182). COX-2 inhibitors or reducing COX-2 expression in 3LL cells, obstructed their capacity to induce arginase I in MDSC (240).
Amino Acid Metabolism
Inhibitors of folate metabolism, thymidine and deoxynucleotide synthesis and elongation of nucleic acid are the so-called antimetabolites and serve as standard chemotherapeutic regimens against many human neoplasms (241). Unfortunately, these agents are linked to toxicity in bone marrow and intestinal epithelium, as these are highly proliferating tissues. Compound 968 (242) and bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide, two specific GLS inhibitors (243), diminish glutamine catabolism and delay tumor growth in models of cancer. Targeting glutamate conversion to α-ketoglutarate by aminotransferases also diminishes tumor growth (244, 245). Replenishing the TCA cycle intermediates by providing substrates, such as glutamine, sustains mitochondrial metabolism in tumor cells (63) (Figure 4).
Mitochondria Respiration
Many types of tumor are highly dependent on OxPhos for their ATP (246–248). Therefore, these cells are probably sensitive to treatments that reduce mitochondrial ATP production. Moreover, inhibiting this mitochondrial ATP production would synergize together with approaches that diminish glycolysis, including inhibitors of the PI3K signaling pathway (249). It has been shown that phenformin (biguanide) inhibits mitochondrial complex I and in that way exerts its anti-tumor effects in experimental cancer models (250). Metformin has also antineoplastic activity (251). This appears to be independent of glycemia (252) and might reflect the ability to preferentially kill cancer stem cells, block mitochondrial respiration, intensify glutamine addiction, or limit inflammatory responses driving tumor growth (253–256). Indeed, its action is to specifically inhibit Mitochondrial complex 1, which in turn activates AMPK as a consequence of ATP decrease (257).
Conclusion
Our understanding of metabolic changes in cancer development has improved significantly over the past years. However, the influence of the hypoxia pathway proteins on the metabolic pathways in tumor cells and the TME is still not entirely known. In a vast amount of physiological as well as pathological situations, hypoxia-induced rewiring permits survival during metabolic stress. Conversely, this drives cancer progression, causing enhanced lethality due to resistance to therapy and greater metastatic potential. Therefore, more research is necessary to better understand hypoxia-induced alterations in cellular metabolism and eventually target these pathways, thereby eliminating malignant cells.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Research in the lab of Ben Wielockx has been supported by grants from the DFG (WI 3291/1-1, 1-2, 3, 5 and SFB/TRR 205 Die Nebenniere: Zentrales Relais in Gesundheit und Krankheit) and Seed grants from the Dresden International Graduate School for Biomedicine and Bioengineering (DIGS-BB, TU-Dresden) and BW is a Heisenberg Professor (DFG).
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
2. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell (2012) 21:297–308. doi:10.1016/j.ccr.2012.02.014
3. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19:1423–37. doi:10.1038/nm.3394
4. Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest (2008) 118:3930. doi:10.1172/JCI36843
5. Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev (2007) 26:225–39. doi:10.1007/s10555-007-9055-1
6. Meneses AM, Wielockx B. PHD2: from hypoxia regulation to disease progression. Hypoxia (Auckl) (2016) 4:53–67. doi:10.2147/HP.S53576
7. Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer (2012) 12:9–22. doi:10.1038/nrc3183
8. Sormendi S, Wielockx B. HIF-pathway proteins: central regulators of tumor immunology. Transl Cancer Res (2016) 5:S1503–8. doi:10.21037/tcr.2016.12.30
9. Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J (2003) 22:4082–90. doi:10.1093/emboj/cdg392
10. Klotzsche-von Ameln A, Muschter A, Mamlouk S, Kalucka J, Prade I, Franke K, et al. Inhibition of HIF prolyl hydroxylase-2 blocks tumor growth in mice through the anti-proliferative activity of transforming growth factorβ. Cancer Res (2011) 71(9):3306–16. doi:10.1158/0008-5472.CAN-10-3838
11. Klotzsche-von Ameln A, Muschter A, Heimesaat MM, Breier G, Wielockx B. HIF prolyl hydroxylase-2 inhibition diminishes tumor growth through matrix metalloproteinase-induced TGFβ activation. Cancer Biol Ther (2012) 13:216–23. doi:10.4161/cbt.13.4.18830
12. Kuchnio A, Moens S, Bruning U, Kuchnio K, Cruys B, Thienpont B, et al. The cancer cell oxygen sensor PHD2 promotes metastasis via activation of cancer-associated fibroblasts. Cell Rep (2015) 12:992–1005. doi:10.1016/j.celrep.2015.07.010
13. Mazzone M, Dettori D, De Oliveira RL, Loges S, Schmidt T, Jonckx B, et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell (2009) 136:839–51. doi:10.1016/j.cell.2009.01.020
14. Branco-Price C, Zhang N, Schnelle M, Evans C, Katschinski DM, Liao D, et al. Endothelial cell HIF-1α and HIF-2α differentially regulate metastatic success. Cancer Cell (2012) 21:52–65. doi:10.1016/j.ccr.2011.11.017
15. Mamlouk S, Wielockx B. Hypoxia-inducible factors as key regulators of tumor inflammation. Int J Cancer (2013) 132:2721–9. doi:10.1002/ijc.27901
16. LaGory EL, Giaccia AJ. The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol (2016) 18:356–65. doi:10.1038/ncb3330
17. Werno C, Menrad H, Weigert A, Dehne N, Goerdt S, Schledzewski K, et al. Knockout of HIF-1α in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis (2010) 31:1863–72. doi:10.1093/carcin/bgq088
18. Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan L-J, et al. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest (2010) 120:2699. doi:10.1172/JCI39506
19. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, Denardo DG, et al. Macrophage expression of hypoxia-inducible factor-1α suppresses T-cell function and promotes tumor progression. Cancer Res (2010) 70:7465–75. doi:10.1158/0008-5472.CAN-10-1439
20. Mamlouk S, Kalucka J, Singh RP, Franke K, Muschter A, Langer A, et al. Loss of prolyl hydroxylase-2 in myeloid cells and T-lymphocytes impairs tumor development. Int J Cancer (2014) 134:849–58. doi:10.1002/ijc.28409
21. Clever D, Roychoudhuri R, Constantinides MG, Askenase MH, Sukumar M, Klebanoff CA, et al. Oxygen sensing by T cells establishes an immunologically tolerant metastatic niche. Cell (2016) 166:1117–31.e1114. doi:10.1016/j.cell.2016.07.032
22. Kumar V, Gabrilovich DI. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology (2014) 143:512–9. doi:10.1111/imm.12380
23. Zeng W, Liu P, Pan W, Singh SR, Wei Y. Hypoxia and hypoxia inducible factors in tumor metabolism. Cancer Lett (2015) 356:263–7. doi:10.1016/j.canlet.2014.01.032
24. Corcoran SE, O’Neill LA. HIF1α and metabolic reprogramming in inflammation. J Clin Invest (2016) 126:3699–707. doi:10.1172/JCI84431
25. Eales K, Hollinshead K, Tennant D. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis (2016) 5:e190. doi:10.1038/oncsis.2015.50
26. Selfridge AC, Cavadas MA, Scholz CC, Campbell EL, Welch LC, Lecuona E, et al. Hypercapnia suppresses the HIF-dependent adaptive response to hypoxia. J Biol Chem (2016) 291:11800–8. doi:10.1074/jbc.M116.713941
27. Chandel NS, Mcclintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia a mechanism of O2 sensing. J Biol Chem (2000) 275:25130–8. doi:10.1074/jbc.M001914200
28. Masson N, Singleton RS, Sekirnik R, Trudgian DC, Ambrose LJ, Miranda MX, et al. The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep (2012) 13:251–7. doi:10.1038/embor.2012.9
29. Boulahbel H, Durán RV, Gottlieb E. Prolyl Hydroxylases as Regulators of Cell Metabolism. Biochem Soc Trans, (2009) 37(1):291–4.
30. Yang M, Soga T, Pollard PJ, Adam J. The emerging role of fumarate as an oncometabolite. Front Oncol (2012) 2:85. doi:10.3389/fonc.2012.00085
31. Ratcliffe PJ. Oxygen sensing and hypoxia signalling pathways in animals: the implications of physiology for cancer. J Physiol (2013) 591:2027–42. doi:10.1113/jphysiol.2013.251470
32. Tannahill G, Curtis A, Adamik J, Palsson-Mcdermott E, Mcgettrick A, Goel G, et al. Succinate is a danger signal that induces IL-1β via HIF-1α. Nature (2013) 496:238. doi:10.1038/nature11986
33. Warburg O. The metabolism of carcinoma cells. J Cancer Res (1925) 9:148–63. doi:10.1158/jcr.1925.148
34. Shestov AA, Liu X, Ser Z, Cluntun AA, Hung YP, Huang L, et al. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. Elife (2014) 3:e03342. doi:10.7554/eLife.03342
35. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab (2008) 7:11–20. doi:10.1016/j.cmet.2007.10.002
36. Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science (2010) 330:1340–4. doi:10.1126/science.1193494
37. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11:85–95. doi:10.1038/nrc2981
38. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11:325–37. doi:10.1038/nrc3038
39. Dang CV. Links between metabolism and cancer. Genes Dev (2012) 26:877–90. doi:10.1101/gad.189365.112
40. Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell (2013) 24:213–28. doi:10.1016/j.ccr.2013.06.014
41. Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol (2015) 17:351. doi:10.1038/ncb3124
42. Gatenby RA, Gawlinski ET. A reaction-diffusion model of cancer invasion. Cancer Res (1996) 56:5745–53.
43. Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513:559. doi:10.1038/nature13490
44. Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle (2010) 9:3506–14. doi:10.4161/cc.9.17.12731
45. Migneco G, Whitaker-Menezes D, Chiavarina B, Castello-Cros R, Pavlides S, Pestell RG, et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: evidence for stromal-epithelial metabolic coupling. Cell Cycle (2010) 9:2412–22. doi:10.4161/cc.9.12.11989
47. Wong N, Ojo D, Yan J, Tang D. PKM2 contributes to cancer metabolism. Cancer Lett (2015) 356:184–91. doi:10.1016/j.canlet.2014.01.031
48. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature (2008) 452:230. doi:10.1038/nature06734
49. Vander Heiden MG, Christofk HR, Schuman E, Subtelny AO, Sharfi H, Harlow EE, et al. Identification of small molecule inhibitors of pyruvate kinase M2. Biochem Pharmacol (2010) 79:1118–24. doi:10.1016/j.bcp.2009.12.003
50. Anastasiou D, Yu Y, Israelsen WJ, Jiang J-K, Boxer MB, Hong BS, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol (2012) 8:839–47. doi:10.1038/nchembio1212-1008b
51. Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell (2011) 145:732–44. doi:10.1016/j.cell.2011.03.054
52. Kim J-W, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab (2006) 3:177–85. doi:10.1016/j.cmet.2006.02.002
53. Chae YC, Vaira V, Caino MC, Tang HY, Seo JH, Kossenkov AV, et al. Mitochondrial Akt regulation of hypoxic tumor reprogramming. Cancer Cell (2016) 30:257–72. doi:10.1016/j.ccell.2016.07.004
54. Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG. TSC2 regulates VEGF through mTOR-dependent and-independent pathways. Cancer Cell (2003) 4:147–58. doi:10.1016/S1535-6108(03)00187-9
55. Mayerhofer M, Valent P, Sperr WR, Griffin JD, Sillaber C. BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor-1α, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamycin. Blood (2002) 100:3767–75. doi:10.1182/blood-2002-01-0109
56. Pollard P, Briere J, Alam N, Barwell J, Barclay E, Wortham N, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1α in tumours which result from germline FH and SDH mutations. Hum Mol Genet (2005) 14:2231–9. doi:10.1093/hmg/ddi227
