 Lihua Duan
Lihua Duan Xiaoquan Rao
Xiaoquan Rao Zachary Braunstein
Zachary Braunstein Amelia C. Toomey4
Amelia C. Toomey4 Jixin Zhong
Jixin Zhong
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 06 December 2017
Sec. Inflammation
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01734
This article is part of the Research Topic Regulation of Inflammation in Chronic Disease View all 48 articles
The inflammatory bowel diseases (IBDs), including Crohn’s disease (CD) and ulcerative colitis (UC), are chronic inflammatory conditions of the gastrointestinal tract and involve a complicated reciprocity of environmental, genetic, and immunologic factors. Despite substantial advances in the foundational understanding of the immunological pathogenesis of IBD, the detailed mechanism of the pathological progression in IBD remains unknown. In addition to Th1/Th2 cells, whose role in IBD has been previously well defined, recent evidence indicates that Th17 cells and Tregs also play a crucial role in the development of IBD. Diets which contain excess sugars, salt, and fat may also be important actors in the pathogenesis of IBD, which may be the cause of high IBD incidence in western developed and industrialized countries. Up until now, the reason for the variance in prevalence of IBD between developed and developing countries has been unknown. This is partly due to the increasing popularity of western diets in developing countries, which makes the data harder to interpret. The enterocrinins glucagon-like peptides (GLPs), including GLP-1 and GLP-2, exhibit notable benefits on lipid metabolism, atherosclerosis formation, plasma glucose levels, and maintenance of gastric mucosa integrity. In addition to the regulation of nutrient metabolism, the emerging role of GLPs and their degrading enzyme dipeptidyl peptidase-4 (DPP-4) in gastrointestinal diseases has gained increasing attention. Therefore, here we review the function of the DPP-4/GLP axis in IBD.
Inflammatory bowel diseases (IBDs), including Crohn’s disease (CD) and ulcerative colitis (UC), are chronic intestinal inflammatory conditions that might be caused by environmental, genetic, and immunological imbalances (1–3). The clinical treatments for these diseases are very limited and inefficient (4, 5). To develop novel therapeutic strategies for IBD, enormous research has been focused on exploring the detailed mechanism of IBD pathophysiology. Animal models, including trinitrobenzene sulfonic acid (TNBS)-induced experimental colitis, dextran sulfate salt (DSS)-induced colitis, and a number of genetic mouse models (such as IL-10−/−), have been established to study the underlying mechanisms (6).
It is well accepted that dysregulated immune response plays a critical role in colitis (7–10). Tumor necrosis factor-α (TNF-α) is a well-studied cytokine that is implicated in the pathological progression of human IBD. Inhibition of TNF-α activity by anti-TNF-α antibody has been widely used as a clinical treatment for IBD. Studies also indicate a profound role of the Th17/Treg axis in the pathogenesis of IBD (11, 12). Therefore, the immune suppressive drugs which can inhibit the effector T cells immune response and promoting Treg expansion are also being used in IBD patients. However, not all patients exhibit an effective response to this therapy (13, 14). In addition, serious side effects, including infection, anaphylaxis, and malignancy, have been observed during these treatments (15). Therefore, alternative therapeutics are imperative for the treatment of IBD.
Glucagon-like peptides (GLPs), including GLP-1 and GLP-2, are secreted by the endocrine cells in the gut up on nutrient uptake (16–18). Through stimulating the islet β cells to secret insulin, inhibiting gastric emptying, and reducing food ingestion, GLP-1 plays a crucial role in lowering blood glucose and controlling body weight (19, 20). Therefore, GLP-1 was used in human subjects with type 2 diabetes, especially in obese patients with type 2 diabetes (21, 22). In contrast, GLP-2 is used as a therapy for intestinal injury and short bowel syndrome due to its effects of promoting mucosal epithelium expansion, and crypt cell proliferation and improving intestinal adaptation and nutrient absorption (23–27). Because GLPs are degraded by dipeptidyl peptidase-4 (DPP-4) very quickly, resulting in very short half-lives (minutes) in vivo (28–31), the DPP-4 inhibitors have recently gained increasing attention (19, 21).
The role of incretin hormones in bowel disease has not been demonstrated until recently (32, 33). In DSS-induced colitis, the severity of intestinal injury was increased in GLP-1R−/− mice (34). In consistency with this, administration of GLP-2 led to significant improvements in animal weight loss and intestinal inflammation in IL-10-deficient mice, a spontaneous colitis mouse model (35). Here, we will discuss in-depth the actions of DPP-4/GLP axis in IBD.
Glucagon-like peptide-1 exerts pleiotropic function through binding to the GLP-1 receptor and is involved in the development and progression of many diseases (17, 18). The GLP-1 receptor is widely expressed in many organs and tissues, including the endocrine pancreas, gastrointestinal tract, heart, and central nervous system. More recent work has shown that a defect in cellular response to GLP-1, akin to insulin resistance, in combination with a diminishment of GLP-1, has a predominant role in the pathogenesis of patients with T2DM. Exogenous administration of pharmacological doses of GLP-1 receptor agonists have been shown to restore β-cell sensitivity to insulin and induce the secretion of insulin. Impaired incretin response is associated with insulin resistance in both non-diabetic and diabetic individuals (36, 37).
The first two amino-acid residues in the N-terminus of GLP-1 are His–Ala, which causes its susceptibility to DPP-4 degradation. The N-terminal His–Ala residues of GLP-1 are rapidly cleaved by DPP-4 expressed on surrounding tissues, resulting in the inactivation of GLP-1 (38). Exenatide, liraglutide, dulaglutide, albiglutide, and lixisenatide are structurally modified GLP-1 analogs used in the clinical setting, exhibiting relative resistance to the cleavage by DPP-4, and a long-circulating half-life (39). Exogenous GLP-1 administration potently inhibits gastric emptying in rodent and human studies, which favors body weight loss (40). Diabetic patients are prone to develop cardiac disorders; the actions of GLP-1 on cardiac function were investigated (41). Since GLP-1 receptor is widely expressed in the brain, the role of GLP-1 in central nervous system, beyond its regulatory function on glycemic control, was explored (42, 43). Expectedly, GLP-1 possesses a protective effect on neuronal damage by reducing ibotenic acid-induced depletion of choline acetyltransferase immunoreactivity (44). GLP-1 receptor-deficient mice were shown to have defects in cognitive function (45), synaptic plasticity, and memory formation (46), which are recovered by transferring the GLP-1R gene in the hippocampus (47). These data reveal that GLP-1 may have pleiotropic functions in a multitude of diseases. The actions of GLP-1 in IBD will be discussed below.
