 Aura Muntasell1*
Aura Muntasell1* Mariona Cabo1
Mariona Cabo1 Sonia Servitja1,2Ignasi Tusquets1,2María Martínez-García1,2Ana Rovira1,2
Sonia Servitja1,2Ignasi Tusquets1,2María Martínez-García1,2Ana Rovira1,2 Federico Rojo3Joan Albanell1,2,4
Federico Rojo3Joan Albanell1,2,4 Miguel López-Botet1,4
Miguel López-Botet1,4
- 1Hospital del Mar Medical Research Institute (IMIM), Barcelona, Spain
- 2Department of Oncology, Hospital del Mar-CIBERONC, Barcelona, Spain
- 3Hospital Fundación Jiménez Díaz, Madrid, Spain
- 4Univ. Pompeu Fabra, Barcelona, Spain
Overexpression of the human epidermal growth factor receptor 2 (HER2) defines a subgroup of breast tumors with aggressive behavior. The addition of HER2-targeted antibodies (i.e., trastuzumab, pertuzumab) to chemotherapy significantly improves relapse-free and overall survival in patients with early-stage and advanced disease. Nonetheless, considerable proportions of patients develop resistance to treatment, highlighting the need for additional and co-adjuvant therapeutic strategies. HER2-specific antibodies can trigger natural killer (NK) cell-mediated antibody-dependent cellular cytotoxicity and indirectly enhance the development of tumor-specific T cell immunity; both mechanisms contributing to their antitumor efficacy in preclinical models. Antibody-dependent NK cell activation results in the release of cytotoxic granules as well as the secretion of pro-inflammatory cytokines (i.e., IFNγ and TNFα) and chemokines. Hence, NK cell tumor suppressive functions include direct cytolytic killing of tumor cells as well as the regulation of subsequent antitumor adaptive immunity. Albeit tumors with gene expression signatures associated to the presence of cytotoxic lymphocyte infiltrates benefit from trastuzumab-based treatment, NK cell-related biomarkers of response/resistance to HER2-specific therapeutic antibodies in breast cancer patients remain elusive. Several variables, including (i) the configuration of the patient NK cell repertoire; (ii) tumor molecular features (i.e., estrogen receptor expression); (iii) concomitant therapeutic regimens (i.e., chemotherapeutic agents, tyrosine kinase inhibitors); and (iv) evasion mechanisms developed by progressive breast tumors, have been shown to quantitatively and qualitatively influence antibody-triggered NK cell responses. In this review, we discuss possible interventions for restoring/enhancing the therapeutic activity of HER2 therapeutic antibodies by harnessing NK cell antitumor potential through combinatorial approaches, including immune checkpoint blocking/stimulatory antibodies, cytokines and toll-like receptor agonists.
Introduction
Breast cancer is a major health-care problem worldwide, with an estimated 1.67 million women diagnosed annually.1 Human epidermal growth factor receptor 2 (HER2, also known as ErbB2 or HER2/neu) is a transmembrane receptor with tyrosine kinase activity, capable of activating several pro-survival intracellular signaling pathways (1). HER2 overexpression occurs in approximately 15–20% of breast tumors and is associated with aggressive disease and decreased survival (2). Addition of HER2-targeted therapeutic monoclonal antibodies (mAb) to chemotherapy improved overall survival in patients with early-stage and advanced disease (3). Currently, two complementary anti-HER2 therapeutic mAbs, trastuzumab, and pertuzumab, and the antibody-drug trastuzumab-emtansine (T-DM1) are approved for clinical use. Combination of chemotherapy with dual HER2 targeting with trastuzumab and pertuzumab are the prevailing therapeutic approaches for HER2+ tumors in the neoadjuvant setting and in the first-line treatment of metastatic disease; trastuzumab and lapatinib (a dual EGFR/HER2 tyrosine kinase inhibitor small molecule) can also be used in refractory patients with advanced disease (4, 5); T-DM1 has been approved for treating advanced HER2+ breast cancer patients with progressive disease following trastuzumab/pertuzumab and chemotherapy regimens (6). Despite significant improvement in the clinical outcome of HER2+ breast cancer since the introduction of these anti-HER2 drugs, there are patients with early disease that eventually relapse and disease progression inevitably occurs due to de novo or acquired resistance to treatment in metastatic patients (7). Potential tumor cell-intrinsic mechanisms of resistance to anti-HER2 mAb treatment have been identified, yet their clinical relevance remains uncertain (8).
All currently approved anti-HER2 mAbs are immunoglobulins (Ig) of the G1 subclass (IgG1) and, in addition to block HER2 oncogenic signaling, share the capability of triggering antitumor immune function by engaging specific receptors expressed by immune cells (FcγR family, Box 1) through their constant domain (Fc). Several publications indicate that NK and tumor-specific T lymphocytes significantly influence disease development and response to treatment with anti-HER2 mAbs (9–12). In addition to considerable data supporting the importance of T cells in immunosurveillance (9), a role for NK cell function in preventing early tumor development and metastatic spread is being increasingly appreciated (13, 14).
Box 1. Antibody structure and FcγR family.
Antibodies (Abs) or immunoglobulins (Ig) display two functionally different domains: a variable Fab region which determines specificity and affinity for a particular antigen and a constant region or Fc fragment which can engage a diversity of cellular receptors in immune cells. Immunoglobulins of the G subclass (IgG) can interact with distinct FcγR family members, respectively, displaying activating and inhibitory signaling capacity. Human activating FcγRs include FcγRI (CD64), FcγRIIA (CD32A), FcγRIIC (CD32C), and FcγRIIIA (CD16A), whereas FcγRIIB (CD32B) is the counterpart with inhibitory function. FcγR in mouse includes FcγRI, FcγRIII, and FcγRIV with stimulatory potential and the inhibitory FcγRIIB. Human NK cells primarily express FcγRIIIA in the absence of inhibitory FcγR; B cells exclusively express the inhibitory FcγRIIB; human dendritic cells express both the activating and the inhibitory forms of FcγRII A and B. Distinct monocyte/macrophage subpopulations have been shown to express diverse combinations of activating and inhibitory FcγR, including FcγRI, FcγRIIA, FcγRIIB, and FcγRIIIA. It is nowadays recognized that the Fc fragment of therapeutic antibodies elicits several of their effector mechanisms. Engagement of activating FcγR results in antibody-dependent cellular cytotoxicity and phagocytosis (ADCC and ADCP). With the exception of FcγRI, remaining FcγR show intermediate/low affinity for IgG and will bind to immune complexes or IgG-coated targets, resulting in receptor crosslinking and triggering of cellular responses. Human IgG2 and IgG4 isotypes display a poor interaction with FcγR whilst human IgG1 and IgG3 interact more strongly (15, 16).
In this review, current understanding of antitumor immune responses driven by anti-HER2 mAbs will be discussed from the NK cell perspective, integrating a conceptual framework for the combinatorial use of anti-HER2 antibodies and several immunotherapy approaches enhancing NK cell function/survival in breast cancer.
Regulation of NK Cell Antitumor Function
Natural killer cells are cytotoxic members of the innate lymphocyte cell family, important in the defense against virus-infected and transformed cells. NK cell activation leads to the polarized release of cytolytic molecules, such as granzyme B and perforin stored in preformed granules, causing target cell death (14, 17, 18). NK cells can also trigger perforin-independent apoptosis by FasL- and TRAIL-mediated engagement of death-inducing receptors on target cells (19). Time-lapse imaging has revealed that a single activated NK cell can make serial contacts with multiple targets and kill an average of four tumor cells in vitro (20, 21). In addition, activated NK cells secrete IFNγ, TNFα, and chemokines (i.e., MIP1α, MIP1β, RANTES), boosting the recruitment of other immune effectors and the development of subsequent antitumor T cell immunity (14, 17, 18).
The importance of NK cell function for early tumor immune surveillance is supported by studies showing increased cancer risk in individuals with low NK cell activity (22), including several genetically predisposed cases (i.e., NKG2D haplotypes LNK1/LNK1) (23). On the other hand, correlation between tumor NK cell density/function and prognosis has been reported for a number of cancer types (e.g., colorectal, hepatocellular, gastric carcinomas, lung adenocarcinoma, and renal cancer), supporting their importance for metastasis control in vivo (13, 24, 25).
Natural killer cell activation is regulated by an array of germ-line encoded surface receptors with stimulatory or inhibitory function. NK cells use inhibitory receptors to prevent the killing of healthy cells, whereas crosslinking of activating receptors is required to initiate an immune response against transformed cells (26). NKG2D, NKp46 and NKp30, together with the co-stimulatory molecule DNAM-1, are considered the main activating receptors involved in direct tumor cell recognition (27–29). NKG2D recognizes stress-induced self-molecules, such as MICA/B and the ULBP family, upregulated in most neoplastic cell types (30); natural cytotoxicity receptors (NKp30 and NKp46) can recognize self-molecules exposed in damaged cells (i.e., BAT3, MLL5) or induced by inflammatory stimuli (i.e., B7-H6) (31, 32); and DNAM-1 specifically recognizes CD155 (PVR) and CD112 (Nectin-2), overexpressed in a variety of tumor types (33). NK cell tolerance to self depends on inhibitory receptors specific for HLA class I molecules (HLA-I), which suppress NK cell activation against healthy cells expressing normal levels of surface HLA-I. Downregulation of surface HLA-I expression, in some virus-infected and transformed cells, allows for rapid NK cell responses against these targets (34). HLA-I specific NK cell receptors comprise killer cell immunoglobulin-like receptors (KIRs; Box 2) specific for distinct sets of HLA-I molecules (HLA-A, -B, -C); the CD94/NKG2A receptor specific for the HLA-I class Ib molecule HLA-E; and the leukocyte immunoglobulin-like receptor B1 (LILRB1) interacting with a broad spectrum of HLA-I molecules, including HLA-G. KIR and NKG2 receptor families also include members with activating function which, in some cases, can interact with HLA-I molecules albeit with lower affinity than their inhibitory counterparts (i.e., KIR2DS1 and CD94/NKG2C) (18).
Box 2. KIR receptors and their ligands.
The KIR receptor family includes six inhibitory receptors (KIR2DL1, KIR2DL2, KIR2DL3, KIR3DL1, KIR3DL2, and KIR2DL5), six activating receptors (KIR2DS1, KIR2DS2, KIR2DS3, KIR2DS4, KIR2DS5, and KIR3DS1), and one, KIR2DL4, harbouring an ambiguous signaling motif. Inhibitory KIRs are characterized by a long cytoplasmatic tail containing ITIM motifs whereas activating KIRs have a short cytoplasmic tail and interact with DAP-12 for transducing stimulatory signals. Inhibitory KIR recognize specific epitopes on HLA-A, -B, and -C molecules, determined by polymorphisms within residues 77–83 of the α1 helix. KIR2DL2/L3 and KIR2DL1 respectively recognize the C1 and C2 epitopes, found in mutually exclusive subsets of HLA-C alleles. KIR3DL1 binds to the Bw4 epitope, carried by subsets of HLA-A and HLA-B alleles whereas KIR3DL2 interacts with the A3/11 epitope, restricted to HLA-A3 and A11 molecules. The HLA class I specificity of activating KIRs is still a matter of study. KIR2DS1 has been shown to recognize the C2-epitope, whereas KIR2DS4 can interact with groups C1 and C2 HLA-C alleles and HLA-A11. Inhibitory KIR2DL1, KIR2DL2/L3, and KIR3DL1 are highly polymorphic. Allelic variants display distinct avidity and/or specificity of the ligand-binding site, level of cell-surface expression, and signal transduction capacity. Combinations of particular KIR and HLA class I have been associated to differential susceptibility to a wide range of diseases (e.g., infectious and autoimmune syndromes) and can influence hematopoietic cell transplantation outcomes (34–36).
Besides direct recognition, FcγRIIIA (CD16A) triggers NK cell activation against antibody-opsonized cells by a mechanism known as antibody-dependent cell-mediated cytotoxicity (ADCC). NK cells and certain T lymphocyte subsets (i.e., γδ T cells) are the only immune cells expressing the activating CD16A, in the absence of other members of the FcγR family with inhibitory function (15) (Box 1). Among all activating NK cell receptors, CD16A was described as the only one capable of triggering resting NK cell activation in the absence of co-stimulation (37) and of increasing the killing frequency per NK cell (38).
