
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 13 December 2017
Sec. Inflammation
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01806
This article is part of the Research Topic Sexual Dimorphism of the Immune Inflammatory Response in Infectious and Non-infectious Diseases View all 9 articles
 Mustapha Chamekh1*
Mustapha Chamekh1* Maud Deny1
Maud Deny1 Marta Romano2
Marta Romano2 Nicolas Lefèvre3,4Francis Corazza3Jean Duchateau1
Nicolas Lefèvre3,4Francis Corazza3Jean Duchateau1 Georges Casimir1,4
Georges Casimir1,4
It is widely acknowledged that males and females exhibit contrasting degrees of susceptibility to infectious and non-infectious inflammatory diseases. This is particularly observed in respiratory diseases where human males are more likely to be affected by infection-induced acute inflammations compared to females. The type and magnitude of the innate immune inflammatory response play a cardinal role in this sex bias. Animal models mimicking human respiratory diseases have been used to address the biological factors that could explain the distinct outcomes. In this review, we focus on our current knowledge about experimental studies investigating sex-specific differences in infection-induced respiratory diseases and we provide an update on the most important innate immune mechanisms that could explain sex bias of the inflammatory response. We also discuss whether conclusions drawn from animal studies could be relevant to human.
Respiratory infections caused by a wide range of microbial pathogens are the most common diseases affecting humans worldwide leading to high rates of hospitalization in childhood and elderly. Human susceptibility to pneumoniae is clearly affected by sex as demonstrated by numerous clinical studies (1–4). We have previously reviewed this clinical aspect, so it will not be explored in further detail here (4). It is becoming clear that the severity of inflammatory symptoms vary between males and females according to the pathogen species and the type and magnitude of the inflammatory response triggered along the infection. Although there is an accumulating evidence for sex-specific differences in susceptibility to number of infectious respiratory diseases, the mechanisms at work are still scarce. Studies on murine infection models which are free of many confounders may enhance our understanding of the underlying biological factors. There are few animal studies in general and those affecting lung tissues in particular where sex impact on infection course was taken into account. In this review, we will provide an overview of infectious models used to study the sex effect on the disease severity and we will summarize the most important innate immune factors that could account for sex differences in innate immune inflammatory response. We will discuss their relevance to human and we will highlight the gaps and perspectives regarding future research developments.
Differences in susceptibility to lung infectious diseases between males and females have been shown in mouse models using varying clinically relevant microorganisms. It has been shown that intranasal inoculation of mice with the bacteria Streptococcus pneumoniae, the main etiological agent of pneumonia, results in a higher susceptibility of males compared to females (5). This was correlated with an early sharp increase of the multiplication of the bacteria in the lung, although the overall bacterial burden in males and females was similar. Males exhibited higher inflammatory response with massive infiltration of neutrophils within pulmonary tissues and increased levels of cytokines and chemokines such as IL-17A, CXCL1, and CXCL2. Likewise, in an infectious model of the pathogenic bacteria Klebsiella pneumoniae, male mice were also more susceptible than females (6). The same trend in favor of females was observed in murine infection by Mycoplasma pulmonis using mice of different genetic background (7). Higher mortality was observed in males compared to females and this was associated with dense inflammatory cell infiltrates within pulmonary alveoli in males. However, there was no significant difference in the number of Mycoplasma recovered from the lung or in the serum anti-Mycoplasma IgM response between males and females. Studies in murine models of mycobacterial infections have also shown sex bias with males being more severely affected than females [reviewed in Ref. (8)].
There are, however, some studies reporting that the advantage of females in controlling the infection and inflammation does not apply in certain infectious models. For instance, female mice infected with influenza virus had greatest impairment in the lung physiological function and produced higher levels of interferon (IFN)-γ and MCP-1 in bronchoalveolar lavage (BAL) fluids when compared to male mice, yet, no difference could be detected in the viral titer within the lung or in the BAL inflammatory cell recruitment (9). Female mice were also found to be more affected by a challenge with the opportunistic pathogen Pseudomonas aeruginosa than male mice, in the sense that a higher amount of bacteria and an increased level of expression of TNF-α and CXCL1 were observed within female lung tissues (10). However, these differences in bacteria burden and inflammatory response did not impact the survival rates between males and females and no sex difference could be seen regarding the numbers of polymorphonuclear cells, macrophages, and lymphocytes in the BAL fluids.
