 Andreas Koenig
Andreas Koenig Iwona Buskiewicz
Iwona Buskiewicz Sally A. Huber
Sally A. Huber- Department of Pathology, University of Vermont, Burlington, VT, United States
Sexual bias is a hallmark in various diseases. This review evaluates sexual dimorphism in clinical and experimental coxsackievirus B3 (CVB3) myocarditis, and how sex bias in the experimental disease changes with increased age. Coxsackieviruses are major causes of viral myocarditis, an inflammation of the heart muscle, which is more frequent and severe in men than women. Young male mice infected with CVB3 develop heart-specific autoimmunity and severe myocarditis. Females infected during estrus (high estradiol) develop T-regulatory cells and when infected during diestrus (low estradiol) develop autoimmunity similar to males. During estrus, protection depends on estrogen receptor alpha (ERα), which promotes type I interferon, activation of natural killer/natural killer T cells and suppressor cell responses. Estrogen receptor beta has opposing effects to ERα and supports pro-inflammatory immunity. However, the sexual dimorphism of the disease is significantly ameliorated in aged animals when old females become as susceptible as males. This correlates to a selective loss of the ERα that is required for immunosuppression. Therefore, sex-associated hormones control susceptibility in the virus-mediated disease, but their impact can alter with the age and physiological stage of the individual.
Introduction
Myocarditis is an inflammation of the myocardium usually following microbial infections. Most viruses and many bacteria, fungi, protozoa, and helminths can initiate the disease including Picornaviruses and Adenoviruses (1–4). The clinical disease takes multiple forms including acute, chronic, giant cell, and eosinophilic myocarditis with differences in prognosis and in underlying pathogenesis (2, 3). Acute myocarditis may be severe but self-limiting and many investigators hypothesize that cardiac injury results from direct microbial injury to the heart or to antimicrobial host defense mechanisms including cytokine storms, which directly suppress myocyte contractility and function (3, 5, 6). Chronic myocarditis may last for months or years and result in either cardiac transplantation or death. Underlying mechanism for the chronic form remains controversial with dueling theories of persistent virus infection causing myocardial dysfunction or infection triggering autoimmunity to heart antigens (3, 5). Clinical trials of type 1 interferon (IFN) treatment of virus positive cardiomyopathy patients showed virus clearance from the heart and partial improvement of cardiac function through 12 weeks posttreatment although the improvement waned somewhat by 24 weeks posttreatment (7). Lack of a long-lasting protection with virus clearance could circumstantially favor autoimmune pathogenic mechanisms. Immunosuppression improves long-term survival for giant cell myocarditis patients (8) and has also been shown to be effective in acute myocarditis (9). However, not all clinical trials have shown protection with immunosuppression (10). Mouse models of coxsackievirus B3 (CVB3)-induced myocarditis cause cardiac inflammation, which histologically resemble the clinical disease. While virus infection initiates myocarditis, predominant cardiac damage is immune mediated with strong evidence implicating induction of autoimmunity to heart antigens (3). Factors required for autoimmunity include as follows: (1) the existence of autoreactive lymphocytes; (2) autoreactive cell activation; and (3) the absence of immunosuppression (11). Infections can lead to autoimmunity by several mechanisms. These include antigenic mimicry in which microbial and self-molecules share sufficient similarity to induce cross-reactive immunity (12, 13), and such antigenic mimicry has been described between CVB3 and cardiac proteins (14, 15). Another mechanism is an adjuvant effect similar to immunization of self-proteins in complete Freund’s adjuvant (16, 17). The ability of adjuvants to promote autoimmunity probably results from their ability to activate the innate immune system through pathogen-associated molecular patterns stimulation of toll-like receptors (TLR). Giving specific TLR agonists along with cardiac proteins/apoptotic myocytes is sufficient for myocarditis induction while giving either adjuvant or heart tissue alone is ineffective (18, 19). Virus-induced tissue damage releases large amounts of normally sequestered antigens not normally available to the immune system. Autoimmunity can result from cryptic epitopes (20). Under certain conditions, these cryptic epitopes can become visible to the immune system and initiate pathogenic immune responses. While cryptic epitopes result from naturally occurring protein folding/unfolding or protein–protein interactions, most microbes encode their own proteases, which cleave cellular as well as microbial proteins and might produce peptides distinct from those produced by cellular proteases (21).
Sex-Associated Hormones Control CVB3-Induced Myocarditis Susceptibility
Both the incidence and severity of myocarditis and dilated cardiomyopathy are greater in men than women (22). This is true of enterovirus infections generally (23). CVB3 mouse models of myocarditis infecting young adult animals mimic the male dominance in disease susceptibility with females being largely protected from cardiac inflammation, myocardial injury, and death subsequent to viral infection (22, 24, 25). Testosterone promotes susceptibility as castrated males are protected while exogenous administration of testosterone restores cardiac inflammation. Similarly, estrogen (E2) is protective as ovariectomized females show increased myocarditis compared with intact females and E2 treatment of males is protective (24, 26). E2 levels determine myocarditis resistance since CVB3 infection of females during diestrus when E2 levels are lowest results in myocarditis susceptibility whereas CVB3 infection during estrus is completely protective (27). Sex hormones are well known for their ability to influence innate and adaptive immunity including type 1 IFN response, TLR type and level of activation, antigen presentation [macrophage/dendritic cells (DCs)], and T lymphocyte polarization [reviewed in Ref. (28–33)]. Male and female CVB3-infected mice generate distinct T cell responses to infection with males generating pro-inflammatory and females generating anti-inflammatory or immunosuppressive (T-regulatory) responses (34, 35). Studies by Xiong and colleagues have shown hormonally regulated polarization of macrophage leading to monocytic myeloid suppressor cells in females, and this may result from differences in innate immune responses (cytokine response by natural killer cells) to infection between males and females (36–38).