Natural killer cells also express functional toll-like receptors (TLRs) (i.e., TLR2, TLR3, TLR5, TLR7/8, and TLR9), which sense the presence of microbe-associated molecular patterns (MAMPs) and damage-associated molecular patterns (DAMPs) in the microenvironment, priming NK cell effector function (39, 40).
Overall NK cell antitumor efficacy depends on the combination of activation, effector function, proliferation, and survival, all these modulated by cytokines. IL-2 and IL-15 signaling through STAT5 promotes NK cell survival as well as increased IFNγ secretion, cytotoxicity, and proliferation (41); IL-12 and IL-18 signaling through STAT4 enhances NK cell cytotoxicity and cytokine production whereas type I IFNs (IFNα/β) are strong stimuli regulating NK cell cytotoxicity through the upregulation of perforin and FasL and promoting IFNγ secretion (42, 43). Conversely, TGFβ has been shown to repress the mTOR pathway in NK cells, consequently reducing their proliferation, the abundance of various activating receptors and cytotoxic activity (44).
Similar to T lymphocytes, NK cells can express several activation-induced co-receptors with stimulatory (e.g., CD137, OX40, NKp44) or inhibitory (e.g., PD1, TIGIT) function which constitute yet another layer of regulatory elements for NK cell activation (45).
NK Cell-Mediated ADCC as Mechanism of Action of Anti-HER2 Antibodies
Natural killer cell recognition of HER2-overexpressing target cells involves a number of receptors that can determine natural cytotoxicity upon direct recognition or influence the magnitude of ADCC in the presence of HER2-specific mAbs (Figure 1).
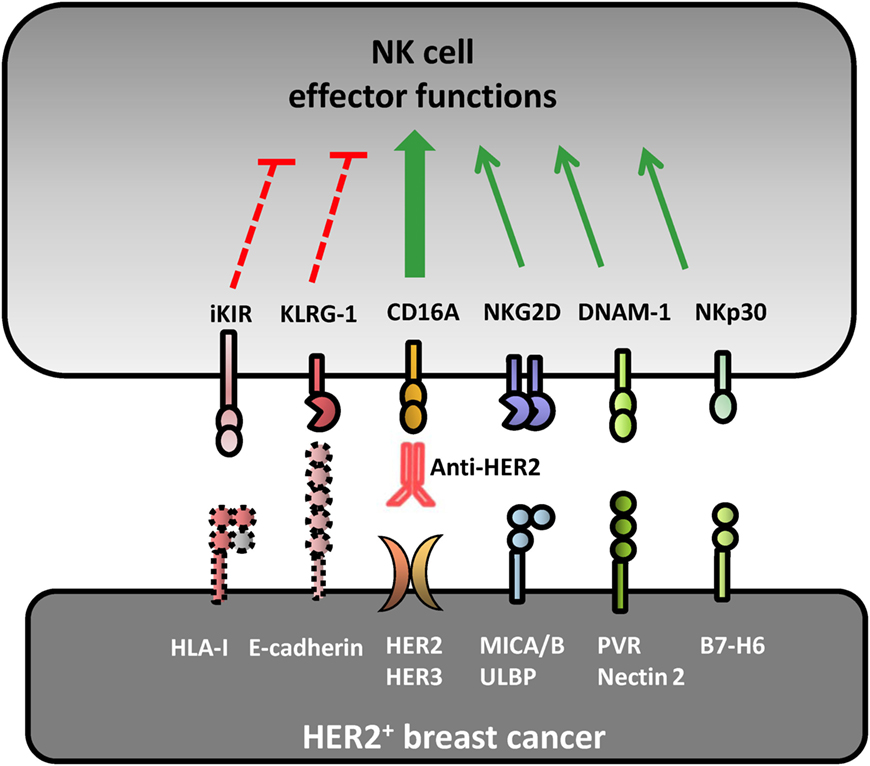
Figure 1. Receptor–ligand pairs involved in natural killer (NK) cell recognition of HER2+ breast cancer cell lines. Several receptor–ligand pairs are involved in the crosstalk between breast cancer (BC) cells and NK lymphocytes. Natural cytotoxicity against HER2+ BC is mainly driven by NKG2D, DNAM-1, and NKp30 activating receptors upon interacting with their cognate ligands MICA/B, PVR/Nectin-2, and B7-H6, respectively. Human epidermal growth factor receptor 2 (HER2)-dependent downregulation of surface HLA-I expression impairs KIR-mediated inhibition facilitating NK cell recognition of BC cell lines. Anti-HER2 therapeutic monoclonal antibodies elicit a strong NK cell-mediated antibody-dependent cell-mediated cytotoxicity response against HER2+ BC cells upon interaction with the activating CD16A receptor. E-cadherin expression can be recognized by KLRG1 inhibitory receptor expressed by some NK cell subsets, modulating their direct and antibody-dependent cytotoxicity.
HER2 signaling was shown to downregulate HLA-I and promote MICA and MICB protein expression in breast cancer cell lines in vitro, enhancing their susceptibility to NKG2D-mediated NK cell recognition and elimination (46–49). Indeed, an inverse relationship between HER2 and HLA-I expression was corroborated by immunohistochemistry (50) and concordant mRNA signatures in HER2+ tumors (51). As a matter of fact, gene expression signatures associated to cytotoxic lymphocytes are enriched in the stroma of good prognosis HER2+ tumors (52), suggesting that HER2+ breast carcinomas might be permissive to NK cell infiltration, at least at early stages of tumor development.
Anti-HER2 therapeutic mAbs introduced a novel ground by which NK cells could contribute to breast tumor control. Preclinical and clinical observations indicate that triggering of NK cell-mediated ADCC is one of the mechanisms accounting for anti-HER2 mAb therapeutic activity (53). Trastuzumab activity against xenografted tumors was severely attenuated in mice deficient in activating FcγR receptors (54) and trastuzumab F(ab′)2 fragments (lacking Fc domain) showed marginal antitumor activity in vivo despite retaining their anti-proliferative and pro-apoptotic effects in vitro (55). More precisely, NK cell depletion abolished anti-HER2 mAb therapeutic activity in preclinical mouse models of HER2+ breast cancer (56–59).
Indirect evidence also points to a significant contribution of NK cells to the clinical success of anti-HER2 mAb in breast cancer patients. Numbers of tumor-infiltrating leukocytes, particularly NK cells, were reported to increase after trastuzumab-docetaxel (60, 61) and T-DM1 treatment (62), suggesting that anti-HER2 mAb promoted NK cell tumor homing or in situ expansion. Remarkably, immune–gene expression signatures reflecting an increased recruitment of activated NK and T cells in breast tumors (i.e., CD8A, CD247, CD3D, GZMA) have been shown to be predictive of clinical benefit from preoperative and adjuvant trastuzumab-based treatment (52, 63, 64). On the other hand, peripheral blood NK cells from patients undergoing complete or partial remission upon trastuzumab plus chemotherapy displayed high ADCC activity in in vitro lysis assays, whereas impaired NK cell-mediated ADCC responses correlated with therapy failure (65, 66). Of note, a number of factors, including the disparity in markers used for precise NK cell enumeration in tumor sections (e.g., CD57, CD56, GzmB) and the absence of standardized functional read-outs, have hindered the development of NK cell-related biomarkers of response to anti-HER2 therapeutic mAbs.
Variables Potentially Modulating NK Cell-Mediated ADCC in HER2+ Breast Cancer
The specific contribution of NK cell-mediated ADCC on the clinical benefit of anti-HER2 mAb in breast cancer patients could be modulated by several NK cell-, tumor cell- and therapy-related variables (Figure 2).
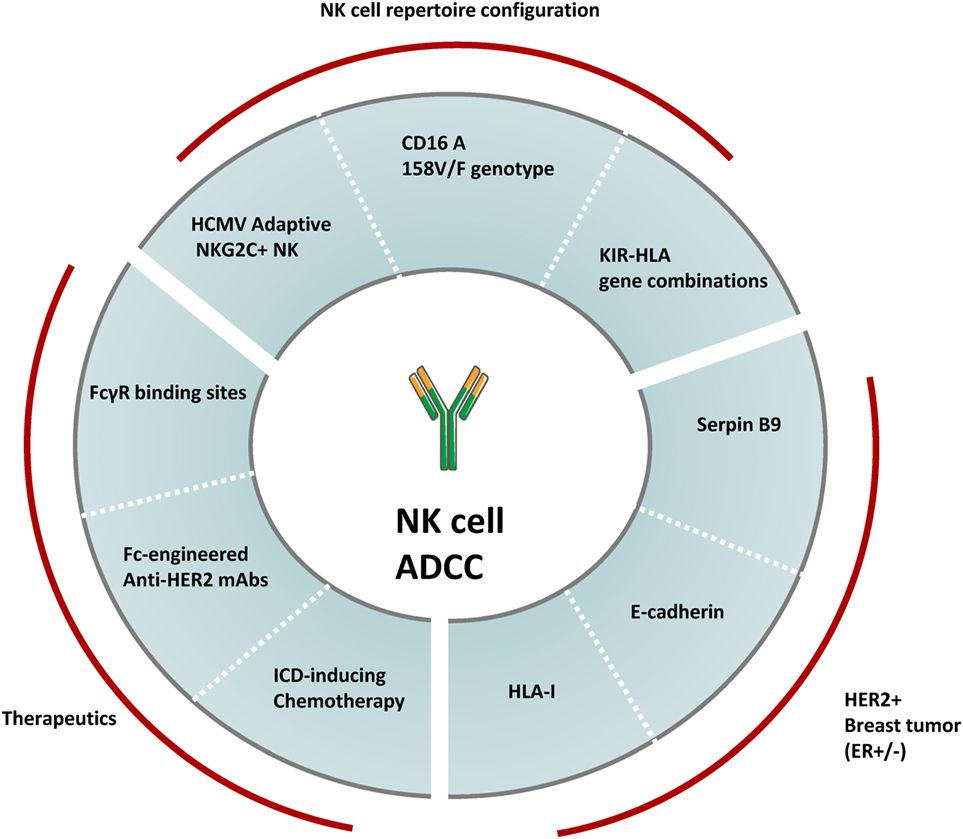
Figure 2. Variables modulating NK cell-mediated ADCC against HER2+ breast cancer. The overall magnitude of NK cell-mediated ADCC induced by anti-HER2 therapeutic monoclonal antibodies can be modulated by several factors including the configuration of the human NK cell repertoire, the heterogeneity in HER2+ breast tumor molecular subtypes and differences in treatment regimens. Factors such as specific KIR-HLA combinations, the CD16A 158V/F genotype and the prevalence of human cytomegalovirus (HCMV) adaptive NKG2C+ NK cells have been shown to modulate the overall NK cell-mediated ADCC potential. A number of tumor molecular features associated to estrogen receptor (ER) co-expression (e.g., expression of Serpin B9, E-cadherin, and HLA-I) can also modulate NK cell-mediated ADCC responses. Finally, the NK cell effector potential against HER2+ breast cancer is also modulated by therapeutic regimens, including the type of HER2-targeting drugs and the combined chemotherapy agents.
Influence of the NK Cell Repertoire Configuration on the Magnitude of ADCC
In healthy adults, approximately 90% of NK cells in peripheral blood belong to the cytotoxic CD56dimCD16+ subpopulation capable of developing ADCC responses. A second major NK cell subpopulation, defined by a CD56bright phenotype and the absence of the CD16A receptor, accounts for 10% of circulating NK cells, prevails in secondary lymphoid organs and lacks ADCC potential. Among CD56dimCD16+ NK cells, several subsets displaying different NK cell receptor combinations are found at variable frequencies. Interindividual variability on the NK cell receptor repertoire is dictated by genetic and environmental factors. Major genetic factors include KIR and HLA-I genotypes. The KIR locus contains a variable number of genes, which together with their allelic diversity, determine the existence of a substantial number of distinct KIR haplotypes distributed in the world population (34, 35). KIR genes are stochastically expressed along NK cell differentiation, generating NK cell clones with discrete KIR combinations (34, 67). Only NK cell clones expressing at least one inhibitory receptor specific for self-HLA-I achieve functional maturity. Thus, KIR–HLA-I interactions contribute to set the overall functional potential in the patient NK cell repertoire (68–70). Whether certain KIR–HLA-I gene combinations can modulate the efficacy of anti-HER2 mAbs in breast cancer patients remains unaddressed, yet associations between distinct paired KIR/KIR-ligands and clinical responses to other tumor antigen-specific mAbs, such as anti-GD2 dinutuximab, anti-CD20 rituximab, or anti-EGFR cetuximab have been reported (71, 72).