Glucagon-like peptide-2 is a 33 amino-acid peptide and that is cleaved by DPP-4 in rodents and humans, but with a half-life that is slightly longer than GLP-1 (17). Unlike GLP-1, which plays a role in glucose homeostasis, GLP-2 primarily exerts a potential effect in intestinal weight gain, mucosal development, and intestinal integrity (17, 27). In view of the above-mentioned effects, GLP-2 treatment reduced intestinal inflammation and improved intestinal healing after injury (48, 49). In addition to the benefits in improving intestinal integrity, GLP-2 also exhibits antimicrobial effects by regulating the synthesis and activity of Paneth cell-produced antimicrobial peptides (50). In addition, GLP-2 reduces bacterial invasion by promoting secretory immunoglobulin A (IgA) expression (51). Because GLP-2 receptor is widely expressed on many tissues and cells, physiological effects of GLP-2 beyond the gut have also been reported. Like GLP-1, GLP-2 also regulates the function of central nervous system (52). Activation of GLP-2 receptors can reduce stress-induced depression (53, 54) and improve memory in animal experiments (55). It also plays a substantial role in bone metabolism via reducing bone reabsorption and improving bone mineral density (56). GLP-2 has been shown to improve liver regeneration and enhance lung recovery in mice (57, 58). Thus, GLP-2 reveals potential effects in and out of the gastrointestinal tract.
Recent studies have demonstrated that GLPs exert inflammation regulatory functions in metabolic disease. Administration of GLP-1 markedly reduced the macrophage infiltration and the production of inflammatory cytokines in the adipose tissue in ob/ob mice (59). GLP-1 has also been shown to regulate invariant natural killer T cells (iNKT) and macrophage function in humans (60, 61). Animal study carried out in Glp1r−/− mice suggested that GLP-1 may play a role in maintaining peripheral Treg numbers and suppressing lymphocyte hyperproliferation (62). Although GLP-2 can also blunt inflammatory cytokine production via inhibition of NF-κB activity and ERK phosphorylation (63), enhanced macrophage accumulation was observed in the colon of colitic mice (35). A recent study reported that GLP-1 controls of gut immunity by regulating the intestinal intraepithelial lymphocyte function, leading to a protective role in the DSS-induced colitis (34). In consistency, GLP-2 treatment also reduced pro-inflammatory cytokine protein levels in the IL-10-deficient mouse model of colitis (35). Taken together, GLPs play a crucial role in inflammation regulation and gut disorders.
Dipeptidyl peptidase-4, a type-II integral transmembrane glycoprotein, is best known for its catalytic function. A soluble form of DPP-4, which lacks the cytoplasmic and transmembrane domain, with preserved catalytic activity is also detected in the plasma (38, 64). Although the mechanism of regulation of DPP-4 expression remains unclear, TNF-α has been implicated (65, 66). The primary substrates for DPP-4 are enterocrinins, such as GLP-1, GIP, and GLP-2, which are responsible for glucose metabolism (17, 39, 67). DPP-4 gene-deficient mice show improved postprandial glucose control and are resistant to the progression of obesity and hyperinsulinemia. Inhibition of DPP-4 enzymatic activity with pharmacological agent administration improves glucose tolerance in wild-type mice, but not in DPP-4 knockout mice (68).
In addition to enterocrinins, some chemokines and cytokines could also be cleaved by DPP-4, such as stromal cell-derived factor-1 (SDF-1, also known as CXCL12), G-CSF, IL-3, GM-CSF, and erythropoietin, thereby allowing DPP-4 to regulate immune responses (69). DPP-4 also exerts non-catalytic functions via interacting with adenosine deaminase (ADA), caveolin-1, fibronectin, and CXCR4 (70, 71). The best-known non-catalytic function is the interaction between DPP-4 and ADA, which can act as a co-stimulatory dyad to promote T-cell activation. Our previous work has demonstrated a role of DPP-4 non-enzymatic function in regulating dendritic cell (DC)/macrophage-mediated adipose tissue inflammation in obesity (64). We also showed that long-term DDP-4 inhibition reduces atherosclerosis and inflammation via effects on macrophage migration (CD11b+, CD11c+, and Ly6Chi) (72, 73). Furthermore, in non-obese diabetic (NOD) mice, DPP-4 inhibitors significantly increased the TGF-β levels and Treg expansion (74). Beyond that, our recent study, as well as others, demonstrated that DPP-4 plays a role in the infection of Middle Eastern respiratory syndrome (MERS) virus (75).
BP-lowering and anti-atherosclerotic effects of GLP-1R agonists have been well demonstrated, while the gastrointestinal effects of GLPs are underappreciated. Here, we will discuss the relationship between GLP-1 and inflammation in the gastrointestinal tract. UC patients with colectomy showed a slower release of GLP-1 in response to intake of glucose (76). Consistently, postprandial GLP-1 response was also impaired in patients with ileostomy (77). Yet it was not known whether the colectomy or inflammatory state affects the GLP-1 release in IBD. Subsequent studies demonstrated that although GLP-1r mRNA levels was reduced in samples harvested from inflamed sites of IBD patients and colitis mice (78), GLP-1 levels were increased in sera of IBD patients when compared with healthy controls (79, 80). The defective GLP-1 release in IBD patients with colectomy might be caused by the loss of the colonic endocrine tissue.