Mechanisms of estrogen signaling in cells have been extensively reviewed in the literature (33, 39–43) and will only be briefly discussed here. Estrogen mediates its effects primarily through two receptors, such as estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ), which bind the hormone with similar affinity but due to slight differences in the ligand binding pockets of the receptors, may promote transcription of distinct sets of genes (40, 43). Most lymphoid cells express either or both types of estrogen receptors [Table 1; reviewed in Ref. (33, 43, 44)]. The estrogen receptor binds to estrogen in the cytosol forming a complex and translocates to the nucleus for direct interaction with estrogen response elements (ERE) in many gene promoters. Various ERE may differ in sequence resulting in differences in estrogen receptor binding affinity and variation in transcriptional activity. Even the same ERE can produce different transcriptional activation between distinct cell types because of variations in specific factors such as coactivators (40). Although ERβ has many similar characteristics to ERα, molecular mechanisms of transcriptional activation may differ between the receptors (45). Alternatively, the estrogen receptors may initiate transcription without directly interacting with DNA through binding to specific transcription factors including NF-κB, SP1, AP-1, or C/EPBβ, which alters their DNA binding or coregulatory factor complex formation and gene activation (46, 47). In studies evaluating ERα–AP-1 and ERβ–AP-1 response elements, evidence demonstrated that these nuclear hormone receptor subtypes can exert opposite transcription controls on gene expression (48). Thus, in certain situations, ERβ can inhibit ERα-induced gene expression (49). Membrane bound ERα/ERβ, which are splice variants of the nuclear forms, and a transmembrane G protein-coupled estrogen receptor (GPER) induce much more rapid signaling compared with the classical nuclear ER isoforms (41, 42).
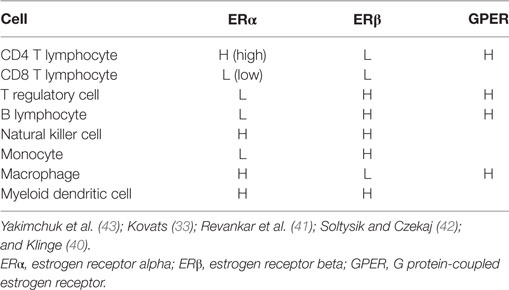
Table 1. Estrogen receptor expression by cells of the immune system.
Studies determined that protection during experimental CVB3-induced myocarditis depends on signaling through ERα as disease susceptibility is increased in ERα knockout mice while infected male mice treated with the specific ERα agonist, propylpyrazoletriol (PPT) were protected (50). By contrast, signaling through ERβ promotes myocarditis as shown by treating female or male mice with the specific ERβ agonist, diarylpropionitrile. No studies were found on GPER in myocarditis, but GPER abrogates cardiac myocyte apoptosis during ischemia/reperfusion injury (51) suggesting a cardioprotective role. The mechanisms of ERα-induced protection are likely to be multifaceted. ERα should increase expression of type 1 IFNs (30), which are crucial to preventing cardiac injury (52). Previous studies have shown that E2 promotes T-regulatory cell activation both through upregulation of FoxP3 and through increased T-regulatory cell proliferation (53, 54). In systemic sclerosis patients, FoxP3(bright) cells were increased in patients with autoantibodies to ERα (55). GPER also attenuates autoimmune diseases and elicits FoxP3 expression (56). In the myocarditis model, infecting young female during estrus but not during diestrus, activates T-regulatory cells (57) as does treating males with the ERα agonist, PPT (50). Signaling through the ERβ prevents T-regulatory cell activation. Unlike prior reports, however, control of T-regulatory cell response is not mediated through direct action on FoxP3 transcription but rather through regulation of natural killer T (NKT) and gamma-delta T (γδT) cell responses (50, 58). Various studies report that NKT cell subpopulations suppress autoimmunity and increase T-regulatory cell numbers in vivo (59–61). By contrast, γδT selectively kill T-regulatory cells and relieve immunosuppression resulting in autoimmunity (62). While PPT treatment of CVB3-infected males induces T-regulatory cells, this fails to occur in NKT knockout mice demonstrating that the effect of E2 signaling must be indirectly mediated through these innate effectors rather than through modulating FoxP3 expression in T-regulatory precursors directly (50). ERα modulates activities of many TLR most notably TLR 2, 7, 8, and 9 (30, 63, 64). By contrast, ERα may inhibit responses dependent on TLR4 (65). CVB3 infection selectively upregulates TLR2 in females but upregulates TLR4 in males (66, 67). Treating males with the specific TLR2 agonist dramatically reduces mortality while treating females with the TLR4 agonist promotes pathogenicity (19, 66). Interaction of NKT cells with immature myeloid DCs triggers tolerogenic maturation of the DC and T-regulatory cell generation while NKT cells interacting with DC in concert with TLR4 stimulation results in pro-inflammatory responses (68).