Collectively, these animal studies demonstrated a sexual dimorphism in the severity of pneumonia caused by various respiratory pathogens. In most cases, the severity of symptoms was found to correlate with a strong innate immune response triggered at the early phase of infection, but not to the overall number of invading microorganisms. Worth noting, the sex bias has been observed in different mouse strains, although most of the studies have used the C57BL/6 background (5–7, 9, 10). The clearance of invasive pathogens relies on the inflammatory and protective immune response whose magnitude should be tightly controlled, yet in males, the induced inflammatory response seems to be excessive and deleterious. These studies are consistent with the finding that human males are a risk factor for a number of infectious diseases. For example, in human tuberculosis, the gender bias is clearly established with a male:female ratio of 1.6:1 reported for 2015 (WHO Global tuberculosis report 2016). As illustrated in a number of infectious respiratory diseases, men are more likely to develop severe airway inflammatory symptoms consisting mainly on polymorphonuclear neutrophil accumulation and elevated expression of cytokines and chemokines including IL-8, TNF-α, and IL-1β (1–4, 11–13). Females generally had more favorable outcome, particularly when virulent pathogens are endowed with high inflammatory potential as is frequently the case of bacterial respiratory pathogens. Whether males or females are more affected relies not only on the pathogen species and its inflammatory potential but also on specific features of the host including age and genetic background.
The immune mechanisms underlying sex differences in susceptibility to infectious inflammatory diseases have not been fully delineated. The cause of this bias is probably multifactorial and includes sexual hormones and genetic background. We will not explore in the current review the role of sex steroids as this has been largely reviewed elsewhere (8, 14–16). Sex differences in many infectious and non-infectious inflammatory diseases are observed in all age groups including premature infants; therefore, one can consider that sexual hormones cannot fully explain this sex bias (4). We will focus on evidence highlighting the potent effect of TLRs and X chromosome-linked genes explored in different inflammatory settings.
The control of the pathogen dissemination relies on an immediate inflammatory response triggered by the innate immune system. The initiation of the innate immunity is achieved through the recognition of molecular structures broadly shared among various microorganisms (pathogen-associated molecular patterns) by specific receptors (pathogen-recognition receptors, PRRs) expressed on innate cells. TLRs are examples of PRRs allowing microbial sensing by immune cells. Upon ligation with specific pathogen motifs, i.e., lipopolysaccharides (LPS) for TLR4 and lipopeptides for TLR2, TLR transmits signals through adaptor proteins like MyD88 which recruit different kinases including IL-1 receptor-associated kinases and, therefore, lead to NF-κB and MAP kinases activation and induction of inflammatory cytokines and chemokines (17). Differences in the level of TLR4 and TLR2 expression between males and females have been suggested as a mechanism that could partially explain sex differences in inflammatory response. For instance, it has been shown that macrophages from male mice expressed higher levels of TLR4 compared to females following LPS endotoxic shock, hence contributing to excessive and deleterious inflammatory cytokine production in males (18, 19). In coxsackievirus infection model, increased expression of TLR4 has been also observed on splenic monocytes, dendritic cells, and CD3+, CD4+ lymphocytes from males compared to females, suggesting its potential implication in the disease severity (20). In this infectious model, resistance of female mice to coxsackievirus was found to correlate with higher TLR2 expression (20). However, there are some divergent reports showing either no difference in TLR4 expression on macrophages between males and females (21) or increased expression of TLR4 in female mice (22). The latter study argued that despite the higher expression of TLR4 in females, the magnitude of the inflammatory response is likely counterbalanced by resident immunomodulatory CD4+ T-lymphocytes that are more prevalent in resting tissues of females than males (22). Whether the level of expression of TLR4 or TLR2 in males and females could have a significant impact in the sex bias of the inflammatory response needs further investigations. On the other hand, studies on endosomal TLR9 and TLR7 have shown their potent implication in sex differences in innate immunity. TLR9 sense non-methylated CpG-containing microbial genomic DNA and induce the production of type I IFNs that are crucial in promoting a protective immunity. A low level of expression of TLR9 was suggested as a factor contributing to the higher susceptibility of female mice to cytomegalovirus infection (23). Since TLR7/8 are among innate genes located on the X chromosome, evidence of their potential contribution will be summarized in the next paragraph.
A number of innate immune genes are located on the X chromosome both in human and mice and this may have significant consequences on their expression in males and females. Because males have one X chromosome, and females have two, one of the X chromosomes is randomly inactivated in females to assure an equal gene expression with males. Consequently, females have mosaic cells expressing two X-linked gene alleles, which is considered as a great advantage to cope with genetic diseases associated with recessive mutations occurring on the X chromosome (24). Among X-linked genes, about 15% escape inactivation and 10% have variable degree of inactivation (25). This may lead to an overexpression of some X-linked genes in females and if innate immune genes are affected by this silencing escape, it can result in a differential innate immune response between males and females. Females have, therefore, an advantageous genetic diversity that may explain their improved survival from number of infectious inflammatory diseases.
