 Melissa Bedard
Melissa Bedard Mariolina Salio
Mariolina Salio Vincenzo Cerundolo
Vincenzo Cerundolo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 December 2017
Sec. Cancer Immunity and Immunotherapy
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01829
This article is part of the Research Topic NKT Cells in Cancer Immunotherapy View all 18 articles
Invariant natural killer T (iNKT) cells are a distinct subset of innate-like lymphocytes bearing an invariant T-cell receptor, through which they recognize lipid antigens presented by monomorphic CD1d molecules. Upon activation, iNKT cells are capable of not only having a direct effector function but also transactivating NK cells, maturing dendritic cells, and activating B cells, through secretion of several cytokines and cognate TCR-CD1d interaction. Endowed with the ability to orchestrate an all-encompassing immune response, iNKT cells are critical in shaping immune responses against pathogens and cancer cells. In this review, we examine the critical role of iNKT cells in antitumor responses from two perspectives: (i) how iNKT cells potentiate antitumor immunity and (ii) how CD1d+ tumor cells may modulate their own expression of CD1d molecules. We further explore hypotheses to explain iNKT cell activation in the context of cancer and how the antitumor effects of iNKT cells can be exploited in different forms of cancer immunotherapy, including their role in the development of cancer vaccines.
The evidence that peptide-specific T cells play an important role in the immune defense against pathogens and cancer progression is compelling (1–3). In the last two decades, it has emerged that in addition to peptide-specific T cells, hereafter referred to as conventional T cells, α/β T cells can also recognize lipids and metabolites of vitamin B2 in the context of monomorphic MHC class I-like molecules (4, 5). Such T cells, hereafter referred to as unconventional T cells, can orchestrate an immune response against pathogens and cancer (4). This review will focus on the description of one of these unconventional T cell populations—invariant natural killer T (iNKT) cells. We will also highlight potential mechanisms of iNKT cell activation in cancer and how these cells can be manipulated for the purpose of cancer immunotherapy.
Invariant natural killer T cells originate from bone marrow-derived progenitors that, like conventional T cells, migrate to the thymus. However, unlike conventional T cells, which are selected by self-peptides presented by MHC class I and II on thymic epithelial cells, iNKT cells are positively selected by CD1d molecules expressed by double-positive (CD4+CD8+) thymocytes (6, 7). Such CD1d molecules present self-lipid ligand(s), not yet fully characterized, and upon expression of the transcription factor PLZF, the thymocytes acquire the iNKT cell effector program (8). iNKT cells subsequently migrate out of the thymus and reach maturity in the periphery (8). Unlike naïve conventional T cells, iNKT cell numbers in humans are high, particularly in the spleen and in the liver, reaching about 1% of total lymphocytes in the latter tissue (9).
Invariant natural killer T cells are considered innate-like lymphocytes as they exhibit characteristics of both innate and adaptive immune cells. Their activation is driven by antigen recognition, a characteristic of conventional adaptive immune cells. However, unlike conventional T cells, iNKT cells bear a semi-invariant TCR that recognizes different lipid antigens presented on monomorphic CD1d molecules. This recognition manner has been likened to a pattern recognition mode (10). iNKT cells further deviate from conventional T cells by their ability to rapidly secrete copious amounts of cytokines, mainly IFN-γ and IL-4, shortly upon activation—a characteristic reminiscent of innate immune responses, and which is imparted at the epigenetic level by their unique developmental program (11–14).
Since iNKT cells are uniquely placed at the interface between innate and adaptive immunity, they have a tremendous influence in shaping immune responses. Cytokine stimulation and cognate interaction between iNKT cells and dendritic cells (DCs), B cells, neutrophils, and macrophages often polarizes these cells toward a pro-inflammatory phenotype (15–25). Similarly, activated iNKT cells can transactivate natural killer cells (26) and enhance stimulation of conventional T cells through their ability to secrete cytokines and mature DCs (16, 18). Although the frequency of iNKT cells in humans ranges from 0.01 to 0.1% in peripheral blood (lower than in mice), this frequency is still orders of magnitude higher than that of naïve peptide-specific T cells (9, 27). In addition, their constitutive expression of CD40L and ability to rapidly secrete cytokines make iNKT cells critical players in immunity, by orchestrating all-encompassing immune responses (9).
There are two primary means of iNKT cell activation: CD1d-dependent and cytokine-driven activation. CD1d molecules are transmembrane proteins that, similar to MHC class I molecules, bind non-covalently to β2-microglobulin. The surface-exposed antigen-binding groove consists of two deep hydrophobic channels that bind the fatty acid tails of lipid antigen, while the head moiety is exposed for recognition by the iNKT-TCR (28, 29). Ceramide-based glycolipids (glycosphingolipids) and glycerol-based lipids (such as membrane phospholipids) are the two main types of iNKT-activating lipids bound to CD1d molecules (30–34). While the most potent iNKT-activating lipid agonists described to date is threitol-6-ceramide (35), the classical iNKT-activating lipid agonist most frequently used in the literature is α-galactosylceramide (αGC), which is derived from a bacterium on the Agelas mauritianus marine sponge (23, 36–38). Analysis of the crystal structure of CD1d monomers with or without αGC, which exploits the full binding capacity of CD1d, allowed for the identification of the hydrogen bonds required to hold the polar head of iNKT cell agonists (29). The presence of both a lipid binding and non-lipid binding molecule in the asymmetric unit of the CD1d crystals has enabled the identification of two different conformations of the antigen-binding groove (29). Using planar lipid bilayers and surface plasmon resonance, the contribution of the length and saturation of the alkyl chains occupying the A′ and F′ channel of human CD1d molecules to the stability of CD1d-lipid complexes and to the affinity of iNKT-TCR binding was further analyzed (39). These results led to the description of a general mechanism by which the length of the lipid chain occupying the F′ channel plays a role in controlling the affinity of lipid-specific CD1d-restricted T cells (39). This concept can be more generally extend to other CD1-restricted cells (40).
In a more physiological context, iNKT cells become activated by microbial or self-lipid antigens bound to CD1d molecules. For example, isoglobotriosylceramide (iGB3), a neutral glycosphingolipid, has been identified as a weak self-lipid antigen for human and murine iNKT cells (41–43), although its role as the only positive-selecting self-lipid in the thymus remains controversial, given that mice lacking the required synthases for iGB3 production maintain an intact iNKT cell repertoire (44, 45). Lysophospholipids and charged glycosphingolipids have been shown to be self-lipid antigens in different contexts (46–48). Self-lipid antigens are weakly immunogenic and iNKT cell activation in this case is often largely driven by IL-12 and IL-18. In a model of hepatitis B infection, it has been shown that viral-induced phospholipases generate lysophospholipids that lead to iNKT cell activation (30, 47).
Cytokine-driven activation is common when lipid antigen is weakly immunogenic (47). Although CD1d-activated iNKT cells can undergo further activation via cytokines secreted from matured DCs, certain cytokines, namely IL-12 and IL-18, are alone sufficient to activate iNKT cells (49, 50). Avidity might play a more important role in iNKT cell activation than previously considered, especially iNKT cell activation by self-lipid antigen repertoire. Alterations in the actin cytoskeleton are evidenced to create CD1d nanoclusters of higher avidity, increasing basal iNKT autoreactivity (51).
The ability of iNKT cells to orchestrate immune responses against cancer is perhaps the most striking example of their role in disease. Work from the laboratory led by Dale Godfrey highlighted the essential role of iNKT cells in tumor immunity by demonstrating that mice lacking iNKT cells were more susceptible to methylcholanthrene-induced sarcomas, consistent with the role of iNKT cells in immunosurveillance (52). This effect was reversed upon iNKT cell reconstitution, an observation that further supports their role in tumor clearance. Although the antitumor effector activity of iNKT cells upon αGC injection was recently confirmed using newly generated Jα18-deficient mice, which bear an otherwise normal T cell repertoire (53), the role of iNKT cells in immunosurveillance of methylcholanthrene-induced sarcomas was called into question in a separate study (54).
Invariant natural killer T cells’ ability to modulate various immune subsets is key to their role in antitumor immune responses. iNKT cells can mature DCs, activate CD4+ T cells, CD8+ T cells, and B cells, and transactivate NK cells (19, 23). In murine models of lung and liver cancers, the antitumor effect of αGC administration was attributed to IFN-γ secretion from iNKT cells and transactivated NK cells, which culminated in NK perforin-mediated cytolysis of tumor cells (23). iNKT cell-derived IFN-γ is also responsible for enhanced activation of tumor antigen-specific CD8+ T cells (19, 55, 56). Additionally IL-12 derived from iNKT cell-matured DCs helped priming of tumor antigen-specific T cells (19, 57).
Invariant natural killer T cells can also augment an antitumor response by diminishing the immunosuppressive activities of immune subsets that promote tumorigenesis. It has been shown that iNKT cells can have a profound effect on the number and function of pro-tumorigenic myeloid populations (22, 58, 59). Tumor-associated macrophages (TAMs), which secrete immunosuppressive molecules such as IL-6 and TGF-β that dampen T-cell responses to MHC-presented tumor antigen, are found in the tumor microenvironment of a variety of cancers, including renal cell carcinomas and neuroblastoma (59). In primary human neuroblastoma samples, iNKT cells specifically killed the tumor-antigen-loaded TAMs rather than neuroblastoma cells, in part relieving the immunosuppressive tumor microenvironment and limiting metastases (59). iNKT cells are also capable of reducing myeloid-derived suppressor cells (MDSC) numbers and immunosuppressive activity (22, 58). These findings beg the question of how iNKT cells remain unaffected by the immunosuppressive microenvironment. It is reported that in patients with head and neck cancer, iNKT cells, unlike conventional T cells, are resistant to hydrogen peroxide produced by CD15+ MDSCs (60). This observation potentially explains their persistent activation and cytotoxic activity within an immunosuppressive tumor microenvironment.