Age and Myocarditis Susceptibility: Loss of Sexual Dimorphism
A systemic review of the world literature in 2010 reported no significant difference in age between men and women when evaluating the proportion of heart failure patients caused by myocarditis (69). However, in a report by Laufer-Perl et al. (70) of 200 pericarditis/myocarditis patients, there was both an increased incidence and younger age for men (73%; 46 ± 19 years of age) compared with women (27%; 60 ± 19 years of age). In this latter study, the women had a lower rate of hospitalization raising the possibility that reduced disease severity in women might partially mask some epidemiological characteristics in this sex missed by evaluating heart failure. Also, when combining data encompassing all forms and etiological causes of myocarditis, distinct pathological mechanisms in each type of myocarditis may be impacted by sex-associated hormones differently. In the experimental CVB3 myocarditis model, the clear sexual dimorphism noted in young (2–4 months old) mice is lost in aged (8–12 months old) female mice, which show high mortality, increased cardiac inflammation and a pro-inflammatory phenotype resembling males (71). While exogenous E2 treatment of young females is protective (57), E2 treatment of old females increased both mortality (68.8% or 11/16 mice) and cardiac inflammation (72). Cardiac virus titers increase corresponding with increases in cardiac inflammation, myocyte injury, and animal mortality. Thus, E2 has the opposite effect in young and old female mice during CVB3-induced myocarditis (Figure 1). When spleens of old (12-month old) female mice were analyzed using Western blot and confirmed by RT-PCR, an approximately 90% reduction in ERα and a twofold increase in ERβ was observed compared with young (2.5-month old) female mice, giving a substantially higher ERα:ERβ ratio of in young than in old females. The trend in males was similar but more modest. A reduced ERα in old females corresponds to abrogated T-regulatory cell activation in these animals (50).
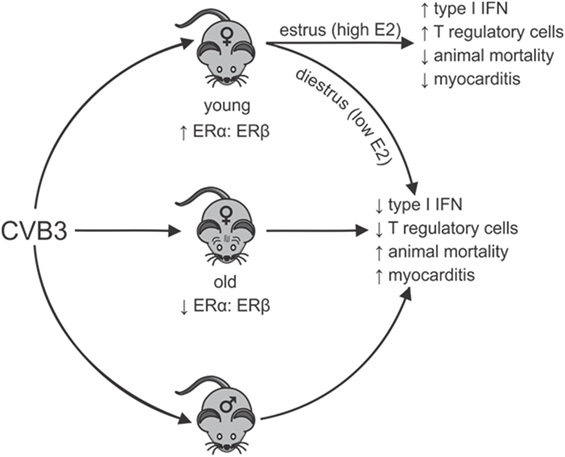
Figure 1. Sexual dimorphism in coxsackievirus B3 (CVB3)-induced myocarditis: dependence on estrogen (E2) concentration and relative estrogen receptor alpha (ERα):estrogen receptor beta (ERβ) expression in lymphoid cells. Young females (age 2–3 months) have high ratios of ERα:ERβ in spleen cells resulting in robust type 1 interferon (IFN) responses to CVB3 infections, especially when virus infection occurs during estrus the time of peak E2. Signaling through ERα promotes T-regulatory cell responses leading to protection from myocarditis and animal mortality. Infection of young females during diestrus when E2 concentrations are low or infection of old females (12 months), which selectively lose ERα but increase ERβ expression in lymphoid cells results in reduced type 1 IFN response to CVB3 and prevents activation of T-regulatory cells. This results in induction of strong pro-inflammatory immunity and increased animal mortality and myocarditis, which is similar to disease observed in males. Thus, there is sexual dimorphism in CVB3-induced disease, but this can be dependent on both the hormonal state of the individual (stage of the estrus cycle when infection occurs) and the age of the individual when infection occurs. The age of the individual could be important as E2 regulation of dendritic cell development is well documented and is ERα dependent. Selective loss of ERα has been shown in the cardiovascular system with age and similar loss in lymphoid cells could affect immunocompetence.
Aging and the Immune System
Aging decreases adaptive immune responses to infection, reduces naïve T cells entering the periphery from the thymus due to thymic involution, and is associated with chronic immune activation and memory cell expansion ultimately leading to immunosenescence and skewering of the immune repertoire at the expense of the ability to respond to new antigens (73–76). Aging affects multiple cells including T cells, B cells, NK cells, DCs, macrophage, neutrophils, and hematopoietic stem cells. DCs are especially important in antigen presentation and ERα but not ERβ has been shown to promote optimal DC differentiation and cytokine production (77). Studies on the effects of aging on DCs have been controversial with some studies reporting no changes in numbers, distribution, turnover, or engraftment ability of aged cells compared with cells from young mice (78); while other studies have found reductions in cDC but not pDC (79). In aged humans, pDC reportedly show both numerical and functional decline in peripheral blood but myeloid DC do not (80). A number of studies report that aged DC from both humans and mice produce less type 1 and type 3 IFNs than cells from young individuals in response to TLR stimulation (81). While divergent reports exist, various studies have found that aging reduces MHC II and co-stimulatory molecule (CD40/CD86), impairs naïve T cell priming and potentially increases the negative co-stimulatory molecule PD-L1, which promotes T-regulatory cell activation (81, 82). Studies in 26-month-old rats show that E2 treatment inhibits maturation and functional activity of DC (83). Aging alters ERα and ERβ expression levels in various tissues most notably in the bone, brain, eye, kidney, and cardiovascular system (84–89). Changes are observed both in humans (84, 86, 90) and in rodents (85, 87, 89). The age-related reduction of ERα in heart and kidney results from increased methylation of the promoter due to oxidative stress (89, 90). Whether reactive oxygen species might similarly reduce ERα in DC is unclear.