The chromosome X contains TLR7/8 encoding genes that are crucial in sensing viral single-strand RNAs (ssRNAs) and inducing a protective type I IFN response. It should be noted that TLR7/8 can also recognize ssRNAs from phagosomal Gram-positive bacteria, as shown in conventional dendritic cells (26). The induction of TLR7/8-dependent type I IFN in the early phase of infection is crucial for the host defense against invasive pathogens by promoting a protective inflammatory response (27). Differences in the overall expression of TLR7/8 on innate cells between males and females have not been reported. A study performed in a humanized mouse model has shown that TLR7 ligation resulted in a higher frequency of IFN-α- and TNF-α-producing plasmacytoid dendritic cells (pDCs) in females compared to males (28). In human, there are very few studies that sought to pinpoint how TLRs can influence the innate immune response in men and women. Plasmacytoid DCs from women were shown to exhibit a higher TLR7-mediated IFN-α production when compared to pDCs from men (29). This was attributed to a stronger activation rather than overexpression of TLR7 in females. Considering the important role of pDCs in inducing a protective immunity to viral pathogens, this may have a significant effect on the disease progression in men and women (30). Increased activation of TLR7-dependent immune response was shown, however, to be associated with chronic inflammations in females like in systemic lupus erythematosus (31). On the other hand, different studies have shown that TLR8 gene polymorphism has sex-specific effects in some infectious diseases, like in human tuberculosis where men are more severely affected than women (32, 33).
The cascade of TLR signaling and NF-κB activation involve the recruitment of kinases including IL-1 receptor-associated kinase-1 (IRAK-1) (34). IRAK-1 is the most studied gene regarding the sex bias of the inflammatory response. Interestingly, IRAK-1 is located on the X chromosome and is considered among genes that escape the X inactivation process, which may favor an enhanced NF-κB-dependent gene transcription in females (25). The chromosome X inactivation escape combined with the chromosome X mosaicism may favor an effective innate immune inflammatory response in females leading to a better outcome of infection-induced acute inflammations. The impact of IRAK-1 mosaicism on sex bias of the inflammatory response has been investigated in mice and human. In a mouse model of inflammatory colitis, IRAK-1 was shown to have a sex-specific role in the evolution of the disease (35). Notably, mosaicism of IRAK-1 expression in mice results in an immune cell deficiency leading to an improved sepsis outcome, as in IRAK-1-deficient mice (36). In human, a variant IRAK-1 haplotype with persistent increase of kinase activity was associated with a strong NF-κB activation and a severe inflammation (37–39). Recently, a study on cord blood cells revealed higher expression of IRAK-1 in female neonates compared to males and this was suggested as an immune advantage for females in infection-induced inflammatory diseases (40). Whether the overexpression of IRAK-1 is accompanied by an increased kinase activity was not shown in this study. Provided this is true, how this finding is an immune advantage for female neonates remains to be reconciled with previous reports arguing that the upregulation of IRAK-1 is associated with a severe inflammation while its downregulation improved sepsis (36, 37). This discrepancy may be related to the particular status of the neonatal innate immune cells compared to adults (41).