57. Lu H, Dalgard CL, Mohyeldin A, Mcfate T, Tait AS, Verma A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J Biol Chem (2005) 280:41928–39. doi:10.1074/jbc.M508718200
58. Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst (2010) 102:932–41. doi:10.1093/jnci/djq187
59. Ye D, Ma S, Xiong Y, Guan K-L. R-2-hydroxyglutarate as the key effector of IDH mutations promoting oncogenesis. Cancer Cell (2013) 23:274–6. doi:10.1016/j.ccr.2013.03.005
60. Wong BW, Kuchnio A, Bruning U, Carmeliet P. Emerging novel functions of the oxygen-sensing prolyl hydroxylase domain enzymes. Trends Biochem Sci (2013) 38:3–11. doi:10.1016/j.tibs.2012.10.004
61. Fogal V, Richardson AD, Karmali PP, Scheffler IE, Smith JW, Ruoslahti E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol Cell Biol (2010) 30:1303–18. doi:10.1128/MCB.01101-09
62. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A (2010) 107:8788–93. doi:10.1073/pnas.1003428107
63. Guo JY, Chen H-Y, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev (2011) 25:460–70. doi:10.1101/gad.2016311
64. Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature (2014) 510:298. doi:10.1038/nature13236
65. Lewis CA, Parker SJ, Fiske BP, Mccloskey D, Gui DY, Green CR, et al. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol Cell (2014) 55:253–63. doi:10.1016/j.molcel.2014.05.008
66. Ackerman D, Simon MC. Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell Biol (2014) 24:472–8. doi:10.1016/j.tcb.2014.06.001
67. de Padua MC, Delodi G, Vucetic M, Durivault J, Vial V, Bayer P, et al. Disrupting glucose-6-phosphate isomerase fully suppresses the “Warburg effect” and activates OXPHOS with minimal impact on tumor growth except in hypoxia. Oncotarget (2017) 8:87623–37. doi:10.18632/oncotarget.21007
68. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer (2007) 7:763. doi:10.1038/nrc2222
69. Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res (2013) 52:585–9. doi:10.1016/j.plipres.2013.08.005
70. Kuemmerle NB, Rysman E, Lombardo PS, Flanagan AJ, Lipe BC, Wells WA, et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol Cancer Ther (2011) 10:427–36. doi:10.1158/1535-7163.MCT-10-0802
71. Griffiths B, Lewis CA, Bensaad K, Ros S, Zhang Q, Ferber EC, et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab (2013) 1:3. doi:10.1186/2049-3002-1-3
72. Ozcan U, Ozcan L, Yilmaz E, Düvel K, Sahin M, Manning BD, et al. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell (2008) 29:541–51. doi:10.1016/j.molcel.2007.12.023
73. Young RM, Ackerman D, Quinn ZL, Mancuso A, Gruber M, Liu L, et al. Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev (2013) 27:1115–31. doi:10.1101/gad.198630.112
74. Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell (2015) 27:57–71. doi:10.1016/j.ccell.2014.12.002
75. Nomura DK, Long JZ, Niessen S, Hoover HS, Ng S-W, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell (2010) 140:49–61. doi:10.1016/j.cell.2009.11.027
76. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature (2012) 481:380. doi:10.1038/nature10602
77. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature (2013) 496:101. doi:10.1038/nature12040
78. Hao Y, Samuels Y, Li Q, Krokowski D, Guan B-J, Wang C, et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat Commun (2016) 7:11971. doi:10.1038/ncomms11971
79. Corn PG, Ricci MS, Scata KA, Arsham AM, Simon MC, Dicker DT, et al. Mxi1 is induced by hypoxia in a HIF-1-dependent manner and protects cells from c-Myc-induced apoptosis. Cancer Biol Ther (2005) 4:1285–94. doi:10.4161/cbt.4.11.2299
80. Gordan JD, Bertout JA, Hu C-J, Diehl JA, Simon MC. HIF-2α promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell (2007) 11:335–47. doi:10.1016/j.ccr.2007.02.006
81. Mullen AR, Wheaton WW, Jin ES, Chen P-H, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumor cells with defective mitochondria. Nature (2012) 481:385. doi:10.1038/nature10642
82. Kim WY, Safran M, Buckley MRM, Ebert BL, Glickman J, Bosenberg M, et al. Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation in vivo. EMBO J (2006) 25:4650–62. doi:10.1038/sj.emboj.7601300
83. Gameiro PA, Yang J, Metelo AM, Pérez-Carro R, Baker R, Wang Z, et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab (2013) 17:372–85. doi:10.1016/j.cmet.2013.02.002
84. Fendt S-M, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM, et al. Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat Commun (2013) 4:2236. doi:10.1038/ncomms3236
85. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. Anendogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature (2011) 478:197. doi:10.1038/nature10491
86. Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2, 3-dioxygenase. Nat Med (2003) 9:1269. doi:10.1038/nm934
87. Wang T, Liu G, Wang R. The intercellular metabolic interplay between tumor and immune cells. Front Immunol (2014) 5:358. doi:10.3389/fimmu.2014.00358
88. Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162:1229–41. doi:10.1016/j.cell.2015.08.016
89. Ho P-C, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162:1217–28. doi:10.1016/j.cell.2015.08.012
90. De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell (2013) 154:651–63. doi:10.1016/j.cell.2013.06.037
91. Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci U S A (2006) 103:5379–84. doi:10.1073/pnas.0601026103
92. Leopold JA, Zhang Y-Y, Scribner AW, Stanton RC, Loscalzo J. Glucose-6-phosphate dehydrogenase overexpression decreases endothelial cell oxidant stress and increases bioavailable nitric oxide. Arterioscler Thromb Vasc Biol (2003) 23:411–7. doi:10.1161/01.ATV.0000056744.26901.BA