Conclusion
Sex-associated hormones determine enterovirus pathogenesis and act through their effects on the innate immune response. This is demonstrated through modulation of type 1 IFN responses, which can impact virus load in infected tissues but also on both the type and level of TLR expression, on the activation of innate effectors including NK, NKT, γδT and macrophage, and on the adaptive immunity including induction of autoimmunity to heart antigens. Androgens promote pro-inflammatory responses. Estrogens promote anti-inflammatory responses at high levels but are pro-inflammatory at low levels. Aged females become more disease susceptible presumably because of the dual effect of lower estradiol and decreased physiological expression of ERα, the protective estrogen receptor that results in a decreased ERα:ERβ ratio. Whether a similar phenomenon occurs in clinical myocarditis is unclear. Few reports fully address age of men and women with myocarditis separately and with consideration of both the various types of myocarditis and the potential etiological mechanisms of each disease form. The acute form presumably resulting from direct virus injury may experience greater benefit from type 1 IFN and hormones promoting this response while chronic myocarditis might have a different pathogenic mechanism.
Author Contributions
All authors listed, have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The work was supported by NHLBI HL108371 and funds from the University of Vermont, Department of Pathology. IB and AK were supported by the NIH grant R21 AR069830.
References
1. Bowles NE, Ni J, Kearney DL, Pauschinger M, Schultheiss HP, McCarthy R, et al. Detection of viruses in myocardial tissues by polymerase chain reaction. Evidence of adenovirus as a common cause of myocarditis in children and adults. J Am Coll Cardiol (2003) 42(3):466–72. doi:10.1016/S0735-1097(03)00648-X
2. Huber SA. Viral myocarditis and dilated cardiomyopathy: etiology and pathogenesis. Curr Pharm Des (2016) 22(4):408–26. doi:10.2174/1381612822666151222160500
3. Rose NR. Viral myocarditis. Curr Opin Rheumatol (2016) 28(4):383–9. doi:10.1097/BOR.0000000000000303
4. Trachtenberg BH, Hare JM. Inflammatory cardiomyopathic syndromes. Circ Res (2017) 121(7):803–18. doi:10.1161/CIRCRESAHA.117.310221
5. Yajima T, Knowlton KU. Viral myocarditis: from the perspective of the virus. Circulation (2009) 119(19):2615–24. doi:10.1161/CIRCULATIONAHA.108.766022
6. Hobai IA, Morse JC, Siwik DA, Colucci WS. Lipopolysaccharide and cytokines inhibit rat cardiomyocyte contractility in vitro. J Surg Res (2015) 193(2):888–901. doi:10.1016/j.jss.2014.09.015
7. Schultheiss HP, Piper C, Sowade O, Waagstein F, Kapp JF, Wegscheider K, et al. Betaferon in chronic viral cardiomyopathy (BICC) trial: effects of interferon-beta treatment in patients with chronic viral cardiomyopathy. Clin Res Cardiol (2016) 105(9):763–73. doi:10.1007/s00392-016-0986-9
8. Cooper LT Jr, Hare JM, Tazelaar HD, Edwards WD, Starling RC, Deng MC, et al. Usefulness of immunosuppression for giant cell myocarditis. Am J Cardiol (2008) 102(11):1535–9. doi:10.1016/j.amjcard.2008.07.041
9. Aziz KU, Patel N, Sadullah T, Tasneem H, Thawerani H, Talpur S. Acute viral myocarditis: role of immunosuppression: a prospective randomised study. Cardiol Young (2010) 20(5):509–15. doi:10.1017/S1047951110000594
10. Mason JW, O’Connell JB, Herskowiz A, Rose NR, McManus BM, Billingham ME, et al. A clinical trial of immunosuppressive therapy for myocarditis. The myocarditis treatment trial investigators. N Engl J Med (1995) 333:269–75. doi:10.1056/NEJM199508033330501
11. Horwitz M, Sarvetnick N. Viruses, host responses, and autoimmunity. Immunol Rev (1999) 169:241–53. doi:10.1111/j.1600-065X.1999.tb01319.x
12. Cunningham MW. Rheumatic fever, autoimmunity, and molecular mimicry: the streptococcal connection. Int Rev Immunol (2014) 33(4):314–29. doi:10.3109/08830185.2014.917411
13. Massilamany C, Huber SA, Cunningham MW, Reddy J. Relevance of molecular mimicry in the mediation of infectious myocarditis. J Cardiovasc Transl Res (2014) 7(2):165–71. doi:10.1007/s12265-013-9519-3
14. Gauntt C, Higdon A, Arizpe H, Tamayo M, Crawley R, Henkel R, et al. Epitopes shared between coxsackievirus B3 (CVB3) and normal heart tissue contribute to CVB3-induced murine myocarditis. Clin Immunol Immunopathol (1993) 68:129–34. doi:10.1006/clin.1993.1108
15. Maisch B, Bauer E, Cirsi M, Kocksiek K. Cytolytic cross-reactive antibodies directed against the cardiac membrane and viral proteins in coxsackievirus B3 and B4 myocarditis. Circulation (1993) 87(Suppl IV):49–65.