miRNAs are small non-coding RNAs of about 22 nucleotides. They have emerged in the last decade as key negative regulators of genes implicated in divers biological processes, through binding to and repressing translation of complementary messenger RNA (42, 43). They are transcribed either in intergenic or intronic regions of the genome. While intronic miRNAs are coregulated with their host genes, intergenic miRNAs have independent transcription units (44–46). The role of miRNAs in modulating the immune response is now well recognized (47–49). The aberrant expression of miRNAs has been associated with a broad range of inflammatory diseases (50–54). Noteworthy, X chromosome is highly enriched in genes encoding miRNAs (10% of total miRNAs) compared to Y chromosome, with an order of density twofold higher than on autosomes both in mice and humans (28). Around 50% of identified X-linked miRNAs are shared between human and mice. For instance, number of reports described the crucial role of X-linked miR223 and miR106a in the differentiation of neutrophils and monocytes, the key cell players of the innate immunity at early stages of infection. Studies on miR-223-deficient mice revealed higher numbers of granulocytes and hypermature neutrophils, indicating that miR-223 negatively regulates granulocyte generation and maturation (55, 56). Furthermore, miR223 KO mice were shown to develop severe inflammatory symptoms upon LPS challenge and increased oxidative burst of neutrophils upon infection with Candida albicans (55). A higher susceptibility to Mycobacterium tuberculosis infection associated with excessive neutrophil accumulation within the lung and tissue damages was also observed in miR223 KO versus wild-type mice (57). In this study, miR223 was shown to target CXCL2, a chemokine involved in neutrophil recruitment. In mice aerosolized with LPS, a time-dependent increase of miR223 was correlated with a reduced expression of TNF-α, CXCL1, and CXCL2 (58). In human, miR223 was shown to be involved in the granulopoiesis (59) and patients suffering from sepsis exhibited a marked decrease of miR223 expression (60). All these data point toward the importance of X-linked miR223 in the control of the immune inflammatory response by regulating neutrophil recruitment. On the other hand, Fontana et al. found miR106a to be functionally involved in the negative regulation of monocyte differentiation and maturation (61). It is, therefore, reasonable to speculate that a potent silencing escape affecting X-linked miRNAs like miR223 and miR106a could have a direct impact on the control of the fate lineage determination of hematopoietic progenitors, and consequently, could significantly impact the magnitude of the innate inflammatory response in males and females. Whether X-linked miRNAs are subjected to silencing escape and exhibit a differential expression profile between males and females under inflammatory settings are interesting ways to explore.