Therefore, these data reveal a link between gut inflammation and GLP-1 expression and brings up an emerging question that how GLP-1 is implicated in IBD. To explore this question, some studies were conducted in experimental animal colitis. In T-cell adoptive transfer-induced colitis, the GLP-1 expression in colonic tissue was significantly diminished in SCID mice with adoptive transfer of CD4+ T cell when compared with control mice (81). Furthermore, in DSS-induced colitis, a considerable increase of GLP-1 was detected in colitic mice with DPP-4 inhibitor treatment (82). Notwithstanding alteration of GLP-1 expression in colitis, the exact role of GLP-1in the development of colitis remains unknown, in terms of being beneficial or detrimental. A recent study showed that the GLP-1 analog liraglutide exerts a significant improvement of disease activity endpoints, including colonic tissues histological changes and colon weight/length ratio, which might be due to its role in reducing inflammatory cytokines and chemokines, such as chemokine (C–C motif) ligand 20 (CCL20), IL-33, and IL-22 (78). As has been previously established, CCL20 is a key chemokine for CCR6 + Th17 cells (83), while IL-33 and IL-22 are the representative cytokines for Th2 and Th17 immune responses, respectively (84, 85). In line with above results, GLP-1 in sterically stabilized phospholipid micelles (GLP-1-SSM), showing a long half-life and resistant to DPP-4, markedly alleviated the development of DSS-induced mice colitis by reducing the expression of pro-inflammatory cytokine IL-1β (86). Moreover, intestinal epithelial architecture in a colitis model with GLP-1-SSM administration was significantly improved. In conclusion, GLP-1 might act as a novel therapeutic tool in ameliorating colonic inflammation.
Regarding the inhibition of enterocyte apoptosis and stimulation of crypt cell proliferation, GLP-2 is thought to be associated with tissue repair during injury or infection (17, 23). Therefore, in chemically induced enteritis (48) or vascular-ischemia reperfusion injury (87–89), GLP-2 shows a protective effect based on reducing epithelial barrier damage and lowering bacterial infection. It stands to reason that GLP-2 might be a potential therapeutic target in IBD, a condition characterized by destruction of the gastrointestinal epithelium. In an adoptive CD4+ T-cell transfer model of colitis, the amount of GLP-2 in colon tissue was also further decreased compared with that in normal mice or SCID mice without CD4+ T-cell adoptive transfer (81). However, these results were not duplicated in human IBD samples. A study showed no changes of GLP-2 levels in fasting plasma between IBD patients and controls, which pinpoints L-cell secretion is not altered in the pathogenesis of IBD (90). Nevertheless, the circulating levels of bioactive GLP-2 (1–33) were markedly increased in CD and UC patients (91). The alteration of GLP-2 (1–33) might be due to an adoptive response to intestinal injury, which promotes mucosal epithelium restoration in a self-repair mechanism. The discrepant data might be the causal agent of the different inflammatory conditions, because an increase in GLP-2-immunoreactive L cells was found in remissive status of colitis. Another reason is probably due to the detection reagent which detects all GLP-2 or bioactive GLP-2 (1–33).
Beyond the promotion of crypt cell proliferation and mucosal integrity, GLP-2 also exerts a distinct role in anti-inflammatory actions. To mimic anti-inflammatory therapeutic approaches in humans, a combination of GLP-2 with aminosalicylates (ASAs) or corticosteroids were administrated into mice with DSS-induced colitis, while no synergistic effect was observed. Interestingly, corticosteroid administration prevented the intestinal weight increase when the mice were treated with corticosteroids and GLP-2 (92), while these treatments exhibited a similar anti-inflammatory effect in colonic tissues. However, in TNBS-induced ileitis and DSS-induced colitis, GLP-2 treatment downregulated expression of inflammatory cytokines, including IFN-γ, TNF-α, and IL-1β, while the anti-inflammatory cytokine IL-10 was increased (93). Another report also showed that GLP-2 alleviates the development of colitis through reducing the pro-inflammatory cytokines in IL-10-deficient mouse model. The level of inducible nitric oxide synthase (iNOS), a marker for classically activated macrophage, was reduced in GLP-2-treated mice (35). This suggests that GLP-2 might alter macrophage polarization.
It is noteworthy that chronic colitis is a risk factor for colon cancer. Interestingly, a few reports have shown that exogenous and endogenous GLP-2 is a potential cancer promoter in mice models, although reduced inflammation was also observed (94, 95). This might be resulted from the strong preference of GLP-2 for epithelium proliferation. Therefore, the surveillance of dysplasia and colon cancer must be vigilant in GLP-2 treatment.
Regarding a catalytic function of DPP-4 on GLP-1 and GLP-2, previous studies have demonstrated that DPP-4 can act as an immune regulator via its expression on immune cells and the ability to cleave biologically active chemokines and cytokines. Hence, DPP-4 involvement in the pathogenesis of colitis has been proposed (96). The involvement of DPP-4 might depend on two major pathways: the catalytic function and non-catalytic function (38, 73, 97). Like GLP-2, DPP-4 inhibitors have a proliferative effect on the colonic epithelium (98). It has also been demonstrated that the protective effects of DPP-4 inhibitors in IBD might be a result of increased levels of GLP-1 (82). Notably, plasma GLP-2 levels were increased in response to DPP-4 inhibitor. Thus, the effect on epithelium expansion induced by DPP-4 inhibitor probably relies on the indirect elevation of GLP-2 expression (99). To investigate the influence of DPP-4 in the pathogenesis of DSS-induced colitis, DPP-4-deficient mice were used in DSS treatment, and an increase of myeloperoxidase (MPO) activity and expression of NF-κB p65 subunit in the colonic tissues was observed. Furthermore, an increase in the percentage of splenic CD8+ cells and NKT cells in CD26-deficient mice was observed (100). In keeping with GLP-2-treated mice, DPP-4-deficient mice also showed a significant increase in macrophages when compared with wild-type mice (101). These data reveal a detrimental role of DPP-4 during the development of colitis. Conversely, DPP-4-deficient rats reveal an apparent diminished disease activity index (DAI) in the low-dose DSS-induced colitis, especially in 1% DSS-induced colitis (102). A similar effect was also investigated in DPP-4 inhibitor anagliptin- and ER-319711-treated mice with DSS-induced colitis (98). In addition to ER-319711, anagliptin administration ameliorated the body weight loss and DAI. Additionally, a significantly lower histological score was observed in the anagliptin-treated group (103), which suggests that inhibition of the DPP-4 activity can facilitate the resolution of mucosal damage. Taken together, these findings suggest a complex and dichotomous biology during the development of IBD, which might be due to its multifunction.