While iNKT cells are best known to potentiate their antitumor effect through enhancing the immunogenic activities of a variety of immune cell subsets, they are capable of themselves recognizing and killing CD1d+ tumor cells. Such is true for the EL4 T-cell lymphoma model, where both in vitro and in vivo iNKT cells directly executed perforin-mediated cytolysis of lymphoma cells in a CD1d-dependent manner (61, 62). Furthermore, in a TRAMP murine model of CD1d+ prostate cancer, iNKT cells directly and predominantly reduce tumorigenesis, to a greater extent than cytotoxic T lymphocytes (63). In addition, in naturally expressing CD1d+ human osteosarcoma cell lines, iNKT cells selectively killed the tumor cells through Fas-FasL interaction, while leaving cocultured CD1d− osteoclasts and CD1d+ mesenchymal stem cells unaffected (64). Glioma and breast cancer cell lines transduced with CD1d are targets of iNKT cell-dependent lysis (65, 66). These results collectively indicate that iNKT cells are capable of directly killing CD1d+ tumors. In the large proportion of cases where solid tumors are CD1d−, tumor-infiltrating CD1d+ myeloid populations might activate iNKT cells either within the tumor or in distal lymphoid tissues enriched in iNKT cells.
Though their activation can contribute greatly to antitumor immune responses, there is a dearth of evidence that iNKT cells are present within the tumor microenvironment, particularly in human solid tumors. This issue might stem from low frequency of iNKT cells in humans, potentially making them difficult to detect by immunohistochemical techniques. While iNKT cell-specific antibodies, including the 6B11 antibody (67), do exist, there are few reports of their use in identifying iNKT cell populations within tumor microenvironments.
Tumor cells use a variety of mechanisms to escape detection and elimination by immune cells. These mechanisms include releasing soluble mediators to dampen antitumor immune responses, notably TGF-β into the microenvironment and inducing T cell anergy and exhaustion (68). Another mechanism involves hindering antigen presentation, often by limiting expression of antigen-presenting molecules in infiltrating myeloid cells or on the tumor cells themselves. MHC class I molecules, which present peptide antigens to CD8+ T cells, are well characterized as a target of such escape mechanisms (68). Similarly, a variety of tumors downregulate CD1d molecules, further emphasizing the important role of iNKT cells in antitumor immunity (Figure 1).
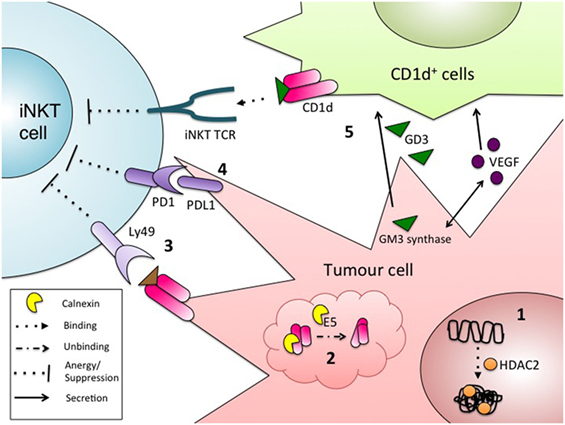
Figure 1. Mechanisms of tumor evasion from invariant natural killer T (iNKT) immunosurveillance. Some tumors cells escape detection by iNKT cells via the regulation of surface CD1d, by: (1) heterochromatin formation at the CD1d locus by histone deacetylases (71, 72); or (2) improper folding and retention of CD1d in the ER (69). Other mechanisms to escape iNKT cell detection include: (3) engagement of surface CD1d with the inhibitory NK receptor Ly49, leading to the induction of iNKT cell anergy (70); (4) inhibitory signaling through PD1/PDL1 between iNKT cells and tumor cells (139); and (5) CD1d-dependent suppression of iNKT cells through presentation of the inhibitory, tumor-derived glycolipid GD3. GD3 production is also driven by the secretion of tumor-derived VEGF (75).
The correlation between reduced CD1d expression and enhanced tumor progression has been reported in a variety of types of CD1d-transduced solid cancers, including breast, cervical, ovarian, prostate, lung, and melanoma (66, 69–72). This observation holds true for many naturally CD1d+ and transduced CD1d+ liquid tumors, such as mantle cell lymphoma, multiple myeloma, and chronic lymphocytic leukemia (61, 71, 73, 74). However, different tumors engage different mechanisms to reduce CD1d surface expression. On the RNA level, modulation of CD1d expression is largely driven by epigenetic changes. Treatment of mantle cell lymphoma cell lines with histone deacetylase inhibitors resulted in enhanced iNKT cell activation upon coculture (71). This observation was attributed to the removal of HDAC2 from the CD1d promoter, resulting in increased CD1d expression (71). Another report, which substantiates these findings, demonstrated that treating human and murine lung cancer and melanoma cell lines with HDAC2 inhibitors induce CD1d expression, although the functional relevance was not investigated (72). CD1d assembly in the endoplasmic reticulum (ER) is another potential target for tumor CD1d downregulation. In a model of HPV-driven cervical cancer, early-infected epithelial cells exhibited reduced CD1d expression compared to uninfected cells. In infected cells, the viral protein E5 inhibited calnexin, resulting in improper folding of CD1d, retention of CD1d molecules in the ER, and subsequent proteasomal degradation (69).
Modulation of iNKT cell function, even when CD1d molecules reach the surface of tumor cells, can contribute to evasion of iNKT surveillance. In the TRAMP murine model of prostate cancer, tumor cells express functional CD1d molecules, but lead to aberrant iNKT-cell activation akin to anergy, likely through the inhibitory receptor Ly49 (70). This phenotype could be rescued by simultaneous stimulation with αGC and IL-12, which likely overrides the inhibitory signal.
Tumor-derived factors can also inhibit trans-CD1d-dependent antigen presentation. When murine CD1d+ fibroblasts were treated with human ascites from ovarian cancer patients, CD1d-dependent iNKT cell activation was markedly reduced, suggesting that a soluble factor released from ovarian tumors could affect CD1d-dependent activation (75). VEGF, a pro-angiogenic and pro-tumorigenic soluble factor, and the suppressive glycolipid antigen GD3, were identified as the factors present in the ascitic fluid inhibiting iNKT cell activation (75). Interestingly, the authors also showed that GD3 synthesis was dependent on VEGF-mediated upregulation of GM3 synthase in the ovarian cancer cells (75).
While these findings illustrate the importance of CD1d in mounting iNKT-driven antitumor immune responses, there exists at least one example where increased CD1d expression and tumor progression are positively correlated (76, 77). Through microarrays, immunohistochemistry, and patient statistics, enhanced CD1d expression was associated with increased malignancy and higher relapse rates in a subset of human renal cell carcinoma, clear cell renal carcinoma (76). This result serves as a rare example of enhanced CD1d expression as a predictor of tumor progression. It is possible that CD1d-dependent activation of suppressive type II iNKT cells, to be discussed later, might contribute to this phenotype (78, 79).
Both tumor cells and tumor-infiltrating immune cells are subject to microenvironmental stress due to nutrient deprivation, hypoxia, or accumulation of toxic products of catabolism (80). This suboptimal environment can lead to upregulation of autophagy, a survival-promoting pathway centered on lysosomal-recycling intracellular material (80). Tumor cells that engage the autophagy pathway become more robust and are able to better persist and metastasize (80). It has been recently shown that in murine bone marrow DCs, deletion of the autophagy regulator protein ATG5 led to increased CD1d-dependent antigen presentation, due to limited CD1d internalization (81). However, it has also been demonstrated that during thymic iNKT cell development, ATG5 is dispensable to CD1d expression (82, 83). More research is required to clarify the role of autophagy in CD1d expression, as perhaps this mechanism is cell and time dependent.
The modulation of CD1d expression in tumor cells provides strong evidence for the critical role of iNKT cells in mounting antitumor immune responses. However, it remains unclear how iNKT cells become activated in the context of cancer, a form of sterile inflammation. The characterization of a growing number of activating stimuli and pathways, some of which might affect lipid antigen presentation, sheds light on a number of mechanisms that might contribute to iNKT sterile activation in cancer (Figure 2).
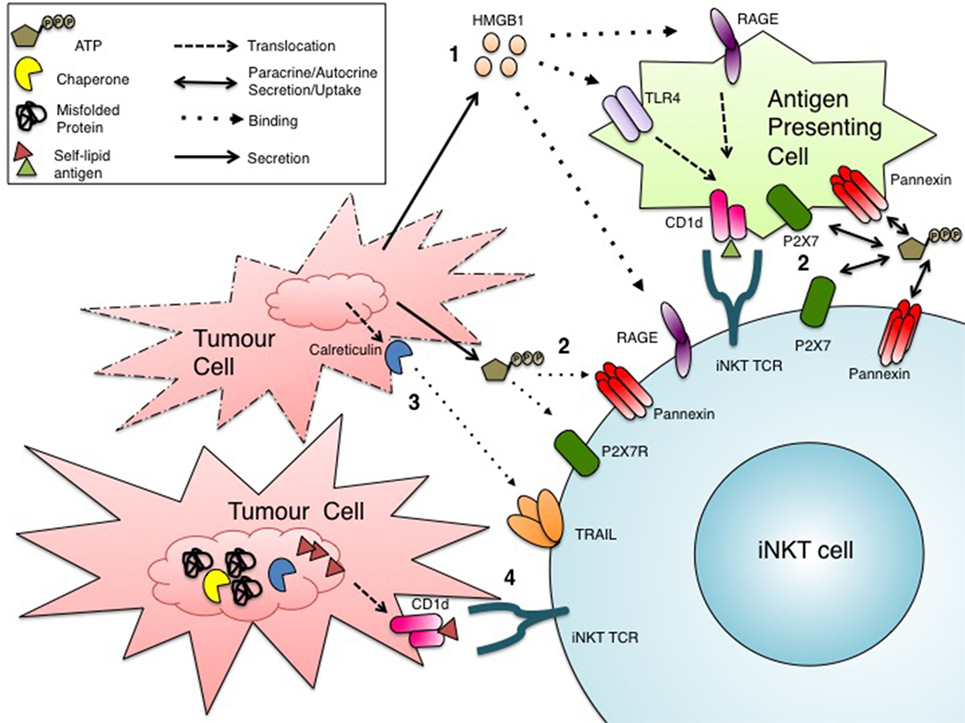
Figure 2. Potential mechanisms of invariant natural killer T (iNKT) cell activation in cancer. Tumor cells subject to drugs or conditions that induce stress might activate iNKT cells through several pathways: (1) secretion or passive release of DAMPs, such as HMGB1, that bind RAGE receptors directly on iNKT cells or TLR4/RAGE receptors on CD1d+ antigen-presenting cells (APCs), leading to the presentation of an immunogenic self-lipid antigen (46, 88, 90); (2) paracrine (from APCs) or autocrine release of ATP into the extracellular environment for uptake by iNKT cells, ultimately leading to iNKT cell activation (94, 95); (3) binding of the surface DAMP calreticulin to TRAIL on iNKT cells (103–110); (4) induction of ER stress in CD1d+ cells, due to the suboptimal physiological tumor microenvironment, might trigger the alternate loading immunogenic self-lipid antigen(s), resulting in enhanced iNKT cell activation (111–113).