Another genetic factor known to modulate antibody-dependent NK cell activation is the CD16A (FcγRIIIA) 158V/F allelic dimorphism encoding for two receptor variants harboring either a phenylalanine (F) or valine (V) at amino acid position 158 in the receptor IgG-binding domain (73). Presence of a V residue defines receptors with high affinity for IgG1 (73). An initial association between the high affinity CD16A 158V/V genotype and complete clinical responses to trastuzumab-based treatment was described in a retrospective analysis of a small cohort of metastatic breast cancer patients (74). Nonetheless, the association of CD16A 158V/F dimorphism with time to relapse and overall survival in larger patient cohorts receiving trastuzumab in adjuvancy remains controversial (75–77). Possible caveats accounting for the different results in these studies have been discussed in critical reviews (9, 78).
Environmental factors challenging the immune system, such as autoimmune or chronic inflammatory diseases and infections, can also shape the configuration of the NK cell compartment. In this regard, infection by human cytomegalovirus (HCMV) promotes, in some individuals, a persistent adaptive expansion of long-lived NK cells hallmarked by the elevated expression of the CD94/NKG2C activating receptor (79–81). Adaptive NKG2C+ NK cells are functionally mature and have been associated with the control of HCMV infection in kidney transplant recipients (82, 83) as well as with protection from leukemia relapse upon hematopoietic stem cell transplantation (84). Remarkably, NKG2C+ NK cells display enhanced effector function upon antibody-driven recognition of virus-infected targets and rituximab-coated B lymphoblastoid cell lines in vitro (85–87).
Influence of HER2 Breast Cancer Molecular Subtypes on NK Cell-Mediated ADCC
Hormone receptor status differentiates two HER2+ breast tumor subgroups with distinct pathological response rate and overall survival upon anti-HER2 mAb treatment (88). The benefit of anti-HER2 therapy is highest in estrogen receptor (ER)-negative tumors and progressively decreases in tumors with increased ER expression (89). Globally, many immune parameters in HER2+ breast tumors (i.e., TILs, CD8+ infiltrate) are inversely correlated with ER or progesterone receptor expression (90), and it is tempting to propose a possible relationship between decreased clinical benefit of ER+ tumors to anti-HER2 mAbs and their increased resistance to NK cell-mediated ADCC. E-cadherin expression associated to ER+ breast carcinomas (91, 92) dampens trastuzumab-dependent ADCC through its specific interaction with the inhibitory killer cell lectin-like receptor G1 (KLRG1) on NK cells in preclinical in vitro and in vivo models (93, 94). Remarkably, resistance to trastuzumab-based treatment has been associated to E-cadherin expression in tumors from patients with HER2+ metastatic breast cancer (94). In addition, estrogens regulate the transcription of SerpinB9/proteinase inhibitor 9, a granzyme B inhibitor shown to decrease the susceptibility of ER+ breast cancer cells to NK and CD8+ T cell cytotoxicity in vitro (95, 96). Estrogens also upregulate HLA-I transcription through a cis-regulatory element in breast cancer cell lines (97–99), potentially modulating their susceptibility to NK cell-mediated ADCC. The relationship between ER and HLA-I expression has been confirmed by HLA-I immunohistochemical score in ER+/HER2+ as compared to ER−/HER2+ tumors (90). Whether other molecular features underlying breast carcinoma heterogeneity (e.g., mutations in PI3K, PTEN, p53, or p95HER2) (8) may modulate the susceptibility to NK cell-mediated ADCC remains uncertain.
Therapeutic Strategies Modulating NK Cell-Mediated ADCC
HER2 dual targeting with trastuzumab in combination with pertuzumab is nowadays the gold standard therapeutic approach for HER2+ breast cancer in the neoadjuvant setting and in the first-line treatment of metastatic disease. Patients that have progressed to prior trastuzumab, pertuzumab, and T-DM1 are treated with lapatinib. Both therapeutic strategies augment the coating of HER2+ tumors with IgG1 increasing the possibilities for NK cell-mediated ADCC antitumor responses. Simultaneous binding of pertuzumab and trastuzumab to HER2 increases the density of FcγR binding sites on HER2+ tumors; lapatinib does so, by preventing HER2 phosphorylation and internalization, hence increasing HER2 availability for trastuzumab (100–103).
Genetic engineering of the antibody Fc domain for optimizing FcγR engagement is one of the current strategies explored for enhancing the clinical success of several tumor antigen-specific mAbs (104). Margetuximab, an Fc-optimized HER2-specific mAb in clinical development, displayed increased binding to CD16A and elicited enhanced ADCC in breast cancer preclinical models (105). Promising single-agent activity of margetuximab has been recently reported for HER2+ breast and gastric cancer patients with advanced disease (106). Results of an ongoing two-arm open-label Phase 3 clinical trial in front of trastuzumab (NCT02492711) will reveal whether margetuximab displays superior efficacy, particularly for patients homozygous for the CD16A 158F/F low affinity genotype, in whom margetuximab showed the highest enhancement of NK cell-mediated ADCC in preclinical studies (105).
In addition to anti-HER2 mAbs, concomitant chemotherapy regimens may significantly impact on NK cell ADCC responses. Several chemotherapeutic agents currently combined or sequentially administered with anti-HER2 mAbs (i.e., anthracyclines, cyclophosphamide, taxanes) elicit a particular type of apoptosis, known as immunogenic cell death (ICD), that is accompanied by the coordinated release of DAMPs (e.g., ATP, and HMGB1) (107). DAMPs released along ICD activate a panel of pattern-recognition receptors (e.g., TLRs, P2RX7) and promote type I IFN release from cancer cells and the secretion of pro-inflammatory cytokines by immune cells (107, 108). Among DAMPs released along ICD, HMGB1 has been shown to enhance NK cell activation and recruitment to the tumor in a TLR2/4-dependent manner in preclinical models (109, 110) whereas type I IFNs have been shown to be necessary for the therapeutic efficacy of anti-HER2 mAb in MMTV-ErbB-2 transgenic mouse model (58). Indeed, a type I IFN signature predicted clinical responses to anthracycline-based chemotherapy in several independent cohorts of patients with breast cancer (108). In addition, in vitro treatment with anthracyclines and taxanes enhanced anti-HER2 mAb-induced ADCC by promoting endoplasmic reticulum-stress and the upregulation of NKG2D-ligands in breast carcinoma cells (111–113). Contravening the traditional view that chemotherapeutic drugs suppress patient immunity, anthracyclines- and taxanes-based treatments associated to enhanced NK cell function in breast cancer patients (60, 113–116).
On the whole, studies integrating information on the patient NK cell repertoire, NK cell receptor ligands on tumor cells and concomitant treatments might shed light on putative resistance mechanisms to anti-HER2 mAbs in HER2+ breast cancer patients.
NK Cell-Mediated ADCC and the Vaccine-Like Effect Induced by Anti-HER2 mAbs
Recent data highlight the importance of a vaccine-like effect by which antitumor mAb treatment facilitates the subsequent development of tumor-specific T cell responses, contributing to tumor elimination (117, 118). Antigen-presenting cells [i.e., macrophages and dendritic cells (DC)] use FcγR-mediated phagocytosis of immune complexes for enhancing tumor antigen processing and presentation, which can result in tumor-specific T-cell immunity (16, 117–119). Certainly, several evidences support the importance of antitumor T cell immunity for the clinical benefit of anti-HER2 mAb in breast cancer patients (115, 120–124).
Tumor cell cytotoxicity and cytokine/chemokine secretion upon antibody-dependent NK cell activation might directly and indirectly contribute to the vaccine-like effect induced by HER2-specific mAbs. On one hand, NK cell tumor cytolytic activity increases the availability of tumor antigen-containing immune complexes for antigen processing and presentation by DC and macrophages present in the tumor microenvironment. Independently of anti-HER2 mAbs, NK cell-DC crosstalk, involving cell–cell contacts and IFNγ, has been shown to prime DC polarization for IL-12 secretion, enhancing cross-presentation of tumor antigens to cytotoxic CD8+ T cells and the polarization of tumor-specific Th1 CD4+ T cells in preclinical models (59, 125–129). Moreover, activated NK cells are presumably capable of selectively killing immature DC while sparing activated DC, owing to their differential levels of surface HLA-I expression (130), thus selecting for immunogenic DC, effective inducers of antitumor T cells (127, 131). In patients, evidence of the participation of NK cell-mediated DC “editing” to the development of tumor-specific T cell immunity remains elusive. On the other hand, anti-HER2 mAb-dependent NK cell activation results in the production of IFNγ and chemokines (MIP1α, MCP-1, RANTES, IL-8) (132), which might contribute to the recruitment and functional polarization of myeloid and T cells with antitumor potential. Noteworthy, coordinated NK and tumor-specific T cell responses have been detected in HER2+ breast cancer patients achieving pathological complete response to trastuzumab (133).
NK Cell Evasion in Breast Cancer
Neoplastic cells can develop a wide array of strategies to subvert NK cell recognition and cytotoxic function along tumor evolution (134, 135). Indeed, NK cell selective pressure contributes to tumor immunoediting leading to the emergence of evasive tumor cell clones (136–139). Generally, strategies hijacking NK cell function can be grouped into four categories: (i) shedding of ligands for NK cell activating receptors from tumor cells which act as decoy molecules leading to NK cell functional impairment (e.g., MICA/B, B7-H6) (140, 141); (ii) upregulation of ligands for inhibitory NK cell receptors (e.g., HLA-I molecules; PD-L1) (142, 143); (iii) dysregulated expression of molecules conferring resistance to NK cell-mediated cytotoxicity (e.g., Bcl-2; Bcl-xL, cFLIP, caspase 8, Fas) (144); and (iv) immune suppressive cytokines (e.g., IL-10, TGFβ) and metabolites (e.g., PGE2, adenosine) leading to NK cell dysfunction (135, 145).
Among all these strategies, increased levels of soluble MICA/B have been described in breast cancer patients (146) as well as overexpression of HLA-E, HLA-G in HER2+ tumors as determined by immunohistochemistry (147, 148). In addition, Fas downregulation in breast tumors has been correlated with shorter patient survival (149). Hence, several NK cell-evading strategies operating along breast tumor progression may hamper the antitumor efficacy of anti-HER2 mAbs.
In concert with the development of an immune suppressive microenvironment in the progressing tumor, NK lymphocytes infiltrating advanced and metastatic breast carcinomas displayed an altered phenotype and reduced cytotoxic potential (150). According to data from distinct tumor types, NK cell infiltrates included high proportions of CD56bright NK cells with increased expression of inhibitory CD94/NKG2A and decreased expression of activating NKp30, NKG2D, and DNAM-1 receptors (150). NK cells isolated from breast tumors also displayed reduced degranulation and IFNγ and TNFα production upon direct or antibody-dependent activation (150). Likewise, stratification of breast cancer patients by local and invasive disease, evidenced a progressive functional impairment of circulating NK cells associated to phenotypic alterations (150). Remarkably, CD16 expression on circulating NK cells was rather preserved, and cytotoxic responses induced by trastuzumab against the HER2+ breast cancer cell line SKBR3 were only affected at low trastuzumab doses in NK cells from patients with locally advanced or metastatic tumors (51, 151).
Enhancing NK Cell-Mediated ADCC through Immunotherapy in HER2 Breast Cancer
Only two mAbs, trastuzumab and pertuzumab and the antibody-drug conjugate T-DM1, are currently approved for breast cancer treatment. Strengthening NK cell-mediated ADCC responses through immunotherapy appears a suitable option for enhancing their clinical efficacy (45, 152, 153). In the following paragraphs, several approaches will be discussed based on data referring to HER2+ breast cancer (Figure 3).
