 Yuedi Wang
Yuedi Wang Feifei Luo2,3
Feifei Luo2,3 Yiwei Chu
Yiwei Chu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 22 December 2017
Sec. Cancer Immunity and Immunotherapy
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01934
In recent years, chimeric antigen receptor (CAR) T-cell therapy has become popular in immunotherapy, particularly after its tremendous success in the treatment of lineage-restricted hematologic cancers. However, the application of CAR T-cell therapy for solid tumors has not reached its full potential because of the lack of specific tumor antigens and inhibitory factors in suppressive tumor microenvironment (TME) (e.g., programmed death ligand-1, myeloid-derived suppressor cells, and transforming growth factor-β). In this review, we include some limitations in CAR design, such as tumor heterogeneity, indefinite spatial distance between CAR T-cell and its target cell, and suppressive TME. We also summarize some new approaches to overcome these hurdles, including targeting neoantigens and/or multiple antigens at once and depleting some inhibitory factors.
Chimeric antigen receptor (CAR) design is based on the signal transduction of T-cell activation (1). The T-cell receptor (TCR) detects antigens presented by antigen-presenting cells (APCs) in the form of the major histocompatibility complex (MHC)–antigen peptide complex (2). TCR binding to the MHC–antigen peptide complex induces a cascade of intracellular events as follows: phosphorylated TCR recruits intracellular second messengers to provide the first signal, and costimulatory molecules (CD28, CD27, CD134, CD137, or ICOS) at the T-cell surface bind to their corresponding receptors (CD80, CD86, CD137L, or ICOSL) on APCs, which further provides the second signal (3). Eventually, T-cells are primed and activated, which subsequently secrete perforin, granzyme, and cytokines, including interleukin 2 (IL-2) and interferon γ (IFN-γ), to defend infection by inducing the apoptosis of target cells.
However, normal T-cells do not efficiently recognize tumors because of the absence of MHC expression and weak immunogenicity of tumors. Investigators first developed chimeric immune receptors in mid-1980s. In 1993, Eshhar et al. modified the T-cell expressing CARs in melanoma treatments, which overcame the issue of MHC restriction and weak immunogenicity (4). Generally, CARs comprise three domains: an extracellular single-chain antibody fragment (scFv), which serves as a target moiety that redirects T-cells to tumor cells by specifically binding to tumor-associated antigens (TAAs); a transmembrane domain and an endodomain, which is often the signal transduction domain comprising a CD3ζ chain and costimulatory factors such as CD28 and 4-1BB (CD137) (5, 6). According to different intracellular domains, CARs are divided into three generations. The intracellular domain of the first generation contains only a CD3ζ chain; the second contains a CD3ζ chain and a costimulatory molecule [CD28, 4-1BB, CD134 (OX40), or ICOS]; and the third contains a CD3ζ chain and two or more different costimulatory molecules (7, 8). Zhang et al. compared CD28 with 4-1BB as a costimulant and demonstrated that 4-1BB was essential for expanding memory CD8+ T-cells and was superior to CD28 in costimulating the generation of CD8+ cytotoxic lymphocytes (9). Therefore, using 4-1BB as a costimulation factor in CAR designs may hold promise for ameliorating exhaustion and improving the effectiveness of CAR T-cell.
There have been an increasing number of CAR T-cell clinical trials for solid tumors because of their unprecedented efficacy for non-solid tumors, particularly anti-CD19 CAR T-cell (Table 1). An important reason for the success of CAR T-cell therapy in leukemia is that because non-solid tumor cells circulate within the blood and lymphatic system, they are more likely to meet adoptive CAR T-cells and induce them into killing activity. However, in solid tumors, it is particularly difficult for CAR T-cells to migrate into tumor sites because of several obstacle layers, such as the extracellular matrix, and the lack of chemokines, which are frequently mismatched with receptors in solid tumors (10). Even if a few CAR T-cells successfully infiltrate the tumor sites, they may become inactivated because of the suppressive tumor microenvironment (TME) (11–14) (Figure 1). Another important reason is that it is extremely difficult to find a specific TAA, such as CD19, in B-cell acute lymphoblastic leukemia (B-ALL) for solid tumors (15). Therefore, CAR T-cells require novel additional modifications for an enhanced antitumor efficiency.
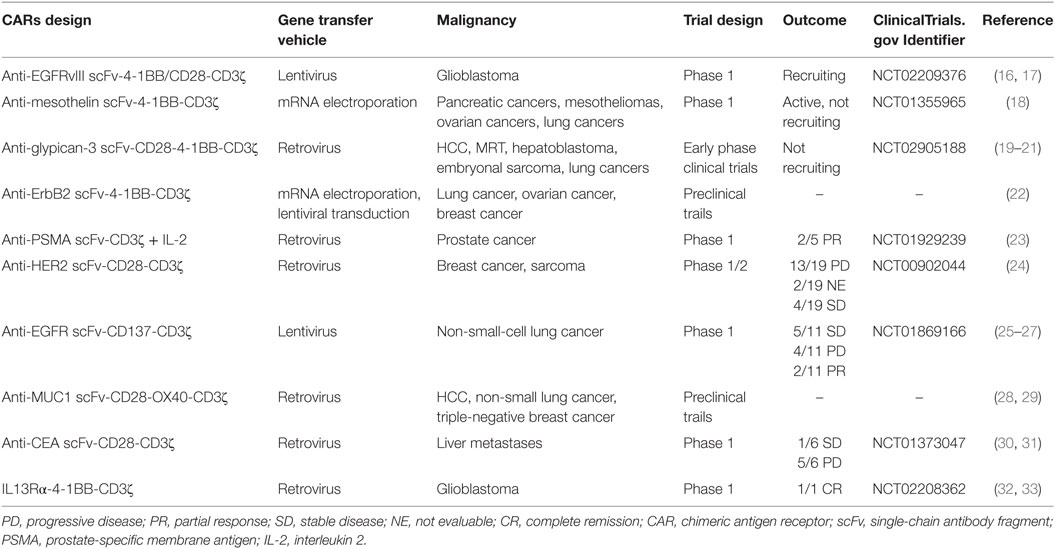
Table 1. Summary of CAR T cell therapy for solid tumor.