At the intersection of chemotherapy and immunotherapy lies a class of drugs that provoke a type of cell death in tumor cells that results in the activation of innate immune cells. This type of cell death is termed immunogenic cell death (ICD). When cancer cells die, for example, by necrosis, they surface-expose or release molecules called danger-associated molecular patterns (DAMPs) that are usually contained within the cell, not on the cell surface or in the extracellular milieu (84). These DAMPs can be recognized by receptors on a variety of immune cells, including toll-like receptors (TLRs), and initiate an immune response (84).
One prototypic DAMP is a protein called high mobility group box 1 (HMGB1). HMGB1, ubiquitously expressed in a variety of cell types, typically resides in the nucleus as a chromatin binding protein (85). However, HMGB1 can be released into the extracellular environment where it behaves as a DAMP (85). Its release may be passive or mediated by an active mechanism. In the passive form, HMGB1 is released from cells dying by necrosis or other forms of ICD (86). In the active form, HMGB1 is secreted from myeloid cells, namely DCs and macrophages (87). In the extracellular milieu, HMGB1 can bind and signal through TLRs 2, 4, and the receptor for advanced glycation end products (RAGE) expressed on innate immune cells, triggering an immune response (86). RAGE is reportedly expressed on iNKT cells and can bind HMGB1, resulting in a Th17 activation profile (88). While the presence of TLRs on iNKT cells is disputed, most CD1d+ cells also bear TLRs (46, 89–91). Viral signaling through TLR7 on human DCs has also been implicated in enhanced de novo synthesis of CD1d molecules (91). Furthermore, engagement of TLR4 on CD1d+ myeloid cells, both murine and human, enhanced loading of self-lipid antigens onto CD1d molecules leading to iNKT cell activation (46, 90). These findings give rise to the possibility that HMGB1 signaling through TLR4 could induce loading of immunogenic self-lipid antigens onto CD1d, thus providing an explanation for iNKT cell activation in cancer.
Another soluble DAMP involved in ICD in cancer is adenosine triphosphate (ATP) (92, 93). ATP can interact with various immunomodulatory receptors and channels on myeloid cells and lymphocytes, a process termed purinergic signaling/regulation (94). Myeloid cells and T cells uptake extracellular ATP through P2X7 or pannexin channels, which can enhance inflammasome activation and amplify TCR-mediated activation, respectively (94). This mode of purinergic signaling might be especially relevant in the context of cancer, where stressed and necrotic tumor cells could release ATP into the tumor microenvironment and in turn augment TCR stimulation in response to weak tumor antigens (94). Like conventional T cells, iNKT cells bear P2XR and pannexins that allow uptake of extracellular ATP, which could provide additional costimulation to CD1d-mediated activation (95). iNKT cells also express two ectonucleotidases, CD39 and CD73 (95). CD39 converts ATP, a pro-inflammatory mediator to ADP, which is in turn converted by CD73 into AMP, an anti-inflammatory mediator (96). This step-wise generation of AMP from ATP is thought to cause a shift toward an immunosuppressive microenvironment, which might be advantageous for tumor progression (96). While the balance between extracellular ATP release and catabolism in the tumor microenvironment is poorly understood, we are gaining insights into mechanisms underlying purinergic signaling in iNKT cells.
The A2 adenosine receptor (A2AR), which binds adenosine to shift immune cells toward an immunosuppressive phenotype, also has a great influence on iNKT cell activation. In a model of concanavalin A-induced hepatitis, which is predominantly iNKT cell dependent, severity of the disease phenotype was dependent on the strength of A2A receptor signaling, with an exaggerated version of the disease seen in A2AR−/− mice, and an abrogation of the disease phenotype in mice treated with an A2AR agonist (97). It was later determined that A2AR exerts control over cytokine secretion in iNKT cells, particularly IL-4 and IL-10 (98).
Adenosine triphosphate is also involved in DC–iNKT cell interactions culminating in the release of inflammatory mediators that promote neutrophil recruitment. In monocyte-derived DC and iNKT cocultures, release of ATP from one or both of these immune cells (the secreting cell type was not identified) induced calcium flux within the DC through P2X7 signaling, which in turn triggered the release of prostaglandin E2 and soluble factors that promote neutrophil recruitment (99, 100). These observations were made in the setting of sterile inflammation, hinting that the influence of ATP in iNKT–DC interactions could hold true in the context of cancer (99). Maturation and stimulation of different immune cell populations by iNKT cells are thought to underlie their ability to induce antitumor immunity. This observation might be explained by the influence of purinergic signaling leading to the recruitment of neutrophils and perhaps other cell types.
Another potentially relevant soluble factor is heat shock protein 70 (Hsp70) an ER-derived chaperone (101). Similar to HMGB1, Hsp70 is upregulated during a variety of stress conditions, and under extreme stress conditions that induce tissue injury or necrotic cell death, Hsp70 is released in the extracellular environment (101). Hsp70, or more specifically the Hsp70-derived 14-amino acid peptide, in combination with either IL-2 or IL-15, enhanced the expression of NK-activating receptors, including their expression on the surface of iNKT cells (102). It remains to be seen whether this observation drives iNKT cell activation in the context of cancer.
Invariant natural killer T cells upregulate a number of different receptors upon activation, including activation markers such as CD69 and CD25, and cytotoxicity molecules including FASL and TNF-related apoptosis-inducing ligand (TRAIL) (103). TRAIL, best known for inducing apoptosis in cells expressing TRAIL receptors such as DR4 and DR5, shares a high degree of homology with FASL in the extracellular binding motif (104). Although FASL-mediated killing is often indiscriminate due to rather ubiquitous expression of FAS on mammalian tissues, TRAIL-mediated cytotoxicity is more selective toward virally infected cells and tumor cells, making it a potential target in immunotherapy (104). In humans, TRAIL is upregulated on iNKT cells upon activation and is consequently able to induce apoptosis is acute myeloid leukemia (AML) cells (105), which bear TRAIL receptors (103). This finding is substantiated in an AML murine model (103). Upon αGC administration, iNKT-derived IFN-γ upregulated TRAIL expression on activated NK cells, which in turn limited the metastasis of liver and lung tumors (106). While TRAIL has a number of well-recognized receptors, a less characterized interaction is its binding and signaling through calreticulin. Under normal circumstances, calreticulin is retained in the ER where it acts as a chaperone. However, under conditions of extreme stress leading to ICD, as is often the case with tumor cells during chemotherapy, calreticulin can be translocated to the surface of dying cells (107). In fact, calreticulin is a marker of ICD and is considered a DAMP. Soluble TRAIL has been found to interact with calreticulin expressed on A375M melanoma cells (107). Furthermore, calreticulin exposure on malignant AML blasts is correlated with increased frequency of T lymphocytes and improved survival—a finding that complements TRAIL+ iNKT cells’ killing of AML cells, although in this cohort of AML patients iNKT cells were not investigated (108). While the link between calreticulin-TRAIL cognate interaction and iNKT-dependent tumor killing requires further corroborating research, it would potentially provide a molecular mechanism for the iNKT cell-mediated antitumor effects in a variety of cancers.
Cells subject to TRAIL-induced killing typically undergo apoptosis, regulated non-inflammatory cell death, or necroptosis, a form of regulated inflammatory cell death due to the release of DAMPs (109). Both apoptosis and necroptosis utilize RIPK1/3 signaling (109). In TRAIL-mediated cytotoxicity, the switch between the two types of death is dictated by the acidity of microenvironment. It is tempting to speculate that in the tumor microenvironment, in which nutrients are in short supply and hypoxia is a hallmark, the consequent acidic surrounding might shift TRAIL-mediated cell death towards necroptosis. Indeed, TRAIL-induced necroptosis can contribute to in vitro killing of human HepG2 liver and HT29 colon cancer cell lines. iNKT cells also induce TRAIL-mediated necroptosis in a ConA model of hepatitis (110). Although iNKT cells exert cytolytic functions via necroptotic signaling in target cells, aspects of the necroptotic signaling pathway are also essential in iNKT cells themselves.
RIPK3, a kinase involved in the transduction of the necroptotic signaling pathway, regulates iNKT cell activation independent of necroptosis, as RIPK3 knockdown iNKT cells exhibited impaired cytokine secretion, including IFN-γ, upon αGC stimulation (110). Furthermore, wild-type mice inoculated with B16 melanoma were able to clear the tumor burden upon administration of αGC, but RIPK3−/− mice were unable to do so, suggesting that RIPK3 is essential in iNKT-mediated anti-tumor responses (110). Further dissection of the mechanism involved illustrated that RIK3 signaling can induce the mitochondrial phosphatase PGAM5, which in turn upregulates NFAT translocation into the nucleus and stimulates the mitochondrial GTPase Drp1 (110). These factors appear to regulate TCR- and cytokine-mediated iNKT cell activation (110). These findings lay the foundation for a new pathway that can be manipulated in therapies centered on enhancing iNKT cell activation.