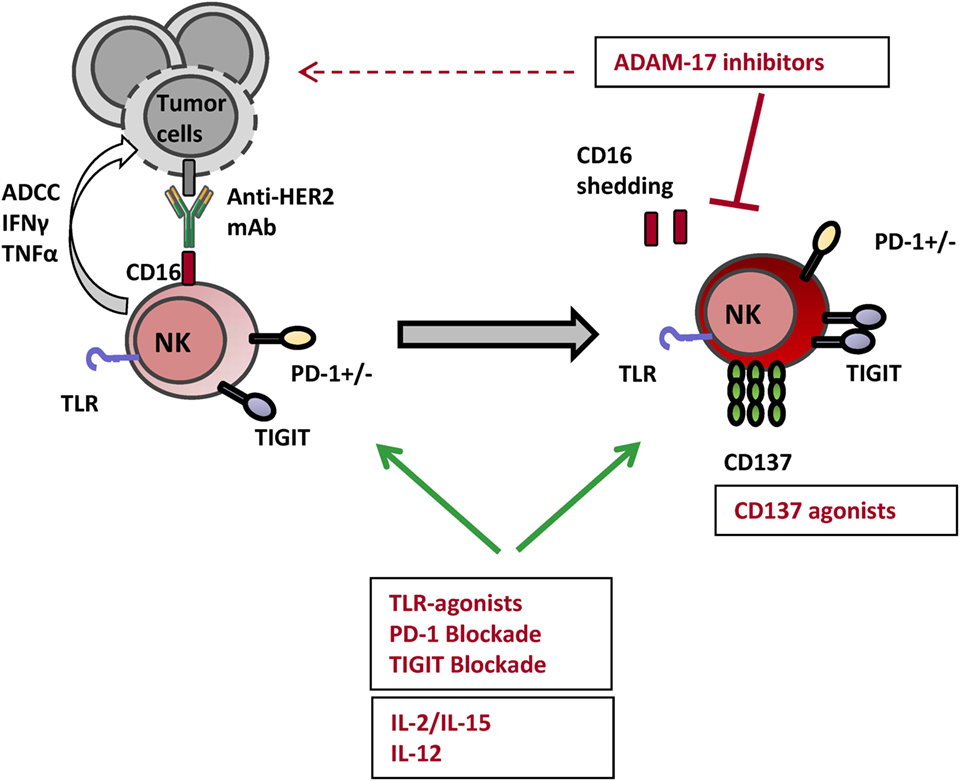
Figure 3. Actionable NK cell checkpoints for enhancing anti-HER2 mAb-induced ADCC responses. Several strategies can be tackled for harnessing NK cell ADCC responses with the objective of enhancing the clinical efficacy of anti-HER2 mAbs. Toll-like receptor (TLR) agonists and cytokines such as IL-2, IL-15, and IL-12 have been shown to lower NK cell activation threshold and enhance their effector potential. Among immune checkpoint modulators targeting surface receptors, anti-TIGIT and anti-PD-1 blocking mAbs as well as anti-CD137 agonist mAbs enhance NK cell-mediated ADCC and survival. Impeding CD16 shedding with A disintegrin and metalloproteinase 17 (ADAM17) inhibitors can be yet another strategy amplifying NK cell-mediated ADCC triggered by HER2-specific therapeutic mAbs.
Immunomodulatory mAbs Targeting Constitutive and Inducible Receptors in NK Cells
Several observations provide the rationale for combinatorial approaches including anti-HER2 mAbs and antibodies targeting surface NK cell receptors or co-receptors with activating and inhibitory function, termed immune checkpoints modulators. Nonetheless and despite promising results in preclinical models, clinical trials combining anti-HER2 mAbs and immune checkpoint-targeting antibodies are currently lacking.
IFNγ secretion by NK cells has been shown to contribute to the tumor adaptive immune resistance response (154) by upregulating the expression of HLA-I and PD-L1 in HER2+ breast cancer cells in vitro and in vivo (58, 155, 156). HLA-I and PD-L1 can be, respectively, recognized by KIR, CD94/NKG2A, LILRB1, and PD-1 inhibitory receptors, modulating the subsequent recognition of transformed cells by NK and T lymphocytes.
Blocking mAbs targeting HLA-I-specific inhibitory receptors with constitutive expression in NK cells include an anti-NKG2A (monalizumab, IPH2201) and an anti-KIR (lirilumab, IPH2101, BMS-986015) (45). Both antibodies are currently in early clinical development being tested for safety and efficacy mostly for the treatment of hematological malignancies.2 No clinical trials combining anti-HER2 mAbs and blocking agents targeting KIR or CD94/NKG2A are being developed, yet the safety and early efficacy of monalizumab and cetuximab (anti-EGFR) combination is being tested for the treatment of head and neck cancer (NCT02643550). Of note, an unexpected NK cell unresponsiveness consequent to treatment with lirilumab associated with treatment limited clinical efficacy in multiple myeloma patients (157, 158) warned about the undesired consequences of chronic targeting of HLA-I-specific NK cell receptors.
An alternative strategy, with unprecedented success as standalone treatment for several cancer types, is the blockade of the immune cell inhibitory PD-1/PD-L1 axis. Though generally considered a T cell co-receptor, PD-1 is also expressed by human exhausted NK cells (159) and circulating PD-1+ NK cell subpopulations were reported to be enriched in individuals with chronic viral infections as well as in cancer patients (159–161). PD-1+ expression is restricted to mature CD56dimCD16+ NK cells and interferes with activation via NKp30, NKp46, or CD16 receptors (159). PD-L1 expression was preferentially detected in HER2+ breast tumors showing a strong cytotoxic local immune response (162) and the numbers of PD-1+ tumor-infiltrating lymphocytes were associated with poor prognosis in HER2+ breast cancer (163, 164). Remarkably, combination of HER2-specific mAbs with blocking antibodies targeting the PD-1/PD-L1 showed greater efficacy in preclinical models (58, 62). These observations support the suitability of combining anti-HER2 mAbs with immunotherapy targeting the PD1/PD-L1 axis. Several clinical trials assessing the benefit of mAbs targeting the PD1–PD1-L axis as monotherapy or in combination with chemotherapy, radiotherapy or hormone therapy are currently being developed for ER+ or triple-negative breast tumors (see text footnote 2); likewise, combinatorial approaches with anti-HER2 mAbs are warranted.
TIGIT, a nectin-binding inhibitory co-receptor showing overlapping ligand specificity with the activating DNAM-1, is another inducible receptor with the capacity to modulate NK cell ADCC responses (165, 166). Both receptors recognize CD155 (also known as PVR) and CD112 (also known as Nectin-2), ubiquitous cell-adhesion molecules (167) overexpressed in HER2+ breast cancer cell lines (51). Besides CD8+ T cells, TIGIT is preferentially expressed on CD16+ NK cells and upregulated upon activation via ADCC (168, 169). TIGIT blockade has been shown to enhance trastuzumab-triggered antitumor response by human NK cells in vitro (169). Currently, an anti-TIGIT blocking mAb (OMP-313M32) is in early clinical development being tested for safety as standalone treatment in patients with locally advanced or metastatic solid tumors (NCT03119428).
Another immune checkpoint shown to synergize with anti-HER2 mAb in xenotransplant models of breast cancer is CD137 (58, 170). CD137 (4-1BB; TNFRSF9) is a co-stimulatory receptor induced in activated leukocytes, originally described for its capacity to enhance antitumor T cell responses (171, 172). CD137 expression following CD16 ligation has been shown in murine and human NK cells (173) and CD137 upregulation has been well documented on ex vivo circulating NK cells from breast and head and neck cancer patients upon tumor antigen-specific mAb infusion (170, 174). Two agonistic anti-CD137 mAb are currently in clinical development (urelumab and utomilumab), being tested alone or in combination with anti-PD-1 mAbs in advanced solid and hematologic tumors (45).
Of note, since NK and some T lymphocyte subsets share many receptor/ligand pairs involved in their functional regulation (e.g., PD-1, TIGIT, 4-1BB/CD137, and CD94/NKG2A), combinations between anti-HER2 therapeutic mAbs and distinct immune checkpoint modulators would promote antitumor immunity by dual targeting T and NK cell functional exhaustion.
Anti-HER2 mAb Combination with Cytokines
Several attempts to potentiate NK cell antitumor function by systemic treatment with recombinant cytokines have also been carried out. Besides their effects on T cells, IL-2, and IL-15 signaling through STAT 5 enhance NK cell antitumor function (41, 175, 176).
IL-2 enhanced NK cell-mediated ADCC triggered by anti-HER2 mAb against breast cancer cell lines in vitro and in vivo (177, 178). However, clinical trials including combined administration of IL-2 with trastuzumab did not show improved disease outcome in metastatic HER2+ breast cancer patients (179, 180). Caveats of systemic IL-2 administration include treatment-associated toxicity, its rapid clearance in vivo and IL-2 pro-tumor effects through the concurrent activation of CD4+ regulatory T cells. Nonetheless, low-dose IL-2 is currently included in a number of clinical trials to support cellular adoptive approaches with combined infusions of NK cells and trastuzumab in HER2+ breast cancer patients (NCT02030561, NCT02843126).
IL-15 is an essential cytokine for human NK cell homeostasis; nonetheless, early clinical assays including systemic IL-15 were withdrawn due to concurrent adverse events and dose-limiting toxicities (181). Similarly, IL-15 enhanced the antitumor activity of trastuzumab, yet causing fatal side effects in a humanized tumor mice model (182). Current research efforts include the development of cytokine variants with extended in vivo half-life and targeted action on precise lymphocyte subsets and tumor sites (i.e., engineered IL-2 “superkine,” IL-15Rα Sushi-Fc fusion protein; IL-15 tri and tetraspecific killer engagers) (183–186).
IL-12 has been shown to enhance the antitumor actions of trastuzumab via the enhancement of NK cell IFN-γ production in mouse models (56, 57). In a clinical trial in which IL-12 was combined with trastuzumab and paclitaxel, increased levels of IFN-γ and several chemokines were detected in sera from patients with clinical benefit, but not in patients with progressive disease (187). Currently, two clinical trials are ongoing including IL-12 and trastuzumab combined treatment (NCT00004074, NCT00028535). Preclinical studies are focused on the development of approaches for targeting cytokine expression in the tumor site to avoid toxicities associated to systemic treatment (i.e., tumor-targeting immunocytokines, gene therapy with loco-regional injections of cytokine-encoding plasmid) (188).
Immunotherapy with TLR Ligands
Toll-like receptor TLR ligands have been shown to improve both the quality and the magnitude of host antitumor innate and adaptive immune responses (189). TLR2, TLR3, TLR8, and TLR9 agonists have been shown to prime NK cell effector function (39, 40) and to synergize with anti-HER2 mAb therapy in a type I and II IFNs-, NK-, and CD8+ T cell-dependent manner in preclinical models (190–192). In the context of breast cancer, TLR ligands are being tested as adjuvants in diverse HER2-peptide vaccination strategies (i.e., TLR9-ligand CpG ODN in NCT00640861; TLR7 agonist imiquimod in NCT02276300; AS15 mixture in NCT02364492, NCT00058526, NCT00140738; TLR3 agonist Hiltonol in NCT01532960), including trastuzumab in some instances (i.e., the TLR9-ligand PF03512676-CpG 7909 or agatolimod-: NCT03512676, NCT00043394, NCT00031278). Strategies for delivering TLR agonists into the tumor site would likely potentiate NK cell-mediated ADCC synergizing with anti-HER2 mAbs antitumor function.
ADAM Inhibitors
One of the consequences of CD16-mediated NK cell activation is the shedding of CD16 extracellular domain by the induced action of the A disintegrin and metalloproteinase 17 (ADAM17), thus limiting subsequent CD16A receptor engagement and NK cell activation (193). Intriguingly, ADAM10 (with constitutive activity) and ADAM17 (inducible) also control the release of ligands for EGFR/HER receptors (194) and promote the shedding of B7-H6 and MICA/B ectodomains, amplified and overexpressed in breast tumors (195, 196) limiting NKp30- and NKG2D-mediated NK cell activation (140). In fact, ADAM10 and ADAM17 levels have been associated with poor responses and shorter relapse-free survival after trastuzumab treatment (197, 198). In this scenario, inhibition of ADAM17/10 could improve NK cell-mediated ADCC triggered by anti-HER2 mAb, preventing CD16 and B7-H6 shedding as well as enhancing HER2 surface availability. ADAM17 specific inhibitor prevented CD16 shedding and improved NK cell-mediated ADCC responses in vitro (199). Two clinical trials tested the combination of an ADAM17 inhibitor (INCB7839) with trastuzumab (NCT01254136, NCT00864175) yet the development of the compound was suspended by the sponsor corporation and no results were published. Currently, the possibility of enhancing NK cell-mediated ADCC by combining ADAM17 inhibitor (INCB7839) and tumor antigen-specific antibodies is being tested in combination with rituximab (NCT02141451).