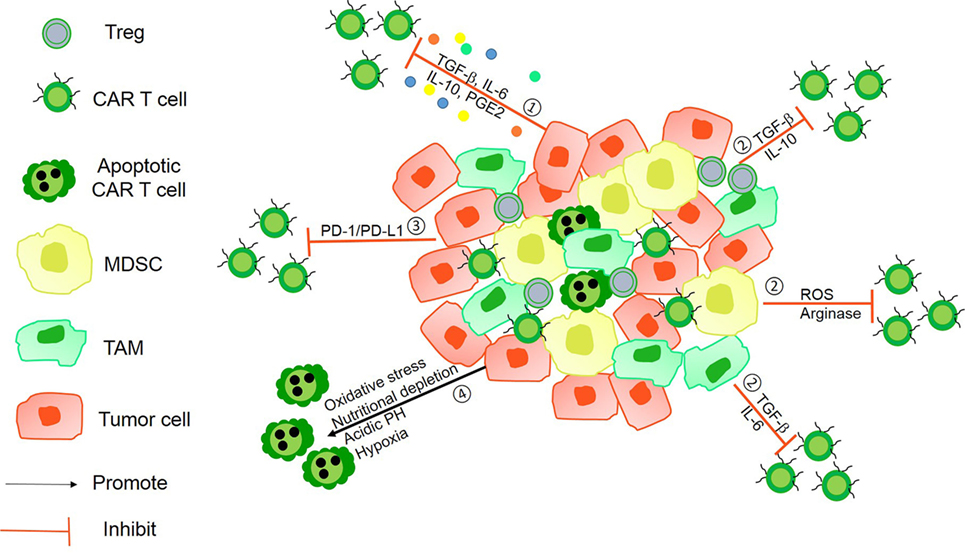
Figure 1. The immunosuppressive mechanisms of tumor microenvironment (TME). ① Tumor-derived soluble factors such as prostaglandin E2 (PGE2) and cytokines such as transforming growth factor-β (TGF-β), IL-6, and IL-10 inhibit chimeric antigen receptor (CAR) T-cells vitality. ② The presence of immunosuppressive immune cells, namely myeloid-derived suppressor cells (MDSCs), Tregs, tumor-associated microphages (TAMs), or neutrophils (TANs), suppress T-cell function via Arg-I, ROS generation, and some soluble inhibitory factors. ③ Tumor cells can utilize the intrinsic negative regulatory mechanisms of T-cells by upregulating surface inhibitory receptors such as programmed death ligand-1 (PD-L1)/PD-L2. ④ The hostile TME makes it difficult for CAR T-cells to survive because of hypoxia, oxidative stress, acidic pH, and nutritional depletion.
Reports of clinical trials of CAR T-cell therapy for solid tumors have shown that most CAR T-cell therapies are stuck at the “on-target, off-tumor” stage (34, 35). The ideal TAAs need to be specifically expressed on tumor cells; however, several TAAs are also expressed on normal cells. For instance, mesothelin is not only overexpressed on mesothelioma but also expressed on peritoneal, pleural, and pericardial surfaces (36). Moreover, most tumor cells remove their immunogenic epitopes of TAAs to escape the attack of the host’s immune system. Therefore, identifying specific and immunogenic tumor antigens is necessary for the treatment of solid tumor.
Investigators may design CARs targeting aberrantly modification of TAAs or tumor-specific oncogenic mutations such as truncated MUC1. For example, Posey et al. recently described a new CAR targeting aberrantly glycosylated tumor-associated cell membrane mucin (MUC1). In this study, they used a second-generation CAR with 4-1BB as a costimulatory molecule, and the binding domain was the scFv region of the high-affinity antibody (5E5) targeting truncated O-glycopeptide epitopes specifically presented on tumor tissues. Thus, these CAR T cells normally did not bind to glycosylated MUC1, but they specifically recognized the Tn glycoform of MUC1 on tumor cells in this case. This study also demonstrated that MUC1–CAR T-cell exhibited no cytotoxicity against normal human primary cells (37).
Investigators are also examining neoantigens specifically expressed on tumor cells (38). Neoantigens are antigens resulting on tumor cells from somatic mutations and are unique to each patient’s cancer, and these mutations may facilitate tumor growth and/or invasion. Using neoantigens as a target can minimize the risk of killing healthy tissue (39). Several investigators have proposed using next-generation sequencing combined with high-throughput immunological screening approaches to identify immunogenic mutations (40, 41). They separated normal cells from tumor cells obtained from patients and subsequently used whole-exome and transcriptome sequencing to identify somatic mutations, which could be presented by APCs, and activate an immune response (42). Verdegaal et al. found that T-cell mediated neoantigen immunoediting and the loss of expression of T-cell-recognized neoantigens may result in tumor resistance (43). Therefore, it is imperative to monitor the neoantigen landscape dynamics during adoptive T-cell therapy.
More recently, a new kind of CARs—tandem CARs—has been designed to express two antigen-binding domains; the tandem CAR T-cell is activated only when simultaneously recognizing two different antigens (Figure 2). CARs, engineered to simultaneously target two different antigens, are more specific and safe. For example, Hegde et al. developed a tandem CAR by joining an anti-human epidermal growth factor receptor-2 (HER2) scFv and an IL-13 receptor α2 (IL-13Rα2)-binding IL-13 mutant and used CD28 as a costimulatory factor and CD3ζ chain as a signal transduction domain. The tandem CAR T-cell showed the potential to bind with either HER2 or IL-13Rα2 and to protect against tumor cells. Compared with single CAR T-cell upon encountering HER2 or IL-13Rα2, the activation dynamics of these CAR T-cells were more sustained but not more exhaustible. In a murine glioblastoma model, the tandem CAR T-cells mitigated antigen escape displayed enhanced antitumor efficacy and improved animal survival (44).