Although caution should be made when extrapolating animal studies to human, it is now agreed that the differential outcome frequently observed in infectious respiratory diseases between males and females is likely a consequence of excessive and damaging inflammatory response rather than microbial burden within host tissues. The evidence currently available suggest the potential implication of TLRs and X-linked innate immune genes in this sex bias. However, these studies remain incomplete and sometimes conflicting. More investigations are needed to study not only the actual role of TLR signaling in the sex bias of the innate immune response but also the possible implication of other intracellular PRRs sensing nucleic acids including NOD-like and AIM2-like receptors. As schematically illustrated in Figure 1, some concepts are emerging, yet, the underlying mechanisms remain to be unraveled. Despite the considerable progress in miRNA biology and their role in the regulation of the immune and inflammatory response, their impact on sex differences of inflammatory diseases remains to be elucidated. Sex-biased expression of miRNAs could control directly the differential expression of genes contributing to sexually dimorphic inflammatory response. This stresses the need for systematic studies that aim at determining whether X-linked innate immune genes and miRNAs could be differentially expressed in male and female patients suffering from inflammatory diseases. Also, longitudinal studies are required to contrast the dynamic process of molecular signatures with the evolution of the disease outcome. Such investigations would be a pivotal step toward defining sex-specific biomarkers that are clinically relevant and whose biological function could be explored in experimental models of infection and inflammation.
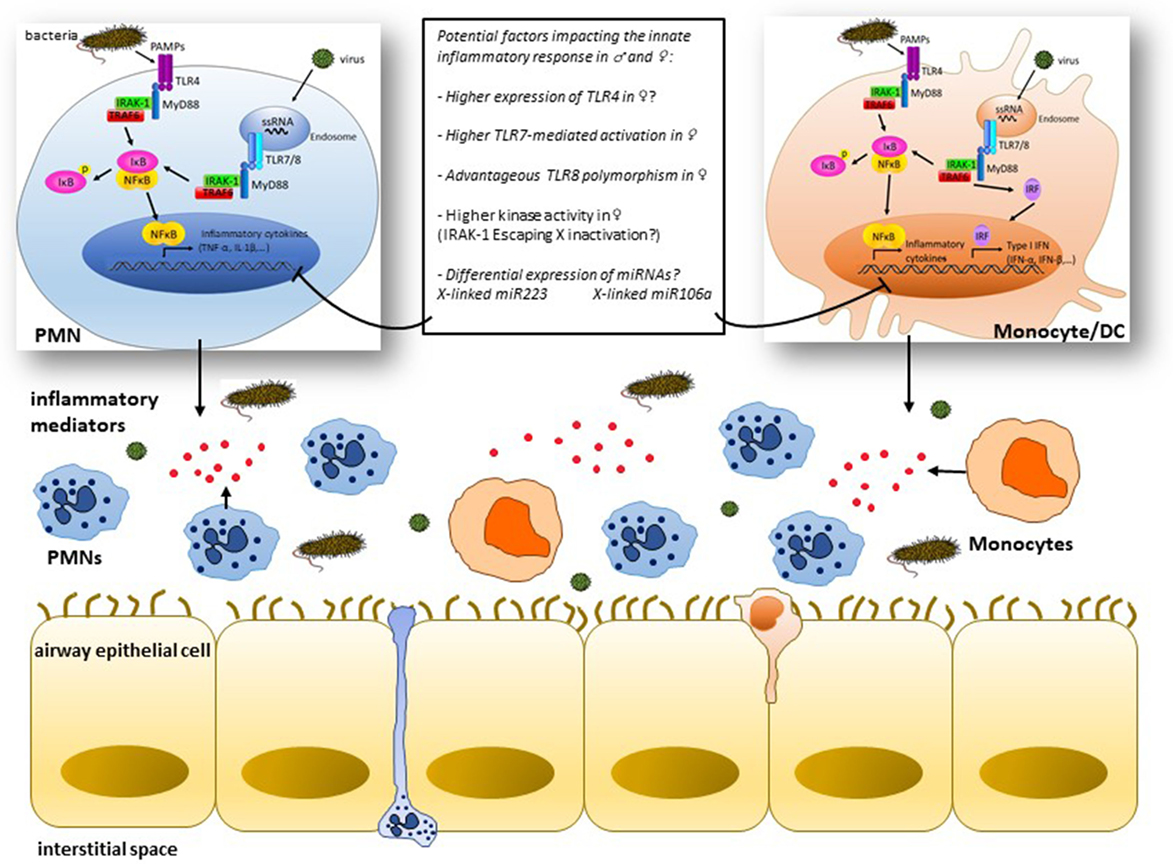
Figure 1. Simplified view of some mechanisms of cell type-specific signaling downstream of toll-like receptors (TLRs) potentially implicated in the sex bias of infection-induced airway inflammation: pathogen-associated molecular patterns (PAMPs) like lipopolysaccharides and single-strand RNA (ssRNA) interact with innate cells, i.e., polymorphonuclear (PMNs) cells and monocytes/dendritic cells through surface expressed TLR4 or endosomal TLR7/8, respectively, leading to the recruitment of adaptor molecules like MyD88 and activation of IL-1 receptor-associated kinase-1 (IRAK-1) among other kinases that culminate through TNF receptor-associated factors (TRAFs) with the activation of the transcription factors NF-κB and interferon regulatory factors (IRFs) and subsequent production of the inflammatory mediators (TNF-α, IL-1β, etc.) and type I interferon (IFN) gene products. Differences in the expression and/or activation of one of the cascade signaling partners between males and females may result in distinct inflammatory responses. Innate genes lying on the X chromosome, e.g., IRAK-1, likely influence the magnitude of the inflammatory response in females through a potent escape from X inactivation process. X chromosome-linked miR223 and miR106a are potentially key miRNAs that may contribute to the contrasting outcome of the inflammatory response in males and females by regulating the innate immune signaling in PMNs and monocytes, respectively.
MC reviewed the literature and drafted the manuscript. MD reviewed the literature and drafted the figure. MR reviewed the literature, reviewed and edited the manuscript. NL, FC, and JD reviewed the manuscript. GC reviewed and edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the Belgian Kids Fund for their valuable support.