16. Israeli E, Agmon-Levin N, Blank M, Shoenfeld Y. Adjuvants and autoimmunity. Lupus (2009) 18(13):1217–25. doi:10.1177/0961203309345724
17. Guimaraes LE, Baker B, Perricone C, Shoenfeld Y. Vaccines, adjuvants and autoimmunity. Pharmacol Res (2015) 100:190–209. doi:10.1016/j.phrs.2015.08.003
18. Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, Watts TH, et al. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med (2003) 9(12):1484–90. doi:10.1038/nm960
19. Frisancho-Kiss S, Davis SE, Nyland JF, Frisancho JA, Cihakova D, Barrett MA, et al. Cutting edge: cross-regulation by TLR4 and T cell Ig mucin-3 determines sex differences in inflammatory heart disease. J Immunol (2007) 178(11):6710–4. doi:10.4049/jimmunol.178.11.6710
20. Mortimer GM, Minchin RF. Cryptic epitopes and functional diversity in extracellular proteins. Int J Biochem Cell Biol (2016) 81(Pt A):112–20. doi:10.1016/j.biocel.2016.10.020
21. Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE, et al. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med (1999) 5(3):320–6. doi:10.1038/6543
22. Fairweather D, Cooper LT Jr, Blauwet LA. Sex and gender differences in myocarditis and dilated cardiomyopathy. Curr Probl Cardiol (2013) 38(1):7–46. doi:10.1016/j.cpcardiol.2012.07.003
23. Moore M, Kaplan M, McPhee J, Bregman D, Klein S. Epidemiologic, clinical and laboratory features of coxsackie B1-B5 infections in the United States, 1970–1979. Public Health Rep (1984) 99:515–22.
24. Huber S, Job L, Auld K. Influence of sex hormones on coxsackie B3 virus infection in Balb/c mice. Cell Immunol (1982) 67(173–189):173–9. doi:10.1016/0008-8749(82)90210-6
25. Fairweather D, Petri MA, Coronado MJ, Cooper LT. Autoimmune heart disease: role of sex hormones and autoantibodies in disease pathogenesis. Expert Rev Clin Immunol (2012) 8(3):269–84. doi:10.1586/eci.12.10
26. Li Z, Yue Y, Xiong S. Distinct Th17 inductions contribute to the gender bias in CVB3-induced myocarditis. Cardiovasc Pathol (2013) 22(5):373–82. doi:10.1016/j.carpath.2013.02.004
27. Schwartz J, Sartini D, Huber S. Myocarditis susceptibility in female mice depends upon ovarian cycle phase at infection. Virology (2004) 330(1):16–23. doi:10.1016/j.virol.2004.06.051
28. Oertelt-Prigione S. The influence of sex and gender on the immune response. Autoimmun Rev (2012) 11(6–7):A479–85. doi:10.1016/j.autrev.2011.11.022
29. Pennell LM, Galligan CL, Fish EN. Sex affects immunity. J Autoimmun (2012) 38(2–3):J282–91. doi:10.1016/j.jaut.2011.11.013
30. Seillet C, Laffont S, Tremollieres F, Rouquie N, Ribot C, Arnal JF, et al. The TLR-mediated response of plasmacytoid dendritic cells is positively regulated by estradiol in vivo through cell-intrinsic estrogen receptor alpha signaling. Blood (2012) 119(2):454–64. doi:10.1182/blood-2011-08-371831
31. Seillet C, Rouquie N, Foulon E, Douin-Echinard V, Krust A, Chambon P, et al. Estradiol promotes functional responses in inflammatory and steady-state dendritic cells through differential requirement for activation function-1 of estrogen receptor alpha. J Immunol (2013) 190(11):5459–70. doi:10.4049/jimmunol.1203312
32. Giefing-Kroll C, Berger P, Lepperdinger G, Grubeck-Loebenstein B. How sex and age affect immune responses, susceptibility to infections, and response to vaccination. Aging Cell (2015) 14(3):309–21. doi:10.1111/acel.12326
33. Kovats S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol (2015) 294(2):63–9. doi:10.1016/j.cellimm.2015.01.018
34. Frisancho-Kiss S, Nyland JF, Davis SE, Frisancho JA, Barrett MA, Rose NR, et al. Sex differences in coxsackievirus B3-induced myocarditis: IL-12Rbeta1 signaling and IFN-gamma increase inflammation in males independent from STAT4. Brain Res (2006) 1126(1):139–47. doi:10.1016/j.brainres.2006.08.003
35. Frisancho-Kiss S, Coronado MJ, Frisancho JA, Lau VM, Rose NR, Klein SL, et al. Gonadectomy of male BALB/c mice increases Tim-3(+) alternatively activated M2 macrophages, Tim-3(+) T cells, Th2 cells and Treg in the heart during acute coxsackievirus-induced myocarditis. Brain Behav Immun (2009) 23(5):649–57. doi:10.1016/j.bbi.2008.12.002
36. Li K, Xu W, Guo Q, Jiang Z, Wang P, Yue Y, et al. Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis. Circ Res (2009) 105(4):353–64. doi:10.1161/CIRCRESAHA.109.195230
37. Liu L, Yue Y, Xiong S. NK-derived IFN-gamma/IL-4 triggers the sexually disparate polarization of macrophages in CVB3-induced myocarditis. J Mol Cell Cardiol (2014) 76:15–25. doi:10.1016/j.yjmcc.2014.07.021
38. Su N, Yue Y, Xiong S. Monocytic myeloid-derived suppressor cells from females, but not males, alleviate CVB3-induced myocarditis by increasing regulatory and CD4(+)IL-10(+) T cells. Sci Rep (2016) 6:22658. doi:10.1038/srep22658
40. Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res (2001) 29(14):2905–19. doi:10.1093/nar/29.14.2905
41. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science (2005) 307(5715):1625–30. doi:10.1126/science.1106943
42. Soltysik K, Czekaj P. Membrane estrogen receptors – is it an alternative way of estrogen action? J Physiol Pharmacol (2013) 64(2):129–42.