Concluding Remarks
Activation of NK cell effector functions by anti-HER2 therapeutic antibodies can directly contribute to tumor control by their direct cytolytic activity against transformed cells, but also indirectly by their effects on the tumor microenvironment, eventually favoring the development of antitumor adaptive immunity. Multiple strategies are being developed for enhancing NK cell-mediated antibody-dependent antitumor activity, while simultaneously targeting other immune cells which contribute to the control of tumor growth and spreading. Understanding which variables underlie breast cancer heterogeneity in terms of lymphocyte infiltration and susceptibility to immune surveillance, as well as how the heterogeneity in the NK cell repertoire can influence on the clinical benefit of HER2-targeting mAbs, will aid in the design of tailored strategies to broaden their therapeutic window.
Author Contributions
All authors have actively contributed to build up the conceptual framework developed in this review and revised the draft written by AM.
Conflict of Interest Statement
Authors individually declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the technical help of Andrea Vera, Gemma Heredia, and Sara Santana.
Funding
The authors are supported by coordinated research projects from Fundación Española contra el Cáncer (GCB15152947MELE) and Proyecto Integrado de Excelencia ISCIII (PIE 2015/00008); ML-B and AM are supported by Worldwide Cancer Research Foundation (15-1146); ML-B by Plan Estatal I + D Retos (SAF2013-49063-C2-1-R; SAF2016-80363-C2-1-R), Spanish Ministry of Economy and Competitiveness (MINECO, FEDER); JA is supported by ISCiii/FEDER (PI15/00146 and CIBERONC) and by Generalitat de Catalunya (2014 SGR 740).
Footnotes
References
1. Moasser MM. The oncogene HER2; its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene (2007) 26:6469–87. doi:10.1038/sj.onc.1210478
2. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science (1987) 235:177–82. doi:10.1126/science.3798106
3. Hortobagyi GN. Trastuzumab in the treatment of breast cancer. N Engl J Med (2005) 353:1734–6. doi:10.1056/NEJMe058196
4. Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet (2012) 379:633–40. doi:10.1016/S0140-6736(11)61847-3
5. Gianni L, Pienkowski T, Im YH, Roman L, Tseng LM, Liu MC, et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol (2012) 13:25–32. doi:10.1016/S1470-2045(11)70336-9
6. Dieras V, Miles D, Verma S, Pegram M, Welslau M, Baselga J, et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol (2017) 18:732–42. doi:10.1016/S1470-2045(17)30312-1
7. Pohlmann PR, Mayer IA, Mernaugh R. Resistance to trastuzumab in breast cancer. Clin Cancer Res (2009) 15:7479–91. doi:10.1158/1078-0432.CCR-09-0636
8. Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. (2012) 17:1–16. doi:10.1615/CritRevOncog.v17.i1.20
9. Bianchini G, Gianni L. The immune system and response to HER2-targeted treatment in breast cancer. Lancet Oncol (2014) 15:e58–68. doi:10.1016/S1470-2045(13)70477-7
10. Callari M, Cappelletti V, D’Aiuto F, Musella V, Lembo A, Petel F, et al. Subtype-specific metagene-based prediction of outcome after neoadjuvant and adjuvant treatment in breast cancer. Clin Cancer Res (2016) 22:337–45. doi:10.1158/1078-0432.CCR-15-0757
11. Luen S, Virassamy B, Savas P, Salgado R, Loi S. The genomic landscape of breast cancer and its interaction with host immunity. Breast (2016) 29:241–50. doi:10.1016/j.breast.2016.07.015
12. Savas P, Salgado R, Denkert C, Sotiriou C, Darcy PK, Smyth MJ, et al. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat Rev Clin Oncol (2016) 13:228–41. doi:10.1038/nrclinonc.2015.215
13. Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell (2017) 32:135–54. doi:10.1016/j.ccell.2017.06.009
14. Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, et al. Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol (2014) 122:91–128. doi:10.1016/B978-0-12-800267-4.00003-1
15. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol (2008) 8:34–47. doi:10.1038/nri2206
16. DiLillo DJ, Ravetch JV. Fc-receptor interactions regulate both cytotoxic and immunomodulatory therapeutic antibody effector functions. Cancer Immunol Res. (2015) 3:704–13. doi:10.1158/2326-6066.CIR-15-0120
17. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol (2008) 9:495–502. doi:10.1038/ni1581
18. Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol (2001) 19:197–223. doi:10.1146/annurev.immunol.19.1.197
19. Wallin RP, Screpanti V, Michaelsson J, Grandien A, Ljunggren HG. Regulation of perforin-independent NK cell-mediated cytotoxicity. Eur J Immunol (2003) 33:2727–35. doi:10.1002/eji.200324070
20. Lopez JA, Brennan AJ, Whisstock JC, Voskoboinik I, Trapani JA. Protecting a serial killer: pathways for perforin trafficking and self-defence ensure sequential target cell death. Trends Immunol (2012) 33:406–12. doi:10.1016/j.it.2012.04.001
21. Vanherberghen B, Olofsson PE, Forslund E, Sternberg-Simon M, Khorshidi MA, Pacouret S, et al. Classification of human natural killer cells based on migration behavior and cytotoxic response. Blood (2013) 121:1326–34. doi:10.1182/blood-2012-06-439851
22. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet (2000) 356:1795–9. doi:10.1016/S0140-6736(00)03231-1
23. Hayashi T, Imai K, Morishita Y, Hayashi I, Kusunoki Y, Nakachi K. Identification of the NKG2D haplotypes associated with natural cytotoxic activity of peripheral blood lymphocytes and cancer immunosurveillance. Cancer Res (2006) 66:563–70. doi:10.1158/0008-5472.CAN-05-2776
24. Delahaye NF, Rusakiewicz S, Martins I, Menard C, Roux S, Lyonnet L, et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat Med (2011) 17:700–7. doi:10.1038/nm.2366
25. Larsen SK, Gao Y, Basse PH. NK cells in the tumor microenvironment. Crit Rev Oncog (2014) 19:91–105. doi:10.1615/CritRevOncog.2014011142
26. Moretta L, Moretta A. Unravelling natural killer cell function: triggering and inhibitory human NK receptors. EMBO J (2004) 23:255–9. doi:10.1038/sj.emboj.7600019
27. Cerboni C, Fionda C, Soriani A, Zingoni A, Doria M, Cippitelli M, et al. The DNA damage response: a common pathway in the regulation of NKG2D and DNAM-1 ligand expression in normal, infected, and cancer cells. Front Immunol (2014) 4:508. doi:10.3389/fimmu.2013.00508
28. Koch J, Steinle A, Watzl C, Mandelboim O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol (2013) 34:182–91. doi:10.1016/j.it.2013.01.003
29. Pende D, Rivera P, Marcenaro S, Chang CC, Biassoni R, Conte R, et al. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res (2002) 62:6178–86.
30. Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol (2016) 16:112–23. doi:10.1038/nri.2015.9
31. Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J Exp Med (2009) 206:1495–503. doi:10.1084/jem.20090681
32. Pogge von Strandmann E, Simhadri VR, von TB, Sasse S, Reiners KS, Hansen HP, et al. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity (2007) 27:965–74. doi:10.1016/j.immuni.2007.10.010
33. Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J Exp Med (2003) 198:557–67. doi:10.1084/jem.20030788
34. Parham P. Taking license with natural killer cell maturation and repertoire development. Immunol Rev (2006) 214:155–60. doi:10.1111/j.1600-065X.2006.00462.x
35. Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol (2002) 20:217–51. doi:10.1146/annurev.immunol.20.092501.134942
36. Parham P. MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol (2005) 5:201–14. doi:10.1038/nri1570
37. Bryceson YT, March ME, Ljunggren HG, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev (2006) 214:73–91. doi:10.1111/j.1600-065X.2006.00457.x
38. Bhat R, Watzl C. Serial killing of tumor cells by human natural killer cells – enhancement by therapeutic antibodies. PLoS One. (2007) 2:e326. doi:10.1371/journal.pone.0000326
39. Della Chiesa M, Marcenaro E, Sivori S, Carlomagno S, Pesce S, Moretta A. Human NK cell response to pathogens. Semin Immunol (2014) 26:152–60. doi:10.1016/j.smim.2014.02.001
40. Roda JM, Parihar R, Carson WE III. CpG-containing oligodeoxynucleotides act through TLR9 to enhance the NK cell cytokine response to antibody-coated tumor cells. J Immunol (2005) 175:1619–27. doi:10.4049/jimmunol.175.3.1619
41. Waldmann TA. The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: implications for cancer therapy. Cancer Immunol Res. (2015) 3:219–27. doi:10.1158/2326-6066.CIR-15-0009
42. Muller L, Aigner P, Stoiber D. Type I interferons and natural killer cell regulation in cancer. Front Immunol (2017) 8:304. doi:10.3389/fimmu.2017.00304
43. Okamura H, Kashiwamura S, Tsutsui H, Yoshimoto T, Nakanishi K. Regulation of interferon-gamma production by IL-12 and IL-18. Curr Opin Immunol (1998) 10:259–64. doi:10.1016/S0952-7915(98)80163-5
44. Viel S, Marcais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, et al. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal (2016) 9:ra19. doi:10.1126/scisignal.aad1884
45. Muntasell A, Ochoa MC, Cordeiro L, Berraondo P, López-Díaz de CA, Cabo M, et al. Targeting NK-cell checkpoints for cancer immunotherapy. Curr Opin Immunol (2017) 45:73–81. doi:10.1016/j.coi.2017.01.003
46. Choudhury A, Charo J, Parapuram SK, Hunt RC, Hunt DM, Seliger B, et al. Small interfering RNA (siRNA) inhibits the expression of the Her2/neu gene, upregulates HLA class I and induces apoptosis of Her2/neu positive tumor cell lines. Int J Cancer (2004) 108:71–7. doi:10.1002/ijc.11497
47. Herrmann F, Lehr HA, Drexler I, Sutter G, Hengstler J, Wollscheid U, et al. HER-2/neu-mediated regulation of components of the MHC class I antigen-processing pathway. Cancer Res (2004) 64:215–20. doi:10.1158/0008-5472.CAN-2522-2
48. Okita R, Mougiakakos D, Ando T, Mao Y, Sarhan D, Wennerberg E, et al. HER2/HER3 signaling regulates NK cell-mediated cytotoxicity via MHC class I chain-related molecule A and B expression in human breast cancer cell lines. J Immunol (2012) 188:2136–45. doi:10.4049/jimmunol.1102237
49. Vertuani S, Triulzi C, Roos AK, Charo J, Norell H, Lemonnier F, et al. HER-2/neu mediated down-regulation of MHC class I antigen processing prevents CTL-mediated tumor recognition upon DNA vaccination in HLA-A2 transgenic mice. Cancer Immunol Immunother (2009) 58:653–64. doi:10.1007/s00262-008-0587-1
50. Inoue M, Mimura K, Izawa S, Shiraishi K, Inoue A, Shiba S, et al. Expression of MHC class I on breast cancer cells correlates inversely with HER2 expression. Oncoimmunology (2012) 1:1104–10. doi:10.4161/onci.21056
51. Mamessier E, Sylvain A, Bertucci F, Castellano R, Finetti P, Houvenaeghel G, et al. Human breast tumor cells induce self-tolerance mechanisms to avoid NKG2D-mediated and DNAM-mediated NK cell recognition. Cancer Res (2011) 71:6621–32. doi:10.1158/0008-5472.CAN-11-0792
52. Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med (2008) 14:518–27. doi:10.1038/nm1764
53. De P, Hasmann M, Leyland-Jones B. Molecular determinants of trastuzumab efficacy: what is their clinical relevance? Cancer Treat Rev (2013) 39:925–34. doi:10.1016/j.ctrv.2013.02.006
54. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med (2000) 6:443–6. doi:10.1038/74704
55. Spiridon CI, Guinn S, Vitetta ES. A comparison of the in vitro and in vivo activities of IgG and F(ab’)2 fragments of a mixture of three monoclonal anti-Her-2 antibodies. Clin Cancer Res (2004) 10:3542–51. doi:10.1158/1078-0432.CCR-03-0549
56. Jaime-Ramirez AC, Mundy-Bosse BL, Kondadasula S, Jones NB, Roda JM, Mani A, et al. IL-12 enhances the antitumor actions of trastuzumab via NK cell IFN-gamma production. J Immunol (2011) 186:3401–9. doi:10.4049/jimmunol.1000328
57. Parihar R, Dierksheide J, Hu Y, Carson WE. IL-12 enhances the natural killer cell cytokine response to Ab-coated tumor cells. J Clin Invest (2002) 110:983–92. doi:10.1172/JCI0215950
58. Stagg J, Loi S, Divisekera U, Ngiow SF, Duret H, Yagita H, et al. Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc Natl Acad Sci U S A (2011) 108:7142–7. doi:10.1073/pnas.1016569108
59. Park S, Jiang Z, Mortenson ED, Deng L, Radkevich-Brown O, Yang X, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell (2010) 18:160–70. doi:10.1016/j.ccr.2010.06.014
60. Arnould L, Gelly M, Penault-Llorca F, Benoit L, Bonnetain F, Migeon C, et al. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br J Cancer (2006) 94:259–67. doi:10.1038/sj.bjc.6602930
61. Gennari R, Menard S, Fagnoni F, Ponchio L, Scelsi M, Tagliabue E, et al. Pilot study of the mechanism of action of preoperative trastuzumab in patients with primary operable breast tumors overexpressing HER2. Clin Cancer Res (2004) 10:5650–5. doi:10.1158/1078-0432.CCR-04-0225
62. Muller P, Kreuzaler M, Khan T, Thommen DS, Martin K, Glatz K, et al. Trastuzumab emtansine (T-DM1) renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. Sci Transl Med (2015) 7:315. doi:10.1126/scitranslmed.aac4925
63. Perez EA, Thompson EA, Ballman KV, Anderson SK, Asmann YW, Kalari KR, et al. Genomic analysis reveals that immune function genes are strongly linked to clinical outcome in the North Central Cancer Treatment Group n9831 Adjuvant Trastuzumab Trial. J Clin Oncol (2015) 33:701–8. doi:10.1200/JCO.2014.57.6298
64. Varadan V, Gilmore H, Miskimen KL, Tuck D, Parsai S, Awadallah A, et al. Immune signatures following single dose trastuzumab predict pathologic response to preoperative trastuzumab and chemotherapy in HER2-positive early breast cancer. Clin Cancer Res (2016) 22:3249–59. doi:10.1158/1078-0432.CCR-15-2021
65. Beano A, Signorino E, Evangelista A, Brusa D, Mistrangelo M, Polimeni MA, et al. Correlation between NK function and response to trastuzumab in metastatic breast cancer patients. J Transl Med (2008) 6:25. doi:10.1186/1479-5876-6-25
66. Varchetta S, Gibelli N, Oliviero B, Nardini E, Gennari R, Gatti G, et al. Elements related to heterogeneity of antibody-dependent cell cytotoxicity in patients under trastuzumab therapy for primary operable breast cancer overexpressing Her2. Cancer Res (2007) 67:11991–9. doi:10.1158/0008-5472.CAN-07-2068
67. Manser AR, Weinhold S, Uhrberg M. Human KIR repertoires: shaped by genetic diversity and evolution. Immunol Rev (2015) 267:178–96. doi:10.1111/imr.12316
68. Anfossi N, Andre P, Guia S, Falk CS, Roetynck S, Stewart CA, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity (2006) 25:331–42. doi:10.1016/j.immuni.2006.06.013
69. Boudreau JE, Liu XR, Zhao Z, Zhang A, Shultz LD, Greiner DL, et al. Cell-extrinsic MHC class I molecule engagement augments human NK cell education programmed by cell-intrinsic MHC class I. Immunity (2016) 45:280–91. doi:10.1016/j.immuni.2016.07.005
70. Goodridge JP, Onfelt B, Malmberg KJ. Newtonian cell interactions shape natural killer cell education. Immunol Rev (2015) 267:197–213. doi:10.1111/imr.12325
71. Erbe AK, Wang W, Reville PK, Carmichael L, Kim K, Mendonca EA, et al. HLA-Bw4-I-80 isoform differentially influences clinical outcome as compared to HLA-Bw4-T-80 and HLA-A-Bw4 isoforms in rituximab or dinutuximab-based cancer immunotherapy. Front Immunol (2017) 8:675. doi:10.3389/fimmu.2017.00675
72. Morales-Estevez C, Haba-Rodriguez J, Manzanares-Martin B, Porras-Quintela I, Rodriguez-Ariza A, Moreno-Vega A, et al. KIR genes and their ligands predict the response to anti-EGFR monoclonal antibodies in solid tumors. Front Immunol (2016) 7:561. doi:10.3389/fimmu.2016.00561
73. Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood (1997) 90:1109–14.
74. Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol (2008) 26:1789–96. doi:10.1200/JCO.2007.14.8957
75. Gavin PG, Song N, Kim SR, Lipchik C, Johnson NL, Bandos H, et al. Association of polymorphisms in FCGR2A and FCGR3A with degree of trastuzumab benefit in the adjuvant treatment of ERBB2/HER2-positive breast cancer: analysis of the NSABP B-31 trial. JAMA Oncol (2017) 3:335–41. doi:10.1001/jamaoncol.2016.4884
76. Hurvitz SA, Betting DJ, Stern HM, Quinaux E, Stinson J, Seshagiri S, et al. Analysis of Fcgamma receptor IIIa and IIa polymorphisms: lack of correlation with outcome in trastuzumab-treated breast cancer patients. Clin Cancer Res (2012) 18:3478–86. doi:10.1158/1078-0432.CCR-11-2294
77. Norton N, Olson RM, Pegram M, Tenner K, Ballman KV, Clynes R, et al. Association studies of Fcgamma receptor polymorphisms with outcome in HER2+ breast cancer patients treated with trastuzumab in NCCTG (Alliance) trial N9831. Cancer Immunol Res. (2014) 2:962–9. doi:10.1158/2326-6066.CIR-14-0059
78. Mellor JD, Brown MP, Irving HR, Zalcberg JR, Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol (2013) 6:1. doi:10.1186/1756-8722-6-1
79. Beziat V, Liu LL, Malmberg JA, Ivarsson MA, Sohlberg E, Bjorklund AT, et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood (2013) 121:2678–88. doi:10.1182/blood-2012-10-459545
80. Guma M, Angulo A, Vilches C, Gomez-Lozano N, Malats N, Lopez-Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood (2004) 104:3664–71. doi:10.1182/blood-2004-05-2058
81. Muntasell A, Lopez-Montanes M, Vera A, Heredia G, Romo N, Penafiel J, et al. NKG2C zygosity influences CD94/NKG2C receptor function and the NK-cell compartment redistribution in response to human cytomegalovirus. Eur J Immunol (2013) 43:3268–78. doi:10.1002/eji.201343773
82. Lopez-Botet M, Vilches C, Redondo-Pachon D, Muntasell A, Pupuleku A, Yelamos J, et al. Dual role of natural killer cells on graft rejection and control of cytomegalovirus infection in renal transplantation. Front Immunol (2017) 8:166. doi:10.3389/fimmu.2017.00166
83. Redondo-Pachon D, Crespo M, Yelamos J, Muntasell A, Perez-Saez MJ, Perez-Fernandez S, et al. Adaptive NKG2C+ NK cell response and the risk of cytomegalovirus infection in kidney transplant recipients. J Immunol (2017) 198:94–101. doi:10.4049/jimmunol.1601236
84. Cichocki F, Cooley S, Davis Z, DeFor TE, Schlums H, Zhang B, et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia (2016) 30:456–63. doi:10.1038/leu.2015.260
85. Costa-Garcia M, Vera A, Moraru M, Vilches C, López-Botet M, Muntasell A. Antibody-mediated response of NKG2Cbright NK cells against human cytomegalovirus. J Immunol (2015) 194:2715–24. doi:10.4049/jimmunol.1402281
86. López-Montañés M, Alari-Pahissa E, Sintes J, Martínez-Rodríguez JE, Muntasell A, López-Botet M. Antibody-dependent NK cell activation differentially targets EBV-infected cells in lytic cycle and bystander B lymphocytes bound to viral antigen-containing particles. J Immunol (2017) 199(2):656–65. doi:10.4049/jimmunol.1601574
87. Moraru M, Black LE, Muntasell A, Portero F, ópez-Botet ML, Reyburn HT, et al. NK cell and Ig interplay in defense against herpes simplex virus type 1: epistatic interaction of CD16A and IgG1 allotypes of variable affinities modulates antibody-dependent cellular cytotoxicity and susceptibility to clinical reactivation. J Immunol (2015) 195:1676–84. doi:10.4049/jimmunol.1500872
88. Vaz-Luis I, Ottesen RA, Hughes ME, Marcom PK, Moy B, Rugo HS, et al. Impact of hormone receptor status on patterns of recurrence and clinical outcomes among patients with human epidermal growth factor-2-positive breast cancer in the National Comprehensive Cancer Network: a prospective cohort study. Breast Cancer Res (2012) 14:R129. doi:10.1186/bcr3324
89. Bhargava R, Dabbs DJ, Beriwal S, Yildiz IA, Badve P, Soran A, et al. Semiquantitative hormone receptor level influences response to trastuzumab-containing neoadjuvant chemotherapy in HER2-positive breast cancer. Mod Pathol (2011) 24:367–74. doi:10.1038/modpathol.2010.209
90. Lee HJ, Kim JY, Park SY, Park IA, Song IH, Yu JH, et al. Clinicopathologic significance of the intratumoral heterogeneity of HER2 gene amplification in HER2-positive breast cancer patients treated with adjuvant trastuzumab. Am J Clin Pathol (2015) 144:570–8. doi:10.1309/AJCP51HCGPOPWSCY
91. da Silva BB, dos Santos AR, Pires CG, Correa-Lima MA, Pereira-Filho JD, dos Santos LG, et al. E-cadherin expression in estrogen receptor-positive and negative breast carcinomas of postmenopausal women. Eur J Gynaecol Oncol (2010) 31:90–3.
92. Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell (2003) 113:207–19. doi:10.1016/S0092-8674(03)00234-4
93. Ito M, Maruyama T, Saito N, Koganei S, Yamamoto K, Matsumoto N. Killer cell lectin-like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J Exp Med (2006) 203:289–95. doi:10.1084/jem.20051986
94. Yamauchi C, Fujii S, Kimura T, Kuwata T, Wada N, Mukai H, et al. E-cadherin expression on human carcinoma cell affects trastuzumab-mediated antibody-dependent cellular cytotoxicity through killer cell lectin-like receptor G1 on natural killer cells. Int J Cancer (2011) 128:2125–37. doi:10.1002/ijc.25803
95. Jiang X, Ellison SJ, Alarid ET, Shapiro DJ. Interplay between the levels of estrogen and estrogen receptor controls the level of the granzyme inhibitor, proteinase inhibitor 9 and susceptibility to immune surveillance by natural killer cells. Oncogene (2007) 26:4106–14. doi:10.1038/sj.onc.1210197
96. Kanamori H, Krieg S, Mao C, Di Pippo VA, Wang S, Zajchowski DA, et al. Proteinase inhibitor 9, an inhibitor of granzyme B-mediated apoptosis, is a primary estrogen-inducible gene in human liver cells. J Biol Chem (2000) 275:5867–73. doi:10.1074/jbc.275.8.5867
97. Hamada K, Gleason SL, Levi BZ, Hirschfeld S, Appella E, Ozato K. H-2RIIBP, a member of the nuclear hormone receptor superfamily that binds to both the regulatory element of major histocompatibility class I genes and the estrogen response element. Proc Natl Acad Sci U S A (1989) 86:8289–93. doi:10.1073/pnas.86.21.8289
98. Rodriguez F, Peran F, Garrido F, Ruiz-Cabello F. Upmodulation by estrogen of HLA class I expression in breast tumor cell lines. Immunogenetics (1994) 39:161–7. doi:10.1007/BF00241256
99. Sim BC, Hui KM. A HLA class I cis-regulatory element whose activity can be modulated by hormones. Int J Cancer (1994) 59:646–56. doi:10.1002/ijc.2910590512
100. Maruyama T, Mimura K, Izawa S, Inoue A, Shiba S, Watanabe M, et al. Lapatinib enhances herceptin-mediated antibody-dependent cellular cytotoxicity by up-regulation of cell surface HER2 expression. Anticancer Res (2011) 31:2999–3005.