1. Gannon CJ, Pasquale M, Tracy JK, McCarter RJ, Napolitano LM. Male gender is associated with increased risk for postinjury pneumonia. Shock (2004) 21:410–4. doi:10.1097/00024382-200405000-00003
2. Gutiérrez F, Masiá M, Mirete C, Soldán B, Rodríguez JC, Padilla S, et al. The influence of age and gender on the population-based incidence of community-acquired pneumonia caused by different microbial pathogens. J Infect (2006) 53:166–74. doi:10.1016/j.jinf.2005.11.006
3. Jensen-Fangel S, Mohey R, Johnsen SP, Andersen PL, Sorensen HT, Ostergaard L. Gender differences in hospitalization rates for respiratory tract infections in Danish youth. Scand J Infect Dis (2004) 36:31–6. doi:10.1080/00365540310017618
4. Casimir GJ, Lefèvre N, Corazza F, Duchateau J. Sex and inflammation in respiratory diseases: a clinical viewpoint. Biol Sex Differ (2013) 4:1–9. doi:10.1186/2042-6410-4-16
5. Kadioglu A, Cuppone AM, Trappetti C, List T, Spreafico A, Pozzi G, et al. Sex-based differences in susceptibility to respiratory and systemic pneumococcal disease in mice. J Infect Dis (2011) 204:1971–9. doi:10.1093/infdis/jir657
6. Mikerov AN, Gan X, Umstead TM, Miller L, Chinchilli VM, Phelps DS, et al. Sex differences in the impact of ozone on survival and alveolar macrophage function of mice after Klebsiella pneumoniae infection. Respir Res (2008) 9(24):1–9. doi:10.1186/1465-9921-9-24
7. Yancey AL, Watson HL, Cartner SC, Simecka JW. Gender is a major factor in determining the severity of Mycoplasma respiratory disease in mice. Infect Immun (2001) 69(5):2865–71. doi:10.1128/IAI.69.5.2865-2871.2001
8. Nhamoyebonde S, Leslie A. Biological differences between the sexes and susceptibility to tuberculosis. J Infect Dis (2014) 15:209. doi:10.1093/infdis/jiu147
9. Larcombe AN, Foong RE, Bozanich EM, Berry LJ, Garratt LW, Gualano RC, et al. Sexual dimorphism in lung function responses to acute influenza A infection. Influenza Other Respir Viruses (2011) 5(5):334–42. doi:10.1111/j.1750-2659.2011.00236.x
10. Guilbault C, Scotland P, Lachance C, Tam M, Keller A, Thompson-Snipes L, et al. Influence of gender and interleukin-10 deficiency on the inflammatory response during lung infection with Pseudomonas aeruginosa in mice. Immunology (2002) 107:297–305. doi:10.1046/j.1365-2567.2002.01508.x
11. Offner PJ, Moore EE, Biffl WL. Male gender is a risk factor for major infections after surgery. Arch Surg (1999) 134:935–40. doi:10.1001/archsurg.134.9.935
12. Migeon BR. Why females are mosaics, X-chromosome inactivation, and sex differences in disease. Gend Med (2007) 4:97–105. doi:10.1016/S1550-8579(07)80024-6
13. Morris JA, Harrison LM. Hypothesis: increased male mortality caused by infection is due to a decrease in heterozygous loci as a result of a single X chromosome. Med Hypotheses (2009) 72:322–4. doi:10.1016/j.mehy.2008.08.027
14. Bouman A, Heineman MJ, Faas MM. Sex hormones and the immune response in humans. Hum Reprod Update (2005) 11(4):411–23. doi:10.1093/humupd/dmi008
15. Fish EN. The X-files in immunity: sex-based differences predispose immune responses. Nat Rev Immunol (2008) 8:737–44. doi:10.1038/nri2394
16. Klein SL, Marriott I, Fish EN. Sex-based differences in immune function and responses to vaccination. Trans R Soc Trop Med Hyg (2015) 109(1):9–15. doi:10.1093/trstmh/tru167
17. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol (2014) 25(5):461. doi:10.3389/fimmu.2014.00461
18. Marriott I, Bost KL, Huet-Hudson YM. Sexual dimorphism in expression of receptors for bacterial lipopolysaccharides in murine macrophages: a possible mechanism for gender-based differences in endotoxic shock susceptibility. J Reprod Immunol (2006) 71(1):12–27. doi:10.1016/j.jri.2006.01.004
19. Marriott I, Huet-Hudson YM. Sexual dimorphism in innate immune responses to infectious organisms. Immunol Res (2006) 34(3):177–92. doi:10.1385/IR:34:3:177
20. Roberts BJ, Dragon JA, Moussawi M, Huber SA. Sex-specific signaling through toll-like receptors 2 and 4 contributes to survival outcome of coxsackievirus B3 infection in C57Bl/6 mice. Biol Sex Differ (2012) 3(1):25. doi:10.1186/2042-6410-3-25