Due to the vital role of GLPs in intestinal healing and anti-inflammatory function, a sound understanding of the production, regulation, and function of GLPs and their degrading enzyme DPP-4 will facilitate the treatment of colitis. The potential mechanisms (Figure 1) of DPP-4/GLP axis in the IBD may include the following: (1) GLPs promote the tissue repair of injured epithelium; (2) GLPs regulate T-cell differentiation and functions (e.g., Treg, effector T cells, and intraepithelial lymphocytes); (3) GLPs and DPP-4 regulate the function of innate immune cells such as macrophages and DCs; and (4) suppression of DPP-4 enzymatic activities by pharmacological inhibitors preserves GLP function. Although most studies in this area mainly were carried out on animal models and there are limited clinical trials, a phase-II clinical trial of teduglutide (a GLP-2 analog) observed a remission rate of 55.6% in CD patients (104). To what extent GLPs and DPP-4 contributes to IBD in humans requires further investigation.
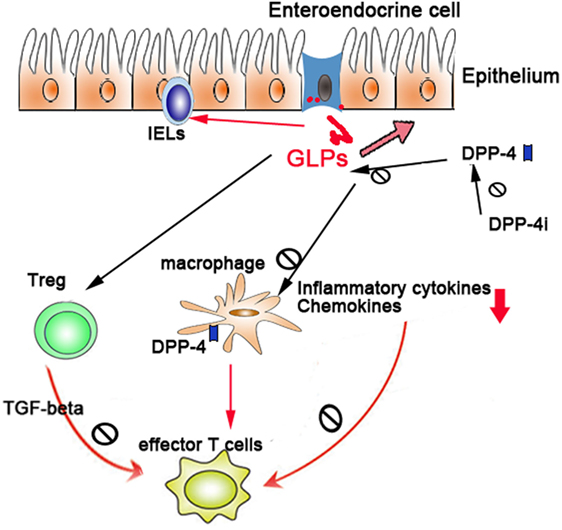
Figure 1. The regulatory role of dipeptidyl peptidase-4 (DPP-4)/glucagon-like peptides (GLPs) in gut immunity: DPP-4/GLP axis regulates gut inflammation through (1) promoting the tissue repair of injured epithelium, (2) regulating the differentiation and functions of Treg and intraepithelial lymphocytes, and (3) regulating the function of macrophages and dendritic cells.
LD and XR reviewed the literature and wrote the first draft. ZB, AT, and JZ reviewed the literature and finalized the manuscript. All authors have read and approved the final manuscript.
JZ is currently receiving a grant from Boehringer Ingelheim (IIS2015-10485). The remaining authors have no conflicts of interest.
This work was supported by grants from NIH (K01 DK105108), American Heart Association (17GRNT33670485), American Association of Immunologists (CIIF-8745), Boehringer Ingelheim (IIS2015-10485), and National Natural Science Foundation of China (81670431 and 81671544).
1. Gensollen T, Iyer SS, Kasper DL, Blumberg RS. How colonization by microbiota in early life shapes the immune system. Science (2016) 352(6285):539–44. doi:10.1126/science.aad9378
2. Uniken Venema WT, Voskuil MD, Dijkstra G, Weersma RK, Festen EA. The genetic background of inflammatory bowel disease: from correlation to causality. J Pathol (2017) 241(2):146–58. doi:10.1002/path.4817
3. Huang Y, Chen Z. Inflammatory bowel disease related innate immunity and adaptive immunity. Am J Transl Res (2016) 8(6):2490–7.
4. Coskun M, Vermeire S, Nielsen OH. Novel targeted therapies for inflammatory bowel disease. Trends Pharmacol Sci (2017) 38(2):127–42. doi:10.1016/j.tips.2016.10.014
5. Korzenik JR, Podolsky DK. Evolving knowledge and therapy of inflammatory bowel disease. Nat Rev Drug Discov (2006) 5(3):197–209. doi:10.1038/nrd1986
6. Elson CO, Sartor RB, Tennyson GS, Riddell RH. Experimental models of inflammatory bowel disease. Gastroenterology (1995) 109(4):1344–67. doi:10.1016/0016-5085(95)90599-5
7. te Velde AA, de Kort F, Sterrenburg E, Pronk I, ten Kate FJ, Hommes DW, et al. Comparative analysis of colonic gene expression of three experimental colitis models mimicking inflammatory bowel disease. Inflamm Bowel Dis (2007) 13(3):325–30. doi:10.1002/ibd.20079
8. Bhan AK, Mizoguchi E, Smith RN, Mizoguchi A. Lessons for human inflammatory bowel disease from experimental models. Curr Opin Gastroenterol (1999) 15(4):285–90. doi:10.1097/00001574-199907000-00002
9. Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol (2003) 3(7):521–33. doi:10.1038/nri1132
10. Neurath MF, Finotto S, Glimcher LH. The role of Th1/Th2 polarization in mucosal immunity. Nat Med (2002) 8(6):567–73. doi:10.1038/nm0602-567