93. Unterluggauer H, Mazurek S, Lener B, Hütter E, Eigenbrodt E, Zwerschke W, et al. Premature senescence of human endothelial cells induced by inhibition of glutaminase. Biogerontology (2008) 9:247–59. doi:10.1007/s10522-008-9134-x
94. Polet F, Feron O. Endothelial cell metabolism and tumour angiogenesis: glucose and glutamine as essential fuels and lactate as the driving force. J Intern Med (2013) 273:156–65. doi:10.1111/joim.12016
95. Avraham-Davidi I, Ely Y, Pham VN, Castranova D, Grunspan M, Malkinson G, et al. ApoB-containing lipoproteins regulate angiogenesis by modulating expression of VEGF receptor 1. Nat Med (2012) 18:967–73. doi:10.1038/nm.2759
96. Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I, et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature (2015) 520:192. doi:10.1038/nature14362
97. Coulon C, Georgiadou M, Roncal C, De Bock K, Langenberg T, Carmeliet P. From vessel sprouting to normalization. Arterioscler Thromb Vasc Biol (2010) 30:2331–6. doi:10.1161/ATVBAHA.110.214106
98. Skuli N, Liu L, Runge A, Wang T, Yuan L, Patel S, et al. Endothelial deletion of hypoxia-inducible factor-2α (HIF-2α) alters vascular function and tumor angiogenesis. Blood (2009) 114:469–77. doi:10.1182/blood-2008-12-193581
99. Skuli N, Majmundar AJ, Krock BL, Mesquita RC, Mathew LK, Quinn ZL, et al. Endothelial HIF-2α regulates murine pathological angiogenesis and revascularization processes. J Clin Invest (2012) 122:1427. doi:10.1172/JCI57322
100. Boidot R, Végran F, Meulle A, Lebreton A, Dessy C, Sonveaux P, et al. Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res (2011) 72:939–48. doi:10.1158/0008-5472.CAN-11-2474
101. De Saedeleer CJ, Copetti T, Porporato PE, Verrax J, Feron O, Sonveaux P. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS One (2012) 7:e46571. doi:10.1371/journal.pone.0046571
102. Sonveaux P, Copetti T, De Saedeleer CJ, Végran F, Verrax J, Kennedy KM, et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One (2012) 7:e33418. doi:10.1371/journal.pone.0033418
103. Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res (2011) 71:2550–60. doi:10.1158/0008-5472.CAN-10-2828
104. Ruan G-X, Kazlauskas A. Lactate engages receptor tyrosine kinases Axl, Tie2 and VEGFR-2 to activate PI3K/Akt and promote angiogenesis. J Biol Chem (2013) 288(29):21161–72. doi:10.1074/jbc.M113.474619
106. Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, et al. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res (2004) 64:8492–5. doi:10.1158/0008-5472.CAN-04-1708
107. Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat M-L, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol (2007) 170:1807–16. doi:10.2353/ajpath.2007.070112
108. Bochet L, Lehuédé C, Dauvillier S, Wang YY, Dirat B, Laurent V, et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res (2013) 73:5657–68. doi:10.1158/0008-5472.CAN-13-0530
109. Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer (2014) 14:430–9. doi:10.1038/nrc3726
110. Madsen CD, Pedersen JT, Venning FA, Singh LB, Moeendarbary E, Charras G, et al. Hypoxia and loss of PHD2 inactivate stromal fibroblasts to decrease tumour stiffness and metastasis. EMBO Rep (2015) 16:1394–408. doi:10.15252/embr.201540107
111. Lemons JM, Feng X-J, Bennett BD, Legesse-Miller A, Johnson EL, Raitman I, et al. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol (2010) 8:e1000514. doi:10.1371/journal.pbio.1000514
112. Santolla MF, Lappano R, De Marco P, Pupo M, Vivacqua A, Sisci D, et al. G protein-coupled estrogen receptor mediates the up-regulation of fatty acid synthase induced by 17β-estradiol in cancer cells and cancer-associated fibroblasts. J Biol Chem (2012) 287:43234–45. doi:10.1074/jbc.M112.417303
113. Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, et al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal (2012) 16:1264–84. doi:10.1089/ars.2011.4243
114. Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, et al. Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging role (s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor microenvironment. Cancer Biol Ther (2010) 10:537–42. doi:10.4161/cbt.10.6.13370
115. Chaudhri VK, Salzler GG, Dick SA, Buckman MS, Sordella R, Karoly ED, et al. Metabolic alterations in lung cancer-associated fibroblasts correlated with increased glycolytic metabolism of the tumor. Mol Cancer Res (2013) 11:579–92. doi:10.1158/1541-7786.MCR-12-0437-T
116. Kim J-W, Evans C, Weidemann A, Takeda N, Lee YS, Stockmann C, et al. Loss of fibroblast HIF-1α accelerates tumorigenesis. Cancer Res (2012) 72:3187–95. doi:10.1158/0008-5472.CAN-12-0534
117. Chiavarina B, Martinez-Outschoorn UE, Whitaker-Menezes D, Howell A, Tanowitz HB, Pestell RG, et al. Metabolic reprogramming and two-compartment tumor metabolism: opposing role (s) of HIF1α and HIF2α in tumor-associated fibroblasts and human breast cancer cells. Cell Cycle (2012) 11:3280–9. doi:10.4161/cc.21643
118. Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res (2012) 72:5130–40. doi:10.1158/0008-5472.CAN-12-1949
119. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol (2005) 5:641. doi:10.1038/nri1668
120. Ino Y, Yamazaki-Itoh R, Oguro S, Shimada K, Kosuge T, Zavada J, et al. Arginase II expressed in cancer-associated fibroblasts indicates tissue hypoxia and predicts poor outcome in patients with pancreatic cancer. PLoS One (2013) 8:e55146. doi:10.1371/journal.pone.0055146
121. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity (2015) 43:435–49. doi:10.1016/j.immuni.2015.09.001
122. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi:10.1016/j.immuni.2011.09.021
123. Wang R, Green DR. Metabolic reprogramming and metabolic dependency in T cells. Immunol Rev (2012) 249:14–26. doi:10.1111/j.1600-065X.2012.01155.x
124. Maclver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259–83. doi:10.1146/annurev-immunol-032712-095956
125. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest (2015) 125:194. doi:10.1172/JCI76012
126. Dang EV, Barbi J, Yang H-Y, Jinasena D, Yu H, Zheng Y, et al. Control of TH 17/Treg balance by hypoxia-inducible factor 1. Cell (2011) 146:772–84. doi:10.1016/j.cell.2011.07.033
127. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208:1367–76. doi:10.1084/jem.20110278
128. Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev (2012) 249:43–58. doi:10.1111/j.1600-065X.2012.01152.x
129. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, Maciver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186:3299–303. doi:10.4049/jimmunol.1003613
130. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20:61–72. doi:10.1016/j.cmet.2014.05.004
131. Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol (2008) 38:2438–50. doi:10.1002/eji.200838289
132. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumor suppression. Nat Rev Cancer (2009) 9:563. doi:10.1038/nrc2676
133. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30:832–44. doi:10.1016/j.immuni.2009.04.014
134. Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, et al. T cell exit from quiescence and differentiation into Th2 cells depend on raptor-mTORC1-mediated metabolic reprogramming. Immunity (2013) 39:1043–56. doi:10.1016/j.immuni.2013.09.015
135. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20:1327–33. doi:10.1038/nm.3704
136. Lee J, Walsh MC, Hoehn KL, James DE, Wherry EJ, Choi Y. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J Immunol (2014) 192:3190–9. doi:10.4049/jimmunol.1302985
137. Nakamura H, Makino Y, Okamoto K, Poellinger L, Ohnuma K, Morimoto C, et al. TCR engagement increases hypoxia-inducible factor-1α protein synthesis via rapamycin-sensitive pathway under hypoxic conditions in human peripheral T cells. J Immunol (2005) 174:7592–9. doi:10.4049/jimmunol.174.12.7592
138. Chisolm DA, Weinmann AS. TCR-signaling events in cellular metabolism and specialization. Front Immunol (2015) 6:292. doi:10.3389/fimmu.2015.00292
139. Noman MZ, Buart S, Van Pelt J, Richon C, Hasmim M, Leleu N, et al. The cooperative induction of hypoxia-inducible factor-1α and STAT3 during hypoxia induced an impairment of tumor susceptibility to CTL-mediated cell lysis. J Immunol (2009) 182:3510–21. doi:10.4049/jimmunol.0800854
140. Bailey SR, Nelson MH, Himes RA, Li Z, Mehrotra S, Paulos CM. Th17 cells in cancer: the ultimate identity crisis. Front Immunol (2014) 5:276. doi:10.3389/fimmu.2014.00276
141. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38:225–36. doi:10.1016/j.immuni.2012.10.020
142. Barsoum IB, Koti M, Siemens DR, Graham CH. Mechanisms of hypoxia-mediated immune escape in cancer. Cancer Res (2014) 74:7185–90. doi:10.1158/0008-5472.CAN-14-2598
143. Chen JL-Y, Lucas JE, Schroeder T, Mori S, Wu J, Nevins J, et al. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet (2008) 4:e1000293. doi:10.1371/journal.pgen.1000293
144. Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell (2010) 39:171–83. doi:10.1016/j.molcel.2010.06.022
145. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol (2015) 36:265–76. doi:10.1016/j.it.2015.02.008
146. Platten M, Von Knebel Doeberitz N, Oezen I, Wick W, Ochs K. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front Immunol (2014) 5:673. doi:10.3389/fimmu.2014.00673
147. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab (2016) 24:657–71. doi:10.1016/j.cmet.2016.08.011
148. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi:10.1016/j.immuni.2014.06.008
149. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity (2014) 40:274–88. doi:10.1016/j.immuni.2014.01.006
150. Rodríguez-Prados J-C, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol (2010) 185:605–14. doi:10.4049/jimmunol.0901698
151. Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol (2009) 9:609. doi:10.1038/nri2607
152. Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J Immunol (2010) 184:1200–9. doi:10.4049/jimmunol.0902584
153. O’Neill LA. A critical role for citrate metabolism in LPS signalling. Biochem J (2011) 438:e5–6. doi:10.1042/BJ20111386
154. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab (2006) 4:13–24. doi:10.1016/j.cmet.2006.08.006
155. O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature (2013) 493:346. doi:10.1038/nature11862
156. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab (2012) 15:813–26. doi:10.1016/j.cmet.2012.04.023
158. Mills C. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol (2012) 32(6):463–88. doi:10.1615/CritRevImmunol.v32.i6.10
159. Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M, et al. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol (2010) 40:824–35. doi:10.1002/eji.200939889
160. Cairo G, Recalcati S, Mantovani A, Locati M. Iron trafficking and metabolism in macrophages: contribution to the polarized phenotype. Trends Immunol (2011) 32:241–7. doi:10.1016/j.it.2011.03.007
161. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature (1999) 399:271–5. doi:10.1038/20459
162. Park J, Lee SE, Hur J, Hong EB, Choi J-I, Yang J-M, et al. M-CSF from cancer cells induces fatty acid synthase and PPARβ/δ activation in tumor myeloid cells, leading to tumor progression. Cell Rep (2015) 10:1614–25. doi:10.1016/j.celrep.2015.02.024
163. Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi:10.1038/ncomms12150
164. Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+ Ly6C+ granulocytes. Proc Natl Acad Sci U S A (2010) 107:21248–55. doi:10.1073/pnas.1015855107
165. Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, et al. Identification of a CD11b+/Gr-1+/CD31+ myeloid progenitor capable of activating or suppressing CD8+ T cells. Blood (2000) 96:3838–46.
166. Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1-bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat (2010) 123:39–49. doi:10.1007/s10549-009-0622-8
167. Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol (2003) 170:270–8. doi:10.4049/jimmunol.170.1.270
168. Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest (2006) 116:2777. doi:10.1172/JCI28828
169. Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev (2008) 222:180–91. doi:10.1111/j.1600-065X.2008.00608.x
170. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12:253. doi:10.1038/nri3175
171. Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest (2015) 125:3365–76. doi:10.1172/JCI80006
172. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64:5839–49. doi:10.1158/0008-5472.CAN-04-0465
173. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res (2009) 69:1553–60. doi:10.1158/0008-5472.CAN-08-1921
174. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res (2010) 70:68–77. doi:10.1158/0008-5472.CAN-09-2587
175. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med (1999) 189:1363–72. doi:10.1084/jem.189.9.1363
176. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol (2006) 177:7303–11. doi:10.4049/jimmunol.177.10.7303
177. Grohmann U, Bronte V. Control of immune response by amino acid metabolism. Immunol Rev (2010) 236:243–64. doi:10.1111/j.1600-065X.2010.00915.x
178. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a novel mechanism of CD8+ T cell tolerance in cancer. Nat Med (2007) 13:828. doi:10.1038/nm1609
179. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol (2009) 182:5693–701. doi:10.4049/jimmunol.0900092
180. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood (2010) 116:5738–47. doi:10.1182/blood-2010-06-287839
181. Arina A, Bronte V. Myeloid-derived suppressor cell impact on endogenous and adoptively transferred T cells. Curr Opin Immunol (2015) 33:120–5. doi:10.1016/j.coi.2015.02.006
182. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res (2015) 3:1236–47. doi:10.1158/2326-6066.CIR-15-0036
183. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn J-I, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med (2010) 207(11):2439–53. doi:10.1084/jem.20100587
184. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med (2014) 211(5):781–90. doi:10.1084/jem.20131916
185. Ye X-Z, Yu S-C, Bian X-W. Contribution of myeloid-derived suppressor cells to tumor-induced immune suppression, angiogenesis, invasion and metastasis. J Genet Genomics (2010) 37:423–30. doi:10.1016/S1673-8527(09)60061-8
186. Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell (2012) 150:165–78. doi:10.1016/j.cell.2012.04.042
187. Erler JT, Bennewith KL, Nicolau M, Dornhöfer N, Kong C, Le Q-T, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature (2006) 440:1222. doi:10.1038/nature04695
188. Sceneay J, Chow MT, Chen A, Halse HM, Wong CS, Andrews DM, et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res (2012) 72:3906–11. doi:10.1158/0008-5472.CAN-11-3873
189. Cox TR, Rumney RM, Schoof EM, Perryman L, Høye AM, Agrawal A, et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature (2015) 522:106–10. doi:10.1038/nature14492
190. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol (2011) 11:519. doi:10.1038/nri3024
191. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell (2009) 16:183–94. doi:10.1016/j.ccr.2009.06.017
192. Schmidt H, Bastholt L, Geertsen P, Christensen IJ, Larsen S, Gehl J, et al. Elevated neutrophil and monocyte counts in peripheral blood are associated with poor survival in patients with metastatic melanoma: a prognostic model. Br J Cancer (2005) 93:273. doi:10.1038/sj.bjc.6602702
193. Bellocq A, Antoine M, Flahault A, Philippe C, Crestani B, Bernaudin J-F, et al. Neutrophil alveolitis in bronchioloalveolar carcinoma: induction by tumor-derived interleukin-8 and relation to clinical outcome. Am J Pathol (1998) 152:83.