21. Eisenmenger SJ, Wichmann MW, Angele P, Faist E, Hatz R, Chaudry IH, et al. Differences in the expression of LPS-receptors are not responsible for the sex-specific immune response after trauma and hemorrhagic shock. Cell Immunol (2004) 230(1):17–22. doi:10.1016/j.cellimm.2004.08.002
22. Scotland RS, Stables MJ, Madalli S, Watson P, Gilroy DW. Sex differences in resident immune cell phenotype underlie more efficient acute inflammatory responses in female mice. Blood (2011) 118(22):5918–27. doi:10.1182/blood-2011-03-340281
23. Traub S, Demaria O, Chasson L, Serra F, Desnues B, Alexopoulou L. Sex bias in susceptibility to MCMV infection: implication of TLR9. PLoS One (2012) 7(9):e45171. doi:10.1371/journal.pone.0045171
24. Spolarics Z. The X-files of inflammation: cellular mosaicism of X-linked polymorphic genes and the female advantage in the host response to injury and infection. Shock (2007) 27:597–604. doi:10.1097/SHK.0b013e31802e40bd
25. Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature (2005) 434(7031):400–4. doi:10.1038/nature03479
26. Mancuso G, Gambuzza M, Midiri A, Biondo C, Papasergi S, Akira S, et al. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat Immunol (2009) 10(6):587–94. doi:10.1038/ni.1733
27. Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol (2007) 178(5):3126–33. doi:10.4049/jimmunol.178.5.3126
28. Laffont S, Rouquié N, Azar P, Seillet C, Plumas J, Aspord C, et al. X-chromosome complement and estrogen receptor signaling independently contribute to the enhanced TLR7-mediated IFN-α production of plasmacytoid dendritic cells from women. J Immunol (2014) 193(11):5444–52. doi:10.4049/jimmunol.1303400
29. Berghöfer B, Frommer T, Haley G, Fink L, Bein G, Hackstein H. TLR7 ligands induce higher IFN-alpha production in females. J Immunol (2006) 177:2088–96. doi:10.4049/jimmunol.177.4.2088
30. Meier A, Chang JJ, Chan ES, Pollard RB, Sidhu HK, Kulkarni S, et al. Sex differences in the toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med (2009) 15:955–9. doi:10.1038/nm.2004
31. Guo Y, Chai Q, Zhao Y, Li P, Qiao J, Huang J. Increased activation of toll-like receptors-7 and -8 of peripheral blood mononuclear cells and upregulated serum cytokines in patients with pediatric systemic lupus erythematosus. Int J Clin Exp Med (2015) 8(11):20472–80.
32. Davila S, Hibberd ML, Hari Dass R, Wong HE, Sahiratmadja E, Bonnard C, et al. Genetic association and expression studies indicate a role of toll-like receptor 8 in pulmonary tuberculosis. PLoS Genet (2008) 4(10):e1000218. doi:10.1371/journal.pgen.1000218
33. Salie M, Daya M, Lucas LA, Warren RM, van der Spuy GD, van Helden PD, et al. Association of toll-like receptors with susceptibility to tuberculosis suggests sex-specific effects of TLR8 polymorphisms. Infect Genet Evol (2015) 34:221–9. doi:10.1016/j.meegid.2015.07.004
34. Kawagoe T, Sato S, Matsushita K, Kato H, Matsui K, Kumagai Y, et al. Sequential control of toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat Immunol (2008) 9(6):684–91. doi:10.1038/ni.1606
35. Berglund M, Thomas JA, Fredin MF, Melgar S, Hörnquist EH, Hultgren OH. Gender dependent importance of IRAK-1 in dextran sulfate sodium induced colitis. Cell Immunol (2009) 259(1):27–32. doi:10.1016/j.cellimm.2009.05.009
36. Chandra R, Federici S, Németh ZH, Csóka B, Thomas JA, Donnelly R, et al. Cellular mosaicism for X-linked polymorphisms and IRAK1 expression presents a distinct phenotype and improves survival following sepsis. J Leukoc Biol (2014) 95(3):497–507. doi:10.1189/jlb.0713397
37. Arcaroli J, Silva E, Maloney JP, He Q, Svetkauskaite D, Murphy JR, et al. Variant IRAK-1 haplotype is associated with increased nuclear factor-κB activation and worse outcomes in sepsis. Am J Respir Crit Care Med (2006) 173:1335–41. doi:10.1164/rccm.200603-341OC
38. Toubiana J, Courtine E, Pene F, Viallon V, Asfar P, Daubin C, et al. IRAK1 functional genetic variant affects severity of septic shock. Crit Care Med (2010) 38:2287–94. doi:10.1097/CCM.0b013e3181f9f9c7
39. Jacob CO, Zhu J, Armstrong DL, Yan M, Han J, Xin J, et al. Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A (2009) 106(15):6256–61. doi:10.1073/pnas.0901181106
40. O’Driscoll DN, De Santi C, McKiernan PJ, McEneaney V, Molloy EJ, Greene CM. Expression of X-linked toll-like receptor 4 signaling genes in female vs. male neonates. Pediatr Res (2017) 81(5):831–7. doi:10.1038/pr.2017.2