11. Siakavellas SI, Bamias G. Role of the IL-23/IL-17 axis in Crohn’s disease. Discov Med (2012) 14(77):253–62.
12. Omenetti S, Pizarro TT. The Treg/Th17 Axis: a dynamic balance regulated by the gut microbiome. Front Immunol (2015) 6:639. doi:10.3389/fimmu.2015.00639
13. Levin AD, Wildenberg ME, van den Brink GR. Mechanism of action of anti-TNF therapy in inflammatory bowel disease. J Crohns Colitis (2016) 10(8):989–97. doi:10.1093/ecco-jcc/jjw053
14. Palladino MA, Bahjat FR, Theodorakis EA, Moldawer LL. Anti-TNF-alpha therapies: the next generation. Nat Rev Drug Discov (2003) 2(9):736–46. doi:10.1038/nrd1175
15. Gisbert JP, Marin AC, Chaparro M. The risk of relapse after anti-TNF discontinuation in inflammatory bowel disease: systematic review and meta-analysis. Am J Gastroenterol (2016) 111(5):632–47. doi:10.1038/ajg.2016.54
16. Brubaker PL, Drucker DJ. Structure-function of the glucagon receptor family of G protein-coupled receptors: the glucagon, GIP, GLP-1, and GLP-2 receptors. Receptors Channels (2002) 8(3–4):179–88. doi:10.1080/10606820213687
17. Drucker DJ. Biological actions and therapeutic potential of the glucagon-like peptides. Gastroenterology (2002) 122(2):531–44. doi:10.1053/gast.2002.31068
18. Marathe CS, Rayner CK, Jones KL, Horowitz M. Glucagon-like peptides 1 and 2 in health and disease: a review. Peptides (2013) 44:75–86. doi:10.1016/j.peptides.2013.01.014
19. Gallwitz B. Glucagon-like peptide-1-based therapies for the treatment of type 2 diabetes mellitus. Treat Endocrinol (2005) 4(6):361–70. doi:10.2165/00024677-200504060-00005
20. Young A. Inhibition of gastric emptying. Adv Pharmacol (2005) 52:99–121. doi:10.1016/S1054-3589(05)52006-4
21. Gutzwiller JP, Drewe J, Goke B, Schmidt H, Rohrer B, Lareida J, et al. Glucagon-like peptide-1 promotes satiety and reduces food intake in patients with diabetes mellitus type 2. Am J Physiol (1999) 276(5 Pt 2):R1541–4.
22. Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest (1998) 101(3):515–20. doi:10.1172/JCI990
23. Drucker DJ. Glucagon-like peptides: regulators of cell proliferation, differentiation, and apoptosis. Mol Endocrinol (2003) 17(2):161–71. doi:10.1210/me.2002-0306
24. Suri M, Turner JM, Sigalet DL, Wizzard PR, Nation PN, Ball RO, et al. Exogenous glucagon-like peptide-2 improves outcomes of intestinal adaptation in a distal-intestinal resection neonatal piglet model of short bowel syndrome. Pediatr Res (2014) 76(4):370–7. doi:10.1038/pr.2014.97
25. Jeppesen PB, Sanguinetti EL, Buchman A, Howard L, Scolapio JS, Ziegler TR, et al. Teduglutide (ALX-0600), a dipeptidyl peptidase IV resistant glucagon-like peptide 2 analogue, improves intestinal function in short bowel syndrome patients. Gut (2005) 54(9):1224–31. doi:10.1136/gut.2004.061440
26. Jeppesen PB. Clinical significance of GLP-2 in short-bowel syndrome. J Nutr (2003) 133(11):3721–4.
27. Martin GR, Wallace LE, Hartmann B, Holst JJ, Demchyshyn L, Toney K, et al. Nutrient-stimulated GLP-2 release and crypt cell proliferation in experimental short bowel syndrome. Am J Physiol Gastrointest Liver Physiol (2005) 288(3):G431–8. doi:10.1152/ajpgi.00242.2004
28. Mentlein R, Gallwitz B, Schmidt WE. Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7-36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem (1993) 214(3):829–35. doi:10.1111/j.1432-1033.1993.tb17986.x
29. Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes (1995) 44(9):1126–31. doi:10.2337/diab.44.9.1126
30. Brubaker PL, Crivici A, Izzo A, Ehrlich P, Tsai CH, Drucker DJ. Circulating and tissue forms of the intestinal growth factor, glucagon-like peptide-2. Endocrinology (1997) 138(11):4837–43. doi:10.1210/endo.138.11.5482
31. Hartmann B, Harr MB, Jeppesen PB, Wojdemann M, Deacon CF, Mortensen PB, et al. In vivo and in vitro degradation of glucagon-like peptide-2 in humans. J Clin Endocrinol Metab (2000) 85(8):2884–8. doi:10.1210/jcem.85.8.6717
32. Zietek T, Waldschmitt N, Rath E. Role of incretin hormones in bowel diseases. Endocr Dev (2017) 32:49–73. doi:10.1159/000475731
33. Zietek T, Rath E. Inflammation meets metabolic disease: gut feeling mediated by GLP-1. Front Immunol (2016) 7:154. doi:10.3389/fimmu.2016.00154
34. Yusta B, Baggio LL, Koehler J, Holland D, Cao X, Pinnell LJ, et al. GLP-1R agonists modulate enteric immune responses through the intestinal intraepithelial lymphocyte GLP-1R. Diabetes (2015) 64(7):2537–49. doi:10.2337/db14-1577
35. Ivory CP, Wallace LE, McCafferty DM, Sigalet DL. Interleukin-10-independent anti-inflammatory actions of glucagon-like peptide 2. Am J Physiol Gastrointest Liver Physiol (2008) 295(6):G1202–10. doi:10.1152/ajpgi.90494.2008
36. Rask E, Olsson T, Soderberg S, Johnson O, Seckl J, Holst JJ, et al. Impaired incretin response after a mixed meal is associated with insulin resistance in nondiabetic men. Diabetes Care (2001) 24(9):1640–5. doi:10.2337/diacare.24.9.1640
37. Holst JJ, Knop FK, Vilsboll T, Krarup T, Madsbad S. Loss of incretin effect is a specific, important, and early characteristic of type 2 diabetes. Diabetes Care (2011) 34(Suppl 2):S251–7. doi:10.2337/dc11-s227
38. Zhong J, Maiseyeu A, Davis SN, Rajagopalan S. DPP4 in cardiometabolic disease: recent insights from the laboratory and clinical trials of DPP4 inhibition. Circ Res (2015) 116(8):1491–504. doi:10.1161/CIRCRESAHA.116.305665
39. Gupta A, Jelinek HF, Al-Aubaidy H. Glucagon like peptide-1 and its receptor agonists: their roles in management of Type 2 diabetes mellitus. Diabetes Metab Syndr (2017) 11(3):225–30. doi:10.1016/j.dsx.2016.09.003
40. Burcelin R, Gourdy P. Harnessing glucagon-like peptide-1 receptor agonists for the pharmacological treatment of overweight and obesity. Obes Rev (2017) 18(1):86–98. doi:10.1111/obr.12465
41. Waldrop G, Zhong J, Peters M, Rajagopalan S. Incretin-based therapy for diabetes: what a cardiologist needs to know. J Am Coll Cardiol (2016) 67(12):1488–96. doi:10.1016/j.jacc.2015.12.058
42. Muscogiuri G, DeFronzo RA, Gastaldelli A, Holst JJ. Glucagon-like peptide-1 and the central/peripheral nervous system: crosstalk in diabetes. Trends Endocrinol Metab (2017) 28(2):88–103. doi:10.1016/j.tem.2016.10.001
43. Holt MK, Trapp S. The physiological role of the brain GLP-1 system in stress. Cogent Biol (2016) 2(1):1229086. doi:10.1080/23312025.2016.1229086
44. Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther (2002) 302(3):881–8. doi:10.1124/jpet.102.037481
45. Li PC, Liu LF, Jou MJ, Wang HK. The GLP-1 receptor agonists exendin-4 and liraglutide alleviate oxidative stress and cognitive and micturition deficits induced by middle cerebral artery occlusion in diabetic mice. BMC Neurosci (2016) 17(1):37. doi:10.1186/s12868-016-0272-9
46. Cai HY, Wang ZJ, Holscher C, Yuan L, Zhang J, Sun P, et al. Lixisenatide attenuates the detrimental effects of amyloid beta protein on spatial working memory and hippocampal neurons in rats. Behav Brain Res (2017) 318:28–35. doi:10.1016/j.bbr.2016.10.033
47. During MJ, Cao L, Zuzga DS, Francis JS, Fitzsimons HL, Jiao X, et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med (2003) 9(9):1173–9. doi:10.1038/nm919
48. Boushey RP, Yusta B, Drucker DJ. Glucagon-like peptide 2 decreases mortality and reduces the severity of indomethacin-induced murine enteritis. Am J Physiol (1999) 277(5 Pt 1):E937–47.