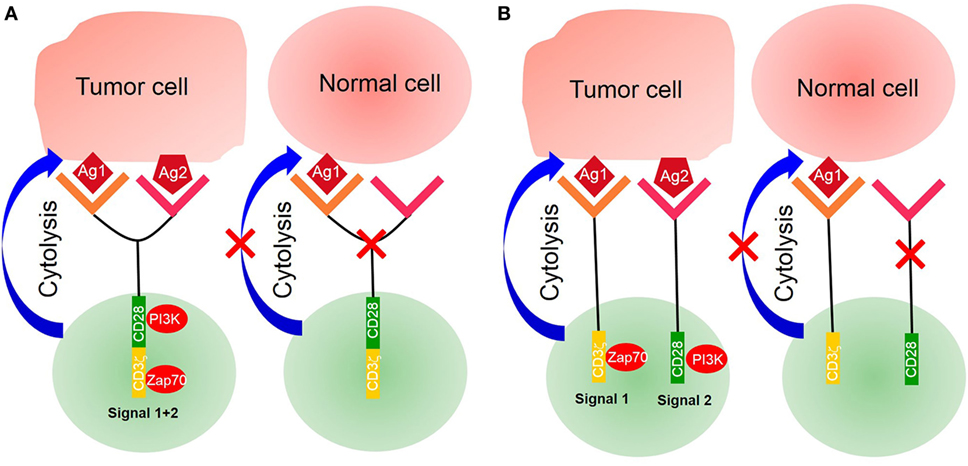
Figure 2. Tandem chimeric antigen receptor (CAR) T-cell. (A) The extracellular binding domain of CARs comprises two different tumor-associated antigen (TAA)-specific single-chain antibody fragments (scFvs) linked to the intracellular signaling domains derived from the CD3ζ chain and CD28 or CD137. CAR T-cell activated by two separate TAAs binding with TAA-specific scFv. (B) CD3ζ chain of CARs was designed with costimulatory factors. A single CAR structure contains two scFvs—one linked CD3ζ chain, providing the first signal, and another linked costimulatory factors, providing the second signal. Only activating double signals activate T-cells.
Another tandem CAR has been designed to deliver two separate scFvs—one linked to a CD3ζ chain providing the first signal and another to a costimulatory molecule providing the second signal. The expression of target antigens alone is insufficient to trigger T-cell activation. Only two antigens that are simultaneously expressed on a target cell can activate a CAR T-cell and induce an antitumor function (45). For example, investigators presented an approach to render CAR T-cells specific for prostate tumors even in the absence of a truly tumor-restricted antigen. In their work, they used two prostate tumor antigens—prostate-specific membrane antigen (PSMA) and prostate stem cell antigen (PSCA)—and demonstrated that CAR T-cell destroyed tumor cells expressing both PSMA and PSCA. In a murine syngeneic model, it was also shown that CAR T-cells were activated and protected against tumors simultaneously expressing PSCA and PSMA (46, 47). However, under the pressure of antigen-specific T-cells, tumor cells generate new mutations with a loss of antigen expression accompanied by resistance. Investigators need to monitor the antigen landscape dynamics to enhance CAR T-cell therapy.
Sensitivity is another challenge of CAR T-cell therapy for the treatment of solid tumors. The spatial distance between T-cells and their target cells also plays a key role in T-cell activation and signal transduction. It is essential for T-cell activation that immune receptor tyrosine-based activation motifs were phosphorylated by the lymphocyte-specific kinase (Lck) of the Src family (48). However, Lck is originally inhibited on T-cells so that it does not exhibit phosphorylation activity. It is activated by the protein tyrosine phosphatases CD45 and CD148, leading to downstream signal transduction (49, 50). Some studies have indicated that the distance between T-cell and APC is approximately 15 nm during the formation of an immunological synapse (51). This spatial distance excludes the phosphatases CD45 and CD148 from the immunological synapse because they have ectodomains that are longer than 15 nm (52, 53). Previous studies have demonstrated that the exclusion of CD45 from the cell–cell interphase is both necessary and sufficient for the formation of T-cell synapse (54, 55). The spatial distance between CAR T-cell and its target tumor cell may be equally important (Figure 3). However, it depends on entirely different structural elements, including the spatial structure of scFv, CAR position on the membrane, and location of the antigen on the target cell. Several studies have demonstrated that the identical epitope activates CAR T-cell with different levels of efficiency when expressed at different positions on the membrane. For example, Hombach et al. have explored the impact of a defined epitope position on the efficacy of CAR T-cell activation. They demonstrated that T-cell activation is more efficient when targeting the membrane proximal epitope than distal epitopes, indicating that the position of the targeted epitope has a major impact on the efficacy of T-cell activation (56). Hudecek et al. have also confirmed that the length and composition of IgG-derived extracellular spacer domains influence the function of CAR T-cells and that extracellular spacer domains lacking intrinsic signaling function are decisive in CAR design for an optimal in vivo activity (57). Thus, it might be an attractive strategy to enhance the sensitivity of CAR T-cell therapy by controlling the spatial distance in future research.