Cancer cells are subject to rapid cell division, leading to a reduction in available nutrients in the microenvironment and accumulation of nascent and/or mutated proteins. These suboptimal conditions, both intrinsic and extrinsic to the cell, compromise ER homeostasis and trigger the unfolded protein response (UPR) (111). Additionally, the UPR is likely triggered in immune cells in the suboptimal tumor microenvironment. UPR activation in malignant and infiltrating immune cells would alter lipid biosynthetic pathways (112). UPR activation might lead to sorting of self-lipid antigens onto CD1d complexes on CD1d+ tumor or surrounding immune cells—which in conjunction with inflammatory cytokines might become immunogenic. It has been shown that the microsomal triglyceride transfer protein (MTTP) lies at the intersection between the UPR and CD1d–lipid complex formation (113). MTTP forms a heterodimer with protein disulfide isomerase (PDI) and transfers different lipid antigens onto assembling CD1d complexes in the ER. Importantly, PDIs are upregulated during UPR activation (112, 113). It is thus possible that ER-stressed CD1d+ cells exhibit altered self-lipid loading, such that immunogenic self-lipid antigens are presented to and activate iNKT cells in the context of cancer and other forms of sterile inflammation (113).
While iNKT cells are generally thought to augment antitumor immune responses, there exist subsets of NKT cells that exhibit a regulatory phenotype, which in fact might hinder antitumor responses and promote tumor progression. One of the earliest reports suggesting the presence of regulatory NKT cells identified that IL-13 secreted from NKT cells could signal through the IL4R-STAT6 pathways in cytotoxic T lymphocytes and consequently hinder their immunosurveillance of colon carcinoma and fibrosarcoma tumors (114). NKT cell-derived IL-13 can further drive impaired tumor immunosurveillance by inducing TGF-β secretion from a population of myeloid cells (115). These findings led to the identification of a Vα14Jα18− CD1d-restricted NKT population, dubbed type II NKT cells, which regulate and suppress antitumor immunity independent of IL-4, in contrast with the better-characterized Vα14Jα18+ CD1d-restricted iNKT population, or type I iNKT cells, that augment antitumor immunity (79). However, in a murine model of osteosarcoma, CD1d-restricted NKT cells activated an immunoregulatory pathway independent of IL-13, IL4R-STAT6 signaling, and TGF-β, suggesting the existence of an alternative mechanism of NKT-mediated immunoregulation or different subsets of immunoregulatory NKT cells in different tumors (116). The regulatory contribution of type II NKT cells compared to classical Tregs was explored in a murine model of colorectal and renal cancers (117). This work indicated that type II NKT cells and classical Treg cells were equally essential in suppressing antitumor responses (117).
The different cytokine profiles between type I and type II iNKT cells were better characterized in a murine model of B-cell lymphoma, where type I iNKT secreted primarily IFN-γ, and type II iNKT cells secreted TGF-β and IL-13 (78). Furthermore, a balance between the two subsets allowed for adequate tumor immunosurveillance, as demonstrated by the enhanced mortality of tumor-bearing mice that are deficient in type I NKT cells, but retain type II NKT cells (78).
In exploring αGC-induced anergy of iNKT cells, a distinct subset of regulatory iNKT cells was identified, now termed NKT10 cells (118). As the name suggests, this population of NKT cells secretes IL-10 upon antigenic stimulation and is able to increase the tumor burden in mice challenged with B16 melanoma cells (118). In line with these results, a recent report noted that the absence of iNKT cells correlated with reduced number of intestinal polyps in a murine model of colorectal cancer (119). It was shown that IL-10 producing iNKT cells—reminiscent of the NKT10 cell subset—were enriched within polyps (119). Furthermore, these cells lack the NKT cell transcription factor PLZF, in keeping with recent findings that the PLZF is absent in regulatory iNKT cells in adipose tissue, where they secrete IL-2 and IL-10, control the number of classical Tregs, and promote M2 polarization of adipose-resident macrophages (120).
Given their essential role in antitumor responses, iNKT cells are suitable targets for cancer immunotherapy research. Most studies have mainly focused on the adjuvant behavior of iNKT cells, in particular efficient methods of αGC delivery, often in combination with tumor antigens, to trigger an all-encompassing immune response against the tumor. However, more recent studies have focused on harnessing iNKT cells in new, promising cancer immunotherapies.
Although αGC is a naturally occurring iNKT cell agonist, which enhances the adjuvant effect of iNKT cells in cancer, there is a focus on identifying stronger iNKT cell agonists, either by modifying αGC or identifying novel molecules based on medicinal chemistry programs. Such efforts are supported by the structural knowledge of CD1d bound to αGC and of the iNKT-TCR either in isolation or during cognate interaction with CD1d–lipid complexes (29, 121–123). In screening a panel of αGC analogs for enhanced iNKT cell activation, several iNKT cell agonists were characterized that produce a strong Th1 response from iNKT cells. One of such compounds features an aromatic ring (or, more specifically, a phenyl ring) within the acyl tail (124–127) and is currently entering clinical trials as a vaccine adjuvant (128).
Since the polar head group of CD1d-bound lipids is key for recognition by the iNKT-TCR, cellular enzymes that might either catabolize some iNKT-cell agonists or redirect them away from lipid–antigen presentation pathways might in part drive suboptimal iNKT cell responses. This observation has fueled work that led to the identification of a novel class of iNKT cell agonists that possess non-carbohydrate structures coupled to the ceramide moiety (129). One of these compounds, threitol-ceramide (ThrCer), which was shown to be very efficient in augmenting antigen-specific T cell responses and minimizing iNKT cell overstimulation and iNKT cell-dependent DC lysis, is capable of rectifying the deficiencies of αGC (130). Recent results have shown that incorporating the head group of ThrCer into a conformationally more restricted six-membered ring results in significantly more potent non-glycosidic analogs. In particular, Thr-6-Cer (IMM60) was found to promote strong antitumor responses and to induce a more prolonged stimulation of iNKT cells than does the canonical αGC, achieving an enhanced T-cell response at lower concentrations compared with αGC both in vitro, using human iNKT cell lines, and in vivo, using C57BL/6 mice (35). The synthetic non-glycolipid IMM60 is currently entering clinical trials in melanoma and non-small-cell lung cancer patients in combination with anti-PD1 blocking antibodies. In addition, given that the coupling of iNKT cell agonists with PLGA nanoparticles enhances their immune adjuvant potential by orders of magnitude (131), a phase I clinical trial in ovarian cancer and prostate cancer patients will be carried out with IMM60 conjugated to PLGA nanoparticles with full length NY-ESO-1 protein.
Efficient delivery of potent lipid iNKT cell agonists is essential in manipulating the adjuvant effects of iNKT cells. Optimizing delivery methods and combining the stimulatory lipid with tumor-specific antigens are critical to ensure that the adjuvanted immune response is targeted predominantly toward the tumor. In that vein, the use of exosomes as a means of codelivering αGC and ovalbumin has proved highly successful in reducing the tumor burden and increasing survival in mice inoculated with OVA-expressing melanoma compared with injection of soluble αGC and OVA together (132). Since exosomes naturally bear surface markers to direct them to a particular destination—and for this reason are utilized by breast cancer cells themselves to create a “metastatic niche” at a location of future metastasis (133)—they make excellent conduits for delivery of this potential “cancer vaccine” directly to the tumor site, while perhaps protecting the contents from degradation during delivery. Synthetic nanoparticle delivery systems also hold great promise. In a construct similar to exosomes, delivery of αGC or TLR 3 and 7/8 agonists polyI:C and R848, and OVA in biodegradable poly(lactic-co-glycolic acid) nanoparticles proved efficient in stimulating CD8+ antigen-specific T cell responses against OVA-B16 independent of CD4+ T cell help (131). Encapsulation of the contents was essential, as injection of a mixture of αGC, TLR ligands, and OVA did not induce a comparable antitumor T cell response (131).
As previously mentioned, a major pitfall in using glycolipid antigens, specifically αGC, as adjuvants in cancer immunotherapy is the induction of iNKT cell anergy, as defined by reduced IFN-γ secretion upon secondary exposure. The delivery of αGC, typically as an injection of free lipid particles that might be taken up and presented by a variety of antigen-presenting cells, might contribute to this issue. An alternate delivery method would involve the intravenous injection of autologous DCs preloaded with αGC. In a clinical study involving five late-stage cancer patients, injection of αGC-loaded DCs led to the robust proliferation of iNKT cells, sustained IFN-γ secretion, and enhanced antigen-specific CD8+ T-cell expansion ex vivo, as compared to injection of unpulsed DCs (134, 135). It is suggested that the extent of anergy induction is dependent on the type of antigen-presenting cells that present αGC, with B cells reportedly inducing a higher degree of anergy than DCs (135, 136). In addition to the augmented Th1 response upon injection of αGC-loaded DCs (137), perhaps upon secondary stimulation with αGC-pulsed DCs, anergic responses would be reduced. Alternatively, targeting iNKT cell agonists to DCs through nanoparticle formulations has been shown to overcome iNKT cell anergy (138).
The recent emergence of monoclonal antibody therapies to checkpoint regulators has revolutionized the field of cancer immunotherapy, particularly antibodies targeting the PD1–PD1L axis and CTLA4. While these therapies are studied predominantly in the context of CD8+ cytotoxic T cells, iNKT cells are not exempt from their influence. Much like conventional T cells, iNKT cells upregulate PD1 on their cell surface upon activation as means of eventually resolving the immune response (139). Blockade of PD1 using anti-PD1 antibodies injected simultaneously with αGC results in iNKT cell activation and prevents iNKT cell anergy, a common occurrence after potent αGC stimulation (139–141). In fact, blockade of PD1 during αGC-mediated iNKT cell activation in a B16 melanoma mouse model leads to a persistent antimetastatic immune response (139).