49. Bulut K, Meier JJ, Ansorge N, Felderbauer P, Schmitz F, Hoffmann P, et al. Glucagon-like peptide 2 improves intestinal wound healing through induction of epithelial cell migration in vitro-evidence for a TGF-beta-mediated effect. Regul Pept (2004) 121(1–3):137–43. doi:10.1016/j.regpep.2004.04.014
50. Lee SJ, Lee J, Li KK, Holland D, Maughan H, Guttman DS, et al. Disruption of the murine Glp2r impairs Paneth cell function and increases susceptibility to small bowel enteritis. Endocrinology (2012) 153(3):1141–51. doi:10.1210/en.2011-1954
51. Chen J, Dong JT, Li XJ, Gu Y, Cheng ZJ, Cai YK. Glucagon-like peptide-2 protects impaired intestinal mucosal barriers in obstructive jaundice rats. World J Gastroenterol (2015) 21(2):484–90. doi:10.3748/wjg.v21.i2.484
52. Brubaker PL, Drucker DJ. Minireview: glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology (2004) 145(6):2653–9. doi:10.1210/en.2004-0015
53. Sasaki-Hamada S, Nakamura R, Nakao Y, Akimoto T, Sanai E, Nagai M, et al. Antidepressant-like effects exerted by the intranasal administration of a glucagon-like peptide-2 derivative containing cell-penetrating peptides and a penetration-accelerating sequence in mice. Peptides (2016) 87:64–70. doi:10.1016/j.peptides.2016.11.013
54. Sasaki-Hamada S, Yuri Y, Hoshi M, Oka JI. Immunohistochemical determination of the site of antidepressant-like effects of glucagon-like peptide-2 in ACTH-treated mice. Neuroscience (2015) 294:156–65. doi:10.1016/j.neuroscience.2015.03.010
55. Iwai T, Jin K, Ohnuki T, Sasaki-Hamada S, Nakamura M, Saitoh A, et al. Glucagon-like peptide-2-induced memory improvement and anxiolytic effects in mice. Neuropeptides (2015) 49:7–14. doi:10.1016/j.npep.2014.11.001
56. Henriksen DB, Alexandersen P, Hartmann B, Adrian CL, Byrjalsen I, Bone HG, et al. Four-month treatment with GLP-2 significantly increases hip BMD: a randomized, placebo-controlled, dose-ranging study in postmenopausal women with low BMD. Bone (2009) 45(5):833–42. doi:10.1016/j.bone.2009.07.008
57. El-Jamal N, Erdual E, Neunlist M, Koriche D, Dubuquoy C, Maggiotto F, et al. Glugacon-like peptide-2: broad receptor expression, limited therapeutic effect on intestinal inflammation and novel role in liver regeneration. Am J Physiol Gastrointest Liver Physiol (2014) 307(3):G274–85. doi:10.1152/ajpgi.00389.2012
58. Arda-Pirincci P, Oztay F, Bayrak BB, Yanardag R, Bolkent S. Teduglutide, a glucagon-like peptide 2 analogue: a novel protective agent with anti-apoptotic and anti-oxidant properties in mice with lung injury. Peptides (2012) 38(2):238–47. doi:10.1016/j.peptides.2012.09.030
59. Lee YS, Park MS, Choung JS, Kim SS, Oh HH, Choi CS, et al. Glucagon-like peptide-1 inhibits adipose tissue macrophage infiltration and inflammation in an obese mouse model of diabetes. Diabetologia (2012) 55(9):2456–68. doi:10.1007/s00125-012-2592-3
60. Lynch L, Hogan AE, Duquette D, Lester C, Banks A, LeClair K, et al. iNKT cells induce FGF21 for thermogenesis and are required for maximal weight loss in GLP1 therapy. Cell Metab (2016) 24(3):510–9. doi:10.1016/j.cmet.2016.08.003
61. Ebinger M, Jehle DR, Fussgaenger RD, Fehmann HC, Jehle PM. Glucagon-like peptide-1 improves insulin and proinsulin binding on RINm5F cells and human monocytes. Am J Physiol Endocrinol Metab (2000) 279(1):E88–94.