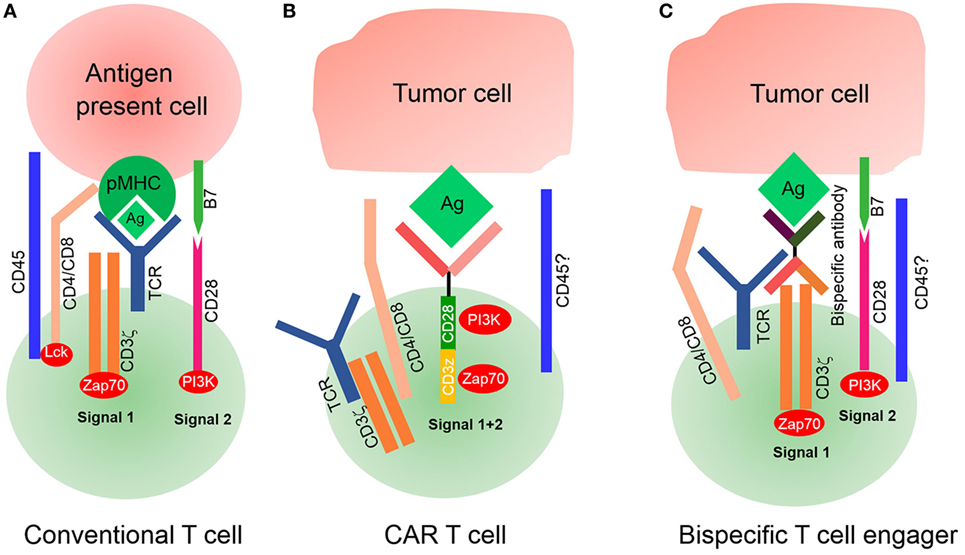
Figure 3. Signaling of conventional T-cell and chimeric antigen receptor (CAR) T-cell. (A) Conventional T-cell activation is initiated when T-cell receptor (TCR) interacts with pMHC for the formation of an immunological synapse. The spatial distance between T-cells and antigen-presenting cells (APCs) is approximately 15 nm, which physically excludes CD45 from the synapse because of its large ectodomain. CD4/CD8 molecules bind with major histocompatibility complex (MHC)I/II to recruit lymphocyte-specific kinase (Lck) phosphorylated by CD45, which then activates Zap70 to provide signal 1. Costimulatory molecules such as CD28 bind with their ligands on APCs to deliver signal 2 for complete T-cell activation. (B) Modified CAR T-cells recognize tumor cells by their tumor-associated antigens (TAAs) in a non-MHC restrictive manner. The spatial distance between CAR T-cells and target tumor cells is unknown, nor it is known whether this distance is small enough to physically exclude phosphatase CD45 from the synapse. It is also unknown whether CARs interact with endogenous TCR/CD3ζ or CD4/CD8 coreceptors. (C) Bispecific T-cell engagers (BiTEs) can secrete bispecific antibodies, one of which can recognize TAAs and another ligates with the intrinsic TCR–CD3 complex, but it is unknown if CD4/CD8 T-cells participate because of deficient MHC expression on tumor cells. Endogenous TCR/CD3ζ delivered signal 1 upon BiTEs ligation with target-expressing cells by secreting bispecific antibodies, and signal 2 is delivered by an intrinsic costimulatory molecule on BiTEs and its receptor lies on tumor cells. The spatial distance between BiTEs and tumor cells is also uncontrollable; therefore, it is also unknown if CD45 is excluded from the synapse.
Previous studies have mainly focused on using exogenous activation elements, instead of intrinsic TCR, to remove MHC molecule restrictions. Recently, investigators developed a novel CAR T-like cell, known as bispecific T-cell engager (BiTE). This novel concept involves the use of a transgenic T-cell that can secrete T-cell-dependent bispecific antibodies, including two different scFv, one for tumor-specific antigens and another for T-cell specific antigens (often for TCR or CD3) (58). Because of its structure, the secreted scFv can link tumor cells with T-cells by acting as a bridge to activate intrinsic TCR/CD3 complex of BiTEs, but it is unknown whether CD4 or CD8 molecules of T-cells participate in this process because of the lack of MHC expression on tumor cells. A combination of endogenous TCR/CD3 and secreted scFv is sufficient to deliver signal 1, while intrinsic costimulatory molecules deliver signal 2. Luo et al. have developed BiTEs that are capable of secreting bispecific antibodies against CD3 and HER2, demonstrating an excellent antitumor effect. Interestingly, they have also highlighted that bispecific antibodies secreted by BiTEs affect the bystander T-cells not transfected with αHER2/CD3 RNA (59). However, the second activation signal for BiTEs, derived from intrinsic costimulatory agonists, has not yet been defined. Investigators need to administer an exogenous second activation signal to enhance the efficiency of BiTEs.
Another key factor influencing the sensitivity of CAR T-cell therapy is the T-cell intrinsic negative regulatory mechanism (60). For example, CAR T-cells successfully transferred into solid tumors often upregulated inhibitory factors, such as programmed death-1, cytotoxic T lymphocyte-associated antigen-4, T-cell immunoglobulin domain and mucin domain-3 (TIM3), and lymphocyte activation gene-3, that specifically bind to ligands on tumor cells to attenuate the antitumor efficacy. Combined immunotherapeutic strategy is promising for improving the sensitivity of CAR T-cell therapy (61, 62). Suarez et al. employed CAR T-cells targeting carbonic anhydrase IX (CAIX) expressed on metastatic clear cell renal cell carcinoma (ccRCC) in combination with programmed death ligand-1 (PD-L1) antibodies (63). In their design, CAR T-cells were engineered to secrete PD-L1 antibodies, which confirmed that local antibody delivery not only prevented T-cell exhaustion but also recruited NK cells into tumor sites. In a humanized mice model of ccRCC, tumor growth diminished five times and tumor weight reduced 50–80% compared with those in the anti-CAIX CAR T-cells alone. Moreover, increasing research groups have devoted their research into combinatorial immunotherapy besides combining with checkpoint inhibitors. For example, Junghans et al. have used anti-PSMA CAR T-cell combined with IL-2 for the treatment of prostate cancer, and they found that the clinical responses to CAR T-cells were restrained by low plasma IL-2. Therefore, a moderate dose of administered IL-2 is necessary to enhance CAR T-cell efficiency. This report also presented a unique example of the critical impact of the pharmacodynamics of drug–drug interactions on the efficacy of their coapplication (23). Curran et al. have established an approach to enhance CAR T-cells by expressing CD40 ligand (CD40L). T-cells modified to constitutively express CD40L (CD40L-modified T-cells) demonstrated an enhanced proliferation and secretion of pro-inflammatory cytokines in vitro. This research also showed that CD40L-modified CAR T-cells induce dendritic cell maturation and secretion of the pro-inflammatory cytokine IL-12 to enhance antitumor effects (64). Thus, in future experimental designs, investigators may consider infusing CAR T-cells with immune checkpoint inhibitors, cytokines, or other costimulatory molecules.