43. Yakimchuk K, Jondal M, Okret S. Estrogen receptor alpha and beta in the normal immune system and in lymphoid malignancies. Mol Cell Endocrinol (2013) 375(1–2):121–9. doi:10.1016/j.mce.2013.05.016
44. Aristimuno C, Teijeiro R, Valor L, Alonso B, Tejera-Alhambra M, de Andres C, et al. Sex-hormone receptors pattern on regulatory T-cells: clinical implications for multiple sclerosis. Clin Exp Med (2012) 12(4):247–55. doi:10.1007/s10238-011-0172-3
45. Tremblay GB, Tremblay A, Copeland NG, Gilbert DJ, Jenkins NA, Labrie F, et al. Cloning, chromosomal localization, and functional analysis of the murine estrogen receptor beta. Mol Endocrinol (1997) 11(3):353–65. doi:10.1210/mend.11.3.9902
46. Cvoro A, Tzagarakis-Foster C, Tatomer D, Paruthiyil S, Fox MS, Leitman DC. Distinct roles of unliganded and liganded estrogen receptors in transcriptional repression. Mol Cell (2006) 21(4):555–64. doi:10.1016/j.molcel.2006.01.014
47. Cui J, Shen Y, Li R. Estrogen synthesis and signaling pathways during aging: from periphery to brain. Trends Mol Med (2013) 19(3):197–209. doi:10.1016/j.molmed.2012.12.007
48. Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, et al. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science (1997) 277(5331):1508–10. doi:10.1126/science.277.5331.1508
49. Matthews J, Gustafsson JA. Estrogen signaling: a subtle balance between ER alpha and ER beta. Mol Interv (2003) 3(5):281–92. doi:10.1124/mi.3.5.281
50. Huber S. ERbeta and ERalpha differentially regulate NKT and Vgamma4+ T-cell activation and T-regulatory cell response in coxsackievirus B3 infected mice. J Clin Cell Immunol (2015) 6(6):1–9. doi:10.4172/2155-9899.1000372
51. Li WL, Xiang W, Ping Y. Activation of novel estrogen receptor GPER results in inhibition of cardiocyte apoptosis and cardioprotection. Mol Med Rep (2015) 12(2):2425–30. doi:10.3892/mmr.2015.3674
52. Buskiewicz IA, Koenig A, Roberts B, Russell J, Shi C, Lee SH, et al. c-FLIP-short reduces type I interferon production and increases viremia with coxsackievirus B3. PLoS One (2014) 9(5):e96156. doi:10.1371/journal.pone.0096156
53. Polanczyk MJ, Hopke C, Huan J, Vandenbark AA, Offner H. Enhanced FoxP3 expression and Treg cell function in pregnant and estrogen-treated mice. J Neuroimmunol (2005) 170(1–2):85–92. doi:10.1016/j.jneuroim.2005.08.023
54. Prieto GA, Rosenstein Y. Oestradiol potentiates the suppressive function of human CD4 CD25 regulatory T cells by promoting their proliferation. Immunology (2006) 118(1):58–65. doi:10.1111/j.1365-2567.2006.02339.x
55. Giovannetti A, Maselli A, Colasanti T, Rosato E, Salsano F, Pisarri S, et al. Autoantibodies to estrogen receptor alpha in systemic sclerosis (SSc) as pathogenetic determinants and markers of progression. PLoS One (2013) 8(9):e74332. doi:10.1371/journal.pone.0074332
56. Brunsing RL, Owens KS, Prossnitz ER. The G protein-coupled estrogen receptor (GPER) agonist G-1 expands the regulatory T-cell population under TH17-polarizing conditions. J Immunother (2013) 36(3):190–6. doi:10.1097/CJI.0b013e31828d8e3b
57. Huber SA. Coxsackievirus B3-induced myocarditis: infection of females during the estrus phase of the ovarian cycle leads to activation of T regulatory cells. Virology (2008) 378(2):292–8. doi:10.1016/j.virol.2008.05.015