Another emerging T cell-based cancer immunotherapy centers on the chimeric antigen receptor (CAR) T cell therapy. CAR T cell therapy works on the principle that genetically engineered CD8+ T cells expressing TCRs specific for a tumor antigen fused to their native CD3 domain or modified with the endodomain of a costimulatory molecule can become activated and expand into a population of tumor-specific CD8+ cytotoxic T cells (142). This approach has recently been applied to iNKT cells (143). A CAR specific for GD2 ganglioside, an abundant neuroblastoma antigen, was expressed in primary human iNKT cells (143). CAR.GD2 iNKT cells took on a Th1 profile and localized directly in the tumor site when transplanted in NSG mice (143). CAR.GD2 iNKT cells were highly cytotoxic against neuroblastoma cells, and when fused with CD28 and 41BB endodomains, they increased long-term survival in a murine model of the disease (143). With conventional T cells, a frequent adverse effect of CAR therapy in NSG mice is that adoptive transfer of the engineered T cells can induce graft-versus-host disease (GVHD) (143). However, there is no evidence of GVHD in in vivo models utilizing CAR.GD2 iNKT cells (143). For this reason, CAR iNKT cells might become an alternative to conventional T cells as vectors for CAR therapy. So far, however, CAR iNKT cell therapy has not been translated into clinical trials due to a poor understanding of the mechanisms underlying their in vivo proliferation and persistence (144). There have been no clear markers to differentiate effector and memory iNKT cells (144). Recently, a subset of iNKT cells that express the adhesion marker CD62L (also found in naïve and central memory T cells) has been identified (144). As expected, this population rapidly expands and can persist upon stimulation (144). In iNKT cells transduced to express a CD19.CAR, it was the CD62L+ population that achieved persistent activation and proliferation in vivo and was responsible for lymphoma and neuroblastoma regression (144).
Recent results have indicated that therapies harnessing iNKT cells seem generally well tolerated by mice and humans. There are still many unanswered questions in the field of iNKT cell therapies that demand full investigation, such as the optimal route of administration, formulation of dosing intervals, etc. Although preclinical studies in animal models may help answer these questions, ultimately, appropriately designed clinical trials in humans will guide protocol optimization. Our ability to manipulate these cells in antitumor therapeutics is critically dependent on our understanding of iNKT cell biology, including the factors that activate and regulate these cells during sterile and non-sterile conditions; the strong immunomodulatory ability of iNKT cells begs the question as to whether their activation in cancer patients, in combination with immune check point inhibitors, can enhance the frequency and quality of neo-antigen tumor-specific CD8+ and CD4+ T cell responses. The identification, optimization, formulation, and clinical use of iNKT cell agonists that promote Th1 immune responses should be a high priority in future clinical trials.
MB wrote the manuscript, while MS and VC contributed to the writing and editing of the text.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the UK Medical Research Council (MRC Human Immunology Unit) and Cancer Research UK (Program Grant #C399/A2291) and by the NIHR Biomedical Research Centre, Oxford.
1. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases – elimination, equilibrium and escape. Curr Opin Immunol (2014) 27:16–25. doi:10.1016/j.coi.2014.01.004
2. Tian Y, Sette A, Weiskopf D. Cytotoxic CD4 T cells: differentiation, function, and application to dengue virus infection. Front Immunol (2016) 7:531. doi:10.3389/fimmu.2016.00531
3. Das S, Chowdhury R, Ghosh S, Das S. A recombinant protein of Salmonella typhi induces humoral and cell-mediated immune responses including memory responses. Vaccine (2017) 35(35 Pt B):4523–31. doi:10.1016/j.vaccine.2017.07.035
4. Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB. The burgeoning family of unconventional T cells. Nat Immunol (2015) 16(11):1114–23. doi:10.1038/ni.3298
5. Salio M, Cerundolo V. Regulation of lipid specific and vitamin specific non-MHC restricted T cells by antigen presenting cells and their therapeutic potentials. Front Immunol (2015) 6:388. doi:10.3389/fimmu.2015.00388
6. Griewank K, Borowski C, Rietdijk S, Wang N, Julien A, Wei DG, et al. Homotypic interactions mediated by Slamf1 and Slamf6 receptors control NKT cell lineage development. Immunity (2007) 27(5):751–62. doi:10.1016/j.immuni.2007.08.020
7. Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol (2013) 13(2):101–17. doi:10.1038/nri3369
9. Berzins SP, Smyth MJ, Baxter AG. Presumed guilty: natural killer T cell defects and human disease. Nat Rev Immunol (2011) 11(2):131–42. doi:10.1038/nri2904
10. Jones EY, Salio M, Cerundolo V. T cell receptors get back to basics. Nat Immunol (2007) 8(10):1033–5. doi:10.1038/ni1007-1033
11. Coquet JM, Chakravarti S, Kyparissoudis K, McNab FW, Pitt LA, McKenzie BS, et al. Diverse cytokine production by NKT cell subsets and identification of an IL-17-producing CD4-NK1.1- NKT cell population. Proc Natl Acad Sci U S A (2008) 105(32):11287–92. doi:10.1073/pnas.0801631105
12. Wesley JD, Tessmer MS, Chaukos D, Brossay L. NK cell-like behavior of Valpha14i NK T cells during MCMV infection. PLoS Pathog (2008) 4(7):e1000106. doi:10.1371/journal.ppat.1000106
13. Stetson DB, Mohrs M, Reinhardt RL, Baron JL, Wang ZE, Gapin L, et al. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J Exp Med (2003) 198(7):1069–76. doi:10.1084/jem.20030630
14. Beyaz S, Kim JH, Pinello L, Xifaras ME, Hu Y, Huang J, et al. The histone demethylase UTX regulates the lineage-specific epigenetic program of invariant natural killer T cells. Nat Immunol (2017) 18(2):184–95. doi:10.1038/ni.3644
15. Kawakami K, Yamamoto N, Kinjo Y, Miyagi K, Nakasone C, Uezu K, et al. Critical role of Valpha14+ natural killer T cells in the innate phase of host protection against Streptococcus pneumoniae infection. Eur J Immunol (2003) 33(12):3322–30. doi:10.1002/eji.200324254
16. Nieuwenhuis EE, Matsumoto T, Exley M, Schleipman RA, Glickman J, Bailey DT, et al. CD1d-dependent macrophage-mediated clearance of Pseudomonas aeruginosa from lung. Nat Med (2002) 8(6):588–93. doi:10.1038/nm0602-588
17. Park SH, Kyin T, Bendelac A, Carnaud C. The contribution of NKT cells, NK cells, and other gamma-chain-dependent non-T non-B cells to IL-12-mediated rejection of tumors. J Immunol (2003) 170(3):1197–201. doi:10.4049/jimmunol.170.3.1197
18. Schmieg J, Yang G, Franck RW, Van Rooijen N, Tsuji M. Glycolipid presentation to natural killer T cells differs in an organ-dependent fashion. Proc Natl Acad Sci U S A (2005) 102(4):1127–32. doi:10.1073/pnas.0408288102
19. Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, et al. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol (2003) 171(10):5140–7. doi:10.4049/jimmunol.171.10.5140
20. Barral P, Polzella P, Bruckbauer A, van Rooijen N, Besra GS, Cerundolo V, et al. CD169(+) macrophages present lipid antigens to mediate early activation of iNKT cells in lymph nodes. Nat Immunol (2010) 11(4):303–12. doi:10.1038/ni.1853
21. Barral P, Eckl-Dorna J, Harwood NE, De Santo C, Salio M, Illarionov P, et al. B cell receptor-mediated uptake of CD1d-restricted antigen augments antibody responses by recruiting invariant NKT cell help in vivo. Proc Natl Acad Sci U S A (2008) 105(24):8345–50. doi:10.1073/pnas.0802968105
22. De Santo C, Salio M, Masri SH, Lee LY, Dong T, Speak AO, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest (2008) 118(12):4036–48. doi:10.1172/JCI36264
23. Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med (2003) 198(2):267–79. doi:10.1084/jem.20030324
24. Galli G, Nuti S, Tavarini S, Galli-Stampino L, De Lalla C, Casorati G, et al. CD1d-restricted help to B cells by human invariant natural killer T lymphocytes. J Exp Med (2003) 197(8):1051–7. doi:10.1084/jem.20021616
25. Galli G, Pittoni P, Tonti E, Malzone C, Uematsu Y, Tortoli M, et al. Invariant NKT cells sustain specific B cell responses and memory. Proc Natl Acad Sci U S A (2007) 104(10):3984–9. doi:10.1073/pnas.0700191104
26. Arora P, Baena A, Yu KO, Saini NK, Kharkwal SS, Goldberg MF, et al. A single subset of dendritic cells controls the cytokine bias of natural killer T cell responses to diverse glycolipid antigens. Immunity (2014) 40(1):105–16. doi:10.1016/j.immuni.2013.12.004