Furthermore, Nishio and Dotti developed a special combinational therapy that employed CAR T-cells combined with oncolytic viruses (OVs) that resulted in a remarkable antitumor effect compared with that using CAR T-cell or OV alone. Their research confirmed that OV supports T-cell function by remaining toxic to tumor cells without damaging or compromising CAR T-cell activities, even at high concentrations of OVs (65). They demonstrated that tumor cells infected by OVs become more susceptible to the lytic effects of CAR T-cells. In turn, the faster lysis of tumor cells facilitates the spread of the virus, which enhances CAR T-cells in solid tumor (66). Several studies have demonstrated that tumor-derived soluble factors and immunosuppressive immune cells in TME limit the sensitivity of CAR T-cells (67). These studies have suggested the importance of blocking the inhibitory factors of TME in CARs design (68). Within TME, various suppressive surveilling immune cells such as myeloid-derived suppressor cells (MDSCs), regulatory T-cells (Tregs), tumor-associated microphages (TAMs), or neutrophils (TANs) with M2 and N2 phenotypes present a barrier against antitumor immunity (69, 70). MDSCs, M2 TAMs, and N2 TANs are also well-known producers of transforming growth factor-β (TGF-β), IL-10, reactive oxygen/nitrogen species, nitric oxide synthase (NOS), and arginase (ARG) (71, 72). TGF-β is a critical cytokine in embryogenesis and tissue homeostasis. TGF-β can induce a large and diverse set of responses, ranging from the induction of tissue growth and morphogenesis in the embryo to the activation of cellular cytostatic and death processes in epithelial cells. However, in tumor tissues, increasing studies have confirmed that TGF-β prevents antitumor effects by inhibiting CD8 cytotoxic T lymphocytes and boosts tumor cell migration and proliferation (73). ARG and NOS are critical for l-arginine metabolism, which plays an important role in tumor immunity (74). The presence of l-arginine promotes T-cell effector function and memory T-cell differentiation. However, in several solid tumors, various suppressive surveilling immune cells overexpress one or both of these enzymes and lead to T-cell dysfunction because of arginine deficits within TME. Thus, the manipulation of tumor-derived soluble factors and immunosuppressive immune cell activity in tumor sites may enhance the efficacy of CAR T-cell therapies.
Chimeric antigen receptor T-cells attack target cells by recognizing TAAs expressed on tumor cells. However, most TAAs are not only highly expressed on tumor cells but also shared with normal cells. Thus, the risk of “on target/off tumor” toxicity is a major obstacle for the development of CAR T-cell therapies for solid tumors. Several CAR T-cell therapies have resulted in life-threatening and fatal adverse events due to tumor lysis syndrome and cytokine storm. For example, Lamers et al. have evaluated the on-target toxicity of treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T-cells. Common toxicity criteria grade 2–4 liver enzyme disturbances were observed in 4 of 12 patients at 1–2 × 109 total cell dose, which can be prevented by pre-treatment with an anti-CAIX monoclonal antibody (35, 75). Morgan et al. have reported a serious adverse event in phase I clinical trials of an anti-HER2 CAR. In the third-generation (CD28.4-1BB.ζ) anti-HER2 CAR trial, a colon cancer patient with lung and liver metastases was intravenously administered with 1 × 1010 CAR T-cells. Within 15 min, the patient developed acute respiratory distress and died 5 days after treatment. They speculated that lung epithelial cells expressing HER2 at low levels were recognized by the administered cells and triggered a cytokine storm (34).
Ultimately, safety is closely dependent on specificity. In CAR design, we need to choose TAAs that are highly expressed on tumor cells but not expressed (or expressed at low levels) on normal cells as targets. Thus far, nearly all TAAs in the treatment of solid tumors have been expressed on normal tissues, particularly in bystander regions. The antitumor efficiency is associated with the dose of CAR T-cells, with high doses potentially increasing the risk of toxicity; therefore, it is difficult to balance safety and efficiency. Thus, researchers are advocating that suicide gene systems may ameliorate these toxicity profiles.
The herpes simplex virus–thymidine kinase (HSV-TK) suicide gene system has been most extensively tested in cell and gene therapy to eliminate the potential side effects of transduced cells (76–78). The HSV-TK gene has been successfully transferred into various cell lines to confer lethal sensitivity to the anti-herpes drug, ganciclovir, and its efficacy has been demonstrated both in vitro and in vivo (79). Another alternative suicide gene system is the inducible caspase 9 (iCasp9) gene, often used together with the small-molecule, AP1903 (80). The iCasp9 gene comprises an intracellular domain of the human caspase9 protein and a pro-apoptotic molecule fused to a drug-binding domain derived from human FK506-binding protein (81). This allows for dimerization and activation of apoptosis upon ligation with a dimerizer drug. This allows for dimerization and activation of apoptosis upon ligation with a dimerizer drug. The presence of AP1903 produces cross-linking of the drug-binding domains of the iCasp9 protein, which, in turn, dimerizes caspase 9 to activate the downstream executioner caspase 3 and results in cellular apoptosis (82). Diaconu et al. generated a novel CD19-specific CAR-modified T-cell (CD19.CAR Ts) selectively modulated by an iCasp9-based suicide gene. They demonstrated that the iCasp9 suicide gene depletes CD19.CAR T-cells in a dose-dependent manner in cases of cytokine release syndrome or complete deletion on demand, granting normal B cell reconstitution. In humanized mouse models, data also confirmed that low doses of AP1903 provide a specific containment for CD19.CAR T-cell expansion and cytokine release (83). Currently, CAR T-cells for solid tumors controlled by a specific safety switch are under study.
Cancer presents a real threat to human health, and the advent of CAR T-cell represents the dawn of anti-cancer therapies, particularly for non-solid tumors. However, CAR T-cell therapy for solid tumors faces some challenges. The three main hurdles in the application of CAR T-cell therapies to solid tumors have been the identification of specific TAAs, limited trafficking of CAR T-cells to solid tumor sites, and immunosuppressive effect of TME. Here, we focus on CAR design to address the third problem of enhancing the specificity, sensitivity, and safety of CAR T-cells.