41. Kollmann TR, Levy O, Montgomery RR, Goriely S. Innate immune function by toll-like receptors: distinct responses in newborns and the elderly. Immunity (2012) 37(5):771–83. doi:10.1016/j.immuni.2012.10.014
42. Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature (2008) 455(7209):58–63. doi:10.1038/nature07228
43. Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol (2008) 9:839–45. doi:10.1038/ni.f.209
44. Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, et al. The microprocessor complex mediates the genesis of microRNAs. Nature (2004) 432(7014):235–40. doi:10.1038/nature03120
45. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell (2004) 116(2):281–97. doi:10.1016/S0092-8674(04)00045-5
46. Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature (2007) 448(7149):83–6. doi:10.1038/nature05983
47. Taganov KD, Boldin MP, Baltimore D. MicroRNAs and immunity: tiny players in a big field. Immunity (2007) 26(2):133–7. doi:10.1016/j.immuni.2007.02.005
48. Lodish HF, Zhou B, Liu G, Chen CZ. Micromanagement of the immune system by microRNAs. Nat Rev Immunol (2008) 8(2):120–30. doi:10.1038/nri2252
49. Gantier MP, Sadler AJ, Williams BR. Fine-tuning of the innate immune response by microRNAs. Immunol Cell Biol (2007) 85(6):458–62. doi:10.1038/sj.icb.7100091
50. O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol (2010) 10:111–22. doi:10.1038/nri2708
51. Dai R, Ansar Ahmed S. MicroRNA, a new paradigm for understanding immunoregulation, inflammation, and autoimmune diseases. Transl Res (2011) 157:163–79. doi:10.1016/j.trsl.2011.01.007
52. Alam MM, O’Neill LA. MicroRNAs and the resolution phase of inflammation in macrophages. Eur J Immunol (2011) 41(9):2482–5. doi:10.1002/eji.201141740
53. Shen N, Liang D, Tang Y, de Vries N, Tak PP. MicroRNAs – novel regulators of systemic lupus erythematosus pathogenesis. Nat Rev Rheumatol (2012) 8(12):701–9. doi:10.1038/nrrheum.2012.142
54. Du C, Liu C, Kang J, Zhao G, Ye Z, Huang S, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol (2009) 10(12):1252–9. doi:10.1038/ni.1798
55. Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature (2008) 451:1125–9. doi:10.1038/nature06607
56. Haneklaus M, Gerlic M, O’Neill LAJ, Masters SL. miR-223: infection, inflammation and cancer. J Intern Med (2013) 274(3):215–26. doi:10.1111/joim.12099
57. Dorhoi A, Iannaccone M, Farinacci M, Faé KC, Schreiber J, Moura-Alves P, et al. MicroRNA-223 controls susceptibility to tuberculosis by regulating lung neutrophil recruitment. J Clin Invest (2013) 123:4836–48. doi:10.1172/JCI67604
58. Moschos SA, Williams AE, Perry MM, Birrell MA, Belvisi MG, Lindsay MA. Expression profiling in vivo demonstrates rapid changes in lung microRNA levels following lipopolysaccharide-induced inflammation but not in the anti-inflammatory action of glucocorticoids. BMC Genomics (2007) 8:240. doi:10.1186/1471-2164-8-240
59. Zardo G, Ciolfi A, Vian L, Starnes LM, Billi M, Racanicchi S, et al. Polycombs and microRNA-223 regulate human granulopoiesis by transcriptional control of target gene expression. Blood (2012) 119:4034–46. doi:10.1182/blood-2011-08-371344
60. Wang JF, Yu ML, Yu G, Bian JJ, Deng XM, Wan XJ, et al. Serum miR-146a and miR-223 as potential new biomarkers for sepsis. Biochem Biophys Res Commun (2010) 394(1):184–8. doi:10.1016/j.bbrc.2010.02.145
Keywords: infectious respiratory diseases, sex bias, inflammatory response, X-linked innate genes, miRNAs
Citation: Chamekh M, Deny M, Romano M, Lefèvre N, Corazza F, Duchateau J and Casimir G (2017) Differential Susceptibility to Infectious Respiratory Diseases between Males and Females Linked to Sex-Specific Innate Immune Inflammatory Response. Front. Immunol. 8:1806. doi: 10.3389/fimmu.2017.01806
Received: 30 September 2017; Accepted: 30 November 2017;
Published: 13 December 2017
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Alessandra Mortellaro, Singapore Immunology Network (A*STAR), SingaporeCopyright: © 2017 Chamekh, Deny, Romano, Lefèvre, Corazza, Duchateau and Casimir. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mustapha Chamekh, bWNoYW1la2hAdWxiLmFjLmJl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.