27. Jenkins MK, Moon JJ. The role of naive T cell precursor frequency and recruitment in dictating immune response magnitude. J Immunol (2012) 188(9):4135–40. doi:10.4049/jimmunol.1102661
28. Zajonc DM, Cantu C III, Mattner J, Zhou D, Savage PB, Bendelac A, et al. Structure and function of a potent agonist for the semi-invariant natural killer T cell receptor. Nat Immunol (2005) 6(8):810–8. doi:10.1038/ni1224
29. Koch M, Stronge VS, Shepherd D, Gadola SD, Mathew B, Ritter G, et al. The crystal structure of human CD1d with and without alpha-galactosylceramide. Nat Immunol (2005) 6(8):819–26. doi:10.1038/ni1225
30. Brennan PJ, Tatituri RV, Brigl M, Kim EY, Tuli A, Sanderson JP, et al. Invariant natural killer T cells recognize lipid self antigen induced by microbial danger signals. Nat Immunol (2011) 12(12):1202–11. doi:10.1038/ni.2143
31. Cox D, Fox L, Tian R, Bardet W, Skaley M, Mojsilovic D, et al. Determination of cellular lipids bound to human CD1d molecules. PLoS One (2009) 4(5):e5325. doi:10.1371/journal.pone.0005325
32. Fox LM, Cox DG, Lockridge JL, Wang X, Chen X, Scharf L, et al. Recognition of lyso-phospholipids by human natural killer T lymphocytes. PLoS Biol (2009) 7(10):e1000228. doi:10.1371/journal.pbio.1000228
33. Muindi K, Cernadas M, Watts GF, Royle L, Neville DC, Dwek RA, et al. Activation state and intracellular trafficking contribute to the repertoire of endogenous glycosphingolipids presented by CD1d [corrected]. Proc Natl Acad Sci U S A (2010) 107(7):3052–7. doi:10.1073/pnas.0915056107
34. Yuan W, Kang SJ, Evans JE, Cresswell P. Natural lipid ligands associated with human CD1d targeted to different subcellular compartments. J Immunol (2009) 182(8):4784–91. doi:10.4049/jimmunol.0803981
35. Jukes JP, Gileadi U, Ghadbane H, Yu TF, Shepherd D, Cox LR, et al. Non-glycosidic compounds can stimulate both human and mouse iNKT cells. Eur J Immunol (2016) 46(5):1224–34. doi:10.1002/eji.201546114
36. Kitamura H, Iwakabe K, Yahata T, Nishimura S, Ohta A, Ohmi Y, et al. The natural killer T (NKT) cell ligand alpha-galactosylceramide demonstrates its immunopotentiating effect by inducing interleukin (IL)-12 production by dendritic cells and IL-12 receptor expression on NKT cells. J Exp Med (1999) 189(7):1121–8. doi:10.1084/jem.189.7.1121
37. Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, et al. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science (1997) 278(5343):1623–6. doi:10.1126/science.278.5343.1623
38. Morita M, Motoki K, Akimoto K, Natori T, Sakai T, Sawa E, et al. Structure-activity relationship of alpha-galactosylceramides against B16-bearing mice. J Med Chem (1995) 38(12):2176–87. doi:10.1021/jm00012a018
39. McCarthy C, Shepherd D, Fleire S, Stronge VS, Koch M, Illarionov PA, et al. The length of lipids bound to human CD1d molecules modulates the affinity of NKT cell TCR and the threshold of NKT cell activation. J Exp Med (2007) 204(5):1131–44. doi:10.1084/jem.20062342
40. Van Rhijn I, Godfrey DI, Rossjohn J, Moody DB. Lipid and small-molecule display by CD1 and MR1. Nat Rev Immunol (2015) 15(10):643–54. doi:10.1038/nri3889
41. Mattner J, Debord KL, Ismail N, Goff RD, Cantu C III, Zhou D, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature (2005) 434(7032):525–9. doi:10.1038/nature03408
42. Zhou D, Mattner J, Cantu C III, Schrantz N, Yin N, Gao Y, et al. Lysosomal glycosphingolipid recognition by NKT cells. Science (2004) 306(5702):1786–9. doi:10.1126/science.1103440
43. Porubsky S, Speak AO, Salio M, Jennemann R, Bonrouhi M, Zafarulla R, et al. Globosides but not isoglobosides can impact the development of invariant NKT cells and their interaction with dendritic cells. J Immunol (2012) 189(6):3007–17. doi:10.4049/jimmunol.1201483
44. Porubsky S, Speak AO, Luckow B, Cerundolo V, Platt FM, Grone HJ. Normal development and function of invariant natural killer T cells in mice with isoglobotrihexosylceramide (iGb3) deficiency. Proc Natl Acad Sci U S A (2007) 104(14):5977–82. doi:10.1073/pnas.0611139104
45. Speak AO, Salio M, Neville DC, Fontaine J, Priestman DA, Platt N, et al. Implications for invariant natural killer T cell ligands due to the restricted presence of isoglobotrihexosylceramide in mammals. Proc Natl Acad Sci U S A (2007) 104(14):5971–6. doi:10.1073/pnas.0607285104
46. Paget C, Mallevaey T, Speak AO, Torres D, Fontaine J, Sheehan KC, et al. Activation of invariant NKT cells by toll-like receptor 9-stimulated dendritic cells requires type I interferon and charged glycosphingolipids. Immunity (2007) 27(4):597–609. doi:10.1016/j.immuni.2007.08.017
47. Zeissig S, Murata K, Sweet L, Publicover J, Hu Z, Kaser A, et al. Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nat Med (2012) 18(7):1060–8. doi:10.1038/nm.2811
48. Gumperz JE, Roy C, Makowska A, Lum D, Sugita M, Podrebarac T, et al. Murine CD1d-restricted T cell recognition of cellular lipids. Immunity (2000) 12(2):211–21. doi:10.1016/S1074-7613(00)80174-0
49. Leite-De-Moraes MC, Hameg A, Arnould A, Machavoine F, Koezuka Y, Schneider E, et al. A distinct IL-18-induced pathway to fully activate NK T lymphocytes independently from TCR engagement. J Immunol (1999) 163(11):5871–6.
50. Tyznik AJ, Tupin E, Nagarajan NA, Her MJ, Benedict CA, Kronenberg M. Cutting edge: the mechanism of invariant NKT cell responses to viral danger signals. J Immunol (2008) 181(7):4452–6. doi:10.4049/jimmunol.181.7.4452
51. Torreno-Pina JA, Manzo C, Salio M, Aichinger MC, Oddone A, Lakadamyali M, et al. The actin cytoskeleton modulates the activation of iNKT cells by segregating CD1d nanoclusters on antigen-presenting cells. Proc Natl Acad Sci U S A (2016) 113(6):E772–81. doi:10.1073/pnas.1514530113
52. Crowe NY, Smyth MJ, Godfrey DI. A critical role for natural killer T cells in immunosurveillance of methylcholanthrene-induced sarcomas. J Exp Med (2002) 196(1):119–27. doi:10.1084/jem.20020092
53. Dashtsoodol N, Shigeura T, Ozawa R, Harada M, Kojo S, Watanabe T, et al. Generation of novel Traj18-deficient mice lacking Valpha14 natural killer T cells with an undisturbed T cell receptor alpha-chain repertoire. PLoS One (2016) 11(4):e0153347. doi:10.1371/journal.pone.0153347
54. Kammertoens T, Qin Z, Briesemeister D, Bendelac A, Blankenstein T. B-cells and IL-4 promote methylcholanthrene-induced carcinogenesis but there is no evidence for a role of T/NKT-cells and their effector molecules (Fas-ligand, TNF-alpha, perforin). Int J Cancer (2012) 131(7):1499–508. doi:10.1002/ijc.27411
55. Moreno M, Molling JW, von Mensdorff-Pouilly S, Verheijen RH, Hooijberg E, Kramer D, et al. IFN-gamma-producing human invariant NKT cells promote tumor-associated antigen-specific cytotoxic T cell responses. J Immunol (2008) 181(4):2446–54. doi:10.4049/jimmunol.181.4.2446
56. Silk JD, Hermans IF, Gileadi U, Chong TW, Shepherd D, Salio M, et al. Utilizing the adjuvant properties of CD1d-dependent NK T cells in T cell-mediated immunotherapy. J Clin Invest (2004) 114(12):1800–11. doi:10.1172/JCI22046
57. Fujii S, Shimizu K, Hemmi H, Steinman RM. Innate Valpha14(+) natural killer T cells mature dendritic cells, leading to strong adaptive immunity. Immunol Rev (2007) 220:183–98. doi:10.1111/j.1600-065X.2007.00561.x
58. De Santo C, Arscott R, Booth S, Karydis I, Jones M, Asher R, et al. Invariant NKT cells modulate the suppressive activity of IL-10-secreting neutrophils differentiated with serum amyloid A. Nat Immunol (2010) 11(11):1039–46. doi:10.1038/ni.1942
59. Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest (2009) 119(6):1524–36. doi:10.1172/JCI37869
60. Liu D, Song L, Wei J, Courtney AN, Gao X, Marinova E, et al. IL-15 protects NKT cells from inhibition by tumor-associated macrophages and enhances antimetastatic activity. J Clin Invest (2012) 122(6):2221–33. doi:10.1172/JCI59535
61. Gorini F, Azzimonti L, Delfanti G, Scarfo L, Scielzo C, Bertilaccio MT, et al. Invariant NKT cells contribute to chronic lymphocytic leukemia surveillance and prognosis. Blood (2017) 129(26):3440–51. doi:10.1182/blood-2016-11-751065
62. Bassiri H, Das R, Guan P, Barrett DM, Brennan PJ, Banerjee PP, et al. iNKT cell cytotoxic responses control T-lymphoma growth in vitro and in vivo. Cancer Immunol Res (2014) 2(1):59–69. doi:10.1158/2326-6066.CIR-13-0104
63. Bellone M, Ceccon M, Grioni M, Jachetti E, Calcinotto A, Napolitano A, et al. iNKT cells control mouse spontaneous carcinoma independently of tumor-specific cytotoxic T cells. PLoS One (2010) 5(1):e8646. doi:10.1371/journal.pone.0008646
64. Fallarini S, Paoletti T, Orsi Battaglini N, Lombardi G. Invariant NKT cells increase drug-induced osteosarcoma cell death. Br J Pharmacol (2012) 167(7):1533–49. doi:10.1111/j.1476-5381.2012.02108.x