194. Atzpodien J, Reitz M. Peripheral blood neutrophils as independent immunologic predictor of response and long-term survival upon immunotherapy in metastatic renal-cell carcinoma. Cancer Biother Radiopharm (2008) 23:129–34. doi:10.1089/cbr.2007.0429
195. Reid MD, Basturk O, Thirabanjasak D, Hruban RH, Klimstra DS, Bagci P, et al. Tumor-infiltrating neutrophils in pancreatic neoplasia. Mod Pathol (2011) 24:1612. doi:10.1038/modpathol.2011.113
196. Caruso RA, Bellocco R, Pagano M, Bertoli G, Rigoli L, Inferrera C. Prognostic value of intratumoral neutrophils in advanced gastric carcinoma in a high-risk area in northern Italy. Mod Pathol (2002) 15:831. doi:10.1097/01.MP.0000020391.98998.6B
197. Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell (2003) 112:645–57. doi:10.1016/S0092-8674(03)00154-5
198. Jun HS, Weinstein DA, Lee YM, Mansfield BC, Chou JY. Molecular mechanisms of neutrophil dysfunction in glycogen storage disease type Ib. Blood (2014) 123:2843–53. doi:10.1182/blood-2013-05-502435
199. Thompson AR, Elks PM, Marriott HM, Eamsamarng S, Higgins KR, Lewis A, et al. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood (2014) 123:366–76. doi:10.1182/blood-2013-05-500207
200. Yoshida N, Granger DN, Anderson DC, Rothlein R, Lane C, Kvietys PR. Anoxia/reoxygenation-induced neutrophil adherence to cultured endothelial cells. Am J Physiol (1992) 262:H1891–8. doi:10.1152/ajpheart.1992.262.6.H1891
201. Lodhi IJ, Wei X, Yin L, Feng C, Adak S, Abou-Ezzi G, et al. Peroxisomal lipid synthesis regulates inflammation by sustaining neutrophil membrane phospholipid composition and viability. Cell Metab (2015) 21:51–64. doi:10.1016/j.cmet.2014.12.002
202. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi:10.1126/science.1092385
203. Azevedo EP, Rochael NC, Guimarães-Costa AB, De Souza-Vieira TS, Ganilho J, Saraiva EM, et al. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril-and phorbol 12-myristate 13-acetate-induced neutrophil extracellular trap (NET) formation. J Biol Chem (2015) 290:22174–83. doi:10.1074/jbc.M115.640094
204. Rodríguez-Espinosa O, Rojas-Espinosa O, Moreno-Altamirano MMB, López-Villegas EO, Sánchez-García FJ. Metabolic requirements for neutrophil extracellular traps formation. Immunology (2015) 145:213–24. doi:10.1111/imm.12437
205. Cools-Lartigue J, Spicer J, Mcdonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest (2013) 123:3446. doi:10.1172/JCI67484
206. Cedervall J, Zhang Y, Huang H, Zhang L, Femel J, Dimberg A, et al. Neutrophil extracellular traps accumulate in peripheral blood vessels and compromise organ function in tumor-bearing animals. Cancer Res (2015) 75:2653–62. doi:10.1158/0008-5472.CAN-14-3299
207. Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer (2010) 10:267. doi:10.1038/nrc2817
208. Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov (2011) 10:671. doi:10.1038/nrd3504
209. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi:10.1126/science.1160809
210. Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature (2012) 491:364. doi:10.1038/nature11706
211. White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer (2012) 12:401. doi:10.1038/nrc3262
212. Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H, et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther (2012) 11:1672–82. doi:10.1158/1535-7163.MCT-12-0131
213. Cao X, Fang L, Gibbs S, Huang Y, Dai Z, Wen P, et al. Glucose uptake inhibitor sensitizes cancer cells to daunorubicin and overcomes drug resistance in hypoxia. Cancer Chemother Pharmacol (2007) 59:495–505. doi:10.1007/s00280-006-0291-9
214. Dwarakanath B, Jain V. Targeting glucose metabolism with 2-deoxy-D-glucose for improving cancer therapy. Future Oncol (2009) 5:581–5. doi:10.2217/fon.09.44
215. Yoo BC, Ku JL, Hong SH, Shin YK, Park SY, Kim HK, et al. Decreased pyruvate kinase M2 activity linked to cisplatin resistance in human gastric carcinoma cell lines. Int J Cancer (2004) 108:532–9. doi:10.1002/ijc.11604
216. Martinez-Balibrea E, Plasencia C, Ginés A, Martinez-Cardús A, Musulén E, Aguilera R, et al. A proteomic approach links decreased pyruvate kinase M2 expression to oxaliplatin resistance in patients with colorectal cancer and in human cell lines. Mol Cancer Ther (2009) 8:771–8. doi:10.1158/1535-7163.MCT-08-0882
217. Li SL, Ye F, Cai WJ, Hu HD, Hu P, Ren H, et al. Quantitative proteome analysis of multidrug resistance in human ovarian cancer cell line. J Cell Biochem (2010) 109:625–33. doi:10.1002/jcb.22413
218. Guo W, Zhang Y, Chen T, Wang Y, Xue J, Zhang Y, et al. Efficacy of RNAi targeting of pyruvate kinase M2 combined with cisplatin in a lung cancer model. J Cancer Res Clin Oncol (2011) 137:65–72. doi:10.1007/s00432-010-0860-5
219. Shi HS, Li D, Zhang J, Wang YS, Yang L, Zhang HL, et al. Silencing of pkm2 increases the efficacy of docetaxel in human lung cancer xenografts in mice. Cancer Sci (2010) 101:1447–53. doi:10.1111/j.1349-7006.2010.01562.x
220. Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, et al. Warburg effect in chemosensitivity: targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer (2010) 9:33. doi:10.1186/1476-4598-9-33
221. Lu C-W, Lin S-C, Chen K-F, Lai Y-Y, Tsai S-J. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem (2008) 283:28106–14. doi:10.1074/jbc.M803508200
222. Lu C-W, Lin S-C, Chien C-W, Lin S-C, Lee C-T, Lin B-W, et al. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am J Pathol (2011) 179:1405–14. doi:10.1016/j.ajpath.2011.05.050
223. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi:10.1038/ncomms7692
224. Gomez-Pinillos A, Ferrari AC. mTOR signaling pathway and mTOR inhibitors in cancer therapy. Hematol Oncol Clin North Am (2012) 26:483–505. doi:10.1016/j.hoc.2012.02.014
225. Zaytseva YY, Valentino JD, Gulhati P, Evers BM. mTOR inhibitors in cancer therapy. Cancer Lett (2012) 319:1–7. doi:10.1016/j.canlet.2012.01.005
226. Yue W, Yang CS, Dipaola RS, Tan X-L. Repurposing of metformin and aspirin by targeting AMPK-mTOR and inflammation for pancreatic cancer prevention and treatment. Cancer Prev Res (2014) 7:388–97. doi:10.1158/1940-6207.CAPR-13-0337