101. Scaltriti M, Verma C, Guzman M, Jimenez J, Parra JL, Pedersen K, et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene (2009) 28:803–14. doi:10.1038/onc.2008.432
102. Scheuer W, Friess T, Burtscher H, Bossenmaier B, Endl J, Hasmann M. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res (2009) 69:9330–6. doi:10.1158/0008-5472.CAN-08-4597
103. Yamashita-Kashima Y, Iijima S, Yorozu K, Furugaki K, Kurasawa M, Ohta M, et al. Pertuzumab in combination with trastuzumab shows significantly enhanced antitumor activity in HER2-positive human gastric cancer xenograft models. Clin Cancer Res (2011) 17:5060–70. doi:10.1158/1078-0432.CCR-10-2927
104. Nimmerjahn F, Ravetch JV. Antibodies, Fc receptors and cancer. Curr Opin Immunol (2007) 19:239–45. doi:10.1016/j.coi.2007.01.005
105. Nordstrom JL, Gorlatov S, Zhang W, Yang Y, Huang L, Burke S, et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcgamma receptor binding properties. Breast Cancer Res (2011) 13:R123. doi:10.1186/bcr3069
106. Bang YJ, Giaccone G, Im SA, Oh DY, Bauer TM, Nordstrom JL, et al. First-in-human phase 1 study of margetuximab (MGAH22), an Fc-modified chimeric monoclonal antibody, in patients with HER2-positive advanced solid tumors. Ann Oncol (2017) 28:855–61. doi:10.1093/annonc/mdx002
107. Pol J, Vacchelli E, Aranda F, Castoldi F, Eggermont A, Cremer I, et al. Trial watch: immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology (2015) 4:e1008866. doi:10.1080/2162402X.2015.1008866
108. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med (2014) 20:1301–9. doi:10.1038/nm.3708
109. Parodi M, Pedrazzi M, Cantoni C, Averna M, Patrone M, Cavaletto M, et al. Natural killer (NK)/melanoma cell interaction induces NK-mediated release of chemotactic high mobility group box-1 (HMGB1) capable of amplifying NK cell recruitment. Oncoimmunology (2015) 4:e1052353. doi:10.1080/2162402X.2015.1052353
110. Qiu Y, Yang J, Wang W, Zhao W, Peng F, Xiang Y, et al. HMGB1-promoted and TLR2/4-dependent NK cell maturation and activation take part in rotavirus-induced murine biliary atresia. PLoS Pathog (2014) 10:e1004011. doi:10.1371/journal.ppat.1004011
111. Di MM, Sfondrini L, Regondi V, Varchetta S, Oliviero B, Mariani G, et al. Taxanes enhance trastuzumab-mediated ADCC on tumor cells through NKG2D-mediated NK cell recognition. Oncotarget (2016) 7:255–65. doi:10.18632/oncotarget.6353
112. Feng H, Dong Y, Wu J, Qiao Y, Zhu G, Jin H, et al. Epirubicin pretreatment enhances NK cell-mediated cytotoxicity against breast cancer cells in vitro. Am J Transl Res. (2016) 8:473–84.
113. Kubo M, Morisaki T, Matsumoto K, Tasaki A, Yamanaka N, Nakashima H, et al. Paclitaxel probably enhances cytotoxicity of natural killer cells against breast carcinoma cells by increasing perforin production. Cancer Immunol Immunother (2005) 54:468–76. doi:10.1007/s00262-004-0617-6
114. Carson WE III, Shapiro CL, Crespin TR, Thornton LM, Andersen BL. Cellular immunity in breast cancer patients completing taxane treatment. Clin Cancer Res (2004) 10:3401–9. doi:10.1158/1078-0432.CCR-1016-03
115. Kono K, Sato E, Naganuma H, Takahashi A, Mimura K, Nukui H, et al. Trastuzumab (Herceptin) enhances class I-restricted antigen presentation recognized by HER-2/neu-specific T cytotoxic lymphocytes. Clin Cancer Res (2004) 10:2538–44. doi:10.1158/1078-0432.CCR-03-0424
116. Tsavaris N, Kosmas C, Vadiaka M, Kanelopoulos P, Boulamatsis D. Immune changes in patients with advanced breast cancer undergoing chemotherapy with taxanes. Br J Cancer (2002) 87:21–7. doi:10.1038/sj.bjc.6600347
117. Clynes R. Antitumor antibodies in the treatment of cancer: Fc receptors link opsonic antibody with cellular immunity. Hematol Oncol Clin North Am (2006) 20:585–612. doi:10.1016/j.hoc.2006.02.010
118. DiLillo DJ, Ravetch JV. Differential Fc-receptor engagement drives an anti-tumor vaccinal effect. Cell (2015) 161:1035–45. doi:10.1016/j.cell.2015.04.016
119. Gall VA, Philips AV, Qiao N, Clise-Dwyer K, Perakis AA, Zhang M, et al. Trastuzumab increases HER2 uptake and cross-presentation by dendritic cells. Cancer Res (2017) 77(19):5374–83. doi:10.1158/0008-5472.CAN-16-2774
120. Datta J, Rosemblit C, Berk E, Showalter L, Namjoshi P, Mick R, et al. Progressive loss of anti-HER2 CD4+ T-helper type 1 response in breast tumorigenesis and the potential for immune restoration. Oncoimmunology (2015) 4:e1022301. doi:10.1080/2162402X.2015.1022301
121. Datta J, Berk E, Xu S, Fitzpatrick E, Rosemblit C, Lowenfeld L, et al. Anti-HER2 CD4(+) T-helper type 1 response is a novel immune correlate to pathologic response following neoadjuvant therapy in HER2-positive breast cancer. Breast Cancer Res (2015) 17:71. doi:10.1186/s13058-015-0584-1
122. Knutson KL, Clynes R, Shreeder B, Yeramian P, Kemp KP, Ballman K, et al. Improved survival of HER2+ breast cancer patients treated with trastuzumab and chemotherapy is associated with host antibody immunity against the HER2 intracellular domain. Cancer Res (2016) 76:3702–10. doi:10.1158/0008-5472.CAN-15-3091
123. Mittal D, Caramia F, Michiels S, Joensuu H, Kellokumpu-Lehtinen PL, Sotiriou C, et al. Improved treatment of breast cancer with anti-HER2 therapy requires interleukin-21 signaling in CD8+ T cells. Cancer Res (2016) 76:264–74. doi:10.1158/0008-5472.CAN-15-1567
124. Taylor C, Hershman D, Shah N, Suciu-Foca N, Petrylak DP, Taub R, et al. Augmented HER-2 specific immunity during treatment with trastuzumab and chemotherapy. Clin Cancer Res (2007) 13:5133–43. doi:10.1158/1078-0432.CCR-07-0507
125. Adam C, King S, Allgeier T, Braumuller H, Luking C, Mysliwietz J, et al. DC-NK cell cross talk as a novel CD4+ T-cell-independent pathway for antitumor CTL induction. Blood (2005) 106:338–44. doi:10.1182/blood-2004-09-3775
126. Mailliard RB, Son YI, Redlinger R, Coates PT, Giermasz A, Morel PA, et al. Dendritic cells mediate NK cell help for Th1 and CTL responses: two-signal requirement for the induction of NK cell helper function. J Immunol (2003) 171:2366–73. doi:10.4049/jimmunol.171.5.2366
127. Moretta A. Natural killer cells and dendritic cells: rendezvous in abused tissues. Nat Rev Immunol (2002) 2:957–64. doi:10.1038/nri956
128. Srivastava RM, Lee SC, Andrade Filho PA, Lord CA, Jie HB, Davidson HC, et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res (2013) 19:1858–72. doi:10.1158/1078-0432.CCR-12-2426
129. Wong JL, Mailliard RB, Moschos SJ, Edington H, Lotze MT, Kirkwood JM, et al. Helper activity of natural killer cells during the dendritic cell-mediated induction of melanoma-specific cytotoxic T cells. J Immunother (2011) 34:270–8. doi:10.1097/CJI.0b013e31820b370b
130. Ferlazzo G, Tsang ML, Moretta L, Melioli G, Steinman RM, Munz C. Human dendritic cells activate resting natural killer (NK) cells and are recognized via the NKp30 receptor by activated NK cells. J Exp Med (2002) 195:343–51. doi:10.1084/jem.20011149
131. Morandi B, Mortara L, Chiossone L, Accolla RS, Mingari MC, Moretta L, et al. Dendritic cell editing by activated natural killer cells results in a more protective cancer-specific immune response. PLoS One. (2012) 7:e39170. doi:10.1371/journal.pone.0039170
132. Roda JM, Parihar R, Magro C, Nuovo GJ, Tridandapani S, Carson WE III. Natural killer cells produce T cell-recruiting chemokines in response to antibody-coated tumor cells. Cancer Res (2006) 66:517–26. doi:10.1158/0008-5472.CAN-05-2429
133. Muraro E, Comaro E, Talamini R, Turchet E, Miolo G, Scalone S, et al. Improved natural killer cell activity and retained anti-tumor CD8(+) T cell responses contribute to the induction of a pathological complete response in HER2-positive breast cancer patients undergoing neoadjuvant chemotherapy. J Transl Med (2015) 13:204. doi:10.1186/s12967-015-0567-0
134. Cantoni C, Huergo-Zapico L, Parodi M, Pedrazzi M, Mingari MC, Moretta A, et al. NK cells, tumor cell transition, and tumor progression in solid malignancies: new hints for NK-based immunotherapy? J Immunol Res. (2016) 2016:4684268. doi:10.1155/2016/4684268
135. Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol (2014) 44:1582–92. doi:10.1002/eji.201344272
136. Gross E, Sunwoo JB, Bui JD. Cancer immunosurveillance and immunoediting by natural killer cells. Cancer J (2013) 19:483–9. doi:10.1097/PPO.0000000000000005
137. Guillerey C, Smyth MJ. NK cells and cancer immunoediting. Curr Top Microbiol Immunol (2016) 395:115–45. doi:10.1007/82_2015_446
138. O’Sullivan T, Saddawi-Konefka R, Vermi W, Koebel CM, Arthur C, White JM, et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J Exp Med (2012) 209:1869–82. doi:10.1084/jem.20112738
139. Wang B, Wang Q, Wang Z, Jiang J, Yu SC, Ping YF, et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res (2014) 74:5746–57. doi:10.1158/0008-5472.CAN-13-2563
140. Schlecker E, Fiegler N, Arnold A, Altevogt P, Rose-John S, Moldenhauer G, et al. Metalloprotease-mediated tumor cell shedding of B7-H6, the ligand of the natural killer cell-activating receptor NKp30. Cancer Res (2014) 74:3429–40. doi:10.1158/0008-5472.CAN-13-3017
141. Zhang J, Basher F, Wu JD. NKG2D ligands in tumor immunity: two sides of a coin. Front Immunol (2015) 6:97. doi:10.3389/fimmu.2015.00097
142. Algarra I, Garcia-Lora A, Cabrera T, Ruiz-Cabello F, Garrido F. The selection of tumor variants with altered expression of classical and nonclassical MHC class I molecules: implications for tumor immune escape. Cancer Immunol Immunother (2004) 53:904–10. doi:10.1007/s00262-004-0517-9
143. Wischhusen J, Waschbisch A, Wiendl H. Immune-refractory cancers and their little helpers – an extended role for immunetolerogenic MHC molecules HLA-G and HLA-E? Semin Cancer Biol (2007) 17:459–68. doi:10.1016/j.semcancer.2007.07.005
144. Zhang L, Fang B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther (2005) 12:228–37. doi:10.1038/sj.cgt.7700792
145. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol (2016) 17:1025–36. doi:10.1038/ni.3518
146. Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICA in malignant diseases. Int J Cancer (2006) 118:684–7. doi:10.1002/ijc.21382
147. de Kruijf EM, Sajet A, van Nes JG, Natanov R, Putter H, Smit VT, et al. HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J Immunol (2010) 185:7452–9. doi:10.4049/jimmunol.1002629
148. Sun J, Tao H, Li X, Wang L, Yang J, Wu P, et al. Clinical significance of novel costimulatory molecule B7-H6 in human breast cancer. Oncol Lett (2017) 14:2405–9. doi:10.3892/ol.2017.6417
149. Reimer T, Koczan D, Müller H, Friese K, Thiesen HJ, Gerber B. Tumour Fas ligand:Fas ratio greater than 1 is an independent marker of relative resistance to tamoxifen therapy in hormone receptor positive breast cancer. Breast Cancer Res (2002) 4:R9. doi:10.1186/bcr456
150. Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest (2011) 121:3609–22. doi:10.1172/JCI45816
151. Mamessier E, Pradel LC, Thibult ML, Drevet C, Zouine A, Jacquemier J, et al. Peripheral blood NK cells from breast cancer patients are tumor-induced composite subsets. J Immunol (2013) 190:2424–36. doi:10.4049/jimmunol.1200140
152. Battella S, Cox MC, Santoni A, Palmieri G. Natural killer (NK) cells and anti-tumor therapeutic mAb: unexplored interactions. J Leukoc Biol (2016) 99:87–96. doi:10.1189/jlb.5VMR0415-141R
153. Kohrt HE, Houot R, Marabelle A, Cho HJ, Osman K, Goldstein M, et al. Combination strategies to enhance antitumor ADCC. Immunotherapy (2012) 4:511–27. doi:10.2217/imt.12.38
154. Ribas A. Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov (2015) 5:915–9. doi:10.1158/2159-8290.CD-15-0563
155. Chaganty BK, Lu Y, Qiu S, Somanchi SS, Lee DA, Fan Z. Trastuzumab upregulates expression of HLA-ABC and T cell costimulatory molecules through engagement of natural killer cells and stimulation of IFNgamma secretion. Oncoimmunology (2015) 5:e1100790. doi:10.1080/2162402X.2015.1100790
156. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep (2017) 19:1189–201. doi:10.1016/j.celrep.2017.04.031
157. Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, et al. Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with myeloma. Clin Cancer Res (2016) 22:5211–22. doi:10.1158/1078-0432.CCR-16-1108
158. Korde N, Carlsten M, Lee MJ, Minter A, Tan E, Kwok M, et al. A phase II trial of pan-KIR2D blockade with IPH2101 in smoldering multiple myeloma. Haematologica (2014) 99:e81–3. doi:10.3324/haematol.2013.103085
159. Beldi-Ferchiou A, Lambert M, Dogniaux S, Vely F, Vivier E, Olive D, et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget (2016) 7:72961–77. doi:10.18632/oncotarget.12150
160. Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood (2010) 116:2286–94. doi:10.1182/blood-2010-02-271874
161. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: a phenotypic and functional characterization. J Allergy Clin Immunol (2017) 139:335–46. doi:10.1016/j.jaci.2016.04.025
162. Sabatier R, Finetti P, Mamessier E, Adelaide J, Chaffanet M, Ali HR, et al. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget (2015) 6:5449–64. doi:10.18632/oncotarget.3216
163. Muenst S, Schaerli AR, Gao F, Daster S, Trella E, Droeser RA, et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res Treat (2014) 146:15–24. doi:10.1007/s10549-014-2988-5
164. Tsang JY, Au WL, Lo KY, Ni YB, Hlaing T, Hu J, et al. PD-L1 expression and tumor infiltrating PD-1+ lymphocytes associated with outcome in HER2+ breast cancer patients. Breast Cancer Res Treat (2017) 162:19–30. doi:10.1007/s10549-016-4095-2
165. Blake SJ, Dougall WC, Miles JJ, Teng MW, Smyth MJ. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res (2016) 22:5183–8. doi:10.1158/1078-0432.CCR-16-0933
166. Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol (2014) 15:431–8. doi:10.1038/ni.2850
167. Mandai K, Rikitake Y, Mori M, Takai Y. Nectins and nectin-like molecules in development and disease. Curr Top Dev Biol (2015) 112:197–231. doi:10.1016/bs.ctdb.2014.11.019
168. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A (2009) 106:17858–63. doi:10.1073/pnas.0903474106
169. Xu F, Sunderland A, Zhou Y, Schulick RD, Edil BH, Zhu Y. Blockade of CD112R and TIGIT signaling sensitizes human natural killer cell functions. Cancer Immunol Immunother (2017). doi:10.1007/s00262-017-2031-x
170. Kohrt HE, Houot R, Weiskopf K, Goldstein MJ, Scheeren F, Czerwinski D, et al. Stimulation of natural killer cells with a CD137-specific antibody enhances trastuzumab efficacy in xenotransplant models of breast cancer. J Clin Invest (2012) 122:1066–75. doi:10.1172/JCI61226
171. Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med (1997) 3:682–5. doi:10.1038/nm0697-682
172. Sanmamed MF, Pastor F, Rodriguez A, Perez-Gracia JL, Rodriguez-Ruiz ME, Jure-Kunkel M, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD130. Semin Oncol (2015) 42:640–55. doi:10.1053/j.seminoncol.2015.05.014
173. Lin W, Voskens CJ, Zhang X, Schindler DG, Wood A, Burch E, et al. Fc-dependent expression of CD137 on human NK cells: insights into “agonistic” effects of anti-CD137 monoclonal antibodies. Blood (2008) 112:699–707. doi:10.1182/blood-2007-11-122465
174. Kohrt HE, Colevas AD, Houot R, Weiskopf K, Goldstein MJ, Lund P, et al. Targeting CD137 enhances the efficacy of cetuximab. J Clin Invest (2014) 124:2668–82. doi:10.1172/JCI73014
175. Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol (2016) 17:816–24. doi:10.1038/ni.3470
176. Gotthardt D, Putz EM, Grundschober E, Prchal-Murphy M, Straka E, Kudweis P, et al. STAT5 is a key regulator in NK cells and acts as a molecular switch from tumor surveillance to tumor promotion. Cancer Discov (2016) 6:414–29. doi:10.1158/2159-8290.CD-15-0732
177. Carson WE, Parihar R, Lindemann MJ, Personeni N, Dierksheide J, Meropol NJ, et al. Interleukin-2 enhances the natural killer cell response to herceptin-coated Her2/neu-positive breast cancer cells. Eur J Immunol (2001) 31:3016–25. doi:10.1002/1521-4141(2001010)31:10<3016::AID-IMMU3016>3.0.CO;2-J
178. Zhu EF, Gai SA, Opel CF, Kwan BH, Surana R, Mihm MC, et al. Synergistic innate and adaptive immune response to combination immunotherapy with anti-tumor antigen antibodies and extended serum half-life IL-2. Cancer Cell (2015) 27:489–501. doi:10.1016/j.ccell.2015.03.004
179. Mani A, Roda J, Young D, Caligiuri MA, Fleming GF, Kaufman P, et al. A phase II trial of trastuzumab in combination with low-dose interleukin-2 (IL-2) in patients (PTS) with metastatic breast cancer (MBC) who have previously failed trastuzumab. Breast Cancer Res Treat (2009) 117:83–9. doi:10.1007/s10549-008-0251-7
180. Repka T, Chiorean EG, Gay J, Herwig KE, Kohl VK, Yee D, et al. Trastuzumab and interleukin-2 in HER2-positive metastatic breast cancer: a pilot study. Clin Cancer Res (2003) 9:2440–6.
181. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol (2015) 33:74–82. doi:10.1200/JCO.2014.57.3329
182. Wege AK, Weber F, Kroemer A, Ortmann O, Nimmerjahn F, Brockhoff G. IL-15 enhances the anti-tumor activity of trastuzumab against breast cancer cells but causes fatal side effects in humanized tumor mice (HTM). Oncotarget (2017) 8:2731–44. doi:10.18632/oncotarget.13159
183. Kim PS, Kwilas AR, Xu W, Alter S, Jeng EK, Wong HC, et al. IL-15 superagonist/IL-15RalphaSushi-Fc fusion complex (IL-15SA/IL-15RalphaSu-Fc; ALT-803) markedly enhances specific subpopulations of NK and memory CD8+ T cells, and mediates potent anti-tumor activity against murine breast and colon carcinomas. Oncotarget (2016) 7:16130–45. doi:10.18632/oncotarget.7470
184. Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature (2012) 484:529–33. doi:10.1038/nature10975
185. Schmohl JU, Felices M, Todhunter D, Taras E, Miller JS, Vallera DA. Tetraspecific scFv construct provides NK cell mediated ADCC and self-sustaining stimuli via insertion of IL-15 as a cross-linker. Oncotarget (2016) 7:73830–44. doi:10.18632/oncotarget.12073
186. Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, et al. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res (2016) 22:3440–50. doi:10.1158/1078-0432.CCR-15-2710
187. Bekaii-Saab TS, Roda JM, Guenterberg KD, Ramaswamy B, Young DC, Ferketich AK, et al. A phase I trial of paclitaxel and trastuzumab in combination with interleukin-12 in patients with HER2/neu-expressing malignancies. Mol Cancer Ther (2009) 8:2983–91. doi:10.1158/1535-7163.MCT-09-0820
188. Lasek W, Zagozdzon R, Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother (2014) 63:419–35. doi:10.1007/s00262-014-1523-1
189. Toussi DN, Massari P. Immune adjuvant effect of molecularly-defined toll-like receptor ligands. Vaccines (Basel). (2014) 2:323–53. doi:10.3390/vaccines2020323
190. Charlebois R, Allard B, Allard D, Buisseret L, Turcotte M, Pommey S, et al. PolyI:C and CpG synergize with anti-ErbB2 mAb for treatment of breast tumors resistant to immune checkpoint inhibitors. Cancer Res (2017) 77:312–9. doi:10.1158/0008-5472.CAN-16-1873
191. Lu H, Yang Y, Gad E, Wenner CA, Chang A, Larson ER, et al. Polysaccharide krestin is a novel TLR2 agonist that mediates inhibition of tumor growth via stimulation of CD8 T cells and NK cells. Clin Cancer Res (2011) 17:67–76. doi:10.1158/1078-0432.CCR-10-1763
192. Lu H, Dietsch GN, Matthews MA, Yang Y, Ghanekar S, Inokuma M, et al. VTX-2337 is a novel TLR8 agonist that activates NK cells and augments ADCC. Clin Cancer Res (2012) 18:499–509. doi:10.1158/1078-0432.CCR-11-1625
193. Grzywacz B, Kataria N, Verneris MR. CD56(dim)CD16(+) NK cells downregulate CD16 following target cell induced activation of matrix metalloproteinases. Leukemia (2007) 21:356–9. doi:10.1038/sj.leu.2404499
194. Duffy MJ, Mullooly M, O’Donovan N, Sukor S, Crown J, Pierce A, et al. The ADAMs family of proteases: new biomarkers and therapeutic targets for cancer? Clin Proteomics (2011) 8(1):9. doi:10.1186/1559-0275-8-9
195. Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, et al. Tumor-associated MICA is shed by ADAM proteases. Cancer Res (2008) 68:6368–76. doi:10.1158/0008-5472.CAN-07-6768
196. Xu Z, Shen J, Wang MH, Yi T, Yu Y, Zhu Y, et al. Comprehensive molecular profiling of the B7 family of immune-regulatory ligands in breast cancer. Oncoimmunology (2016) 5:e1207841. doi:10.1080/2162402X.2016.1207841
197. Duffy MJ, Crown J, Mullooly M. ADAM10 and ADAM17: new players in trastuzumab tesistance. Oncotarget (2014) 5:10963–4. doi:10.18632/oncotarget.2794
198. Feldinger K, Generali D, Kramer-Marek G, Gijsen M, Ng TB, Wong JH, et al. ADAM10 mediates trastuzumab resistance and is correlated with survival in HER2 positive breast cancer. Oncotarget (2014) 5:6633–46. doi:10.18632/oncotarget.1955
Keywords: natural killer cells, breast cancer, human epidermal growth factor receptor 2, trastuzumab, pertuzumab, antibody-dependent cell-mediated cytotoxicity, immunotherapy
Citation: Muntasell A, Cabo M, Servitja S, Tusquets I, Martínez-García M, Rovira A, Rojo F, Albanell J and López-Botet M (2017) Interplay between Natural Killer Cells and Anti-HER2 Antibodies: Perspectives for Breast Cancer Immunotherapy. Front. Immunol. 8:1544. doi: 10.3389/fimmu.2017.01544
Received: 29 September 2017; Accepted: 30 October 2017;
Published: 13 November 2017
Edited by:
Jose A. Garcia-Sanz, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Daniel Olive, Institut national de la santé et de la recherche médicale, FranceKerry S. Campbell, Fox Chase Cancer Center, United States
Copyright: © 2017 Muntasell, Cabo, Servitja, Tusquets, Martínez-García, Rovira, Rojo, Albanell and López-Botet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aura Muntasell, YW11bnRhc2VsbEBpbWltLmVz