65. Dhodapkar KM, Cirignano B, Chamian F, Zagzag D, Miller DC, Finlay JL, et al. Invariant natural killer T cells are preserved in patients with glioma and exhibit antitumor lytic activity following dendritic cell-mediated expansion. Int J Cancer (2004) 109(6):893–9. doi:10.1002/ijc.20050
66. Hix LM, Shi YH, Brutkiewicz RR, Stein PL, Wang CR, Zhang M. CD1d-expressing breast cancer cells modulate NKT cell-mediated antitumor immunity in a murine model of breast cancer metastasis. PLoS One (2011) 6(6):e20702. doi:10.1371/journal.pone.0020702
67. Exley MA, Hou R, Shaulov A, Tonti E, Dellabona P, Casorati G, et al. Selective activation, expansion, and monitoring of human iNKT cells with a monoclonal antibody specific for the TCR alpha-chain CDR3 loop. Eur J Immunol (2008) 38(6):1756–66. doi:10.1002/eji.200737389
68. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology (2007) 121(1):1–14. doi:10.1111/j.1365-2567.2007.02587.x
69. Miura S, Kawana K, Schust DJ, Fujii T, Yokoyama T, Iwasawa Y, et al. CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: a possible mechanism for immune evasion by HPV. J Virol (2010) 84(22):11614–23. doi:10.1128/JVI.01053-10
70. Nowak M, Arredouani MS, Tun-Kyi A, Schmidt-Wolf I, Sanda MG, Balk SP, et al. Defective NKT cell activation by CD1d+ TRAMP prostate tumor cells is corrected by interleukin-12 with alpha-galactosylceramide. PLoS One (2010) 5(6):e11311. doi:10.1371/journal.pone.0011311
71. Tiper IV, Webb TJ. Histone deacetylase inhibitors enhance CD1d-dependent NKT cell responses to lymphoma. Cancer Immunol Immunother (2016) 65(11):1411–21. doi:10.1007/s00262-016-1900-z
72. Yang PM, Lin PJ, Chen CC. CD1d induction in solid tumor cells by histone deacetylase inhibitors through inhibition of HDAC1/2 and activation of Sp1. Epigenetics (2012) 7(4):390–9. doi:10.4161/epi.19373
73. Spanoudakis E, Hu M, Naresh K, Terpos E, Melo V, Reid A, et al. Regulation of multiple myeloma survival and progression by CD1d. Blood (2009) 113(11):2498–507. doi:10.1182/blood-2008-06-161281
74. Zaborsky N, Gassner FJ, Asslaber D, Reinthaler P, Denk U, Flenady S, et al. CD1d expression on chronic lymphocytic leukemia B cells affects disease progression and induces T cell skewing in CD8 positive and CD4CD8 double negative T cells. Oncotarget (2016) 7(31):49459–69. doi:10.18632/oncotarget.10372
75. Tiper IV, Temkin SM, Spiegel S, Goldblum SE, Giuntoli RL II, Oelke M, et al. VEGF potentiates GD3-mediated immunosuppression by human ovarian cancer cells. Clin Cancer Res (2016) 22(16):4249–58. doi:10.1158/1078-0432.CCR-15-2518
76. Chong TW, Goh FY, Sim MY, Huang HH, Thike AA, Lim WK, et al. CD1d expression in renal cell carcinoma is associated with higher relapse rates, poorer cancer-specific and overall survival. J Clin Pathol (2015) 68(3):200–5. doi:10.1136/jclinpath-2014-202735
77. Vyth-Dreese FA, Sein J, van de Kasteele W, Dellemijn TA, van den Bogaard C, Nooijen WJ, et al. Lack of anti-tumour reactivity despite enhanced numbers of circulating natural killer T cells in two patients with metastatic renal cell carcinoma. Clin Exp Immunol (2010) 162(3):447–59. doi:10.1111/j.1365-2249.2010.04274.x
78. Renukaradhya GJ, Khan MA, Vieira M, Du W, Gervay-Hague J, Brutkiewicz RR. Type I NKT cells protect (and type II NKT cells suppress) the host’s innate antitumor immune response to a B-cell lymphoma. Blood (2008) 111(12):5637–45. doi:10.1182/blood-2007-05-092866
79. Terabe M, Swann J, Ambrosino E, Sinha P, Takaku S, Hayakawa Y, et al. A nonclassical non-Valpha14Jalpha18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J Exp Med (2005) 202(12):1627–33. doi:10.1084/jem.20051381
80. Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer (2007) 7(12):961–7. doi:10.1038/nrc2254
81. Keller CW, Loi M, Ewert S, Quast I, Theiler R, Gannage M, et al. The autophagy machinery restrains iNKT cell activation through CD1D1 internalization. Autophagy (2017) 13(6):1025–36. doi:10.1080/15548627.2017.1297907
82. Salio M, Puleston DJ, Mathan TS, Shepherd D, Stranks AJ, Adamopoulou E, et al. Essential role for autophagy during invariant NKT cell development. Proc Natl Acad Sci U S A (2014) 111(52):E5678–87. doi:10.1073/pnas.1413935112
83. Pei B, Zhao M, Miller BC, Vela JL, Bruinsma MW, Virgin HW, et al. Invariant NKT cells require autophagy to coordinate proliferation and survival signals during differentiation. J Immunol (2015) 194(12):5872–84. doi:10.4049/jimmunol.1402154
84. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol (1994) 12:991–1045. doi:10.1146/annurev.iy.12.040194.005015
85. Muller S, Ronfani L, Bianchi ME. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med (2004) 255(3):332–43. doi:10.1111/j.1365-2796.2003.01296.x
86. Yamazaki T, Hannani D, Poirier-Colame V, Ladoire S, Locher C, Sistigu A, et al. Defective immunogenic cell death of HMGB1-deficient tumors: compensatory therapy with TLR4 agonists. Cell Death Differ (2014) 21(1):69–78. doi:10.1038/cdd.2013.72
87. Dumitriu IE, Bianchi ME, Bacci M, Manfredi AA, Rovere-Querini P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J Leukoc Biol (2007) 81(1):84–91. doi:10.1189/jlb.0306171
88. Sharma AK, LaPar DJ, Stone ML, Zhao Y, Kron IL, Laubach VE. Receptor for advanced glycation end products (RAGE) on iNKT cells mediates lung ischemia-reperfusion injury. Am J Transplant (2013) 13(9):2255–67. doi:10.1111/ajt.12368
89. Askenase PW, Itakura A, Leite-de-Moraes MC, Lisbonne M, Roongapinun S, Goldstein DR, et al. TLR-dependent IL-4 production by invariant Valpha14+Jalpha18+ NKT cells to initiate contact sensitivity in vivo. J Immunol (2005) 175(10):6390–401. doi:10.4049/jimmunol.175.10.6390
90. Salio M, Speak AO, Shepherd D, Polzella P, Illarionov PA, Veerapen N, et al. Modulation of human natural killer T cell ligands on TLR-mediated antigen-presenting cell activation. Proc Natl Acad Sci U S A (2007) 104(51):20490–5. doi:10.1073/pnas.0710145104
91. Raftery MJ, Winau F, Giese T, Kaufmann SH, Schaible UE, Schonrich G. Viral danger signals control CD1d de novo synthesis and NKT cell activation. Eur J Immunol (2008) 38(3):668–79. doi:10.1002/eji.200737233
92. Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L, et al. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med (2010) 182(6):774–83. doi:10.1164/rccm.201003-0359OC
93. Rodrigues RJ, Tome AR, Cunha RA. ATP as a multi-target danger signal in the brain. Front Neurosci (2015) 9:148. doi:10.3389/fnins.2015.00148
94. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature (2014) 509(7500):310–7. doi:10.1038/nature13085
95. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol (2016) 16(3):177–92. doi:10.1038/nri.2016.4
96. Bastid J, Regairaz A, Bonnefoy N, Dejou C, Giustiniani J, Laheurte C, et al. Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res (2015) 3(3):254–65. doi:10.1158/2326-6066.CIR-14-0018
97. Subramanian M, Kini R, Madasu M, Ohta A, Nowak M, Exley M, et al. Extracellular adenosine controls NKT-cell-dependent hepatitis induction. Eur J Immunol (2014) 44(4):1119–29. doi:10.1002/eji.201343866
98. Nowak M, Lynch L, Yue S, Ohta A, Sitkovsky M, Balk SP, et al. The A2aR adenosine receptor controls cytokine production in iNKT cells. Eur J Immunol (2010) 40(3):682–7. doi:10.1002/eji.200939897
99. Xu X, Pocock GM, Sharma A, Peery SL, Fites JS, Felley L, et al. Human iNKT cells promote protective inflammation by inducing oscillating purinergic signaling in monocyte-derived DCs. Cell Rep (2016) 16(12):3273–85. doi:10.1016/j.celrep.2016.08.061
100. Felley LE, Sharma A, Theisen E, Romero-Masters JC, Sauer JD, Gumperz JE. Human invariant NKT cells induce IL-1beta secretion by peripheral blood monocytes via a P2X7-independent pathway. J Immunol (2016) 197(6):2455–64. doi:10.4049/jimmunol.1600790
101. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol (2010) 10(12):826–37. doi:10.1038/nri2873
102. Hromadnikova I, Li S, Kotlabova K, Dickinson AM. Influence of in vitro IL-2 or IL-15 alone or in combination with Hsp 70 derived 14-mer peptide (TKD) on the expression of NK cell activatory and inhibitory receptors on peripheral blood T cells, B cells and NKT cells. PLoS One (2016) 11(3):e0151535. doi:10.1371/journal.pone.0151535
103. Nieda M, Nicol A, Koezuka Y, Kikuchi A, Lapteva N, Tanaka Y, et al. TRAIL expression by activated human CD4(+)V alpha 24NKT cells induces in vitro and in vivo apoptosis of human acute myeloid leukemia cells. Blood (2001) 97(7):2067–74. doi:10.1182/blood.V97.7.2067
104. Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer (2008) 8(10):782–98. doi:10.1038/nrc2465
105. Fisher-Owens SA, Gansky SA, Platt LJ, Weintraub JA, Soobader MJ, Bramlett MD, et al. Influences on children’s oral health: a conceptual model. Pediatrics (2007) 120(3):e510–20. doi:10.1542/peds.2006-3084