227. Li W, Saud SM, Young MR, Chen G, Hua B. Targeting AMPK for cancer prevention and treatment. Oncotarget (2015) 6:7365. doi:10.18632/oncotarget.3629
228. Alli PM, Pinn ML, Jaffee EM, Mcfadden JM, Kuhajda FP. Fatty acid synthase inhibitors are chemopreventive for mammary cancer in neu-N transgenic mice. Oncogene (2005) 24:39. doi:10.1038/sj.onc.1208174
229. Seguin F, Carvalho M, Bastos D, Agostini M, Zecchin K, Alvarez-Flores M, et al. The fatty acid synthase inhibitor orlistat reduces experimental metastases and angiogenesis in B16-F10 melanomas. Br J Cancer (2012) 107:977. doi:10.1038/bjc.2012.355
230. Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell (2005) 8:311–21. doi:10.1016/j.ccr.2005.09.008
231. Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, et al. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res (2007) 67:8180–7. doi:10.1158/0008-5472.CAN-07-0389
232. Yalcin A, Clem B, Makoni S, Clem A, Nelson K, Thornburg J, et al. Selective inhibition of choline kinase simultaneously attenuates MAPK and PI3K/AKT signaling. Oncogene (2010) 29:139. doi:10.1038/onc.2009.317
233. Clem BF, Clem AL, Yalcin A, Goswami U, Arumugam S, Telang S, et al. A novel small molecule antagonist of choline kinase-α that simultaneously suppresses MAPK and PI3K/AKT signaling. Oncogene (2011) 30:3370. doi:10.1038/onc.2011.51
234. Cao Z, Fan-Minogue H, Bellovin DI, Yevtodiyenko A, Arzeno J, Yang Q, et al. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res (2011) 71:2286–97. doi:10.1158/0008-5472.CAN-10-3367
235. Menendez JA, Lupu R, Colomer R. Inhibition of tumor-associated fatty acid synthase hyperactivity induces synergistic chemosensitization of HER-2/neu-overexpressing human breast cancer cells to docetaxel (taxotere). Breast Cancer Res Treat (2004) 84:183–95. doi:10.1023/B:BREA.0000018409.59448.60
236. Vazquez-Martin A, Colomer R, Brunet J, Menendez JA. Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin™) by transcriptionally inhibiting ‘HER2 super-expression’ occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int J Oncol (2007) 31:769–76. doi:10.3892/ijo.31.4.769
237. Liu H, Liu Y, Zhang J-T. A new mechanism of drug resistance in breast cancer cells: fatty acid synthase overexpression-mediated palmitate overproduction. Mol Cancer Ther (2008) 7:263–70. doi:10.1158/1535-7163.MCT-07-0445
238. Durán RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell (2012) 47:349–58. doi:10.1016/j.molcel.2012.05.043
239. Kamata S, Kishimoto T, Kobayashi S, Miyazaki M, Ishikura H. Possible involvement of persistent activity of the mammalian target of rapamycin pathway in the cisplatin resistance of AFP-producing gastric cancer cells. Cancer Biol Ther (2007) 6:1036–43. doi:10.4161/cbt.6.7.4253
240. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med (2005) 202:931–9. doi:10.1084/jem.20050715
241. Chabner BA, Roberts TG Jr. Chemotherapy and the war on cancer. Nat Rev Cancer (2005) 5:65. doi:10.1038/nrc1529
242. Wang J-B, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell (2010) 18:207–19. doi:10.1016/j.ccr.2010.08.009
243. Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab (2012) 15:110–21. doi:10.1016/j.cmet.2011.12.009
244. Thornburg JM, Nelson KK, Clem BF, Lane AN, Arumugam S, Simmons A, et al. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res (2008) 10:R84. doi:10.1186/bcr2154
245. Qing G, Li B, Vu A, Skuli N, Walton ZE, Liu X, et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell (2012) 22:631–44. doi:10.1016/j.ccr.2012.09.021
246. Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell (2012) 22:547–60. doi:10.1016/j.ccr.2012.08.014
247. Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell (2013) 23:302–15. doi:10.1016/j.ccr.2013.02.003
248. Vazquez F, Lim J-H, Chim H, Bhalla K, Girnun G, Pierce K, et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell (2013) 23:287–301. doi:10.1016/j.ccr.2012.11.020
249. Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med (2008) 14:1351–6. doi:10.1038/nm.1890
250. Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature (2014) 508:108. doi:10.1038/nature13110
251. Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest (2010) 120:2355. doi:10.1172/JCI40671
252. Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care (2006) 29:254–8. doi:10.2337/dc06-0997
253. Buzzai M, Jones RG, Amaravadi RK, Lum JJ, Deberardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res (2007) 67:6745–52. doi:10.1158/0008-5472.CAN-06-4447
254. Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res (2009) 69:7507–11. doi:10.1158/0008-5472.CAN-09-2994
255. Fendt S-M, Bell EL, Keibler MA, Davidson SM, Wirth GJ, Fiske B, et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res (2013) 73:4429–38. doi:10.1158/0008-5472.CAN-13-0080
256. Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc Natl Acad Sci U S A (2013) 110:972–7. doi:10.1073/pnas.1221055110
257. Pelletier J, Roux D, Viollet B, Mazure NM, Pouyssegur J. AMP-activated protein kinase is dispensable for maintaining ATP levels and for survival following inhibition of glycolysis, but promotes tumour engraftment of Ras-transformed fibroblasts. Oncotarget (2015) 6:11833–47. doi:10.18632/oncotarget.3738
Keywords: oxygen sensors, hypoxia, immunity, hypoxia-inducible factor prolyl hydroxylases, glycolysis, lactate
Citation: Sormendi S and Wielockx B (2018) Hypoxia Pathway Proteins As Central Mediators of Metabolism in the Tumor Cells and Their Microenvironment. Front. Immunol. 9:40. doi: 10.3389/fimmu.2018.00040
Received: 30 September 2017; Accepted: 05 January 2018;
Published: 29 January 2018
Edited by:
Yongsheng Li, Third Military Medical University, ChinaReviewed by:
Carlos Alfaro, Universidad de Navarra, SpainJacques Pouyssegur, Université Côte d’Azur, France
Copyright: © 2018 Sormendi and Wielockx. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ben Wielockx, YmVuLndpZWxvY2t4QHR1LWRyZXNkZW4uZGU=