Several approaches to overcome the solid TME are discussed in this review. Researchers may combine CAR T-cell therapy with checkpoint inhibitors or design CARs targeting immune checkpoints (84). Investigators may also design CARs targeting TME concluding hypoxia, nutrient starvation, metabolism, stroma, and cytokine networks (85–89). For example, indoleamine 2,3-dioxygenase (IDO) is an intracellular enzyme expressed on tumor and myeloid cells, which blocks the proliferation and survival of CAR T-cells; therefore, it is feasible to develop a CAR targeting IDO or to combine CAR T-cells and IDO inhibitors for tumor treatment (90). Generating CARs that are capable of recognizing multiple antigens is also an effective alternative to address the hurdles of TAA identification. Researchers have also identified neoantigens specifically expressed on tumor cells as potential targets. Several groups have also demonstrated that the successful use of chemokine receptors matched with tumor cell chemokines can attract CAR T-cells to tumor sites (91).
The remarkable success of CAR T therapy in hematological malignancies has propelled the development of CAR T therapy in solid tumors (92, 93). A better understanding of tumorigenesis and tumor progression will drive advances of future cancer treatment and provide hope for preventing cancer.
YW, FL, JY, CZ, and YC conceived the theme of this review and wrote the manuscript.
The authors did not receive any payment or services from a third party for any aspect of the submitted work. The authors also declare that they have no other relative affiliation or financial involvement with any organization or entity or other relationships/activities that may influence the submitted work.
The authors would like to thank Enago (www.enago.cn) for the English language review.
This work was supported by the National Natural Science Foundation of China (31400772, 81730045, and 91527305), the National Key Research and Development Program for Precision Medicine (2017YFC0909800), and National Science and Technology Major Project of China (2017ZX10203207).
1. Srivastava S, Riddell SR. Engineering CAR-T cells: design concepts. Trends Immunol (2015) 36(8):494–502. doi:10.1016/j.it.2015.06.004
2. Malissen B, Bongrand P. Early T cell activation: integrating biochemical, structural, and biophysical cues. Annu Rev Immunol (2015) 33:539–61. doi:10.1146/annurev-immunol-032414-112158
3. Malissen B, Gregoire C, Malissen M, Roncagalli R. Integrative biology of T cell activation. Nat Immunol (2014) 15(9):790–7. doi:10.1038/ni.2959
4. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A (1993) 90(2):720–4. doi:10.1073/pnas.90.2.720
5. Ramos CA, Heslop HE, Brenner MK. CAR-T cell therapy for lymphoma. Annu Rev Med (2016) 67:165–83. doi:10.1146/annurev-med-051914-021702
6. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev (2014) 257(1):107–26. doi:10.1111/imr.12131
7. Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood (2014) 124(7):1070–80. doi:10.1182/blood-2013-10-535245
8. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med (2015) 21(6):581–90. doi:10.1038/nm.3838
9. Zhang H, Snyder KM, Suhoski MM, Maus MV, Kapoor V, June CH, et al. 4-1BB is superior to CD28 costimulation for generating CD8+ cytotoxic lymphocytes for adoptive immunotherapy. J Immunol (2007) 179(7):4910–8. doi:10.4049/jimmunol.179.7.4910
10. Gilham DE, Debets R, Pule M, Hawkins RE, Abken H. CAR-T cells and solid tumors: tuning T cells to challenge an inveterate foe. Trends Mol Med (2012) 18(7):377–84. doi:10.1016/j.molmed.2012.04.009
11. Krneta T, Gillgrass A, Chew M, Ashkar AA. The breast tumor microenvironment alters the phenotype and function of natural killer cells. Cell Mol Immunol (2016) 13(5):628–39. doi:10.1038/cmi.2015.42
12. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14(10):1014–22. doi:10.1038/ni.2703
13. Newick K, O’Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med (2017) 68:139–52. doi:10.1146/annurev-med-062315-120245
14. Zhang BL, Qin DY, Mo ZM, Li Y, Wei W, Wang YS, et al. Hurdles of CAR-T cell-based cancer immunotherapy directed against solid tumors. Sci China Life Sci (2016) 59(4):340–8. doi:10.1007/s11427-016-5027-4
15. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol (2012) 12(4):269–81. doi:10.1038/nri3191
16. Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med (2015) 7(275):275ra22. doi:10.1126/scitranslmed.aaa4963
17. Maus MV. Designing CAR T cells for glioblastoma. Oncoimmunology (2015) 4(12):e1048956. doi:10.1080/2162402X.2015.1048956
18. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res (2014) 2(2):112–20. doi:10.1158/2326-6066.CIR-13-0170
19. Li W, Guo L, Rathi P, Marinova E, Gao X, Wu MF, et al. Redirecting T cells to glypican-3 with 4-1BB zeta chimeric antigen receptors results in Th1 polarization and potent antitumor activity. Hum Gene Ther (2017) 28(5):437–48. doi:10.1089/hum.2016.025
20. Li K, Pan X, Bi Y, Xu W, Chen C, Gao H, et al. Adoptive immunotherapy using T lymphocytes redirected to glypican-3 for the treatment of lung squamous cell carcinoma. Oncotarget (2016) 7(3):2496–507. doi:10.18632/oncotarget.6595
21. Gao H, Li K, Tu H, Pan X, Jiang H, Shi B, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res (2014) 20(24):6418–28. doi:10.1158/1078-0432.CCR-14-1170
22. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res (2015) 75(17):3596–607. doi:10.1158/0008-5472.CAN-15-0159
23. Junghans RP, Ma Q, Rathore R, Gomes EM, Bais AJ, Lo AS, et al. Phase I trial of anti-PSMA designer CAR-T cells in prostate cancer: possible role for interacting interleukin 2-T cell pharmacodynamics as a determinant of clinical response. Prostate (2016) 76(14):1257–70. doi:10.1002/pros.23214
24. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol (2015) 33(15):1688–96. doi:10.1200/JCO.2014.58.0225
25. Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci (2016) 59(5):468–79. doi:10.1007/s11427-016-5023-8
26. Sui X, Kong N, Zhu M, Wang X, Lou F, Han W, et al. Cotargeting EGFR and autophagy signaling: a novel therapeutic strategy for non-small-cell lung cancer. Mol Clin Oncol (2014) 2(1):8–12. doi:10.3892/mco.2013.187
27. Han W, Pan H, Chen Y, Sun J, Wang Y, Li J, et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One (2011) 6(6):e18691. doi:10.1371/journal.pone.0018691
28. Maher J, Wilkie S, Davies DM, Arif S, Picco G, Julien S, et al. Targeting of tumor-associated glycoforms of MUC1 with CAR T cells. Immunity (2016) 45(5):945–6. doi:10.1016/j.immuni.2016.10.014
29. Beatson R, Tajadura-Ortega V, Achkova D, Picco G, Tsourouktsoglou TD, Klausing S, et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat Immunol (2016) 17(11):1273–81. doi:10.1038/ni.3552
30. Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res (2015) 21(14):3149–59. doi:10.1158/1078-0432.CCR-14-1421
31. Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA, et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother (2015) 64(7):817–29. doi:10.1007/s00262-015-1692-6
32. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med (2016) 375(26):2561–9. doi:10.1056/NEJMoa1610497
33. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res (2015) 21(18):4062–72. doi:10.1158/1078-0432.CCR-15-0428
34. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18(4):843–51. doi:10.1038/mt.2010.24
35. Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther (2013) 21(4):904–12. doi:10.1038/mt.2013.17
36. Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov (2016) 6(2):133–46. doi:10.1158/2159-8290.CD-15-0583
37. Posey AD Jr, Clausen H, June CH. Distinguishing truncated and normal MUC1 glycoform targeting from Tn-MUC1-specific CAR T cells: specificity is the key to safety. Immunity (2016) 45(5):947–8. doi:10.1016/j.immuni.2016.10.015
38. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science (2015) 350(6266):1387–90. doi:10.1126/science.aad1253
39. Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med (2016) 22(1):26–36. doi:10.1038/nm.4015
40. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med (2016) 22(4):433–8. doi:10.1038/nm.4051
41. Tran E, Robbins PF, Rosenberg SA. ‘Final common pathway’ of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol (2017) 18(3):255–62. doi:10.1038/ni.3682
42. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science (2015) 348(6236):803–8. doi:10.1126/science.aaa3828
43. Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature (2016) 536(7614):91–5. doi:10.1038/nature18945
44. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest (2016) 126(8):3036–52. doi:10.1172/JCI83416
45. Schneider D, Xiong Y, Wu D, Nlle V, Schmitz S, Haso W, et al. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J Immunother Cancer (2017) 5:42. doi:10.1186/s40425-017-0246-1
46. Themeli M, Sadelain M. Combinatorial antigen targeting: ideal T-cell sensing and anti-tumor response. Trends Mol Med (2016) 22(4):271–3. doi:10.1016/j.molmed.2016.02.009
47. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol (2013) 31(1):71–5. doi:10.1038/nbt.2459
48. McNeill L, Salmond RJ, Cooper JC, Carret CK, Cassady-Cain RL, Roche-Molina M, et al. The differential regulation of Lck kinase phosphorylation sites by CD45 is critical for T cell receptor signaling responses. Immunity (2007) 27(3):425–37. doi:10.1016/j.immuni.2007.07.015
49. Hermiston ML, Zikherman J, Zhu JW. CD45, CD148, and Lyp/Pep: critical phosphatases regulating Src family kinase signaling networks in immune cells. Immunol Rev (2009) 228(1):288–311. doi:10.1111/j.1600-065X.2008.00752.x
50. Chang VT, Fernandes RA, Ganzinger KA, Lee SF, Siebold C, McColl J, et al. Initiation of T cell signaling by CD45 segregation at ‘close contacts’. Nat Immunol (2016) 17(5):574–82. doi:10.1038/ni.3392
51. Cho JH, Kim HO, Ju YJ, Kye YC, Lee GW, Lee SW, et al. CD45-mediated control of TCR tuning in naive and memory CD8+ T cells. Nat Commun (2016) 7:13373. doi:10.1038/ncomms13373
52. Cordoba SP, Choudhuri K, Zhang H, Bridge M, Basat AB, Dustin ML, et al. The large ectodomains of CD45 and CD148 regulate their segregation from and inhibition of ligated T-cell receptor. Blood (2013) 121(21):4295–302. doi:10.1182/blood-2012-07-442251
53. Irles C, Symons A, Michel F, Bakker TR, van der Merwe PA, Acuto O. CD45 ectodomain controls interaction with GEMs and Lck activity for optimal TCR signaling. Nat Immunol (2003) 4(2):189–97. doi:10.1038/ni877
54. Davis SJ, van der Merwe PA. The kinetic-segregation model: TCR triggering and beyond. Nat Immunol (2006) 7(8):803–9. doi:10.1038/ni1369
55. James JR, Vale RD. Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature (2012) 487(7405):64–9. doi:10.1038/nature11220
56. Hombach AA, Schildgen V, Heuser C, Finnern R, Gilham DE, Abken H. T cell activation by antibody-like immunoreceptors: the position of the binding epitope within the target molecule determines the efficiency of activation of redirected T cells. J Immunol (2007) 178(7):4650–7. doi:10.4049/jimmunol.178.7.4650
57. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res (2015) 3(2):125–35. doi:10.1158/2326-6066.CIR-14-0127
58. Zhukovsky EA, Morse RJ, Maus MV. Bispecific antibodies and CARs: generalized immunotherapeutics harnessing T cell redirection. Curr Opin Immunol (2016) 40:24–35. doi:10.1016/j.coi.2016.02.006
59. Luo F, Qian J, Yang J, Deng Y, Zheng X, Liu J, et al. Bifunctional alphaHER2/CD3 RNA-engineered CART-like human T cells specifically eliminate HER2(+) gastric cancer. Cell Res (2016) 26(7):850–3. doi:10.1038/cr.2016.81
60. Chen N, Morello A, Tano Z, Adusumilli PS. CAR T-cell intrinsic PD-1 checkpoint blockade: a two-in-one approach for solid tumor immunotherapy. Oncoimmunology (2017) 6(2):e1273302. doi:10.1080/2162402X.2016.1273302