62. Hadjiyanni I, Siminovitch KA, Danska JS, Drucker DJ. Glucagon-like peptide-1 receptor signalling selectively regulates murine lymphocyte proliferation and maintenance of peripheral regulatory T cells. Diabetologia (2010) 53(4):730–40. doi:10.1007/s00125-009-1643-x
63. Xie S, Liu B, Fu S, Wang W, Yin Y, Li N, et al. GLP-2 suppresses LPS-induced inflammation in macrophages by inhibiting ERK phosphorylation and NF-kappaB activation. Cell Physiol Biochem (2014) 34(2):590–602. doi:10.1159/000363025
64. Zhong J, Rao X, Deiuliis J, Braunstein Z, Narula V, Hazey J, et al. A potential role for dendritic cell/macrophage-expressing DPP4 in obesity-induced visceral inflammation. Diabetes (2013) 62(1):149–57. doi:10.2337/db12-0230
65. Duvnjak L, Blaslov K, Perkovic MN, Cuca JK. Dipeptidyl peptidase-4 activity might be a link between tumour necrosis factor alpha and insulin resistance in type 1 diabetes. Endocrine (2016) 53(2):453–8. doi:10.1007/s12020-016-0899-4
66. Salgado FJ, Vela E, Martin M, Franco R, Nogueira M, Cordero OJ. Mechanisms of CD26/dipeptidyl peptidase IV cytokine-dependent regulation on human activated lymphocytes. Cytokine (2000) 12(7):1136–41. doi:10.1006/cyto.1999.0643
67. Zhong J, Rao X, Rajagopalan S. An emerging role of dipeptidyl peptidase 4 (DPP4) beyond glucose control: potential implications in cardiovascular disease. Atherosclerosis (2013) 226(2):305–14. doi:10.1016/j.atherosclerosis.2012.09.012
68. Marguet D, Baggio L, Kobayashi T, Bernard AM, Pierres M, Nielsen PF, et al. Enhanced insulin secretion and improved glucose tolerance in mice lacking CD26. Proc Natl Acad Sci U S A (2000) 97(12):6874–9. doi:10.1073/pnas.120069197
69. Broxmeyer HE, Hoggatt J, O’Leary HA, Mantel C, Chitteti BR, Cooper S, et al. Dipeptidylpeptidase 4 negatively regulates colony-stimulating factor activity and stress hematopoiesis. Nat Med (2012) 18(12):1786–96. doi:10.1038/nm.2991
70. Kameoka J, Tanaka T, Nojima Y, Schlossman SF, Morimoto C. Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science (1993) 261(5120):466–9. doi:10.1126/science.8101391
71. Wagner L, Klemann C, Stephan M, von Horsten S. Unravelling the immunological roles of dipeptidyl peptidase 4 (DPP4) activity and/or structure homologue (DASH) proteins. Clin Exp Immunol (2016) 184(3):265–83. doi:10.1111/cei.12757
72. Shah Z, Kampfrath T, Deiuliis JA, Zhong J, Pineda C, Ying Z, et al. Long-term dipeptidyl-peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation (2011) 124(21):2338–49. doi:10.1161/CIRCULATIONAHA.111.041418
73. Zhong J, Rajagopalan S. Dipeptidyl peptidase-4 regulation of SDF-1/CXCR4 axis: implications for cardiovascular disease. Front Immunol (2015) 6:477. doi:10.3389/fimmu.2015.00477
74. Tian L, Gao J, Hao J, Zhang Y, Yi H, O’Brien TD, et al. Reversal of new-onset diabetes through modulating inflammation and stimulating beta-cell replication in nonobese diabetic mice by a dipeptidyl peptidase IV inhibitor. Endocrinology (2010) 151(7):3049–60. doi:10.1210/en.2010-0068
75. Coleman CM, Sisk JM, Halasz G, Zhong J, Beck SE, Matthews KL, et al. CD8+ T cells and macrophages regulate pathogenesis in a mouse model of MERS-CoV disease. J Virol (2016) 91(1). doi:10.1128/JVI.01825-16
76. Palnaes Hansen C, Andreasen JJ, Holst JJ. The release of gastric inhibitory peptide, glucagon-like peptide-I, and insulin after oral glucose test in colectomized subjects. Scand J Gastroenterol (1997) 32(5):473–7. doi:10.3109/00365529709025084
77. Robertson MD, Livesey G, Morgan LM, Hampton SM, Mathers JC. The influence of the colon on postprandial glucagon-like peptide 1 (7-36) amide concentration in man. J Endocrinol (1999) 161(1):25–31. doi:10.1677/joe.0.1610025
78. Bang-Berthelsen CH, Holm TL, Pyke C, Simonsen L, Sokilde R, Pociot F, et al. GLP-1 induces Barrier protective expression in Brunner’s glands and regulates colonic inflammation. Inflamm Bowel Dis (2016) 22(9):2078–97. doi:10.1097/MIB.0000000000000847
79. Keller J, Beglinger C, Holst JJ, Andresen V, Layer P. Mechanisms of gastric emptying disturbances in chronic and acute inflammation of the distal gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol (2009) 297(5):G861–8. doi:10.1152/ajpgi.00145.2009
80. Keller J, Binnewies U, Rosch M, Juul Holst J, Beglinger C, Andresen V, et al. Gastric emptying and disease activity in inflammatory bowel disease. Eur J Clin Invest (2015) 45(12):1234–42. doi:10.1111/eci.12542
81. Schmidt PT, Hartmann B, Bregenholt S, Hoist JJ, Claesson MH. Deficiency of the intestinal growth factor, glucagon-like peptide 2, in the colon of SCID mice with inflammatory bowel disease induced by transplantation of CD4+ T cells. Scand J Gastroenterol (2000) 35(5):522–7. doi:10.1080/003655200750023796
82. Yazbeck R, Sulda ML, Howarth GS, Bleich A, Raber K, von Horsten S, et al. Dipeptidyl peptidase expression during experimental colitis in mice. Inflamm Bowel Dis (2010) 16(8):1340–51. doi:10.1002/ibd.21241
83. Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med (2007) 204(12):2803–12. doi:10.1084/jem.20071397
84. Duan L, Chen J, Zhang H, Yang H, Zhu P, Xiong A, et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3(+) regulatory T-cell responses in mice. Mol Med (2012) 18:753–61. doi:10.2119/molmed.2011.00428
85. Duan L, Chen J, Gong F, Shi G. The role of IL-33 in rheumatic diseases. Clin Dev Immunol (2013) 2013:924363. doi:10.1155/2013/924363
86. Anbazhagan AN, Thaqi M, Priyamvada S, Jayawardena D, Kumar A, Gujral T, et al. GLP-1 nanomedicine alleviates gut inflammation. Nanomedicine (2017) 13(2):659–65. doi:10.1016/j.nano.2016.08.004
87. Guan L, Gong D, Tian N, Zou Y. Uncoupling protein 2 involved in protection of glucagon-like peptide 2 in small intestine with ischemia-reperfusion injury in mice. Dig Dis Sci (2005) 50(3):554–60. doi:10.1007/s10620-005-2474-3
88. Zhang W, Zhu W, Zhang J, Li N, Li J. Protective effects of glucagon-like peptide 2 on intestinal ischemia-reperfusion rats. Microsurgery (2008) 28(4):285–90. doi:10.1002/micr.20491
89. Rajeevprasad R, Alavi K, Schwartz MZ. Glucagonlike peptide-2 analogue enhances intestinal mucosal mass and absorptive function after ischemia-reperfusion injury. J Pediatr Surg (2000) 35(11):1537–9. doi:10.1053/jpsu.2000.18301
90. Schmidt PT, Ljung T, Hartmann B, Hare KJ, Holst JJ, Hellstrom PM. Tissue levels and post-prandial secretion of the intestinal growth factor, glucagon-like peptide-2, in controls and inflammatory bowel disease: comparison with peptide YY. Eur J Gastroenterol Hepatol (2005) 17(2):207–12. doi:10.1097/00042737-200502000-00012
91. Xiao Q, Boushey RP, Cino M, Drucker DJ, Brubaker PL. Circulating levels of glucagon-like peptide-2 in human subjects with inflammatory bowel disease. Am J Physiol Regul Integr Comp Physiol (2000) 278(4):R1057–63.