106. Smyth MJ, Cretney E, Takeda K, Wiltrout RH, Sedger LM, Kayagaki N, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J Exp Med (2001) 193(6):661–70. doi:10.1084/jem.193.6.661
107. de Bruyn M, Wiersma VR, Helfrich W, Eggleton P, Bremer E. The ever-expanding immunomodulatory role of calreticulin in cancer immunity. Front Oncol (2015) 5:35. doi:10.3389/fonc.2015.00035
108. Fucikova J, Truxova I, Hensler M, Becht E, Kasikova L, Moserova I, et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood (2016) 128(26):3113–24. doi:10.1182/blood-2016-08-731737
109. Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F, et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ (2012) 19(12):2003–14. doi:10.1038/cdd.2012.90
110. Kang YJ, Bang BR, Han KH, Hong L, Shim EJ, Ma J, et al. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat Commun (2015) 6:8371. doi:10.1038/ncomms9371
111. Bettigole SE, Glimcher LH. Endoplasmic reticulum stress in immunity. Annu Rev Immunol (2015) 33:107–38. doi:10.1146/annurev-immunol-032414-112116
112. Dougan SK, Salas A, Rava P, Agyemang A, Kaser A, Morrison J, et al. Microsomal triglyceride transfer protein lipidation and control of CD1d on antigen-presenting cells. J Exp Med (2005) 202(4):529–39. doi:10.1084/jem.20050183
113. Wang S, Chen Z, Lam V, Han J, Hassler J, Finck BN, et al. IRE1alpha-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab (2012) 16(4):473–86. doi:10.1016/j.cmet.2012.09.003
114. Terabe M, Matsui S, Noben-Trauth N, Chen H, Watson C, Donaldson DD, et al. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat Immunol (2000) 1(6):515–20. doi:10.1038/82771
115. Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med (2003) 198(11):1741–52. doi:10.1084/jem.20022227
116. Terabe M, Khanna C, Bose S, Melchionda F, Mendoza A, Mackall CL, et al. CD1d-restricted natural killer T cells can down-regulate tumor immunosurveillance independent of interleukin-4 receptor-signal transducer and activator of transcription 6 or transforming growth factor-beta. Cancer Res (2006) 66(7):3869–75. doi:10.1158/0008-5472.CAN-05-3421
117. Izhak L, Ambrosino E, Kato S, Parish ST, O’Konek JJ, Weber H, et al. Delicate balance among three types of T cells in concurrent regulation of tumor immunity. Cancer Res (2013) 73(5):1514–23. doi:10.1158/0008-5472.CAN-12-2567
118. Sag D, Krause P, Hedrick CC, Kronenberg M, Wingender G. IL-10-producing NKT10 cells are a distinct regulatory invariant NKT cell subset. J Clin Invest (2014) 124(9):3725–40. doi:10.1172/JCI72308
119. Wang Y, Sedimbi S, Lofbom L, Singh AK, Porcelli SA, Cardell SL. Unique invariant natural killer T cells promote intestinal polyps by suppressing TH1 immunity and promoting regulatory T cells. Mucosal Immunol (2017). doi:10.1038/mi.2017.34
120. Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat Immunol (2015) 16(1):85–95. doi:10.1038/ni.3047
121. Gadola SD, Koch M, Marles-Wright J, Lissin NM, Shepherd D, Matulis G, et al. Structure and binding kinetics of three different human CD1d-alpha-galactosylceramide-specific T cell receptors. J Exp Med (2006) 203(3):699–710. doi:10.1084/jem.20052369
122. Borg NA, Wun KS, Kjer-Nielsen L, Wilce MC, Pellicci DG, Koh R, et al. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature (2007) 448(7149):44–9. doi:10.1038/nature05907
123. Wun KS, Cameron G, Patel O, Pang SS, Pellicci DG, Sullivan LC, et al. A molecular basis for the exquisite CD1d-restricted antigen specificity and functional responses of natural killer T cells. Immunity (2011) 34(3):327–39. doi:10.1016/j.immuni.2011.02.001
124. Hung JT, Huang JR, Yu AL. Tailored design of NKT-stimulatory glycolipids for polarization of immune responses. J Biomed Sci (2017) 24(1):22. doi:10.1186/s12929-017-0325-0
125. Chang YJ, Huang JR, Tsai YC, Hung JT, Wu D, Fujio M, et al. Potent immune-modulating and anticancer effects of NKT cell stimulatory glycolipids. Proc Natl Acad Sci U S A (2007) 104(25):10299–304. doi:10.1073/pnas.0703824104
126. Aspeslagh S, Li Y, Yu ED, Pauwels N, Trappeniers M, Girardi E, et al. Galactose-modified iNKT cell agonists stabilized by an induced fit of CD1d prevent tumour metastasis. EMBO J (2011) 30(11):2294–305. doi:10.1038/emboj.2011.145
127. Huang JR, Tsai YC, Chang YJ, Wu JC, Hung JT, Lin KH, et al. alpha-Galactosylceramide but not phenyl-glycolipids induced NKT cell anergy and IL-33-mediated myeloid-derived suppressor cell accumulation via upregulation of egr2/3. J Immunol (2014) 192(4):1972–81. doi:10.4049/jimmunol.1302623
128. Tefit JN, Crabe S, Orlandini B, Nell H, Bendelac A, Deng S, et al. Efficacy of ABX196, a new NKT agonist, in prophylactic human vaccination. Vaccine (2014) 32(46):6138–45. doi:10.1016/j.vaccine.2014.08.070
129. Tashiro T, Sekine-Kondo E, Shigeura T, Nakagawa R, Inoue S, Omori-Miyake M, et al. Induction of Th1-biased cytokine production by alpha-carba-GalCer, a neoglycolipid ligand for NKT cells. Int Immunol (2010) 22(4):319–28. doi:10.1093/intimm/dxq012
130. Silk JD, Salio M, Reddy BG, Shepherd D, Gileadi U, Brown J, et al. Cutting edge: nonglycosidic CD1d lipid ligands activate human and murine invariant NKT cells. J Immunol (2008) 180(10):6452–6. doi:10.4049/jimmunol.180.10.6452
131. Dolen Y, Kreutz M, Gileadi U, Tel J, Vasaturo A, van Dinther EA, et al. Co-delivery of PLGA encapsulated invariant NKT cell agonist with antigenic protein induce strong T cell-mediated antitumor immune responses. Oncoimmunology (2016) 5(1):e1068493. doi:10.1080/2162402X.2015.1068493
132. Gehrmann U, Hiltbrunner S, Georgoudaki AM, Karlsson MC, Naslund TI, Gabrielsson S. Synergistic induction of adaptive antitumor immunity by codelivery of antigen with alpha-galactosylceramide on exosomes. Cancer Res (2013) 73(13):3865–76. doi:10.1158/0008-5472.CAN-12-3918
133. Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature (2015) 527(7578):329–35. doi:10.1038/nature15756
134. Chang DH, Osman K, Connolly J, Kukreja A, Krasovsky J, Pack M, et al. Sustained expansion of NKT cells and antigen-specific T cells after injection of alpha-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J Exp Med (2005) 201(9):1503–17. doi:10.1084/jem.20042592
135. Carreno LJ, Saavedra-Avila NA, Porcelli SA. Synthetic glycolipid activators of natural killer T cells as immunotherapeutic agents. Clin Transl Immunology (2016) 5(4):e69. doi:10.1038/cti.2016.14
136. Parekh VV, Wilson MT, Olivares-Villagomez D, Singh AK, Wu L, Wang CR, et al. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J Clin Invest (2005) 115(9):2572–83. doi:10.1172/JCI24762
137. Fujii S, Shimizu K, Kronenberg M, Steinman RM. Prolonged IFN-gamma-producing NKT response induced with alpha-galactosylceramide-loaded DCs. Nat Immunol (2002) 3(9):867–74. doi:10.1038/ni827
138. Thapa P, Zhang G, Xia C, Gelbard A, Overwijk WW, Liu C, et al. Nanoparticle formulated alpha-galactosylceramide activates NKT cells without inducing anergy. Vaccine (2009) 27(25–26):3484–8. doi:10.1016/j.vaccine.2009.01.047
139. Durgan K, Ali M, Warner P, Latchman YE. Targeting NKT cells and PD-L1 pathway results in augmented anti-tumor responses in a melanoma model. Cancer Immunol Immunother (2011) 60(4):547–58. doi:10.1007/s00262-010-0963-5
140. Parekh VV, Lalani S, Kim S, Halder R, Azuma M, Yagita H, et al. PD-1/PD-L blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant NKT cells. J Immunol (2009) 182(5):2816–26. doi:10.4049/jimmunol.0803648
141. Chang WS, Kim JY, Kim YJ, Kim YS, Lee JM, Azuma M, et al. Cutting edge: programmed death-1/programmed death ligand 1 interaction regulates the induction and maintenance of invariant NKT cell anergy. J Immunol (2008) 181(10):6707–10. doi:10.4049/jimmunol.181.10.6707
142. Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer (2016) 16(9):566–81. doi:10.1038/nrc.2016.97
143. Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood (2014) 124(18):2824–33. doi:10.1182/blood-2013-11-541235
Keywords: invariant natural killer T cells, CD1d molecules, tumor immunology, innate immune response, lipid antigens
Citation: Bedard M, Salio M and Cerundolo V (2017) Harnessing the Power of Invariant Natural Killer T Cells in Cancer Immunotherapy. Front. Immunol. 8:1829. doi: 10.3389/fimmu.2017.01829
Received: 01 September 2017; Accepted: 04 December 2017;
Published: 18 December 2017
Edited by:
Paolo Dellabona, Scientific Institute San Raffaele (IRCCS), ItalyReviewed by:
Sergio Abrignani, Istituto Nazionale Genetica Molecolare (INGM), ItalyCopyright: © 2017 Bedard, Salio and Cerundolo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincenzo Cerundolo, dmluY2Vuem8uY2VydW5kb2xvQGltbS5veC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.