58. Liu W, Moussawi M, Roberts B, Boyson JE, Huber SA. Cross-regulation of T regulatory-cell response after coxsackievirus B3 infection by NKT and gammadelta T cells in the mouse. Am J Pathol (2013) 183(2):441–9. doi:10.1016/j.ajpath.2013.04.015
59. Sonoda KH, Faunce DE, Taniguchi M, Exley M, Balk S, Stein-Streilein J. NK T cell-derived IL-10 is essential for the differentiation of antigen-specific T regulatory cells in systemic tolerance. J Immunol (2001) 166(1):42–50. doi:10.4049/jimmunol.166.1.42
60. La Cava A, Van Kaer L, Fu Dong S. CD4+CD25+ Tregs and NKT cells: regulators regulating regulators. Trends Immunol (2006) 27(7):322–7. doi:10.1016/j.it.2006.05.003
61. Liu W, Huber SA. Cross-talk between cd1d-restricted nkt cells and gammadelta cells in t regulatory cell response. Virol J (2011) 8:32. doi:10.1186/1743-422X-8-32
62. Huber S. γδ T lymphocytes kill T regulatory cells through CD1d. Immunology (2010) 131(2):202–9. doi:10.1111/j.1365-2567.2010.03292.x
63. Cunningham MA, Wirth JR, Naga O, Eudaly J, Gilkeson GS. Estrogen receptor alpha binding to ERE is required for full Tlr7- and Tlr9-induced inflammation. SOJ Immunol (2014) 2(1):1–7. doi:10.15226/soji.2014.00107
64. Young NA, Wu LC, Burd CJ, Friedman AK, Kaffenberger BH, Rajaram MV, et al. Estrogen modulation of endosome-associated toll-like receptor 8: an IFNalpha-independent mechanism of sex-bias in systemic lupus erythematosus. Clin Immunol (2014) 151(1):66–77. doi:10.1016/j.clim.2014.01.006
65. Fan MJ, Huang-Liu R, Shen CY, Ju DT, Lin YM, Pai P, et al. Reduction of TLR4 mRNA stability and protein expressions through inhibiting cytoplasmic translocation of HuR transcription factor by E(2) and/or ERalpha in LPS-treated H9c2 cardiomyoblast cells. Chin J Physiol (2014) 57(1):8–18. doi:10.4077/CJP.2014.BAC197
66. Roberts BJ, Dragon JA, Moussawi M, Huber SA. Sex-specific signaling through toll-like receptors 2 and 4 contributes to survival outcome of coxsackievirus B3 infection in C57Bl/6 mice. Biol Sex Differ (2012) 3(1):25. doi:10.1186/2042-6410-3-25
67. Roberts BJ, Moussawi M, Huber SA. Sex differences in TLR2 and TLR4 expression and their effect on coxsackievirus-induced autoimmune myocarditis. Exp Mol Pathol (2013) 94(1):58–64. doi:10.1016/j.yexmp.2012.06.005
68. Caielli S, Conforti-Andreoni C, Di Pietro C, Usuelli V, Badami E, Malosio ML, et al. On/off TLR signaling decides proinflammatory or tolerogenic dendritic cell maturation upon CD1d-mediated interaction with invariant NKT cells. J Immunol (2010) 185(12):7317–29. doi:10.4049/jimmunol.1000400
69. Cooper LT Jr, Keren A, Sliwa K, Matsumori A, Mensah GA. The global burden of myocarditis: part 1: a systematic literature review for the global burden of diseases, injuries, and risk factors 2010 study. Glob Heart (2014) 9(1):121–9. doi:10.1016/j.gheart.2014.01.007
70. Laufer-Perl M, Havakuk O, Shacham Y, Steinvil A, Letourneau-Shesaf S, Chorin E, et al. Sex-based differences in prevalence and clinical presentation among pericarditis and myopericarditis patients. Am J Emerg Med (2017) 35(2):201–5. doi:10.1016/j.ajem.2016.10.039
71. Lyden D, Olszewski J, Feran M, Job L, Huber S. Coxsackievirus B3-induced myocarditis. Effects of sex steroids on viremia and infectivity of cardiocytes. Am J Pathol (1987) 126:432–8.
72. Lyden D. The Effects of Sex Steroids on Viral Infection and Cellular Immunity Associated with Coxsackie-Induced Myocarditis in BALB/c Mice. Ph.D. Burlington: University of Vermont (1986).