61. Serganova I, Moroz E, Cohen I, Moroz M, Mane M, Zurita J, et al. Enhancement of PSMA-directed CAR adoptive immunotherapy by PD-1/PD-L1 blockade. Mol Ther Oncolytics (2017) 4:41–54. doi:10.1016/j.omto.2016.11.005
62. John LB, Kershaw MH, Darcy PK. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology (2013) 2(10):e26286. doi:10.4161/onci.26286
63. Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget (2016) 7(23):34341–55. doi:10.18632/oncotarget.9114
64. Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther (2015) 23(4):769–78. doi:10.1038/mt.2015.4
65. Nishio N, Dotti G. Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. Oncoimmunology (2015) 4(2):e988098. doi:10.4161/21505594.2014.988098
66. Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res (2014) 74(18):5195–205. doi:10.1158/0008-5472.CAN-14-0697
67. Scarfo I, Maus MV. Current approaches to increase CAR T cell potency in solid tumors: targeting the tumor microenvironment. J Immunother Cancer (2017) 5:28. doi:10.1186/s40425-017-0230-9
68. Irving M, Vuillefroy de Silly R, Scholten K, Dilek N, Coukos G. Engineering chimeric antigen receptor T-cells for racing in solid tumors: don’t forget the fuel. Front Immunol (2017) 8:267. doi:10.3389/fimmu.2017.00267
69. Tang H, Qiao J, Fu YX. Immunotherapy and tumor microenvironment. Cancer Lett (2016) 370(1):85–90. doi:10.1016/j.canlet.2015.10.009
70. Huang Y, Rajappa P, Hu W, Hoffman C, Cisse B, Kim JH, et al. A proangiogenic signaling axis in myeloid cells promotes malignant progression of glioma. J Clin Invest (2017) 127:1826–38. doi:10.1172/JCI86443
71. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi:10.1038/nm.3394
72. Pergamo M, Miller G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther (2017) 24(3):100–5. doi:10.1038/cgt.2016.65
73. Huang CY, Wang H, Liao W, Han F, Li YQ, Chen SW, et al. Transforming growth factor beta is a poor prognostic factor and inhibits the favorable prognostic value of CD8+ CTL in human hepatocellular carcinoma. J Immunother (2017) 40(5):175–86. doi:10.1097/CJI.0000000000000166
74. Anderson KG, Stromnes IM, Greenberg PD. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell (2017) 31(3):311–25. doi:10.1016/j.ccell.2017.02.008
75. Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol (2006) 24(13):e20–2. doi:10.1200/JCO.2006.05.9964
76. Chendeb M, Schneider R, Davidson I, Fadloun A. Selective elimination of long INterspersed element-1 expressing tumour cells by targeted expression of the HSV-TK suicide gene. Oncotarget (2017) 8(24):38239–50. doi:10.18632/oncotarget.16013
77. Ciceri F, Bonini C, Stanghellini MT, Bondanza A, Traversari C, Salomoni M, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol (2009) 10(5):489–500. doi:10.1016/S1470-2045(09)70074-9
78. Kuo WY, Hwu L, Wu CY, Lee JS, Chang CW, Liu RS. STAT3/NF-kappaB-regulated lentiviral TK/GCV suicide gene therapy for cisplatin-resistant triple-negative breast cancer. Theranostics (2017) 7(3):647–63. doi:10.7150/thno.16827
79. Greco R, Oliveira G, Stanghellini MT, Vago L, Bondanza A, Peccatori J, et al. Improving the safety of cell therapy with the TK-suicide gene. Front Pharmacol (2015) 6:95. doi:10.3389/fphar.2015.00095
80. Xie X, Zhao X, Liu Y, Zhang J, Matusik RJ, Slawin KM, et al. Adenovirus-mediated tissue-targeted expression of a caspase-9-based artificial death switch for the treatment of prostate cancer. Cancer Res (2001) 61(18):6795–804.
81. Zhou X, Di Stasi A, Brenner MK. iCaspase 9 suicide gene system. Methods Mol Biol (2015) 1317:87–105. doi:10.1007/978-1-4939-2727-2_6
82. Nor JE, Hu Y, Song W, Spencer DM, Nunez G. Ablation of microvessels in vivo upon dimerization of iCaspase-9. Gene Ther (2002) 9(7):444–51. doi:10.1038/sj.gt.3301671
83. Diaconu I, Ballard B, Zhang M, Chen Y, West J, Dotti G, et al. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol Ther (2017) 25(3):580–92. doi:10.1016/j.ymthe.2017.01.011
84. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest (2016) 126(8):3130–44. doi:10.1172/JCI83092
85. Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther (2013) 21(8):1611–20. doi:10.1038/mt.2013.110
86. Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity (2016) 44(2):380–90. doi:10.1016/j.immuni.2016.01.021
87. Howie D, Waldmann H, Cobbold S. Nutrient sensing via mTOR in T cells maintains a tolerogenic microenvironment. Front Immunol (2014) 5:409. doi:10.3389/fimmu.2014.00409
88. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114(8):1537–44. doi:10.1182/blood-2008-12-195792
89. Zhang Y, Ertl HC. Starved and asphyxiated: how can CD8(+) T cells within a tumor microenvironment prevent tumor progression. Front Immunol (2016) 7:32. doi:10.3389/fimmu.2016.00032
90. Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood (2015) 125(25):3905–16. doi:10.1182/blood-2015-01-621474
91. Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res (2011) 17(14):4719–30. doi:10.1158/1078-0432.CCR-11-0351
92. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol (2016) 13(6):394. doi:10.1038/nrclinonc.2016.65
Keywords: chimeric antigen receptor T-cell, immunotherapy, solid tumor, adoptive T-cell therapy, tumor microenvironment
Citation: Wang Y, Luo F, Yang J, Zhao C and Chu Y (2017) New Chimeric Antigen Receptor Design for Solid Tumors. Front. Immunol. 8:1934. doi: 10.3389/fimmu.2017.01934
Received: 30 August 2017; Accepted: 15 December 2017;
Published: 22 December 2017
Edited by:
Malini Mukherjee, Baylor College of Medicine, United StatesReviewed by:
Amorette Barber, Longwood University, United StatesCopyright: © 2017 Wang, Luo, Yang, Zhao and Chu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yiwei Chu, eWl3ZWljaHVAZnVkYW4uZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.