92. L’Heureux MC, Brubaker PL. Glucagon-like peptide-2 and common therapeutics in a murine model of ulcerative colitis. J Pharmacol Exp Ther (2003) 306(1):347–54. doi:10.1124/jpet.103.051771
93. Sigalet DL, Wallace LE, Holst JJ, Martin GR, Kaji T, Tanaka H, et al. Enteric neural pathways mediate the anti-inflammatory actions of glucagon-like peptide 2. Am J Physiol Gastrointest Liver Physiol (2007) 293(1):G211–21. doi:10.1152/ajpgi.00530.2006
94. Trivedi S, Wiber SC, El-Zimaity HM, Brubaker PL. Glucagon-like peptide-2 increases dysplasia in rodent models of colon cancer. Am J Physiol Gastrointest Liver Physiol (2012) 302(8):G840–9. doi:10.1152/ajpgi.00505.2011
95. Iakoubov R, Lauffer LM, Trivedi S, Kim YI, Brubaker PL. Carcinogenic effects of exogenous and endogenous glucagon-like peptide-2 in azoxymethane-treated mice. Endocrinology (2009) 150(9):4033–43. doi:10.1210/en.2009-0295
96. Pucar LB, Detel D, Varljen J. [Dipeptidyl peptidase IV in inflammatory bowel diseases (DPP IV/CD26)]. Arh Hig Rada Toksikol (2012) 63(1):75–100. doi:10.2478/10004-1254-63-2012-2185
97. Fatima S, Ruter J, Niess JH, Klapp BF, Arck PC, Hildebrandt M. CD26-/DPP IV-positive lymphocytes in murine acute experimental colitis. Adv Exp Med Biol (2003) 524:345–50. doi:10.1007/0-306-47920-6_41
98. Ban H, Bamba S, Imaeda H, Inatomi O, Kobori A, Sasaki M, et al. The DPP-IV inhibitor ER-319711 has a proliferative effect on the colonic epithelium and a minimal effect in the amelioration of colitis. Oncol Rep (2011) 25(6):1699–703. doi:10.3892/or.2011.1223
99. Sakanaka T, Inoue T, Yorifuji N, Iguchi M, Fujiwara K, Narabayashi K, et al. The effects of a TGR5 agonist and a dipeptidyl peptidase IV inhibitor on dextran sulfate sodium-induced colitis in mice. J Gastroenterol Hepatol (2015) 30(Suppl 1):60–5. doi:10.1111/jgh.12740
100. Detel D, Buljevic S, Pucar LB, Kucic N, Pugel EP, Varljen J. Influence of CD26/dipeptidyl peptidase IV deficiency on immunophenotypic changes during colitis development and resolution. J Physiol Biochem (2016) 72(3):405–19. doi:10.1007/s13105-016-0491-7
101. Detel D, Pugel EP, Pucar LB, Buljevic S, Varljen J. Development and resolution of colitis in mice with target deletion of dipeptidyl peptidase IV. Exp Physiol (2012) 97(4):486–96. doi:10.1113/expphysiol.2011.061143
102. Iwaya H, Fujii N, Hagio M, Hara H, Ishizuka S. Contribution of dipeptidyl peptidase IV to the severity of dextran sulfate sodium-induced colitis in the early phase. Biosci Biotechnol Biochem (2013) 77(7):1461–6. doi:10.1271/bbb.130105
103. Mimura S, Ando T, Ishiguro K, Maeda O, Watanabe O, Ujihara M, et al. Dipeptidyl peptidase-4 inhibitor anagliptin facilitates restoration of dextran sulfate sodium-induced colitis. Scand J Gastroenterol (2013) 48(10):1152–9. doi:10.3109/00365521.2013.832366
Keywords: incretin, inflammatory bowel diseases, dipeptidyl peptidase-4, glucagon-like peptide-1, liraglutide
Citation: Duan L, Rao X, Braunstein Z, Toomey AC and Zhong J (2017) Role of Incretin Axis in Inflammatory Bowel Disease. Front. Immunol. 8:1734. doi: 10.3389/fimmu.2017.01734
Received: 25 September 2017; Accepted: 23 November 2017;
Published: 06 December 2017
Edited by:
Kai Fang, University of California, Los Angeles, United StatesReviewed by:
Maria Angela Sortino, Università degli Studi di Catania, ItalyCopyright: © 2017 Duan, Rao, Braunstein, Toomey and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jixin Zhong, aml4aW4uemhvbmdAY2FzZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.