73. Linton PJ, Dorshkind K. Age-related changes in lymphocyte development and function. Nat Immunol (2004) 5(2):133–9. doi:10.1038/ni1033
74. Arnold CR, Wolf J, Brunner S, Herndler-Brandstetter D, Grubeck-Loebenstein B. Gain and loss of T cell subsets in old age – age-related reshaping of the T cell repertoire. J Clin Immunol (2011) 31(2):137–46. doi:10.1007/s10875-010-9499-x
75. Solana R, Tarazona R, Gayoso I, Lesur O, Dupuis G, Fulop T. Innate immunosenescence: effect of aging on cells and receptors of the innate immune system in humans. Semin Immunol (2012) 24(5):331–41. doi:10.1016/j.smim.2012.04.008
76. Pereira BI, Akbar AN. Convergence of innate and adaptive immunity during human aging. Front Immunol (2016) 7:445. doi:10.3389/fimmu.2016.00445
77. Douin-Echinard V, Laffont S, Seillet C, Delpy L, Krust A, Chambon P, et al. Estrogen receptor alpha, but not beta, is required for optimal dendritic cell differentiation and [corrected] CD40-induced cytokine production. J Immunol (2008) 180(6):3661–9. doi:10.4049/jimmunol.180.6.3661
78. Tan SY, Cavanagh LL, d’Advigor W, Shackel N, Fazekas de St Groth B, Weninger W. Phenotype and functions of conventional dendritic cells are not compromised in aged mice. Immunol Cell Biol (2012) 90(7):722–32. doi:10.1038/icb.2011.104
79. Pereira LF, de Souza AP, Borges TJ, Bonorino C. Impaired in vivo CD4+ T cell expansion and differentiation in aged mice is not solely due to T cell defects: decreased stimulation by aged dendritic cells. Mech Ageing Dev (2011) 132(4):187–94. doi:10.1016/j.mad.2011.03.005
80. Jing Y, Shaheen E, Drake RR, Chen N, Gravenstein S, Deng Y. Aging is associated with a numerical and functional decline in plasmacytoid dendritic cells, whereas myeloid dendritic cells are relatively unaltered in human peripheral blood. Hum Immunol (2009) 70(10):777–84. doi:10.1016/j.humimm.2009.07.005
81. Wong C, Goldstein DR. Impact of aging on antigen presentation cell function of dendritic cells. Curr Opin Immunol (2013) 25(4):535–41. doi:10.1016/j.coi.2013.05.016
82. Kitazawa Y, Fujino M, Wang Q, Kimura H, Azuma M, Kubo M, et al. Involvement of the programmed death-1/programmed death-1 ligand pathway in CD4+CD25+ regulatory T-cell activity to suppress alloimmune responses. Transplantation (2007) 83(6):774–82. doi:10.1097/01.tp.0000256293.90270.e8
83. Stojic-Vukanic Z, Nacka-Aleksic M, Bufan B, Pilipovic I, Arsenovic-Ranin N, Djikic J, et al. 17beta-Estradiol influences in vitro response of aged rat splenic conventional dendritic cells to TLR4 and TLR7/8 agonists in an agonist specific manner. Int Immunopharmacol (2015) 24(1):24–35. doi:10.1016/j.intimp.2014.11.008
84. Ogueta SB, Schwartz SD, Yamashita CK, Farber DB. Estrogen receptor in the human eye: influence of gender and age on gene expression. Invest Ophthalmol Vis Sci (1999) 40(9):1906–11.
85. Wilson ME, Rosewell KL, Kashon ML, Shughrue PJ, Merchenthaler I, Wise PM. Age differentially influences estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) gene expression in specific regions of the rat brain. Mech Ageing Dev (2002) 123(6):593–601. doi:10.1016/S0047-6374(01)00406-7
86. Batra GS, Hainey L, Freemont AJ, Andrew G, Saunders PT, Hoyland JA, et al. Evidence for cell-specific changes with age in expression of oestrogen receptor (ER) alpha and beta in bone fractures from men and women. J Pathol (2003) 200(1):65–73. doi:10.1002/path.1332
87. Novensa L, Novella S, Medina P, Segarra G, Castillo N, Heras M, et al. Aging negatively affects estrogens-mediated effects on nitric oxide bioavailability by shifting ERalpha/ERbeta balance in female mice. PLoS One (2011) 6(9):e25335. doi:10.1371/journal.pone.0025335
88. Foster TC. Role of estrogen receptor alpha and beta expression and signaling on cognitive function during aging. Hippocampus (2012) 22(4):656–69. doi:10.1002/hipo.20935
89. Pereira-Simon S, Xia X, Catanuto P, Elliot S. Oxidant stress and mitochondrial signaling regulate reversible changes of ERalpha expression and apoptosis in aging mouse glomeruli and mesangial cells. Endocrinology (2012) 153(11):5491–9. doi:10.1210/en.2012-1379
Keywords: aging and immunocompetence, mouse models, T-regulatory cells and innate immunity, estrogen receptor alpha, estrogen receptor alpha:beta ratios and immune competence, sex bias in coxsackievirus B3 myocarditis
Citation: Koenig A, Buskiewicz I and Huber SA (2017) Age-Associated Changes in Estrogen Receptor Ratios Correlate with Increased Female Susceptibility to Coxsackievirus B3-Induced Myocarditis. Front. Immunol. 8:1585. doi: 10.3389/fimmu.2017.01585
Received: 13 September 2017; Accepted: 03 November 2017;
Published: 16 November 2017
Edited by:
Georges Jacques Casimir, Free University of Brussels, BelgiumReviewed by:
Rui Li, University of Pennsylvania, United StatesAdriana Maggi, Università degli Studi di Milano, Italy
Cyril Seillet, Walter and Eliza Hall Institute of Medical Research, Australia
Copyright: © 2017 Koenig, Buskiewicz and Huber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sally A. Huber, c2FsbHkuaHViZXJAdXZtLmVkdQ==